
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermal expansion of lithiated silicon (Li13Si4 and Li7Si3) anodes: a powder neutron diffraction study
Atia
Azad
 *a,
Katherine
Bateman
a,
Matthew
Irvine
a,
Aaron B.
Naden
a,
Stewart A. M.
Dickson
a,
Ronald I.
Smith
b,
Richard K. B.
Gover
c and
John T. S.
Irvine
a
*a,
Katherine
Bateman
a,
Matthew
Irvine
a,
Aaron B.
Naden
a,
Stewart A. M.
Dickson
a,
Ronald I.
Smith
b,
Richard K. B.
Gover
c and
John T. S.
Irvine
a
aSchool of Chemistry, University of St Andrews, North Haugh, St Andrews, KY16 9ST, UK. E-mail: jtsi@st-andrews.ac.uk
bISIS Neutron and Muon Source STFC Rutherford Appleton Laboratory, Harwell Campus, Didcot, Oxfordshire OX11 0QX, UK
cAWE, Reading, Berkshire, RG7 4PR, UK
First published on 24th March 2025
Abstract
Whilst at room temperature structural changes on lithiation of the silicon electrode are hard to study due to formation of amorphous phases, at high temperatures, used in thermal batteries, clear phase changes linked to voltage plateaux are observed. Here we report results from a galvanostatic discharge of a FeS2/LiCl–KCl/Li13Si4 cell at 500 °C. The cell discharge showed a flat voltage plateau between Li13Si4 and Li7Si3 which indicates that both phases are in equilibrium and are line phases. In situ powder neutron diffraction study of two lithiated phases of silicon, Li13Si4 and Li7Si3, was performed. The two phases were heated from room temperature to 500 °C. This showed the phases did not become amorphous and did not undergo phase changes, with both phases being remarkably stable. The variation with temperature of the unit cell parameters was linear. The volumetric thermal expansion coefficient of Li13Si4 is 99.3 × 10−6 °C−1 and Li7Si3 is 106 × 10−6 °C−1. The volumetric thermal expansion of the two phases is significantly larger than that of silicon and closer to the thermal expansion of lithium metal. Thus, the LixSi electrode is mechanically closer to lithium than to silicon, and it can be considered as silicon clusters embedded within the lithium array rather than the silicon lattice hosting lithium.
Introduction
Silicon is a promising negative electrode material for lithium-ion batteries because of its high theoretical capacity that is ten times higher than graphite. Silicon suffers from large volume change of up to 400% (ref. 1) when lithium is inserted2 at room temperature which leads to contact loss of electrode and capacity loss in the first cycle. Lithium reacts with silicon by forming an alloy which breaks the silicon matrix.3 The lithiation mechanism of silicon at room temperature has been widely discussed in the literature; however, there is no clear consensus. Limthongkul et al. reported solid-state amorphisation occurs during lithiation, forming silicon and amorphous LixSi which are separated by a sharp reaction front with a thickness in the nanometre scale.4At room temperature, the lithiation of silicon, during the cycling of a crystalline silicon electrode vs. a lithium electrode in a coin cell, results in one broad voltage plateau.5 Approximately 3500 mA h g−1 is observed from the discharge of silicon at room temperature.5 This is close to the theoretical capacity of silicon = 3580 mA h g−1 with a fully lithiated state of Li15Si4. Obrovac and Christensen first reported that crystalline Li15Si4 forms when the potential is less than 0.05 V vs. Li.5 The study observed the voltage plateaux of coin cells containing crystalline silicon vs. lithium electrode that were stopped at various states of charge for ex situ X-ray diffraction studies.5 From the X-ray diffraction data collected at room temperature, crystalline Li15Si4 peaks were identified.5 Li15Si4 is a metastable phase.6 The structure of Li15Si4 was established using single-crystal X-ray diffraction at room temperature by Zeilinger et al. who synthesised the phase from equilibrated melts and isolated the phase using isothermal centrifugation.6
Because the cycling of a silicon electrode at room temperature involves the formation of amorphous phases, nuclear magnetic resonance (NMR) studies are available in the literature.7–10 Key et al. did room temperature 7Li and 29Si NMR studies where silicon clusters were found as intermediate units during cycling of crystalline silicon vs. lithium in coin cells.7,8
The formation of amorphous phases during lithiation is difficult to study with X-ray diffraction. On the other hand, clear phase changes are observed at high temperature, in thermal batteries, linked to voltage plateaux. Thermal batteries are an established primary battery technology that are used for specialised applications such as aircraft emergency power supplies, space flights, and borehole drilling.11 Thermal batteries are implemented when a constant power needs to be drawn over a period of time. Li13Si4 is the most common anode material for thermal batteries whereas Li7Si3 cannot provide the power needed for high-rate pulsing due to a lower lithium diffusion in the phase.12
Wen and Huggins reported that the electrochemical alloying of lithium and silicon at high temperature (415 °C) follows the equilibrium Li–Si phase diagram with four distinct voltage plateaux, which corresponds to the formation of Li12Si7, Li7Si3, Li13Si4, and Li21Si5.13 Li13Si4 and Li7Si3 are reported to be stable phases that congruently melt at 722 and 752 °C, respectively, whilst Li22Si5 and Li12Si7 incongruently melt at 628 and 648 °C, respectively.14,15
The transition from Li13Si4 to Li7Si3 has a voltage change of 44 mV to 158 mV with respect to lithium,13 and the transition from Li7Si3 to Li12Si7 has a voltage change of 158 mV to 288 mV with respect to lithium.13 The voltages and unit cell volumes divided by the total number of atoms of the Li–Si phases are shown in Fig. 1. Fig. 1 shows the unit cell volume decreases as silicon becomes more lithiated until the ratio is Li13Si4 (Li3.25Si). The unit cell volume expands with further lithiation until the ratio is Li21Si5 (Li4.2Si).
 | ||
| Fig. 1 Coulometric titration curve for lithium–silicon phases at 415 °C adapted from a study by Wen and Huggins.13 The unit cell volume per total number of atoms of Li–Si phases, and Si are shown. | ||
The structures of the four thermodynamically stable Li–Si phases at high temperature (Li21Si5, Li13Si4, Li7Si3 and Li12Si7) are shown in Fig. 2. The coulometric titration was measured using a three electrode cell (Al, “LiAl”/LiCl–KCl/LixSi).13 Al, “LiAl” represents a saturated solid solution of lithium in aluminium and an intermediate LiAl phase. Li15Si4 was not observed at 415 °C, so it only exists in ambient conditions.
 | ||
| Fig. 2 Thermodynamically stable phases in the lithium–silicon system.13 The green atoms are lithium, and the blue atoms are silicon. The phases have complex structures and are structurally different to elemental lithium or silicon. The structures were visualised on VESTA.16 | ||
Li21Si5
Li21Si5 (416 atoms per unit cell) is a lithium rich phase. Li21Si5 has 12 Li sites and 4 Si sites.17 It was previously thought to be Li22Si5, but a single crystal X-ray diffraction study showed it has a composition of Li21Si5.17 The unit cell is shown in Fig. 3. The crystal data from Nesper et al.17 is shown in Table 1. The synthesis of Li21Si5 was described briefly in the literature. A stoichiometric mixture of lithium and silicon was sealed in a niobium ampoule. The ampoule was heated to 727 °C.17 The crystals were collected after a slow cooling. The structure was determined by a least-squares refinement with 67 parameters and 356 reflections.17 | ||
| Fig. 3 Primitive unit cell of Li21Si5. The unit cell is cubic and made of 416 atoms. There are no partially occupied sites. The figure only shows the array of lithium atoms in green. | ||
| Space group | Cubic F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m 3m |
| a (Å) | 18.7100(20) |
| V (Å3) | 6549.699 |
| Radiation | Mo Kα |
| T (K) | 298 |
| Diffractometer | P1 four circle |
| Theoretical specific capacity (mA h g−1) | 4008 |
Li13Si4
Li13Si4 is a lithium rich phase. Li13Si4 has 38 atoms per unit cell. Li13Si4 has 8 Li and 2 Si sites.19 The unit cell is shown in Fig. 4. | ||
| Fig. 4 The primitive unit cell of Li13Si4. The unit cell is orthorhombic and is made from 38 atoms. There are two partially occupied lithium sites, highlighted with purple in the figure, which are located next to each other and can be considered a split site. The partially occupied lithium sites have a 0.84/0.16 ratio. The figure only shows the array of lithium atoms. | ||
The single crystal X-ray diffraction data reported by Zeilinger et al.19 is shown in Table 2. Zeilinger et al. synthesised Li13Si4 from a mixture of lithium rods and silicon powder. The synthesis was carried out in an argon glovebox. The mixture was placed into a tantalum ampoule which was arc welded inside the glovebox.20 The tantalum ampoule was heated in a muffle furnace. The sample was heated to 750 °C for 30 minutes.5 It was cooled to 500 °C for 1 hour.20 The ampoule was transferred back into the glovebox and the final single crystals were collected. The single crystals were sealed in a 0.3 mm diameter glass capillary for X-ray diffraction.19 The structure was determined by a least-squares refinement with 60 parameters and 2429 reflections.19
| Space group | Orthorhombic Pbam |
| a (Å) | 7.9488(4) |
| b (Å) | 15.1248(8) |
| c (Å) | 4.4661(2) |
| V (Å3) | 536.932 |
| Radiation | Mo Kα |
| T (K) | 100 |
| Diffractometer | Bruker APENXII CCD |
| Theoretical specific capacity (mA h g−1) | 3101 |
Li7Si3
Li7Si3 (21 atoms per unit cell) is a violet phase due to its distinct colour. The dark violet colour was also observed after synthesising Li7Si3 in this work. Li7Si3 has 3 Li and 1 Si sites.21 The unit cell is shown in Fig. 5.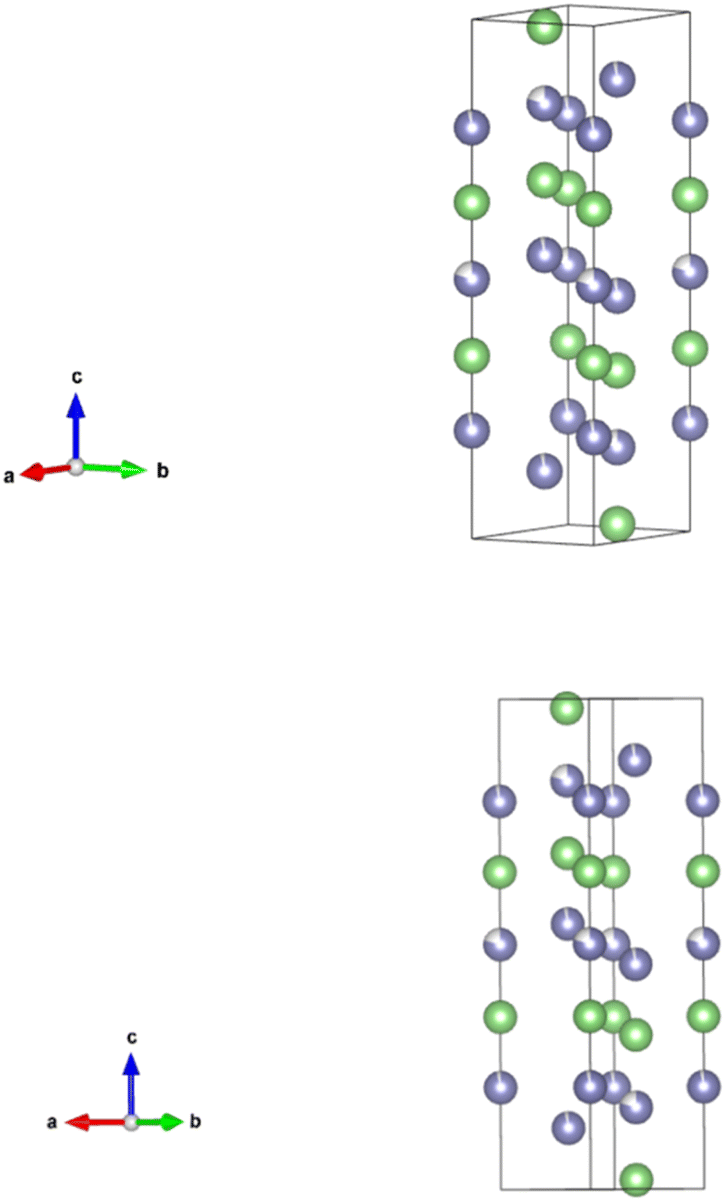 | ||
| Fig. 5 The primitive unit cell of Li7Si3. The unit cell is trigonal and is made of 21 atoms. There are two partially occupied lithium sites, highlighted with purple in the figure, with 0.95 and 0.8 occupancy. The figure only shows the array of lithium atoms. | ||
The ICSD database has only one study that determined the structure of Li7Si3 or Li2.33Si. The crystal data is shown in Table 3.
| Space group | Trigonal R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m m |
| a (Å) | 4.4350(10) |
| b (Å) | 4.4350(10) |
| c (Å) | 18.1340(30) |
| V (Å3) | 308.895 |
| Radiation | X-ray |
| T (K) | 293 |
| Diffractometer | N/A |
| Theoretical specific capacity (mA h g−1) | 2226 |
Li12Si7
Li12Si7 (152 atoms per unit cell) is a silicon rich phase. Li12Si7 has 13 Li and 9 Si sites.22 The unit cell is shown in Fig. 6.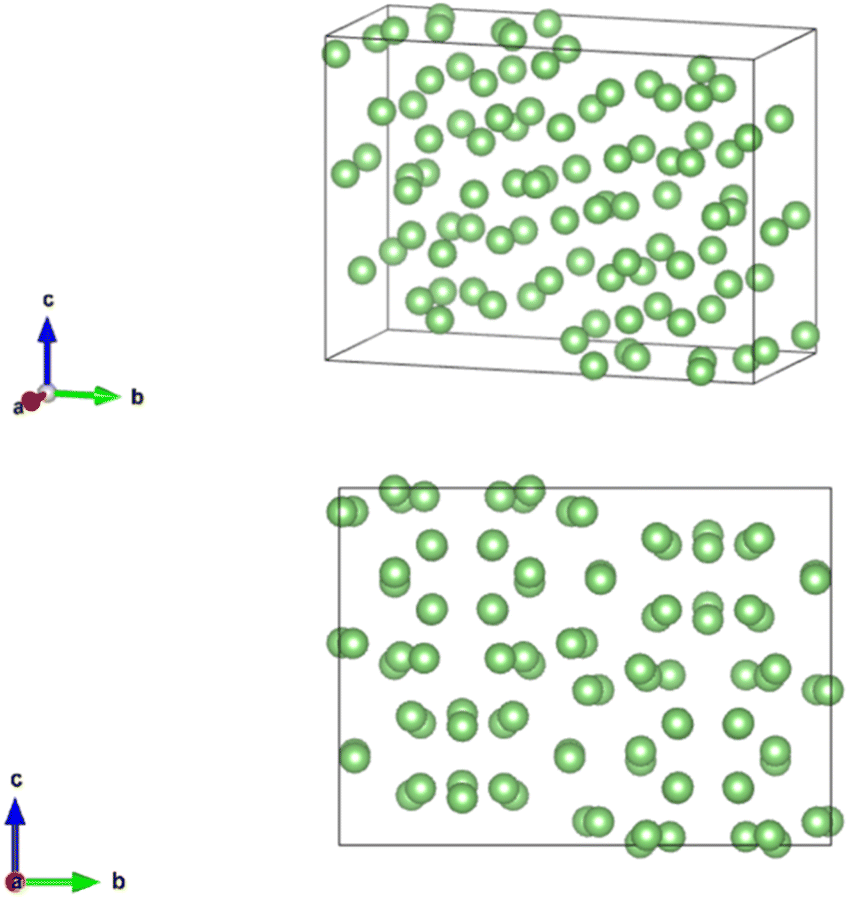 | ||
| Fig. 6 The primitive unit cell of Li12Si7. The unit cell is orthorhombic and made of 152 atoms. There are no partially occupied sites. The figure only shows the array of lithium atoms in green. | ||
The synthesis of Li12Si7 is described briefly in the literature. The sample was prepared by mixing lithium and silicon and placing the mixture in a sealed tantalum ampoule. The ampoule was heated to 997 °C.22 The Li12Si7 X-ray diffraction data is shown in Table 4.
| Space group | Orthorhombic Pnma |
| a (Å) | 8.6100(20) |
| b (Å) | 19.7380(40) |
| c (Å) | 18.1340(30) |
| V (Å3) | 308.895 |
| Radiation | X-ray |
| T (K) | 293 |
| Diffractometer | N/A |
| Theoretical specific capacity (mA h g−1) | 1636 |
This work explores if Li13Si4 and Li7Si3 undergo phase changes or become an amorphous phase on heating, and explores the thermal expansion of both phases which gives insights into the mechanical behaviour of the amorphous LixSi phase that forms during lithiation of silicon at room temperature. To the best of our knowledge, the current literature does not include a high-temperature neutron diffraction study of LixSi phases. Neutron diffraction is ideal for studying materials composed of lithium in the presence of heavier atoms because neutrons scatter from atomic nuclei, unlike X-ray scattering which depends on the number of electrons.23 In this work, we report a galvanostatic cell discharge to identify voltage plateaux and high-temperature structural characterisation of Li13Si4 and Li7Si3 from time-of-flight (TOF) powder neutron diffraction data collected at the ISIS Neutron Source, Rutherford Appleton Laboratory, UK. The bonding environments are also discussed.
Experimental
Ex situ synthesis
The handling and storage of materials were carried out in an argon glovebox (PureLab HE, Ar atmosphere, H2O less than 0.5 ppm and O2 levels less than 1 ppm). Starting materials were lithium metal (Li; granular 4–10 mesh 99%) and silicon powder (Si; 325 mesh 99%). Li13Si4 was purchased from Albemarle. Li7Si3 was synthesised ex situ.A solid-state synthesis method was adapted from literature.7,17,24–26 In this work, 0.5 g of stoichiometric ratio of lithium and silicon were mixed by hand with a mortar and pestle in the glovebox for 15 minutes. The mixture was placed in a tantalum crucible (melting point = 3017 °C) and transferred into a silica glass test tube attached to a valve fitting with an O-ring. The silica glass test tube had dimensions of 9 mm inner diameter and 12 mm outer diameter.
The silica tube was connected to a vacuum pump. The tube was evacuated (gauge pressure = 10−5 mbar) and sealed into a silica glass ampoule using a high-temperature flame. Prior to use, the silica glass and tantalum were wiped clean with ethanol and dried in an oven at 80 °C.
The ampoule was placed in a tube furnace. The temperature was increased at a rate of 5 °C per minute and kept at 190 °C for 2 hours to allow the lithium to melt close to its melting point. The temperature slowly increased to 500 °C at a rate of 2 °C min−1. The sample remained at this temperature for 12 hours. The reaction was stopped, and the temperature decreased at a rate of 5 °C per minute to below 180.5 °C and the ampoule was moved away from the hot zone of the furnace. Once cooled, the ampoule was cut open in the glovebox to obtain the final powder. The alloying of lithium and silicon is complex due to the inert conditions required for synthesis.
Powder X-ray diffraction
X-ray diffraction patterns were obtained at room temperature from the diffractometer (PANalytical Empyrean). The Bragg–Brentano focusing geometry was used. The diffractometer has CuKα1 radiation with wavelength of λ = 1.54 Å. The diffractometer has a X'celerator RTMS detector. Moisture sensitive samples were placed in a zero-background silicon substrate holder and had a protective Kapton film covering the holder. The diffraction range was 5 to 90° 2θ and the data was collected for 60 minutes. The resulting diffraction pattern shows the peak positions (x-axis) and relative peak intensities (y-axis) of the sample.The phases were identified by comparing the experimental diffraction pattern with a reference pattern from a database. In this work, the Inorganic Crystal Structure Database (ICSD) was used to find reference patterns from crystallographic information files (CIF). The experimental and reference patterns were compared by superimposing them on the same plot in CrystalDiffract. The X-ray diffraction patterns were indexed and refined using WinXPOW to determine phase purity.
Soft X-ray emission spectroscopy
Soft X-ray emission spectroscopy was performed on the JEOL JSM-IT800 FEG-SEM. Soft X-ray emission spectroscopy allows the mapping of lighter elements such as lithium ions because the X-rays of lighter elements have lower energies. Soft X-ray emissions are produced from the electronic transitions inside the sample.27 It is a characterisation technique that probes the energy state of bonding electrons27 by measuring the difference in the peak shape.27 The acceleration voltage was 5 kV and beam current was 60 nA. The nominal energy resolution was 0.3 eV.Electrochemical discharge
A cell was made from 0.15 g of Li13Si4, 0.05 g LiCl–KCl–MgO (anode); 0.2 g LiCl–KCl (electrolyte) mixed with 35% wt. MgO (separator); and 0.15 g FeS2, 0.05 g LiCl–KCl–MgO (cathode) were pressed into three different pellets. The two electrode pellets were 13 mm in diameter. The electrolyte/separator pellet was 15 mm in diameter. The pellets were loaded into a Swagelok type cell which allowed electrochemical testing to be performed at 500 °C ex situ inside the argon glovebox. The testing rig consists of a vertical tube furnace with a platform to hold the cell. Solartron analytical software was used to collect the data. The raw data was plotted on XM Studio ECS software. The cell was tested galvanostatically with slow discharge (current = 10 mA) to identify the voltage plateaus.Powder neutron diffraction
Powder neutron diffraction data were collected on the Polaris high flux, medium resolution diffractometer at the ISIS Neutron and Muon Source, UK. The high intensity of the incident neutron beam and a large solid angle of detector coverage enables it to collect good quality data in short periods of time from small samples.Polaris's multiple detector banks allow the collection of diffraction patterns of varying Δd/d Bragg peak resolutions over a series of partially overlapping d-spacings. The banks at lower 2θ angles access the longest d-spacings but have lower resolution. The banks at higher 2θ angles have the best resolution but measure to progressively smaller maximum d-spacings.
For this study, the best compromise between required d-spacing range and peak resolution was achieved in detector bank 3 (average 2θ angle = 52°). The dimensions of the incident beam were decreased to 10 mm wide × 14 mm high to prevent the beam scattering from the steel and ceramic components in the sample holder. This would contaminate the diffraction patterns with unwanted Bragg reflections.
A thin pair of vanadium plates held the sample in place in the neutron beam. Vanadium has a low neutron scattering length (b = −0.39 fm). This means the intensities of its Bragg reflections are low relative to those from the samples, so they make a negligible contribution to the overall diffraction pattern.
2g of Li13Si4 and Li7Si3 were weighed. The samples were pressed into pellets of 20 mm diameter in an argon glovebox using an electronically controlled uniaxial hydraulic press. The pellets were made using a pressure of 3 tonnes for 3 minutes. The pellets were sealed, under argon, in aluminium bags and transported to the ISIS Neutron and Muon Source.
The pellets were placed into an electrochemical testing rig designed by the University of St. Andrews.28 The furnace temperature was monitored using a thermocouple located close to the sample.
Neutron data analysis
Rietveld Refinement was carried out on the neutron diffraction patterns using known unit cell parameters and atom positions from the ICSD database. The CIF files from the database provide initial approximations to fit the calculated pattern with the experimental pattern. The objective of the refinement is to find the unit cell parameters at different temperatures and to find trends in the parameters which may indicate phase transitions. The General Structure Analysis System (GSAS) II29 and EXPGUI graphical interface were used for refinement.30Results and discussion
Phase identification
Li13Si4 and Li7Si3 powder were characterised by X-ray diffraction. Fig. 7 shows that the Li13Si4 and Li7Si3 patterns agree with the reference patterns by Zeilinger et al.19 and von Schnering et al.,21 respectively. The amorphous broad peaks around 20° 2θ are due to the Kapton film covering the samples during the measurements. The synthesised Li7Si3 had unreacted silicon in the first batch, and this is likely because a small quantity of silicon powder fell outside of the tantalum foil during loading. | ||
| Fig. 7 Ex situ X-ray diffraction patterns of Li13Si4 and Li7Si3 recorded from 5 to 90° 2θ at room temperature. The experimental patterns are compared with the reference patterns of Li13Si4 and Li7Si3, respectively. | ||
The peaks in Fig. 7 were indexed using WinXPOW. A peak list for each diffraction pattern was made. All peaks above 10% relative intensity were indexed for Li13Si4 which indicates the phase had no significant impurities. The unit cell was determined to have a = 7.985(6) Å, b = 15.168(7) Å, c = 4.4771(21) Å and V = 542.2(7) Å3 after refinement.
In Fig. 7, all peaks above 10% relative intensity for the second batch of Li7Si3 (orange graph) were indexed which indicates this phase did not have significant impurities. The unit cell was determined to have a = 4.438(3) Å, c = 18.140(13) Å, and V = 309.42(24) Å3 after refinement.
In Fig. 7, the first batch of Li7Si3 (blue graph) had impurities of Si and Li13Si4 after indexing peaks with 10% relative intensity on WinXPOW. 7 Peaks remained unindexed which indicates impurities. The two batches from Fig. 7 were mixed to make a 2 g pellet for the neutron diffraction study.
Cell discharge
The galvanostatic discharge at 500 °C shows two distinct voltage plateaux (Fig. 8). The voltage plateaux show Li13Si4 is an ideal negative electrode for high-capacity thermal batteries. The Li13Si4/Li7Si3 plateau is flat with a constant voltage which means Li13Si4 and Li7Si3 are in equilibrium and both phases are line phases. The change in voltage is determined by the transitions in the FeS2 positive electrode. | ||
| Fig. 8 Battery discharge profile for a FeS2/LiCl–KCl/Li13Si4 cell discharged galvanostatically at 500 °C using a current of 10 mA. The voltage changes are associated with phase transitions in FeS2. | ||
Fig. 8 shows the cell gives a voltage of 1.9 V which is close to the 1.8 V reported by Guidotti and Masset.11 The two voltage plateaux shown in Fig. 9 also align with the voltage plateaux reported by Guidotti and Masset.11
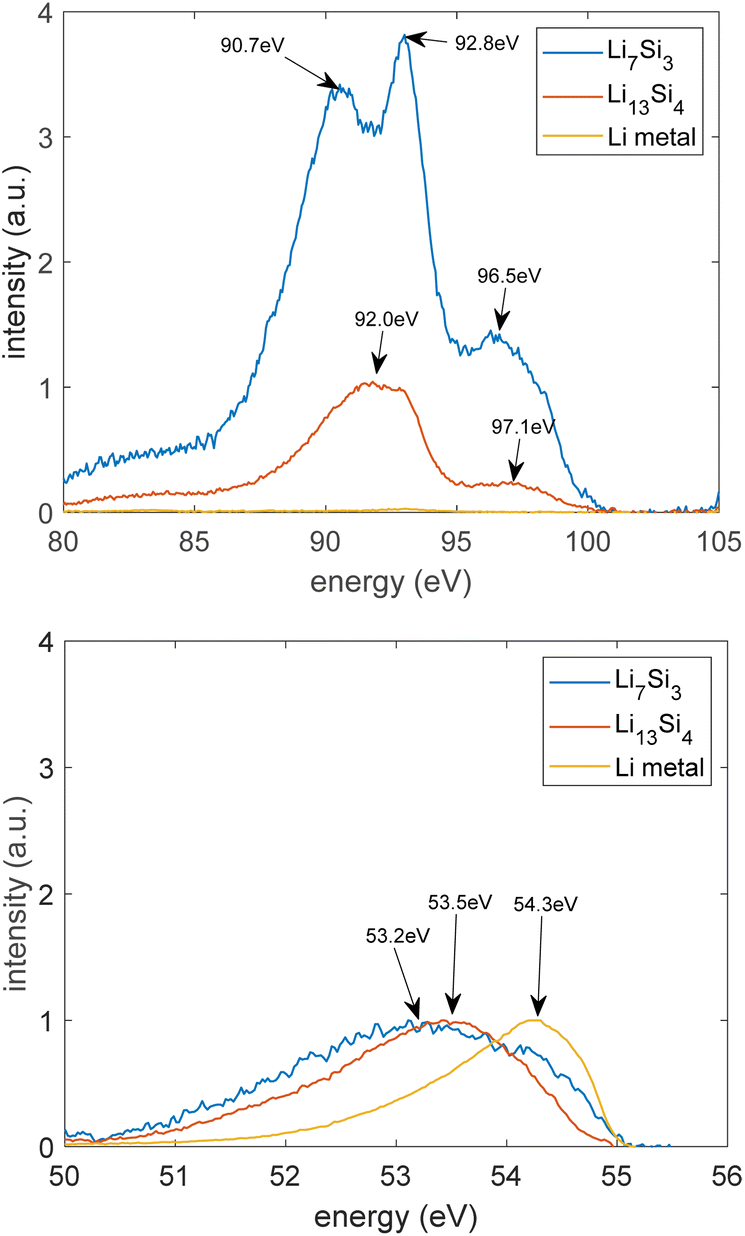 | ||
| Fig. 9 Lithium K-emission and silicon L-emission spectra obtained from lithium–silicon phases. | ||
As the transition of Li13Si4 to Li7Si3 occurs, the emf of the electrode is −0.15 V vs. Li–Al at 415 °C.13 The transition from FeS2 to Li3Fe2S4 has an emf of +1.75 V vs. Li–Al at 415 °C.31 The full cell potential is Ecell = Ecathode − Eanode = +1.75 V − (−0.15 V) = +1.90 V. The positive cell potential indicates that the overall redox reaction is spontaneous.
Bonding environments
Fig. 9 presents the results of the soft X-ray emission spectra where a lithium K-emission (2s to 1s shell) occurs, and a silicon L-emission (3p to 2p shell) occurs. Fig. 9 shows the background subtracted spectra that were normalised by the maximum intensity of the lithium K peak. Lithium has a single asymmetric spectral peak. This is because of the electronic transition from the p states of the valence bands to the 1s state of the atom.32 Crystalline silicon gives three peaks. This is because of the electronic transition from the valence bands to the 2p vacancy of the atom.32The lithium and silicon peak shapes are different due to the different bonding environments in the phases. From Fig. 9, the lithium peak in Li13Si4 and Li7Si3 occurs at lower energies than pure lithium because Si clusters are embedded in Li arrays. Li7Si3 gives three silicon peaks, like crystalline Si, whereas Li13Si4 gives two silicon peaks because Li7Si3 is a more silicon rich phase and contains more Si dimers.
Lin et al. reported that the Li peak shifted to a lower energy from 53.6 to 53.3 eV after lithiation of a single crystal silicon.33 They also reported that after lithiation of silicon, two Si peaks disappear and one Si peak shifts to a higher energy from 91.5 to 91.7 eV.33 These changes indicate a transition from amorphous Li13Si4 to crystalline Li15Si4.33 The energies shown in Fig. 9 are within the range of energies reported by Lin et al.33 These results suggest the similarity of Li13Si4 and Li7Si3 to crystalline Li15Si4 which forms during lithiation of the silicon electrode at room temperature.
Unit cell size on heating
The neutron diffraction patterns of Li13Si4 and Li7Si3 are shown in Fig. 10. Fig. 10 shows neither phase becomes amorphous as the temperature increases to 500 °C. The phases are stable at high temperature compared to the lithium electrode which melts at 180.5 °C and the lithiated silicon electrode that becomes electrochemically amorphous at room temperature.4 Lithium and silicon bond well with each other because the phases do not lose crystallinity at high temperature. The complex structured background in the diffraction patterns arises from scattering by the amorphous silica glass tube of the rig. The background was modelled by Chebyshev polynomials with ten coefficients. The structure models of Li13Si4 and Li7Si3 were refined to obtain the best fit to the data collected in the low angle detector bank (number 3) over the TOF region of 2500 to 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ms (0.75 to 4.45 Å). The isotropic atomic displacement parameters (Uiso), unit cell parameters, background parameters, instrument parameters and sample parameters were refined. From Rietveld refinement of the structure models, the unit cell parameters were obtained for Li13Si4 and Li7Si3.
000 ms (0.75 to 4.45 Å). The isotropic atomic displacement parameters (Uiso), unit cell parameters, background parameters, instrument parameters and sample parameters were refined. From Rietveld refinement of the structure models, the unit cell parameters were obtained for Li13Si4 and Li7Si3.
 | ||
| Fig. 10 Neutron diffraction data recorded between room temperature and 500 °C. The x-axis shows time-of-flight (TOF). | ||
As a starting point for Rietveld refinement, atomic coordinates and unit cell parameters were taken from Zeilinger et al.19 for Li13Si4 and von Schnering et al.21 for Li7Si3. The initial structure refinements were carried out using multiple data sets collected at 500 °C summed into single diffraction patterns to improve counting statistics on the data points and give a more stable refinement. After convergence, the resulting structures were used as starting models for sequential refinements of the structures at lower temperatures (450 °C to room temperature), which were single run data sets. Sequential refinement involves refining the background, hydrostatic strain parameters and isotropic atomic displacement parameters. Fig. 11 and 12 show the unit cell parameters as a function of temperature for Li13Si4 and Li7Si3, respectively.
 | ||
| Fig. 11 Unit cell parameters a, b, c and cell volume, V, derived from the neutron diffraction data as Li13Si4 was heated from room temperature to 500 °C. The error bars are shown on the measured (blue) points. | ||
 | ||
| Fig. 12 Unit cell parameter a, b, c and cell volume, V, derived from the neutron diffraction data as Li7Si3 was heated from room temperature to 500 °C. The error bars are shown on the measured (blue) points. | ||
The neutron data indicates that there are no phase changes between room temperature and 500 °C. Both phases did not become amorphous at 500 °C. The unit cells for both phases expand as the temperature increases.
The lattice parameters for Li13Si4 in the literature by Zeilinger et al.19 are smaller (a = 7.9488 Å, b = 15.1248 Å, c = 4.4661 Å and V = 536.93 Å3) than that observed in this work once the extrapolated lattice parameters were calculated at 100 K/−173.15 °C (a = 7.9922 Å, b = 15.2433 Å, c = 4.4763 Å and V = 545.09 Å3). The structural model of Li13Si4 reported by Zeilinger et al. was from a single crystal X-ray diffraction study at 100 K/−173.15 °C.19 On the other hand, the lattice parameters for Li7Si3 by von Schnering et al.21 are larger (a = 4.435 Å, c = 18.134 Å and V = 308.9 Å3) than that observed in this work, although the lattice parameter a at room temperature matches very well with that measured by von Schnering et al. Once again, the structural model of Li7Si3 was from an X-ray diffraction study at room temperature which explains the differences observed after Rietveld refinement. The differences in unit cell parameters between this work and that of literature is likely because of instrumental differences or disordering caused by the unit cells expanding.
The expansion of the unit cells is significant. Li13Si4 has 1.8% expansion in a, 0.95% expansion in b, and 1.9% expansion in c. The unit cell volume expands by 4.7%. Li7Si3 has 1.8% expansion in a, and 1.4% expansion in c. The unit cell volume for Li7Si3 expands by 5.1%.
Thermal expansion
The variation with temperature of the lattice parameters is linear for Li13Si4 and Li7Si3. Therefore, constant values of the thermal expansion coefficients of a, b and c were determined. Linear thermal expansion coefficient is calculated using34α = (1/L0) × (dL/dT). L0 is the initial value of a, b, or c in Å and dL = dT is the rate of change in a, b, or c with respect to the change in temperature (the gradient of the straight line). The unit of thermal expansion coefficient is °C−1 or K−1. Table 5 presents the thermal expansion coefficients of Li13Si4 and Li7Si3.| Phase | α a 10−6 (K−1) | α b 10−6 (K−1) | α c 10−6 (K−1) |
|---|---|---|---|
| Li13Si4 | 37.9 | 19.9 | 39.9 |
| Li7Si3 | 37.2 | 37.2 | 29.8 |
The linear thermal expansion coefficient of silicon at room temperature is reported to be approximately 2.6 × 10−6 °C−1.35 At 500 °C, it is reported to be approximately 4 × 10−6 °C−1.35 The thermal expansion of Li13Si4 and Li7Si3 is significantly larger than that of silicon. The volumetric thermal expansion coefficient of silicon at 500 °C, assuming isotropic expansion,36 is αV = 3(4 × 10−6 °C−1) = 12 × 10−6 °C−1. The volumetric expansion of Li13Si4 is αV =(1/V0) (ΔV/ΔT) = 0.0552/555.912 = 99.3 × 10−6 °C−1. The volumetric expansion of Li7Si3 is αV = 0.0327/307.786 = 106 × 10−6 °C−1. The volumetric expansion of both phases is significant compared to that of silicon. The volumetric thermal expansion coefficient of lithium is approximately 47 × 10−6 °C−1 at room temperature.37 This means Li13Si4 and Li7Si3 are mechanically closer to lithium metal than silicon. Lithium metal is highly ductile, and ductility can mitigate fracturing. Basu et al. reported the stress–strain curves of an amorphous silicon electrode at room temperature using Grand Canonical Monte Carlo and molecular dynamic simulations.38 The authors reported that LixSi with x = 2.5 to 3.5, which are similar compositions to Li7Si3 and Li13Si4, is ductile during lithiation.38
Li13Si4 and Li7Si3 can be considered as Li arrays with Si clusters embedded (Si5 rings, Si4 stars and Si2 dimers). Li13Si4 and Li7Si3 are both Zintl-like phases with Sinm− polyanions called Zintl ions. Zintl phases have delocalised bonding in clusters.39 All Zintl phases show remarkable stability because of closed shells.39 There is covalent interactions in the anionic part (Si clusters) of the compound and ionic interaction between the cation and anion species (Si clusters and Li array).39 Planar Si5 rings, Si4 stars and Si2 dimers stabilise the structure more than isolated Si atoms found in Li15Si4. This is summarised in Fig. 13.
 | ||
| Fig. 13 The lithiation of silicon, adapted from the work of Limthongkul et al.4 Planar Si5, Si4 stars and Si2 dimers stabilise the structure more than isolated Si atoms found in Li15Si4. | ||
The findings of this work are also connected to the discharge mechanisms in thermal batteries. It is recommended to carry out simultaneous electrochemical and neutron diffraction measurements of thermal battery cells with Li13Si4 and Li7Si3 as the anode active materials. The Polaris diffractometer together with the St Andrews conductivity rig can provide simultaneous data collection from neutron diffraction and electrochemical potentiostatic discharge at 500 °C. This may lead to new discharge mechanisms at the anode side which will advance the understanding of processes occurring inside a thermal battery. Previous work by the St Andrews research group studied NiS2 and CoS2 cathodes and new discharge mechanisms were proposed.40,41
Conclusions
Li13Si4 and Li7Si3 can be made from solid state synthesis. The galvanostatic cell discharge showed a flat voltage plateau indicating that Li13Si4 and Li7Si3 are in equilibrium and both phases are line phases. The powder neutron diffraction study of Li13Si4 and Li7Si3 was performed for the first time. Li13Si4 and Li7Si3 neutron diffraction data confirmed that within the temperature range of 20 °C and 500 °C, there were no phase changes and no amorphous phase formed. The volumetric thermal expansion of Li13Si4 and Li7Si3 is significant and closer to lithium metal than to silicon which suggests the LixSi electrode is mechanically closer to lithium metal and can be considered as having silicon clusters within a lithium array rather than the silicon lattice hosting lithium.Data availability
Data for this article, including X-ray diffraction, cell discharge, and scanning electron microscopy are available at the University of St Andrews repository at https://doi.org/10.17630/e44eae73-ca6e-4562-a9f6-0e06865174c1. Data files collected at the ISIS Neutron and Muon Source may be obtained from https://doi.org/10.5286/ISIS.E.RB1910457-1.Author contributions
Atia Azad: conceptualisation, data curation, formal analysis, investigation, methodology, validation, visualisation, writing – original draft. Katherine Bateman: investigation, data curation, formal analysis, validation, visualisation. Matthew Irvine: investigation, methodology. Aaron B. Naden: resources, visualisation. Stewart A. M. Dickson: conceptualisation, investigation, methodology. Ronald I. Smith: data curation, resources, writing – review & editing. Richard K. B. Gover: conceptualisation, funding acquisition, supervision. John T. S. Irvine: conceptualisation, funding acquisition, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by AWE. Neutron beamtime at the ISIS Neutron and Muon Source (RB 1910457) source was provided by the UK Science and Technology Facilities Council (STFC). The authors would like to thank the EPSRC Light Element Analysis Facility Grant EP/T019298/1 and the EPSRC Strategic Equipment Resource Grant EP/R023751/1 for funding the scanning electron microscopy facilities at the University of St Andrews.Notes and references
- J. E. Cloud, Y. Wang, X. Li, T. S. Yoder, Y. Yang and Y. Yang, Inorg. Chem., 2014, 53, 11289–11297 CrossRef CAS PubMed.
- A. S. Cattaneo, S. Dupke, A. Schmitz, J. P. Badillo, M. Winter, H. Wiggers and H. Eckert, Solid State Ionics, 2013, 249–250, 41–48 CrossRef CAS.
- M. T. McDowell, S. W. Lee, W. D. Nix and Y. Cui, Adv. Mater., 2013, 25, 4966–4985 CrossRef CAS PubMed.
- P. Limthongkul, Y. Il Jang, N. J. Dudney and Y. M. Chiang, Acta Mater., 2003, 51, 1103–1113 CrossRef CAS.
- M. N. Obrovac and L. Christensen, Electrochem. Solid-State Lett., 2004, 7, A93–A96 CrossRef CAS.
- M. Zeilinger, V. Baran, L. Van Wüllen, U. Häussermann and T. F. Fässler, Chem. Mater., 2013, 25, 4113–4121 CrossRef CAS.
- B. Key, R. Bhattacharyya, M. Morcrette, V. Seznéc, J.-M. Tarascon and C. P. Grey, J. Am. Chem. Soc., 2009, 131, 9239–9249 CrossRef CAS PubMed.
- B. Key, M. Morcrette, J.-M. Tarascon and C. P. Grey, J. Am. Chem. Soc., 2010, 133, 503–512 CrossRef PubMed.
- S. Dupke, T. Langer, F. Winter, R. Pöttgen, M. Winter and H. Eckert, Solid State Nucl. Magn. Reson., 2015, 65, 99–106 CrossRef CAS PubMed.
- A. Kuhn, S. Dupke, M. Kunze, S. Puravankara, T. Langer, R. Pöttgen, M. Winter, H. D. Wiemhöfer, H. Eckert and P. Heitjans, J. Phys. Chem. C, 2014, 118, 28350–28360 CrossRef CAS.
- R. A. Guidotti and P. Masset, J. Power Sources, 2006, 161, 1443–1449 CrossRef CAS.
- R. A. Guidotti and P. J. Masset, J. Power Sources, 2008, 183, 388–398 CrossRef CAS.
- C. J. Wen and R. A. Huggins, J. Solid State Chem., 1981, 37, 271–278 CrossRef CAS.
- H. Okamoto, J. Phase Equilib. Diffus., 2009, 30, 118–119 CrossRef CAS.
- C. van der Marel, G. J. B. Vinke and W. van der Lugt, Anim. Reprod. Sci., 1985, 9, 95–98 CrossRef.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2008, 41, 653–658 CrossRef CAS.
- R. Nesper and H. G. Von Schnering, J. Solid State Chem., 1987, 70, 48–57 CrossRef CAS.
- J. Y. Kwon, J. H. Ryu and S. M. Oh, Electrochim. Acta, 2010, 55, 8051–8055 CrossRef CAS.
- M. Zeilinger and T. F. Fässler, Acta Crystallogr., Sect. E:Struct. Rep. Online, 2013, 69, i81–i82 CrossRef CAS PubMed.
- M. Zeilinger, I. M. Kurylyshyn, U. Ha and T. F. Fa, Chem. Mater., 2013, 4623–4632 CrossRef CAS.
- H. G. Von Schnering, R. Nesper, K.-F. Tebbe and J. Curda, Chem. Informationsdienst, 1980, 11, 1–8 Search PubMed.
- H. G. von Schnering, R. Nesper, J. Curda and K.-F. Tebbe, Angew. Chem., Int. Ed., 1980, 19, 1033–1034 CrossRef.
- J. L. Payne, K. Giagloglou, G. Carins, C. J. Crouch, J. D. Percival, R. I. Smith, R. Gover and J. T. Irvine, Front. Energy Res., 2018, 6, 1–15 CrossRef.
- S. Iwamura, H. Nishihara, Y. Ono, H. Morito, H. Yamane, H. Nara, T. Osaka and T. Kyotani, Sci. Rep., 2015, 5, 1–8 Search PubMed.
- S. Dupke, T. Langer, R. Pöttgen, M. Winter, S. Passerini and H. Eckert, Phys. Chem. Chem. Phys., 2012, 14, 6496–6508 RSC.
- B. Han, M. J. Piernas-Muñoz, F. Dogan, J. Kubal, S. E. Trask, I. D. Bloom, J. T. Vaughey and B. Key, J. Electrochem. Soc., 2019, 166, A2396–A2402 CrossRef.
- Y. Yamamoto, T. Murano, H. Onodera, N. Erdman, R. Matsuda and A. Matsuda, Microsc. Microanal., 2020, 26, 68–70 CrossRef.
- G. J. Irvine, F. Demmel, H. Y. Playford, G. Carins, M. O. Jones and J. T. S. Irvine, Chem. Mater., 2022, 34, 9934–9944 CrossRef CAS.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- Z. Tomczuk and D. R. Vissers, J. Electrochem. Soc., 1986, 133, 2505–2509 CrossRef CAS.
- L. Andrey, J. Electrochem. Soc., 2019, 166, A5362–A5368 CrossRef.
- H. Lin, K. Uosaki and H. Noguchi, Appl. Surf. Sci., 2021, 569, 1–7 CrossRef.
- R. K. B. Gover, N. D. Withers, S. Allen, R. L. Withers and J. S. O. Evans, J. Solid State Chem., 2002, 166, 42–48 CrossRef CAS.
- M. Okaji, Int. J. Thermophys., 1988, 9, 1101–1109 CrossRef CAS.
- W. Callister and D. Rethwisch, in Materials Science and Engineering: an Introduction, Hachette Livre – Département Pratique, 10th edn, 2018, pp. 703–704 Search PubMed.
- E. Mantysalo, Phys. Lett., 1965, 16, 17–18 CrossRef.
- S. Basu, N. Koratkar and Y. Shi, Acta Mater., 2019, 175, 11–20 CrossRef CAS.
- A. V. Mudring and J. D. Corbett, J. Am. Chem. Soc., 2004, 126, 5277–5281 CrossRef CAS PubMed.
- J. L. Payne, J. D. Percival, K. Giagloglou, C. J. Crouch, G. M. Carins, R. I. Smith, R. Comrie, R. K. B. Gover and J. T. S. Irvine, ChemElectroChem, 2017, 4, 1916–1923 CrossRef CAS.
- J. L. Payne, J. D. Percival, K. Giagloglou, C. J. Crouch, G. M. Carins, R. I. Smith, R. K. B. Gover and J. T. S. Irvine, J. Electrochem. Soc., 2019, 166, A2660–A2664 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |