
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polynorbornene copolymers combining flexible ether side chains and rigid hydrophobic segments for AEMWE†
Linus
Hager
 *ab,
Timo
Maron
ab,
Trung Ngo
Thanh
c,
Julian
Stonawski
ab,
Andreas
Hutzler
a,
Thomas
Böhm
a,
Peter
Strasser
c,
Simon
Thiele
ab and
Jochen
Kerres
*ad
*ab,
Timo
Maron
ab,
Trung Ngo
Thanh
c,
Julian
Stonawski
ab,
Andreas
Hutzler
a,
Thomas
Böhm
a,
Peter
Strasser
c,
Simon
Thiele
ab and
Jochen
Kerres
*ad
aForschungszentrum Jülich GmbH, Helmholtz Institute Erlangen-Nürnberg for Renewable Energy (IEK-11), Cauerstr. 1, Erlangen, 91058, Germany. E-mail: l.hager@fz-juelich.de; j.kerres@fz-juelich.de
bDepartment of Chemical and Biological Engineering, Friedrich Alexander Universität Erlangen-Nürnberg, Egerlandstr. 3, Erlangen, 91058, Germany
cDepartment of Chemistry, Chemical Engineering Division, Technical University of Berlin, Straße des 17. Juni, Berlin, 19623, Germany
dChemical Resource Beneficiation Faculty of Natural Sciences, North-West University, Potchefstroom, 2520, South Africa
First published on 17th February 2025
Abstract
In this study, we report the synthesis of copolymers incorporating an ether-sidechain containing (EM) and a sterically hindered hydrophobic, anthracene-derived norbornene monomer (AM). Using this copolymer system, we developed both block and statistical copolymers, with the latter demonstrating excellent potential for the fabrication of robust anion exchange membranes (AEMs) when blended with poly(oxindolebiphenylene) (POB). Stable membranes with controllable swelling and water uptake properties were obtained. The composition of the blend was systematically varied to evaluate the effects of different POB ratios on membrane conductivity, stability, and performance in anion exchange membrane water electrolysis (AEMWE). The blend membrane comprising 10 wt% POB and 90 wt% of the statistical copolymer EM0.54-q-co-AM0.46-h demonstrated excellent AEMWE performance, delivering a current density of 2.0 A cm−2 at 1.91 V without the use of platinum group metal (PGM) catalysts, and under a dry cathode configuration.
Introduction
The most significant disadvantage of renewable energy sources is their intermittency due to time and location dependency. Consequently, energy storage solutions are required.1 Hydrogen, as a multi-purpose energy carrier independent of its location, offers a promising approach to solving the problem of fluctuating energy supply by renewable energy sources.2 Water electrolysis utilizing electric power from non-fossil energy sources is necessary to store energy in hydrogen. Besides the two mature technologies, alkaline water electrolysis (AWE) and proton exchange membrane water electrolysis (PEMWE), anion exchange membrane water electrolysis (AEMWE) is the newest technology. Here, the zero-gap architecture of PEMWE, resulting in high current densities, is combined with the advantages of AWE, such as noble metal-free electrocatalysts and materials for bipolar plates.3–7 In many AEMWE studies, the electrolyte is supplied to both the anode and the cathode to enhance the cell performance. However, supplying the electrolyte only to the anode offers the advantage of producing highly pure and dry hydrogen gas. This is also the preferred operation mode for electrolyzers on an industrial scale. Notably, dry cathode mode AEMWE is also beneficial when impure water feeds (e.g., seawater) are utilized, as demonstrated by Frisch et al.8 For operating an AEMWE electrolyzer under dry cathode mode, improved water management, especially water transport from anode to cathode, where the H2O splitting reaction occurs, is necessary.8–10 Thus, novel polymers allowing efficient water diffusion through the membrane need to be developed. Here, ether-containing side chains may facilitate water uptake and transport of hydroxide ions and water through the membrane from the anode to the cathode to ensure sufficient water supply for the water-splitting reaction at the cathode. Ether-containing moieties were already introduced into AEMs to construct nano-assembled structures, creating ion-conducting pathways by increasing local hydrophilicity and hydrogen-bond interactions.11–14 Thus, within this study, we also introduced ether chains into our polymers to exploit these benefits.Quaternized polymers are essential in electrochemical energy applications like AEMWE since these polymers are used to manufacture hydroxide conducting membranes and ionomers in the catalyst layers.3,15 However, this membrane type performs worse than proton-exchange membranes in electrochemical applications such as water electrolysis and fuel cells due to lower ionic conductivities and decreased chemical stability of the quaternary ammonium groups in highly alkaline media.16 Furthermore, aryl-ether bridges in the polymer backbone (e.g., poly(phenylene oxide)-based membranes) could lead to backbone degradation by hydroxide attacks.17 Therefore, polynorbornenes represent an interesting material class for tailored membranes for alkaline energy applications like AEMWE.18 First, the synthesis of polynorbornenes via ring-opening metathesis polymerization (ROMP) is fast, controllable, resource-efficient, and accepts a wide range of functional groups, including cationic groups necessary as conducting moiety in AEMWE.19,20 Second, the polymer backbone is heteroatom-free, increasing chemical stability. Third, monomers could easily be synthesized via Diels–Alder reactions. Diels–Alder reactions are very economical since no by-products are produced, and the polymer and membrane properties can be adjusted according to the desired properties and the target application.21–24 Polynorbornenes have been investigated as anion exchange membranes (AEMs), primarily for applications in alkaline fuel cells. In 2024, Chen et al. demonstrated using hydrogenated block copolymers crosslinked with triimidazolium groups in AEM fuel cells (AEMFCs).25 Additionally, clustered polynorbornene-based AEMs were synthesized using a tri-functional crosslinking reagent, exhibiting promising performance in AEMFCs.26 Sun et al. explored the impact of the polynorbornene backbone—comparing vinyl-addition polymerization with ring-opening metathesis polymerization (ROMP)—on the alkaline stability and AEMFC performance.27 Meanwhile, Li et al. investigated the influence of various quaternary ammonium groups on polynorbornene-based AEM performance using a backbone structure similar to that of Chen et al.25,28 Their findings identified N-methylpyrrolidinium and N-methylpiperidinium groups as the most effective among the studied quaternary ammonium functionalities regarding AEMFC performance.28 Finally, vinyl-addition polymerized polynorbornene-copolymers have been utilized by Lehmann et al.29 as membranes for AEMFC and by Leonard et al.30 as ionomers for AEMFC and AEMWE applications. When used as an ionomer, it was found that polynorbornenes are superior to ionomers comprising aromatic moieties due to their lower adsorption on the catalyst surface.30 The group of Noonan also contributed to developing vinyl-addition polymerized polynorbornenes for AEMFC application. For statistical polynorbornene copolymers functionalized with trimethylammonium and phosphonium cations, an improved AEMFC performance was found for the trimethylammonium-comprising membranes.31 A study comparing pentablock polynorbornene copolymers wtih trimethylammonium and phosphonium cations found lower water uptake and higher hydroxide conductivity for the pentablock utilizing the trimethylammonium cations.32 The group of Kohl has contributed extensively to this field, reporting on trimethylammonium-functionalized polynorbornenes prepared via vinyl-addition polymerization and ROMP for AEMFC applications while emphasizing the preparation of block copolymers.33–35 More recently, the group of Kohl applied vinyl-addition polymerized polynorbornene-based AEMs in anion exchange membrane water electrolysis (AEMWE) under dry-cathode conditions. They focused on the effects of KOH concentrations and the addition of inert electrolytes on AEMWE performance rather than on membrane optimization.36–38 Overall, the application of polynorbornene-based AEMs in water electrolysis remains underreported in the literature, highlighting the relevance of the present study.
Due to the superior conductivity reached by ether-based polynorbornene AEMs reported by Price et al., we adapted their synthesis. Still, we simplified it significantly by attaching the bromine directly to the ethylene glycol spacer instead of two additional steps to increase the spacer length further.39 However, in the work of Price et al., the excessive swelling caused by the ether linkages limited the applicability of the ether-containing polynorbornenes.39 Consequently, an anthracene-containing norbornene comonomer, selected to limit water uptake, was introduced in our study. This effect is attributed to the hydrophobic nature of the incorporated phenyl units, which introduce steric hindrance and reduce the overall hydrophilicity of the polymer matrix.23,40,41 Thus, it was aimed to combine the best of both monomers: the high conductivity of ether-based monomers and the decreased swelling caused by the anthracene-based comonomer.39,40,42 To emphasize the specific contributions of the ether-containing monomer, we compared it to an analogous polymer incorporating alkyl chains in place of ether chains. Moreover, a novel reinforcement strategy is introduced. In the literature, poly(oxindolebiphenylene) has already been demonstrated to be an extraordinary alkaline-stable polymer.14,43–45 Blending the synthesized copolymers with the highly alkaline stable poly(oxindolebiphenylene) yielded robust membranes with excellent conductivities, superior alkaline stability, and mechanical properties.
Moreover, we compared statistical and block-copolymers with the same monomer composition regarding microstructure and membrane properties to evaluate the ideal polymer architecture for potential application in AEMWE. Finally, we demonstrate the excellent performance of the polynorbornene blend membranes in an AEMWE cell with dry cathode mode and with a completely PGM-free cell design, representing the desired industrial operating conditions of electrolyzers.
Results and discussion
Polymer synthesis
The monomers were obtained by Diels–Alder reactions (Fig. S1†) with slightly adapted procedures from the literature.39,40 Fig. S2a† shows the 1H NMR spectrum of the EM with the signal assignment to the protons of the target compound with 13C, 1H–13C HSQC, and 1H–1H COSY NMR experiments (Fig. S3–S5†). The 1H-NMR spectrum of the AM is shown in Fig. S2b,† whereby the spectrum is identical to the literature.23 No signals of anthracene or other by-products or reactants are detected, indicating sufficient monomer purity. Additionally, we synthesized a norbornene monomer comprising a butyl side chain (BM) with a bromine group, without ether moieties, for comparison to demonstrate the advantages of incorporating ether side chains (Fig. S1d,† BM). The synthesis was conducted according to a previously reported procedure,21 and the 1H NMR spectrum (Fig. S6†) was on par with the literature.21For polymer synthesis, we have chosen ROMP due to the control over the molecular weight, the well-established catalyst systems, the living character of the polymerization enabling the synthesis of defined copolymers or block-copolymers, and the superior properties of AEMs based on ROMP polymers, which have already been reported in the literature.33,40–42,46–49
In the present study, we focused on investigating the influence of microstructure on polymer and membrane properties while maintaining a fixed ratio of the charged ether-based norbornene monomer (EM) to the hydrophobic anthracene-derived monomer (AM) of 54![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :46, corresponding to a theoretical ion exchange capacity (IEC) of 2.00 mmol g−1. Fig. 1a schematically describes the statistical copolymerization of both monomers utilizing a 3rd generation Grubbs catalyst. Moreover, block copolymers were prepared by sequential polymerization of both monomers to investigate the influence of nanostructure and phase separation on the membrane properties. The same polymerization procedure was applied for the ether-free monomer (BM, Fig. S8†) to obtain the analogous BM-co-AM with the same IEC as the EM-based copolymer.
:46, corresponding to a theoretical ion exchange capacity (IEC) of 2.00 mmol g−1. Fig. 1a schematically describes the statistical copolymerization of both monomers utilizing a 3rd generation Grubbs catalyst. Moreover, block copolymers were prepared by sequential polymerization of both monomers to investigate the influence of nanostructure and phase separation on the membrane properties. The same polymerization procedure was applied for the ether-free monomer (BM, Fig. S8†) to obtain the analogous BM-co-AM with the same IEC as the EM-based copolymer.
 | ||
| Fig. 1 (a) Synthesis of the precursor polymer (EM-co-AM) via ROMP utilizing the 3rd generation Grubbs catalyst with a 54:46 molar ratio of EM/AM in the feed (IECtheo = 2.00 mmol g−1). (b) Hydrogenation of the double bonds with p-toluenesulfonyl hydrazide. (c) Quaternization with trimethylamine. | ||
The unsaturated precursor polymer (EM-co-AM) was hydrogenated (Fig. 1b) to ensure the chemical stability of the backbone. For the unsaturated polymers, significant degradation was observed when immersed in 1 M KOH at evaluated temperature, consistent with recent literature regarding the stability of unsaturated polymers applied as anion exchange membranes.27 After obtaining the saturated EM-co-AM-h, the bromine groups were quaternized in a Menschutkin reaction with trimethylamine to obtain the positively charged EM-q-co-AM-h (Fig. 1c).
Via
1H NMR spectroscopy, the build-in-ratio of the EM and the AM was calculated utilizing the resonance signal of the double bond protons (EM-co-AM signal b, Fig. 2a) and the signal corresponding to the anthracene unit (EM-co-AM signal a, Fig. 2a). Consequently, 54 mol% of the EM was incorporated in the copolymer, matching the targeted value of 54 mol%, corresponding to a final IEC of 2.00 mmol g−1 and highlighting the excellent controllability of this type of polymerization. A lower build-in ratio was targeted for the ether-free BM to achieve an IEC of 2.00 mmol g−1. Via1H NMR spectroscopy, a build-in ratio of 45 mol% was calculated for BM0.45-co-AM0.55 (Fig. S9†). Fig. 3a depicts the GPC curve for a 54:46 statistical copolymer EM0.54-co-AM0.46 before the hydrogenation step with Pn(theo.) = [M]/[Kat.] = 183 corresponding to a molecular weight of 50000 g mol−1 matching the measured Mn of 49000 g mol−1. A dispersity below 1.10 showcases the well-controlled copolymerization of the EM and AM via ROMP. Notably, the GPC analysis of the ether-free BM0.45-co-AM0.55 (Fig. S14†) revealed a broad molecular weight distribution with a dispersity of 1.79, indicating no ideal polymerizability of the BM via ROMP compared to the EM/AM monomer combination which is consistent with recent literature on the BM/AM system.27,50 Nevertheless, the measured molecular weight of 49000 g mol−1 matches the expected molecular weight of 50000 g mol−1.
 | ||
| Fig. 2 (a) 1H-NMR spectrum of the unsaturated EM0.54-co-AM0.46 measured in CDCl3 (b) 1H-NMR spectrum after the hydrogenation reaction (EM0.54-co-AM0.46-h) with p-toluenesulfonyl hydrazide showcasing significant peak sharping due to the loss of cis/trans isomerism measured in CDCl3 (c) 1H-NMR spectrum of the quaternized EM0.54-q-co-AM0.46-h measured in DMSO-d6. | ||
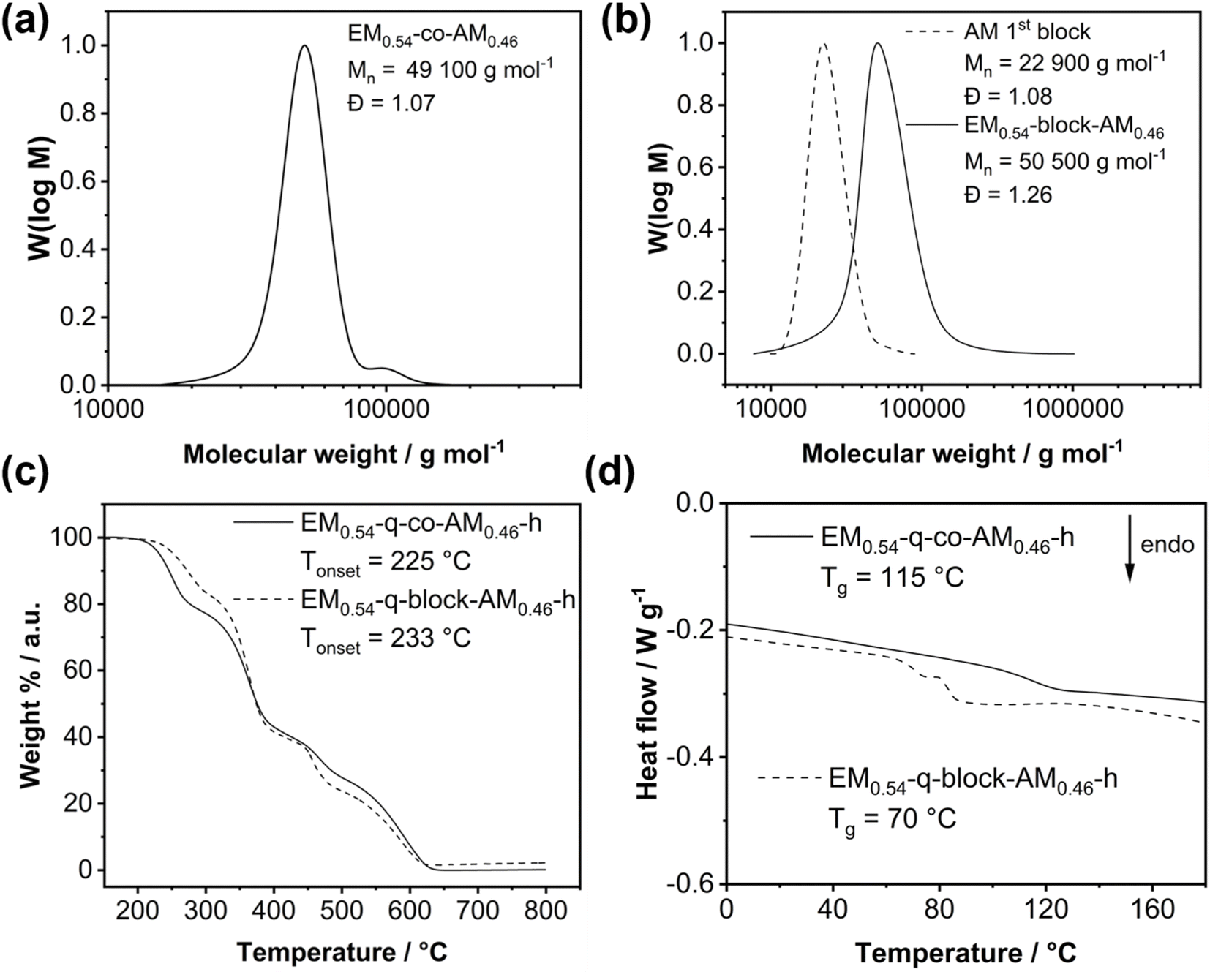 | ||
| Fig. 3 (a) GPC curve of a statistical copolymer with a 54:46 ratio of EM:AM in the feed. (b) GPC curves of the first block (AM, dashed) compared to the GPC curve after addition of the second monomer (EM). (c) TGA measurements under synthetic air atmosphere of the statistical copolymer compared to the block copolymer after hydrogenation and quaternization with a heating rate of 20 K min−1. (d) DSC analysis (2nd heating curve, 10 K min−1 heating rate) of the statistical and block copolymer after hydrogenation and quaternization. | ||
After the hydrogenation step, the double bond signal vanishes, verifying the complete conversion of the unsaturated ROMP polymer to the saturated EM-co-AM-h (Fig. 2b). Notably, a significant peak sharping occurred after the hydrogenation step, probably due to the absence of cis/trans isomerism in the saturated copolymer. Moreover, we checked for changes in the polymer's molecular weight after hydrogenation (Fig. S12†) via GPC and found slight changes in molecular weight and increased dispersity. We speculate that the change in the GPC curve is due to a different hydrodynamic volume of the polymer chains since the methylene spacer in the saturated polynorbornene enables more rotational freedom and, consequently, different chain entanglement motifs. Furthermore, different interactions with the solvent due to the absence of the double bond can also influence the GPC measurement. Notably, sufficiently high molecular weight is necessary for membrane formation. Since only a slight molecular weight decrease was measured after the double bond hydrogenation, this reaction step was proven applicable for the synthesis route toward ether-containing anion exchange membranes. Furthermore, the 1H-NMR spectra shown in Fig. 2 indicate no other changes in the polymer composition after hydrogenation. Notably, the same hydrogenation procedure was applicable for the ether-free polymer, and complete conversion to the saturated polymer (BM0.45-co-AM0.55-h) was proven (Fig. S9†).
In the quaternized copolymer, a new signal at 3.1 ppm corresponding to the protons of the TMA group arises (EM-q-co-AM-h, signal d, Fig. 2c), which proves conversion to the trimethylammonium substituted copolymer. Identical reaction conditions were applied for BM0.45-co-AM0.55-h to yield the TMA-functionalized BM0.45-q-co-AM0.55-h (Fig. S8†).
In the present study, we applied a yet uninvestigated monomer combination to compare a statistical copolymer with a block-copolymer with the same ratio of both monomers in the copolymer (54 mol% EM). Thus, we also checked the polymerization time until the final molecular weight and complete monomer conversion were reached to ensure smooth block copolymerization (Fig. S17–S19†). In both cases, complete conversion is achieved after 5 min polymerization time. Thus, it was proven that both monomers could be polymerized independently and controlled, a prerequisite for preparing block copolymers.
The block copolymer was prepared by sequential polymerization of both monomers, whereby the AM was used to prepare the first block. The block copolymer's final targeted degree of polymerization was 183, corresponding to 84 repeating units of the AM (22800 g mol−1) and 99 repeating units (27200 g mol−1) for the EM. Fig. 3b highlights that both blocks' theoretically expected molecular weight and measured values perfectly match. A monomodal GPC curve after adding the second monomer proves no parallel polymerization of both monomers, whereby a slight increase in dispersity was observed. This may likely be due to phase separation effects between the more polar EM and the highly hydrophobic AM, resulting in a changed hydrodynamic behavior.
Before applying the novel copolymers as anion exchange membranes, we investigated the thermal properties of the quaternized polymers. Most importantly, the quaternized polymers show comparable thermal stability (Fig. 3c) until 225 °C and 233 °C, respectively, enabling their use in low-temperature electrochemical energy applications such as anion exchange membrane water electrolysis (AEMWE). Overall, the TGA curves show a similar shape with slight differences in the onset points for the respective degradation steps, probably originating from the different connection patterns in the statistical copolymer, where EM–AM bonds appear statistically. In contrast, only one EM–AM bond exists under ideal polymerization kinetics in the block copolymer. Consequently, this would also affect the thermal stability of the respective bonds and, hence, the thermal degradation behavior of the polymer. For the BM0.45-q-co-AM0.55-h, we found a slightly higher thermal stability, whereby this polymer was stable until 276 °C (Fig. S15†).
Notably, glass transitions for the statistical and the block-copolymer were revealed utilizing differential scanning calorimetry (Fig. 3d). Due to the softening effect of the ether bonds, a comparatively low glass transition of 115 °C for the statistical copolymer was obtained. Typically, the glass transition is significantly higher (around 200 °C) for cationic polymers8 or sometimes above the decomposition temperature due to the charges limiting chain mobility. The glass transition temperature of the EM0.54-q-co-AM0.46-h could be beneficial for preparing membrane electrode assemblies (MEA) for AEMWE via the decal method.51 The softening effect of the ether-chains becomes evident by comparing the Tg of EM0.54-q-co-AM0.46-h (115 °C) with the Tg of BM0.45-q-co-AM0.55-h (181 °C, Fig. S16†).
Two separate glass transition temperatures for both blocks are expected for block copolymers.52
However, in the present case, only one glass transition at 70 °C was obtained (Fig. 3d, dashed curve), and no second glass transition for the anthracene block could be measured within the thermal stability window of the block copolymer. The glass transition for the anthracene block is expected to be above 200 °C,23 where the TGA already revealed polymer decomposition, explaining why only one glass transition was measured in the DSC measurement until 200 °C. This observation was substantiated by performing dynamic mechanical analysis (DMA). The DMA measurement for EM0.54-q-block-AM0.46-h is presented in Fig. 4. The glass transition temperature of the block copolymer is distinctly observed at 73 °C, indicated by a peak in tanδ and a reduction in the storage modulus by an order of magnitude (Fig. 4). No second glass transition temperature was detected, corroborating the DSC analysis and suggesting that the second glass transition temperature may lie above the decomposition temperature.
 | ||
| Fig. 4 Dynamic mechanical analysis (2 K min−1 heating rate, 1 Hz oscillation frequency) of EM0.54-q-block-AM0.46-h revealing only one glass transition temperature. | ||
Blend membrane preparation with poly(oxindolebiphenylene)
After assessing the thermo-physical properties of the copolymers, membranes were prepared. Initial attempts to use pure EM0.54-q-co-AM0.46-h resulted in excessive swelling and gel formation in water. Consequently, no analysis of pure EM0.54-q-co-AM0.46-h membranes was carried out since, during membrane preparation, these films need to be removed from the glass substrates by immersion in water. This was impossible due to excessive swelling of EM0.54-q-co-AM0.46-h. To improve this, we blended the copolymer with a second polymer, following a strategy from our previous work that had shown improved membrane stability and performance.53,54Poly(oxindolebiphenylene) (POB) was chosen due to its reported stability in an alkaline environment (Fig. S11†),14,45,55 the possibility of preparing high molecular weight polymers, and its excellent film-forming properties.
The optimum blend composition was determined by analyzing different ratios of EM0.54-q-co-AM0.46-h/POB. Specifically, blends containing 5 wt%, 10 wt%, 15 wt%, 20 wt%, and 30 wt% POB were prepared to find the best balance between high ionic conductivity, adequate water uptake, and sufficient mechanical strength. We applied POB with a high molecular weight of 165000 g mol−1 (Fig. S11†) as a matrix polymer to ensure sufficient mechanical support for the norbornene-based anion exchange polymers. The polymer must have superior mechanical properties and balanced hydrophobicity/hydrophilicity when used as a stabilizing blend component. On the one hand, the stabilizing polymer must be miscible with the highly hydrophilic cationic polymer, making a particular polarity necessary. On the other hand, the stabilizing matrix polymer must provide sufficient mechanical strength and limit excess water uptake.
The EM0.54-q-co-AM0.46-h showed excellent miscibility with POB, and homogeneous, transparent blend membranes could be obtained (Fig. S20a†). We assume the attractive interactions between the two polymers result from ionic crosslinking with the deprotonated POB if the membrane is immersed in an alkaline solution or hydrogen bonds if the membrane is not alkaline treated (Fig. 5a). The block copolymer showed a different behavior. Interestingly, it formed stable membranes without adding POB, showing lower water uptake and a lower swelling ratio than the statistical copolymer. The anthracene-containing phase within the hydrophilic–hydrophobic block copolymer limits the dimensional swelling and stabilizes the polymer when immersed in water.
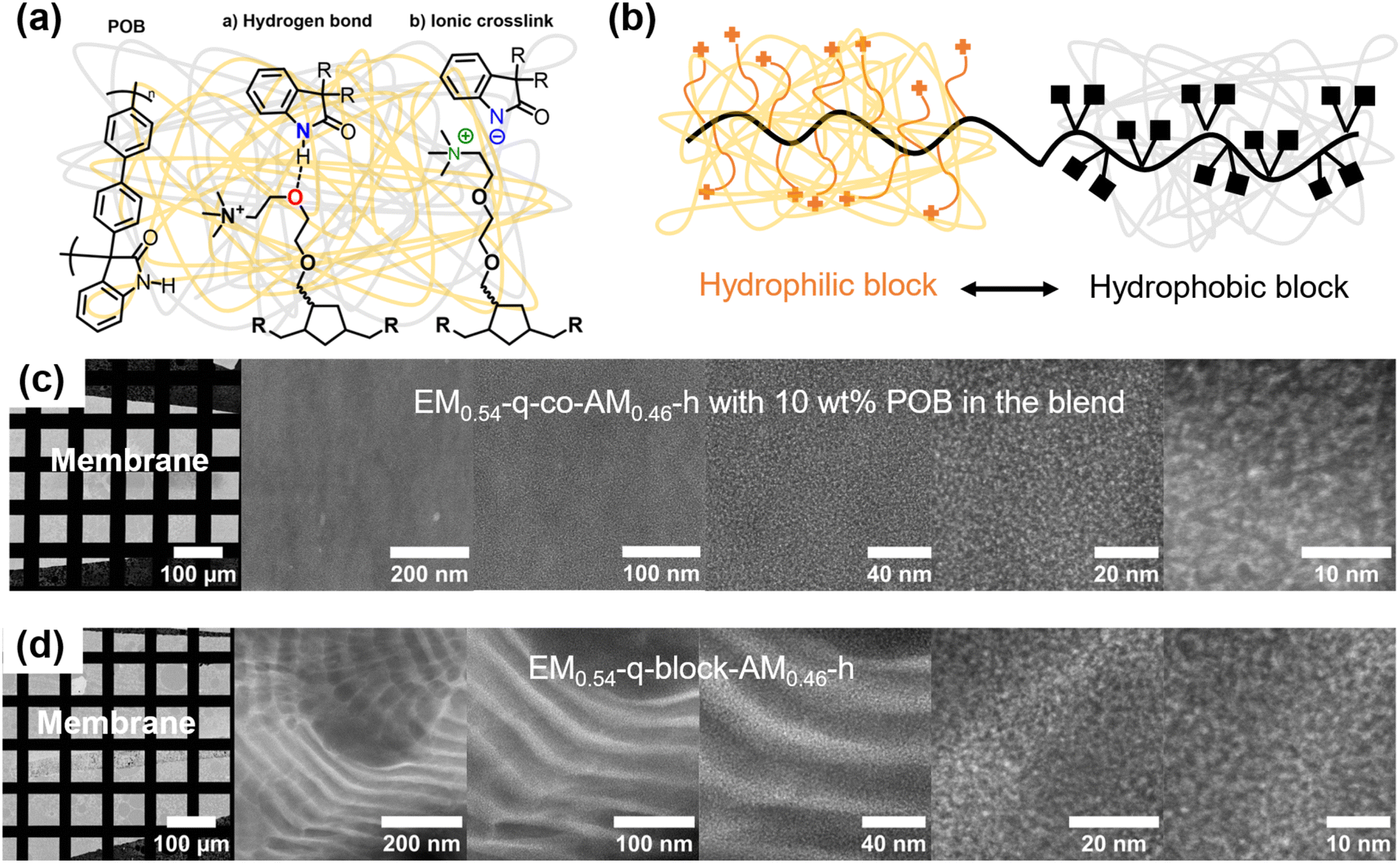 | ||
| Fig. 5 (a) Polymer structure of POB and attractive interactions between EM0.54-q-co-AM0.46-h and POB in neutral and alkaline conditions. (b) Schematical representation of the phase separation in the hydrophilic–hydrophobic EM0.54-q-block-AM0.46-h. (c) HAADF-STEM images of EM0.54-q-co-AM0.46-h blended with 10 wt% POB showing structures in the size of 3.5 nm roughly corresponding to the length of the ether-containing sidechain. (d) HAADF-STEM images of the EM0.54-q-block-AM0.46-h with nano-phase separation on the scale of 20 nm representing lamellar block copolymer morphology. | ||
Cl− or Br− membranes could be treated with hot water without gelation, and mechanically stable membranes without POB reinforcement could be obtained (Fig. 5b). These membranes visually appeared slightly opaque compared to the statistical copolymer (Fig. S20b†). However, when the block copolymer membranes encounter KOH solutions, excessive swelling causes gelation and membrane distortion, making further membrane characterization impossible. Consequently, blends of EM0.54-q-block-AM0.46-h with POB were also attempted to increase the mechanical integrity of the block copolymer membranes. Compared to the statistical copolymer, the block copolymer showed worse miscibility with POB since segregation of the polymers was observed after the addition of the POB solution to an EM0.54-q-block-AM0.46-h solution, and no homogeneous membranes could be obtained. It could be speculated that the decreased miscibility results from the phase separation of the block copolymer since the highly hydrophobic anthracene block does not have attractive interactions with the POB. Moreover, the excessive swelling observed in the diblock copolymer membrane may be attributed to its diblock structure. When exposed to KOH solutions, the hydrophilic block undergoes significant swelling, while the hydrophobic block remains unaffected. This differential swelling likely generates mechanical stress at the interface of the two blocks, potentially affecting the membrane's structural integrity.
The homogeneity of the membranes on a molecular level was investigated with HAADF-STEM, and the results are depicted in Fig. 5c and d. To gain a mass-thickness contrast, the membranes were stained with Na2WO4 to exchange the counter-ions in the membrane with WO42−. More electrons are scattered towards higher angles at the WO42− predominantly accumulating at the positively charged TMA groups. Consequently, in the HAAD-STEM images shown in Fig. 5c and d, the bright areas correspond to the WO42− rich region predominantly consisting of the charged polymer or blend part, respectively. One homogeneous phase can be observed for a representative blend comprising 90 wt% EM0.54-q-co-AM0.46-h and 10 wt% POB. The phase separation of the positively charged TMA group from the polynorbornene backbone by the ether linkages can be observed as bright spots at large magnifications. By fitting the radial profile of the fast Fourier transformed micrograph (FFT), an average structure size of 3.5 nm was calculated, roughly corresponding to the length of the ether side chains with the attached TMA cation. Overall, the blend membrane is homogeneous on a molecular level, proving the miscibility of POB and EM0.54-q-co-AM0.46-h. Importantly, for the block-copolymer, two nanophase separations are observed (Fig. 5d). A lamellar block copolymer morphology was observed, characterized by lamellae with a uniform thickness of approximately 20 nm. Moreover, comparable to the statistical copolymer, the phase separation between the backbone and the cationic group is seen at a length scale of roughly 3.5 nm.
Homogeneous blends are characterized by a single glass transition temperature (Tg).56 To investigate this phenomenon, DMA and DSC measurements were conducted on a blend comprising 10 wt% POB and 90 wt% EM0.54-q-co-AM0.46-h, with results compared to measurements of pure POB and EM0.54-q-co-AM0.46-h. A single Tg was observed in DSC and DMA analyses for the blend. DMA revealed a distinct maximum in the loss modulus/tanδ and a sharp drop in the storage modulus, indicating the glass-to-rubber transition (Fig. 6a). The overall DMA curve shape was consistent with previous analyses of positively charged polymers in the literature.57 The Tg of the blend (152 °C by DSC, 160 °C by DMA, Fig. 6a and b) exceeds the Tg of pure EM0.54-q-co-AM0.46-h (115 °C), likely due to the influence of the rigid POB component. POB did not exhibit a Tg within the temperature range of 450 °C (Fig. S22†). In homogeneous blends, the Tg is intermediate between the individual components. The higher Tg of the blend compared to pure EM0.54-q-co-AM0.46-h can be attributed to the POB component, which likely has a Tg exceeding 450 °C.43 The rigid POB component effectively increases the Tg of the homogeneous blend.
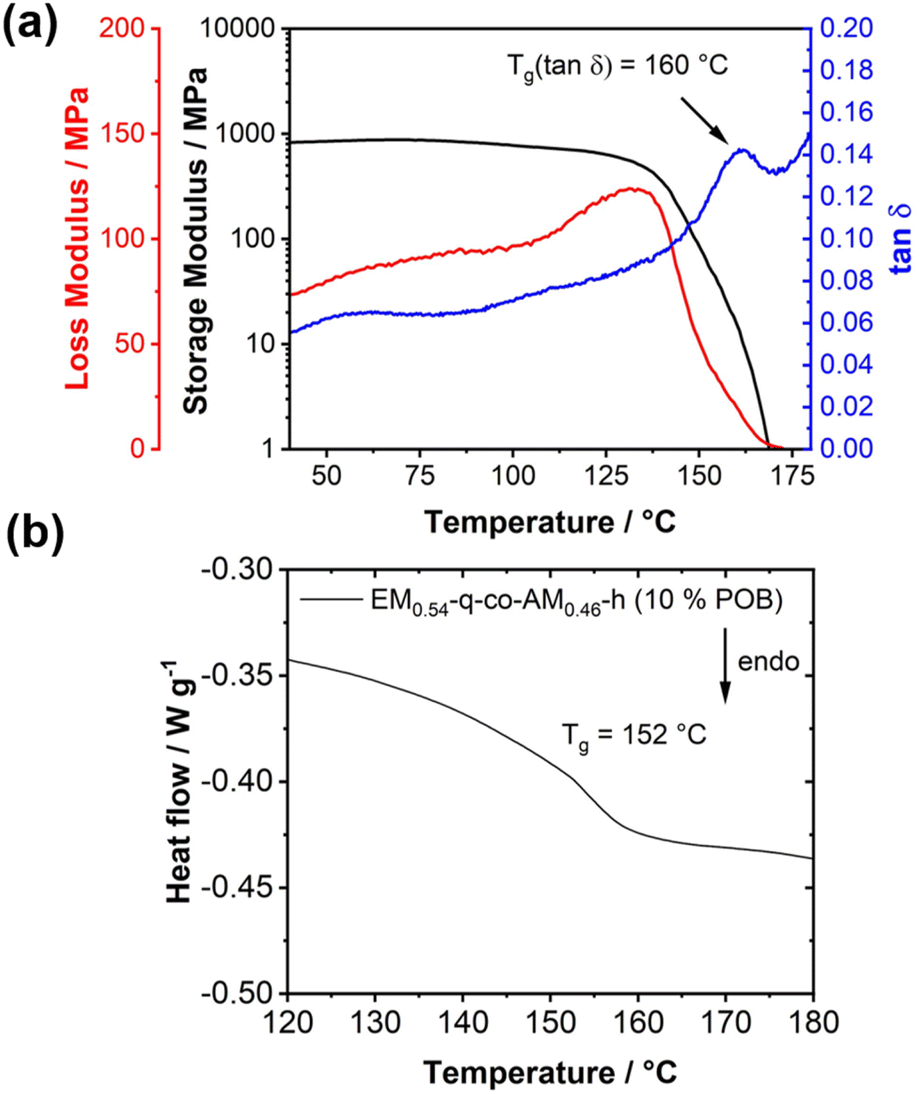 | ||
| Fig. 6 (a) Dynamic mechanical analysis of EM0.54-q-co-AM0.46-h revealing only one glass transition temperature (2 K min−1 heating rate, 1 Hz oscillation frequency). (b) DSC analysis (2nd heating curve, 10 K min−1 heating rate) of a blend membrane consisting of 90 wt% EM0.54-q-co-AM0.46-h and 10 wt% POB. | ||
In the next step, we systematically studied the influence of the POB content on the membrane properties for blends of POB with EM0.54-q-co-AM0.46-h and compared it to the pure block-copolymer since no blend membranes could be prepared here.
Fig. 7a shows the ionic conductivity depending on the POB content in the blend. Decreasing the POB content from 20 wt% to 5 wt% leads to a successive increase in Cl− and OH− conductivity (Fig. 7a). The increase in conductivity is accompanied by a higher water uptake and swelling ratio, which is depicted in Fig. 7c and d. Notably, when the POB content is decreased from 10 wt% to 5 wt%, a substantial increase in water uptake and swelling occurs, whereas only a moderate increase in conductivity was measured (Fig. 7a). The ion exchange capacity of the membranes with different POB contents follows a similar trend. From an initial value of 1.1 mmol g−1 for 30 wt% POB in the blend, the ion exchange capacity increases to 1.5 mmol g−1 for 5 wt% POB. Interestingly, for 10 wt% POB in the blend, the identical IEC was measured since, considering the overall mass of the membrane, a difference in POB content of 5 wt% will not significantly affect the ion exchange capacity. The analogous membranes containing ether-free alkyl chains (BM0.45-q-co-AM0.55-h) and 10 wt% POB demonstrated lower conductivity compared to those with ether side chains, as shown in Fig. S21† (5 mS cm−1 at 70 °C). The significantly lower conductivity of the ether-free membrane can be attributed to its reduced water uptake (WU = (19 ± 1)% and SRL = (4 ± 1)%) compared to the analogous ether-based membrane (WU = (98 ± 2)% and SRL = (33 ± 10)%). Notably, the ion exchange capacities of both membranes were comparable, measured at (1.40 ± 0.01) mmol g−1 for the 90 wt% BM0.45-q-co-AM0.55-h/10 wt% POB membrane and (1.41 ± 0.08) mmol g−1 for the 90 wt% EM0.54-q-co-AM0.46-h/10 wt% POB membrane by titration. The higher conductivity observed in the ether-based membrane, despite identical IEC and POB content, underscores the benefit of incorporating highly hydrophilic ether chains into anion exchange membranes to enhance conductivity.
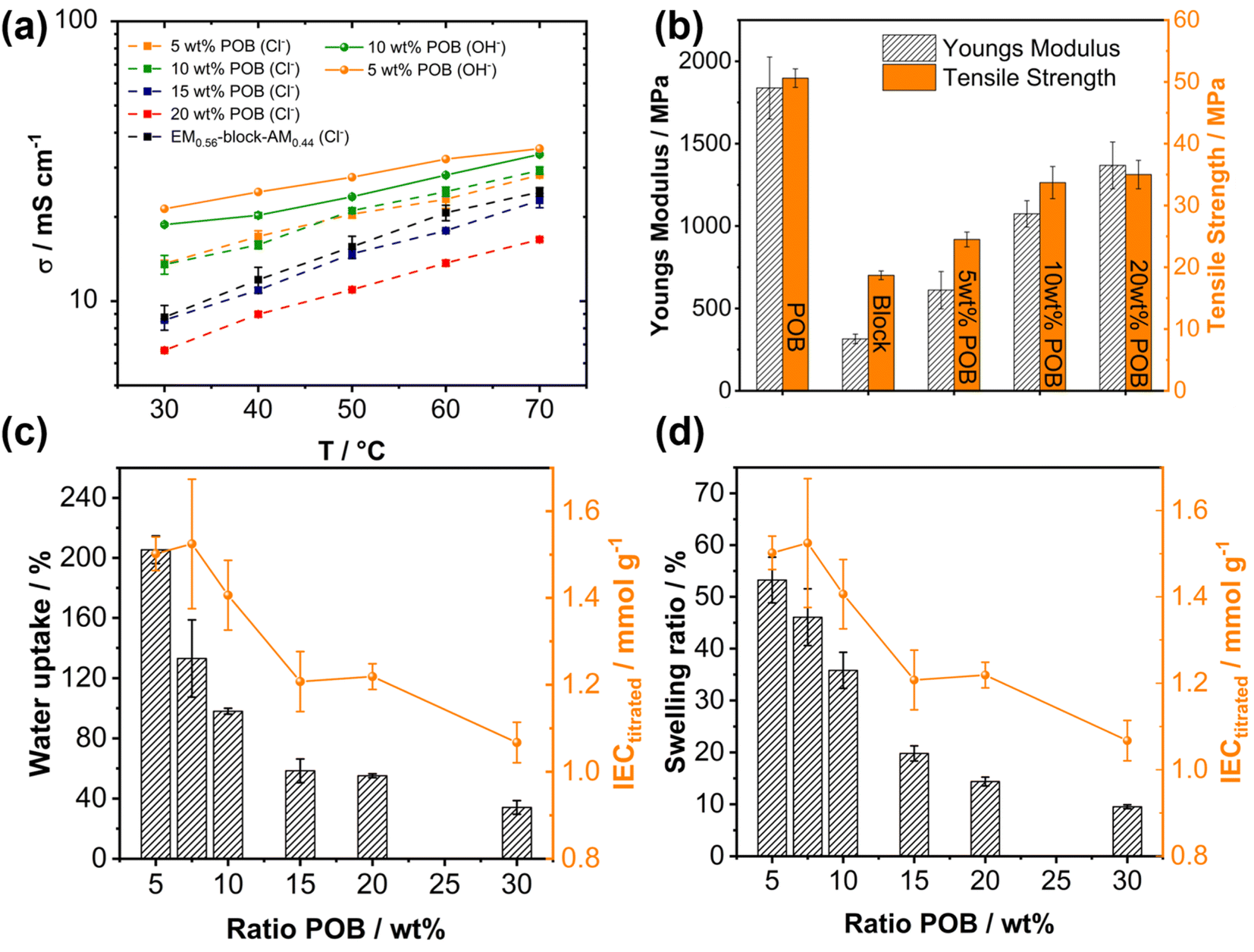 | ||
| Fig. 7 (a) Ionic conductivity for the blend membranes consisting of EM0.54-q-co-AM0.46-h and varying amounts of POB and chloride conductivity for a block-copolymer EM0.54-q-block-AM0.46-h. (b) Tensile properties (Youngs modulus and tensile strength) of exemplary blend membranes, the pure block-copolymer and pure POB. (c) Water uptake of blend membranes consisting of EM0.54-q-co-AM0.46-h and varying amounts of POB in the blend and the corresponding IEC of the blend membranes. (d) Swelling ratio of blend membranes consisting of EM0.54-q-co-AM0.46-h and varying amounts of POB in the blend and the corresponding IEC of the blend membranes. | ||
Furthermore, mechanical strength is sacrificed for POB contents below 10 wt%, assessed by stress–strain curves of membranes with various POB contents (Fig. 7b). The mechanical properties clearly show the benefit of adding a defined amount of POB to the blend membranes. Pure POB is a strong material with a comparatively high Young's Modulus of 1838 MPa and tensile strength of 51 MPa. The strain at break of the blend membranes and POB is identical at 6% within the measurement error (Fig. S23†). Notably, the block copolymer exhibited a significantly higher elongation at break of 23% (Fig. S23†), indicating a higher ductility of the block copolymer than the blends. The significant difference between the blends and the block copolymer could be explained by the stiff nature of the POB component in the blends, which comprises a rigid aromatic polymer backbone with hydrogen bond interactions.43 Since no POB is present in the membranes comprised of the block copolymer, the purely aliphatic backbone of EM0.54-q-co-AM0.46-h results in a higher ductility.
The pure block copolymer shows more elastic properties, resulting in softer membranes with a Young's Modulus of (315 ± 29) MPa and tensile strength of (18.7 ± 0.7) MPa but too low mechanical strength to withstand the swelling in KOH. Since no pure membranes of the statistical copolymer could be obtained, no direct comparison is possible. However, by comparing blend membranes with varying POB contents, the tensile strength and Young's modulus improved by increasing the POB ratio from 5 wt% to 20 wt% (Fig. 7b). Again, like the swelling and water uptake analysis results, a more significant difference between the membranes with 5 and 10 wt% POB in the blend was measured compared to those with 10 and 20 wt% POB. An increase in tensile strength was observed as the poly(oxindolebiphenylene) (POB) content increased from 5 wt% to 10 wt%, with values rising from (24.5 ± 1.2) MPa to (33.7 ± 2.6) MPa. However, further increasing the POB content from 10 wt% to 20 wt% did not result in a statistically significant difference in tensile strength, with values of (33.7 ± 2.6) MPa for 10 wt% and (35.0 ± 2.3) MPa for 20 wt%, remaining within the margin of measurement error. The Young's Modulus of blend membranes containing 10 wt% POB is slightly lower than those containing 20 wt% POB.
Finally, a comparison of the mechanical properties of the ether-free membrane (10 wt% POB and 90 wt% BM0.45-q-co-AM0.55-h) with the ether-containing membrane (10 wt% POB and 90 wt% EM0.54-q-co-AM0.46-h) reveals distinct differences (Table S1†). The ether-containing membrane exhibits a softer, more ductile material, as evidenced by a lower Young's modulus (1074 MPa vs. 1670 MPa), slightly lower tensile strength (33.7 MPa vs. 38.2 MPa), and a higher strain at break (5.7% vs. 2.3%). These mechanical differences can be attributed to the softening effect of the ether chains, which enhance ductility. In contrast, the ether-free membrane demonstrates a brittle nature, further highlighting the impact of the ether-side chains on mechanical behavior.
To conclude, from an analysis of ionic conductivity, swelling, and water uptake studies combined with mechanical testing, a content of 10 wt% POB in the blend with a statistical copolymer EM0.54-q-co-AM0.46-h was identified as the most promising membrane material for a more detailed study of the electrochemical performance in an AEMWE device. The membrane without ether chains was excluded from further evaluation in AEMWE due to the low conductivity and brittleness of the membranes.
Before the application test in an AEMWE device, the chemical stability of the 90 wt% EM0.54-q-co-AM0.46-h/10 wt% POB blend membrane had to be assessed since the harsh conditions of hot KOH often could lead to cationic group degradation, e.g., by Hofmann elimination or nucleophilic substitution reactions. The membranes were immersed in 1 M KOH at 85 °C for different time intervals to study the stability under conditions similar to the target application (1 M KOH, 60–80 °C). The stability was investigated by analyzing the aged membranes with 1H-NMR spectroscopy (Fig. 8a) and conductivity measurements (Fig. 8b). To better assign each signal to the respective blend component, the spectra of pure EM0.54-q-co-AM0.46-h and POB are compared. By taking the signal at 4.08 ppm, corresponding to the protons next to the anthracene moieties in the copolymer, and the signal at 7.57 ppm originating from the protons of an aromatic ring of the POB the ratio of both components was calculated and compared for the pristine membrane and the membrane aged for 4 weeks in 1 M KOH at 85 °C. 10 wt% POB in the blend corresponds to a molar ratio of 10.7 mol% POB. For the pristine membrane, the molar ratio obtained from 1H NMR analysis is 10.3 mol% POB content in the blend, and after 4 W in 1 M KOH at 85 °C the ratio is identical with 10.3 mol% POB content, proving that the POB content in the blend does not change after aging the membranes.
 | ||
| Fig. 8 (a) 1H NMR spectra of the pure EM0.54-q-co-AM0.46-h (black), pure POB (grey) the pristine blend consisting of 10 wt% POB and 90 wt% EM0.54-q-co-AM0.46-h (green) and after treating the respective membrane with 1 M KOH at 85 °C for 4 weeks (yellow). (b) Ionic conductivity after certain time intervals in 1 M KOH at 85 °C. | ||
Notably, the integral of the TMA group at 3.15 ppm is 10.82 for the pristine sample and 10.45 for the sample aged in 1 M KOH at 85 °C for 4 weeks, indicating only a marginal loss of the TMA group during the stability test. The remaining signals in the aromatic and aliphatic regions appear unchanged, proving no backbone or side-chain degradation during the aging procedure. By checking the ionic conductivity of different samples aged for various time intervals in 85 °C hot 1 M KOH, the results of the NMR analysis are confirmed. Here, an unchanged ionic conductivity was also measured for two different blend membranes (5 and 10 wt% POB), indicating that the TMA group is comparatively stable under the investigated conditions. These results are on par with results reported in the literature, where the TMA cation was already reported to be chemically resistant to hydroxide attacks in 1 M KOH.58–60 Most importantly, we did not observe a signal corresponding to Hoffman elimination products in the double bond region, indicating no degradation via this pathway and underlining the high stability of the TMA cation.
Notably, a recent publication reported an adverse effect of poly(oxindolebiphenylene) (POB) and poly(biphenyl 4-imidazole carboxaldehyde) (PB4Im) on the alkaline stability of blend membranes composed of these materials and poly(aryl piperidine) polymers.61 The enhancing effect of POB and PB4Im on the degradation of poly(aryl piperidine) polymers was attributed to the increased uptake of hydroxide ions, which results from the deprotonation of POB and PB4Im. As the content of POB and PB4Im in the blend membrane increases, the concentration of hydroxide ions within the membrane also rises. This elevated hydroxide concentration accelerates degradation reactions, particularly Hoffmann eliminations, leading to more significant polymer breakdown and compromising the membrane's alkaline stability.61 Interestingly, we did not observe the same effect for our blend system (Fig. 8). It can be speculated that the hydrophilicity of the ether-containing segments counteracts the increased hydroxide uptake by promoting water adsorption, thereby reducing the overall hydroxide concentration within the membrane. Moreover, due to the direct attachment of the trimethylammonium group to the hydrophilic ether chains, the solvation of the hydroxide ions surrounding the ammonium group is enhanced. As a result, the degradation processes are not accelerated in the POB/EM0.54-q-co-AM0.46-h blend system, preserving its stability despite the presence of POB. This balance between hydroxide uptake and water absorption may explain the absence of significant degradation in this copolymer blend under alkaline conditions.
As an accelerated stress test, the stability test was conducted in 4 M KOH instead of the typical 1 M KOH. No decrease in conductivity was observed during the first week (Fig. S24†). However, after 2 weeks, significant degradation was detected, as evidenced by a reduction in the integral of the TMA groups in the 1H NMR spectrum from 10.83 to 6.07, corresponding to a 44% degradation of the TMA groups (Fig. S24a†). After 4 weeks, 67% of the TMA groups have been degraded, and the membrane's solubility decreased, indicating crosslinking. It should be noted, however, that these test conditions are extremely harsh. Consequently, current research focuses on diluted KOH solutions with concentrations below 1 M.6,62,63 TMA groups are known to exhibit increased degradation under elevated temperatures in concentrated KOH but remain stable in 1 M KOH, consistent with our observations.58,64–66
To summarize, a blend membrane comprising 10 wt% POB and 90 wt% EM0.54-q-co-AM0.46-h showed superior ionic conductivity while retaining the excellent mechanical properties of the POB component combined with a decent water uptake and swelling ratio, necessary for sufficient water transport from anode to cathode in an AEMWE device operating with dry cathode. Furthermore, this blend membrane showed high resistance to degradation when treated with hot 1 M KOH for 4 weeks, indicating applicability in AEMWE.
PGM-free AEMWE with dry cathode
After confirmation of sufficient ionic conductivity, suitable water uptake, and chemical stability in 1 M KOH at elevated temperature, the membrane with the most balanced mechanical strength, water uptake, and conductivity was tested in a water electrolyzer. Based on the ex situ investigations, a membrane with 10 wt% of POB as a reinforcer and 90 wt% EM0.54-q-co-AM0.46-h as an ion-conducting polymer was selected. One main benefit of alkaline electrolyzers compared to acidic systems is the electrochemical stability of non-PGM electrocatalysts. NiFe-LDH is accepted as an anode catalyst, which makes using the expensive iridium unnecessary.3 Platinum is still commonly used on the cathode side for AEMWE, even though there are alternatives based on more abundant elements.8 Moreover, to combine the advantages of PEMWE and AWE, running an AEMWE cell under dry cathode mode without supplying KOH as an electrolyte on the cathode side is the preferred operation mode to get pure hydrogen.Here, the applicability of the ether-containing polynorbornenes to a PGM-free, dry-cathode AEMWE cell (Fig. 9a) design is demonstrated, and the results are shown in (Fig. 9).
 | ||
| Fig. 9 (a) Schematical representation of the dry cathode cell setup with NiFe-LDH as oxygen evolution reaction (OER) catalyst and CoP as hydrogen evolution reaction (HER) catalyst and the transport of hydroxide ions from cathode to anode and most importantly under dry cathode mode, water transport from anode to supply the cathode with sufficient water as feed for the water splitting reaction. (b) Polarization curve of a membrane with 10 wt% POB and 90 wt% EM0.54-q-co-AM0.46-h in the blend (thickness = 50 μm) and high frequency resistance compared to AF2-HLF8-25X (thickness = 25 μm), measured at 60 °C. (c) Constant current hold at 1 A cm−2 and 60 °C for 120 h and impedance scans after 20 h each for the AF2-HLF8-25X and 10 wt% POB and 90 wt% EM0.54-q-co-AM0.46-h under dry cathode mode (asym) and additionally for 10 wt% POB and 90 wt% EM0.54-q-co-AM0.46-h with 1 M KOH on anode and cathode (sym, purple curve). | ||
The cathode comprised a custom-made nickel phosphide felt (Ni@NiP-felt) with 4 mg cm−2 CoP/C as HER catalyst with 10 wt% of a custom-made ionomer (PBPIm+).8 For dry cathode operation, no supporting electrolyte was applied at the cathode, but it was purged with 100 mL min−1 of dry N2 gas. NiFe with 2 mg cm−2 loading and 10 wt% of the PBIm+ ionomer on the anode side was used on a self-made nickel sulfide felt (Ni@NiS-felt). Furthermore, 1 M KOH was used as an electrolyte on the anode side. The operating temperature of the cell was 60 °C. A current density of 2.0 A cm−2 was achieved at a voltage of 1.91 V, demonstrating an industrially relevant current density at a comparatively low voltage.
Furthermore, enough water is transported from the anode to the cathode, ensuring sufficient wetting of the cathode to let the water-splitting reaction occur, characterized by no mass-transport characteristics in the polarization curve (Fig. 9b). Comparable performance to AF2-HLF8-25X was measured (Fig. 9b) for our 90 wt% EM0.54-q-co-AM0.46-h/10 wt% POB membrane, reflecting similar HFR values of 99.7 mΩ cm−2 and 99.5 mΩ cm−2, respectively. Notably, the self-synthesized membrane was 50 μm thick and the commercial membrane only 25 μm, indicating a higher intrinsic conductivity of the norbornene membrane and demonstrating the excellent applicability of the norbornene membrane in dry cathode mode AEMWE. The present study's PGM-free, dry-cathode AEMWE test results were compared to recent publications on AEMWE utilizing polynorbornene-based membranes (Table 1). Notably, the results from previous reports all contained PGM materials, either on the cathode only (entries 1, 2, and 3, Table 1) or on the cathode and anode (entry 4, Table 1). Specifically, in the present study, at a voltage of 1.79 V, a current density of 1.0 A cm−2 was reached, which is better compared to the results of Wang et al. using a vinylic-addition polymerized polycyclic polynorbornene membrane which was UV-crosslinked and quaternized with TMA (entry 1, Table 1).67 Compared to studies using comparable vinyl-addition polymerized polynorbornenes with bromobutyl side chains (entry 2, Table 1) and operating the AEWME under dry cathode mode, the performance of our cell system is auspicious considering a fully PGM-free cell in our study.38 Here, it should be mentioned that in the study of Park et al., 0.01 M NaOH was used as an electrolyte, which will influence cell performance negatively due to a higher ohmic resistance.38 Finally, compared to commercially available polynorbornene membranes (entries 3 and 4, Table 1) used in dry-cathode mode AEMWE with PGM catalysts, the AEMWE design utilized in this study offered comparable performance.
| Study | Anode catalyst | Cathode catalyst | Dry cathode | Membrane | Feed | Cell temperature | Cell voltage | Current density | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | NiFe | Pt/C | No | D-2-H | 1 M KOH | 60 °C | 2.00 V | 1.3 A cm−2 | 67 |
| 2 | NiFe2O4 | Pt3Ni | Yes | 75 mol% BrBuNB and 25 mol% BuNB | 0.01 M NaOH | 60 °C | 2.10 V | 1.0 A cm−2 | 38 |
| 3 | NiFe2O4 | Pt3Ni | Yes | GT75-5 (Xergy Inc.) | 1 M KOH | 60 °C | 1.75 V | 1.0 A cm−2 | 36 |
| 4 | IrO2 | Pt3Ni | Yes | XION™ composite-72–10CL-30 | 0.3 M KOH | 60 °C | 1.75 V | 1.0 A cm−2 | 37 |
| 5 | NiFe | CoP | Yes | 90 wt% EM0.54-q-co-AM0.46-h/10 wt% POB | 1 M KOH | 60 °C | 1.79 V | 1.0 A cm−2 | This work |
The in situ stability of the membrane was investigated with a constant current hold for 5 days at 1 A cm−2 (Fig. 9c). Here, we observed a higher voltage increase for the MEA setup utilizing the self-synthesized membrane (1.35 mV h−1) compared to the commercial reference (0.45 mV h−1). The constant HFR observed for both membranes (Fig. 9c) suggests the absence of membrane degradation, further supported by their excellent ex situ KOH stability. However, this does not rule out the possibility of other degradation pathways that could impact cell performance and longevity. Further investigation is necessary to identify potential degradation mechanisms beyond membrane deterioration. We hypothesize that the different membrane-electrode interfaces for the woven reinforced AF2-HLF8-25X and the self-synthesized blend membrane will influence the dry cathode, leading to different humidity and local pH on the cathode. At this point, it is worth noting that a lower humidity on the cathode side may lead to increased catalyst layer degradation (e.g., ionomer) due to less solvated hydroxides since these weakly solvated hydroxides are known to be very reactive nucleophiles.68–70
To investigate the origins of cell degradation, a constant current hold for 120 hours at 1 A cm−2 with 1 M KOH on both the anode and cathode sides was performed for the self-synthesized membrane (purple curve, symm, Fig. 9c). All other test conditions were maintained identically. Compared to the dry cathode measurement, the degradation rate improved to 0.75 mV h−1 (with break-in) or 0.51 mV h−1 (without break-in). Notably, under testing with a liquid electrolyte circulating on both sides, the high-frequency resistance (HFR) remained constant at 100 mΩ cm2 throughout the testing period after the break-in (Fig. S25†). While the degradation rate for the wet cathode mode is initially higher during the first 20 hours of testing, it decreases steadily over the 120-hour testing period, ultimately resulting in a lower overall degradation rate compared to the dry cathode mode. Specifically, during the last 20 hours of testing, the degradation rate for the wet cathode mode was 0.38 mV h−1 (purple curve, Fig. 9c), significantly lower than 0.75 mV h−1 for the commercial reference under dry cathode mode (blue curve, Fig. 9c) and 1.25 mV h−1 for the self-synthesized membrane under dry cathode mode (green curve, Fig. 9c). These findings suggest that cathode conditioning is pivotal in cell degradation during the tested period, influenced by local conditions such as pH and humidity. For the wet cathode mode, the highest initial degradation rate of 2.83 mV h−1 during the first 20 hours correlates with rapid cathode conditioning, stabilizing to 0.38 mV h−1 during the final testing phase. Interestingly, the 50 μm thick self-synthesized membranes degradation rate under dry cathode mode also improved during testing, with the degradation rate decreasing from 1.69 mV h−1 in the first 20 hours to 1.25 mV h−1 in the last 20 hours, indicating slower cathode conditioning under lower humidity. The commercial reference membrane under dry cathode mode showed intermediate conditioning behavior, with degradation improving from 1.50 mV h−1 in the first 20 hours to 0.745 mV h−1 in the last 20 hours (blue curve, Fig. 9). These findings yield three key conclusions regarding AEMWE cell endurance: (1) a stable HFR may indicate the absence of membrane degradation. However, other factors could influence this observation. For instance, degradation at the membrane-electrode interface could lead to cell performance loss without affecting HFR. Additionally, the loss of ionic groups and conductivity within the membrane might be masked by the presence of 1 M KOH, which continues to provide ionic conductivity, potentially concealing underlying degradation mechanisms. However, this is expected only to have a minor effect since the overall cell resistance is dominated by the membrane and not the 1 M KOH due to the way higher conductivity of 1 M KOH at 60 °C (322 mS cm−1 for 1 M KOH vs. 30 mS cm−1 for our membrane at 60 °C in pure water).37,71 (2) Impact of cathode-side humidity: the break-in process is strongly influenced by cathode-side humidity, with higher degradation rates linked to humidity differences. This suggests that the higher degradation rate observed for the self-synthesized membrane than the commercial reference arises from differences in cathode humidity. (3) Electrode conditioning: during the initial 120 hours, electrode conditioning reduces degradation rates, a trend consistent with the literature on long-term AEMWE operation. For example, Moreno-González et al. reported a degradation rate of 0.918 mV h−1 during the first 150 hours, with a decreasing degradation rate over time attributed to electrode conditioning.72
Next to the dry cathode, additional effects on the anode side could cause increased degradation of the norbornene membrane. Recently, norbornene oxidation under anodic potentials was reported.73,74 Norbornene oxidation yields ketones and carboxylic acids, whereby the latter will lower the local pH at the anode-membrane interface, causing performance loss without HFR increase.74 Since norbornene moieties are only present in the self-synthesized membrane, this could also explain the difference between the commercial reference and the norbornene membrane. To contextualize the measured degradation rates of our system, it is essential to compare them with previous studies. Notably, Fortin et al. reported significantly higher degradation rates ranging from 2.39 mV h−1 to 11.9 mV h−1 when using first-generation Aemion membranes and PGM-based electrodes.75 They also observed a higher degradation rate for thicker (50 μm) membranes than thinner ones (25 μm), which is on par with our observations.75 Here, our results are very competitive (0.75 mV h−1 for wet cathode, 1.35 mV h−1 for dry cathode), and we have demonstrated significant progress, considering the absence of PGM catalysts and a novel membrane concept, which is still not fully optimized. Moreover, Wang et al. observed a degradation rate between 1.65 mV h−1 and 2.85 mV h−1 for a polycyclic polynorbornene membrane in AEMWE with 0.1 M KOH at 60 °C within 75 h, which also highlights that the degradation observed in our system is superior to recent literature on comparable membranes. Nevertheless, investigating the origin of the different voltage increases for both cells will be part of future research. Finding optimized membrane–ionomer combinations is crucial for ideal cell performance, primarily if the cell is operated in dry-cathode mode.
Summary and conclusion
In conclusion, a new anion exchange blend membrane was synthesized using a polynorbornene copolymer bearing highly hydrophilic ether side chains (EM) functionalized with a trimethylammonium group and a hydrophobic comonomer (AM) comprising anthracene units. Block copolymers were synthesized and compared to statistical copolymers by utilizing controlled ring-opening metathesis polymerization (ROMP) to investigate the influence of polymer architecture on membrane properties. We used polymers with a constant IEC of 2.00 mmol g−1 corresponding to a build-in ratio of 54 mol% EM (EM0.54-q-co-AM0.46-h and EM0.54-q-block-AM0.46-h) to study only the polymer architecture and blend composition. Moreover, to highlight the specific contributions of ether chains, the copolymers were compared to analogous materials with alkyl chains instead of ethers (BM0.45-q-co-AM0.55-h).Excellent control over molecular weight was demonstrated, and the efficient preparation of block copolymers was shown. The target molecular weight for all polymers was 50000 g mol−1, which was also achieved for the statistical and the block copolymers. Notably, complete hydrogenation of the polymers yielded the saturated polymer quantitatively, which is vital regarding alkaline stability.
Interestingly, there were significant differences between the statistical and the block copolymers. Statistical copolymers did not yield stable membranes on their own. Still, due to excellent miscibility with poly(oxindole biphenylene), mechanically robust blend membranes could be obtained ranging from 5 wt% POB to 20 wt% POB in the blend. With HAADF-STEM, the nanophase separation of the blend membranes was investigated. An average structure size of 3.5 nm was calculated for the statistical copolymer, corresponding to the nanophase separation between the hydrophobic norbornene backbone and the TMA groups. The block copolymers showed two different structure sizes. One on the same length scale as the statistical copolymers (3.5 nm) arising from the ether-side chains and lamellar patterns with roughly 20 nm thickness, corresponding to the phase separation between the hydrophilic ether-containing block and the hydrophobic anthracene block. The block copolymers formed stable membranes, but no further characterization could be carried out due to excessive swelling in KOH-containing solutions.
The membranes with 10 wt% POB in the blend showed the best compromise between high ionic conductivity (33 mS cm−1 mixed OH−/HCO3−, 70 °C) water uptake (98%), dimensional stability (36%), and mechanical properties (Young's modulus 1074 MPa, tensile strength 33.7 MPa). Incorporating ether groups increased conductivity and ductility compared to an analogous membrane with alkyl chains (5 mS cm−1 at 70 °C) instead of ether. The membrane with 10 wt% POB in the blend also showed ex situ alkaline stability for four weeks in 1 M KOH, as proved by 1H-NMR spectroscopy and conductivity measurements.
Finally, the performance of this membrane (90 wt% EM0.54-q-co-AM0.46-h/10 wt% POB, d = 50 μm) in a fully PGM-free electrolyzer operating in dry-cathode mode was demonstrated and compared to AF2-HLF8-25X (d = 25 μm) as a commercial reference. Dry-cathode mode AEMWE is vital in terms of hydrogen purity, and thus, it is necessary to demonstrate the membrane applicability under these conditions. For 90 wt% EM0.54-q-co-AM0.46-h/10 wt% POB, a current density of 2.0 A cm−2 at 1.91 V was reached, comparable to the commercial reference. At this point, it is worth noting that the commercial reference had half of the thickness compared to the self-synthesized membrane, which will reflect in the ohmic resistance of the cell. A constant current hold at 1 A cm−2 for 5 days gave a first impression of the stability of the setup. Here, for both the commercial reference and the self-synthesized membrane, the HFR stayed constant at 100 mΩ cm−2, indicating no relevant decrease in the membrane conductivity. A higher voltage increase of 1.35 mV h−1 compared to AF2-HLF8-25X (0.45 mV h−1) was observed for the self-synthesized membrane, indicating different cell degradation for both systems. However, since the overall increase in voltage over time is also influenced by the electrodes, the membrane-electrode interface, and the humidity on the cathode, it is hard to elucidate the origin of the different degradation behavior. With additional measurements with 1 M KOH circulating on the cathode side, the degradation rate improved from 1.35 mV h−1 to 0.75 mV h−1 for the self-synthesized membrane, indicating that cathode humidity and electrode conditioning dominate the degradation in the investigated time scale. The investigation of this phenomenon will be part of future research. Moreover, we plan to exploit the superior thermal transition of the EM0.54-q-co-AM0.46-h polymer to prepare membrane-electrode assemblies via the decal method to improve the adhesion of the catalyst layer to the membrane and to enhance the cell performance and stability further. Moreover, further improvements could involve the development of next-generation ionomers dedicated to dry-cathode mode operation. It is also planned to functionalize the brominated copolymers with other tertiary amines, including diamines for simultaneous quaternization and covalent crosslinking, to improve further the alkaline stability of the anion-exchange (blend) membranes.
Data availability
The data supporting this article have been included in the ESI.†Author contributions
Linus Hager: conceptualization, methodology, investigation, data curation, visualization, writing – original draft. Timo Maron: investigation, methodology, writing – review & editing. Trung Ngo Thanh: investigation, data curation, writing – review & editing. Julian Stonawski: writing – review & editing. Andreas Hutzler: investigation, writing – review & editing. Thomas Böhm: investigation, writing – review & editing. Peter Strasser: supervision, writing – review & editing, funding acquisition. Simon Thiele: supervision, writing – review & editing. Jochen Kerres: supervision, conceptualization, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung, BMBF) in the collaborative research projects H2Meer (grant number: 03SF0611A), H2Mare (grant number: 03HY302Q) and SeaEly (03SF0663D) is thankfully acknowledged by the authors.Notes and references
- M. M. Rahman, A. O. Oni, E. Gemechu and A. Kumar, Assessment of energy storage technologies: a review, Energy Convers. Manag., 2020, 223, 113295 Search PubMed.
- D. Ferrero, M. Gamba, A. Lanzini and M. Santarelli, Power-to-gas hydrogen: techno-economic assessment of processes towards a multi-purpose energy carrier, Energy Procedia, 2016, 101, 50–57 CrossRef CAS.
- N. Chen and Y. M. Lee, Anion exchange polyelectrolytes for membranes and ionomers, Prog. Polym. Sci., 2021, 113, 101345 CrossRef CAS.
- F. Dionigi, T. Reier, Z. Pawolek, M. Gliech and P. Strasser, Design criteria, operating conditions, and nickel-iron hydroxide catalyst materials for selective seawater electrolysis, ChemSusChem, 2016, 9, 962–972 Search PubMed.
- S. Gottesfeld, D. R. Dekel, M. Page, C. Bae, Y. Yan, P. Zelenay and Y. S. Kim, Anion exchange membrane fuel cells: current status and remaining challenges, J. Power Sources, 2018, 375, 170–184 Search PubMed.
- D. Henkensmeier, M. Najibah, C. Harms, J. Žitka, J. Hnát and K. Bouzek, Overview: state-of-the art commercial membranes for anion exchange membrane water electrolysis, J. Electrochem. Energy Convers. Storage, 2021, 18, 024001 CrossRef CAS.
- X. Peng, T. J. Omasta, E. Magliocca, L. Wang, J. R. Varcoe and W. E. Mustain, Nitrogen-doped carbon–CoOx nanohybrids: a precious metal free cathode that exceeds 1.0 W cm−2 peak power and 100 h life in anion-exchange membrane fuel cells, Angew. Chem., Int. Ed., 2019, 131, 1058–1063 CrossRef.
- M. L. Frisch, T. N. Thanh, A. Arinchtein, L. Hager, J. Schmidt, S. Brückner, J. Kerres and P. Strasser, Seawater electrolysis using all-PGM-free catalysts and cell components in an asymmetric feed, ACS Energy Lett., 2023, 8, 2387–2394 CrossRef CAS.
- Q. Xu, L. Zhang, J. Zhang, J. Wang, Y. Hu, H. Jiang and C. Li, Anion exchange membrane water electrolyzer: electrode design, lab-scaled testing system and performance evaluation, EnergyChem, 2022, 4, 100087 Search PubMed.
- N. Chen, S. Y. Paek, J. Y. Lee, J. H. Park, S. Y. Lee and Y. M. Lee, High-performance anion exchange membrane water electrolyzers with a current density of 7.68 A cm−2 and a durability of 1000 hours, Energy Environ. Sci., 2021, 14, 6338–6348 RSC.
- L. Ma, M. Hussain, L. Li, N. A. Qaisrani, L. Bai, Y. Jia, X. Yan, F. Zhang and G. He, Octopus-like side chain grafted poly(arylene piperidinium) membranes for fuel cell application, J. Membr. Sci., 2021, 636, 119529 Search PubMed.
- B. Yang and C. Zhang, Facilitated belt for ion transport by comb-shape poly(terphenyl piperidone) carried with macrocyclic crown ether as anchor in anion exchange membrane, J. Power Sources, 2024, 602, 234324 CrossRef CAS.
- J. Zhang, W. Ma, T. Yin, S. Chen, X. Zhang, N. Li and L. Liu, Oligo(ethylene glycol)-grafted poly(terphenyl indole piperidinium) with high water diffusivity for anion exchange membrane fuel cells, J. Membr. Sci., 2024, 694, 122424 Search PubMed.
- M. Makrygianni, S. Aivali, Y. Xia, M. R. Kraglund, D. Aili and V. Deimede, Polyisatin derived ion-solvating blend membranes for alkaline water electrolysis, J. Membr. Sci., 2023, 669, 121331 CrossRef CAS.
- N. Du, C. Roy, R. Peach, M. Turnbull, S. Thiele and C. Bock, Anion-exchange membrane water electrolyzers, Chem. Rev., 2022, 122, 11830–11895 CrossRef CAS PubMed.
- K. M. Hugar, H. A. Kostalik and G. W. Coates, Imidazolium cations with exceptional alkaline stability: a systematic study of structure-stability relationships, J. Am. Chem. Soc., 2015, 137, 8730–8737 CrossRef CAS PubMed.
- A. D. Mohanty, S. E. Tignor, J. A. Krause, Y.-K. Choe and C. Bae, Systematic alkaline stability study of polymer backbones for anion exchange membrane applications, Macromolecules, 2016, 49, 3361–3372 CrossRef CAS.
- Z. He, G. Wang, C. Wang, L. Guo, R. Wei, G. Song, D. Pan, R. Das, N. Naik, Z. Hu and Z. Guo, Overview of anion exchange membranes based on ring opening metathesis polymerization (ROMP), Polym. Rev., 2021, 61, 689–713 CrossRef CAS.
- W. You, K. M. Hugar and G. W. Coates, Synthesis of alkaline anion exchange membranes with chemically stable imidazolium cations: unexpected cross-linked macrocycles from ring-fused ROMP monomers, Macromolecules, 2018, 51, 3212–3218 CrossRef CAS.
- K. J. T. Noonan, K. M. Hugar, H. A. Kostalik, E. B. Lobkovsky, H. D. Abruña and G. W. Coates, Phosphonium-functionalized polyethylene: a new class of base-stable alkaline anion exchange membranes, J. Am. Chem. Soc., 2012, 134, 18161–18164 CrossRef CAS PubMed.
- S. Martínez-Arranz, A. C. Albéniz and P. Espinet, Versatile route to functionalized vinylic addition polynorbornenes, Macromolecules, 2010, 43, 7482–7487 CrossRef.
- Z. Yang and C. D. Han, Synthesis of hydrogenated functional polynorbornene (HFPNB) and rheology of HFPNB-based miscible blends with hydrogen bonding, Polymer, 2008, 49, 5128–5136 CrossRef CAS.
- M. Hong, L. Cui, S. Liu and Y. Li, Synthesis of novel cyclic olefin copolymer (COC) with high performance via effective copolymerization of ethylene with bulky cyclic olefin, Macromolecules, 2012, 45, 5397–5402 CrossRef CAS.
- S. Martínez-Arranz, E. Sánchez-Pérez, J. A. La Molina de Torre, I. Pérez-Ortega and A. C. Albéniz, p-Bromoaryl- and ω-bromoalkyl-VA-PNBs: suitable starting materials for the functionalization of vinylic addition polynorbornenes via palladium-catalyzed cross-coupling reactions, RSC Adv., 2016, 6, 105878–105887 RSC.
- S. Chen, A. Zhang, X. He and D. Chen, Hydrogenated diblock copoly(norbornene)s bearing triimidazolium anion exchange membranes with enhanced alkaline stability for fuel cells, Int. J. Hydrogen Energy, 2024, 50, 1282–1292 CrossRef.
- S. Chen, A. Zhang, X. He and D. Chen, Towards high conductivity and stability diblock poly(norbornene)s-based anion exchange membranes crosslinked with alkyl quaternary ammonium cationic clusters, J. Membr. Sci., 2024, 700, 122706 CrossRef CAS.
- X. Sun, D. Cao, M. Liu, B. Wang, D. Song, L. Pan, N. Li and Y. Li, Remarkable impact of chain backbone on the performance of poly(norbornene derivatives)-based anion exchange membranes, J. Membr. Sci., 2024, 703, 122830 CrossRef CAS.
- Q. Li, X. He, L. Feng, W. Zhang, C. Tang, J. Ye and D. Chen, The effect of various functional groups on the morphology and performances of crosslinked addition-type poly(norbornene)s-based anion exchange membranes, J. Appl. Polym. Sci., 2024, 1–16 Search PubMed.
- M. Lehmann, D. Leonard, J. Zheng, L. He, X. Tang, X. C. Chen, K. H. Lim, S. Maurya, Y. S. Kim and T. Saito, Quaternized polynorbornene random copolymers for fuel cell devices, ACS Appl. Energy Mater., 2023, 6, 1822–1833 CrossRef CAS.
- D. P. Leonard, M. Lehmann, J. M. Klein, I. Matanovic, C. Fujimoto, T. Saito and Y. S. Kim, Phenyl-free polynorbornenes for potential anion exchange ionomers for fuel cells and electrolyzers, Adv. Energy Mater., 2023, 13, 2203488 Search PubMed.
- J. C. Gaitor, A. C. Yang-Neyerlin, D. Markovich, B. P. Fors, G. W. Coates, L. F. Kourkoutis, B. S. Pivovar, T. Kowalewski and K. J. T. Noonan, Comparing ammonium and tetraaminophosphonium anion-exchange membranes derived from vinyl-addition polynorbornene copolymers, ACS Appl. Energy Mater., 2024, 7, 1517–1526 CrossRef CAS PubMed.
- R. Selhorst, J. Gaitor, M. Lee, D. Markovich, Y. Yu, M. Treichel, C. Olavarria Gallegos, T. Kowalewski, L. F. Kourkoutis, R. C. Hayward and K. J. T. Noonan, Multiblock copolymer anion-exchange membranes derived from vinyl addition polynorbornenes, ACS Appl. Energy Mater., 2021, 4, 10273–10279 CrossRef CAS.
- W. Chen, M. Mandal, G. Huang, X. Wu, G. He and P. A. Kohl, Highly conducting anion-exchange membranes based on cross-linked poly(norbornene): ring opening metathesis polymerization, ACS Appl. Energy Mater., 2019, 2, 2458–2468 CrossRef CAS.
- M. Mandal, G. Huang, N. U. Hassan, X. Peng, T. Gu, A. H. Brooks-Starks, B. Bahar, W. E. Mustain and P. A. Kohl, The importance of water transport in high conductivity and high-power alkaline fuel cells, J. Electrochem. Soc., 2020, 167, 54501 CrossRef CAS.
- G. Huang, M. Mandal, X. Peng, A. C. Yang-Neyerlin, B. S. Pivovar, W. E. Mustain and P. A. Kohl, Composite poly(norbornene) anion conducting membranes for achieving durability, water management and high power (3.4 w/cm2) in hydrogen/oxygen alkaline fuel cells, J. Electrochem. Soc., 2019, 166, F637–F644 CrossRef CAS.
- H. Park and P. A. Kohl, Alkaline membrane water electrolysis performance improvement from high ionic strength anolyte, J. Power Sources, 2023, 585, 233643 CrossRef CAS.
- N. U. Hassan, Y. Zheng, P. A. Kohl and W. E. Mustain, KOH vs. deionized water operation in anion exchange membrane electrolyzers, J. Electrochem. Soc., 2022, 169, 44526 CrossRef CAS.
- H. Park, C. Li and P. A. Kohl, Performance of anion exchange membrane water electrolysis with high ionic strength electrolyte, J. Electrochem. Soc., 2024, 171, 24506 CrossRef CAS.
- S. C. Price, X. Ren, A. M. Savage and F. L. Beyer, Synthesis and characterization of anion-exchange membranes based on hydrogenated poly(norbornene), Polym. Chem., 2017, 8, 5708–5717 RSC.
- D. Cao, F. Yang, W. Sheng, Y. Zhou, X. Zhou, Y. Lu, F. Nie, N. Li, L. Pan and Y. Li, Polynorbornene-based anion exchange membranes with hydrophobic large steric hindrance arylene substituent, J. Membr. Sci., 2022, 641, 119938 CrossRef CAS.
- M. Treichel, C. Tyler Womble, R. Selhorst, J. Gaitor, T. M. S. K. Pathiranage, T. Kowalewski and K. J. T. Noonan, Exploring the effects of bulky cations tethered to semicrystalline polymers: the case of tetraaminophosphoniums with ring-opened polynorbornenes, Macromolecules, 2020, 53, 8509–8518 CrossRef CAS.
- J. C. Gaitor, M. Treichel, T. Kowalewski and K. J. T. Noonan, Suppressing water uptake and increasing hydroxide conductivity in ring-opened polynorbornene ion-exchange materials via backbone design, ACS Appl. Polym. Mater., 2022, 4, 8032–8042 CrossRef CAS.
- X. Hu, B. Hu, C. Niu, J. Yao, M. Liu, H. Tao, Y. Huang, S. Kang, K. Geng and N. Li, An operationally broadened alkaline water electrolyser enabled by highly stable poly(oxindole biphenylene) ion-solvating membranes, Nat. Energy, 2024, 9, 401–410 CrossRef CAS.
- S. Gjoshi, P. Loukopoulou, M. Plevova, J. Hnat, K. Bouzek and V. Deimede, Cycloaliphatic quaternary ammonium functionalized poly(oxindole biphenyl) based anion-exchange membranes for water electrolysis: stability and performance, Polymers, 2023, 16, 99 CrossRef PubMed.
- Y. Yuan, X. Du, H. Zhang, H. Wang and Z. Wang, Poly(isatin biphenylene) polymer containing ferrocenium derivatives for anion exchange membrane fuel cell, J. Membr. Sci., 2022, 642, 119986 CrossRef CAS.
- D. Cao, X. Sun, H. Gao, L. Pan, N. Li and Y. Li, Crosslinked polynorbornene-based anion exchange membranes with perfluorinated branch chains, Polymers, 2023, 15, 1073 CrossRef CAS PubMed.
- X. He, J. Zou, Y. Wen, B. Wu, X. Zang, J. Deng, Z. Qin, G. Yang, J. Xu and D. Chen, Preparation and performance of bisimidazole cationic crosslinked addition-type polynorbornene-based anion exchange membrane, Int. J. Hydrogen Energy, 2022, 47, 69–80 Search PubMed.
- S. Huang, X. He, C. Cheng, F. Zhang, Y. Guo and D. Chen, Facile self-crosslinking to improve mechanical and durability of polynorbornene for alkaline anion exchange membranes, Int. J. Hydrogen Energy, 2020, 45, 13068–13079 CrossRef CAS.
- W. Wang, D.-F. Cao, X.-W. Sun, L. Pan, Z. Ma and Y.-S. Li, The influence of various cationic group on polynorbornene based anion exchange membranes with hydrophobic large steric hindrance arylene substituent, Chin. J. Polym. Sci., 2023, 41, 278–287 CrossRef CAS.
- J. Stonawski, F. Junginger, A. Münchinger, L. Hager, S. Thiele and J. Kerres, Fluorine-free polynorbornene membranes based on a sterically hindered pyridine for vanadium redox flow batteries, ACS Appl. Polym. Mater., 2024, 6, 13512–13517 CrossRef CAS.
- S. Koch, L. Metzler, S. K. Kilian, P. A. Heizmann, F. Lombeck, M. Breitwieser and S. Vierrath, Toward scalable production: catalyst-coated membranes (CCMs) for anion-exchange membrane water electrolysis via direct bar coating, Adv. Sustainable Syst., 2023, 7, 2200332 CrossRef CAS.
- H. Daimon, H. Okitsu and J. Kumanotani, Glass transition behaviors of random and block copolymers and, Polym. J., 1975, 7, 460–466 CrossRef CAS.
- L. Hager, M. Hegelheimer, J. Stonawski, A. T. S. Freiberg, C. Jaramillo-Hernández, G. Abellán, A. Hutzler, T. Böhm, S. Thiele and J. Kerres, Novel side chain functionalized polystyrene/O-PBI blends with high alkaline stability for anion exchange membrane water electrolysis (AEMWE), J. Mater. Chem. A, 2023, 11, 22347–22359 RSC.
- L. Hager, M. Schrodt, M. Hegelheimer, J. Stonawski, P. Leuaa, C. Chatzichristodoulou, A. Hutzler, T. Böhm, S. Thiele and J. Kerres, Cationic groups in polystyrene/O-PBI blends influence performance and hydrogen crossover in AEMWE, Chem. Commun., 2025, 61, 149–152 RSC.
- R. Ren, S. Zhang, H. A. Miller, F. Vizza, J. R. Varcoe and Q. He, Facile preparation of novel cardo poly(oxindolebiphenylylene) with pendent quaternary ammonium by superacid-catalysed polyhydroxyalkylation reaction for anion exchange membranes, J. Membr. Sci., 2019, 591, 117320 CrossRef CAS.
- J. H. Kim, J. Jang, D.-Y. Lee and W.-C. Zin, Thickness and composition dependence of the glass transition temperature in thin homogeneous polymer blend films, Macromolecules, 2002, 35, 311–313 Search PubMed.
- R. Narducci, J.-F. Chailan, A. Fahs, L. Pasquini, M. L. Di Vona and P. Knauth, Mechanical properties of anion exchange membranes by combination of tensile stress-strain tests and dynamic mechanical analysis, J. Polym. Sci. Part B: Polym. Phys., 2016, 54, 1180–1187 CrossRef CAS.
- D. Pan, S. Chen and P. Jannasch, Alkali-stable anion exchange membranes based on poly(xanthene), ACS Macro Lett., 2023, 12, 20–25 CrossRef CAS PubMed.
- W.-H. Lee, Y. S. Kim and C. Bae, Robust hydroxide ion conducting poly(biphenyl alkylene)s for alkaline fuel cell membranes, ACS Macro Lett., 2015, 4, 814–818 CrossRef CAS PubMed.
- W.-H. Lee, E. J. Park, J. Han, D. W. Shin, Y. S. Kim and C. Bae, Poly(terphenylene) anion exchange membranes: the effect of backbone structure on morphology and membrane property, ACS Macro Lett., 2017, 6, 566–570 CrossRef CAS PubMed.
- M. He, Y. Huang, F. Wang, Z. Lv, X. Hu, X. Wang, M. Liu, B. Hu, Q. Chen, H. Li, J. Zheng and N. Li, Insight into alkaline stability of N-heteroatom on N-dimethylpiperidinium based anion exchange membranes (AEMs) for alkaline water electrolysis, J. Membr. Sci., 2023, 688, 122109 CrossRef CAS.
- D. Li, E. J. Park, W. Zhu, Q. Shi, Y. Zhou, H. Tian, Y. Lin, A. Serov, B. Zulevi, E. D. Baca, C. Fujimoto, H. T. Chung and Y. S. Kim, Highly quaternized polystyrene ionomers for high performance anion exchange membrane water electrolysers, Nat. Energy, 2020, 5, 378–385 Search PubMed.
- M. S. Cha, J. E. Park, S. Kim, S.-H. Han, S.-H. Shin, S. H. Yang, T.-H. Kim, D. M. Yu, S. So, Y. T. Hong, S. J. Yoon, S.-G. Oh, S. Y. Kang, O.-H. Kim, H. S. Park, B. Bae, Y.-E. Sung, Y.-H. Cho and J. Y. Lee, Poly(carbazole)-based anion-conducting materials with high performance and durability for energy conversion devices, Energy Environ. Sci., 2020, 13, 3633–3645 RSC.
- A. Allushi, T. H. Pham, J. S. Olsson and P. Jannasch, Ether-free polyfluorenes tethered with quinuclidinium cations as hydroxide exchange membranes, J. Mater. Chem. A, 2019, 7, 27164–27174 RSC.
- L. Tian, W. Ma, S. Tuo, F. Wang and H. Zhu, Novel polyaryl isatin polyelectrolytes with flexible monomers for anion exchange membrane fuel cells, J. Membr. Sci., 2024, 690, 122172 Search PubMed.
- Y. Ma, C. Hu, G. Yi, Z. Jiang, X. Su, Q. Liu, J. Y. Lee, S. Y. Lee, Y. M. Lee and Q. Zhang, Durable multiblock poly(biphenyl alkylene) anion exchange membranes with microphase separation for hydrogen energy conversion, Angew. Chem., Int. Ed., 2023, 62, e202311509 CrossRef CAS PubMed.
- T. Wang, Y. Wang and W. You, Polycyclic norbornene based anion-exchange membranes with high ionic conductivity and chemical stability, J. Membr. Sci., 2024, 702, 122747 CrossRef CAS.
- D. R. Dekel, M. Amar, S. Willdorf, M. Kosa, S. Dhara and C. E. Diesendruck, Effect of water on the stability of quaternary ammonium groups for anion exchange membrane fuel cell applications, Chem. Mater., 2017, 29, 4425–4431 CrossRef CAS.
- D. R. Dekel, S. Willdorf, U. Ash, M. Amar, S. Pusara, S. Dhara, S. Srebnik and C. E. Diesendruck, The critical relation between chemical stability of cations and water in anion exchange membrane fuel cells environment, J. Power Sources, 2018, 375, 351–360 Search PubMed.
- S. Willdorf-Cohen, A. Zhegur-Khais, J. Ponce-González, S. Bsoul-Haj, J. R. Varcoe, C. E. Diesendruck and D. R. Dekel, Alkaline stability of anion-exchange membranes, ACS Appl. Energy Mater., 2023, 6, 1085–1092 CrossRef CAS PubMed.
- R. J. Gilliam, J. W. Graydon, D. W. Kirk and S. J. Thorpe, A review of specific conductivities of potassium hydroxide solutions for various concentrations and temperatures, Int. J. Hydrogen Energy, 2007, 32, 359–364 Search PubMed.
- M. Moreno-González, P. Mardle, S. Zhu, B. Gholamkhass, S. Jones, N. Chen, B. Britton and S. Holdcroft, One year operation of an anion exchange membrane water electrolyzer utilizing Aemion+® membrane: minimal degradation, low H2 crossover and high efficiency, J. Power Sources, 2023, 19, 100109 Search PubMed.
- E. J. Park, P. Jannasch, K. Miyatake, C. Bae, K. Noonan, C. Fujimoto, S. Holdcroft, J. R. Varcoe, D. Henkensmeier, M. D. Guiver and Y. S. Kim, Aryl ether-free polymer electrolytes for electrochemical and energy devices, Chem. Soc. Rev., 2024, 53, 5704–5780 RSC.
- J. Nikl, K. Hofman, S. Mossazghi, I. C. Möller, D. Mondeshki, F. Weinelt, F.-E. Baumann and S. R. Waldvogel, Electrochemical oxo-functionalization of cyclic alkanes and alkenes using nitrate and oxygen, Nat. Commun., 2023, 14, 4565 CrossRef CAS PubMed.
- P. Fortin, T. Khoza, X. Cao, S. Y. Martinsen, A. Oyarce Barnett and S. Holdcroft, High-performance alkaline water electrolysis using Aemion™ anion exchange membranes, J. Power Sources, 2020, 451, 227814 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details on materials, synthesis and characterization. See DOI: https://doi.org/10.1039/d4ta07753a |
| This journal is © The Royal Society of Chemistry 2025 |