
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic study of PFAS adsorption using a QPPTA linked viologen-modified covalent organic framework†
Amin
Zarei‡
a,
Leila
Khazdooz‡
a,
Amin
Zadehnazari
a,
Saeed
Amirjalayer§
b,
Matthew A.
Addicoat
c,
Ahmadreza
Khosropour
a and
Alireza
Abbaspourrad
 *a
*a
aDepartment of Food Science, College of Agriculture & Life Sciences, Cornell University, Stocking Hall, Ithaca, New York, 14853, USA. E-mail: Alireza@cornell.edu
bUniversity of Münster, Institute for Solid State Theory, Center for Nanotechnology and Center for Multiscale Theory and Computation, Wilhelm-Klemm-Straße 10, 48149 Münster, Germany
cSchool of Science and Technology, Nottingham Trent University, NG11 8NS Nottingham, UK
First published on 3rd February 2025
Abstract
A novel viologen-based covalent organic framework (VZ-COF) featuring hydrogen-bond acceptor sites, positively charged functional groups, and hydrophobic segments was synthesized to adsorb per- and polyfluoroalkyl substances (PFAS) using a new aromatic tetraamine linker (QPPTA) and Zincke salt. Including building blocks that offer multiple types of interactions cumulatively increases the capacity of COFs to adsorb PFAS. QPPTA provides two different kinds of intermolecular interactions: hydrophobic interactions through aromatic regions and hydrogen bonding via nitrogen atoms. The Zincke salt provides electrostatic interactions through positively charged functional groups. This combination of functionalities in VZ-COF enabled competitive PFAS adsorption efficiency. VZ-COF was tested against a series of PFAS and was found to adsorb all at >90%, and adsorption of perfluorooctane sulfonic acid (PFOS) and perfluorooctanoic acid (PFOA) was >99%. The adsorption capacity of VZ-COF was found to be 1000 mg g−1 and was unaffected by salts, pH variations, and complex organic acids, such as humic and tannic acids. VZ-COF also effectively removed PFOS and PFOA, reducing 1 μg L−1 concentrations to less than 1.6 ng L−1 and 4.1 ng L−1 respectively, even at extremely low concentrations (less than 3.8 ng L−1) in tap water. A short column of VZ-COF was used for flow adsorption of tap water to preconcentrate PFAS from tap water to detect and quantify very low concentrations. VZ-COF can be regenerated and reused with little change in adsorption efficiency.
1. Introduction
Engineering covalent organic frameworks (COF) from organic building blocks allows for the precise control of functionality which then allows for designed interactions with guest species. This functionality can be tailored to address specific scientific and technological needs, such as adsorption, catalysis, sensing, biomedical applications, and single-layer material design.Cationic COFs, featuring positively charged building blocks or pendant groups, represent a pivotal subclass of ionic COFs with versatile applications.1,2 Their structures position them as proficient adsorbents for the removal of toxic species.3–9 The synthesis of cationic COFs involves either the direct utilization of predesigned cationic monomers or the post-synthetic modification of neutral COFs.1,2
Per- and polyfluoroalkyl substances (PFAS), a class of engineered chemicals extensively used in industrial applications for decades, pose a distinctive challenge due to their robust C–F bonds, imparting chemical and thermal stability that resists degradation and metabolization.10–12 This stability, coupled with hydrophilic–lipophilic properties, contributes to their persistence in the environment and accumulation in human tissues, earning the label forever chemicals.13,14 Recent research underscores the environmental and health risks associated with PFAS accumulation necessitating urgent research into water purification and PFAS removal.12–16
Solid adsorbents have emerged as a preferred technique for PFAS removal owing to their simplicity, high efficiency, and economic feasibility.10,17 Solid state sorbents such as anion-exchange resins, metal–organic frameworks (MOFs), and COFs, among others, have been explored for PFAS removal.12,18–38 Despite their large specific surface areas, suitable pore sizes, and functional groups, these materials often encounter limitations such as slow removal rates, low capacity, poor regeneration ability, and low chemical stability. Other challenges arise from variations in pH and the presence of salts.
To address these challenges effectively, researchers have designed adsorbents with specialized multi-functional groups such as positive charges,28 fluorocarbon chains to enhance hydrophobicity,39 and heteroatoms capable of forming hydrogen bonds19 to increase PFAS adsorption efficiency. Another critical factor that is likely to enhance PFAS adsorption efficiency is increasing the water wettability of COF surfaces by increasing the number of hydrophilic groups within the framework. Enhancing water wettability significantly improves water penetration into the pores of porous adsorbents, allowing PFAS molecules to access internal adsorption sites more efficiently and thereby improving adsorption efficiency. Furthermore, increased wettability reduces surface tension, facilitating interactions between PFAS molecules, water, and the adsorbent surface, which enhances the exposure of PFAS molecules to adsorption sites.28 Additionally, fine-tuning the pore size of the adsorbent is vital to improve the selectivity of PFAS adsorption while concurrently mitigating the co-adsorption of organic interferences, such as humic and tannic acids.28 Furthermore, ensuring the stability of the adsorbent is essential for maintaining its structural integrity throughout the regeneration and reusability processes.
Although the use of COFs for PFAS removal has shown promising results,18–21,40,41 their adsorption capacity remains relatively low. For example, the maximum adsorption capacities of two hydrophobic COFs, COF-TpDt and olefin-linked 2D COF, for PFOA are 13.7 and 19.3 mg g−1, respectively, highlighting their low adsorption capacity.40,41 The incorporation of hydrogen-bonding sites into a fluorinated-squaramide-COF, a hydrophobic COF, obviously enhances its adsorption capacity to 370 mg g−1.19 Similarly, hollow V-COF, another hydrophobic COF, exhibits a low capacity of 53 mg g−1 for PFOA adsorption.20 However, functionalization of this COF with amine groups, which act as weak bases and provide both hydrogen-bond donor and acceptor functionalities, enables the synthesis of cystamine-grafted hollow COFs with a significantly improved capacity of 577 mg g−1.20 Furthermore, the introduction of positive charges into the COF enhances electrostatic interactions with PFOA, increasing the adsorption capacity of methylated TAPB-BP-allyl COF to 769 mg g−1.42 Despite this, the presence of positive charges alone is insufficient for the highly efficient removal of PFAS from real drinking water samples. Therefore, to improve the performance of COFs for the removal of diverse PFAS, the frameworks must be functionalized with a variety of groups to strengthen electrostatic and hydrophobic interactions while facilitating hydrogen bonding.
Incorporating positive charges into the COF skeleton increases electrostatic interactions with PFAS and improves the capacity.42 The presence of positive charges alone, however, is insufficient for highly efficient PFAS removal from real drinking water samples. Therefore, in addition to positive charges, the COFs must be modified with diverse functional groups to enhance hydrophobic interactions and facilitate hydrogen bonding.
Taking into account these considerations, we prepared a novel viologen-based COF, VZ-COF, using a new aromatic tetraamine linker, (4,4′,4′′,4′′′-(quinoxalino[2′,3′:9,10]phenanthro[4,5-abc]phenazine-6,7,15,16-etrayl)tetraaniline, QPPTA) and 1,1′-bis(2,4-dinitrophenyl)-4,4′-bipyridinium dichloride (Zincke salt) via the Zincke reaction (Scheme 1). The viologen building blocks in the VZ-COF structure give the COF a region of positive charge of +28.1 mV, as revealed by zeta potential measurements. Furthermore, the incorporation of nitrogen atoms in the quinoxaline rings of QPPTA introduces potential hydrogen bonding sites within the VZ-COF framework, while the pyrene and phenyl subunits of the QPPTA linker contribute to the formation of hydrophobic regions within the VZ-COF structure. This combination of functionalities enhances PFAS adsorption and makes VZ-COF a highly efficient adsorbent for PFAS. VZ-COF remains unaffected by changes in pH, as well as interference from salts, humic acid, and tannic acid. Additionally, it can be easily regenerated and reused.
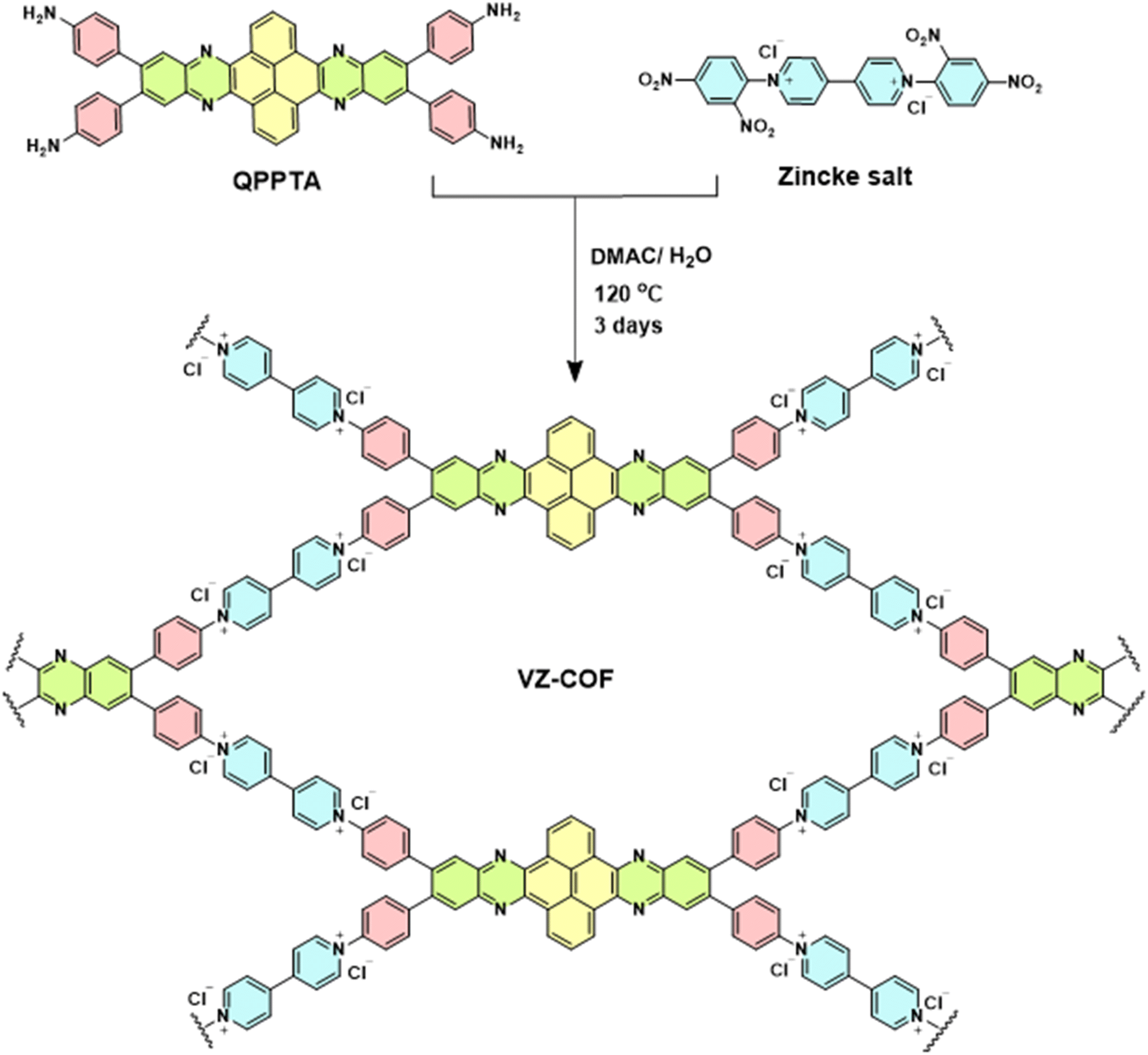 | ||
| Scheme 1 Synthesis of VZ-COF using the Zincke reaction. | ||
2. Results and discussion
2.1 Synthesis and characterization
QPPTA was synthesized through a two-step reaction process. The first step involved the synthesis of 6,7,15,16-tetrabromoquinoxalino[2′,3′:9,10]phenanthro[4,5-abc]phenazine (TBQPP) from pyrene-4,5,9,10-tetraone and 4,5-dibromobenzene-1,2-diamine. The second step was a Suzuki reaction between TBQPP and 4-aminophenylboronic acid pinacol ester. QPPTA was characterized using Fourier-transform infrared (FT-IR) spectroscopy, proton-1 and carbon-13 nuclear magnetic resonance spectroscopy, direct analysis in real-time of flight mass spectrometry (DART-TOF-MS), and ultraviolet/visible (UV/Vis) spectroscopy (Fig. S1–S8†). The synthesis of VZ-COF was investigated via the Zincke reaction involving the amine groups of the QPPTA monomer and Zincke salt under one-pot solvothermal conditions. The optimal reaction conditions, balancing crystallinity and product yield, required the use of a solvent mixture of dimethyl acetamide/water (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 120 °C for 72 hours.
:1) at 120 °C for 72 hours.
Powder X-ray diffraction (PXRD) of VZ-COF showed peaks at 4.23°, 5.84°, 7.91°, 8.62° and 24.86°, which were identified as the (110), (130), (220), (022), and (006) facets, respectively (Fig. 1, black curve). An eclipsed AA-stacking model produced a PXRD pattern (Fig. 1, blue curve) consistent with the observed experimental pattern. PXRD patterns derived from Pawley refinements (Fig. 1, red curves) confirmed the peak assignments, as indicated by their minimal differences (Fig. 1, yellow curve). The unit cell parameters were determined to be a = 25.0 Å, b = 56.7 Å, and c = 22.0 Å, with α = β = γ = 90°, in the space group P1. The refinement showed satisfactory agreement with the experimental data, with Rp = 8.6% and Rwp = 11.0%. The COF has a d-spacing of 3.87 Å, calculated using the Bragg equation from the last peak in the pattern. Hence, it comprises triclinic pores and 1D channels that are spatially distinct and not intertwined, without any interchannel penetration or entanglement.43
 | ||
| Fig. 1 Crystallinity and unit cell parameters of VZ-COF. | ||
The chemical structure of VZ-COF was investigated via FT-IR analysis to identify functional groups (Fig. 2a). A noticeable disappearance of the band at 1535 cm−1 corresponding to the asymmetric stretching vibration of NO2 of the Zincke salt, along with a diminished intensity of the band at 1340 cm−1 for the symmetric stretching vibration of NO2, confirmed the successful condensation between QPPTA and the Zincke salt.3,8 Further supporting evidence for the Zincke reaction was revealed in the weakened intensity of bands at 3420 cm−1 which corresponds to the asymmetric stretching vibration of NH2 and 3340 cm−1 which is the symmetric stretching vibration of NH2 in the end groups of VZ-COF compared to that of the QPPTA monomer.3,8 Additionally, a broad band centered at 1608 cm−1 was attributed to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N pyridinium and quinoxaline rings within the VZ-COF structure.3,8
N pyridinium and quinoxaline rings within the VZ-COF structure.3,8
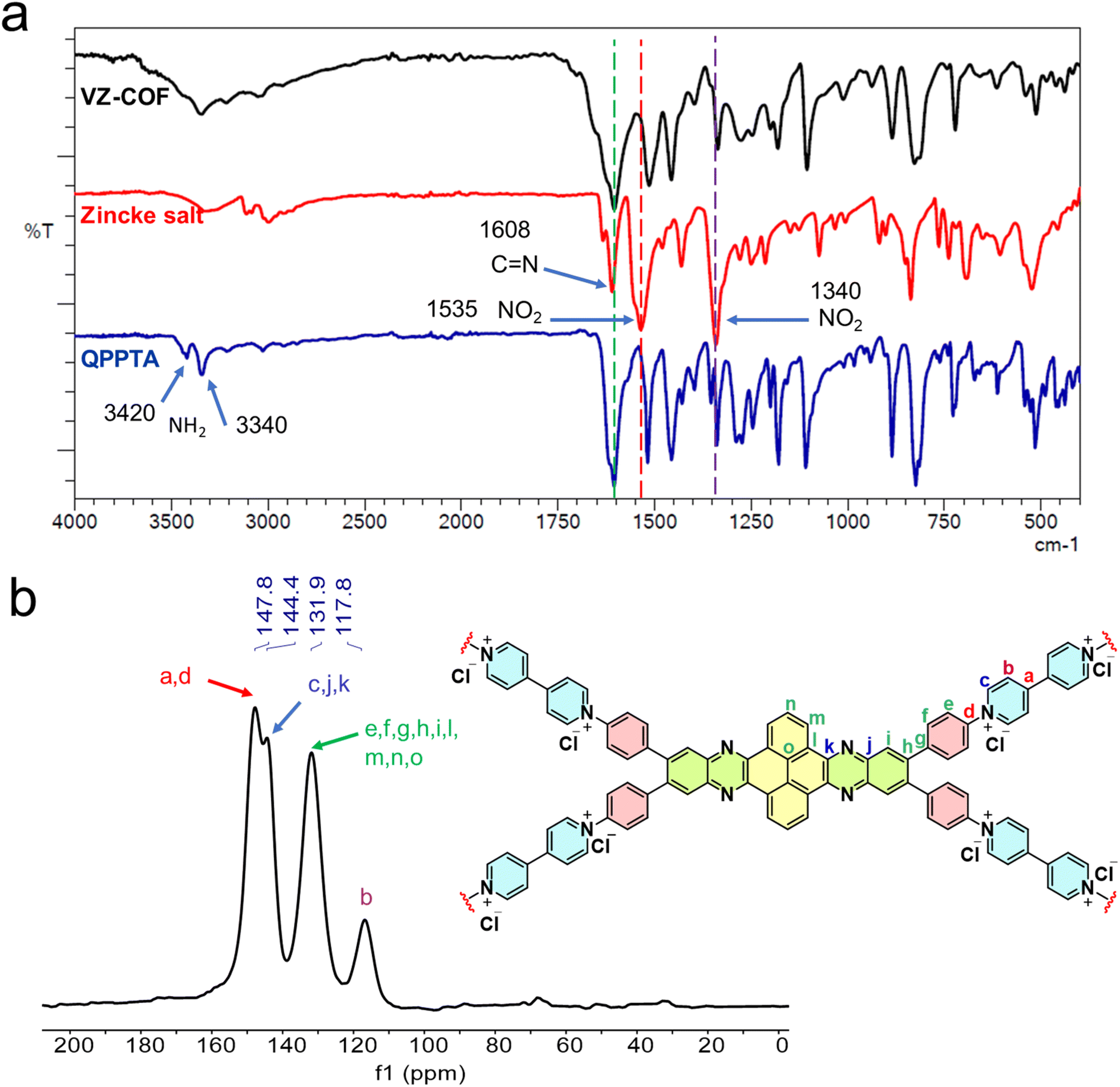 | ||
| Fig. 2 (a) FT-IR spectra of VZ-COF, Zincke salt, and QPPTA. (b) 13C CP/MAS ssNMR of VZ-COF. | ||
Solid-state 13C cross-polarization magic-angle spinning (13C CP/MAS) NMR spectroscopy was also used to determine the chemical structure of the COF (Fig. 2b). The predominant signals in the spectra were identified within the 118–148 ppm range, indicating the overlap of different sp2 hybridized carbon atoms within the aromatic and heteroaromatic rings of the VZ-COF. The presence of multiple signals within this matches the building blocks used in the synthesis of the COF.3,8
X-ray photoelectron spectroscopy (XPS) was used to understand the surface composition of VZ-COF, confirming the presence of C, N, O, and Cl in the COF structure (Fig. 3). The high-resolution XPS was deconvoluted for each element. The deconvolution of C 1s spectrum shows a large peak for carbon atoms with a binding energy of 284.5 eV, confirming the C–C and CC configurations of the aromatic rings. Another large peak at 285.9 eV corresponded to the C–N and CN carbons (Fig. 3b). The deconvoluted spectra of N 1s showed various N-containing functional groups, including NC, –NH2, +NC, and –NO2 (Fig. 3c). The binding energies for NC and +NC were found to be 398.8 and 402.0 eV, respectively.44 Peaks at 400.2 and 405.7 eV were attributed to amine and nitro end groups, respectively, which are proposed to be located at the edges or existing as defects in the lattice structures of the VZ-COF.44 Likewise, a peak at 532.1 eV, signifying the presence of oxygen, was assigned to the NO2 end groups or may be present due to trapped moisture or solvents on the surface (Fig. 3d).44 Peaks at 196.6, 198.2, and 268.1 eV were identified as Cl 2p3/2, Cl 2p1/2, and Cl 2s, respectively, confirming the presence of chloride anions (Fig. 3e and f).45 In addition to FT-IR and 13C CP/MAS NMR, the XPS analysis also corroborated the existence of viologen linkers in the VZ-COF.
 | ||
| Fig. 3 (a) High-resolution XPS analysis of VZ-COF. Deconvoluted profiles of (b) C 1s, (c) N 1s, (d) O 1s, (e) Cl 2p, and (f) Cl 2s. | ||
The surface charge of the VZ-COF particles was measured, revealing a zeta potential of +28.1 mV attributed to the presence of viologen units in its backbone (Fig. S9†).
The porosity of VZ-COF was investigated through N2 sorption measurements at 77 K, and the surface area was determined using the Brunauer–Emmett–Teller (BET) technique, based on adsorption–desorption isotherms. The BET surface area for VZ-COF was determined to be 147.4 m2 g−1 (Fig. 4a), accompanied by the pore-size distribution concentrated around 3.1 nm (Fig. S10†). Ionic COFs, in comparison to most neutral COFs, often have lower surface area due to the influence of counter ions, for example, chloride ions, which may obstruct some of the material's pores.3
 | ||
| Fig. 4 (a) N2 adsorption and desorption isotherms of VZ-COF. (b) TGA thermograms of VZ-COF and QPPTA. (c) UV-vis DRS spectrum of VZ-COF. (d) The optical band gap value of VZ-COF, obtained from the Kubelka–Munk-transformed reflectance spectra. | ||
To further study its long-term chemical stability, VZ-COF was exposed to aqueous solutions of HCl (pH 2) and NaOH (pH 12) for 24 hours at room temperature (∼25 °C). PXRD patterns affirmed the preservation of crystallinity (Fig. S11†), while FT-IR spectra indicated no changes in functional groups, indicating structural integrity (Fig. S12†). Furthermore, SEM imaging revealed no observable changes in surface morphology or particle shape after treatment (Fig. S13†).
The thermal stability of VZ-COF was investigated by thermogravimetric analysis (TGA) conducted under a nitrogen atmosphere and compared with the TGA of the monomer (QPPTA) to further confirm the synthesis of VZ-COF (Fig. 4b). Because the viologen linker is charged, water and polar solvents can be trapped within the resulting COF's porous structures. Consequently, elevated temperatures are required to release these trapped impurities.3,44,46 In the case of VZ-COF, the observed 4.7% mass loss around 150 °C is attributed to the evaporation of trapped moisture or solvent. Notably, 90% of the as-synthesized material remained stable up to 415 °C. The results indicate that VZ-COF demonstrated remarkable thermal stability, retaining around 55% of its initial weight to 800 °C. In contrast, the monomer exhibited nearly complete decomposition at 800 °C. The thermal stability observed in VZ-COF can be attributed to the incorporation of fully aromatic and heteroaromatic rigid building blocks into the structural framework alongside the interlayer interactions that stabilize the VZ-COF structure.
Ultraviolet-visible diffuse reflectance spectroscopy (UV-vis DRS) was used to examine the optical characteristics of VZ-COF (Fig. 4c). A broad semiconductor-like absorption peak was observed in the visible region with a maximum absorption around 585 nm and an absorption edge adjacent to 682 nm. This confirms the extended π-delocalization within the VZ-COF structure, consistent with its dark brown color. Applying the Kubelka–Munk equation, the optical band gap (Eg) was calculated to be 2.04 eV (Fig. 4d), indicating the semiconducting nature of VZ-COF.47 The optical activity can be attributed to the presence of viologen groups, aromatic, and fused aromatic rings, which enhance the π-conjugation of donor–acceptor pairs in the material's skeleton.
The morphology of the VZ-COF was analyzed using scanning electron microscopy (SEM). The SEM images depicted irregular spongy particle shapes, characterized by a granular and notably rough surface (Fig. 5a and b). Despite the lack of distinct shapes in the SEM images, high-resolution transmission electron microscopy (HRTEM) images showed a continuous crystalline arrangement extending nearly perpendicular to the observable patterns from one edge to another. This distinctive structure is noticeable across various areas of a particle when placed side by side (Fig. 5c). The presence of a triclinic array of spots in the fast Fourier transform (FFT) pattern signifies the appearance of crystalline characteristics within the VZ-COF particles (Fig. 5d). All FFT spots were identified and indexed along the [110] zone axis of the COF pores.48
 | ||
| Fig. 5 (a and b) SEM images of VZ-COF. (c and d) HRTEM images of VZ-COF and the top inset in (d) shows the FFT of the highlighted region. | ||
2.2 Adsorption of PFOA
VZ-COF was used to adsorb PFAS from contaminated water. The initial focus involved testing its ability to remove perfluorooctanoic acid (PFOA) from an aqueous solution prepared using Milli-Q water. First, 10 mg of vacuum-dried VZ-COF was diluted with 10 mL of Milli-Q water to produce a 1000 mg L−1 suspension of the adsorbent. The resulting suspension was sonicated for 2 min at room temperature. Then, the mixture was stirred for 2 hours at room temperature to make a homogeneous dispersion of the small particles (average size was 730 nm, Fig. S14†). To prepare VZ-COF and PFOA solution, 3 mL of the COF suspension (1000 mg L−1), 2 mL of an aqueous PFOA solution (10 mg L−1), and 15 mL of Milli-Q water were mixed.The initial concentration of PFOA was 1.0 mg L−1, while the corresponding concentration of the suspended VZ-COF in water was 150 mg L−1. After 2 h at room temperature (∼25 °C), the PFOA remaining in the solution was quantified using LC-MS. The removal efficiency for PFOA was found to be nearly quantitative (∼99.6%) (the instrumental detection limit for PFOA was 0.1 μg L−1). Further investigations were done to analyze the time-dependent and kinetic aspects of PFOA removal within 2 h (Fig. S15 and S16†). The kinetics of the PFOA removal were fitted with the linear pseudo-second-order kinetic model. The resulting rate constant for PFOA removal with COF was determined to be 807 g mg−1 h−1, showing near quantitative removal within 10 minutes.
The adsorption capacity of VZ-COF for PFOA removal from water was investigated. The adsorption isotherm was designed using 150 mg L−1 of the COF and a range of PFOA solution concentrations (0.2–800 mg L−1) (Fig. S17†). The adsorption isotherm exhibited an excellent fit with the linear Langmuir model and the experimental data with a correlation coefficient of approximately 0.99 (Fig. S18†). According to the Langmuir model, the estimated maximum adsorption capacity (Qm) for VZ-COF was determined to be 1000 mg g−1. This value is higher than the adsorption capacities reported for most other COFs used for removing PFOA from water (Table S1†). The affinity coefficient (KL) was calculated to be 1.5 × 104 L mol−1, indicating a high affinity between both COFs and PFOA. Due to a higher correlation coefficient, the linear Langmuir model exhibited a superior fit when compared to the Freundlich isotherm (Fig. S19†).
As a supplementary experiment, energy-dispersive X-ray spectroscopy (EDS) and FT-IR were used to confirm the presence of PFOA following adsorption onto VZ-COF. A uniform distribution of fluorine was observed in the EDS mapping of the surface of the VZ-COF after PFAS adsorption (Fig. S20†). FT-IR analysis of the adsorbed PFOA on VZ-COF showed a prominent band at 1690 cm−1, corresponding to the stretching vibration of the carbonyl functional group of PFOA (Fig. S21†). Additionally, two bands appearing around 1196 and 1137 cm−1 were attributed to the stretching vibrations of CF3 and CF2 groups of PFOA, respectively.49
The mechanisms and parameters influencing the adsorption performance of VZ-COF toward PFAS were investigated. While electrostatic and hydrophobic interactions between PFAS and adsorbents are the two main mechanisms that control PFAS adsorption, the surface area, charge, porosity, shape, and particle size of the sorbents also play critical roles in the affinity and ability of the solid adsorbents towards PFAS molecules.10–12 The zeta potential of +28.1 mV attributed to the presence of viologen units, confirms electrostatic interactions as a mechanism for PFAS removal by VZ-COF.
Porosity plays an important factor influencing PFAS adsorption, especially at different PFAS concentration levels. At higher concentrations of PFAS, self-aggregated structures of micelles and hemi-micelles can form on the positively charged VZ-COF surface.12 This process may lead to the obstruction of specific adsorption sites within the microporous structures lowering the adsorption efficiency and capacity of the adsorbent.12 The incorporation of mesoporous structures within solid adsorbents may address this issue and improve the capacity. In VZ-COF, the pore width distribution is ∼3.1 nm, which is sufficiently large to allow the entry of PFOA molecules with dimensions of 1.1 × 0.36 × 0.36 nm.28 This pore size is well-suited to accommodate the molecular geometry of PFOA,40 facilitating its access to the interior adsorption sites of VZ-COF. Surface area is another critical factor in PFAS removal from contaminated water. Compared to other cationic COFs used for PFAS removal,21,22 our direct synthetic protocol provides higher surface area, particularly relative to cationic COFs made via post-synthetic modification.21 VZ-COF exhibited mesoporous structures and a suitable surface area to enhance removal efficiency. Through sonication, the particle size was reduced to 730 nm and therefore VZ-COF was better dispersed in water. Breaking up the agglomerates and reducing particle size enhance the colloidal dispersion of the COF and makes its active sites more accessible. The particle size still allowed for filtration and facilitated the removal of the PFAS during the regeneration and reusability studies of VZ-COF because the PFAS adsorption sites were more accessible.
VZ-COF was designed with fused aromatic rings, which are hydrolytically more stable, addressing the challenge of some adsorbents that have hydrolysable fluorocarbon linkers that may release linkers to the environment.19,30,39 Additionally, these aromatic linkers increase the size of the hydrophobic domains between the polar viologen subunits which increases the hydrophobicity of VZ-COF enhancing the removal of PFAS. The measured contact angle of VZ-COF is approximately 68° (Fig. S22†), classifying it as a weakly hydrophobic material.50
Using Density Functional Theory (DFT) calculations at the B3LYP+D3BJ/def2-TZVP level, as implemented in the ORCA package,51–55 the high performance of the VZ-COF toward the adsorption of PFAS molecules was found to be due to its framework structure, which facilitates both hydrogen bonding and hydrophobic interactions between PFAS molecules and the COF's inner surface. The strong binding energy of 72.72 kJ mol−1 per molecule arises from the interaction between the acid head group and unfunctionalized nitrogen atoms in the quinoxaline rings of the VZ-COF (indicated in green), as well as the hydrophobic interactions between the fluorinated backbone and the aromatic groups (indicated in purple) (Fig. 6). The structural details of the optimized geometry of a single PFAS molecule positioned at the edge of the inner surface of the VZ-COF are provided in Table S2.† Theoretically, the pore dimensions allow four PFAS molecules to occupy these energetically favorable adsorption sites (Fig. 6) resulting in high adsorption capacity.
 | ||
| Fig. 6 Ball-and-stick representation of the optimized geometry of a single PFOA molecule at the edge of the inner surface of VZ-COF (carbon: black; nitrogen: blue; chlorine: orange; fluorine: ochre; oxygen: red; hydrogen: white). The full pore structure is superimposed to indicate the four energetically equivalent adsorption sites. | ||
Electrostatic interactions work with the hydrophobicity of VZ-COF to adsorb PFAS from water. To study the effect of the electrostatic interactions, the adsorption of PFOA using VZ-COF at different pH levels was investigated. Adjustments to the solution pH were made using 0.1 M solutions of HCl and NaOH. The initial concentrations of VZ-COF and PFOA were 150 mg L−1 and 0.20 mg L−1, respectively (Fig. 7a). After 1 h at room temperature, the best PFOA removal efficiency was observed at pH 7 which is attributed to the high electrostatic interactions and is consistent with the zeta potential of VZ-COF at pH 7 (+28.1 mV). At pH levels of 2, 3, 5, and 9, there was minimal change in the adsorption efficiency of VZ-COF for PFOA from Milli-Q water. However, a clear decrease in adsorption efficiency was observed at pH 11 and 12 where the surface charge was negative which caused electrostatic repulsion between the surface of the COF and the PFOA.
 | ||
| Fig. 7 (a) Effect of pH on the adsorption of PFOA using VZ-COF ([PFOA]0 = 0.20 mg L−1; [VZ-COF] = 150 mg L−1) after 60 min. (b) Effect of inorganic ions, tannic and humic acids on the adsorption of PFOA usingVZ-COF ([PFOA]0 = 0.20 mg L−1; [VZ-COF] = 150 mg L−1) after 75 min. (c) Adsorption of different PFAS spiked in Milli-Q water usingVZ-COF in single-solute batch experiments after 45 min (initial concentration of each PFAS was 30 μg L−1; [VZ-COF] = 150 mg L−1). (d) Competitive adsorption of different PFAS spiked in Milli-Q water using VZ-COF in multiple-solute batch experiments after 45 min (initial concentration of each PFAS was 30 μg L−1; [VZ-COF] = 150 mg L−1). (e) Competitive adsorption of different PFAS added in tap water using two concentrations of VZ-COF in multiple-solute batch experiments after 45 min (initial concentration of each PFAS was 1 μg L−1; [VZ-COF] = 150 mg L−1 and 450 mg L−1). (f) Reusability of VZ-COF in PFOA removal by consecutive adsorption/desorption experiments. All measurements were carried out in triplicate. Note: ✶ indicates that the concentrations are below the detection limits and the standard error bars are not presented in those cases. | ||
The zeta potential of VZ-COF was measured at various pH levels (Fig. S23†). Upon increasing the pH from 7 to 9, the corresponding zeta potential decreased from +28.1 to +14.0 mV. Despite this decrease, the positive surface charge remained considerable, and no distinct change was observed in the PFOA removal efficiency. However, increasing the pH to 11 dropped the surface charge to −22.4 mV at which point the PFOA removal efficiency also decreased to 28%. At pH 11, the electrostatic interactions between the anionic PFOA and the original positively-charged surface of the VZ-COF were lost as the surface charge became more negative and the VZ-COF and PFOA repelled each other. The concentration of excess hydroxide ions at pH 11 is 0.001 M, ∼2000 times higher than that of PFOA. Consequently, the competition between hydroxide and PFOA on the surface of VZ-COF adversely affects the number of sites available for PFOA adsorption lowering the removal efficiency. At pH 12, the zeta potential of VZ-COF measured −43.6 mV, resulting in a reduction of PFOA removal efficiency to 15%. The concentration of OH− ions at pH 12 is 0.01 M, which is ∼20000 times higher than that of PFOA. This indicates a lack of electrostatic interactions between PFOA and VZ-COF at this pH. Based on these results, electrostatic interactions are a major driving force for PFOA adsorption at pH 7, and when the electrostatic interactions are absent at high pH levels, the hydrophobic interactions of PFOA and VZ-COF become the predominant mechanism of adsorption.
Decreasing the pH, below pH 7, did not have a major effect on the removal of PFOA. The surface charges of VZ-COF at pH levels of 5, 3, and 2 were measured to be +28.7, +33.1, and +22.6 mV respectively (Fig. S23†). Decreasing the pH allows the unfunctionalized nitrogen atoms of quinoxaline rings within the VZ-COF structure to act as weak bases and gradually become protonated, increasing the positive surface charge. This enhancement can facilitate interactions with PFOA, particularly at neutral and acidic pH levels, thereby improving removal efficiency compared to alkaline pH.29,41 The zeta potential analysis confirmed that maximum protonation occurred at pH 3, resulting in the highest positive surface charge. Adjusting to pH 2, the zeta potential decreased to +22.6 mV. After protonation of the most unfunctionalized nitrogen atoms, the pH decrease led to an increase in the ionic strength of the aqueous solution, thereby causing a reduction in the static charge on the surface.22 As pH decreases with the addition of HCl solution, the anionic forms of PFOA may undergo partial protonation, thereby reducing electrostatic interactions between PFOA and VZ-COF. Additionally, the increased concentration of chloride ions in the solution may compete with PFOA, decreasing the interactions of PFOA with the COF. For instance, at pH 2, the concentration of Cl− ions is 0.01 M, which is ∼20000 times higher than that of PFOA. However, at this pH level, the PFOA removal percentage remains at 98.1%, indicating a slight change in the COF's removal efficiency. The increase in positive surface charge, combined with hydrophobic interactions and the presence of unfunctionalized nitrogen atoms, prevents a decline in the VZ-COF adsorption of PFOA from water at lower pH levels.
VZ-COF was assessed for its ability to remove PFOA under real-world conditions by evaluating the potential interference of inorganic ions, humic and tannic acids (Fig. 7b). Common inorganic anions found in groundwater, including chloride, nitrate, sulfate, and chromate, were used in this study. Specifically, NaCl, NaNO3, Na2SO4, and K2CrO4 were used as the sources of these inorganic anions. To show the selectivity of VZ-COF in the presence of organic interferences, humic acid and tannic acid were used. In each experiment, 150 mg L−1 of VZ-COF, 0.20 mg L−1 of PFOA, and 2.0 mg L−1 of the inorganic ions, humic acid or tannic acid were added to Milli-Q water. A control experiment was carried out, in which similar amounts of PFOA and VZ-COF were added to Milli-Q water without adding any humic acid, tannic acid or inorganic ions. The results showed a small decrease in removal efficiency from >99% to 97% when the SO42− concentration was increased to 10 times that of PFOA. This could be attributed to the strong competitive adsorption of divalent anions with PFOA.56 Similarly, the presence of CrO42− resulted in a slight reduction in PFOA removal efficiency from >99% to 98%. Other pollutants showed no significant impact on PFOA removal efficiency.
Expanding the scope, we added PFOA, VZ-COF, and a mixture of these anions, humic acid, and tannic acid into Milli-Q water. Each anion and complex organic acid was maintained at a concentration of 1.0 mg L−1, while the concentrations of PFOA and VZ-COF were 0.20 mg L−1 and 150 mg L−1 respectively. Despite the total concentration of inorganic anions, humic acid, and tannic acid reaching 6.0 mg L−1, which was 30 times higher than that of PFOA, only a slight, <4%, decrease in removal efficiency was observed.
Finally, we used tap water from Ithaca, NY in place of Milli-Q water to assess PFOA removal. Tap water analysis was reported in the annual drinking water quality report of Ithaca, NY, (specifically Tables 1–4 in the report for 2024).57 PFOA and VZ-COF were present at concentrations of 0.20 mg L−1 and 150 mg L−1, respectively. A decrease in removal efficiency of less than 5% affirms the ability of the VZ-COF to remove PFOA from tap water (Fig. 7b).
2.3 Adsorption of other PFAS compounds
VZ-COF was also tested against other PFAS compounds that are known to be present in contaminated water effluent. In addition to PFOA, perfluorobutanoic acid (PFBA), perfluorohexanoic acid (PFHA), perfluoro-2-methyl-3-oxahexanoic acid (GenX), perfluorooctane sulfonic acid (PFOS), and perfluorodecanoic acid (PFDA) were chosen as representative PFAS to evaluate (Fig. 7c). The removal of each PFAS was first performed separately in Milli-Q water with each PFAS initially present at a concentration of 30 μg L−1 and a VZ-COF concentration of 150 mg L−1. After a 45 minute exposure period at room temperature (∼25 °C), the PFAS remaining in the solution was quantified using LC-MS/MS. PFOA, PFOS, and PFDA, PFAS with longer carbon chains, exhibited remarkable removal efficiency by the COF, surpassing 99%. This confirms that the hydrophobic sites in VZ-COF enhance its affinity for longer-chain PFAS. Among these, PFOS had the highest adsorption result, and it was not detected in the solution after exposure to the COF (detection limit of PFOS was 1.6 ng L−1). This may be attributed to the higher acidity of PFOS58 which increases its interaction with the unfunctionalized nitrogen atoms of the quinoxaline subunits in the VZ-COF. Although the removal efficiencies for GenX and PFHA were slightly lower, the observed range of 95% to 97% confirms the effectiveness of VZ-COF toward PFAS with shorter carbon chains. Finally, PFBA, a leachable short-chain anionic PFAS, exhibited approximately 90% adsorption, which is in agreement with other studies using COFs where removal efficiencies range from 23% to 90%.19,20,41To further investigate the efficiency of VZ-COF in removing PFAS contaminants, we studied the competitive adsorption of PFBA, PFHA, GenX, PFOA, PFOS, and PFDA (Fig. 7d). Each PFAS was present at a concentration of 30 μg L−1 in Milli-Q water, for a total PFAS concentration of 180 μg L−1, while the concentration of VZ-COF was maintained at 150 mg L−1. The PFAS mixture was exposed to VZ-COF for 45 minutes. The high adsorption of VZ-COF resulted in negligible decreases in removal percentages for each PFAS compound in the mixture solution.
Consequently, VZ-COF maintains a consistently high overall removal efficiency. The results validated that long-chain PFAS, particularly PFOS, exhibited a higher preference for adsorption onto the adsorbent compared to the shorter-chain variants, thereby confirming the presence of hydrophobic interactions between longer-chain PFAS and VZ-COF.
A PFAS competitive adsorption study was also carried out in tap water. PFBA, PFHA, GenX, PFOA, PFOS, and PFDA were added to tap water from Ithaca, NY, such that the concentration of each PFAS was 1.0 μg L−1, for a total PFAS concentration of 6.0 μg L−1, and the VZ-COF concentration was 150 mg L−1. A PFAS concentration of 6.0 μg L−1 represents the average PFAS concentration in the wastewater effluent of sewage plants worldwide (approximately 5.7 μg L−1).59 The mixture was then exposed to the COF for 45 minutes (Fig. 7e). VZ-COF removed PFOS and PFDA such that these compounds were not detected in the solution after exposure to the COF (detection limits of PFOS and PFDA were 1.6 ng L−1 and 2.2 ng L−1 respectively). For PFOA the removal efficiency was measured at around >99%, resulting in a notable reduction in PFOA concentration from 1.0 μg L−1 to 6.3 ng L−1. In the case of PFHA, GenX, and PFBA, the removal efficiencies were observed at ∼94%, ∼87%, and ∼61% respectively.
When the concentration of VZ-COF was increased from 150 mg L−1 to 450 mg L−1 the concentrations of PFOA, in addition to PFOS and PFDA, were also not detected in the solution (the detection limit of PFOA was 4.1 ng L−1). Additionally, the removal efficiencies for PFHA, GenX, and PFBA increased to ∼98%, ∼97%, and ∼88%, respectively (detection limits of PFHA, GenX, and PFBA were 1.4 ng L−1, 2.4 ng L−1, and 8.4 ng L−1). The surface zeta potential of VZ-COF decreased from +28.1 mV in Milli-Q water to +2.6 mV in tap water; this is attributed to the competition of the solutes in tap water, which decreases the electrostatic interactions between PFAS and the COF. VZ-COF removed PFOS and PFOA reducing their levels to below the U.S. Environmental Protection Agency's advisory limit of 4 ng L−1.38 This observation also supports the role of the hydrophobic and unfunctionalized nitrogen atoms as synergistic binding sites in VZ-COF, enhancing PFAS removal from tap water.
2.4 Reusability of the VZ-COF
We also investigated the reusability of VZ-COF through consecutive adsorption/desorption experiments. Following previously established procedures for regenerating cationic COFs and polymers,22,28 desorption experiments were done by washing the COF with a mixture of methanol and 1 w/v% aqueous NaCl. The quantities of adsorbed and desorbed PFOA were measured in each cycle, and the results indicated sustained high efficiency (>99%) of VZ-COF for PFOA adsorption even after four cycles (Fig. 7f).The crystallinity of VZ-COF was evaluated after four use cycles as a measure of assessing the structural stability of VZ-COF. The PXRD patterns were unchanged, confirming the retention of the COF's crystallinity (Fig. S24†). Additionally, FT-IR spectra showed no alterations in functional groups, thereby suggesting the sustained integrity of the structure (Fig. S25†). SEM imaging also showed no observable changes in the surface morphology or particle shape during the reusability process (Fig. S26†).
Finally, to demonstrate the role of an adsorbent in removing PFAS at extremely low concentrations, a 50 mg sample of VZ-COF was packed into a short column fitted with a sintered glass filter (4–8 μ size). Then ∼6.6 L of locally sourced tap water from Ithaca, NY, was passed through the column (Fig. S27†), after which VZ-COF was separated from the filter. The desorption step involved pouring the VZ-COF into 100 mL of a solution composed of HPLC-grade methanol (90 mL) and 1 w/v% aqueous NaCl in Milli-Q water (10 mL). The suspension was then sonicated for 1 minute at 25 °C. Subsequently, the suspension was filtered through a sintered glass filter (4–8 μm pore size) and washed with methanol (3 × 20 mL). The filtrate was then evaporated under vacuum, and the residual solid was dissolved in 15 mL of Milli-Q water, followed by analysis via LC-MS/MS.
Prior to treatment only perfluoropropionic acid (PFPrA) was detected at a concentration of 210 ng L−1 in the tap water, which decreased to 150 ng L−1 after passing through VZ-COF. Following PFAS desorption from the COF and concentration of the filtrate to 15 mL, LC-MS/MS analysis of the sample revealed that 15 PFAS were present (Table 1). Although the real concentration of PFAS in Ithaca's tap water falls below detection limits, using our VZ-COF sorbent and preconcentration method, we were able to detect and quantify the PFAS in tap water. These findings underscore the ability of VZ-COF to remove a diverse array of PFAS, even below their detection limits from tap water.
| Entry | PFAS | Abbreviation | Eluent concentrationa (ng L−1) | Detection limit (ng L−1) | Calculated amountb (ng L−1) |
|---|---|---|---|---|---|
| a Concentrations of PFASs after elution from VZ-COF (the eluent volume was 15 mL). b Calculated amount of PFASs in tap water. | |||||
| 1 | Hexafluoropropylene oxide dimer acid | GenX | 4.9 ± 0.18 | 2.4 | 0.01 |
| 2 | N-Ethyl perfluorooctanesulfonamidoacetic acid | NEtFOSAA | 3.9 ± 0.14 | 3.5 | 0.01 |
| 3 | Perfluorobutanesulfonic acid | PFBS | 31 ± 2.8 | 3.6 | 0.07 |
| 4 | Perfluorobutanoic acid | PFBA | 24 ± 2.0 | 8.4 | 0.05 |
| 5 | Perfluorodecanesulfonic acid | PFDS | 3.8 ± 0.42 | 2.6 | 0.01 |
| 6 | Perfluorodecanoic acid | PFDA | 9.5 ± 0.49 | 2.2 | 0.02 |
| 7 | Perfluoroheptanesulfonic acid | PFHpS | 60 ± 4 | 1.6 | 0.14 |
| 8 | Perfluoroheptanoic acid | PFHpA | 68 ± 5.8 | 3.2 | 0.15 |
| 9 | Perfluorohexanesulfonic acid | PFHxS | 100 ± 8 | 1.6 | 0.23 |
| 10 | Perfluorohexanoic acid | PFHxA | 59 ± 4.0 | 1.4 | 0.13 |
| 11 | Perfluorononanesulfonic acid | PFNS | 5.6 ± 0.42 | 1.9 | 0.01 |
| 12 | Perfluorononanoic acid | PFNA | 22 ± 2.3 | 2.2 | 0.05 |
| 13 | Perfluorooctanesulfonic acid | PFOS | 1700 ± 42 | 1.6 | 3.86 |
| 14 | Perfluorooctanoic acid | PFOA | 680 ± 18 | 4.1 | 1.55 |
| 15 | Perfluoropentanesulfonic acid | PFPeS | 4.3 ± 0.28 | 2.9 | 0.01 |
3. Conclusion
VZ-COF, a novel viologen-based COF, was synthesized using QPPTA as a new multi-functional monomer via the Zincke reaction. VZ-COF showed high stability, a highly positive surface charge, appropriate surface area, porosity, and pore size. The combined effects of electrostatic interactions, along with the presence of hydrogen bonding sites and hydrophobic interactions, synergistically enhanced the efficacy of VZ-COF for PFAS removal, even at low concentrations. In practical applications, VZ-COF demonstrated exceptional efficacy in reducing the concentration of PFOA and PFOS from 1.0 μg L−1 to less than 4.0 ng L−1, thereby surpassing the advisory level set by the U.S. Environmental Protection Agency (EPA). Its successful application in real tap water and simulated wastewater samples, rapid kinetic adsorption, high capacity, and ease of reusability confirm its practical application for removing PFAS from polluted water sources. Ongoing investigations in our lab aim to synthesize new adsorbents with multi-functional groups to further enhance PFAS removal efficiency.Data availability
Source data for figures are freely available at https://zenodo.org/records/13785934.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project received support from the Cornell Center for Materials Research (CCMR), funded by the National Science Foundation under grant DMR-1719875. We are also thankful for the opportunity to use the NMR facility at Cornell University, which is supported by the NSF through MRI grant number CHE-1531632. The authors certify that generative AI was not used in preparing this article. Non-generative AI, such as spelling and grammar checkers in Office 365 and Google Docs, and citation managing software, was used. All instances when non-generative AI was used were reviewed by the authors and editor.References
- X. Liang, Y. Tian, Y. Yuan and Y. Kim, Adv. Mater., 2021, 33, 2105647 CrossRef CAS PubMed.
- P. Zhang, Z. Wang, P. Cheng, Y. Chen and Z. Zhang, Coord. Chem. Rev., 2021, 438, 213873 CrossRef CAS.
- X.-R. Chen, C.-R. Zhang, W. Jiang, X. Liu, Q.-X. Luo, L. Zhang, R.-P. Liang and J.-D. Qiu, Sep. Purif. Technol., 2023, 312, 123409 CrossRef CAS.
- C. Du, X. Chen, H. Wu, Z. Pan, C. Chen, G. Zhong and C. Cai, RSC Adv., 2023, 13, 24064–24070 RSC.
- C.-H. Yang, J.-S. Chang and D.-J. Lee, Chemosphere, 2020, 253, 126736 CrossRef CAS PubMed.
- S. I. G. P. Mohamed, T. Zhang, Z. Jiang, A. M. Rappe and S. Nejati, J. Phys. Chem. Lett., 2022, 13, 10030–10034 CrossRef CAS PubMed.
- A. Hassan, M. M. R. Mollah, S. Das and N. Das, J. Mater. Chem. A, 2023, 11, 17226–17236 RSC.
- T. Skorjanc, D. Shetty, F. Gándara, L. Ali, J. Raya, G. Das, M. A. Olson and A. Trabolsi, Chem. Sci., 2020, 11, 845–850 RSC.
- H.-Z. Li, C. Yang, H.-L. Qian and X.-P. Yan, Sep. Purif. Technol., 2023, 306, 122704 CrossRef CAS.
- C. T. Vu and T. Wu, Crit. Rev. Environ. Sci. Technol., 2022, 52, 90–129 CrossRef CAS.
- S. Yin and D. Villagrán, Sci. Total Environ., 2022, 831, 154939 CrossRef CAS PubMed.
- P. S. Pauletto and T. J. Bandosz, J. Hazard. Mater., 2022, 425, 127810 CrossRef CAS PubMed.
- H. Brunn, G. Arnold, W. Körner, G. Rippen, K. G. Steinhäuser and I. Valentin, Environ. Sci. Eur., 2023, 35, 1–50 CrossRef.
- M. G. Evich, M. J. Davis, J. P. McCord, B. Acrey, J. A. Awkerman, D. R. Knappe, A. B. Lindstrom, T. F. Speth, C. Tebes-Stevens and M. J. Strynar, Science, 2022, 375, eabg9065 CrossRef CAS PubMed.
- S. E. Fenton, A. Ducatman, A. Boobis, J. C. DeWitt, C. Lau, C. Ng, J. S. Smith and S. M. Roberts, Environ. Toxicol. Chem., 2021, 40, 606–630 CrossRef CAS PubMed.
- E. Panieri, K. Baralic, D. Djukic-Cosic, A. Buha Djordjevic and L. Saso, Toxics, 2022, 10, 44 CrossRef CAS PubMed.
- E. Gagliano, M. Sgroi, P. P. Falciglia, F. G. Vagliasindi and P. Roccaro, Water Res., 2020, 171, 115381 CrossRef CAS PubMed.
- W. Ji, L. Xiao, Y. Ling, C. Ching, M. Matsumoto, R. P. Bisbey, D. E. Helbling and W. R. Dichtel, J. Am. Chem. Soc., 2018, 140, 12677–12681 CrossRef CAS PubMed.
- J. Huang, Y. Shi, G. Huang, S. Huang, J. Zheng, J. Xu, F. Zhu and G. Ouyang, Angew. Chem., Int. Ed., 2022, 61, e202206749 CrossRef CAS PubMed.
- J. Huang, Y. Shi, J. Xu, J. Zheng, F. Zhu, X. Liu and G. Ouyang, Adv. Funct. Mater., 2022, 32, 2203171 CrossRef CAS.
- A. N. Zeppuhar, D. S. Rollins, D. L. Huber, E. A. Bazan-Bergamino, F. Chen, H. A. Evans and M. K. Taylor, ACS Appl. Mater. Interfaces, 2023, 15, 52622–52630 CAS.
- W. Wang, Z. Zhou, H. Shao, S. Zhou, G. Yu and S. Deng, Chem. Eng. J., 2021, 412, 127509 CrossRef CAS.
- S. Tang, X. Qin, Y. Lv, K. Hu and S. Zhao, Appl. Surf. Sci., 2022, 601, 154224 CrossRef CAS.
- P. McCleaf, S. Englund, A. Östlund, K. Lindegren, K. Wiberg and L. Ahrens, Water Res., 2017, 120, 77–87 CrossRef CAS PubMed.
- C. C. Murray, R. E. Marshall, C. J. Liu, H. Vatankhah and C. L. Bellona, J. Water Process Eng., 2021, 44, 102342 CrossRef.
- W. Wang, X. Mi, Z. Zhou, S. Zhou, C. Li, X. Hu, D. Qi and S. Deng, J. Colloid Interface Sci., 2019, 557, 655–663 CrossRef CAS PubMed.
- T. Tamanna, P. J. Mahon, R. K. Hockings, H. Alam, M. Raymond, C. Smith, C. Clarke and A. Yu, Appl. Sci., 2023, 13, 6263 CrossRef CAS.
- X. Liu, C. Zhu, J. Yin, J. Li, Z. Zhang, J. Li, F. Shui, Z. You, Z. Shi and B. Li, Nat. Commun., 2022, 13, 2132 CrossRef CAS PubMed.
- D. Abdullatif, A. Khosropour, A. Khojastegi, I. Mosleh, L. Khazdooz, A. Zarei and A. Abbaspourrad, ACS Appl. Polym. Mater., 2022, 5, 412–419 CrossRef.
- C. Ching, Y. Ling, B. Trang, M. Klemes, L. Xiao, A. Yang, G. Barin, W. R. Dichtel and D. E. Helbling, Water Res., 2022, 209, 117938 CrossRef CAS PubMed.
- J. M. Steigerwald, S. Peng and J. R. Ray, ACS ES&T Eng., 2023, 3, 520–532 Search PubMed.
- M. Ateia, A. Alsbaiee, T. Karanfil and W. Dichtel, Environ. Sci. Technol. Lett., 2019, 6, 688–695 CrossRef CAS.
- V. Alonso-de-Linaje, M. C. Mangayayam, D. J. Tobler, V. Rives, R. Espinosa and K. N. Dalby, Environ. Technol. Innovation, 2021, 21, 101231 CrossRef CAS.
- R. Mukhopadhyay, B. Sarkar, K. N. Palansooriya, J. Y. Dar, N. S. Bolan, S. J. Parikh, C. Sonne and Y. S. Ok, Adv. Colloid Interface Sci., 2021, 297, 102537 CrossRef CAS PubMed.
- P.-H. Chang, C.-Y. Chen, R. Mukhopadhyay, W. Chen, Y.-M. Tzou and B. Sarkar, J. Colloid Interface Sci., 2022, 623, 627–636 CrossRef CAS PubMed.
- R. Li, S. Alomari, T. Islamoglu, O. K. Farha, S. Fernando, S. M. Thagard, T. M. Holsen and M. Wriedt, Environ. Sci. Technol., 2021, 55, 15162–15171 CrossRef CAS PubMed.
- R. Li, S. Alomari, R. Stanton, M. C. Wasson, T. Islamoglu, O. K. Farha, T. M. Holsen, S. M. Thagard, D. J. Trivedi and M. Wriedt, Chem. Mater., 2021, 33, 3276–3285 CrossRef CAS.
- K. Li, J. Hu, Q. Gu, J. He, Y.-K. Peng and Z. Xu, ACS Appl. Mater. Interfaces, 2023, 15, 35107–35116 CrossRef CAS PubMed.
- I. M. Manning, N. Guan Pin Chew, H. P. Macdonald, K. E. Miller, M. J. Strynar, O. Coronell and F. A. Leibfarth, Angew. Chem., 2022, 134, e202208150 CrossRef.
- W. Wang, Y. Jia, S. Zhou and S. Deng, J. Hazard. Mater., 2023, 460, 132522 CrossRef CAS PubMed.
- A. Zarei, A. Khosropour, L. Khazdooz, S. Amirjalayer, A. Khojastegi, A. Zadehnazari, Y. Zhao and A. Abbaspourrad, ACS Appl. Mater. Interfaces, 2024, 16, 9483–9494 CrossRef CAS PubMed.
- N. Zorigt, A. Zarei, F. Auras, L. Khazdooz, A. Khosropour and A. Abbaspourrad, Small, 2024, 2406805 Search PubMed.
- P. Wang, Q. Xu, Z. Li, W. Jiang, Q. Jiang and D. Jiang, Adv. Mater., 2018, 30, 1801991 CrossRef PubMed.
- X.-R. Chen, C.-R. Zhang, X. Liu, R.-P. Liang and J.-D. Qiu, Chem. Commun., 2023, 59, 9521–9524 RSC.
- Z. Shi, K. G. Neoh and E. T. Kang, Biomaterials, 2005, 26, 501–508 CrossRef CAS PubMed.
- M. Ding, L. Chen, Y. Xu, B. Chen, J. Ding, R. Wu, C. Huang, Y. He, Y. Jin and C. Xia, Chem. Eng. J., 2020, 380, 122581 CrossRef CAS.
- S. Fu, E. Jin, H. Hanayama, W. Zheng, H. Zhang, L. Di Virgilio, M. A. Addicoat, M. Mezger, A. Narita and M. Bonn, J. Am. Chem. Soc., 2022, 144, 7489–7496 CrossRef CAS PubMed.
- L. Pascual, R. J. Martín-Palma, A. R. Landa-Cánovas, P. Herrero and J. M. Martínez-Duart, Appl. Phys. Lett., 2005, 87, 251921 CrossRef.
- R. M. Silverstein, F. X. Webster, D. Kiemle and D. L. Bryce, Spectrometric Identification of Organic Compounds, Wiley, 8th edn, 2014 Search PubMed.
- J. Drelich, E. Chibowski, D. D. Meng and K. Terpilowski, Soft Matter, 2011, 7, 9804–9828 RSC.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- Z. Du, S. Deng, Y. Chen, B. Wang, J. Huang, Y. Wang and G. Yu, J. Hazard. Mater., 2015, 286, 136–143 CrossRef CAS PubMed.
- Annual Drinking Water Quality Report for the City of Ithaca, https://www.cityofithaca.org/DocumentCenter/View/15429, last accessed February 14, 2025 Search PubMed.
- A. Maimaiti, S. Deng, P. Meng, W. Wang, B. Wang, J. Huang, Y. Wang and G. Yu, Chem. Eng. J., 2018, 348, 494–502 CrossRef CAS.
- S. Y. Wee and A. Z. Aris, npj Clean Water, 2023, 6, 57 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Physical methods used for characterization, detailed synthetic information, experimental details for adsorption and desorption, supporting data tables for structural data, and supporting figures and tables. See DOI: https://doi.org/10.1039/d4ta06827k |
| ‡ These authors contributed equally to this work. |
| § Present address: Interdisciplinary Center for Scientific Computing, University of Heidelberg, Im Neuenheimer Feld 205A, 69120 Heidelberg, Germany. |
| This journal is © The Royal Society of Chemistry 2025 |