
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New insight into designing a thick-sintered cathode for Li-ion batteries: the impact of excess lithium in LiCoO2 on its electrode performance†
Shinichi
Takeno
a,
Taiki
Suematsu
a,
Ryusei
Kunisaki
a,
Gen
Hasegawa
 b,
Ken
Watanabe
*c,
Naoaki
Kuwata
b,
Kazutaka
Mitsuishi
b,
Tsuyoshi
Ohnishi
b,
Kazunori
Takada
b,
Kohichi
Suematsu
c and
Kengo
Shimanoe
c
b,
Ken
Watanabe
*c,
Naoaki
Kuwata
b,
Kazutaka
Mitsuishi
b,
Tsuyoshi
Ohnishi
b,
Kazunori
Takada
b,
Kohichi
Suematsu
c and
Kengo
Shimanoe
c
aDepartment of Molecular and Material Sciences, Interdisciplinary Graduate School of Engineering Sciences, Kyushu University, Kasuga, Fukuoka, 816-8580, Japan
bNational Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba 305-0044, Japan
cDepartment of Advanced Materials Science and Engineering, Faculty of Engineering Sciences, Kyushu University, Kasuga, Fukuoka, 816-8580, Japan. E-mail: watanabe.ken.331@m.kyushu-u.ac.jp
First published on 6th December 2024
Abstract
Increasing the capacity of Li-ion batteries is one of the critical issues that must be addressed. A thick and dense electrode using an active material sintered disk is expected to have a high capacity because the volume of the active material is 100% in the cathode. This study focused on LiCoO2, the most well-known active material for the cathode, to improve the properties of the sintered cathode. We investigated the impact of excess Li on various properties. We found that the degree of c-axis orientation in the sintered disk decreased as excess Li increased. In addition, results of 7Li-MAS-NMR suggest the presence of defects resulting from excess Li when the Li excess reached 5.1% or more. The discharge capacity of the LiCoO2 sintered cathode increased as the amount of excess Li increased, and a maximum discharge capacity of 11.2 mA h cm−2 was obtained when the Li excess amount was 7.3%. This result was attributed to the significant improvement in the Li-ion conductivity of LiCoO2 by both the decrease in the degree of c-axis orientation and the introduction of defects due to excess Li. Notably, introducing defects derived from excess Li enhances the Li-ion conductivity. Thus, tuning the amount of excess Li for the LiCoO2 sintered cathode was crucial in enhancing its electrochemical performance as an electrode.
Introduction
Li-ion batteries are widely used as power sources for mobile applications and electric vehicles, and there is a strong demand for highly capacitive batteries to realize a carbon-neutral society.1 The capacity of Li-ion batteries depends on the loading amount of active materials in electrodes. Therefore, to achieve high capacity, efforts to increase the amount of active material in electrodes are underway. One approach to increase the amount of active material is to increase the thickness of electrodes. Thick electrodes result in a higher amount of active material per unit area. Therefore, thick electrodes can achieve high capacity. To increase the thickness of electrodes, methods such as using foam current collectors to shorten the electronic conduction path,2,3 formation of electrodes using 3D printing,4 composite electrodes using active materials and solid electrolytes,5 bilayer electrodes,6 electrodes with a conductive agent/binder composite,7 electrode preparation by a dry electrode coating process8 and phase-inversion method9 have been attempted.Another approach is to increase the volume ratio of the active material in the electrode. Park et al. reported an all-in-one multi-layered cathode–separator–anode monolith structure with slurry that functions as electrochemically active glue and has a high capacity of 44.5 mA h.10 Generally, composite cathodes of active material, a conductive additive, binder, and organic electrolyte are widely used for Li-ion batteries. Therefore, the amount of active material in the electrode is limited. To overcome this limitation, Yamada et al. proposed a sintered cathode that consists of a highly densified LiCoO2 disk.11 Since the electrode does not contain electrolytes or conductive additives, it can be composed only of active material, resulting in high capacity. Furthermore, if the active material can be co-sintered with the oxide-based electrolyte, the sintered high-capacity cathode is suitable for a high-performance cathode of the co-sintered solid-state battery.12–15
When we design the sintered cathode, there are two key factors: the mixed Li-ion and electronic conductivity of LiCoO2, the most representative active material for the cathode,16 and the interfacial resistance between LiCoO2 and electrolyte. Regarding interfacial resistance, it has been shown that the interface resistance between the solid electrolyte, Li3PO4, and LiCoO2 can be reduced to 8.6 Ω cm2 in thin-film batteries.17 In addition, Ohnishi et al. suggested that negligibly low LiCoO2/Li3PO4 interface resistance can be achieved by controlling sputtering conditions during interface formation.18 Therefore, even a low surface area is expected to be sufficient to achieve low resistance. Therefore, the most critical key is increasing the mixed conductivity of Li ions and electrons in LiCoO2.
As for the mixed conductivity of LiCoO2, utilizing the anisotropic conduction derived from the crystal structure of LiCoO2 is a promising approach. LiCoO2 exhibits rapid electronic/Li-ion conduction pathways along the c-plane, while conduction in the c-axis direction is significantly low.19 Yamada et al. reported the cathode properties using an oriented sintered disk of LiCoO2. A sintered cathode with (110)-orientation, the fast Li-ion conduction pathway, has a discharge capacity of 102.3 mA h g−1 at 1/3 C with a thickness of 130 μm. Many similar studies have also been conducted in epitaxial thin films. Among them, Kawashima et al. have demonstrated high-speed charge–discharge of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 C in (104)-oriented epitaxial thin films.20 On the other hand, despite low electronic/Li-ion conductivity along the c-axis, several research groups have reported that c-axis-oriented epitaxial thin films can operate as batteries. The grain boundary diffusion of Li ions21 often explains these phenomena. Hasegawa et al. suggested that antisite Li defects, which Li occupies at Co sites, act as a conduction path along the c-axis direction and enhance the Li-ion diffusion based on DFT calculation.22 According to their idea, controlling not only the orientation of the LiCoO2 sintered cathode but also the defects caused by excess Li can enhance the Li-ion conductivity and improve battery performance.
000 C in (104)-oriented epitaxial thin films.20 On the other hand, despite low electronic/Li-ion conductivity along the c-axis, several research groups have reported that c-axis-oriented epitaxial thin films can operate as batteries. The grain boundary diffusion of Li ions21 often explains these phenomena. Hasegawa et al. suggested that antisite Li defects, which Li occupies at Co sites, act as a conduction path along the c-axis direction and enhance the Li-ion diffusion based on DFT calculation.22 According to their idea, controlling not only the orientation of the LiCoO2 sintered cathode but also the defects caused by excess Li can enhance the Li-ion conductivity and improve battery performance.
The enhancement of battery performance with the excess Li was previously demonstrated using the battery with liquid electrolyte and LiCoO2 powder.23,24 However, the mechanism is still under discussion. Levasseur et al. reported that 7Li MAS NMR measurements for LiCoO2 with or without Li-excess, calcined at 900 °C, showed an evident local structural change in the sample with Li-excess.25 They suggested the existence of Li defects, which are substituted for the Co site (antisite Li), and oxygen vacancies compensate for the antisite Li. This model was used in the previous calculation by Hasegawa et al. and may serve as a diffusion path in the c-axis direction.22
On the other hand, Murakami et al. examined the state of excess Li in LiCoO2 calcined at 800 °C.26 Based on various investigations, they stated that excess Li exists in their sample as a defect pair of the low spin Co2+ and interstitial Li. Since this interstitial Li may also work as a Li-ion conductive carrier, it may improve Li-ion conductivity. These defect models differ depending on the heat treatment conditions. However, in any case, defects derived from excess Li are thought to contribute to the improvement of Li ion conductivity in LiCoO2. Furthermore, it has also been reported that excess Li promotes grain growth27 and changes the direction of grain growth,28 which is expected to bring about unique changes in the orientation and microstructure of the sintered disk.
In this study, we aim to improve the electrode performance of the LiCoO2 sintered cathode and investigate the effects of excess Li on its microstructure and electrical properties.
Experimental
Preparation of the LiCoO2 sintered disk
LiCoO2 with excess Li of 0%, 1.0%, 2.0%, 3.0%, 4.1%, 5.1%, 6.2%, 7.3%, 8.3%, and 12.8% powder was synthesized by the amorphous malic acid precursor method.12,29DL-malic acid (C4H6O5, 99%, Fujifilm Wako Pure Chemical Corp. Japan), lithium nitrate (LiNO3, 99.9%, Fujifilm Wako Pure Chemical Corp. Japan), and cobalt nitrate (Co(NO3)2·6H2O, 99.5%, Fujifilm Wako Pure Chemical corp. Japan) were dissolved in distilled water. The pH of the mixed solution was adjusted to 3 with aqueous ammonia (28%). The solution was evaporated to dryness and heated at 400 °C until the reactive ignition became unobservable. The powder was calcined at 850 °C for 10 hours, and LiCoO2 powder was obtained. LiCoO2 powder was grounded and ball-milled at 450 rpm for 20 hours with isopropanol as the solvent. After ball milling, the solvent was evaporated and ground in a mortar. The fine powder was press-formed into a disk shape and pressed again by cold isostatic pressing. The obtained disks were sintered at 1000 °C for 15 hours. The disk was covered with LiCoO2 powder to avoid the evaporation of Li and contamination of other elements during sintering. In all compositions, the relative density of sintered disks achieved more than 92%.Material characterization
The crystal structure of the LiCoO2 sintered disk was evaluated using X-ray diffraction (XRD: MiniFlex600, RIGAKU, Japan) with Cu Kα as an X-ray source. All samples can be assigned to the layered rock salt structure (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), as shown in Fig. S1.† To evaluate the c-axis orientation degree for the sintered disk, we defined the c-axis orientation factor (f003) as the following equation.
m), as shown in Fig. S1.† To evaluate the c-axis orientation degree for the sintered disk, we defined the c-axis orientation factor (f003) as the following equation.I104 and I003 are the diffraction peak intensity for 104 and 003, respectively. The constant value 0.66 is calculated from the ideal I104/I003 based on ICSD 51381.

The change in the local structure around Li was evaluated by 7Li magic angle spinning NMR (MAS-NMR) using an ECA-400 spectrometer (JEOL Ltd, Japan). The resonance frequency of the 7Li nucleus was 155.4 MHz. A 1 mol L−1 LiCl aqueous solution was used as the chemical shift reference at 0 ppm. A 3.2 mm MAS probe (HXMAS probe; JEOL) and a 3.2 mm zirconia sample tube were used. The MAS spinning rate was 20 kHz. The width of the π/2 pulse was 2.8 μs. The pulse-recycling period was kept longer than 5 s to confirm spin recovery. A single-pulse sequence obtained the NMR spectra. Scanning transmission electron microscope (STEM) observation was conducted using a JEM-ARM200F (JEOL Ltd, Japan). Focused ion beam milling was used to prepare the specimens for STEM observation.
The oxidation state of Co ions was evaluated by electron spin resonance (ESR, ESR 5000, Bruker, Germany). LCO sintered disk grinding powder was obtained and dried in a vacuum at 200 °C, and approximately 50 mg was used for measurement. The field was swept from 100 to 600 mT in 60 s. The modulation amplitude was 0.2 mT at a modulation frequency of 100 kHz. Microwave power was 10 mW.
Electrical properties and battery performance
The electric conductivity of the LiCoO2 sintered disk was evaluated by the DC polarization method with the ion-blocking electrode. The surface of the sintered disk was polished with the lapping film sheet (3 M) up to 280 μm in thickness. The Au electrode for ion blocking was deposited on both sides of the sintered disk by sputtering. The DC polarization method was carried out using an HJ1001SD8 (Hokuto Denko Co., Ltd, Japan) in galvanostatic mode. The details of the experimental conditions are provided in Fig. S1.†Cathode performance was evaluated using an organic electrolyte-based cell with a Li-metal anode. Firstly, both sides of the sintered disk without any carbon or conductive additives were polished up to 180 μm in thickness. The Au current collector was deposited on one side of the polished disk by sputtering. Then, the sample was transferred to an Ar-filled glovebox, and the test cell was fabricated, as shown in Fig. S2.† Here, the loading amount is 82–93 mg cm−2. The charge–discharge performance of the fabricated cell was evaluated by galvanostatic charging/discharging with a constant current of 0.03 C using an HJ1001SD8 (Hokuto Denko Co., Ltd, Japan). The upper and lower cut-off voltages were set at 4.2 and 3.0 V (vs. Li+/Li). The non-blocking cell was used to evaluate the Li-ion conductivity for the sintered disk before and after charging at 4.2 V. A symmetric cell consisting of Li|liquid electrolyte|LiCoO2 sintered disk|liquid electrolyte|Li, as shown in Fig. S3,† was assembled. The electrical resistance of the cell was evaluated by the DC polarization method at 25 °C. The Li-ion conductivity was calculated using the slope of the thickness dependence of the total resistance for the cell. Experimental conditions are provided in Fig. S2.†
Results and discussion
Firstly, to reveal the effect of excess Li on the crystal orientation of the sintered LiCoO2 disk, XRD was conducted on the surface of the LiCoO2 sintered disks. For all samples, the diffraction peaks can be assigned to NaFeO2-type LiCoO2, as shown in Fig. S1.†Fig. 1a shows the dependence of the c-axis orientation factor (f003) on the Li-excess amount. As the amount of Li excess increased from 0% to 5.1%, the value of f003 gradually decreased, although the rapid drop at 3.0% was confirmed. Then, f003 rapidly decreases with the increase in the Li excess amount. In the case without excess Li (stoichiometric composition), f003 was 71%, and the value was the highest among all samples we tested. On the other hand, in the range from 7.3% to 12.8%, these values were almost constant at approximately 15%. In this study, since uniform powders are sintered without applying pressure, there is no driving force to orient in the direction of thickness during sintering. Therefore, the mechanism of orientation by the crystal growth during sintering is unlikely, and the orientation may be caused by pressing for molding. For plate-like particles, the basal plane is preferentially oriented toward applying pressure. Then, the crystal orientation before and after sintering was examined. Fig. 1b shows f003 before and after sintering for Li excess amounts of 0% and 12.8%, respectively. The f003 of 0% LiCoO2 before and after sintering is almost the same as that of 67% and 71%, respectively. This result indicates that it is already preferentially oriented toward the c-axis at the molding. Similarly, in the case of 12.8%, the f003 before and after sintering was 22% and 15%, respectively. The f003 after molding is directly related to the orientation of the sintered compact. Ceder et al. reported the effect of the excess Li on the crystal growth from DFT calculations.28 They reported that the c-plane preferentially grows in the stoichiometric LiCoO2 because the surface energy of (003) is the lowest. In contrast, the crystal growth becomes isotropic with excess Li because excess Li increases the surface energy of (003). Therefore, it is considered that the grain growth of LiCoO2 during the first calcination is altered by excess Li, resulting in a particle shape-dependent change in the degree of c-axis orientation under pressing for molding. Unfortunately, we observed no big difference in the particle shape between 0% and 12.8% from the SEM observation shown in Fig. S4.† Thus, further investigation into the orientation mechanism is needed. | ||
| Fig. 1 (a) The c-axis orientation factor for the sintered LiCoO2 as a function of Li-excess amount. (b) The comparison of the c-axis orientation factor of 0% and 12.8% Li excess LiCoO2 disks before and after sintering at 1000 °C. | ||
To reveal the local structure change in LiCoO2 by adding excess Li, 7Li-MAS-NMR was carried out. Fig. 2a shows 7Li MAS NMR spectra for LiCoO2 prepared with different Li-excess amounts. In the 5.1–8.3% Li-excess samples, minor peaks around 3, −6, and −16 ppm were observed. Those minor peaks agreed with the previous results,25 indicating the formation of Li-excess-related defects. Two different models were proposed, and their heat treatment conditions were different. Levasseur et al. performed high-temperature heat treatment and proposed the formation of antisite Li-oxygen vacancy couples, as shown in Fig. 2b.25 In our case, the sintering temperature was 1000 °C, similar to that reported by Levasseur et al. Thus, we believe that the antisite Li and oxygen vacancy couple is formed after sintering.
 | ||
| Fig. 2 (a) 7Li-MAS-NMR spectra for the sintered LiCoO2 with different Li-excess amounts. The crystal structure model of LiCoO2: (b) interstitial Li and (c) antisite Li, respectively. | ||
Next, the effect of excess Li on the electronic conductivity of the LiCoO2 sintered disk was investigated. Fig. 3a shows the electronic conductivity of the LiCoO2 sintered disks as a function of the Li excess amount, and correlations between current and voltage are shown in Fig. S5.† The dependency of electronic conductivity on Li excess amount can be divided into two parts. In the range from 0% to 4.1%, the electronic conductivity drastically increased with an increase in excess Li. The conductivity reached 2 × 10−3 S cm−1 when the Li-excess amount was 4.1%. In contrast, the conductivity decreased when the Li-excess amount became larger than 4.1%. Fig. 3b shows the relationship between the electronic conductivity and the c-axis orientation factor (f003). As seen in Fig. 3b, in the f003 range from 60% to 80% without the local structure change, the electronic conductivity strongly depends on the c-axis orientation degree of the sintered disk. In contrast, the electronic conductivity of the sintered disk, in which 7Li-MAS-NMR detected the local structure change, decreased despite decreasing the c-axis orientation degree. This tendency means that the excess Li-related defect affects the electronic conduction in the LiCoO2. There are two possible excess Li-related defects: the pair of the antisite Li and oxygen vacancy25 and the pair of the interstitial Li and low spin Co2+.26 In our case, LiCoO2 was sintered at 1000 °C, which was similar to the one reported by Levasseur et al.25 According to their model, the defect in our LiCoO2 sintered disk is likely to be the antisite Li and oxygen vacancy pair. LCO is a p-type semiconductor, and Co4+, which has unpaired electrons in d orbitals, seems to contain a charge carrier. Therefore, it is considered that the electronic conductivity of LCO depends on the amount of Co4+. To evaluate Co4+ in LCO, we conducted ESR measurements. Fig. 3(c and d) show the ESR spectra of LCO of 1.0%, 4.1%, and 7.3% Li-excess and the relationship between the electronic conductivity and the maximum value of the peak in ESR spectra (Imax). The peak appeared at g ≈ 2.13 for all spectra, and the electronic conductivity strongly depends on Imax. Mukai et al. reported this peak can be assigned to unpaired electrons for Co4+ in a low-spin state.30 Compared with 1.0% Li excess, the maximum value of the EPR peak of 4.1% excess was larger. It was indicated that an increase in Co4+ was due to charge compensation of the excess Li. On the other hand, in the case of 7.3% Li-excess with antisite Li, the maximum value of the peak was lower than that of 4.1% without antisite Li. These results indicate that the antisite Li was compensated for not only by Co4+ but also by oxygen vacancy, resulting in a decrease in the electronic conductivity with the formation of the antisite Li. There is another possible reason for the decrease in electronic conductivity. That is the formation of an impurity, an electronic insulator, at the grain boundary. From STEM observation, as shown in Fig. S6,† there is no impurity at the grain boundary. Therefore, the formation of the antisite Li related to the excess Li is a reasonable reason for the decrease in the electronic conductivity with an increase in excess Li.
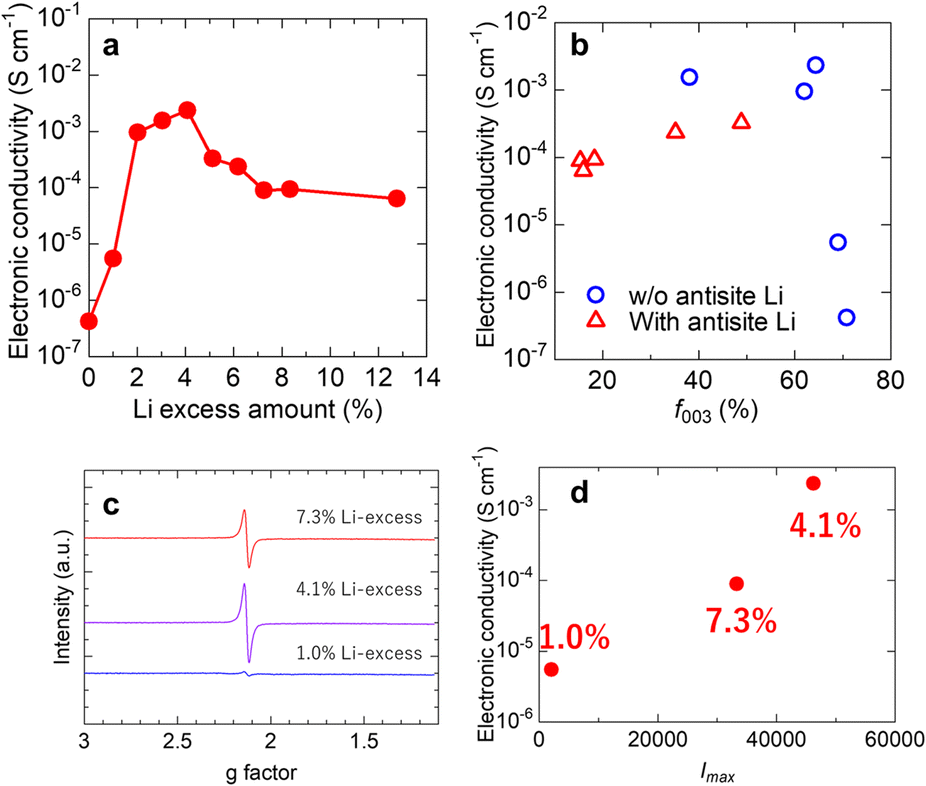 | ||
| Fig. 3 The dependence of electronic conductivity on (a) the Li excess amount in LiCoO2 sintered disk and (b) the c-axis orientation factor f003. (c) ESR spectra of LCO with excess Li. (d) The relationship between electronic conductivity and the maximum value of the peak in ESR spectra (Imax). | ||
The charge–discharge properties for the LiCoO2 sintered cathode prepared in the composition of 1.0%, 3.0%, and 7.3% Li-excess are shown in Fig. 4a–c. For all samples, the charge capacity was more than 120 mA h g−1, and no significant difference was observed. On the other hand, the discharge capacity drastically increased as the amount of Li-excess increased. Fig. 4d shows the dependence of the discharge capacity for the first three cycles on the Li excess amount. It is clear that the discharge capacity drastically increases with an increase in the Li excess amount to 5.1%. In addition, the discharge capacity is almost constant in the Li excess amount range of more than 5.1%. 7.3% excess Li exhibits the highest discharge capacity of 135.8 mA h g−1 and 11.2 mA h cm−2. Fig. S7† shows the relationship between cycle number and discharge capacities of charge–discharge tests at 0.1 C using LCO sintered disks with a thickness of approximately 130 μm. The capacity retention was improved by excess Li addition. As mentioned before, the excess Li affects the c-axis orientation degree of the disk and forms the anti-site Li defect. Thus, this tendency should be related to both of them. Fig. 4e shows the discharge capacity of the LiCoO2 sintered cathode as a function of the c-axis orientation factor f003. Here, the red triangle and the blue rectangle show the LiCoO2 disk with or without the Li-excess-related defect detected by 7Li-MAS-NMR, respectively. The discharge capacity of LiCoO2 without Li-excess-related defects depends on the c-axis orientation degree, indicating that the relaxation of c-axis orientation for the sintered disk causes the initial increase in the discharge capacity. In contrast, these values do not depend on the c-axis orientation degree for the samples with Li-excess-related defects. It should be noted that, as seen in region (i) shown in Fig. 4e, although the c-axis orientation degree of 3.0% is lower than that of 5.1%, 5.1% Li excess exhibits a higher discharge capacity of 125 mA h g−1 than 3.0%. These results clearly indicate that not only the c-axis orientation degree but also the Li-excess-related defects affect the electrode performance. From the electronic conductivity measurement results, 5.1% exhibits lower conductivity than 3.0%. Thus, the higher discharge capacity of 5.1% than 3.0% is likely related to the Li-ion conductivity. To confirm whether the Li-ion conductivity can be increased by introducing the Li-excess-related defects, the Li-ion conductivities of 3.0% and 5.1% Li-excess LiCoO2 disks were evaluated. Fig. 4f compares the Li-ion conductivity of 3.0%,4.1%, 5.1%, and 7.3% Li-excess LiCoO2 disks, and correlations between current and voltage are shown in Fig. S8.† The Li-ion conductivity of 3.0% was 2.7 × 10−6 S cm−1, higher than that of 4.1% (8.4 × 10−8 S cm−1). As Fig. 1a shows, f003 of 3.0% is lower than that of 4%. Therefore, it was found that the f003 decreases due to excess Li, contributing to the increase of Li-ion conductivity. The Li-ion conductivity of 5.1% was 5.2 × 10−6 S cm−1, higher than that of 3.0%. Therefore, it was found that the Li-excess-related defect enhances the Li-ion conductivity of the LiCoO2 sintered disk. This result agrees with the prediction calculated by DFT, suggesting the antisite Li probably works as the diffusion pathway across the CoO6 layer.22 Moreover, it was reported that the Li-ion diffusion coefficient of LiCoO2 increased with an increase in the charging state.22,31Fig. 4g shows the Li-ion conductivity of LiCoO2 at 4.2 V after charging under CCCV mode, and correlations between current and voltage are shown in Fig. S9.† For all samples, the Li-ion conductivity at 4.2 V was much higher than that before charging, reaching 10−4–10−5 S cm−1. In addition, a rate of increase in Li-ion conductivity before and after charging agrees with the results of the diffusion coefficient.22,31 These results strongly indicate that the increase in Li-ion conductivity through the charging process enhances the charging behavior for all samples, resulting in similar charge capacities. Fig. 4h shows the correlation between charge–discharge capacity at the 1st cycle and Li-ion conductivity before charging. Although the charge capacity is almost constant, the discharge capacity depends on the Li-ion conductivity before charging. As mentioned, Li-ion conductivity drastically decreases as the discharge proceeds during lithiation due to the decrease in Li vacancy. This trend suggests that to enhance the discharge capacity through the thick and dense LiCoO2 sintered pellet, the improvement in the Li-ion conductivity for full lithiation of LiCoO2 is crucial.
 | ||
| Fig. 4 The charge–discharge curves for sintered disks of (a) 1.0%, (b) 3.0%, and (c) 7.3% Li-excess LiCoO2. The dependence of the discharge capacity of LiCoO2 sintered disks on (d) Li excess amount and (e) the c-axis orientation factor f003. (f) The Li-ion conductivity for 3.0%, 4.1%, 5.1% and 7.3% Li excess LiCoO2 sintered disks. (g) The Li-ion conductivity for 3.0%, 4.1%, 5.1% and 7.3% Li excess LiCoO2 sintered disks after CCCV charge to 4.2 V. (h) The correlation between the charge–discharge capacity of the 1st cycle and Li-ion conductivity before charging. | ||
These results show that excess Li affects the electrode properties of the LiCoO2 sintered cathode, and this tendency can be explained by the increase in Li-ion conduction through the LiCoO2 sintered disk. This means it is a crucial design factor when the LiCoO2 sintered cathode is applied to the Li-ion battery. Moreover, in the case of the co-sintered solid-state battery, the excess Li is added to the electrolyte to prevent Li-loss during sintering at high temperatures. Thus, more precise tuning of the amount of excess Li in LiCoO2 for the cathode of the co-sintered solid-state battery will be strongly required to realize the high-performance battery.
Conclusion
This study investigated the effect of excess Li in the LiCoO2 thickly and densely sintered cathode without conductive carbon additives on the microstructure, the local structure, electrical properties, and battery performance to enhance the electrode performance of thick, sintered LiCoO2 cathodes for Li-ion batteries. Four key findings followed.(1) The degree of c-axis orientation of the sintered disk decreases with an increase in the Li-excess amount.
(2) 7Li-MAS-NMR detects the formation of the defect-related excess Li.
(3) LiCoO2 with excess Li exhibited superior electrode properties compared to the stoichiometric version.
(4) The Li-ion conductivity increases with an increase in Li-excess amount.
Notably, the highest discharge capacity of 135.8 mA h g−1 and 11.2 mA h cm−2 was achieved when the Li-excess amount was 7.3%. This outstanding battery performance can be attributed to improving the Li-ion conductivity by decreasing the c-axis orientation and introducing the antisite Li defects. Therefore, our presented results strongly highlight the importance of tuning the excess Li in the LiCoO2 sintered cathode for highly capacitive Li-ion and solid-state batteries.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Shinichi Takeno conducted the experiment and drafted the manuscript. Taiki Suematsu and Ryusei Kunisaki carried out the experiment. Ken Watanabe created the idea, conducted the experiment, and performed supervision and editing. Gen Hasegawa and Naoaki Kuwata conducted the NMR experiment. Kazutaka Mitsuishi conducted STEM observation. Tsuyoshi Ohnishi and Kazunori Takada supported the cell fabrication experiment and performed the discussion. Koichi Sematsu and Kengo Simanoe performed the discussion and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Japan Science and Technology (JST), the Advanced Low Carbon Technology Research and Development Program, Specially Promoted Research for Innovative Next Generation Battery (ALCA-SPRING) project, Grant Number JPMJAL1301, Green technologies of excellence (GteX) Program Japan, Grant number JPMJGX23S22, the establishment of university fellowships towards the creation of science technology innovation, Grant Number JPMJFS2132, and JSPS KAKENHI Grant Number 22K04739. We thank the National Institute for Materials Science (NIMS) Battery Research Platform for preparing TEM samples.Notes and references
- L. Matthew, L. Jun, C. Zhongwei and A. Khalil, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- G. F. Yang, K. Y. Song and S. K. Joo, RSC Adv., 2015, 5, 16702 RSC.
- M. Fritsch, G. Standke, C. Heubner, U. Langklotz and A. Michaelis, J. Energy Storage, 2018, 16, 125–132 CrossRef.
- T. S. Wei, B. Y. Ahn, J. Grotto and J. A. Lewis, Adv. Mater., 2018, 30, 1 Search PubMed.
- Y. Li, S. Song, H. Kim, K. Nomoto, H. Kim, X. Sun, S. Hori, K. Suzuki, N. Matsui, M. Hirayama, T. Mizoguchi, T. Saito, T. Kamiyama and R. Kanno, Science, 2023, 381, 50 CrossRef CAS PubMed.
- M. Chouchane, W. Yao, A. Cronk, M. Zhang and Y. S. Meng, ACS Energy Lett., 2024, 9, 1480–1486 CrossRef CAS.
- X. Shen, H. Yu, L. Ben, W. Zhao, Q. Wang, G. Cen, R. Qiao, Y. Wu and X. Huang, J. Energy Chem., 2024, 90, 133–143 CrossRef CAS.
- W. Yao, M. Chouchane, W. Li, S. Bai, Z. Liu, L. Li, A. X. Chen, B. Sayahpour, R. Shimizu, G. Raghavendran, M. A. Schroeder, Y. T. Chen, D. H. S. Tan, B. Sreenarayanan, C. K. Waters, A. Sichler, B. Gould, D. J. Kountz, D. J. Lipomi, M. Zhang and Y. S. Meng, Energy Environ. Sci., 2023, 16, 1620–1630 RSC.
- Y. Zhang, Y. Xiao, L. Chen and S. Hu, J. Mater. Chem. A, 2024, 12, 16537–16545 RSC.
- S. H. Park, N. K. Lee, J. H. Han, S. H. Eo, Y. Park, K. C. Choi and Y. J. Lee, J. Mater. Chem. A, 2024, 12, 25056–25066 RSC.
- H. Yamada, T. S. Suzuki, T. Uchikoshi, M. Hozumi, T. Saito and Y. Sakka, APL Mater., 2013, 1, 042110 CrossRef.
- K. Watanabe, A. Tashiro, Y. Ichinose, S. Takeno, K. Suematsu, K. Mitsuishi and K. Shimanoe, J. Ceram. Soc., 2022, 130, 416 CrossRef CAS.
- N. Hayashi, K. Watanabe and K. Shimanoe, J. Mater. Chem. A, 2023, 11, 2042–2053 RSC.
- N. Hayashi, K. Watanabe, T. Ohnishi, K. Takada and K. Shimanoe, J. Mater. Chem. A, 2023, 11, 15681–15690 RSC.
- N. Hayashi, K. Watanabe and K. Shimanoe, J. Mater. Chem. A, 2024, 12, 5269–5281 RSC.
- K. Mizushima, P. C. Jonnes, P. J. Wiseman and J. B. Goodenough, Mater. Res. Bull., 1980, 15, 783 CrossRef CAS.
- M. Haruta, S. Shiraki, T. Suzuki, A. Kumatani, T. Ohsawa, Y. Takagi, R. Shimizu and T. Hitosugi, Nano Lett., 2015, 15, 1498 CrossRef CAS PubMed.
- T. Ohnishi and K. Takada, ACS Omega, 2022, 7, 21199 CrossRef CAS PubMed.
- Y. Takahashi, Y. Gotoh, J. Akimoto, S. Mizuta, K. Tokiwa and T. Watanabe, J. Solid State Chem., 2002, 164, 1 CrossRef CAS.
- K. Kawashima, T. Ohnishi and K. Takada, ACS Appl. Energy Mater., 2020, 3, 11803 CrossRef CAS.
- H. Xia and L. Lu, Electrochim. Acta, 2007, 52, 7014 CrossRef CAS.
- G. Hasegawa, N. Kuwata, Y. Tanaka, T. Miyazaki, N. Ishigaki, K. Takada and J. Kawamura, Phys. Chem. Chem. Phys., 2021, 23, 2438 RSC.
- N. Imanishi, M. Fujii, A. Hirano, Y. Takeda, M. Inaba and Z. Ogumi, Solid State Ionics, 2001, 140, 45 CrossRef CAS.
- M. Hirooka, T. Okumura and K. Ariyoshi, J. Electrochem. Soc., 2023, 17, 100506 CrossRef.
- S. Levasseur, M. Ménétrier, Y. Shao-Horn, L. Gautier, A. Audemer, G. Demazeau, A. Largeteau and C. Delmas, Chem. Mater., 2003, 15, 348–354 CrossRef CAS.
- M. Murakami, Y. Noda, Y. Koyama, K. Takegoshi, H. Arai, Y. Uchimoto and Z. Ogumi, J. Phys. Chem. C, 2014, 118, 15375 CrossRef CAS.
- T. Nakamura and A. Kajiyama, J. Eur. Ceram. Soc., 1999, 19, 871–874 CrossRef CAS.
- D. Kramer and G. Ceder, Chem. Mater., 2009, 21, 3799 CrossRef CAS.
- Y. Teraoka, H. Kakebayashi, I. Moriguchi and S. Kagawa, Chem. Lett., 1991, 20, 673 CrossRef.
- K. Mukai, Y. Aoki, D. Andreica, A. Amato, I. Watanabe, S. R. Giblin and J. Sugiyama, Phys. Rev. B:Condens. Matter Mater. Phys., 2014, 89, 094406 CrossRef.
- K. Dokko, M. Mohamedi, Y. Fujita, T. Itoh, M. Nishizawa, M. Umeda and I. Uchida, J. Electrochem. Soc., 2001, 148, A422 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta07377k |
| This journal is © The Royal Society of Chemistry 2025 |