
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Operando monitoring the redox activity of sodium vanadium titanium phosphate electrodes in organic and aqueous electrolytes by magnetometry†
Benedikt
Huemer‡
a,
Anna
Jodlbauer‡
b,
Martin
Wilkening
 bc,
Heinz
Krenn
d,
Peter
Knoll
d,
Roland
Würschum
a,
Ilie
Hanzu
bc and
Stefan
Topolovec
*a
bc,
Heinz
Krenn
d,
Peter
Knoll
d,
Roland
Würschum
a,
Ilie
Hanzu
bc and
Stefan
Topolovec
*a
aInstitute of Materials Physics, Graz University of Technology, NAWI Graz, Graz, Austria. E-mail: stefan.topolovec@tugraz.at
bInstitute for Chemistry and Technology of Materials, Graz University of Technology, NAWI Graz, Graz, Austria
cALISTORE – ERI European Research Institute, CNRS FR3104, Hub de l'Energie, Rue Baudelocque, F-80039 Amiens, France
dInstitute of Physics, University of Graz, NAWI Graz, Graz, Austria
First published on 17th December 2024
Abstract
A sol–gel method was used for the synthesis of composite electrodes consisting of sodium vanadium titanium phosphate (NVTP) and carbon. X-ray diffraction studies confirm that the NVTP possesses the NASICON crystalline structure, and cell tests indicate that the electrodes exhibit good electrochemical performance. Temperature-dependent magnetic susceptibility measurements reveal the presence of the V3+ and Ti4+ oxidation states in the pristine Na2VTi(PO4)3. The change in oxidation states during de-/sodiation was monitored continuously by means of operando magnetometry measurements in both organic (1 M NaPF6 in EC:PC) and aqueous electrolyte (1 M Na2SO4). The variation of the magnetic susceptibility demonstrates that in both types of electrolytes, the V3+/V4+redox couple is active in the high potential regime, whereas the Ti3+/Ti4+ couple operates in the low potential regime, and that no significant side reactions occur. Furthermore, it is shown that the two redox couples are distinctly separated and occur in a consecutive manner. Additionally, the results of our operando study indicate that both redox couples contribute approximately equally to the total capacity of NVTP.
1 Introduction
In recent years, phosphates with sodium super ionic conductor (NASICON) structure have been a subject of great interest for their application as electrodes in sodium-ion batteries.1–3 The most prominent example is Na3V2(PO4)3 (NVP), which is used as a cathode because of its relatively high redox potential of about 3.4 V vs. Na+/Na, assigned to the V3+/V4+ redox couple.2–4 On the other hand, NaTi2(PO4)3 (NTP) exhibits a relatively low redox potential of 2.1 V vs. Na+/Na attributed to the Ti3+/Ti4+ redox couple, which makes it a suitable anode material.1,2,5 The combination of NVP and NTP in a full cell is especially attractive because the overall cell voltage allows the use of aqueous electrolytes.6 Compared to organic electrolytes, such electrolytes are non-toxic, non-flammable, less expensive and more environmentally friendly, making aqueous sodium-ion batteries particularly appealing for future large-scale stationary energy storage systems.7,8 However, the application of NVP in aqueous electrolytes is limited by the rapid degradation caused by the dissolution of vanadium.6,9In 2015 Mason et al.10 showed, that this degradation can be prevented by substituting half of the vanadium with titanium, and, that the resulting sodium vanadium titanium phosphate (Na2VTi(PO4)3, NVTP) still exhibits a high potential redox couple. Further studies demonstrated, that multiple redox plateaus emerge for NVTP in organic11–16 as well as aqueous electrolytes,17–22 and, that symmetric full cells can be constructed using NVTP.11,13,14,17–21 Such symmetric NVTP cells have also recently been investigated for their potential use as electrochemical desalination devices.23,24In situ X-ray absorption spectroscopy measurements revealed that in organic electrolyte, the V3+/V4+ redox couple is active in the high potential regime, while Ti3+/Ti4+ is redox active at low potentials.11 Furthermore the redox activity of vanadium at high potentials and of titanium at low potentials was observed by ex situ X-ray photoelectron spectroscopy measurements in organic16 as well as aqueous electrolytes.17,19,21,23
One limitation of these spectroscopic techniques is that the spectra are recorded only at specific charging states. Continuous monitoring of the oxidation states by an operando technique would be particularly advantageous for electrodes containing multiple redox centers, such as NVTP, as it would allow for a more precise distinction between the various redox regimes. For instance, the behavior in the transition region could be resolved. Furthermore, in the individual studies mentioned above, these techniques were either applied solely in organic or aqueous electrolyte. A direct comparison of the redox activities in both aqueous and organic electrolytes is still lacking.
In this study we conducted operando magnetic susceptibility measurements on NVTP electrodes in organic and aqueous electrolytes. This technique enables the distinction of different redox couples based on the changing magnetic susceptibility, as well as a quantitative assessment if all electrons participate in these redox reactions.25–29 This quantification is of prime interest in the context of NVTP in aqueous electrolytes, since several studies reported parasitic side reactions in the low potential regime.18,20 We show in this work that the V3+/V4+ and Ti3+/Ti4+ redox regimes are well separated in both electrolytes, with both redox couples contributing approximately equally to the total capacity. Moreover, we elucidate that the charge compensation processes of NVTP are identical in both types of electrolyte, and that in both cases most of the charge is consumed for the oxidation/reduction of the NVTP and not for side reactions. As magnetometry is an integral method, our measurements also clearly show that the changes in the oxidation states occur uniformly throughout the bulk of the electrodes.
2 Experimental
2.1 Material synthesis
Sodium vanadium titanium phosphate carbon composite (NVTP@C) was prepared by a sol–gel synthesis procedure.24 Citric acid monohydrate (Sigma Aldrich, 99.5–102![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) %), acting as the gel forming agent, ammonium metavandate (Sigma Aldrich, ≥99%) and sodium dihydrogen phosphate (Sigma Aldrich, ≥99%) were dissolved in 200 ml deionized water. The molar ratio between the reactants can be described by 1:1:3 (corresponding to 0.04 M:0.04 M:0.12 M in concentration units). The solution was heated up to 353 K and stirred for 7 hours at this temperature. While stirring the solution turned dark blue indicating a reduction process from V4+ to V3+. A second solution was prepared where glacial acetic acid (Sigma Aldrich, ≥99%) and titanium isopropoxide (Sigma Aldrich, >97%) in a molar ratio of 1:1 (0.04 M:0.04 M in concentration units) were dissolved in pure ethanol. This mixture was then stirred for 1 hour at room temperature. Both solutions were poured together and heated to 373 K until the solvents have fully evaporated, leaving a gel in the beaker. Drying the gel over night at 333 K yielded a powder which was grinded by hand using an agate mortar. Thermal treatment was performed in a two step program on the fine powder using a quartz synthesis tube inserted into a tube furnace. In the first step, the powder was heated to 623 K with a heating rate of 3 K min−1 followed by a holding time of 5 hours, then a second heating step was carried out heating with the same rate up to 1223 K and holding this temperature for 12 hours. Finally, the sample was cooled down to room temperature by the natural cooling ramp of the oven. The whole thermal treatment was carried out strictly under nitrogen atmosphere. During the synthesis the citric acid converts into conductive carbon resulting in a NVTP@C composite. The obtained powder was again grounded by hand in an agate mortar before it was used for further analysis.
%), acting as the gel forming agent, ammonium metavandate (Sigma Aldrich, ≥99%) and sodium dihydrogen phosphate (Sigma Aldrich, ≥99%) were dissolved in 200 ml deionized water. The molar ratio between the reactants can be described by 1:1:3 (corresponding to 0.04 M:0.04 M:0.12 M in concentration units). The solution was heated up to 353 K and stirred for 7 hours at this temperature. While stirring the solution turned dark blue indicating a reduction process from V4+ to V3+. A second solution was prepared where glacial acetic acid (Sigma Aldrich, ≥99%) and titanium isopropoxide (Sigma Aldrich, >97%) in a molar ratio of 1:1 (0.04 M:0.04 M in concentration units) were dissolved in pure ethanol. This mixture was then stirred for 1 hour at room temperature. Both solutions were poured together and heated to 373 K until the solvents have fully evaporated, leaving a gel in the beaker. Drying the gel over night at 333 K yielded a powder which was grinded by hand using an agate mortar. Thermal treatment was performed in a two step program on the fine powder using a quartz synthesis tube inserted into a tube furnace. In the first step, the powder was heated to 623 K with a heating rate of 3 K min−1 followed by a holding time of 5 hours, then a second heating step was carried out heating with the same rate up to 1223 K and holding this temperature for 12 hours. Finally, the sample was cooled down to room temperature by the natural cooling ramp of the oven. The whole thermal treatment was carried out strictly under nitrogen atmosphere. During the synthesis the citric acid converts into conductive carbon resulting in a NVTP@C composite. The obtained powder was again grounded by hand in an agate mortar before it was used for further analysis.
2.2 XRD and TGA measurements
X-ray measurements were performed using a Rigaku Miniflex 600 diffractometer (2θ Bragg–Brentano geometry, Cu Kα) with a D/teX Ultra silicon strip detector. The data was recorded in a 2θ range from 10°–60° with a step size of 0.02.
For the thermogravimetric analysis (TGA) a Netzsch STA 449 F1 Jupiter thermal analyzer was used. The measurements were conducted in a temperature range from 303–1223 K.
2.3 Cell tests
For electrochemical cell tests slurries were prepared containing 80 wt% NVTP@C, 5 wt% carbon black (Super C, from TimCal) and 15 wt% PVDF binder dispersed in NMP. This composition of the slurry was taken over from the work of Shrivastava et al.24 The slurries were mixed in a planetary mill (Fritsch Pulverisette 7) for 15 minutes followed by a 5 minutes break for a total of four cycles. The dispersion was then blade coated (coating gap: 200 μm) on Al foil (Goodfellow, 50 μm thick) and the substrates were dried at 333 K for 12 hours. Prior to coating the slurry, the Al foil was etched by using 5 wt% KOH solution in water, followed by rinsing it with deionized water and drying. Electrodes were punched out with a diameter of 8.5 mm and their weight was determined. Prior to this, they were dried at 333 K in a vacuum oven (Büchi) for 24 hours to remove possible solvent residues. For the cell assembly three-electrode Swagelok cells were used stacking the dried electrodes, a glass fibre separator (Whatman GF/B) and sodium metal as counter and reference electrodes on top of each other. As an electrolyte a mixture of EC:PC 1:1 (vol. ratio) containing 1 M of NaPF6 was used. The cells were electrochemically characterized by cyclic voltammetry as well as galvanostatic cycling with potential limitation (GCPL) using a Biologic MPG-2 potentiostat.
2.4 Ex situ magnetic characterization
Temperature dependent magnetic susceptibility measurements were performed with a SQUID magnetometer (Quantum Design, MPMPS-XL-7) for the pristine NVTP@C powder. The powder was filled into a polycarbonate capsule which was then mounted in a SQUID sample holder straw. The susceptibility measurements were performed at an applied magnetic field of 5000 Oe in a temperature range of 300–5 K. The background signal arising from the polycarbonate capsule was corrected.2.5 Operando SQUID magnetometry measurements
For the operando SQUID magnetometry measurements during electrochemical charging/discharging a cell design established in previous works25–27 was applied. As described in detail in these references, this cell enables a continuous tracking of the changes of the magnetic susceptibility of the working electrode, which is in our case the NVTP@C electrode. A description and schematic of the general measurement principle of the SQUID magnetometer, as well as sketches of the electrochemical cell design used for the operando measurements, are provided in Section S1 of the ESI.†The NVTP@C electrodes were prepared using the same slurry as for the cell tests (see above). Either Al (ChemPUR, 99.999%) or Au wires (ChemPUR, 99.9%) with a diameter of 0.25 mm were used as substrate for the electrode material. Approximately 0.5 cm of one end of the wires were pressed to form a flat surface and this flat part was then dipped into the slurry. After drying the slurry overnight at ambient conditions the coated part of the wires was pressed again slightly, to ensure a good contact between the substrate and the electrode material. Afterwards the electrodes were dried at 383 K in a vacuum oven (Pfeiffer Vacuum HiCube/Carbolite) for 24hours to remove any residual solvent. The mass of the electrode material was determined by weighing the wires before and after the dip coating.
Operando magnetometry measurements were conducted for cells with organic electrolyte (1 M NaPF6 in EC:PC 1:1 vol%) as well as for cells with aqueous electrolyte (1 M Na2SO4). For the cells with organic electrolyte, the NVTP@C electrodes dip-coated on Al were used as working electrode. Metallic sodium pressed onto the tail ends of Cu wires (ChemPur, 99.9%, 0.25 mm diameter) served as counter and reference electrodes. For the aqueous electrolyte, symmetric cells were assembled with NVTP@C electrodes dip-coated on Au as working and counter electrodes. Here it was ensured, that the counter electrode contained more electrode material. A Ag wire (ChemPur, 99.995%, 0.25 mm diameter) coated on one end with AgCl was used as Ag/AgCl quasi reference electrode. Each electrode wire was inserted in a small polyethylene tube to ensure that only the parts with the electrode material were in contact with the electrolyte and to prevent short circuits in the cell. Long PE tubes (length: 17.5cm, diameter: 5mm) and NMR tubes (Rotilabo, length: 17.8 cm, diameter: 4.95 mm) were used as cell compartments for the organic and aqueous electrolyte cells, respectively. The electrodes were positioned in the tubes in such a way that the working electrode was approximately in the middle and the other two electrodes approximately 6 cm above. In this way it was ensured that only the working electrode causes a magnetic flux change in the SQUID pickup coils during the susceptibility measurements. The cells with organic electrolyte were assembled in an Ar filled glove box. The aqueous electrolyte was purged with Ar gas before the assembly. All cells were closed with a polypropylene plug which had feedthroughs for the wires and which were sealed with epoxy resin.
The operando magnetometry measurements were performed at 300 K and an applied magnetic field of 5000 Oe. The NVTP@C electrodes were charged and discharged with a constant current density of ±12 mA g−1.
3 Results and discussion
3.1 Characterization of pristine NVTP@C powder by XRD and TGA
In Fig. 1, the XRD pattern of the synthesized powder and a comparison with the reference patterns of Na3V2(PO4)3 and NaTi2(PO4)3 are shown. The measurement results indicate that the reflexions of our sample match very well with the reflexions of the Na3V2(PO4)3 reference sample. This is expected as we wanted to incorporate Ti into the Na3V2(PO4)3 host structure. The structure of the compound can be described by space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c which belongs to the NASICON structure class. Accordingly the reflexion positions can be indexed at 14.3°, 20.1°, 20.6°, 23.9°, 28.9°, 32.2°, and 36.0° to certain lattice planes (see Fig. 1).11 The crystal structure can be described by [TiO6]/[VO6] octahedra and [PO4] tetrahedra which are corner-sharing to build up a 3-dimensional framework where the Na+ ions are located at interstitial sites 6b and 18b.19,30
c which belongs to the NASICON structure class. Accordingly the reflexion positions can be indexed at 14.3°, 20.1°, 20.6°, 23.9°, 28.9°, 32.2°, and 36.0° to certain lattice planes (see Fig. 1).11 The crystal structure can be described by [TiO6]/[VO6] octahedra and [PO4] tetrahedra which are corner-sharing to build up a 3-dimensional framework where the Na+ ions are located at interstitial sites 6b and 18b.19,30
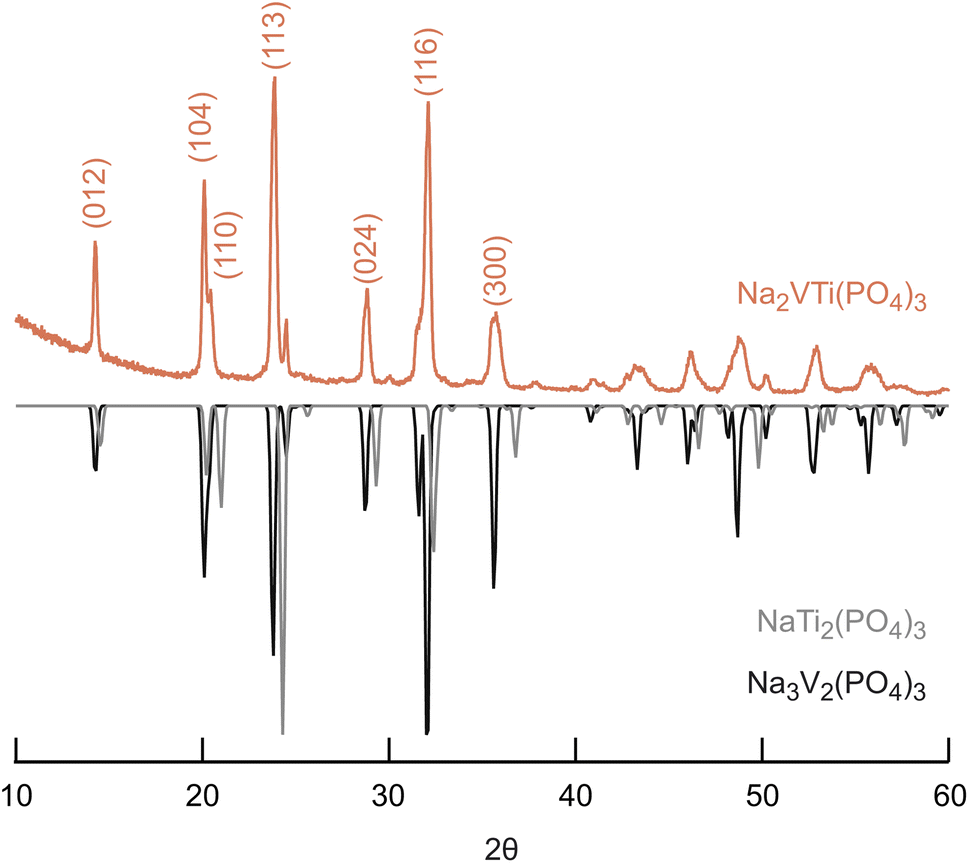 | ||
| Fig. 1 XRD pattern of Na2VTi(PO4)3 with reference patterns of NaTi2(PO4)3 (ICSD 29079) and Na3V2(PO4)3 (ICSD 248140); the main reflexes are indexed according to the crystal structure Rc. | ||
The carbon content in the pristine NVTP@C powder was determined to be about 5 wt% by TGA measurements (see Section S2 in ESI† for details). This value coincides quite well with literature values for other NVTP@C composites prepared by sol–gel synthesis.11,13,19,23 Since the TGA revealed a water content of about 3.5 wt%, the NVTP fraction in the pristine powder can be estimated to be about 91.5 wt%.
3.2 Cell test
To gain insight on the redox activity of the synthesized NVTP@C material, cyclic voltammetry was performed. In Fig. 2(a) the cyclic voltammogram between 1.75 V and 4 V at a scan rate of 0.1 mV s−1 is shown. As expected, the measurement yields two reversible redox peak couples, the one in the range of 2.1–2.3 V can be attributed to the redox reaction of Ti3+/Ti4+ while the other peak between 3.3–3.4 V stands for the V3+/V4+ reaction.1–5 In Fig. 2(b) the influence of different scan rates is presented. With increasing scan rate the oxidation and reduction peaks shift slightly towards higher potentials, however even at scan rates as high as 5 mV s−1 the two different peaks are still clearly visible. | ||
| Fig. 2 (a) Cyclic voltammetry of the NVTP@C electrode at a scan rate of 0.1 mV s−1. (b) Cyclic voltammetry of the same electrode at different scan rates between 0.1 mV s−1 and 5 mV s−1. Note the different scales of the ordinate axis. | ||
We conducted galvanostatic cycling with potential limitation in order to characterize the cycling performance of our material. Initially the potential range 1.75–4 V was examined (see Fig. 3(a)). The coulombic efficiency reaches nearly 100% for all cycles and without any visible decay. Cycling the cell at 0.1C resulted in a capacity as high as 105.7 mA h g−1 for the first discharge. This value coincides within a few percent with literature values of NVTP@C obtained under similar conditions11,13 and therefore shows that the electrochemical properties of our NVTP@C are consistent with literature. The discrepancy between the obtained capacity and the theoretical one of NVTP (124.8 mA h g−1) can be explained partly by the reduced NVTP content in our NVTP@C sample. However, the lower value also indicates that not all active material particles can be accessed and that there are some kinetic limitations. Increasing the cycling rates leads to smaller capacities, but even at a cycling rate as high as 10C still 5.5 mA h g−1 could be reached. Capacity retention can be checked by cycling again at 0.1C, this results in a value of 86 mA h g−1 for cycle number 69. This capacity value at the end of the measurement indicates only a slow capacity fade. However, the fact that the capacity at each cycling rate is not totally constant can be a hint towards slow decay of the material.
 | ||
| Fig. 3 Cycling protocol of the NVTP@C electrode. (a) Electrode cycled at different C-rates in a potential range of 1.75–4 V: specific capacity and coulombic efficiency are plotted against cycle number. (b) The same electrode cycled with a C-rate of 0.1 C in different potential ranges to gain insight into the capacities for the two different redox couples. The potential ranges chosen for the experiment are labeled in the figure. Note that the first five cycles shown in (b) correspond to the last cycles shown in (a), and that the deviations of the coulombic efficiency from around 100% are due to a change of the C-rate or the potential range. | ||
After the 70 cycles seen in Fig. 3(a) the potential range was changed to 4–2.5 V for five cycles to obtain the capacity caused by the redox reaction of V3+/V4+ (see Fig. 3(b)). Lastly the same was done for Ti3+/Ti4+ in a range of 2.5–1.75 V. The cycling was performed with a cycling rate of 0.1C. For the higher potential range, namely the V3+/V4+ redox couple we obtained capacities of 43 mA h g−1, the cycling of the Ti3+/Ti4+ redox couple yielded 30 mA h g−1. An overview of the different capacities is given in Table 1.
| Potential range/V | Redox couple | Capacity/mA h g−1 |
|---|---|---|
| 1.75–4 | Ti3+/Ti4+, V3+/V4+ | 88 |
| 2.5–4 | V3+/V4+ | 43 |
| 1.75–2.5 | Ti3+/Ti4+ | 30 |
3.3 Ex situ magnetic characterization
Fig. 4(a) depicts the temperature dependence of the magnetic susceptibility of the pristine NVTP@C powder. Since carbon exhibits a small diamagnetic susceptibility,31 its contribution to the total magnetic susceptibility can be deemed negligible. The molar susceptibility normalized to the amount of NVTP in the composite was obtained by considering the NVTP weight fraction of about 91.5% determined by the TGA analysis (see above).
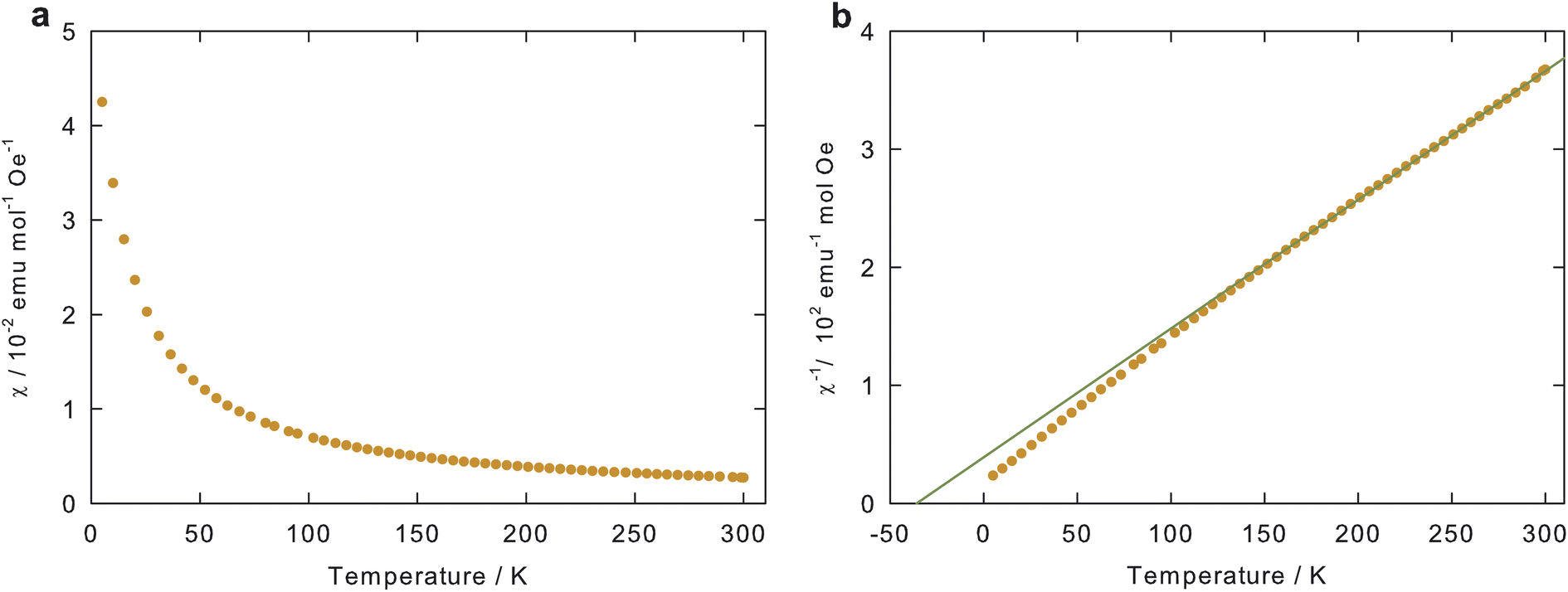 | ||
| Fig. 4 (a) Molar and (b) inverse molar magnetic susceptibility of pristine NVTP@C powder as a function of temperature. The straight line in (b) shows a linear fit for the temperature range of 150–300 K according to the Curie–Weiss law (see eqn (1)). Note that as described in the text the magnetic susceptibility is normalized to the amount of NVTP in the NVTP@C composite, since the magnetic contribution of carbon is negligible. | ||
The molar susceptibility in Fig. 4(a) exhibits the typical temperature dependence of localized paramagnetic magnetic moments, like it is expected for the 3d transition metal ions in NVTP. Therefore, the susceptibility can be described by the Curie–Weiss law:32
 | (1) |
To determine the effective magnetic moment μeff and the Curie–Weiss temperature ΘCW, the inverse susceptibility was plotted as a function of temperature (see Fig. 4(b)). Thereafter, a straight line was fitted to the linear regime of this plot (150–300 K). This procedure yielded values of ΘCW = −36 K and μeff = 2.71μB.
Considering that the paramagnetic susceptibility arises from the 3d transition metal ions in NVTP, the theoretical expected value of μeff can be calculated by
 | (2) |
| V3+ | V4+ | Ti3+ | Ti4+ | |
|---|---|---|---|---|
| S | 1 | 1/2 | 1/2 | 0 |
| μ i /μB | 2.83 | 1.73 | 1.73 | 0 |
| c i | μ eff/μB | χ mol/10−3 emu mol−1 Oe−1 | ||||
|---|---|---|---|---|---|---|
| NaVTi(PO4)3 | 0 | 1 | 0 | 1 | 1.73 | 1.11 |
| Na2VTi(PO4)3 | 1 | 0 | 0 | 1 | 2.83 | 2.98 |
| Na3VTi(PO4)3 | 1 | 0 | 1 | 0 | 3.32 | 4.09 |
3.4 Operando SQUID magnetometry measurements
Fig. 5 summarizes all the results of the operando SQUID magnetometry measurements. It is evident from this figure that, for all experiments, a systematic variation of the magnetic susceptibility χ (gray dots) occurred while charging and discharging the NVTP@C with constant current (see green lines for potential profiles). Prior to examining the individual measurements, we will initially discuss which changes of the magnetic susceptibility can be theoretically expected with sodiation and desodiation of the NVTP@C electrodes. As the magnetic signal of the carbon is negligible small and the de-/sodiation will exclusively occur at the NVTP, it is sufficient to consider the magnetic susceptibility of NVTP alone. As can be seen from the eqn (1) the susceptibility of NVTP is proportional to μeff2, which depends on the concentration of the transition metal ions and their corresponding ionic magnetic moments (see eqn (2)). Supposing now that the V3+/V4+ redox couple is active for the desodiation of Na2VTi(PO4)3 to NaVTi(PO4)3, the corresponding change of V3+ to V4+ would result in a decrease of the susceptibility χmol (see Table 2). Conversely, the sodiation of Na2VTi(PO4)3 to Na3VTi(PO4)3 with the Ti4+ to Ti3+ redox transition active, is expected to result in an increase of the magnetic susceptibility. Due to the different ionic magnetic moments of the vanadium and titanium oxidation states, the second reaction results in a smaller susceptibility change. If we consider now that the operando cells are charged/discharged with a constant current, thus that the change of the concentrations of the oxidation states ci occurs continuously, linear variations of χmol are awaited, whose slopes will be characteristic for the active redox couple.¶ These theoretical expected changes of χmol are indicated in Fig. 5 by orange dotted lines for the redox couple of V3+/V4+ and by blue ones for the Ti3+/Ti4+ redox couple.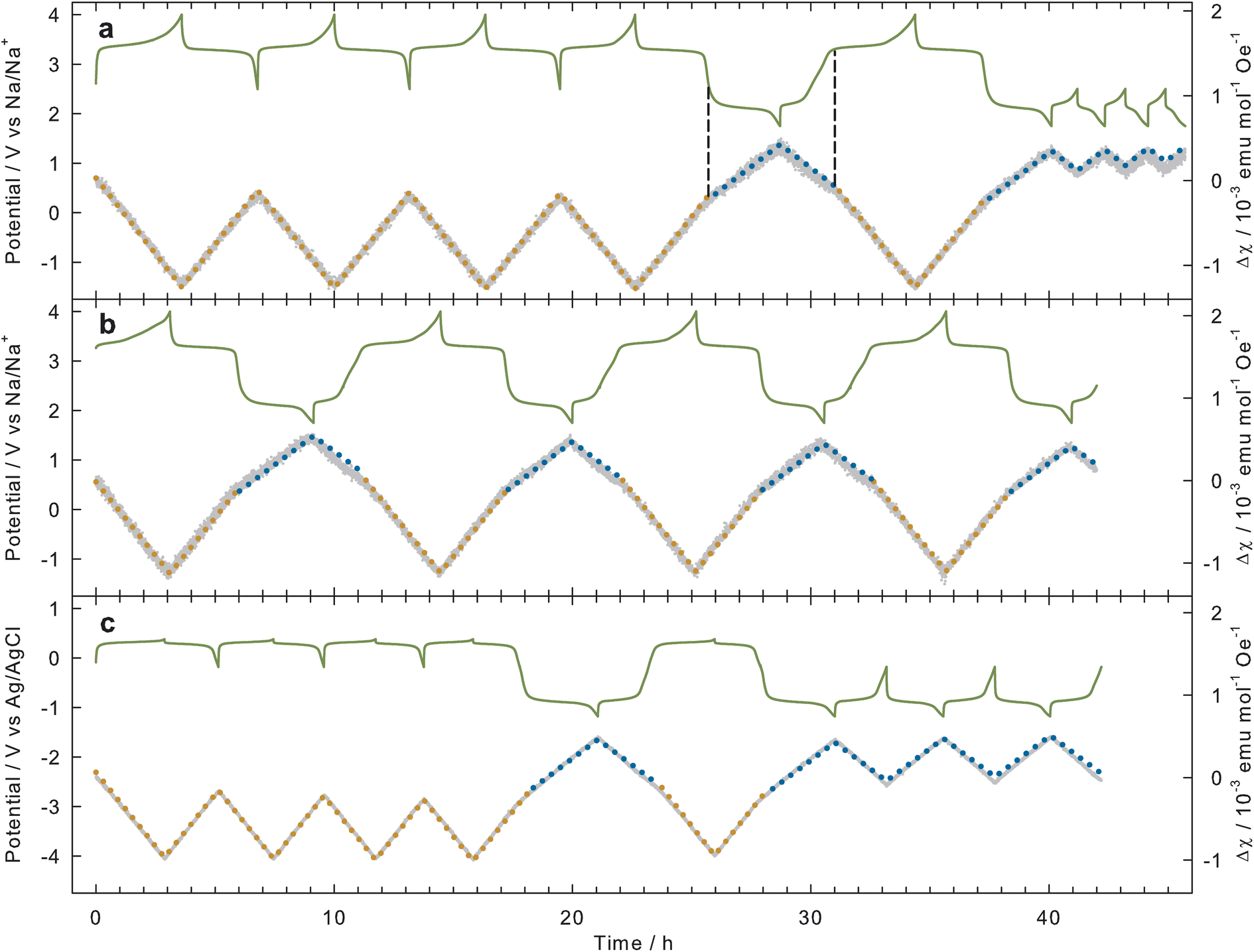 | ||
| Fig. 5 Potential (green line) and change in magnetic susceptibility Δχ (gray dots) of NVTP@C charged and discharged with 12 mA g−1. (a) Organic electrolyte (1 M NaPF6 in EC:PC) cycled first between 2.5–4 V, then between 1.75–4 V, and, finally between 1.75–2.5 V. (b) Organic electrolyte (1 M NaPF6 in EC:PC) cycled between 1.75–4 V. (c) Aqueous electrolyte (1 M Na2SO4) cycled first between −0.18–0.38 V, then between −1.18–0.38 V, and, finally between −1.18 to −0.18 V. The dotted lines indicate the theoretically expected change of χmol for the V3+/V4+ (orange) and the Ti3+/Ti4+ (blue) redox couples in NVTP. The black dashed lines in (a) exemplary show the correlation between the kink in the susceptibility data and the potentials. | ||
Fig. 5(a) shows the results for a cell with organic electrolyte. In this experiment, the cell was first cycled four times in the potential range of 2.5–4 V, followed by a cycle encompassing the entire potential range of 1.75–4 V. Subsequently, three cycles were conducted within the potential range of 1.75–2.5 V.|| In the initial potential range the magnetic susceptibility exhibits a linear decrease during charging and a subsequent increase with the same slope during discharging. As shown by the orange dotted lines the slopes align with the theoretically expected slopes of the V3+/V4+ redox reaction, indicating that this redox couple is indeed active within this potential range and that no significant side reactions occur. The fact that the slope of the susceptibility change is constant during the whole charging/discharging process, further reveals that the oxidation/reduction takes places uniformly throughout the bulk of the NVTP. Upon extending the lower potential limit to 1.75 V after the fourth cycle, a transition to a reduced slope of the magnetic susceptibility can be observed in the discharge cycle once the potential is below 2.5 V. In the subsequent charging cycle, the magnetic susceptibility initially decreases with this reduced slope, followed by a transition to the higher slope. The ratio of the reduced slope to the total susceptibility change is approximately equal in the discharging and charging cycles. As shown by the blue dotted lines, the reduced slope in the low potential region coincide well with the expected slope for the Ti3+/Ti4+ redox couple. This proves that this redox couple becomes active in this potential range, and again that no considerable side reactions are occurring. In the charging cycle the transition between the two regimes occurs at a higher potential than in the discharging cycles, as indicated by the black dashed lines. The activity of the Ti3+/Ti4+ redox couple within the low potential range is further demonstrated by the final cycles in the potential range of 1.75–2.5 V. In these cycles, the susceptibility consistently exhibits the slope expected for this redox transition.
The final cycles in the low potential range of 1.75–2.5 V where the Ti3+/Ti4+ redox couple is active, are notably shorter in comparison to the cycles with the active V3+/V4+ redox couple in the high potential range of 2.5–4 V, which were performed at the beginning of the measurement. The same outcome was observed in the cell tests, where a diminished capacity was determined for the low potential range in comparison to the high potential range (see Table 1). Since, as discussed above, the transition between the two redox regimes occurs at higher potentials in the charging cycle than in the discharging cycle (see black dashed lines in Fig. 5(a)), it can be concluded that the assumed lower capacity in the low potential range arises, because in the charging cycle the oxidation of Ti3+ to Ti4+ is only partially completed when the cut-off potential of 2.5 V is reached. To further investigate this charge–discharge asymmetry of the potential-capacity profiles additional measurements were performed with a second organic cell, where we always cycled in the entire potential range of 1.75–4 V (see Fig. 5(b)). Once more, a distinct separation between the V3+/V4+ redox activity within the high potential range and the Ti3+/Ti4+ activity within the low potential range is evident, as indicated by the different slopes of the susceptibility change. At the boundary between the two regimes a distinct kink appears in the susceptibility data, which reveals that even in the transition region the two reactions occur consecutively. This kink between the two slopes is observed in all charging and discharging cycles at approximately Δχ = 0, which demonstrates a high reversibility for both redox couples. For each charge/discharge half-cycle the susceptibility undergoes a change with both individual slopes for almost the same duration. This suggests that both redox couples contribute about equally to the total capacity when cycling within the entire potential range, and supports the notion that the different capacities for the V3+/V4+ and Ti3+/Ti4+ regimes observed in the cell tests are primarily due to the selected cut-off potentials.
Finally, the same measurement procedure as for the first cell was applied to a cell with aqueous electrolyte (see Fig. 5(c)). Here the potential limits of the entire potential range (−1.18–0.38 V) were adapted to the different electrolyte and reference electrode. In addition, the cut-off potential separating the high (−0.18–0.38 V) and the low potential (−1.18 to −0.18 V) ranges was adjusted, to ensure a better adaptation to the two different redox regimes. As can be seen from the susceptibility variation, the same behavior as for the organic electrolyte occurs here. The V3+/V4+ redox couple is active in the high potential regime, while the Ti3+/Ti4+ couple operates in the low potential regime. Again, the two redox couples are well separated from each other, as indicated by the rather sharp transition between the different slopes, and, both of them contribute about equally to the total capacity. The good agreement between the measured and the theoretically expected susceptibility variations, implying that most of the electrons are involved in the oxidation/reduction of the NVTP and not in parasitic side reactions, is particularly noteworthy in the low potential regime, where the Ti3+/Ti4+ redox couple is active. This is because a parasitic side reaction, which was assumed to be mainly the oxygen reduction reaction, has been reported in several studies in this regime.18,20 Since we found indications for the occurrence of this side reaction when we did not purge the aqueous electrolyte with argon (not shown), we can assume that the careful argon purging performed for the aqueous operando cells has largely suppressed this unwanted reaction.
4 Conclusions
In conclusion, magnetometry measurements have been used to gain a detailed insight into the bulk oxidation states of the transition metals in NVTP@C electrodes and their variation during de-/sodiation in both organic and aqueous electrolytes. Temperature-dependent magnetic susceptibility measurements revealed that in the pristine state the Na2VTi(PO4)3 in the NVTP@C powder prepared by a sol–gel synthesis procedure exhibits a good stoichiometry and that the V3+ and Ti4+ oxidation states are present. The systematic variations of the magnetic susceptibility occurring during charging/discharging of the electrodes showed that the V3+/V4+ redox couple, which is active in the high potential regime, and the Ti3+/Ti4+ redox couple, which becomes active at low potentials, are well separated from each other and occur one after the other when cycling over the whole potential range. In addition, these measurements revealed that there were no significant side reactions in either type of electrolyte and provided compelling evidence that the analogous charge compensation processes take place in both. Furthermore, it was demonstrated that both redox couples contribute about equally to the total capacity of NVTP and that the potential ranges must be carefully chosen to allow the individual capacities to be determined by electrochemical experiments alone. Overall, it was shown that operando magnetometry studies can provide additional insights into the charge compensation processes of electrode materials comprising multiple redox centers, and are therefore a very valuable complement to spectroscopic techniques.Data availability
All data supporting the findings of this study are available within the paper or the ESI.†Author contributions
Benedikt Huemer: investigation, formal analysis, writing – review & editing. Anna Jodlbauer: investigation, writing – original draft, visualization. Martin Wilkening: supervision. Heinz Krenn: resources. Peter Knoll: resources. Roland Würschum: writing – review & editing, supervision. Ilie Hanzu: conceptualization, writing – review & editing, supervision. Stefan Topolovec: conceptualization, formal analysis, writing – original draft, visualization, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank L. Winkler and A. Maric (Institute of Physics, University of Graz) for their support with the liquid helium infrastructure.References
- R. Rajagopalan, Z. Zhang, Y. Tang, C. Jia, X. Ji and H. Wang, Understanding crystal structures, ion diffusion mechanisms and sodium storage behaviors of NASICON materials, Energy Storage Mater., 2021, 34, 171–193 CrossRef.
- R. Thirupathi, V. Kumari, S. Chakrabarty and S. Omar, Recent progress and prospects of NASICON framework electrodes for Na-ion batteries, Prog. Mater. Sci., 2023, 137, 101128 CrossRef.
- Q. Huang, Z. Hu, K. Chen, Z. Zeng, Y. Sun, Q. Kong, W. Feng, K. Wang, Z. Li, Z. Wu, T. Chen and X. Guo, Partial modification strategies of NASICON-type Na3V2(PO4)3 materials for cathodes of sodium-ion batteries: progress and perspectives, ACS Appl. Energy Mater., 2023, 6, 2657–2679 CrossRef.
- Y. Xu, Q. Wei, C. Xu, Q. Li, Q. An, P. Zhang, J. Sheng, L. Zhou and L. Mai, Layer-by-layer Na3V2(PO4)3 embedded in reduced graphene oxide as superior rate and ultralong-life sodium-ion battery cathode, Adv. Energy Mater., 2016, 6, 1600389 CrossRef.
- G. Pang, P. Nie, C. Yuan, L. Shen, X. Zhang, H. Li and C. Zhang, Mesoporous NaTi2(PO4)3/CMK-3 nanohybrid as anode for long-life Na-ion batteries, J. Mater. Chem. A, 2014, 2, 20659–20666 RSC.
- Q. Zhang, C. Liao, T. Zhai and H. Li, A high rate 1.2 V aqueous sodium-ion battery based on all NASICON structured NaTi2(PO4)3 and Na3V2(PO4)3, Electrochim. Acta, 2016, 196, 470–478 CrossRef.
- D. Bin, F. Wang, A. G. Tamirat, L. Suo, Y. Wang, C. Wang and Y. Xia, Progress in aqueous rechargeable sodium-ion batteries, Adv. Energy Mater., 2018, 8, 1703008 CrossRef.
- H. Zhang, X. Tan, H. Li, S. Passerini and W. Huang, Assessment and progress of polyanionic cathodes in aqueous sodium batteries, Energy Environ. Sci., 2021, 14, 5788–5800 RSC.
- D. Tediashvili, J. Pilipavičius, J. Juodkazytė and L. Vilčiauskas, On the degradation of vanadium-based phosphate framework electrode materials in aqueous environments, J. Electrochem. Soc., 2023, 170, 120529 CrossRef.
- C. W. Mason and F. Lange, Aqueous ion battery systems using sodium vanadium phosphate stabilized by titanium substitution, ECS Electrochem. Lett., 2015, 4, A79–A82 CrossRef.
- D. Wang, X. Bie, Q. Fu, D. Dixon, N. Bramnik, Y.-S. Hu, F. Fauth, Y. Wei, H. Ehrenberg, G. Chen and F. Du, Sodium vanadium titanium phosphate electrode for symmetric sodium-ion batteries with high power and long lifespan, Nat. Commun., 2017, 8, 15888 CrossRef PubMed.
- F. Lalère, V. Seznec, M. Courty, J. N. Chotard and C. Masquelier, Coupled X-ray diffraction and electrochemical studies of the mixed Ti/V-containing NASICON: Na2TiV(PO4)3, J. Mater. Chem. A, 2018, 6, 6654–6659 RSC.
- H. Zhang, S. Jeong, B. Qin, D. Vieira Carvalho, D. Buchholz and S. Passerini, Towards high-performance aqueous sodium-ion batteries: stabilizing the solid/liquid interface for NASICON-type Na2VTi(PO4)3 using concentrated electrolytes, ChemSusChem, 2018, 11, 1382–1389 CrossRef PubMed.
- W. Gao, R. Zhan, M. K. Aslam, D. Liu, J. Jiang and M. Xu, Na3TiV(PO4)3/C nanoparticles for sodium-ion symmetrical and full batteries, Energy Storage, 2019, 1, e74 CrossRef.
- A. Gilankar, A. Mitra, J. Singh, S. Das and S. B. Majumder, Investigations on different strategies towards improving the electrochemical properties of Na2VTi (PO4)3 for symmetrical sodium-ion batteries, J. Alloys Compd., 2021, 851, 156813 CrossRef.
- W. Zhang, Z. Xu, H. Li, M. Xu, S. Wang, Z. Li, A. Wang, L. Zhang, L. He, S. Li, B. Zhu, Z. Zhang and Y. Lai, All-climate and air-stable NASICON-Na2TiV(PO4)3 cathode with three-electron reaction toward high-performance sodium-ion batteries, Chem. Eng. J., 2022, 433, 133542 CrossRef.
- J. Dong, G. Zhang, X. Wang, S. Zhang and C. Deng, Cross-linked Na2VTi(PO4)3@C hierarchical nanofibers as high-performance bi-functional electrodes for symmetric aqueous rechargeable sodium batteries, J. Mater. Chem. A, 2017, 5, 18725–18736 RSC.
- H. Wang, T. Zhang, C. Chen, M. Ling, Z. Lin, S. Zhang, F. Pan and C. Liang, High-performance aqueous symmetric sodium-ion battery using NASICON-structured Na2VTi(PO4)3, Nano Res., 2018, 11, 490–498 CrossRef.
- L. Shen, H. Yang, Y. Jiang, J. Ma, T. Sun, M. Zhang and N. Zhu, NASICON-structured Na2VTi(PO4)3@C for symmetric aqueous rechargeable Na-Ion Batteries with long lifespan, ACS Sustain. Chem. Eng., 2021, 9, 3490–3497 CrossRef.
- M. Petrulevičienė, J. Pilipavičius, J. Juodkazytė, D. Gryaznov and L. Vilčiauskas, Electrochemical performance of NASICON-structured Na3−xV2−xTix(PO4)3 (0.0 < x < 1.0) as aqueous Na-ion battery positive electrodes, Electrochim. Acta, 2022, 424, 140580 CrossRef.
- Q. Y. Meng, J. C. Shao, X. R. Dou and H. Z. Chi, N-Containing Na2VTi(PO4)3/C for aqueous sodium-ion batteries, Small, 2024, 20, 2308483 CrossRef.
- J. C. Shao, Q. Y. Meng, X. R. Dou and H. Z. Chi, Manipulating the microstructure of Na2TiV(PO4)3 for aqueous Na ion storage, Electrochim. Acta, 2024, 473, 143468 CrossRef.
- S. Vafakhah, M. Saeedikhani, M. Tanhaei, S. Huang, L. Guo, S. Y. Chiam and H. Y. Yang, An energy efficient bi-functional electrode for continuous cation-selective capacitive deionization, Nanoscale, 2020, 12, 22917 RSC.
- A. Shrivastava, V. Q. Do and K. C. Smith, Efficient, selective sodium and lithium removal by faradaic deionization using symmetric sodium titanium vanadium phosphate intercalation electrodes, ACS Appl. Mater. Interfaces, 2022, 14, 30672–30682 CrossRef.
- S. Topolovec, H. Kren, G. Klinser, S. Koller, H. Krenn and R. Würschum, Operando magnetometry on LixCoO2 during charging/discharging, J. Solid State Electrochem., 2016, 20, 1491 CrossRef.
- G. Klinser, S. Topolovec, H. Kren, S. Koller, W. Goessler, H. Krenn and R. Würschum, Continuous monitoring of the bulk oxidation states in LixNi1/3Mn1/3Co1/3O2 during charging and discharging, Appl. Phys. Lett., 2016, 109, 213901 CrossRef.
- G. Klinser, R. Zettl, M. Wilkening, H. Krenn, I. Hanzu and R. Würschum, Redox processes in sodium vanadium phosphate cathodes - insights from operando magnetometry, Phys. Chem. Chem. Phys., 2019, 21, 20151–20155 RSC.
- H. Nguyen and R. J. Clément, Rechargeable batteries from the perspective of the electron spin, ACS Energy Lett., 2020, 5, 3848–3859 CrossRef.
- X. Li, L. Zhang, H. Liu, Q. Li and Y. Hou, Magnetic measurements applied to energy storage, Adv. Energy Mater., 2023, 13, 2300927 CrossRef.
- G. Chen, Q. Huang, T. Wu and L. Lu, Polyanion sodium vanadium phosphate for next generation of sodium-ion batteries - a review, Adv. Funct. Mater., 2020, 30, 2001289 CrossRef.
- P. Stamenov and J. M. D. Coey, Magnetic susceptibility of carbon - experiment and theory, J. Magn. Magn. Mater., 2005, 290–291, 279–285 CrossRef.
- S. Mugiraneza and A. M. Hallas, Tutorial: a beginner's guide to interpreting magnetic susceptibility data with the Curie-Weiss law, Commun. Phys., 2022, 5, 95 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta06628f |
| ‡ These authors contributed equally to this work. |
| § Note that theoretically one could construct a case where two simultaneous redox reactions could yield the same slope as the formal oxidation states. However, as pointed out in Section S3 of the ESI, it is highly unlikely that this case actually occurs. |
| ¶ Note that slight deviations from the linear behavior would occur in the case that the Curie–Weiss temperature ΘCW changes with the charging state. |
| || It should be noted that due to the higher internal resistance of the operando cells in comparison to conventional cells typically used for cell tests (e.g. Swagelok cells), minor potential discrepancies may arise, due to higher ohmic drops. |
| This journal is © The Royal Society of Chemistry 2025 |