
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing the electrochemical catalytic performance of novel bifunctional oxygen vacancy-enriched silver niobate (AgNbO3) through electrochemical activation†
Deepak Rajaram
Patil
a,
Harish S.
Chavan
a,
Ah-yeong
Lee
a,
Geon
Lee
b,
Jungho
Ryu
 b,
Younggon
Son
a and
Kiyoung
Lee
*a
b,
Younggon
Son
a and
Kiyoung
Lee
*a
aDepartment of Chemistry and Chemical Engineering, Inha University, 100 Inha-ro, Michuhol-gu, 22212 Incheon, Republic of Korea. E-mail: kiyoung@inha.ac.kr
bSchool of Materials Science and Engineering, Yeungnam University, Gyeongsan, Gyeongbuk 38541, Republic of Korea
First published on 15th January 2025
Abstract
Introducing oxygen vacancies has emerged as a powerful strategy to enhance the electrocatalytic activity of materials for the oxygen evolution reaction (OER). This approach enhances active site exposure, improves conductivity, and facilitates mass transport, thereby significantly boosting performance. This study explores the synthesis of silver niobate (AgNbO3, ANO) enriched with oxygen vacancies; a modification known to enhance its catalytic properties. Here, ANO was synthesized with abundant oxygen vacancies using a solid-state method followed by aerosol deposition (AD) onto Ni foam substrates. The AD process rapidly produces polymeric binder-free dense ceramic films with strong interfacial adhesion, crucial for efficient electron transfer and enhanced electrocatalyst performance. X-ray photoelectron spectroscopy (XPS) and Raman spectroscopy studies confirmed the presence of oxygen vacancies, pivotal for augmenting ANO's bifunctional activity. This included achieving low overpotentials of 276 mV for the OER and 179 mV for the hydrogen evolution reaction (HER). Impressively, the ANO‖ANO water electrolyser (full cell) demonstrated a low working voltage of 1.69 V at 10 mA cm−2, showcasing its efficacy for water splitting. The long-term durability of ANO‖ANO full cell testing confirmed a minimal voltage increase (0.01 V) after 10 hours, highlighting ANO's robust catalytic stability. Overall, this study highlights the efficacy of oxygen vacancy-rich ANO for enhancing electrocatalytic performance in water splitting, positioning it as a promising candidate for sustainable energy conversion technologies.
1. Introduction
The advancement in green energy technologies represents a promising strategy for mitigating the energy crisis and controlling environmental pollution. Recently, high-energy-density hydrogen (H2) has been produced from water without generating toxic byproducts. Electrochemical water splitting technology is one of the easiest methods for producing clean hydrogen energy. The dissociation of water into molecular hydrogen (H2) and oxygen (O2) is achieved through the half-cell reactions of the cathodic hydrogen evolution reaction (HER) and the anodic oxygen evolution reaction (OER), respectively.1–5 The OER is regarded as the most crucial step for achieving high efficiency in water splitting, as it involves a sluggish four-electron transfer process. Therefore, to surpass the theoretical electrolysis potential of 1.23 V, a suitable electrocatalyst is essential.6,7 Iridium/ruthenium (Ir/Ru) oxides8,9 and platinum on carbon (Pt/C)10 exhibit exceptional performance for the OER and HER, respectively. However, their high cost hinders their scalability for widespread application. Therefore, significant efforts have been taken to develop nonprecious metal-based materials such as metal oxides/hydroxides, metal-nitrates, metal-sulfates, spinels, and perovskites.Perovskite oxides (ABO3) are emerging as highly cost-effective and versatile materials compared to traditional noble metals due to their compositional and structural adaptability, which allows for various substitutions at the A and B sites in the perovskite structure.11–15 This adaptability results in diverse redox active sites and oxygen vacancies, directly impacting their physicochemical and catalytic behaviour. This unique configuration enables perovskite oxides to exhibit adjustable phase and electronic structures, making them exceptional catalysts for the HER and OER in alkaline solutions. Extensive research has focused on engineering perovskite oxide electrocatalysts to achieve abundant exposed active sites, high intrinsic activity, chemical stability, and superior conductivity and electron transfer efficiency. These efforts involve meticulous manipulation of crystallinity, coordination numbers, oxidation states, defect structures, and morphologies, surface reconstructions, and the development of heterostructures within perovskite oxides.11,16,17 However, most perovskite oxides suffer from poor conductivity and electron transfer efficiency, greatly hindering the movement of electrons from the electrocatalyst to the support electrode, which in turn reduces their overall efficiency and increases the overpotential. Additionally, most of these oxides are monofunctional and suited only to the OER. Employing a single bifunctional catalyst for the OER and HER could streamline the water-splitting process and reduce costs. Yet, this approach is challenging, as the material must effectively catalyze both the OER and HER while maintaining a low overall overpotential and achieving the highest possible current density.
Introducing oxygen vacancies has emerged as a highly effective strategy to enhance OER electrocatalytic activities. This approach is widely recognized for its ability to increase the exposure of active sites, improve conductivity, and facilitate mass transfer, thereby significantly boosting OER performance.17–19 For instance, Bai et al.18 reported amorphous ternary oxide composites, FeNiCoOx/CoOx, enriched with oxygen vacancies. The synergistic effect of multi-metal atom interactions, combined with the high exposure of active sites due to the oxygen vacancies and amorphous structure, endows the FeNiCoOx/CoOx electrocatalyst with remarkable catalytic performance with the lowest overpotential of 221 mV at a current density of 100 mA cm−2. Similarly, Li et al.20 synthesized a series of Bi0.15Sr0.85Co1−xFexO3−δ perovskites, discovering that introducing oxygen vacancies promoted the formation of reconstructed active layers. This advancement resulted in a remarkable overpotential of 290 mV at a current density of 10 mA cm−2. Kang et al.21 developed Co-based catalysts rich in oxygen vacancies, demonstrating enhanced OER performance due to accelerated kinetics and improved charge transfer.
Recently, silver niobate (AgNbO3) ceramic, a distinguished member of the perovskite oxide family, has attracted considerable attention for energy storage applications owing to its high recoverable energy density and exceptional temperature stability.22–24 However, when AgNbO3 (ANO) ceramic is prepared under conventional solid-state reaction conditions in an air atmosphere, the volatilization of Ag during the high temperature sintering process leads to the formation of oxygen vacancies. However, these vacancies increase the material's conductivity, making it unsuitable for energy storage applications.22,25 Conversely, these same oxygen vacancies in ANO can significantly alter the coordination environment around metal active centres, thereby exposing additional active sites and enhancing the intrinsic catalytic activity for the OER and HER, thereby boosting conductivity and mass transfer for catalytic applications.
In this study, we synthesized ANO with a high concentration of oxygen vacancies using a conventional solid-state method. Subsequently, the aerosol deposition (AD) technique was employed to fabricate electrocatalyst films on Ni foam from ANO ceramic powder. Remarkably, the AD process requires only a few minutes to produce highly dense ceramic films with thicknesses ranging from 1 to 10 μm and robust interfacial adhesion to the Ni foam.25,26 This strong interfacial bonding is crucial for ensuring efficient electron transfer from the film to the Ni foam substrate, significantly enhancing the overall performance of the electrocatalyst. Unlike conventional electrocatalyst fabrication techniques that rely on polymeric binders, the AD method produces binder-free electrocatalyst films, enhancing performance and purity. The presence of oxygen vacancies was confirmed by X-ray photoelectron spectroscopy (XPS). The ANO electrocatalyst exhibited remarkable bifunctional activity due to the oxygen vacancy-derived exposed active sites, with low overpotentials of 276 mV for the OER and 179 mV for the HER. More importantly, the ANO‖ANO full cell demonstrated an impressively low voltage of 1.69 V at a current density of 10 mA cm−2, making it a highly suitable candidate for electrocatalytic water splitting.
2. Experimental section
To prepare ANO ceramic powder, stoichiometric amounts of Ag2O (99%) and Nb2O5 (99.9%) from Sigma-Aldrich, USA, were accurately measured and mixed. The mixture was ball-milled for 24 hours in high-density polyethene (HDPE) bottles with 5 mm zirconia balls in ethanol solution. Post-milling, the powder underwent calcination at 950 °C for 9 hours in an air atmosphere. Subsequently, the calcined powders were further milled for 24 hours and then dried at 80 °C using a rotary evaporator. The resulting dried powder was finely ground and passed through a 125 μm sieve, yielding high-quality ANO ceramic powder. Prior to AD, the NF substrate underwent a thorough cleaning process involving sequential ultrasonic baths for 10 minutes each in diluted HCl, acetone and ethanol. The AD process for fabricating ANO thick films involved mixing calcined and ball-milled ANO granules with a carrier gas (medical-grade dried air, 5 L min−1 flow rate) in a powder chamber to form an aerosol. This aerosol was then sprayed through a de Laval-type nozzle (20 mm slit length, 0.5 mm throat width) onto a moving Ni foam substrate in an evacuated chamber (2.3 Torr). The photograph of the as-deposited ANO film on the Ni foam substrate fabricated by the AD technique is shown in Fig. S1.† The comprehensive synthesis methodology of ceramic films using the AD technique has been elucidated in a separate publication.26The morphologies of the ANO electrocatalysts were examined using field-emission scanning electron microscopy (FESEM; S-4300, Hitachi). Their elemental composition and distribution were determined via energy-dispersive X-ray spectrometry (EDX; EX-250, Horiba). The analysis was conducted at the Inha University Core Facility Center for Sustainable Energy. High-resolution X-ray diffraction (XRD; X'Pert PRO MRD, Phillips) was employed to identify the crystalline phases, and X-ray photoelectron spectroscopy (XPS; K-Alpha, Thermos Scientific) was utilized to analyze the surface elemental composition.
The AD electrodes were cut into 1 cm2 sections to assess the electrocatalytic activity. These electrodes were evaluated using a three-electrode configuration, which included a working electrode, a platinum wire counter electrode, and a saturated calomel reference electrode (SCE). All electrochemical measurements were carried out in a 1 M KOH electrolyte, and potentials were converted to the reversible hydrogen electrode (RHE) scale using the standard formula. A potentiostat (VSP, Bio-Logics) electrochemical workstation was employed for these measurements. Electrode activation was achieved via cyclic voltammetry (CV) until a stable overpotential was reached, followed by linear sweep voltammetry (LSV) within a potential window of 0 to 0.6 V vs. SCE at a scan rate of 5 mV s−1. Electrode stability was assessed using chronopotentiometry (CP) at a constant current density of 10 mA cm−2. The Tafel equation (η = b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log|j| + a) was utilized to derive Tafel curves from the LSV data, relating the overpotential (η) to the current density (j) and the Tafel slope (b). Electrochemical impedance spectroscopy (EIS) was used to measure the interface charge transfer resistance across a frequency range of 1 Hz to 10 kHz. The full cell was assembled using ANO as both the anode and cathode, with 1 M KOH as the electrolyte.
log|j| + a) was utilized to derive Tafel curves from the LSV data, relating the overpotential (η) to the current density (j) and the Tafel slope (b). Electrochemical impedance spectroscopy (EIS) was used to measure the interface charge transfer resistance across a frequency range of 1 Hz to 10 kHz. The full cell was assembled using ANO as both the anode and cathode, with 1 M KOH as the electrolyte.
3. Results and discussion
Fig. 1a illustrates the schematic of the dynamic crystal structure of ANO, showcasing its transformation with and without oxygen vacancies. At room temperature, ANO typically crystallizes in an orthorhombic perovskite structure. This structure is characterized by a framework where silver ions (Ag+) occupy the A-site and niobium ions (Nb5+) reside at the B-site in the ABO3 perovskite. In ANO, the silver ions at the A-site are surrounded by oxygen atoms in a 12-coordinated arrangement, which forms a dodecahedral configuration. This means each silver ion is bonded to 12 oxygen atoms in the crystal lattice. On the other hand, niobium ions at the B-site are coordinated with oxygen atoms in a typical NbO6 octahedral geometry. Each niobium ion is bonded to six oxygen atoms arranged around it in an octahedral fashion. The NbO6 octahedra share edges and corners with neighbouring octahedra and dodecahedra, creating a robust and interconnected framework within the crystal lattice. This arrangement not only provides structural stability to ANO but also influences its physical and chemical properties. However, during annealing of ANO at 950 °C for 9 hours in an air atmosphere, Ag is easily volatilized due to the low melting point resulting in the formation of cation vacancies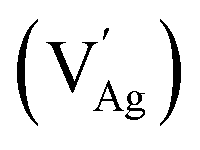 . This phenomenon is accompanied by the creation of oxygen vacancies
. This phenomenon is accompanied by the creation of oxygen vacancies  to preserve electrical balance within the material. This can be described by using the following equation:22
to preserve electrical balance within the material. This can be described by using the following equation:22 | (1) |
 | ||
| Fig. 1 (a) Schematic representation of the crystal structure of AgNbO3 (ANO), highlighting the creation of oxygen vacancies. (b) XRD patterns of ANO powder, the ANO film deposited on Ni foam, and Ni foam. FESEM images of (c) ANO powder and (d) the ANO film on Ni foam. | ||
Fig. 1b presents the XRD patterns of ANO sintered powder, the AD film, and bare Ni foam. The AD film reveals distinct XRD peaks corresponding to both ANO (PDF # 70-4738) and the Ni foam substrate (PDF # 65-2865). These peaks are well-defined and match the orthorhombic perovskite structure of ANO.22–24 However, compared to the sintered powder, the XRD peaks of the AD film are relatively weak and broad. This broadening and intensity reduction are attributed to the high kinetic energy particle collisions of ANO particles to the Ni foam during the deposition process, which leads to a decrease in grain size and less crystallized film structure.25Fig. 1c and d display the FESEM images of the ANO powder and the AD film, respectively. The FESEM images of the ANO powder reveal well-defined microparticles with irregular shapes. In contrast, the AD film displays a uniform and dense packing of nanocrystallites. During deposition, the crystalline particles are fractured into nanoscale particles, reducing crystallinity and particle size. This nanoscale architecture of the ANO film is advantageous for enhancing electrocatalytic performance. The EDX spectrum of the ANO electrode is presented in Fig. S2(a),† with the detailed elemental composition tabulated in Fig. S2(b).† Analysis reveals that the atomic concentration of Ag is notably lower than that of Nb, suggesting an Ag deficiency in the ANO material. This observation further supports the hypothesis of oxygen vacancy formation within the sample.
XPS was utilized to examine the surface composition and presence of oxygen vacancies in the AD-deposited ANO electrocatalyst. Fig. 2 presents the XPS spectra of the AD-deposited ANO electrocatalyst. Fig. 2a highlights the high-resolution Ag 3d spectrum, which features two distinct peaks at 367 eV and 373 eV corresponding to Ag 3d5/2 and Ag 3d3/2 respectively. These peaks are further deconvoluted into Ag0 and Ag+ states. The Ag0 peaks are centred at 367.78 eV and 373.88 eV, while the Ag+ peaks are centred at 367.18 eV and 373.18 eV.27,28 The greater intensity of the Ag0 peak compared to the Ag+ peak indicates a substantial reduction of silver from its oxidized state (Ag+) to its metallic state (Ag0), likely due to the reduction of Ag during the annealing of ANO in air. The deconvoluted Nb 3d spectrum is shown in Fig. 2b, with Nb 3d5/2 and Nb 3d3/2 peaks located at 206.9 eV and 209.7 eV respectively, indicating that Nb is in the 5+ valence state in ANO.29 The high-resolution O 1s spectrum, depicted in Fig. 2c, reveals multiple oxygen species. The peak at 529.38 eV signifies the presence of lattice oxygen atoms binding with Ag–O, while the peak at 531.25 eV indicates the presence of oxygen vacancies within the ANO electrocatalyst.25,29 Additionally, the peak at 531.88 eV is attributed to absorbed water (H2O). The intensity of the oxygen vacancy peak is significantly higher than that of lattice oxygen, suggesting a substantial amount of oxygen vacancies in the electrocatalysts. These vacancies are likely generated during the annealing of ANO to maintain charge neutrality following the reduction of Ag from a higher to a lower oxidation state. The XPS results confirm that the oxygen vacancies in ANO only alter the valence of Ag, indicating that the Nb does not contribute to the OER activity. The atomic concentration of Ag and Nb was determined from the survey spectrum (Fig. S3†). The atomic concentration of Ag is lower than that of Nb, suggesting an Ag deficiency in the ANO material, consistent with that in EDX results. The presence of oxygen vacancies in ANO was further examined using electron paramagnetic resonance (EPR), a powerful technique capable of detecting surface-active sites through the characteristic signal of unpaired electrons. Specifically, the signal observed at g = 2.003 corresponds to molecular oxygen from air occupying oxygen vacancies on the surface. In the present case, the distinct signal observed at g = 2.006 confirms the existence of oxygen vacancies in ANO, which likely originate from the formation of surface defects during the synthesis process (Fig. 2d). The concentration of oxygen vacancies in ANO was quantified to be 1.84 × 1014 g−1.
 | ||
| Fig. 2 High-resolution spectra of (a) Ag 3d, (b) Nb 3d, and (c) O 1s. (d) EPR data of AgNbO3. | ||
The electrochemical properties of ANO electrocatalysts deposited via aerosol deposition (AD) were evaluated in a 1 M KOH electrolyte using a three-electrode system. Prior to assessing the oxygen evolution reaction (OER) activity, the electrocatalysts underwent activation through cyclic voltammetry, ranging from 0 to 1.65 V vs. RHE, at a scan rate of 100 mV s−1. This activation step is crucial for metal oxide electrocatalysts as it facilitates the reconstruction of a catalytically favorable surface by removing adsorbed contaminants or passivating layers that may impede catalytic efficiency.30,31 Additionally, the cyclic potential induces oxidation and reduction processes on the surface, thereby exposing more active sites or arranging these sites into a more ordered structure.30 Fig. S4(a)† illustrates the evolution of cyclic voltammetry (CV) curves over multiple cycles. Notably, the current density at the redox peak increased progressively with continued cycling, reaching stabilization after approximately 2000 cycles. Concurrently, the cut-off current density at 1.65 V vs. RHE for the OER also showed a gradual increase. This rise in oxidation peak intensity and current density is indicative of the continuous generation of electrochemically active sites on ANO. The redox peaks observed in the CV curves are attributed to the Ni present in the Ni foam substrate. As previously discussed, ANO features a high amount of oxygen vacancies where oxygen atoms are missing from the lattice, creating vital active sites for reactions. During the initial CV cycles, hydroxyl groups (OH−) from the electrolyte adsorb onto the outer surface of ANO at these vacancies. The OH− ions then start to fill the oxygen vacancies. The OH− groups lose additional protons and electrons, transforming into oxygen intermediates (O2−), which are more stable and closer to integrating into the lattice structure. These O2− intermediates are further incorporated into the lattice, effectively filling the oxygen vacancies in the outer surface layer. As the CV cycling progresses, water molecules and hydroxyl groups continue to diffuse from the outer surface to the sub-surface layer, encountering and filling additional vacancies. With ongoing cycling, oxygen ions from the outer and sub-surface layers migrate deeper into the bulk layer, addressing vacancies within the lattice. Over extended CV cycling, a dynamic equilibrium between vacancy formation and healing is established, which maintains catalytic activity and ensures long-term stability.30,31 To elucidate the mechanism of oxygen vacancy filling by oxygen ions, we measured the Raman spectra in the range of 50–800 cm−1 after every 500 cycles, as shown in Fig. S4(b).† The Raman peaks at approximately 579 cm−1 and 529 cm−1 correspond to the v1 and v2 modes, respectively, primarily associated with the NbO6 octahedron stretching vibration modes.32 Multiple peaks within the range of 100–250 cm−1 are indicative of the transitional modes associated with Ag cations and the v5 mode at ∼258 cm−1 is related to NbO6 octahedral tilting and Nb5+ cation displacement.32 It is well-documented that oxygen vacancies can distort the Nb–O octahedron, resulting in broadening of Raman peaks.33 As illustrated in Fig. S4(b),† the Raman spectrum of the ANO material prior to CV cycling exhibits broadened and diffuse characteristic peaks. This broadening signifies an increased degree of local structural disorder, attributed to the presence of oxygen vacancies. The emergence of both cation and oxygen vacancies disrupts long-range order, resulting in a more disordered local structure.33 With increasing CV cycling, the initially broad peaks in the Raman spectrum evolve into distinct characteristic peaks of ANO, becoming progressively sharper and more intense. This transformation indicates that the oxygen vacancies are being progressively filled by oxide ions throughout the cycling process.
To validate the necessity of the CV cycling process for oxygen vacancy-rich ANO, we synthesized the state-of-the-art electrocatalyst RuO2 on Ni foam by using the drop-casting method. We conducted CV cycling activation of RuO2 and Ni foam at a scan rate of 100 mV s−1 as shown in Fig. S4(c) and (d),† respectively. Notably, the RuO2 electrocatalyst required only 50 cycles to achieve activation and stabilization. In contrast, for the Ni foam, the current density at the redox peak progressively increased up to 1000 cycles and declined with further cycling. This increase in current density is attributed to the conversion of Ni into higher oxidation states. However, it is noteworthy that the cut-off current density at 1.65 V vs. RHE for Ni foam remains significantly smaller than that observed for ANO, highlighting a clear difference in performance at this voltage. This stark contrast suggests that the extended CV cycling required for ANO is likely due to the progressive healing of oxygen vacancies within the material. The rapid stabilization of RuO2 underscores the unique challenge and significance of vacancy dynamics in ANO, affirming the critical role of sustained CV cycling in its optimal activation and performance.
The OER activity of the ANO electrocatalyst was rigorously evaluated using linear sweep voltammetry (LSV) in 1 M KOH, with benchmark comparisons to RuO2. As depicted in Fig. 3a, the iR-corrected LSV curves for ANO across different CV cycles reveal a progressive increase in overpotential (at 10 mA cm−2) and current density, stabilizing at around 2000 cycles. Upon reaching stabilization, ANO demonstrated an impressively low overpotential of 276 mV at 10 mA cm−2. This performance notably surpasses that of the benchmark RuO2 (360 mV) electrocatalyst. Further analysis of the reaction kinetics, through the determination of Tafel slopes, underscored the superior efficiency of ANO. As shown in Fig. 3b, ANO exhibited a remarkably low Tafel slope of 40.4 mV dec−1, significantly lower than that of RuO2 (96.42 mV dec−1) and Ni foam (118.97 mV dec−1) electrocatalysts. This low overpotential and Tafel slope highlight the rapid kinetic advantages of oxygen vacancy-rich ANO, affirming its exceptional potential for efficient water electrolysis. Moreover, electrochemical impedance spectroscopy (EIS) measurements were performed to elucidate the charge transfer kinetics during the OER test (Fig. 3c and Table S1†). The charge transfer resistance of the ANO electrocatalyst consistently decreased with each cycle, indicating enhanced efficiency in filling oxygen vacancies with hydroxyl ions. This phenomenon effectively enhances electrical conductivity and facilitates accelerated charge transfer between the electrode and electrolyte during the OER. Furthermore, the electrochemical double-layer capacitance (Cdl) values, which were used to assess the electrochemically active surface area (ECSA) of catalysts, were meticulously derived from cyclic voltammetry (CV) data measured at different scan rates of 20, 40, 60, 80, 100 and 120 mV s−1 (Fig. S5(a)†). ANO exhibited significantly higher Cdl and ECSA values of 16.8 mF cm−2 and 420 cm2, respectively, (Fig. S5(b)†) underscoring its abundant active sites favourable to superior OER activity. In addition, the ANO electrocatalyst showed excellent stability in 1 M KOH during chronoamperometry (CP) testing at a high current density of 100 mA cm−2 (Fig. S6†). Impressively, it maintained durability over 75 hours of continuous operation. Notably, the OER performance of the ANO electrocatalyst not only demonstrates exceptional efficiency but also exceeds that of previously reported perovskite oxide-based electrocatalysts, as depicted in Fig. 3d.34–47
 | ||
| Fig. 3 OER performance of the ANO film in 1 M KOH. (a) LSV curves of ANO after various CV cycles, Ni foam and RuO2. (b) Tafel plots illustrating the kinetics of the OER, (c) EIS spectra at 0.5 V, highlighting the charge transfer resistance, (d) comparison of the OER activity of the ANO film with various perovskite oxide electrocatalysts. | ||
XPS spectra were recorded to thoroughly investigate the electronic structure of Ag, Nb, and O in ANO before and after OER activity testing. Fig. 4a presents the high-resolution comparative XPS spectra of Ag 3d, showcasing the Ag0 and Ag+ states before and after the OER. Post-OER activity testing, the intensity of the Ag0 peaks significantly diminishes, indicating a reduction in the relative content of Ag0 and an increase in the oxidation state to Ag+. Additionally, a slight positive shift in the Ag peaks is observed following the OER process. Fig. 4b illustrates the comparative Nb 3d spectra before and after OER activity testing. No peak intensity or position changes are detected, suggesting that Nb does not directly contribute to the OER activity. In the O 1s spectra (Fig. 4c), an increase in the M–O peak intensity and a decrease in the Ov peak intensity are noted. A distinct peak at 530.8 eV emerges after the OER process, indicating the formation of hydroxyl groups. Moreover, a minor positive shift is observed in the O peaks post-OER. The XPS results reveal that oxygen vacancies act as active sites, stabilizing the adsorbed oxygen intermediates and thereby enhancing the OER performance.
 | ||
| Fig. 4 High-resolution XPS spectra of (a) Ag 3d, (b) Nb 3d, and (c) O 1s before and after the OER. (d) Illustration of the mechanism by which oxygen vacancies enhance OER activity in ANO electrocatalysts. | ||
We develop the OER mechanisms of oxygen-vacant sites to gain insights into the enhancement of OER activity. The two primary OER mechanisms are the Absorbate Evolution Mechanism (AEM) and the Lattice Oxygen Oxidation Mechanism (LOM). In the AEM, a metal cation functions as the redox centre (cation redox mechanism), where various reaction intermediates adsorb onto metal active centres. Conversely, in the LOM, oxygen serves as the redox centre (anion redox mechanism), where oxygen ligands are activated and released from the lattice matrix, creating oxygen vacancies. However, in this study, we observe an OH− coupled oxygen vacancy-originated OER mechanism that deviates from the conventional AEM or LOM mechanisms. ANO lacks eg electrons and does not adopt a new valence state, ruling out the possibility of a conventional cation redox mechanism. Similarly, in ANO, the OH− adsorbates are directly incorporated into the site of an oxygen vacancy without the removal of the original oxygen atoms, excluding the possibility of an anion-redox or LOM reaction. In this unique case, direct O–O bond formation occurs due to the OH− adsorbate on the oxygen vacancy sites.19
Fig. 4d illustrates the possible OER mechanism in oxygen vacancy-rich ANO. Initially, Ag adopts a zero-oxidation state (Ag0), fully compensating for the charge imbalance caused by the oxygen vacancies, thereby stabilizing the active sites. This stabilization ensures that the material remains electrically neutral and chemically stable, while maintaining a high concentration of oxygen vacancies essential for the catalytic process. These oxygen vacancies act as catalytically active sites by creating regions of enhanced electron density, making them attractive for hydroxyl ions (OH−) in the reaction medium. Initially, oxygen vacancies in the material attract OH−. The adsorbed OH− ions undergo deprotonation, losing H2O and an electron (e−), converting into oxygen intermediates, O2− ions. This step effectively fills the oxygen vacancies with oxygen atoms. Subsequently, another OH− ion adsorbs onto the newly formed O2− intermediate and results in the formation of OOH− (peroxide) intermediates. The OOH− intermediate undergoes further reactions, losing another proton and electron, resulting in the formation and release of an O2 molecule.19 Ag facilitates this process by enhancing electron transfer and maintaining structural integrity, ensuring that the active sites remain stable and reactive. The synergistic interaction between Ag and oxygen vacancies significantly accelerates the adsorption, transformation, and release of oxygen intermediates, ultimately boosting the OER efficiency. This cyclic process highlights the importance of oxygen vacancies in promoting and sustaining the oxygen evolution reaction.
Perovskite oxides have been extensively researched for their efficacy in catalyzing the OER. However, their potential as catalysts for the HER has received limited attention, constraining their utility as bifunctional catalysts for overall water splitting. To explore the HER capabilities of ANO, we evaluated its performance in 1 M KOH electrolyte using a three-electrode system. For comparison, we measured the HER activity of commercially available 20 wt% Pt/C under identical experimental conditions. Fig. 5a illustrates the LSV curves for ANO and Pt/C electrocatalysts at a scan rate of 5 mV s−1. ANO exhibited an overpotential of −150 mV to achieve a current density of 10 mA cm−2, whereas the benchmark Pt/C catalyst required an overpotential of −50 mV, consistent with reported values. Tafel slopes were obtained to further evaluate the reaction kinetics as illustrated in Fig. 5b. The ANO material exhibited a Tafel slope of 75.83 mV dec−1, signifying its adherence to the Volmer–Heyrovsky mechanism, where adsorption governs the rate-determining step. This Tafel slope for ANO exceeds that of Ni foam but falls short of the values observed for Pt/C electrocatalysts. However, the HER performance of the ANO electrocatalyst not only demonstrates exceptional efficiency but also exceeds that of previously reported perovskite oxide-based electrocatalysts, as depicted in Fig. 5c.37–40,43–45,48–51
 | ||
| Fig. 5 HER performance of the ANO film in 1 M KOH. (a) LSV curves of ANO after various CV cycles, Ni foam and Pt/C. (b) Tafel plots illustrating the kinetics of the HER. (c) Comparison of the HER activity of the ANO film with various perovskite oxide electrocatalysts. (d) LSV curves of the ANO‖ANO full cell, measured before and after 10 hours of stability testing. | ||
The electrocatalytic performance of ANO demonstrates superior capabilities in both the HER and OER, surpassing the bifunctional performance of numerous previously reported perovskite oxide catalysts. This underscores its potential for comprehensive electrocatalytic water-splitting applications. To assess its overall water-splitting activity, we constructed a water electrolyser employing ANO electrodes as both the anode and cathode in a 1 M KOH electrolyte (Fig. S7†). The H2 and O2 bubbles generated throughout the electrolysis process are distinctly visible in Fig. S7.†Fig. 5d shows the LSV curve of the ANO‖ANO full cell before and after 10 h chronopotentiometry. Operating at a current density of 10 mA cm−2, the ANO‖ANO full cell achieves a nominal working voltage of 1.69 V, highlighting its exceptional efficiency in complete water splitting. Furthermore, chrono-potentiometry results reveal that the ANO‖ANO full cell maintains robust catalytic durability, as depicted in the inset of Fig. 5d. After a 10-hour durability test, the corresponding voltage for overall water splitting increases by a mere 0.01 V, further affirming its outstanding long-term catalytic stability.
Vacancy-rich ANO, synthesized via solid-state methods and aerosol deposition (AD), exhibits exceptional bifunctional electrocatalytic performance for the OER and HER due to its high oxygen vacancy concentration. ANO achieves overpotentials of 276 mV for the OER and 179 mV for the HER, outperforming many perovskite oxides and matching advanced electrocatalysts. The robust ANO‖ANO full cell achieves a low working voltage of 1.69 V at 10 mA cm−2 with minimal degradation over 10 hours, underscoring its long-term stability. When compared to hydroxide-based electrocatalysts such as NiFe–LDH and CoFe hydroxides,52 which exhibit lower overpotentials in the range of 200 mV at10 mA cm−2, ANO demonstrates competitive efficiency but slightly higher overpotentials. However, these hydroxides often require additional modifications for stability and bifunctionality. When evaluated against oxide catalysts such as Ba0.5Sr0.5Co0.8Fe0.2O3−δ (ref. 36) and LaNiO3,53 which also leverage oxygen vacancies to improve OER performance and display overpotentials of around 300 mV, ANO holds a distinct advantage in both bifunctionality and long-term stability. Similarly, while transition metal sulfides and nitrides, including Ni0.7Fe0.3S2 and FeNi3N,54 show lower overpotentials, they often struggle with structural stability over prolonged use. Advanced catalysts like MOF-derived systems surpass ANO in the OER, while MXenes outperform in the HER.55 Ir/Ru-based catalysts,56 with near-zero HER overpotentials and outstanding bifunctionality, remain the leading performers in overall electrocatalytic performance. Unfortunately, their high cost and limited scalability present significant challenges for widespread adoption of these catalysts. Emerging catalysts, such as Cr-doped FeNi–P nanoparticles and Rh/NiFe–LDH hybrids,57,58 also offer promising performance enhancements. Cr-doped FeNi–P nanoparticles demonstrate a cell voltage of 1.50 V at 10 mA cm−2, with improved durability and catalytic activity resulting from electronic structure modifications. Rh-doping in NiFe–LDH hybrids has been shown to significantly enhance OER kinetics, with stability exceeding 100 hours, reinforcing their potential for long-term applications in renewable energy conversion technologies. Although these electrocatalysts demonstrate excellent OER performance, they suffer from limitations related to the cost of Rh and the need for further optimization in scaling for large-scale applications. In addition, the full cell value obtained for ANO‖ANO is smaller than or comparable to that of most of the reported noble metal free catalysts such as CoMO LDH,59 VOOH,60 NiSe,61 NiCo2O4,62 and NiFe/NiCo2O4.63 Furthermore, ANO's low-cost synthesis, unique binder-free configuration, and competitive bifunctionality position it as an attractive alternative. To further enhance ANO's electrocatalytic performance, integrating oxygen vacancy engineering with strategies like heteroatom doping or nanoscale structural modifications could reduce overpotentials and improve durability. These enhancements would enable ANO to match or exceed the performance of the most advanced hydroxide and oxide catalysts under industrial conditions, solidifying its role in sustainable hydrogen production.
4. Conclusions
This study highlights the critical role of oxygen vacancies in enhancing the oxygen evolution reaction (OER) activity and overall water-splitting efficiency of AgNbO3. Utilizing a traditional solid-state synthesis method coupled with aerosol deposition, the research produced oxygen vacancy-rich ANO and created dense ceramic films on Ni foam substrates. The AD technique enabled the formation of binder-free films with superior properties, including high density and exceptional interfacial adhesion, crucial for optimizing electron transfer efficiency. Unlike conventional methods that use polymeric binders, AD significantly improved the electrocatalyst's performance and purity. The OER activity of ANO was meticulously investigated through electrochemical activation using XPS and Raman spectroscopy. The presence of oxygen vacancies facilitated the efficient adsorption of OH− ions, thereby promoting the rapid release of O2 bubbles. The ANO electrocatalyst exhibited outstanding bifunctional performance, with low overpotentials of 276 mV for the OER and 179 mV for the hydrogen evolution reaction (HER), demonstrating its effectiveness for comprehensive water-splitting applications. Furthermore, the ANO‖ANO full cell achieved a remarkably low operating voltage of 1.69 V at 10 mA cm−2, underscoring its practical efficacy in water-splitting scenarios. These findings offer valuable insights into how oxygen vacancies can be leveraged to enhance the design of high-efficiency catalysts for accelerating OER kinetics. The oxygen vacancies can be further created by strategically doping cations at both the A and B sites in perovskite. This research opens promising avenues for improving other catalytic reactions and energy storage systems that involve oxygen chemistry.Data availability
All the data will be made available on request.Author contributions
Deepak Patil: conceptualization, writing – original draft, data curation. Harish Chavan: methodology. Ah-yeong Lee: formal analysis, investigation. Geon Lee: methodology, formal analysis. Jungho Ryu: funding acquisition, validation. Younggon Son: formal analysis. Kiyoung Lee: supervision, funding acquisition, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research work at Inha University was supported by the Korea Institute of Energy Technology Evaluation and Planning (KETEP) (No. 20213030040590), and Korea Basic Science Institute (National research Facilities and Equipment Center) grant funded by the Ministry of Education. (2021R1A6C101A404). The work at YU was supported by Yeungnam University Research Grant #224A061007 and National Research Foundation of Korea #RS-2023-NR077116.References
- D. M. F. Santos, C. A. C. Sequeira and J. H. Figueiredo, Quim. Nova, 2013, 36, 1176–1193 CrossRef CAS.
- L. H. Chen, S. Jia, X. Xu, B. Lin, H. Yang, H. Song and L. Wang, Nat. Commun., 2017, 8, 15377 CrossRef PubMed.
- Y. Holade, K. Servat, S. Tingry, T. W. Napporn, H. Remita, D. Cornu and K. B. Kokoh, ChemPhysChem, 2017, 18, 2573–2605 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, 4998 CrossRef PubMed.
- H. Liang, A. N. Gandi, D. H. Anjum, X. Wang, U. Schwingenschlogl and H. N. Alshareef, Nano Lett., 2016, 16, 7718–7725 CrossRef CAS PubMed.
- A. Dutta, A. K. Samantara, S. K. Dutta, B. K. Jena and N. Pradhan, ACS Energy Lett., 2016, 1, 169–174 CrossRef CAS.
- K. Qin, L. Wang, S. Wen, L. Diao, P. Liu, J. Li, L. Ma, C. Shi, C. Zhong, W. Hu, E. Liu and N. Zhao, J. Mater. Chem. A, 2018, 6, 8109–8119 RSC.
- X. K. Kong, K. Xu, C. L. Zhang, J. Dai, S. N. Oliae, L. Y. Li, X. C. Zeng, C. Z. Wu and Z. M. Peng, ACS Catal., 2016, 6, 1487–1492 CrossRef CAS.
- S. Siracusano, N. Van Dijk, E. Payne-Johnson, V. Baglio and A. S. Arico, Appl. Catal., B, 2015, 164, 488–495 CrossRef CAS.
- Y. Li, Y. Sun, Y. Qin, W. Zhang, L. Wang, M. Luo, H. Yang and S. Guo, Adv. Energy Mater., 2020, 10, 1903120 CrossRef CAS.
- H. Sun, J. Dai, W. Zhou and Z. Shao, Energy Fuels, 2020, 34, 10547–10567 CrossRef CAS.
- X. Xu, Y. Zhong and Z. Shao, Trends Chem., 2019, 1(4), 410–424 CrossRef CAS.
- B. Hua, M. Li, W. Pang, W. Tang, S. Zhao, Z. Jin, Y. Zeng, B. Shalchi Amirkhiz and J.-L. Luo, Chem, 2018, 4(12), 2902–2916 CAS.
- C. Sun, J. A. Alonso and J. Bian, Adv. Energy Mater., 2021, 11, 2000459 CrossRef CAS.
- H. Wang, M. Zhou, P. Choudhury and H. Luo, Appl. Mater. Today, 2019, 16, 56–71 CrossRef CAS.
- D. P. Kour, I. Ahmed, Sunny, S. K. Sharma, K. Yadav and Y. K. Mishra, ChemCatChem, 2023, 15, e202300040 CrossRef.
- Y. Wang, R. Yang, Y. Ding, B. Zhang, H. Li, B. Bai, M. Li, Y. Cui, J. Xiao and Z.-S. Wu, Nat. Commun., 2023, 14, 1412 CrossRef CAS PubMed.
- Q. Bai, D. Liu, X. Yan, P. Guo, X. Ding, K. Xiang, X. Tu, Y. Guo and R. Wu, ACS Mater. Lett., 2024, 6, 2948–2956 CrossRef CAS.
- S. Hirai, K. Morita, K. Yasuoka, T. Shibuya, Y. Tojo, Y. Kamihara, A. Miura, H. Suzuki, T. Ohno, T. Matsuda and S. Yagi, J. Mater. Chem. A, 2018, 6, 15102 RSC.
- J. Li, F. Yang, Y. Z. Du, M. Jiang, X. Y. Cai, Q. D. Hu and J. Zhang, Chem. Eng. J., 2023, 451, 138646 CrossRef CAS.
- T. Kang and J. Kim, Appl. Surf. Sci., 2021, 560, 150035 CrossRef CAS.
- J. Zhou, J. Du, L. Chen, Y. Li, L. Xu, Q. Zhao, H. Yang, J. Ding, Z. Sun, X. Hao and X. Wang, J. Eur. Ceram. Soc., 2023, 43, 6059–6068 CrossRef CAS.
- J. Huang, X. Hou, Y. He, S. Gao, Y. Zhou, H. Huang and Q. Zhang, J. Mater. Chem. A, 2022, 10, 16337–16350 RSC.
- D. Feng, H. Du, H. Ran, T. Lu, S. Xia, L. Xu, Z. Wang and C. Ma, J. Solid State Chem., 2022, 310, 123081 CrossRef CAS.
- G. Lee, T. K. Sung and H. Song, J. Korean Ceram. Soc., 2024, 61, 918–927 CrossRef CAS.
- J. Ryu, D.-S. Park, B.-D. Hahn, J.-J. Choi, W.-H. Yoon, K.-Y. Kim and H.-S. Yun, Appl. Catal., B, 2008, 83(1), 1–7 CrossRef CAS.
- T. H. Jeon, D. Monllor-Satoca, G.-h Moon, W. Kim, H.-I. Kim, D. W. Bahnemann, H. Park and W. Cho, Nat. Commun., 2020, 11, 967 CrossRef CAS PubMed.
- Y. Bao and K. Chen, Nano-Micro Lett., 2016, 8(2), 182–192 CrossRef PubMed.
- L. Shu, X. Zhang, W. Li, J. Gao, H. Wang, Y. Huang, Y.-Y. Shan Cheng, Q. Li, L. Liu and J.-F. Li, J. Mater. Chem. A, 2022, 10, 12632–12642 RSC.
- Y. J. Son, S. Kim, V. Leung, K. Kawashima, J. Noh, K. Kim, R. A. Marquez, O. A. Carrasco-Jaim, L. A. Smith, H. Celio, D. J. Milliron, B. A. Korgel and C. B. Mullins, ACS Catal., 2022, 12(16), 10384–10399 CrossRef CAS.
- S. Li, Z. Li, R. Ma, C. Gao, L. Liu, L. Hu, J. Zhu, T. Sun, Y. Tang, D. Liu and J. Wang, Angew. Chem., Int. Ed., 2021, 60, 3773 CrossRef CAS PubMed.
- N. Luo, C. Liao, G. Zhang, X. Chen, Q. Feng, J.-F. Li, S. Zhang and Y. Wei, Nat. Commun., 2020, 11, 4824 CrossRef CAS PubMed.
- W. Chao, J. Du, P. Li, W. Li and T. Yang, Dalton Trans., 2024, 53, 2949 RSC.
- S. Song, J. Zhou, X. Su, Y. Wang, J. Li, L. Zhang, G. Xiao, C. Guan, R. Liu, S. Chen, H.-J. Lin, S. Zhang and J.-Q. Wang, Energy Environ. Sci., 2018, 11(10), 2945–2953 RSC.
- H. Wang, J. Wang, Y. Pi, Q. Shao, Y. Tan and X. Huang, Angew. Chem., Int. Ed., 2019, 58(8), 2316–2320 CrossRef CAS PubMed.
- Q. Luo, D. Lin, W. Zhan, W. Zhang, L. Tang, J. Luo, Z. Gao, P. Jiang, M. Wang, L. Hao and K. Tang, ACS Appl. Energy Mater., 2020, 3(7), 7149–7158 CrossRef CAS.
- C. Y. Wang, L. R. Zeng, W. Guo, C. R. Gong and J. Yang, RSC Adv., 2019, 9, 35646–35654 RSC.
- Y.-N. Zhao, C. Liu, S. Xu, S. kang, M. Wen, C. Wang, N. Mitsuzaki and Z. Chen, Inorg. Chem., 2023, 62, 12590–12599 CrossRef CAS PubMed.
- R. K. Hona, S. B. Kark and F. Ramezanipour, ACS Sustain. Chem. Eng., 2020, 8, 11549–11557 CrossRef CAS.
- J. Wang, Y. Gao, D. J. Chen, J. P. Liu, Z. B. Zhang, Z. P. Shao and F. Ciucci, ACS Catal., 2018, 8, 364–371 CrossRef CAS.
- J. Dai, Y. L. Zhu, Y. C. Yin, H. A. Tahini, D. Q. Guan, F. F. Dong, Q. Lu, S. C. Smith, X. W. Zhang, H. T. Wang, W. Zhou and Z. P. Shao, Small, 2019, 15, 1903120 CrossRef PubMed.
- R. Majee, S. Chakraborty, H. G. Salunke and S. Bhattacharyya, ACS Appl. Energy Mater., 2018, 1, 3342–3350 CrossRef CAS.
- D. W. Ji, C. H. Liu, Y. H. Yao, L. L. Luo, W. C. Wang and Z. D. Chen, Nanoscale, 2021, 13, 9952–9959 RSC.
- Y. L. Zhu, W. Zhou, J. Sunarso, Y. J. Zhong and Z. P. Shao, Adv. Funct. Mater., 2016, 26, 5862–5872 CrossRef CAS.
- J. Dai, Y. L. Zhu, Y. J. Zhong, J. Miao, B. W. Lin, W. Zhou and Z. P. Shao, Adv. Mater. Interfaces, 2019, 6, 1801317 CrossRef.
- D. D. He, G. H. He, H. Q. Jiang, Z. K. Chen and M. H. Huang, Chem. Commun., 2017, 53, 5132–5135 RSC.
- S. G. Chandrappa, P. Moni, D. Chen, G. Karkera, K. R. Prakasha, R. A. Caruso and A. S. Prakash, J. Mater. Chem. A, 2020, 8, 20612–20620 RSC.
- W. Y. Li, Y. M. Zhu, W. Guo, H. W. Xu, C. R. Gong and G. Xue, J. Chem. Sci., 2022, 134, 38 CrossRef CAS.
- X. X. Wang, J. Dai, C. Zhou, D. Q. Guan, X. H. Wu, W. Zhou and Z. P. Shao, ACS Mater. Lett., 2021, 3, 1258–1265 CrossRef CAS.
- J. Hui, C. P. Yan, Y. Shi and Z. Yang, Mater. Lett., 2023, 332, 133532 CrossRef CAS.
- S. B. Karki, A. N. Andriotis, M. Menon and F. Ramezanipour, ACS Appl. Energy Mater., 2021, 4, 12063–12066 CrossRef CAS.
- F. Dionigi, J. Zhu, Z. Zeng, T. Merzdorf, H. Sarodnik, M. Gliech, L. Pan, W. X. Li, J. Greeley and P. Strasser, Angew. Chem., Int. Ed., 2021, 60, 14446–14457 CrossRef CAS PubMed.
- H. Liu, R. Xie, Q. Wang, J. Han, Y. Han, J. Wang, H. Fang, J. Qi, M. Ding, W. Ji, B. He and W. Lü, Adv. Sci., 2023, 10, 2207128 CrossRef CAS PubMed.
- W. Zhang, D. Li, L. Zhang, X. She and D. Yang, J. Energy Chem., 2019, 39, 39–53 CrossRef.
- K. Sharma, V. Hasija, S. Patial, P. Singh, V.-H. Nguyen, A. K. Nadda, S. Thakur, P. N. Tri, C. C. Nguyen, S. Y. Kim, Q. V. Le and P. Raizada, Int. J. Hydrogen Energy, 2023, 48(17), 6560–6574 CrossRef CAS.
- L.-W. Chen and H.-W. Liang, Catal. Sci. Technol., 2021, 11, 4673–4689 RSC.
- Y. Wu, X. Tao, Y. Qing, H. Xu, F. Yang, S. Luo, C. Tian, M. Liu and X. Lu, Adv. Mater., 2019, 31, 1900178 CrossRef PubMed.
- H. Sun, W. Zhang, J.-G. Li, Z. Li, X. Ao, K.-H. Xue, K. K. Ostrikov, J. Tang and C. Wang, Appl. Catal., B, 2021, 284, 119740 CrossRef CAS.
- J. Bao, Z. Wang, J. Xie, L. Xu, F. Lei, M. Guan, Y. Huang, Y. Zhao, J. Xia and H. Li, Inorg. Chem. Front., 2018, 5, 2964–2970 RSC.
- H. Shi, H. Liang, F. Ming and Z. Wang, Angew. Chem., Int. Ed., 2017, 129, 588–592 CrossRef.
- C. Tang, N. Cheng, Z. Pu, W. Xing and X. Sun, Angew. Chem., Int. Ed., 2017, 127, 9483–9487 CrossRef.
- X. Gao, H. Zhang, Q. Li, X. Yu, Z. Hong, X. Zhang, C. Liang and Z. Lin, Angew. Chem., Int. Ed., 2017, 128, 6398–6402 CrossRef.
- C. Xiao, Y. Li, X. Lu and C. Zhao, Adv. Funct. Mater., 2016, 26, 3515–3523 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta06718e |
| This journal is © The Royal Society of Chemistry 2025 |