
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTrimetallic spinel NiCo2−xMoxO4 oxygen evolution catalyst enabling bias-free solar water splitting with inverted perovskite solar cells†
Mahmoud G.
Ahmed‡
 a,
Yi Fei
Phang‡
b,
Ying Fan
Tay
c,
Anupam
Sadhu
a,
Pritish
Mishra
b,
Akhmad Herman
Yuwono
de and
Lydia H.
Wong
*ab
a,
Yi Fei
Phang‡
b,
Ying Fan
Tay
c,
Anupam
Sadhu
a,
Pritish
Mishra
b,
Akhmad Herman
Yuwono
de and
Lydia H.
Wong
*ab
aEnergy Research Institute @ NTU (ERI@N) Nanyang Technological University, Singapore 637553, Singapore. E-mail: lydiawong@ntu.edu.sg
bSchool of Materials Science and Engineering, Nanyang Technological University, Singapore 639798, Singapore
cInstitute of Sustainability for Chemicals, Energy and Environment (ISCE2), Agency for Science, Technology and Research (A*STAR), 1 Pesek Road Jurong Island, Singapore, 627833, Singapore
dAdvanced Materials Research Center (AMRC), Faculty of Engineering, Universitas Indonesia, Indonesia
eDepartment of Metallurgical and Materials Engineering, Universitas Indonesia, Depok 16424, Indonesia
First published on 9th April 2025
Abstract
Efficient spinel oxide catalysts are pivotal for driving the oxygen evolution reaction (OER) for hydrogen production via solar-driven water splitting. Designing trimetallic spinel oxides with high-valence metal ions which leach out to promote surface reconstruction is an effective strategy to maximize active sites for the OER. Herein, we report a trimetallic spinel oxide, NiCo2−xMoxO4 as an efficient OER catalyst, generating 10 mA cm−2 at a low overpotential of 250 mV, and demonstrating stability for over 25 h. Experimental and spectroscopic results indicate that the partial leaching of Mo ions from tetrahedral sites in the electrolyte facilitates Ni2+ oxidation to Ni3+, leading to the formation of an active nickel (oxy)hydroxide with numerous catalytic sites. Furthermore, integrating this spinel oxide in a 2-electrode water electrolyzer coupled with an inverted p-i-n perovskite solar cell enables bias-free solar water splitting with a solar-to-hydrogen efficiency of 8.8%. This work underscores the efficacy of using high-valence metal ions as effective dopants in activating spinel oxide pre-catalysts for the OER, thereby broadening their application in solar-driven water splitting technologies.
Introduction
Sunlight as an endlessly renewable energy source, offers a beacon of hope for humanity in our collective endeavor to surmount the dual challenges of energy scarcity and environmental pollution. Efficient utilization of solar energy is pivotal in this pursuit, and photovoltaic (PV) cells stand out as a key technology for converting solar energy into electricity.1 Recently, hybrid organic–inorganic metal halide perovskites have emerged as extremely promising candidates for the next wave of PV devices.2,3 These materials exhibit unique features, including a high optical absorption coefficient, prolonged carrier lifetime, and ambipolar charge transport, resulting in a remarkable solar-to-electricity conversion efficiency surpassing 26%.3,4 However, due to the intermittent and unpredictable nature of solar radiation, the need to store solar energy in chemical fuels becomes imperative, with hydrogen (H2) emerging as the leading choice.5 Integrated energy conversion–storage systems are proposed as highly effective routes to convert solar energy into chemical fuel.6–8 In particular, the integrated PV cell with a water electrolysis system can efficiently produce green and recyclable hydrogen in a sustainable manner.9Water electrolysis comprises two distinct half-reactions, the oxygen evolution reaction (OER) occurring at the anode and the hydrogen evolution reaction (HER) at the cathode.10 The OER, with its inherent slow kinetics resulting from a four-electron transfer mechanism, is essential in various electrochemical energy conversion and storage devices, including water electrolysis and photoelectrochemical cells.11,12 Consequently, extensive research endeavors have been focused on developing OER catalysts that exhibit high activity, durability, and cost-effectiveness.11,13
Exploring 3d transition metal oxides, particularly Ni and/or Co-based oxides, has garnered significant interest due to their good catalytic activity and stability.14–16 The bimetallic spinel NiCo2O4, in particular, has demonstrated remarkable activity in the OER owing to its favorable electrical conductivity, high stability and possession of rich redox-active sites. Notably, spinel oxides containing Ni and/or Co have been reported to undergo a surface reconstruction process under electro-oxidation conditions leading to the formation of an active Ni and/or Co(oxy)hydroxide phase, respectively.14 This surface reconstruction is identified as a crucial step for maximizing the exposure of active sites, thereby enhancing OER catalysis. For instance, Wang et al. demonstrated that the Ni3+ enrichment on the surface of spinel NiCo2O4 is more active than Co, leading to the formation of active NiOOH for the OER.14 Nevertheless, the performance of NiCo2O4 is still inferior due to the poor intrinsic activity and limited exposed active sites for the OER process. To address this limitation, several strategies have been proposed to activate the surface reconstruction and increase the density of exposed active sites for high OER activity. The formation of multimetallic spinel oxides is an effective strategy to increase the exposure of active sites.17,18 For instance, the trimetallic NiCo2−xFexO4 exhibits high catalytic performance (274 mV at 10 mA cm−2) compared to NiCo2O4 (330 mV), attributed to the porous hollow structure, trimetallic composition and the surface reconstruction phenomenon of Ni2+ to Ni3+OOH.16 Liu et al. have reported that the OER activity of Cr-NiCo2O4 is higher than that of NiCo2O4, attributed to enhanced conductivity, enabling greater accessibility to active sites.19
Recent studies indicate that incorporating high-valence metals like Mo and W can trigger the surface reconstruction of a pre-catalyst into the active phase due to their leaching capabilities in alkaline medium, maximizing the number of active sites. For example, Fan et al. demonstrated that the sacrificial W facilitates the surface transformation of CoFe sulfide to CoFe (oxy)hydroxide, generating abundant active species.20 Similarly, Dürr et al. observed that leaching of Mo ions from the NiMoO4·H2O pre-catalyst promotes the formation of the active NiOOH phase.21 Such observations highlight the benefits of using high-valence metal ions in other catalyst materials to promote the transformation of other pre-catalyst materials into active phases, enhancing the OER catalytic activity.
In the case of the spinel NiCo2O4 oxide as the pre-catalyst, the OER activity is often limited by the low exposure of Ni sites, particularly those in the tetrahedral sites. The incorporation of high-valence Mo6+ in tetrahedral sites introduces a unique advantage. Mo6+ can be selectively leached under alkaline conditions, creating cation vacancies, and significantly increasing the exposure of Ni sites for the OER. Consequently, forming a trimetallic spinel oxide NiCo2−xMoxO4 by incorporating high-valence Mo (i.e., Mo6+) is a promising strategy to generate numerous highly active sites and enhance the catalytic performance.
Herein, we report the synthesis of a trimetallic NiCo2−xMoxO4 spinel oxide by a facile hydrothermal method followed by one annealing step. Notably, the partial leaching of Mo increases the exposure of Ni active sites for the OER and triggers the self-surface reconstruction, forming the active nickel (oxy)hydroxide. The reconstructed spinel NiCo2−xMoxO4 (x = 0.1) oxide demonstrates high catalytic activity on Ni foam with a low overpotential of 250 mV at 10 mA cm−2 and a small Tafel slope of 41.5 mV dec−1. By integrating the highly active OER catalyst (NiCo2−xMoxO4, x = 0.1) into the water electrolysis system, the overall voltage to drive water splitting was only 1.53 V at 10 mA cm−2. The integration of the water electrolysis system with a p-i-n architecture perovskite solar cell enables an unbiased solar water splitting, achieving a solar-to-hydrogen efficiency of 8.8%.
Results and discussion
Bi/trimetallic spinel oxides (NiCo2−xMoxO4) were deposited directly on conductive fluorine-doped tin oxide (FTO) via a straightforward hydrothermal approach followed by annealing at 400 °C (more details are provided in the ESI†). The variation of Mo ratio in the trimetallic spinel oxide NiCo2−xMoxO4 shows a significant effect on the OER catalytic performance as shown in Fig. S1.† NiCo2−xMoxO4 with x = 0.1 demonstrates the highest catalytic performance, characterized by a low overpotential value. X-ray diffraction (XRD) patterns of bimetallic NiCo2O4 and trimetallic NiCo1.9Mo0.1O4 are shown in Fig. 1a. The diffraction peaks of both NiCo2O4 and NiCo1.9Mo0.1O4 matched well with that of the standard cubic spinel NiCo2O4 with a space group of Fc![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (JCPDS no. 20-0781). The XRD pattern of trimetallic NiCo1.9Mo0.1O4 did not show any peak shift, which might be attributed to the low amount of Mo. Fig. 1b shows the polarization curves with iR correction for bimetallic NiCo2O4 and trimetallic NiCo1.9Mo0.1O4 catalysts. Fig. S2† shows the polarization curves of oxides without iR correction. NiCo2O4 achieves a current density of 10 mA cm−2 with an overpotential of 460 mV, whereas NiCo1.9Mo0.1O4 achieves the same current density with only 328 mV (Fig. 1c). The Tafel slope values for these spinel oxides are derived from the polarization curves shown in Fig. 1d. The Tafel slope of trimetallic NiCo1.9Mo0.1O4 (59.6 mV dec−1) is much lower than that of bimetallic NiCo2O4 (176.7 mV dec−1), indicating improved surface reaction kinetics. Generally, a Tafel slope of 120 mV dec−1 or higher suggests that the rate-limiting step (RLS) in the OER process is the adsorption of OH−, as observed with NiCo2O4. In contrast, the lower Tafel slope of NiCo1.9Mo0.1O4, closer to 40 mV dec−1, indicates that the RLS is the formation of MOOH.10 This shift in the RLS is likely responsible for the enhanced OER performance of the trimetallic oxide, which benefits from accelerated reaction kinetics and better OH− adsorption.
m (JCPDS no. 20-0781). The XRD pattern of trimetallic NiCo1.9Mo0.1O4 did not show any peak shift, which might be attributed to the low amount of Mo. Fig. 1b shows the polarization curves with iR correction for bimetallic NiCo2O4 and trimetallic NiCo1.9Mo0.1O4 catalysts. Fig. S2† shows the polarization curves of oxides without iR correction. NiCo2O4 achieves a current density of 10 mA cm−2 with an overpotential of 460 mV, whereas NiCo1.9Mo0.1O4 achieves the same current density with only 328 mV (Fig. 1c). The Tafel slope values for these spinel oxides are derived from the polarization curves shown in Fig. 1d. The Tafel slope of trimetallic NiCo1.9Mo0.1O4 (59.6 mV dec−1) is much lower than that of bimetallic NiCo2O4 (176.7 mV dec−1), indicating improved surface reaction kinetics. Generally, a Tafel slope of 120 mV dec−1 or higher suggests that the rate-limiting step (RLS) in the OER process is the adsorption of OH−, as observed with NiCo2O4. In contrast, the lower Tafel slope of NiCo1.9Mo0.1O4, closer to 40 mV dec−1, indicates that the RLS is the formation of MOOH.10 This shift in the RLS is likely responsible for the enhanced OER performance of the trimetallic oxide, which benefits from accelerated reaction kinetics and better OH− adsorption.
 | ||
| Fig. 1 (a) XRD pattern of NiCo2O4 and NiCo1.9Mo0.1O4 deposited on FTO substrates. (b) Polarization curves of NiCo2O4 and NiCo1.9Mo0.1O4 after iR correction. (c) Overpotential values of NiCo2O4 and NiCo1.9Mo0.1O4 at 10 mA cm−2. (d) Tafel plots of NiCo2O4 and NiCo1.9Mo0.1O4 derived from LSV scans. | ||
The morphology of spinel NiCo2O4 and NiCo1.9Mo0.1O4 is investigated by scanning electron microscopy (SEM) and high-resolution transmission electron microscopy (HR-TEM). SEM images show that NiCo2O4 comprises a hybrid of 2-dimensional (2D) nanosheets and 1D nanowires (Fig. 2a and S3a†). HR-TEM further provides a closer examination of the nanowires (Fig. 2b). The HR-TEM image of NiCo2O4 displays distinct lattice fringes with an interplanar spacing (d) of 0.29 nm, corresponding to the (220) lattice plane of spinel NiCo2O4 (Fig. 2c and d). Additionally, the selected area electron diffraction (SAED) pattern (Fig. S3b†) confirms the polycrystalline nature of spinel NiCo2O4. In contrast, trimetallic NiCo1.9Mo0.1O4 exhibits 3D flower-like microstructures (Fig. S4†) composed of interconnected 1D nanowires as shown in Fig. 2e and f. The variation in the morphology of NiCo2O4 and NiCo1.9Mo0.1O4 can be explained by the decomposition of ammonium molybdate (Mo precursor) to release ammonia which changes the solution pH and thus the degree of supersaturation and the morphology.22 HR-TEM confirms the high crystallinity of NiCo1.9Mo0.1O4 as shown in Fig. 2g and h. Furthermore, energy dispersive X-ray spectroscopy (EDS) verifies the existence of Mo, Co, Ni and O, while the elemental mapping images additionally illustrate their uniform distribution in NiCo1.9Mo0.1O4 (Fig. 2i and S5†). Similarly, EDS mapping of NiCo2O4 reveals a homogeneous elemental distribution (Fig. S6†). The atomic ratio of Ni and Co in NiCo2O4 is determined to be about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (Table S1†). Similarly, in NiCo1.9Mo0.1O4, the atomic ratio of Ni to the sum of Mo and Co is approximately 1:2, aligning with the intended target composition (Table S1†).
:2 (Table S1†). Similarly, in NiCo1.9Mo0.1O4, the atomic ratio of Ni to the sum of Mo and Co is approximately 1:2, aligning with the intended target composition (Table S1†).
 | ||
| Fig. 2 (a and b) SEM and HR-TEM images of NiCo2O4. (c and d) HR-TEM of NiCo2O4 showing the crystal lattice fringes. (e and f) SEM and HR-TEM images of NiCo1.9Mo0.1O4. (g and h) HR-TEM of NiCo1.9Mo0.1O4 showing the crystal lattice fringes. (i) EDS atomic elemental mapping of trimetallic NiCo1.9Mo0.1O4. | ||
Compared to 2D-NiCo2O4, the 3D morphology of NiCo1.9Mo0.1O4 maximizes the exposed active sites at the surface and increases the contact area with the electrolyte, thereby enhancing the OER catalytic performance.23 The active surface area is further evaluated by the measurements of double-layer capacitance (Cdl) (Fig. S7 and S8†). Cdl, which correlates with the active surface area, serves as an indicator for estimating the electrochemical active surface area (ECSA). 3D-NiCo1.9Mo0.1O4 exhibits a significantly higher surface area, approximately twice that of 2D-NiCo2O4. To ensure a fair assessment of the catalyst, the impact of morphology is excluded by normalizing the OER performance with respect to ECSA. The ECSA-normalized specific activity in Fig. S8b† demonstrates that at η = 300 mV, NiCo1.9Mo0.1O4 exhibits a high specific activity (0.01 mA cmECSA−2), which is 3 times higher than that of NiCo2O4 (0.003 mA cmECSA−2). This highlights the effective role of Mo in boosting the catalytic performance of spinel NiCo2O4.
To gain insight into the effect of Mo on the electronic structure of the spinel oxide, we conducted X-ray photoelectron spectroscopy (XPS) measurements on both NiCo2O4 and NiCo1.9Mo0.1O4. Fig. 3a shows the XPS spectra of Co 2p core level, consisting of two characteristic peaks Co 2p3/2 at ≈779.5 eV and Co 2p1/2 at ≈795.0 eV along with their corresponding shakeup satellites. Due to the proximity of 2p1/2 and 2p3/2 peak locations for Co2+ and Co3+, the satellite features and their intensity were analyzed using a series of standard components (i.e., 787 eV for Co(OH)2-like Co2+ and 789.5 eV for Co3O4 with dominant Co3+) as shown in Fig. 3a.24 Therefore, the Co2+ to Co3+ ratio is determined based on their respective compositions. Fig. 3a shows that the Co3+ is the predominant oxidation state in both NiCo2O4 and NiCo1.9Mo0.1O4 (Fig. 3b and S9†). However, in NiCo1.9Mo0.1O4, the Co(OH)2-like Co2+ at 787 eV is slightly decreased indicating more Co3+. The fitting results are summarized in Fig. 3b. Fig. 3c shows that the XPS spectrum of Mo 3d is dominated by the high valence state, i.e., Mo6+ and Mo5+. The partial reduction of Mo6+ to Mo5+ might be explained by the electron exchange occurring between Co2+ and Mo5+ as follows: Mo6+ + Co2+ ↔ Mo5+ + Co3+.25 The cation redistribution of Co2+ and Mo6+ can explain the slight decrease of Co2+ ratios which is induced by the occupancy of Mo6+ in tetrahedral sites in NiCo1.9Mo0.1O4.26,27
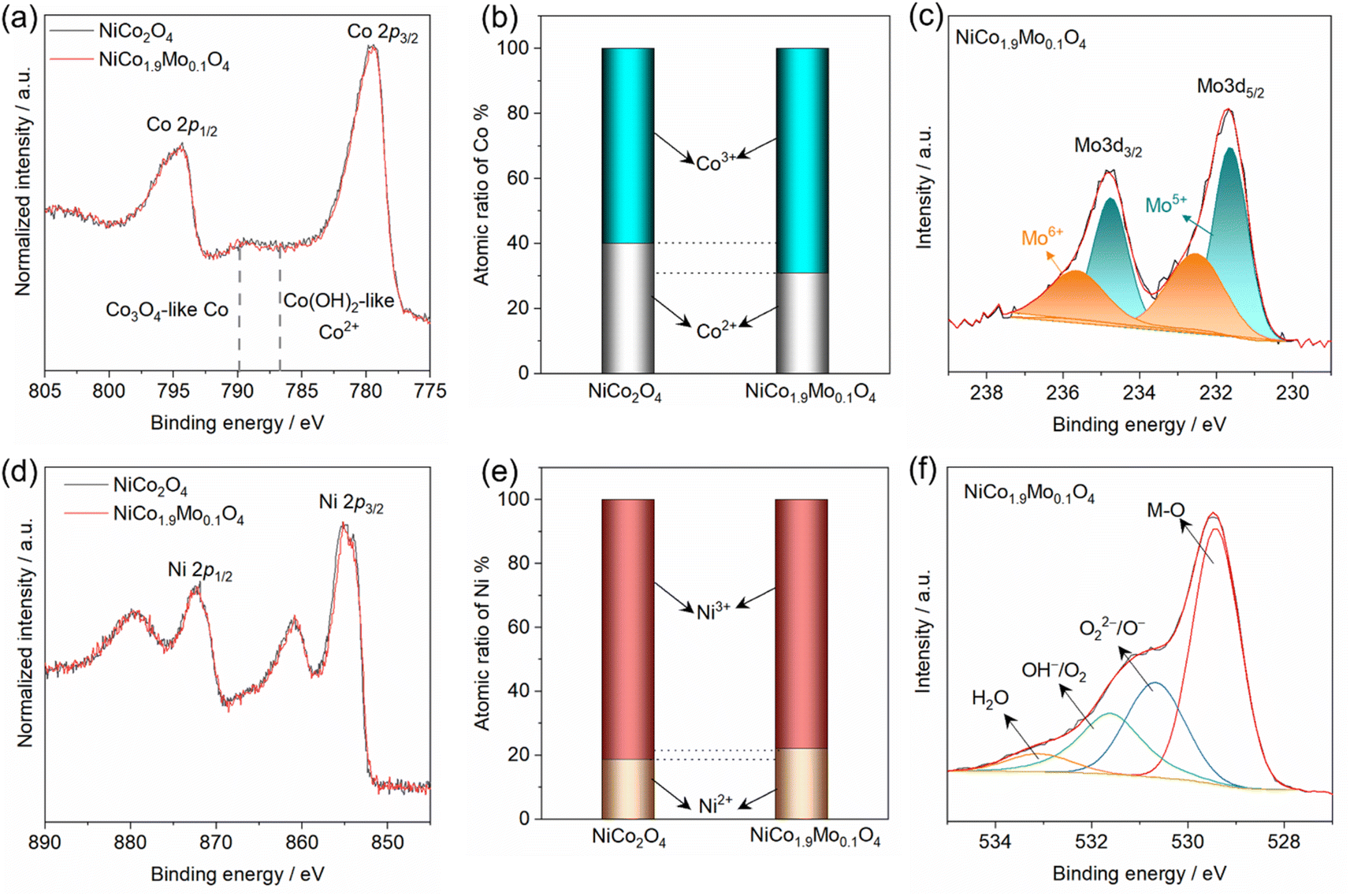 | ||
| Fig. 3 XPS spectra of (a) Co 2p in NiCo2O4 and NiCo1.9Mo0.1O4, (b) atomic ratios of Co2+ and Co3+ in oxides, (c) XPS spectra of Mo 3d in NiCo1.9Mo0.1O4, (d) XPS spectra of Ni 2p in NiCo2O4 and NiCo1.9Mo0.1O4, (e) atomic ratios of Ni2+/Ni3+ in oxides and (f) O 1s XPS in NiCo1.9Mo0.1O4. | ||
The XPS spectra of Ni 2p in NiCo2O4 and NiCo1.9Mo0.1O4 are also shown in Fig. 3d. The spectral line of Ni 2p of NiCo1.9Mo0.1O4 closely resembles that of NiCo2O4, suggesting an insignificant change in Ni2+/Ni3+ ratio in both spinel oxides. The fitting results are further summarized in Fig. 3e and S10.† Additionally, the O 1s XPS spectra for these spinel oxides are shown in Fig. S11.† Comparatively, the O 1s XPS spectrum of NiCo1.9Mo0.1O4 shows negligible change compared to that of NiCo2O4. The O 1s XPS spectra of both oxides can be deconvoluted into 4 peaks as shown in Fig. 3f. These peaks correspond to: lattice oxygen species (M–O) at 529.2 eV, oxygen defects (O22−/O−), hydroxyl group (OH−/O2) at 531.4 eV and molecular water (H2O) at 532.5 eV.28
Spinel oxides are widely recognized as pre-catalysts for the OER, undergoing surface reconstruction under anodic potential to form the true active species.17 This reconstruction involves the pre-oxidation of cations with low oxidation states to higher oxidation states, which is a key step in forming the active (oxy)hydroxide phase. Cyclic voltammetry (CV) is commonly used to both induce and monitor this electrochemical activation, providing insights into the surface changes and formation of active species. The CV profiles of NiCo2O4 and NiCo1.9Mo0.1O4 are shown in Fig. 4a. The polarization curves of NiCo2O4 and NiCo1.9Mo0.1O4 show a distinct redox couple peak at ≈1.45 V vs. RHE, corresponding to the oxidation of Ni species (Ni2+ → Ni3+).29,30 However, in the case of NiCo1.9Mo0.1O4, the CV curve exhibits a significantly higher and broader profile than that of NiCo2O4, suggesting the substantial generation of active Ni species. This indicates that Mo accelerates the self-activation/reconstruction process. Notably, the Ni2+/Ni3+ redox peak gradually increases and stabilizes after the 47th cycle, signifying a stable steady state and indicating the successful completion of activation (Fig. S12†).30 This incremental rise is attributed to the progressive generation of active NiOOH and a growing abundance of accessible catalytically active sites in NiCo1.9Mo0.1O4. Consequently, CV cycling accelerates the reconstruction and self-activation processes, resulting in the exposure of a greater number and density of active sites.
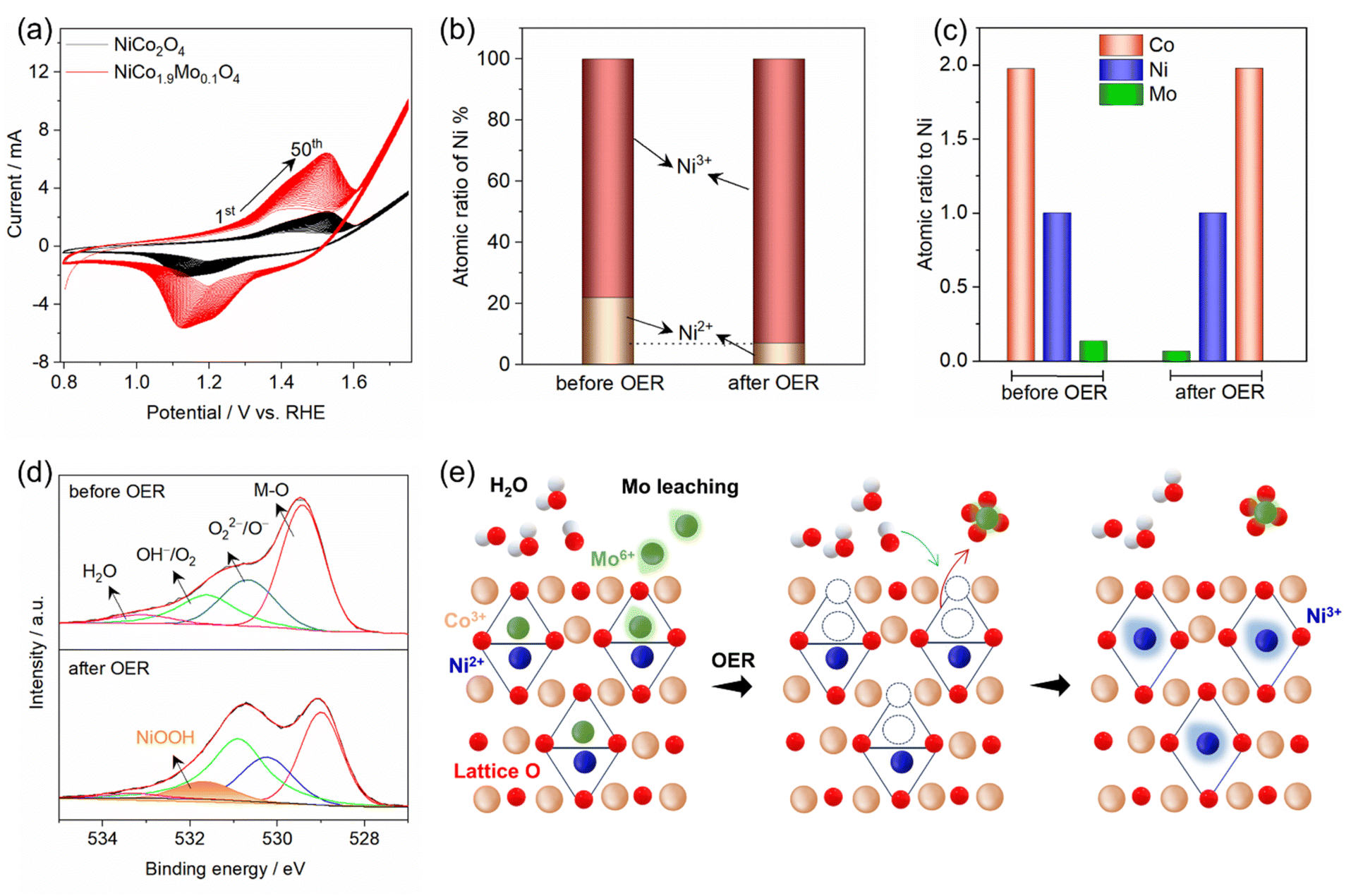 | ||
| Fig. 4 (a) CV measurements of NiCo2O4 and NiCo1.9Mo0.1O4 (50 cycles), (b) atomic ratios of Ni2+ and Ni3+ in NiCo1.9Mo0.1O4 before and after the OER, (c) surface atomic concentration of Co, Ni and Mo, (d) O 1s XPS spectra in NiCo1.9Mo0.1O4 before and after the OER, (e) schematic illustration of surface transformation in NiCo1.9Mo0.1O4 showing Mo6+ leaching, cation vacancies, and NiOOH formation. | ||
XPS was employed to monitor the surface chemical states after the OER to further identify the real active sites in NiCo1.9Mo0.1O4 for the OER. The peaks at 855 eV and 857.6 eV correspond to Ni2+ and Ni3+ states present in NiCo1.9Mo0.1O4 (Fig. S13†). The calculated Ni3+/Ni2+ ratios of NiCo1.9Mo0.1O4 is increased by 2.6-fold after the OER (Fig. 4b). This significant variation in ratios reveals the surface changes of NiCo1.9Mo0.1O4 during the OER process, forming active NiOOH species, contributing to enhanced catalytic activity. Conversely, a minor change is observed in the Co 2p states for NiCo1.9Mo0.1O4 before and after the OER (Fig. S14†). The calculated Co2+/Co3+ ratio was almost the same before and after the OER, as shown in Fig. S15.† This suggests that Co persists as a mixture of Co2+/Co3+ during the OER. Surface atomic ratio calculations further reveal the partial leaching of Mo after the OER, whereas Ni and Co remain unchanged (Fig. 4c).
Furthermore, to determine whether the enhanced OER activity arises from the incorporation of Mo in spinel oxide or the presence of trace Fe impurities in the electrolyte, OER activity was evaluated in 0.1 M ultrapure KOH electrolyte and unpurified KOH containing Fe (<0.0005%). The LSV curves (Fig. S16†) show nearly identical OER activity in both electrolytes, indicating that Fe impurities have no significant effect on the measurements. This further confirms that the enhanced activity of trimetallic NiCo1.9Mo0.1O4 stems from Mo incorporation.
Based on the aforementioned results, Ni2+ ions play a more active role than Co2+ and Co3+ in tetrahedral and octahedral sites, respectively, within spinel oxides.14 This result is supported by the gradual increase of Ni2+/Ni3+ redox pairs from CV curves (Fig. 4a). However, in NiCo2O4, the number of active Ni sites was limited as indicated by the weak anodic peak. In contrast, in NiCo1.9Mo0.1O4, the high valence Mo6+ ions replace the tetrahedral Co2+ ions. Under anodic conditions in an alkaline medium (pH ≈ 14), these Mo6+ ions partially leach out, as predicted by the Pourbaix diagram.31 Surface hydroxylation promotes the dissolution of Mo6+ as MoO42− (Mo6+O3 + OH− → MoO42− + H2O), particularly at high anodic potentials. The partial leaching of Mo6+, along with the loss of lattice oxygen, generates cation and anion vacancies in the tetrahedral sites (Fig. 4e), triggering surface rearrangement, and increasing Ni2+ exposure for the OER as evidenced by the CV curves (Fig. 4a). The highly exposed Ni2+ ions react with OH− ions from the electrolyte, forming a hydroxide layer that is subsequently electro-oxidized to form the active layer of amorphous NiOOH. The formation of NiOOH is confirmed by post-OER XPS, where a new peak appears in the O 1s spectrum (Fig. 4d). Furthermore, HR-TEM imaging (Fig. S17†) provides direct structural evidence of this transformation, revealing a distinct difference between the crystalline spinel oxide and the amorphous NiOOH domains. Fast Fourier-transform (FFT) patterns obtained from the yellow and white squares further confirm the structural changes, with the yellow square (dark regions) reflecting the high crystallinity of the spinel oxide, while the white square (light regions) exhibits the highly disordered nature of amorphous NiOOH. EDS mapping further reveals the distribution of Ni and Co, with Mo retained at ≈60% of its initial concentration after partial leaching (Fig. S18†). The schematic of the proposed mechanism is shown in Fig. 4e.
Nevertheless, the catalytic role of Co ions cannot be dismissed entirely, as the behavior of Ni cation might be affected by the presence of coordinated Co cations and oxygen anions in the surrounding environment. Additionally, Co oxide serves as a structural support, providing a conductive matrix for charge carriers.14
Motivated by the promising performance of NiCo1.9Mo0.1O4/FTO as an effective electrocatalyst, we extended our investigation to include the deposition of NiCo1.9Mo0.1O4 on 3D-interconnected porous and conducting nickel foam (NF). The SEM images of NiCo2O4 and NiCo1.9Mo0.1O4 on NF show nanorods and nanowires, respectively (Fig. S19†). It is noteworthy that the morphology of NiCo1.9Mo0.1O4 remains the same as on FTO (Fig. 2d and S19c†). The OER activity of NiCo2O4 and NiCo1.9Mo0.1O4 on NF in 1 M KOH is shown in Fig. 5a. NiCo1.9Mo0.1O4/NF achieves a current density of 10 and 500 mA cm−2 at a remarkable low overpotential values of 250 and 320 mV, respectively. In contrast, NiCo2O4/NF demonstrated poor catalytic activity for the OER with an overpotential of 330 mV at 10 mA cm−2. The Tafel slope for NiCo1.9Mo0.1O4/NF was 41.5 mV dec−1, which is lower than that of NiCo2O4 (73 mV dec−1) as shown in Fig. S20.† The specific activity of NiCo1.9Mo0.1O4 on NF is still high compared to NiCo2O4 on the same substrate (Fig. S21†). The NiCo1.9Mo0.1O4/NF maintains continuous operation for over 25 h under electrochemical OER conditions with a negligible change in current density (Fig. S22†). Notably, the morphology remains largely unchanged after the stability test (Fig. S23†), demonstrating the high structural stability of the oxide.
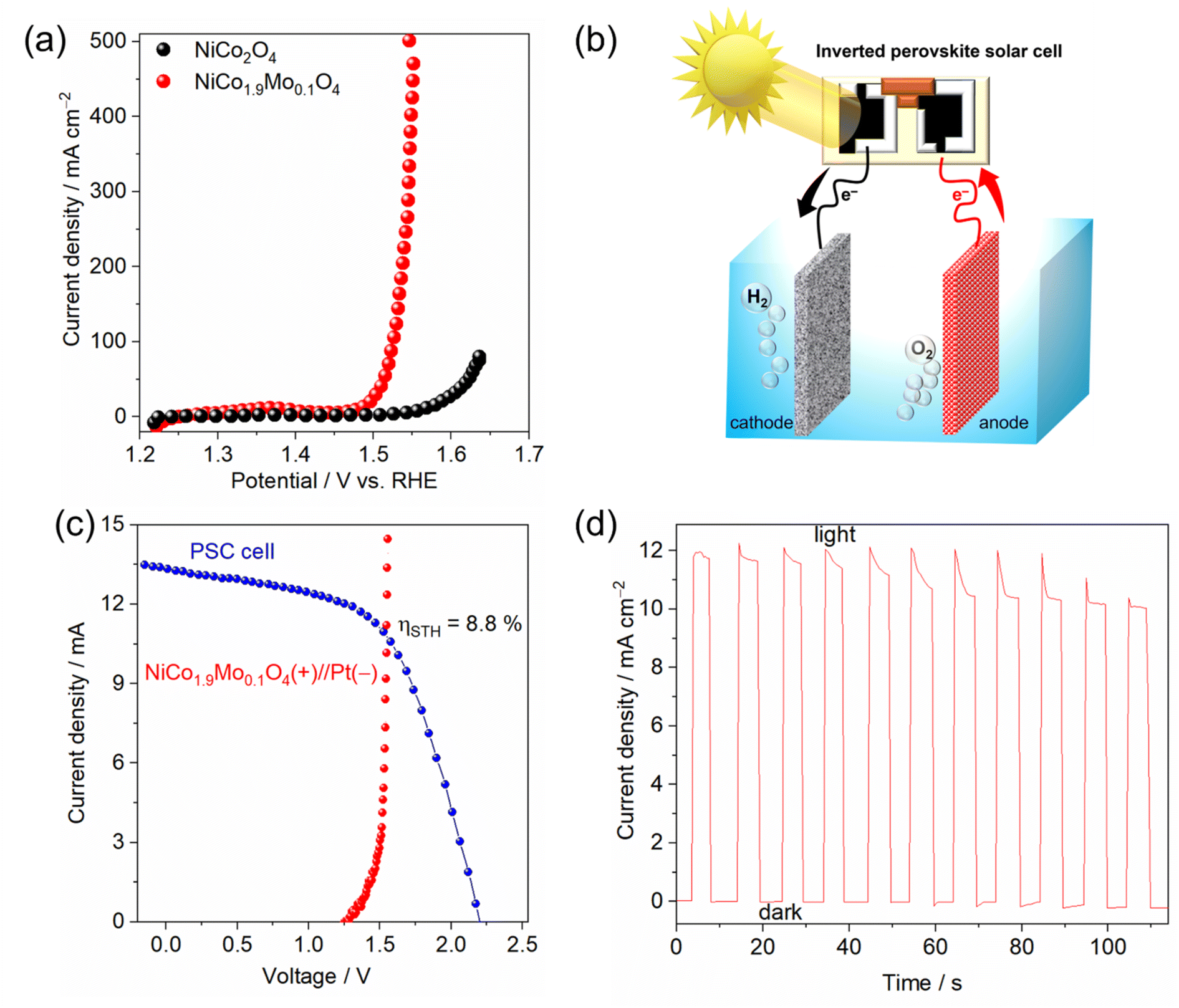 | ||
| Fig. 5 (a) Polarization curves of NiCo2O4/NF and NiCo1.9Mo0.1O4/NF, (b) schematic illustration of the PSC-driven water electrolysis, (c) current–voltage curves of the perovskite solar cell under 1 sun illumination (AM 1.5 G, 100 mW cm−2) and /NF in a two-electrode cell configuration, (d) current–time plot of the water electrolysis system without external bias potential (V = 0) under 1 sun illumination. | ||
Inspired by the high catalytic activity observed in NiCo1.9Mo0.1O4/NF for the OER, we utilized this activity of the OER catalyst in a photovoltaic (PV)–water electrolysis integrated system. The water electrolysis cell consisted of a 2-electrode system, featuring a platinum (Pt) cathode for the HER and KOH as the electrolyte (Fig. 5b). The Pt/NF//NiCo1.9Mo0.1O4/NF cell requires only 1.53 V to generate 10 mA cm−2, surpassing most reported catalysts or even comparable to noble-based catalysts (Table S2†). The solar water splitting system was fabricated by integrating this cell with an inverted perovskite solar cell (Fig. 5b), with its detailed structure shown in Fig. S24a.† The inverted perovskite solar cell demonstrated a short circuit current density (JSC) of 18.12 mA cm−2 and an open-circuit voltage (VOC) of 1.03 V (Fig. S24b†). It achieved a power conversion efficiency (PCE) of ≈13% with a fill factor (FF) of ≈70%. However, since 1.03 V voltage is not sufficient for driving the water splitting reaction, two solar cell devices were connected in series to generate the required voltage. Connecting the series-configured inverted perovskite solar cell and water electrolysis cell with a wire (Fig. 5b and S25†) enabled the PV–water electrolyzer system to drive the overall water splitting without an external bias, operating at a current density of ≈11 mA cm−2 under standard AM 1.5 G sunlight (Fig. 5c, d and ESI Video†). A solar to hydrogen efficiency (STH) of 8.8% is achieved. It should be noted that the STH can be improved to a higher value with the improvement of perovskite solar cell efficiency.
Conclusions
In summary, we have successfully synthesized a trimetallic spinel oxide NiCo2−xMoxO4 with nanowire morphology using a facile hydrothermal method followed by one-step annealing. Electrochemical measurements and electronic structure analyses reveal that the surface of spinel NiCo2−xMoxO4 (x = 0.1) undergoes reconstruction to form the active phase, nickel (oxy)hydroxide. This transformation is induced by partial leaching of Mo cations, increasing the exposure of more active sites to the OER. Consequently, the oxide exhibits high catalytic activity, with a low overpotential of 250 mV at 10 mA cm−2 and high stability for over 25 h in alkaline medium. Additionally, when integrated with an inverted perovskite solar cell, the trimetallic spinel oxide enables bias-free solar water splitting, reaching 8.8% solar-to-hydrogen efficiency. This study highlights the role of metal ions with leaching capabilities to enhance the OER activity of spinel oxides and expand their application for photovoltaic-driven water splitting.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Mahmoud G. Ahmed: conceptualization, writing-original draft, synthesis, characterization of oxides, electrochemical measurements. Yi Fei Phang: synthesis, electrochemical measurements. Y. F. Tay: XPS analysis. Anupam Sadhu: fabrication of the perovskite solar cell device. Pritish Mishra: TEM characterization. Akhmad Herman Yuwono: writing-reviewing and editing; Lydia H. Wong: conceptualization, validation, supervision, administration, writing-reviewing and editing and funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by Singapore Ministry of Education (MOE) AcRF Tier 2 grant (MOE T2EP50120-00081) and Tier 1 grant (2020-T1-001-147 (RG64/20)). Additionally, we would like to acknowledge the Indonesian Endowment Fund for Education (LPDP) on behalf of the Indonesian Ministry of Education, Culture, Research, and Technology, managed under the INSPIRASI Program (Grant No. PRJ-61/LPDP/2022 and 612/E1/KS.06.02/2022).References
- G. K. Singh, Energy, 2013, 53, 1–13 CrossRef.
- A. Sadhu, M. Rai, T. Salim, X. Jin, J. M. R. Tan, S. W. Leow, M. G. Ahmed, S. Magdassi, S. G. Mhaisalkar and L. H. Wong, Adv. Funct. Mater., 2021, 31, 2103807 CrossRef CAS.
- A. S. R. Bati, Y. L. Zhong, P. L. Burn, M. K. Nazeeruddin, P. E. Shaw and M. Batmunkh, Commun. Mater., 2023, 4, 2 CrossRef CAS.
- NREL, https://www.nrel.gov/pv/interactive-cell-efficiency.html, (accessed July, 2024).
- T. J. Jacobsson, V. Fjällström, M. Edoff and T. Edvinsson, Energy Environ. Sci., 2014, 7, 2056–2070 RSC.
- H. L. Tuller, Mater. Renew. Sustain. Energy, 2017, 6, 3 CrossRef PubMed.
- Z. Li, S. Fang, H. Sun, R.-J. Chung, X. Fang and J.-H. He, Adv. Energy Mater., 2023, 13, 2203019 CrossRef CAS.
- J. Chen, C. Dong, H. Idriss, O. F. Mohammed and O. M. Bakr, Adv. Energy Mater., 2020, 10, 1902433 CrossRef CAS.
- B. Weng, F. Xu, C. Wang, W. Meng, C. R. Grice and Y. Yan, Energy Environ. Sci., 2017, 10, 121–128 RSC.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- M. G. Ahmed, Y. F. Tay, X. Chi, M. Zhang, J. M. R. Tan, S. Y. Chiam, A. Rusydi and L. H. Wong, Small, 2023, 19, 2204520 CrossRef CAS PubMed.
- M. G. Ahmed, M. Zhang, Y. F. Tay, S. Y. Chiam and L. H. Wong, ChemSusChem, 2020, 13, 5489–5496 CrossRef CAS PubMed.
- M. G. Ahmed, Y. F. Tay, X. Chi, A. S. Razeen, Y. Fang, M. Zhang, A. Sng, S. Y. Chiam, A. Rusydi and L. H. Wong, Angew. Chem., Int. Ed., 2025, 64, e202416757 Search PubMed.
- H.-Y. Wang, Y.-Y. Hsu, R. Chen, T.-S. Chan, H. M. Chen and B. Liu, Adv. Energy Mater., 2015, 5, 1500091 Search PubMed.
- X. Gao, H. Zhang, Q. Li, X. Yu, Z. Hong, X. Zhang, C. Liang and Z. Lin, Angew. Chem., Int. Ed., 2016, 55, 6290–6294 CrossRef CAS PubMed.
- Y. Huang, S. L. Zhang, X. F. Lu, Z.-P. Wu, D. Luan and X. W. Lou, Angew. Chem., Int. Ed., 2021, 60, 11841–11846 CrossRef CAS PubMed.
- L. Gao, X. Cui, C. D. Sewell, J. Li and Z. Lin, Chem. Soc. Rev., 2021, 50, 8428–8469 RSC.
- M. G. Ahmed, Y. F. Tay, M. Zhang, S. Y. Chiam and L. H. Wong, ACS Mater. Lett., 2024, 4756–4764, DOI:10.1021/acsmaterialslett.4c00857.
- T. Liu and P. Diao, Nano Res., 2020, 13, 3299–3309 CrossRef CAS.
- K. Fan, H. Zou, Y. Ding, N. V. R. A. Dharanipragada, L. Fan, A. K. Inge, L. Duan, B. Zhang and L. Sun, Small, 2022, 18, 2107249 CrossRef CAS PubMed.
- R. N. Dürr, P. Maltoni, H. Tian, B. Jousselme, L. Hammarström and T. Edvinsson, ACS Nano, 2021, 15, 13504–13515 Search PubMed.
- D. T. Phat, P. M. Thao, N. Van Nghia, L. T. Son, T. V. Thu, N. T. Lan, N. Q. Quyen, N. Van Ky and T. Van Nguyen, J. Energy Storage, 2021, 33, 102030 CrossRef.
- P. Sun, Y. Zhou, H. Li, H. Zhang, L. Feng, Q. Cao, S. Liu, T. Wågberg and G. Hu, Appl. Catal., B, 2022, 310, 121354 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- T. Mazari, C. R. Marchal, S. Hocine, N. Salhi and C. Rabia, J. Nat. Gas Chem., 2010, 19, 54–60 CrossRef CAS.
- J. M. Byrne, V. S. Coker, E. Cespedes, P. L. Wincott, D. J. Vaughan, R. A. D. Pattrick, G. van der Laan, E. Arenholz, F. Tuna, M. Bencsik, J. R. Lloyd and N. D. Telling, Adv. Funct. Mater., 2014, 24, 2518–2529 CrossRef CAS.
- G. D. Dwivedi, K. F. Tseng, C. L. Chan, P. Shahi, J. Lourembam, B. Chatterjee, A. K. Ghosh, H. D. Yang and S. Chatterjee, Phys. Rev. B, 2010, 82, 134428 CrossRef.
- C. Yuan, J. Li, L. Hou, X. Zhang, L. Shen and X. W. Lou, Adv. Funct. Mater., 2012, 22, 4592–4597 CrossRef CAS.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS.
- S. Hou, W. Li, S. Watzele, R. M. Kluge, S. Xue, S. Yin, X. Jiang, M. Döblinger, A. Welle, B. Garlyyev, M. Koch, P. Müller-Buschbaum, C. Wöll, A. S. Bandarenka and R. A. Fischer, Adv. Mater., 2021, 33, 2103218 CrossRef CAS PubMed.
- M. Nishimoto, I. Muto, Y. Sugawara and N. Hara, J. Electrochem. Soc., 2019, 166, C3081 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08267b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |