
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling interfacial dynamics of zero-dimensional bismuth-based halide perovskite emitters for electrochemiluminescence applications†
Chun Hong
Mak
ai,
Yaojia
Ai
 b,
Shun Cheung
Cheng
c,
Wenxin
Niu
d,
Minshu
Du
e,
Kuan-Chen
Cheng
flmno,
Guohua
Jia
g,
Xue-Qing
Xu
h,
Zheng
Hu
k,
Chi Chiu
Ko
c,
Guizheng
Zou
*b,
Duu-Jong
Lee
*i and
Hsien-Yi
Hsu
*aj
b,
Shun Cheung
Cheng
c,
Wenxin
Niu
d,
Minshu
Du
e,
Kuan-Chen
Cheng
flmno,
Guohua
Jia
g,
Xue-Qing
Xu
h,
Zheng
Hu
k,
Chi Chiu
Ko
c,
Guizheng
Zou
*b,
Duu-Jong
Lee
*i and
Hsien-Yi
Hsu
*aj
aSchool of Energy and Environment, Department of Materials Science and Engineering, Centre for Functional Photonics (CFP), City University of Hong Kong, Kowloon Tong, Hong Kong, China. E-mail: sam.hyhsu@cityu.edu.hk
bSchool of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, China. E-mail: zouguizheng@sdu.edu.cn
cDepartment of Chemistry, City University of Hong Kong, Kowloon Tong, Hong Kong, China
dState Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, 5625 Renmin Street, Changchun, Jilin 130022, P. R. China
eSchool of Materials Science and Engineering, Northwestern Polytechnical University, Xi'an 710072, Shaanxi, P. R. China
fInstitute of Food Science and Technology, National Taiwan University, Taipei City 106319, Taiwan
gCurtin Institute of Functional Molecules and Interfaces, School of Molecular and Life Sciences, Curtin University, GPO Box U1987, Perth, WA 6845, Australia
hKey Laboratory of Renewable Energy, Guangdong Provincial Key Laboratory of New and Renewable Energy Research and Development, Guangzhou Institute of Energy Conversion, Chinese Academy of Sciences, Guangzhou 510640, P. R. China
iDepartment of Mechanical Engineering, City University of Hong Kong, Kowloon Tong, Hong Kong, China. E-mail: tuclee@cityu.edu.hk
jShenzhen Research Institute of City University of Hong Kong, Shenzhen 518057, P. R. China
kKey Laboratory of Mesoscopic Chemistry of MOE and Jiangsu Provincial Laboratory for Nanotechnology, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China
lInstitute of Biotechnology, National Taiwan University, No. 1, Sec. 4, Roosevelt Rd., Taipei 10617, Taiwan
mDepartment of Optometry, Asia University, 500, Lioufeng Rd., Wufeng, Taichung, Taiwan
nDepartment of Medical Research, China Medical University Hospital, China Medical University, 91, Hsueh-Shih Road, Taichung, Taiwan
oDept of Food Science, Fu Jen Catholic University, 24205, New Taipei City, Taiwan
First published on 31st January 2025
Abstract
Organic–inorganic halide perovskites have emerged as a novel category of optoelectronic materials owing to their exceptional physical and chemical properties. Notably, zero-dimensional (0-D) dimethylammonium bismuth iodide (DMA3BiI6) perovskite is an emerging candidate for electrochemiluminescence (ECL) light-emitting applications. Herein, we design 0-D DMA3BiI6 perovskite emitters and provide a detailed analysis of exciton transport dynamics through temperature-dependent transient photoluminescence (TRPL) and charge transport kinetics by electrochemical ECL techniques. Efficient exciton transport has been substantiated by the reduced activation energy and enhanced electronic coupling. Based on the diffusion coefficient and electron-transfer rate through electrochemical methods, we demonstrate that effective heterogeneous charge transfer at the electrode–electrolyte interface leads to red-shifted ECL emission with the addition of the tripropylamine (TPrA) co-reactant. As a result, the creation of zero-dimensional perovskite emitters paves the way for advancements in the rapidly evolving fields of optoelectronic and biosensing technologies, including but not limited to ECL devices, ECL immunoassays, light-emitting electrochemical cells, organic light-emitting diodes, and perovskite-based light-emitting diodes.
Introduction
Since Bard's pioneering research on inorganic-based silicon quantum dots (QDs) for electrochemiluminescence (ECL) in 2002, the field has seen an extensive exploration of various QDs for their ECL properties.1 Despite this, the ECL efficiencies for most QDs remain substandard (compared with classical organometallic complexes), which has sparked ongoing research into innovative nano-based emitters that have higher efficiency.Metal halide perovskites have high potential in optoelectronic applications like lasers, photodetectors, solar cells, and light-emitting diodes (LEDs), owing to their superior optical and electronic properties, including broad and intense absorption, narrowly focused emission, tunable band gaps, and a high photoluminescence quantum yield (PLQY).2–4 Halide perovskite QDs are noted for their narrow spectral widths and defect-tolerant photophysics, setting them apart from traditional colloidal semiconductor QDs.5–8 Consequently, the investigation into the ECL properties of halide perovskites represents a promising and valuable research direction.
Amid challenges, lead halide perovskite NCs still provide favorable prospects for commercial applications, although lead toxicity remains a barrier to their commercial use. Opportunities for low-toxicity and eco-friendly metal substitutes may gain traction in future commercial settings.9 Furthermore, achieving efficient charge transfer at the electrode/electrolyte interface remains an ongoing challenge, which is related to the electrochemical production rate of cations and anions (i.e., rate of heterogeneous electron transfer).10 Addressing these issues is critical for advancing the use of perovskites in ECL applications.
Bismuth-based hybrid perovskites have aroused widespread interest.11–15 In recent years, dimethylammonium iodide (DMAI) has been used to stabilize the perovskite phase and is widely utilized in various applications.16–19 The approach involves incorporating DMAI into perovskite solar cells and photocatalysts, a process that has been reported to enhance air and thermal stability and improve charge transport properties.20–23 The stable features discovered in bismuth-based hybrid perovskites are promising for other potential applications.
In this study, temperature-dependent transient photoluminescence (TRPL) has been employed to explore electron–hole pair diffusion in DMA3BiI6 emitters. This finding underscores the potential of DMA3BiI6 perovskite as a promising material for ECL applications. Furthermore, DMA3BiI6 emitters exhibit a notably higher diffusion coefficient and electron transfer rate constant at the electrode/electrolyte interface, leading to the generation of red-shifted ECL emission via triplet excited states, a phenomenon known as the T-route, likely arising from efficient heterogeneous charge transfer. Tri-n-propylamine (TPrA) is an effective co-reactant for ECL. Oxidation of TPrA or related amines initially produces the corresponding aminium radical cation, which rapidly deprotonates to form a highly reductive α-amino alkyl radical for creating DMA3BiI6 perovskite anions,24 resulting in enhanced ECL intensity.
Results and discussion
Characterization of DMA3BiI6
Fig. 1a exhibits the XRD pattern of rhombohedral DMA3BiI6, with strong peaks at 11.5°, 12°, 16.7°, 17.7°, 24.9°, 25.2°, 25.5°, 31.46°, and 31.54° ascribed to the (420), (201), (401), (531), (532), (810), (701), (107), and (107) planes, respectively, along with the crystal structural model of DMA3BiI6 (Fig. 1b); the results are in good agreement with previously reported data15,23 and the standard PDF card (PDF# 01-075-7841). Fig. 1c shows the scanning electron microscope (SEM) images. DMA3BiI6 consists of aggregated irregular particles with evidence of 0D nanoscale features. The TEM images (Fig. S4†) reveal the presence of aggregated structures composed of irregularly shaped particles, with 0D nanoscale features observed as discrete, confined nanoparticles distributed on the surfaces of the larger aggregates. Ultraviolet-visible (UV-vis) absorption and steady-state photoluminescence (PL) spectra were used to study the optical properties of DMA3BiI6. The absorption edge of pristine DMA3BiI6 is at approximately 600 nm (Fig. 1d), which aligns with the literature.23 The PL spectrum of DMA3BiI6 perovskites presents a broad emission peak centered at 644 nm (Fig. 1d), consistent with previous reports.15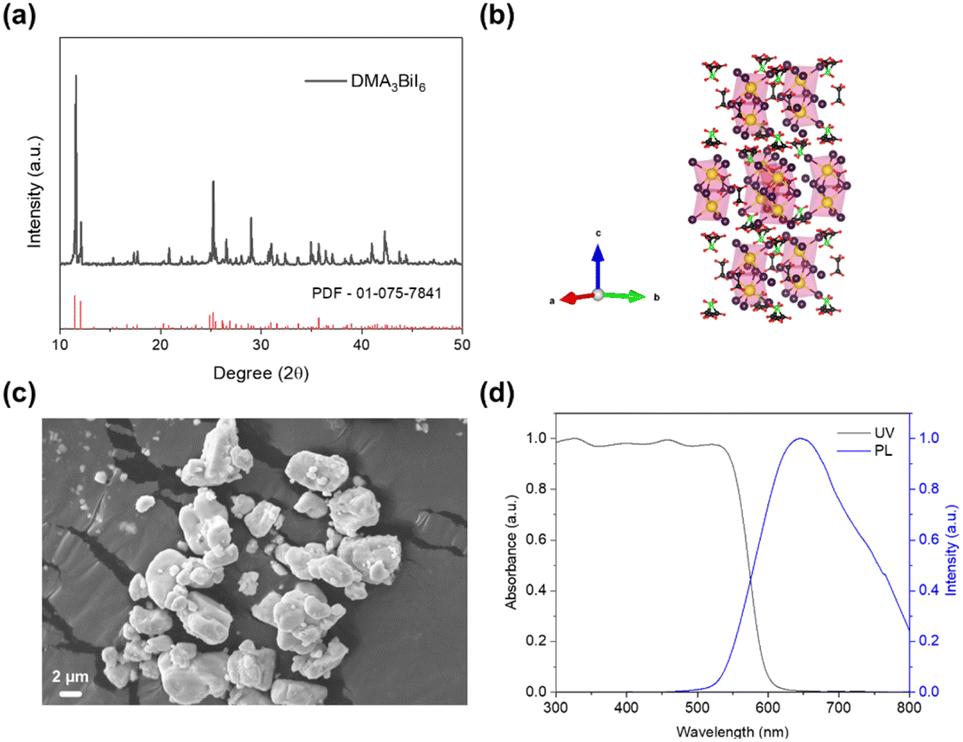 | ||
| Fig. 1 (a) X-ray diffraction (XRD) patterns of DMA3BiI6; (b) crystal structural model of DMA3BiI6. (c) SEM image of DMA3BiI6; (d) normalized UV-vis absorption and steady-state photoluminescence (PL) spectra of DMA3BiI6 with an excitation wavelength at 375 nm. | ||
The early-time transient spectrum was obtained using a fs-TA spectrometer, specifically at t = 0.2 ps post-photo-excitation (as shown in Fig. 2a), revealing an oscillatory triple-peak structure with a pronounced negative band peak and two distinct positive bands. A significant negative band observed around 455 nm is attributable to the combined effects of ground state bleaching (GSB) and a blue-shifted photoinduced absorption (PA). After 0.5 ps, a positive signal emerges at approximately 489 nm, which becomes more pronounced as the PA signal diminishes in the lower-energy region of the spectrum, as shown in Fig. 2a. This phenomenon arises due to exciton absorption, similar to previous findings. This feature is consistent with observations in bismuth-based perovskite materials and indicates a persistent excitonic population.25,26
 | ||
| Fig. 2 (a) Femtosecond (fs)-TA spectrum obtained by exciting the DMA3BiI6 dispersed solution in ethanol at 355 nm. (b) fs-TA kinetic traces of DMA3BiI6 over a timescale between 0 ps and 50 ps. (c) ns-TA spectrum obtained by exciting the DMA3BiI6 dispersed solution in ethanol at 355 nm. (d) ns-TA kinetic traces of DMA3BiI6 at 400 nm and 720 nm. | ||
The need for additional nanosecond transient absorption (ns-TA) measurements (Fig. 2c) arises to capture these longer-lived processes that fs measurements till 2.5 ns might overlook. From the ns-TA spectrum, two peaks were identified: a sharp peak at 400 nm and a broader band centered around 720 nm (Fig. 2c). The bleach kinetics at 400 nm were analyzed by single exponential fitting. The fitted results showed a longish lifetime (τ) of 563 μs at 400 nm and 611 μs at 720 nm. The decay kinetics derived from the ns-TA spectrum observed a long-lived exciton due to the severely inhibited free motion of photogenerated carriers,27 as shown in Fig. 2d, consistent with the zero-dimensional structure, and even more significant electron–phonon coupling in Cs3Bi2I9 reported by Li et al.28
The lifetime of DMA3BiI6 perovskites exhibits a decreasing trend with rising temperatures, as depicted in Fig. 3a and documented in Table 1. The temperature-dependent time-resolved photoluminescence (TRPL) spectral response of DMA3BiI6 emitters (seen in Fig. 3a) allows for the calculation of activation energies for the fast and slow decay phases, which are 5.12 meV and 3.97 meV, respectively. These values were obtained by fitting the PL decay curves with the equation  (Fig. 3, Tables 1 and S1†). The pre-exponential factor from the fitted equation yields temperature-independent electronic coupling matrix elements |HAB|, whose values represent the overlap of the excited-state wave functions between the initial and final sites, are determined to be 0.0062 cm−1 and 0.0045 cm−1.
(Fig. 3, Tables 1 and S1†). The pre-exponential factor from the fitted equation yields temperature-independent electronic coupling matrix elements |HAB|, whose values represent the overlap of the excited-state wave functions between the initial and final sites, are determined to be 0.0062 cm−1 and 0.0045 cm−1.
 | ||
Fig. 3 Temperature-dependent TRPL decay curves of (a) DMA3BiI6 and (b) MA3Bi2I9; Arrhenius plots of (c) DMA3BiI6 and (d) MA3Bi2I9. The red curves correspond to the equation  . . | ||
| Sample | Activation energy (Ea)/meV | Electronic coupling (|HAB|)/cm−1 | |
|---|---|---|---|
| MA3Bi2I9 | τ fast | 5.15 | 0.00631 |
| τ slow | 4.90 | 0.00424 | |
| τ | 4.15 | 0.00409 | |
| DMA3BiI6 | τ fast | 5.12 | 0.00621 |
| τ slow | 3.97 | 0.00449 | |
| τ | 3.80 | 0.00431 |
For comparative purposes, the temperature-dependent TRPL spectra of MA3Bi2I9 are also recorded and shown in Fig. 3b, with the corresponding activation energies for the fast and slow decay components found to be 5.15 meV and 4.90 meV, respectively (as listed in Table 1). These activation energies are higher than those for DMA3BiI6 emitters. Additionally, the electronic coupling element |HAB| for the average decay lifetime of DMA3BiI6 is higher than that of MA3Bi2I9, indicating that the wave functions in DMA3BiI6 are more delocalized within the perovskite material. In contrast, in MA3Bi2I9, the wave function overlap occurs between neighboring molecules. Additionally, the 0D structure of DMA3BiI6 provides certain advantages for charge transfer compared to the layered structure of MA3Bi2I9. This may be related to defect formation in the crystal lattice, possibly due to unreacted bismuth exposed on the surface, which could create defect sites that enhance charge transport pathways in DMA3BiI6.15,29
The proportions of exciton recombination for each photophysical process, denoted as α1 and α2 in Tables S1 and S2,† reveal that the fast decay components in DMA3BiI6 emitters have amplitudes (α1) ranging between approximately 15.95% and 21.00% across all temperatures. In contrast, the fast decay components in MA3Bi2I9 exhibit higher amplitudes, approximately 18.38% to 21.71%. The significant amplitudes of the slow decay component (α2) in both materials suggest that a more dominant non-radiative recombination process happens due to interface defects in both the organic and inorganic perovskite emitters (DMA3BiI6 and MA3Bi2I9). This implies that non-radiative recombination is the primary photophysical process at all temperatures, indicating that most excited electrons and holes in these two types of perovskite emitters favor a faster decay pathway.30
Furthermore, the higher amplitudes observed in MA3Bi2I9 suggest a more substantial non-radiative recombination within its emitting layer. Consequently, the stronger electronic coupling and lower activation energies in DMA3BiI6 contribute to improved exciton transfer and charge recombination, highlighting low-dimensional DMA3BiI6 as a promising material for ECL applications.10,31
Electrochemistry and heterogeneous electron transfer kinetics
The CV tests were conducted to assess the reversibility, diffusion coefficient, number of electrons transferred and the stability of the radical cations and anions of DMA3BiI6. A 2 mM solution of DMA3BiI6 was dispersed in the electrolyte solution for CV analysis. Generally, the CV of DMA3BiI6 in DCM showed one reduction wave and two oxidation waves (Fig. 4a). The CV graph showed an irreversible reduction wave and two oxidation waves at potentials of −0.98 V vs. SCE, +0.30 V vs. SCE, and +0.65 V vs. SCE. The quasi-reversible reduction wave of DMA3BiI6 indicates that the DMA3BiI6 radical anion is unstable. | ||
| Fig. 4 (a) Reduction voltammogram of DMA3BiI6 at various scan rates. (b) Oxidation voltammogram of DMA3BiI6 at various scan rates. (c) Standard cyclic voltammograms (CV) of DMA3BiI6 at a scan rate of 100 mV s−1. (d) Reduction peak current vs. v1/2. (e) First oxidation peak current vs. v1/2. (f) Second oxidation peak current vs. v1/2. All CV measurements were performed with 2 mM DMA3BiI6 in 100 mM TBAPF6 using DCM as the solvent. | ||
The CV measurements for both reduction and oxidation at 10 mV s−1 to 1 V s−1 were performed (Fig. 4a and b) to study the electrochemical reversibility of the reduction and oxidation of DMA3BiI6. Scan-rate-dependent CVs for the first oxidation, first reduction, and second reduction peaks are depicted in Fig. 4d–f, respectively. The peak currents change linearly with the square root of the scan rate for the first oxidation wave (ip,ox), the first reduction wave (ip,red1), and the second reduction wave (ip,red2), supporting that diffusion controls the reaction rates.
From the scan rate studies, as shown in Fig. S2a–c,† the peak current varied linearly with the square root of the scan rate for the first oxidation wave (ip,o) and the first reduction wave (ip,r), confirming that the current is diffusion-controlled. Critical scan rates were determined from the plot of Ep against the log of the scan rate at low and high scan rates as shown in Fig. S2d–f.† The diffusion coefficients, D, of reduction, first oxidation, and second oxidation determined using the Randles–Sevcik equation, listed in Table 2, were found to be 5.74 × 10−5 cm2 s−1, 0.74 × 10−5 cm2 s−1 and 10 × 10−5 cm2 s−1, respectively. The single electron-transfer step in each wave, and the experimental conditions at 25 °C, were assumed for the calculation. The reduction rate constant was determined to be 7.0 × 10−3; the first and second oxidation of DMA3BiI6 showed lower electron transfer rates of 3.9 × 10−3 and 5.7 × 10−3 cm s−1, respectively. For comparison, the kinetic parameters of MA3Bi2I9 were reported (i.e.), reduction and oxidation rate constants were determined to be 3.3 × 10−3 and 4.6 × 10—3. The DMA3BiI6 emitter is a potential material for ECL applications, owing to the efficient heterogeneous electron transfer within the reaction system.
| Reduction | 1st oxidation | 2nd oxidation | |
|---|---|---|---|
| E pa/pc/V vs. SCE | −0.98 | 0.3 | 0.65 |
| E 1/2/V vs. SCE | — | 0.16 | 0.52 |
| 10−5D/cm2 s−1 | 5.74 | 0.74 | 10 |
| α | 0.12 | 0.46 | 0.04 |
| k 0/cm s−1 | 0.007 | 0.0039 | 0.0057 |
ECL
For transient ECL measurements, since DMA3BiI6 serves as the oxidative ECL species, the potential was stepped from the reduction wave at Epc –80 mV to the oxidation wave at Epa +80 mV for the DMA3BiI6. Fast-responsive emission at different ECL intensities was observed in the cathodic and anodic pulses (Fig. 5a and b). The asymmetric ECL transients occur during smaller cathodic pulses compared to anode pulses in the finding since the radical anion (DMA3BiI6˙−) −radical cation (DMA3BiI6˙+) annihilation is not stable in the cathodic pulse as shown by the CV study. Besides, the solution depletes the ECL intensity during the cathodic pulse (Fig. 5a and b). This could be explained by the instability of the radical anion (DMA3BiI6˙−) on the electrode surface. | ||
| Fig. 5 (a) 1st reduction and oxidation potential and (b) 2nd reduction and oxidation potential and (c) 1st reduction and oxidation potential and (d) 2nd reduction and oxidation potential with TPrA as the co-reactant of current (top) and ECL transients (bottom) for the DMA3BiI6/Pt disk electrode (PE) pulsed in DCM between 80 mV past the reduction peak and at 80 mV past the first oxidation potential, respectively. The pulse width is 1 second. | ||
It is possible that the cations diffused far away from the annihilation zone. However, they diffused back to the zone during the next anodic pulse. Therefore, the results indicated a considerable decay in the second anodic pulse after the first anodic pulse. Herein, the inconsistency in ECL intensities at different potentials provides evidence of the instability in forming radical cations. To further enhance the ECL performance, TPrA (tri-n-propylamine) was used as a co-reactant (Fig. 5c and d). TPrA as the co-reactant is first oxidized to a short-lived TPrA radical cation (TPrA˙+), followed by the deprotonation from an α-carbon to produce the strongly reducing intermediate TPrA˙;32 this intermediate then reduces the oxidized DMA3BiI6 cations, thus enhancing ECL emission. The ECL intensity of the first oxidation is weaker than that of the second oxidation. This may be attributed to the enhanced oxidation state of DMA3BiI6 during the second oxidation, allowing more efficient energy transfer, generating a higher population of radical cations (DMA3BiI6+), thus boosting the ECL intensity.
In the cyclic voltammetry-electrochemiluminescence (CV-ECL) plot, two critical regions denote the system's reduction and oxidation potentials under investigation (Fig. 6a and b). The oxidation process unfolds within a voltage range of 0 V to +1.2 V vs. SCE. As the potential is swept towards more positive values, the system undergoes an oxidation reaction, with ECL occurring and its intensity increasing notably once the potential exceeds +0.6 V vs. SCE. ECL is observed during the reduction sweep from 0 V to −1.8 V vs. SCE, with its intensity increasing when the potential drops below −1.00 V vs. SCE. This pronounced ECL intensity increase at specified potentials marks the system's reaction thresholds for oxidation and reduction. The ECL intensity of MA3Bi2I9 is lower than that of DMA3BiI6, as shown in Fig. S3.† It is likely due to the higher degree of non-radiative recombination processes occurring at defect sites in MA3Bi2I9, which reduce the efficiency of exciton generation and radiative decay.
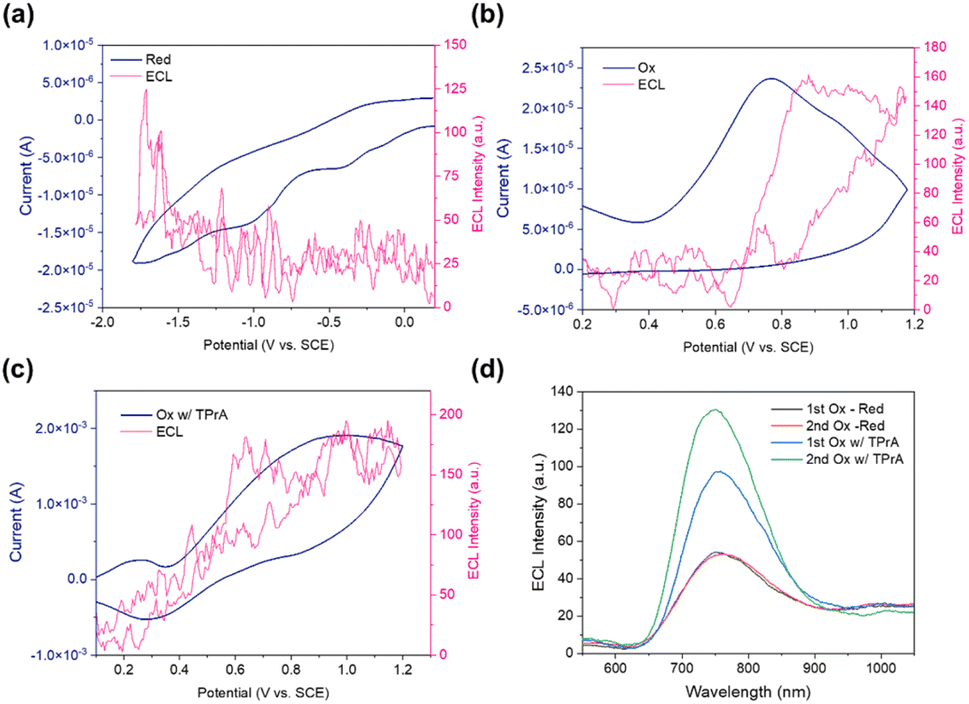 | ||
| Fig. 6 ECL (red curve) – CV (blue curve) simultaneous measurements by applying a pulsing potential from (a) approximately 0.0 V to −1.8 V vs. SCE and (b) approximately +0.2 V to +1.2 V vs. SCE; (c) DMA3BiI6/PE in the presence of 10 mM TPrA as the co-reactant for pulsing potential from approximately +1.2 to +0.1 V vs. SCE; (d) ECL spectrum of first oxidation and reduction potential and second oxidation and reduction potential, first oxidation and reduction potential and second oxidation and reduction potential with TPrA as the co-reactant. DMA3BiI6/PE pulsed in DCM between 80 mV past the reduction peak and 80 mV past the first oxidation potential, respectively. Pulse width is 1 second. | ||
During the cathodic sweep towards more negative potentials, anions at the electrode surface undergo reduction and, subsequently, detach from the electrode. Upon transitioning to the anodic sweep, where the potential shifts towards more positive values, DMA3BiI6 anions can react with DMA3BiI6 cations. This leads to annihilation reactions, during which the energy released as DMA3BiI6 ions return to their ground state is emitted as light. This light emission contributes to the observed ECL signal, thereby providing insights into the electrochemical behavior of the system. If any one of the radical ions is unstable, the co-reactant could be applied. TPrA was employed as a co-reactant with DMA3BiI6 perovskite to stabilize the DMA3BiI6 anion. TPrA could form a potent reducing agent at +1.5 V vs. NHE after being reduced.33 The stronger ECL emission was observed after using TPrA as the co-reactant Fig. 6c. Noteworthy observations were made regarding the ECL intensity, a strong ECL intensity was observed when the potential exceeded +0.2 V vs. SCE. Under this operating procedure, Fig. 6d in the respective study illustrates the ECL behavior with and without the addition of TPrA as a co-reactant. This comparison is crucial for understanding the role of TPrA in the ECL behavior of DMA3BiI6. A more substantial applied bias generally resulted in a stronger ECL emission. This condition was observed under specific circumstances: the ECL spectrum of the first reduction and oxidation potentials and the second reduction and oxidation potentials, both with and without TPrA as a co-reactant. The ECL spectra of the first reduction and oxidation potentials and the second reduction and oxidation potentials exhibited a remarkable similarity. This observation suggests a consistent electrochemical response across different redox states under different applied biases, further highlighting the stability of the DMA3BiI6/Pt disk electrode (PE) under these operating conditions (Fig. 6a and b).
ECL mechanism
There is a 118-nm red shift in the ECL spectrum compared to the fluorescence spectrum (Fig. 7). The ECL emission wavelength was consistent under different conditions (i.e., DMA3BiI6/PE with and without the TPrA co-reactant during first oxidation/second oxidation and reduction pulses). The energy of the excited singlet state is estimated from the fluorescence emission maximum using the equation Es (in eV) = 1239.81/λ (in nm), where λ is the wavelength at maximum emission (i.e., 645 nm).34 The corresponding excited singlet-state energy was calculated to be 1.92 eV. The energy of the annihilation reaction is based on the equation
 | ||
| Fig. 7 UV-vis absorption spectra (black solid line), photoluminescence (PL) spectra (magenta solid line), and ECL spectra (sky blue solid line); ECL spectra of DMA3BiI6/PE with and without TPrA in 0.1 M TBAPF6 in DCM obtained by pulsing between 80 mV past the reduction peak potential and two different anodic potentials, 80 mV past the first oxidation peak potential and 80 mV over the second oxidation peak potential. The excitation wavelength was 375 nm. | ||
The equation showing the differences between the thermodynamic potentials of the first oxidation and the second oxidation with the reduction potential determined and calculated from the cyclic voltammogram, such that  By assumption of the entropy effect, estimated at 0.1 eV, subtracted gives values of 1.12 eV and 1.63 eV. Since the annihilation reaction energy of 1.12 eV and 1.63 eV is lower than the energy required to populate the singlet excited state at 1.92 eV, ECL in DMA3BiI6 is likely processed in an energy-deficient system (T-route), where the triplet–triplet annihilation indirectly enhances the population of singlet excited states (eqn (3) and (4)).
By assumption of the entropy effect, estimated at 0.1 eV, subtracted gives values of 1.12 eV and 1.63 eV. Since the annihilation reaction energy of 1.12 eV and 1.63 eV is lower than the energy required to populate the singlet excited state at 1.92 eV, ECL in DMA3BiI6 is likely processed in an energy-deficient system (T-route), where the triplet–triplet annihilation indirectly enhances the population of singlet excited states (eqn (3) and (4)).
The T-route mechanism is shown as follows:
| DMA3BiI6 + e− → DMA3BiI6− (reduction at electrode) | (1) |
| DMA3BiI6 − e− → DMA3BiI6+ (oxidation at electrode) | (2) |
| DMA3BiI6− + DMA3BiI6+ → 3DMA3BiI6* + DMA3BiI6 (excited triplet state formation) | (3) |
| 3DMA3BiI6* + 3DMA3BiI6* → 1DMA3BiI6* + DMA3BiI6 (excited singlet state formation) | (4) |
Upon the oxidation of DMA3BiI6, in the presence of the TPrA co-reactant, the mechanism could be shown as follows:
| TPrA − e− → [TPrA˙]+ (reaction at electrode) | (5) |
| [TPrA˙]+ → TPrA˙ + H+ (chemical reactions) | (6) |
| DMA3BiI6 + TPrA˙ → DMA3BiI6* + products (excited state species formation) | (7) |
| DMA3BiI6* → DMA3BiI6 + hv (light emission) | (8) |
Conclusions
In summary, we demonstrated that the zero-dimensional (0-D) perovskite DMA3BiI6 exhibits a superior diffusion coefficient and more efficient heterogeneous electron transfer compared to MA3Bi2I9. The systematic characterization of DMA3BiI6 perovskite emitters through X-ray diffraction, scanning electron microscopy, and steady-state photoluminescence has confirmed the formation of rod-like structures with absorption edges and emission peaks consistent with the literature. At the solid–solid interface, the 0-D DMA3BiI6 exhibits higher electronic coupling and lower activation energy than 2-D MA3Bi2I9, as confirmed by using temperature-dependent transient photoluminescence. Photophysical studies have revealed persistent excitonic populations and long-lived excited states, thereby indicating efficient ECL materials. These observed electrochemical phenomena suggest that low-dimensional DMA3BiI6 holds promise as a practical ECL emitter. Furthermore, the T-route ECL mechanism, characterized by a significant red shift from the fluorescence spectrum, confirms the capability of DMA3BiI6 in generating singlet excited states through triplet–triplet annihilation. Our findings present DMA3BiI6 as a viable and promising material for ECL applications, combining the advantages of lead-free composition with superior electro-optical properties. Consequently, the designed DMA3BiI6 perovskite holds great potential for eco-friendly optoelectronics and opens new avenues for the practical application of perovskite-based ECL and LED systems.Data availability
The data that support the findings of this study are available from the corresponding author, H.-Y. Hsu, upon reasonable request.Author contributions
C. H. Mak: conceptualization, investigation, methodology, writing – original draft; Y. Ai and S. C. Cheng: investigation, methodology; W. Niu, M. Du, K–C Cheng, G. Jia, and X-Q Xu: writing – review & editing; C.C. Ko, G. Zou, D-J Lee, and H.-Y. Hsu: supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Research Grants Council of Hong Kong (grant no. CityU 21203518 and F-CityU106/18), Innovation and Technology Commission (grant no. MHP/104/21), Shenzhen Science Technology and Innovation Commission (grant no. JCYJ20210324125612035, R-IND12303 and R-IND12304), City University of Hong Kong (grant no. 9229160, 9360140, 7005289, 7005580, 7005720, 9667213, 9667229, 9680331 and 9678291), and National Natural Science Foundation of China (51901119, 61874165, 51701159, 21974131 and 21833009). X. Xu thanks Guangdong Provincial Science and Technology Plan Project under Guangdong-Hong Kong-Macao Joint Funding Scheme for Collaborative Innovation (2021) [No. 2023A0505010003] and "Transformational Technologies for Clean Energy and Demonstration", Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA 21061001). Z. H. acknowledges the National Key Research and Development Program of China (no. 2021YFA1500900) and the National Natural Science Foundation of China (no. 52071174). C. H. Mak acknowledges the financial support from the Hong Kong Jockey Club under the research work Hong Kong JC STEM Lab for Circular Bio-economy (Project No. 2023-0078).Notes and references
- J. D. Holmes, K. P. Johnston, R. C. Doty and B. A. Korgel, Science, 2000, 287, 1471–1473 CrossRef CAS PubMed.
- Y. Bekenstein, B. A. Koscher, S. W. Eaton, P. Yang and A. P. Alivisatos, J. Am. Chem. Soc., 2015, 137, 16008–16011 CrossRef CAS.
- C. A. Richard, Z. Pan, H.-Y. Hsu, S. Cekli, K. S. Schanze and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2014, 6, 5221–5227 CrossRef CAS.
- K. Liu, S. Huang, Y. Jin, L. Ma, W.-X. Wang and J. C.-H. Lam, J. Hazard. Mater., 2022, 433, 128702 CrossRef CAS PubMed.
- J. Jia, K. Fu, S. Hou, B. Zhang, L. Fu, H.-Y. Hsu and G. Zou, J. Phys. Chem. C, 2019, 123, 29916–29921 CrossRef CAS.
- Y. Huang, M. Fang, G. Zou, B. Zhang and H. Wang, Nanoscale, 2016, 8, 18734–18739 RSC.
- L. Fu, K. Fu, X. Gao, S. Dong, B. Zhang, S. Fu, H.-Y. Hsu and G. Zou, Anal. Chem., 2021, 93, 2160–2165 CrossRef CAS.
- J. Xue, Z. Zhang, F. Zheng, Q. Xu, J. Xu, G. Zou, L. Li and J.-J. Zhu, Anal. Chem., 2017, 89, 8212–8216 CrossRef CAS PubMed.
- Y. Cao and J.-J. Zhu, Front. Chem., 2021, 9, 629830 CrossRef CAS.
- R. Liu, C. H. Mak, X. Han, Y. Tang, G. Jia, K.-C. Cheng, H. Qi, X. Zou, G. Zou and H.-Y. Hsu, J. Mater. Chem. A, 2020, 8, 23803–23811 RSC.
- J. Sheng, Y. He, J. Li, C. Yuan, H. Huang, S. Wang, Y. Sun, Z. Wang and F. Dong, ACS Nano, 2020, 14, 13103–13114 Search PubMed.
- B. W. Park, B. Philippe, X. Zhang, H. Rensmo, G. Boschloo and E. M. Johansson, Adv. Mater., 2015, 27, 6806–6813 Search PubMed.
- J. K. Pious, C. Muthu, S. Dani, A. Saeki and C. Vijayakumar, Chem. Mater., 2020, 32, 2647–2652 CrossRef.
- Y. Guo, G. Liu, Z. Li, Y. Lou, J. Chen and Y. Zhao, ACS Sustain. Chem. Eng., 2019, 7, 15080–15085 Search PubMed.
- Y. Tang, C. H. Mak, R. Liu, Z. Wang, L. Ji, H. Song, C. Tan, F. Barrière and H. Y. Hsu, Adv. Funct. Mater., 2020, 30, 2006919 CrossRef.
- W. Ke, I. Spanopoulos, C. C. Stoumpos and M. G. Kanatzidis, Nature Commun., 2018, 9, 4785 CrossRef.
- H. Meng, Z. Shao, L. Wang, Z. Li, R. Liu, Y. Fan, G. Cui and S. Pang, ACS Energy Lett., 2019, 5, 263–270 CrossRef.
- Y. Tang, C. H. Mak, C. Wang, Y. Fu, F.-F. Li, G. Jia, C.-W. Hsieh, H.-H. Shen, J. C. Colmenares, H. Song, M. Yuan, Y. Chen and H.-Y. Hsu, Small Methods, 2022, 6, 2200326 CrossRef.
- Y. Tang, C. H. Mak, J. Zhang, G. Jia, K.-C. Cheng, H. Song, M. Yuan, S. Zhao, J.-J. Kai, J. C. Colmenares and H.-Y. Hsu, Adv. Mater., 2023, 35, 2207835 Search PubMed.
- A. Pisanu, A. Speltini, P. Quadrelli, G. Drera, L. Sangaletti and L. Malavasi, J. Mater. Chem. C, 2019, 7, 7020–7026 Search PubMed.
- Y. Tang, C. H. Mak, R. Liu, Z. Wang, L. Ji, H. Song, C. Tan, F. Barrière and H. Y. Hsu, Adv. Funct. Mater., 2020, 2006919 Search PubMed.
- G. E. Eperon, K. H. Stone, L. E. Mundt, T. H. Schloemer, S. N. Habisreutinger, S. P. Dunfield, L. T. Schelhas, J. J. Berry and D. T. Moore, ACS Energy Lett., 2020, 5, 1856–1864 CrossRef.
- H. Zhao, K. Chordiya, P. Leukkunen, A. Popov, M. Upadhyay Kahaly, K. Kordas and S. Ojala, Nano Res., 2021, 14, 1116–1125 Search PubMed.
- W. Miao, J.-P. Choi and A. J. Bard, J. Am. Chem. Soc., 2002, 124, 14478–14485 Search PubMed.
- G. M. Paternò, N. Mishra, A. J. Barker, Z. Dang, G. Lanzani, L. Manna and A. Petrozza, Adv. Funct. Mater., 2019, 29, 1805299 Search PubMed.
- A. R. Srimath Kandada and A. Petrozza, Acc. Chem. Res., 2016, 49, 536–544 CrossRef PubMed.
- K. M. McCall, Z. Liu, G. Trimarchi, C. C. Stoumpos, W. Lin, Y. He, I. Hadar, M. G. Kanatzidis and B. W. Wessels, ACS Photonics, 2018, 5, 3748–3762 Search PubMed.
- W.-G. Li, X.-D. Wang, J.-F. Liao, Y. Jiang and D.-B. Kuang, Adv. Funct. Mater., 2020, 30, 1909701 Search PubMed.
- S. Öz, J.-C. Hebig, E. Jung, T. Singh, A. Lepcha, S. Olthof, F. Jan, Y. Gao, R. German and P. H. van Loosdrecht, Sol. Energy Mater. Sol. Cells, 2016, 158, 195–201 CrossRef.
- Z. Wang, L. Wang, Y. Xing, D. Yang, J. Yu, Z. Hao, C. Sun, B. Xiong, Y. Han and J. Wang, ACS Photonics, 2017, 4, 2078–2084 CrossRef.
- B. L. Li, J. Wang, H. L. Zou, S. Garaj, C. T. Lim, J. Xie, N. B. Li and D. T. Leong, Adv. Funct. Mater., 2016, 26, 7034–7056 CrossRef.
- P. J. Smith and C. K. Mann, J. Org. Chem., 1969, 34, 1821–1826 CrossRef CAS.
- W. Miao, Chem. Rev., 2008, 108, 2506–2553 CrossRef CAS.
- A. J. Bard, Electrogenerated Chemiluminescence, CRC Press, 2004 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta07204a |
| This journal is © The Royal Society of Chemistry 2025 |