
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlginate–oligothiophene aerogels as photocatalysts for the degradation of emerging organic contaminants in water†‡
Andrea
Trifoglio§
ac,
Francesca
Tunioli§
ac,
Laura
Favaretto
a,
Massimo
Zambianchi
 a,
Cristian
Bettini
a,
Ilse
Manet
a,
Livia
Mariani
b,
Anna
Barra Caracciolo
b,
Paola
Grenni
b,
Manuele
Di Sante
cd,
Matteo
Di Giosia
cd,
Tainah Dorina
Marforio
cd,
Edoardo Jun
Mattioli
cd,
Matteo
Calvaresi
*cd and
Manuela
Melucci
*a
a,
Cristian
Bettini
a,
Ilse
Manet
a,
Livia
Mariani
b,
Anna
Barra Caracciolo
b,
Paola
Grenni
b,
Manuele
Di Sante
cd,
Matteo
Di Giosia
cd,
Tainah Dorina
Marforio
cd,
Edoardo Jun
Mattioli
cd,
Matteo
Calvaresi
*cd and
Manuela
Melucci
*a
aInstitute for Organic Synthesis and Photoreactivity (ISOF), National Research Council of Italy (CNR), Via P. Gobetti 101, 40129 Bologna, Italy. E-mail: manuela.melucci@isof.cnr.it; matteo.calvaresi3@unibo.it
bWater Research Institute (IRSA), National Research Council (CNR), Via Salaria Km 29, 300 C. P. 10 – 00015 Monterotondo Stazione, Rome, Italy
cDepartment of Chemistry ‘G. Ciamician’, Alma Mater Studiorum – University of Bologna, Via Selmi 2, 40126 Bologna, Italy
dIRCCS Azienda Ospedaliero – Universitaria di Bologna, Preclinical & Translational Research in Oncology Lab (PRO), Bologna, Italy
First published on 8th January 2025
Abstract
V-shaped oligothiophenes bearing a benzothiophene (BT) core and S-oxide or S,S-dioxide moieties are herein proposed as photocatalysts for the degradation of three selected water emerging contaminants. Photocatalyst molecules were designed in order to minimize aggregation in the solid state (V-shape structure), lower the HOMO–LUMO gap (S,S-dioxide moiety functionalization) with respect to conventional oligothiophenes and enhance the photocatalytic performance. The targeted BT molecules were synthesized by the Stille cross-coupling reaction and their ability to generate reactive oxygen species (ROS) upon blue-light irradiation was demonstrated. For practical exploitation, BTs were embedded into an alginate matrix by ionotropic gelation–freeze drying processes to obtain porous aerogels that were dispersed in water and used for the degradation of emerging contaminants. Degradation performance of up to 99% after 2 h on ofloxacin (antibiotic), rhodamine B (dye) and bisphenol A (plastic additive), was observed. In addition, regenerated photocatalysts showed unchanged performance after five reuse cycles. The relationship between the BT molecular structure and ROS production, investigated by QM calculations, highlighted the role of the S,S-dioxide moiety in ROS generation, with ofloxacin antibiotic degradation performance reaching up to 99% for BTO2-T2. Remarkably, ecotoxicity studies showed no toxicity of the photodegradation products toward the Aliivibrio fischeri bioluminescent bacterium, highlighting the potential of BT molecules for water purification.
Introduction
The removal of emerging contaminants (ECs), such as per- and polyfluoroalkyl substances, pesticides, pharmaceuticals, and endocrine disruptors, has emerged as one of the foremost drinking water issues in the past decade.1–3 The allowed limits for some of these contaminants in drinking water are continuously updated to ever-decreasing values,4,5 posing increasingly hard challenges for the development of new remediation technologies since conventional drinking water treatment plants are not designed to remove all ECs.6,7 Conventional treatments for the removal of emerging contaminants are mainly based on membrane technologies (nanofiltration and reverse osmosis),8,9 advanced oxidative processes (AOPs)10 and adsorption.11–13 Adsorption is particularly advantageous in terms of cost and ease of use but it produces wastes to be regenerated or disposed of.14 Despite its widespread use for desalination or point-of-use drinking water treatment, reverse osmosis still has high water rejection rates and high energy consumption.15 AOPs include methods for the generation of hydroxyl radicals, superoxide and other reactive oxygen species (ROS), which are powerful oxidizing agents that react with harmful organic compounds, resulting into contaminant degradation. One of the most widely investigated AOPs is photodegradation, which involves the use of photocatalysts such as TiO2, CdS and ZnO nanoparticles, i.e., materials that absorb light and subsequently generate reactive species destroying contaminants.16–18Due to their high redox potential, reactivity, efficiency, chemical stability, non-toxicity, and low cost, TiO2-based photocatalysts are the most studied and widely employed.19 The non-specific nature of the reactive species created upon irradiation makes TiO2-based photocatalysts capable of degrading different classes of organic contaminants.20–22 On the other hand, the recombination of photo-generated charge carriers, inefficient visible light exploitation, and low adsorption capacity require further optimization to boost photocatalytic potential and enable practical exploitation.23
In recent years, other nanomaterials, such as metal–organic frameworks (MOFs),24 covalent organic frameworks (COFs)25 and graphene nanosheets26 have been proposed as photocatalysts with excellent performance due to their tuneable-by-design functionalities, high surface area and the large number of active sites available for EC degradation.27,28 In particular, MOFs have shown remarkable performance in the degradation of organic dyes such as methylene blue and rhodamine B, e.g., Fe-MOFs are able to degrade them by 100% and 94%, respectively, in 60 minutes under visible light irradiation.29 Zr-based MOFs have also been used to degrade pesticides such as glyphosate, achieving selective degradation of 64% into sarcosine and orthophosphates.30 MOF-C3N4 binary composites have been employed in the degradation of pharmaceuticals such as ofloxacin, demonstrating 96% degradation in 200 minutes.31
Small π-conjugated organic molecules such as porphyrin have also been used as hybrid metal–organic photocatalysts for light-assisted degradation of various organic contaminants in water.32 Overall, porphyrins immobilized in inorganic semiconductors,33 zeolites,34 graphene,35 or porous organic polymers,36 usually exhibit improved physicochemical properties and stability, and they are active in the photodegradation of pesticides, dyes, and pharmaceuticals.37,38 For example, Sun et al. reported the complete degradation of rhodamine B in just 36 minutes with the use of CuPp–TiO2 (CuPp: mono-[4-(2-ethyl-p-hydroxybenzoate)ethoxyl]-10,15,20-tri(phenyl)porphyrin copper(II)).39 The photocatalytic activity of the graphene–TiO2–tetrakis(4-carboxyphenyl)porphyrin (3.0%) nanocomposite was tested on bisphenol A, and a degradation of 85% was observed after 240 minutes of irradiation.40
Here, for the first time, we propose and investigate oligothiophenes entrapped in an alginate matrix, as organic photocatalysts for the degradation of rhodamine B dye (RhB), bisphenol A (BPA) and ofloxacin antibiotic (OFLOX), three contaminants of high environmental relevance in drinking water.41,42 RhB is a common water-soluble organic dye employed in the textile industry and its presence in water bodies has been found with concentrations ranging from ng L−1 to μg L−1.42 Even at very low concentrations (1.0 mg L−1), it imparts a vivid colour to water, but the limit concentration to protect against hazardous effects is 140 μg L−1.43,44 BPA is a plastic additive used primarily in the production of polycarbonate plastics and epoxy resins. It is classified as an endocrine-disrupting chemical and recent research has revealed the widespread presence of BPA in groundwater, categorizing it as a ubiquitous pollutant.45,46 OFLOX is a fluorinated quinolone antibiotic used for human and animal bacterial infections, and it has raised increasing concern due to its widespread use. OFLOX was found in surface water up to 160 μg L−1 and its presence in aquatic environments is associated with the potential to induce antibiotic resistance.47
Branched oligothiophenes (V-shaped) based on the benzo[b]thiophene core (BT) were introduced by G. Barbarella et al. as active layers for organic light emitting diodes.48 The V-shape minimizes aggregation and related fluorescent quenching in the solid state and promotes efficient solid state photoluminescence.48 In addition, the insertion of S-oxide or S,S-dioxide moieties into the aromatic backbone decreases the HOMO–LUMO energy gap.49,50 The capability of oligothiophene molecules to produce reactive oxygen species under light stimulus has been recently exploited for cancer therapy applications.51,52
Here, aiming at EC degradation in water, we studied how the energy gap of the targeted BT materials family affects the generation of radicals. We synthesized a rational class of molecules having the same molecular V-shape and respectively no oxide (BT-T2), S-oxide (BTO-T2) and S,S-dioxide (BTO2-T2) moieties (Scheme 1). T2(BTO2)3 was synthesized with the aim of extending the π system and increasing the number of S,S-dioxide moieties to promote the generation of radicals. Finally, BTO2-Tz2 was synthesized by introducing electron-deficient nitrogen into the system and thus creating a push–pull system, which leads to the lowering of the HOMO–LUMO energy gap.53
 | ||
| Scheme 1 Branched V-shaped (BT-T2) and S-oxide (BTO-T2) or S,S-dioxide (BTO2-T2, T2(BTO2)3, and BTO2-Tz2) molecules engineered for photocatalysis studies. | ||
The optical properties of BTs were investigated by spectroscopy and DFT calculations. The ROS produced upon irradiation were quantified and their generation mechanism was studied. For the technological exploitation of these insoluble compounds in water, the BT molecules were encapsulated in alginate, a safe and water processable matrix,54,55 and used to determine the degradation mechanism toward RhB, BPA and OFLOX as well as their reusability.
Finally, to ensure water safety, we investigated the possible ecotoxicity of the photodegradation by-products of OFLOX in the presence of photocatalysts. This was performed using an aquatic organism, the bacterium Aliivibrio fischeri, to perform an eco-toxicological bioassay.56–58A. fischeri (formerly classified as Vibrio fischeri) is a biosensor59 extremely sensitive to different environmental contaminants60,61 and it is currently used to assess water quality with standardized procedures and ecotoxicological evaluation.62
Experimental section
Synthesis of BT molecules
2,3-Dibromo-benzo[b]thiophene, 2-bromothiazole, 3-methylbenzothiophene, n-butyllithium (n-BuLi), trimethyltin chloride (Me3SnCl), tributyltin chloride (Bu3SnCl), N-bromosuccinimide (NBS), m-chloroperbenzoic acid (mCPBA), boron trifluoride diethyl etherate (BF3·Et2O), tetrabutylammonium bromide (TBAB), N,N-diisopropylethylamine (i-Pr2NEt), tris(dibenzylideneacetone)dipalladium(0)-chloroform adduct (Pd2(dba)3·CHCl3), triphenylarsine (AsPh3), palladium(II) acetate ([Pd(OAc)2]3) were purchased in the highest grade available from Sigma-Aldrich Co, Milan, Italy, and were used without further purification. OFLOX (>99%), RhB (>95%) and BPA (>95%) were purchased from Sigma-Aldrich Co, Milan, Italy, and used as received.The syntheses of 5-tributylstannyl-2,2′-bithiophene (3),49 2,3-di([2,2′-bithiophen]-5-yl)benzo[b]thiophene (BT-T2), 2,3-dibromobenzo[b]thiophene-1,1-dioxide (5), 2,3-di([2,2′-bithiophen]-5-yl)benzo[b]thiophene-1,1-dioxide (BTO2-T2),48 and 2,2-bithiazole63,64 (6) have already been reported.
Unless otherwise noted, all operations were carried out under a dry, oxygen-free nitrogen atmosphere. Organic solvents were dried following standard procedures. Analytical thin-layer chromatography was carried out using a 0.2 mm sheet of silica 60 F254 (Merck, Milan, Italy). Visualization was accomplished using UV light. Preparative column chromatography was performed on glass columns of different sizes hand-packed with silica gel 60 (particle sizes 0.040–0.063 mm, Merck). We referred to ethyl acetate as EA, petroleum ether as PE, and tetrahydrofuran as THF.
The syntheses of 2,3-dibromobenzo[b]thiophene-1-oxide (2), 5-(trimethylstannyl)-2,2′-bithiazole (6), 2-bromo-3-methylbenzo[b]thiophene(11), 2-bromo-3-methylbenzo[b]thiophene-1,1-dioxide (12), 2-([2,2′-bithiophen]-5-yl)-3-methylbenzo[b]thiophene-1,1-dioxide (13), 2-(5′-bromo-[2,2′-bithiophen]-5-yl)-3-methylbenzo[b]thiophene 1,1-dioxide (14), and 3-methyl-2-(5′-(tributylstannyl)-[2,2′-bithiophen]-5-yl)benzo[b]thiophene-1,1-dioxide (8) are reported in Section 1.1, ESI.‡
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15:15 v/v). The isolated product was recrystallized from toluene and pentane to obtain an orange powder (0.1 g, 83% yield); mp 129 °C; DEP-EI-MS m/z 479 [M˙+]; λmax = 430 nm in CH2Cl2; λPL = 571 nm in CH2Cl2. 1H NMR (CDCl3, TMS/ppm) δ 7.96 (d, J = 7.6 Hz, 1H), 7.59 (d, J = 3.6 Hz, 1H), 7.50 (m, 2H), 7.42 (d, J = 7.6 Hz, 1H), 7.30 (d, J = 3.6 Hz, 1H), 7.29 (dd, J = 4.0, 1.2 Hz, 1H), 7.26 (m, 1H), 7.23 (dd, J = 5.2, 1.2 Hz, 1H), 7.16 (d, J = 3.6 Hz, 1H), 7.13 (m, 2H), 7.06 (dd, J = 5.2, 3.6 Hz, 1H), 6.98 (dd, J = 5.2, 3.6 Hz, 1H); 13C NMR (CDCl3, TMS/ppm) δ 143.8, 142.2, 141.1, 140.9, 140.1, 136.5, 136.4, 132.6, 131.6, 131.0, 130.6, 130.4, 130.2, 128.6, 128.0, 127.9, 126.4, 125.5, 125.3, 124.8, 124.6, 124.3, 124.0, 123.9. Anal. calcd for C24H14OS5 (478.68): C, 60.22; H, 2.95. Found: C, 60.64; H, 2.81.
:40 to 0:100 v/v). The isolated product was recrystallized from toluene and pentane to obtain a yellow powder (0.33 g, 67% yield); mp 140 °C; DEP-EI-MS m/z 498 [M˙+]; λmax = 403 nm in CH2Cl2; λPL = 530 nm in CH2Cl2. 1H NMR (CDCl3, TMS/ppm) δ 8.52 (s, 1H), 8.02 (s, 1H), 7.97 (d, J = 3.2 Hz, 1H), 7.91 (m, 1H), 7.87 (d, J = 3.2 Hz, 1H), 7.64 (m, 2H), 7.57 (d, J = 3.2 Hz, 1H), 7.49 (d, J = 3.2, 1H), 7.41 (m, 1H); 13C NMR (CDCl3, TMS/ppm) δ 165.3, 163.9, 160.6, 160.4, 146.2, 145.5, 144.4, 144.4, 135.4, 134.4, 133.8, 132.6, 130.9, 127.3, 126.2, 124.9, 124.0, 122.4, 122.3, 122.0. Anal. calcd for C20H10N4 O2S5 (498.63): C, 48.18; H, 2.02. Found: C, 48.41; H, 2.19.
:15:15 v/v). The isolated product was recrystallized from toluene and pentane to obtain an orange powder (0.1 g, 83% yield); mp 129 °C; DEP-EI-MS m/z 479 [M˙+]; λmax = 430 nm in CH2Cl2; λPL = 571 nm in CH2Cl2. 1H NMR (CDCl3, TMS/ppm) δ 7.96 (d, J = 7.6 Hz, 1H), 7.59 (d, J = 3.6 Hz, 1H), 7.50 (m, 2H), 7.42 (d, J = 7.6 Hz, 1H), 7.30 (d, J = 3.6 Hz, 1H), 7.29 (dd, J = 4.0, 1.2 Hz, 1H), 7.26 (m, 1H), 7.23 (dd, J = 5.2, 1.2 Hz, 1H), 7.16 (d, J = 3.6 Hz, 1H), 7.13 (m, 2H), 7.06 (dd, J = 5.2, 3.6 Hz, 1H), 6.98 (dd, J = 5.2, 3.6 Hz, 1H); 13C NMR (CDCl3, TMS/ppm) δ 143.8, 142.2, 141.1, 140.9, 140.1, 136.5, 136.4, 132.6, 131.6, 131.0, 130.6, 130.4, 130.2, 128.6, 128.0, 127.9, 126.4, 125.5, 125.3, 124.8, 124.6, 124.3, 124.0, 123.9. Anal. calcd for C24H14OS5 (478.68): C, 60.22; H, 2.95. Found: C, 60.64; H, 2.81.
:40 to 0:100 v/v). The isolated product was recrystallized from toluene and pentane to obtain a yellow powder (0.33 g, 67% yield); mp 140 °C; DEP-EI-MS m/z 498 [M˙+]; λmax = 403 nm in CH2Cl2; λPL = 530 nm in CH2Cl2. 1H NMR (CDCl3, TMS/ppm) δ 8.52 (s, 1H), 8.02 (s, 1H), 7.97 (d, J = 3.2 Hz, 1H), 7.91 (m, 1H), 7.87 (d, J = 3.2 Hz, 1H), 7.64 (m, 2H), 7.57 (d, J = 3.2 Hz, 1H), 7.49 (d, J = 3.2, 1H), 7.41 (m, 1H); 13C NMR (CDCl3, TMS/ppm) δ 165.3, 163.9, 160.6, 160.4, 146.2, 145.5, 144.4, 144.4, 135.4, 134.4, 133.8, 132.6, 130.9, 127.3, 126.2, 124.9, 124.0, 122.4, 122.3, 122.0. Anal. calcd for C20H10N4 O2S5 (498.63): C, 48.18; H, 2.02. Found: C, 48.41; H, 2.19.
Preparation of alginate–BT aerogels
BTs were embedded in an alginate (ALG) matrix by ionotropic gelation technique, according to a previously reported method.65 Sodium alginate (50 mg, 1% w/v) and the desired BT (10 mg, 0.2% w/w) were dispersed in 5 mL of ultrapure water and sonicated for 2 h. The suspension was extruded as small drops using a Gilson pipette (1000 μL) into a gently stirred aqueous solution of calcium chloride (CaCl2, 0.2 M). The distance between the pipette tip and the collection bath was approximately 30 mm. To minimize aggregation the obtained hydrogel beads were kept at room temperature under gentle agitation for 12 h. After removing water by filtration, the beads were washed three times with mQ water, and then subjected to freeze-drying to obtain aerogel beads. The total water content of the beads was determined gravimetrically by measuring the difference in weight before and after drying a portion of the beads in an oven at 100 °C until a constant weight was reached and the water content was found to be approximately 93%. The wet beads produced in this study had a size of 3–4 mm, while the final freeze-dried beads had a diameter ranging from 1.5 to 2.5 mm. The BT loading was quantitative, i.e., no trace of BT was found in the water–CaCl2 solution, meaning that the average concentration of BT in the beads was 16.7% (w/w) BT:alginate. In a typical experiment, about 200 beads with 3.5 mm (±0.5 mm) diameter were obtained, meaning that the BT content in a single bead was approximately 50 μg (with an Alg content of 250 μg).
Theoretical calculations
All computations were carried out using the Gaussian16 series of programs.65 The geometry of the oligothiophene molecules was optimized in DMSO using the IEF-PCM solvation model,66 using the DFT Minnesota functional M06-2X in conjunction with the 6-31+G* basis set (M06-2X/6-31+G*). The first excited state (S1) was calculated considering a vertical transition from the optimized ground state (S0) structure, using time-dependent DFT formalism (TD-DFT) with the long-range corrected hybrid functional CAM-B3LYP67 and the 6-31+G* basis set (TD-CAM-B3LYP/6-31+G*). This combination effectively reproduces the photophysical properties of oligothiophene molecules.52,68Optical properties and confocal microscopy
Absorption spectra were recorded on a spectrophotometer PerkinElmer λ950 with a 1 cm quartz cuvette. Reflectance spectra of the beads were recorded on the same spectrophotometer with and integrating sphere and a 1 cm quartz cuvette. Fluorescence spectra were measured on an Edinburgh Spectrofluorimeter FLSP920 with 2 nm steps and 0.5 s dwell time. The slits were set to 2–3 nm for both excitation and emission. Right angle detection was used. All the measurements were carried out at 295 K in quartz cuvettes with a path length of 1 cm. All fluorescence spectra were obtained for air-equilibrated solutions with absorbance less than 0.1 and corrected for the wavelength dependent response of the monochromator/PMT couple. Fluorescence lifetimes were measured in air-equilibrated solutions with a time correlated single photon counting system. A pulsed Hamamatsu 405 nm laser source with a 1 MHz repetition rate was used for excitation and the emission was collected at a right angle at 520 nm (BT-T2, BTO2-Tz2), and 620 nm for the others. Decay profiles were fitted using a multiexponential function with deconvolution of the instrumental response.Fluorescence confocal imaging was performed on an inverted Nikon Ti-E microscope (Nikon Co., Shinjuku, Japan). The confocal fluorescence microscope Nikon A1 is equipped with an argon ion CW laser as well as 405 nm and 485 nm pulsed diode lasers (PicoQuant GmbH, Berlin, Germany). Images were collected using a Nikon Plan Apo VC 60× objective with NA 0.9. Filters were set to register fluorescence in the 500–540, 560–630 nm and 660–740 nm ranges. Fluorescence imaging was completed with differential interference contrast (DIC) images.
Spectral imaging was performed with the Nikon spectral mode with a 32-PMT array detector. Wavelengtth resolution was set to 10 nm per PMT and the membranes were excited either at 488 nm or 405 nm.
Fluorescence lifetime imaging was performed by exciting with a pulsed 405 or 485 nm diode laser and collecting photons with an integrated PicoHarp 300 electronic system (PicoQuant GmbH, Berlin, Germany) for TCSPC measurements. A single-photon avalanche diode detector equipped with a bandpass filter was used as the detector. Histograms of the collected photons consist of 3200 channels, each with 16 ps width. The repetition rate of the pulsed excitation was 20 MHz. The instrument response function of the system is approximately 220 ps. The fluorescence decay fit was performed on the histogram calculated for a region of interest. The fluorescence decay profile was analyzed with a least-squares method, using a biexponential decay function provided by Picoquant SymPhoTime software.
The fitting function used is
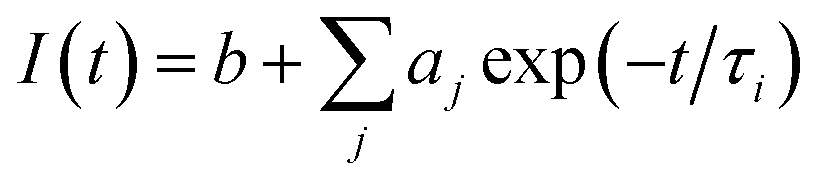 | (1) |
The fractional intensity and the average fluorescence lifetime are calculated according to the following equations:
 | (2) |
 | (3) |
Photodegradation experiments
All the photodegradation experiments were performed using a blue LED stripe (λEM = 461 nm, irradiation power density = 10 mW cm−2, and a distance of about 10 cm) and the set-up is depicted in Fig. 6. 5 mg of photocatalysts (aerogels containing BTs) were added to 3 mL of a solution of RhB, BPA or OFLOX (4 mg L−1 in ultrapure water). Samples were stirred under blue light irradiation for 5 h; during this time 200 μL aliquots were collected every hour and each sample was analysed by HPLC. Reuse tests were performed on Alg–BTO2-T2, Alg–BTO2-Tz2 and Alg–T2(BTO2)3 aerogels. After the first photodegradation experiment, the aerogels were externally dried on filter paper, washed with deionized H2O, and reused in a new photodegradation test (3 mL of a spiked solution at 4 mg L−1). The procedure was repeated for a total of five cycles.Results and discussion
Synthesis of BT molecules and alginate aerogel preparation
The synthesis of BT molecules was carried out via Stille coupling between dibromo derivatives 2 and 5 and the appropriate stannane (Scheme 2a). The reactions were carried out in refluxing toluene using in situ prepared Pd(AsPh3)4 as the catalyst.48 Compounds 2 and 5 were synthesized starting from commercially available 2,3-dibromo-benzo[b]thiophene (1) by reaction with mCPBA and BF3·Et2O (yielding 76% and 90% for 2 and 5, respectively). Compound 4 was synthesized by Stille coupling between dibromo derivative 2 and stannane 3, yielding 83%. Stannyl-derivative 8 was synthesised starting from methyl benzothiophene 10 through a five step procedure. Compound 10 was first reacted with NBS in AcOH/CH2Cl2 to give the brominated adduct 11. Oxidation of by mCPBA in dichloromethane afforded the targeted sulphone derivative 12 in excellent yields (90%). The Stille coupling reaction of compound 12 with distannyl bithiophene 3 afforded 13 in 91% isolated yield. After bromination of compound 13 and subsequent stannylation, Stille coupling with dibromo-benzothiophene-S,S-dioxide 5 afforded V-shaped BT oligomers 7 and 9 in 67% and 76% yields, respectively. BT-T2 and BTO2-T2 were synthesized under already described conditions and obtained in 48% (BT-T2) and 90% (BTO2-T2).48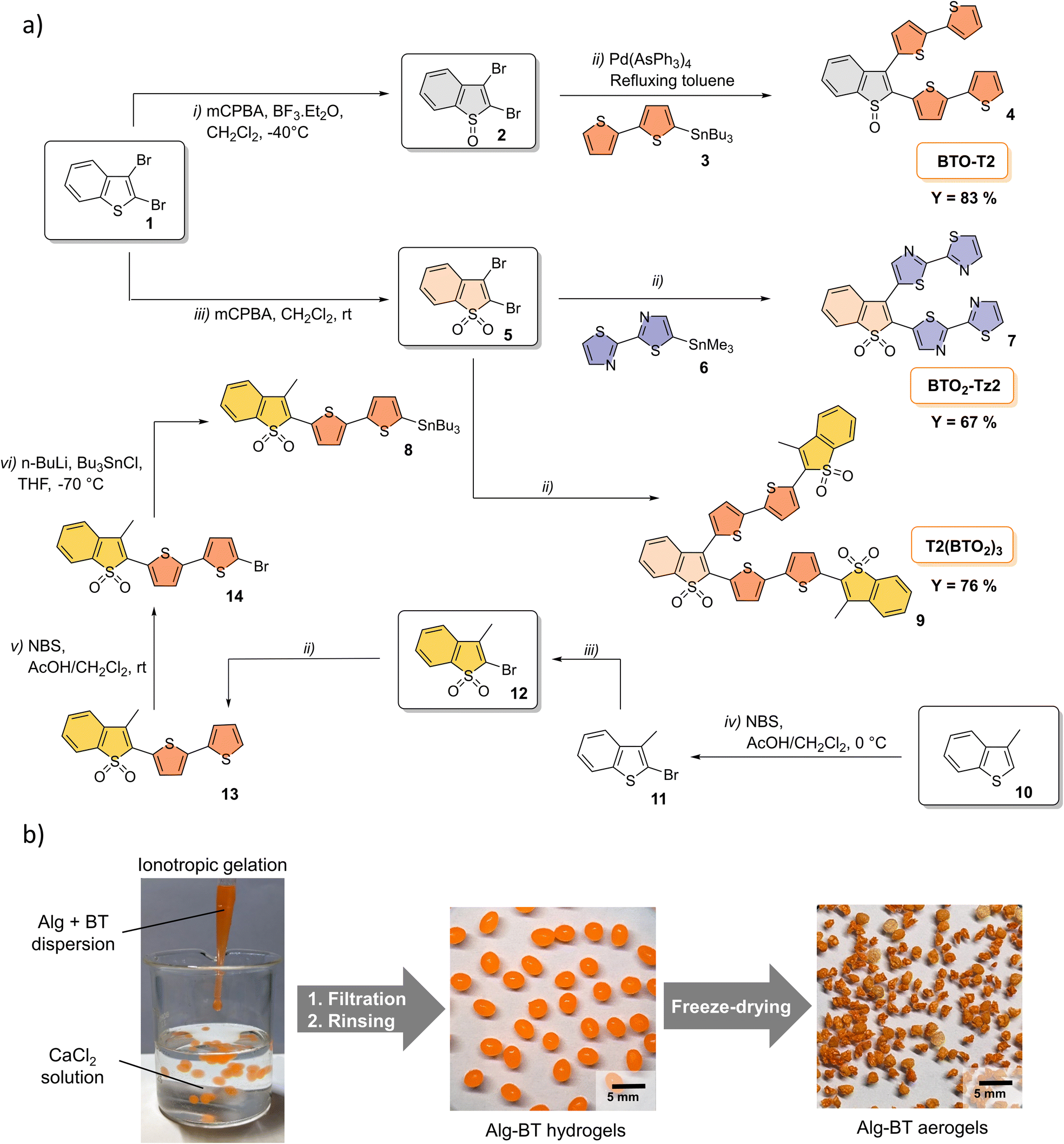 | ||
| Scheme 2 (a) Synthesis of V-shaped BT oligomers. (b) Preparation of BTs–alginate aerogels by the ionotropic gelation method. | ||
Due to the poor solubility of oligothiophenes in water, BT molecules were embedded in an alginate matrix54,69 by the ionotropic gelation method (Scheme 2b). In detail, sodium alginate and BT molecules in powder form (ratio 5:1 w/w) were dispersed in water and sonicated for 2 h to obtain a homogeneous suspension which was added dropwise into a CaCl2 solution. Finally, hydrogels were freeze-dried to produce the corresponding aerogels (Alg–BT, Fig. 1). All experimental details on characterization are reported in Section 1.2, ESI.‡
 | ||
| Fig. 1 Images of BT powders (top), alginate aerogels (middle) and dispersion (bottom, after 5 days from preparation) in water under white light (left) and under UV light with emission at 365 nm (right). | ||
Fig. 1 shows images of BT powders, Alg-beads and their dispersion under normal and UV light. The different powders are mostly yellow, orange and red in colour and show strong emission when exposed to UV irradiation. The reflectance spectra shown in Fig. S1, ESI‡ are in line with the bead colors.
Furthermore, Fig. 1 (bottom line) shows that when Alg–BT aerogels are dispersed in water, there is no leaching from the alginate matrix even after 5 days of contact time, making them suitable candidates for water treatment purposes since they can be dispersed but not dissolved in water. The morphology of the Alg–BT aerogels was studied by scanning electron microscopy (SEM), as shown in Fig. 2. All the samples showed the typical micrometric porous structure of alginate aerogel with a dense skin layer.
 | ||
| Fig. 2 SEM images of (a) pristine alginate aerogel, and (b) alginate–BTO2-T2 aerogel. (c) Confocal fluorescence image and differential interference contrast microscopy (DIC) image of BTO2-T2 beads. | ||
Fig. 2b shows the cross-section of the pristine alginate bead and Alg–BTO2-T2 bead, taken as a case study, demonstrating that the porous structure of pristine alginate is maintained after the ionotropic gelation and freeze-drying processes. The porous structure of Alg–BT can favour the penetration of contaminant molecules into the bead and the concomitant interaction with the photocatalysts. Noteworthily, the ability of alginate hydrogels to adsorb contaminants has already been demonstrated by some of us for alginate–graphene composites and for amino-modified alginate hydrogels.55,70 The morphology was further explored by means of confocal fluorescence and spectral imaging. All beads exhibit emission from the loaded oligothiophene. The images of all beads (Fig. 2c and S2‡) show that BT molecules tend to accumulate in aggregates of μm size on the beads.
Optical properties and confocal microscopy
The optical properties of BT molecules were investigated by UV-vis and fluorescence spectroscopy in CH2Cl2 and PBS with 5% DMSO (Fig. S3, ESI‡). Fig. 3a gives an overview of the optical behaviour of the studied BT molecules in CH2Cl2 solution highlighting the typical fluorescence under blue light irradiation. The absorption spectra of the BT molecules in CH2Cl2 and PBS/DMSO are shown in Fig. 3b and S3a.‡ All BT molecules absorb in the violet–blue range of the visible spectrum and exhibit two distinct peaks corresponding to electronic transitions from the ground state to the first and second excited state. The insertion of S-oxide or S,S-dioxide moieties results in a red-shift of the absorption wavelength, as shown by the λabs of BT-T2 (375 nm), BTO-T2 (430 nm) and BTO2-T2 (430 nm). The extension of the conjugated π-system and the presence of two other S,S-dioxide moieties in T2(BTO2)3 lead to a further red-shift (λabs = 460 nm). The introduction of a push–pull system in BTO2-Tz2, compared to BTO2-T2, leads to a blue-shift (403 nm vs. 430 nm).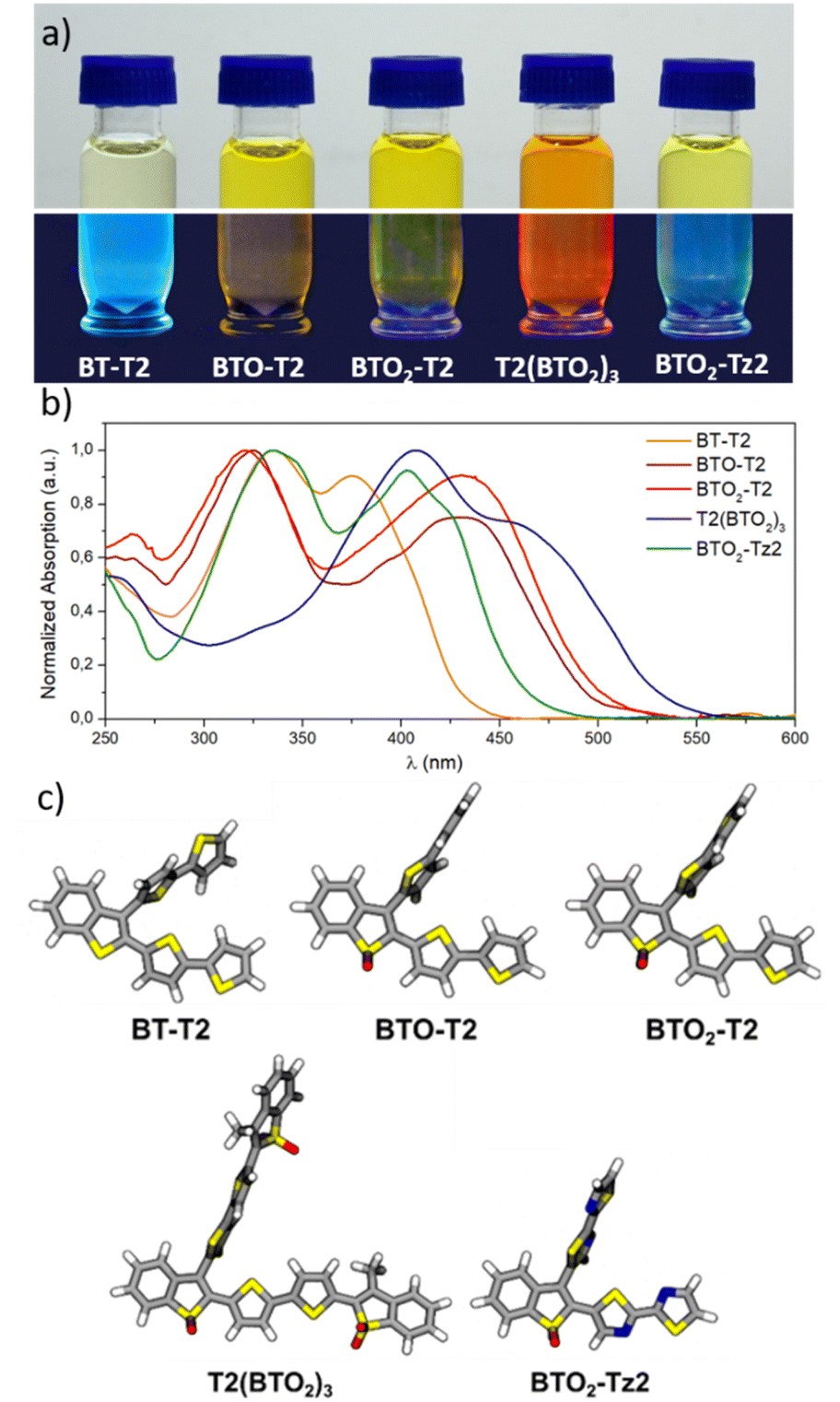 | ||
| Fig. 3 (a) images of BT solution in CH2Cl2 under white light (top) and UV light (λ = 365 nm, bottom), (b) absorption spectra in CH2Cl2 and (c) DFT optimized geometries, for BT-T2, BTO-T2, BTO2-T2, T2(BTO2)3 and BTO2-Tz2. | ||
The spectra calculated at the TD-CAM-B3LYP/6-31+G* level (see computational details) well reproduce the experimental ones (Fig. S4 and Table S2‡), validating the accuracy of the calculations. DFT optimization of the molecules indicates that in the most stable conformation, the BT ring and the thiophene at BT position 2 are conjugated and remain planar while the thiophene substituent at BT position 3 adopts an orthogonal orientation (Fig. 3c). This structural characteristic affects the nature of the S0 → S1 transitions of the different molecules, because they are localized only on the planar core of the molecules, where the π-conjugation is maximized (π–π* transitions). The molecular orbitals (MOs) involved in the electronic transition were calculated (Fig. S5‡). The transitions with the larger oscillator strength (Table S2‡) correspond to the experimental bands with higher molar extinction coefficients. Jablonski diagrams of the thiophene derivatives (Fig. 4) showed that the calculated T1 excited states (Table 1) are at higher energies than the singlet excited state of the oxygen, suggesting that all the compounds, upon intersystem crossing (ISC), can produce singlet oxygen (1O2).
 | ||
| Fig. 4 Jablonski diagrams for BT molecules. | ||
| Structure | S1 (eV) | T1 (eV) |
|---|---|---|
| BT-T2 | 3.45 | 2.07 |
| BTO-T2 | 3.06 | 1.57 |
| BTO2-T2 | 3.06 | 1.51 |
| T2(BTO2)3 | 2.95 | 1.50 |
| BTO2-Tz2 | 3.21 | 1.68 |
The fluorescence spectra of BTs display a large stokes shift, especially in PBS/DMSO. The fluorescence decay is multi-exponential with average lifetimes below 1 ns for all oligothiophenes (Table S3‡). Likely, the dye can exist in multiple conformations/aggregation states displaying different lifetimes. The study was completed by measuring the emission spectra and fluorescence lifetime of the BT-loaded beads on a confocal microscope as well as the lifetime. Except for BTO-T2, the confocal spectra in Fig. S6‡ depend on the selected area in the sample, indicating that the oligothiophenes are likely experiencing different local environments. This lack of homogeneity is also confirmed by the FLIM images where we can discriminate areas with different average fluorescence lifetimes (Fig. S2b‡).
Reactive oxygen species (ROS) generation by BT molecules and Alg–BT aerogels
The ability of the different BT molecules and related alginate aerogels to produce ROS, upon irradiation, was determined by spectrophotometric measurements, as shown in Fig. 5 (experimental details in ESI, Section 3‡). | ||
| Fig. 5 Generation of ROS after 1 hour of irradiation with a blue LED (10 mW cm−2). (a) Quantification of 1O2 production (type II mechanism) using the ABMDMA colorimetric assay by BT molecules. Quantification of the production of hydrogen peroxide (type I mechanism) via the Amplex Red fluorometric assay (b) by BT molecules and (c) by Alg–BT aerogels. | ||
Both the mechanisms of ROS production were investigated, by using (i) the Amplex Red assay to quantify the generation of peroxides (type I mechanism)71–74 and (ii) the 9,10-anthracenediyl-bis(methylene)dimalonic acid (ABMDMA) assay to quantify the production of singlet oxygen (type II mechanism).75–78 The results summarized in Fig. 5a and b show that all the synthesized BT molecules are suitable photosensitizers, capable of producing ROS following both mechanisms. The different efficiencies in ROS generation, upon irradiation, can be ascribed to the different absorption coefficients of the molecules at the wavelength used for irradiation, i.e., 460 nm, with T2(BTO2)3 characterized by the highest absorption coefficient and larger oscillator strength.
Interestingly, the photosensitizing behaviour of Alg–BT aerogels changes substantially with respect to the molecules in solution (Fig. 5c). The possibility of generating ROS via the type II pathway seems hampered; indeed when the molecules are dispersed in the alginate matrix, the production of singlet oxygen is not detected. Differently, the ability of the Alg–BT aerogels to produce ROS via the type I pathway is preserved for all the alginate embedded molecules (Fig. 5c) and the trend observed in solution is almost maintained, with a better performance of BTO2-Tz2 compared to solution.
This effect is not due to the reaction of 1O2 with the alginate matrix, because when the experiment is repeated with thiophenes in solution, in the presence of empty alginate beads or alginate in solution, the production of singlet oxygen is unaffected. The dispersion of the BTs in the alginate matrix affects their photophysical properties. Oligothiophene molecules exhibit different fluorescence spectra according to the selected area on the beads, indicating that their photophysical behaviour is influenced by the alginate matrix. In addition, confocal imaging shows that the thiophene dyes tend to accumulate in the pores of the alginate beads.
Aggregation is known to drastically reduce the singlet oxygen production efficiency79 primarily due to triplet–triplet annihilation. However, the persistence of the type I mechanism could be attributed to the specific chemical environment. Electron-rich environments may facilitate the photoactivation switch from type II to type I mechanisms. This effect can be observed when photosensitizers are dispersed in micelles80 or conjugated to proteins.81 A sacrificial electron donor is generally required to activate the type I mechanism.82 The environment inside the beads is extremely electron rich, due to the presence of the carboxylate groups in the alginate matrix. We investigated computationally the possibility to form an anion radical, in the case of BTO2-Tz2 (Fig. S7‡). The excited state of BTO2-Tz2 can receive an electron from the carboxylates, forming the anion radical (Fig. S8‡), while the alginate undergoes decarboxylation. The production of a CO2 molecule is the driving force (entropy-driven reaction) of the process and makes it exothermic. In this case, the formation of ROS via the type I mechanism, is due to an electron transfer process involving the ground state of the oxygen molecule rather than the short-lived 1O2. In fact, negatively charged radicals can react with oxygen, eventually leading to H2O2 formation in an aqueous environment.83
Photodegradation experiments
All Alg–BT aerogels were tested for the photodegradation of the three different ECs, i.e. RhB, BPA and OFLOX (Fig. 6a), under blue visible light with λmax = 460 nm, and the experimental set-up is shown in Fig. 6b–d. A solution of the given contaminant was gently stirred in the presence of Alg–BT aerogels for a total of 5 h and samples were collected every hour to perform HPLC analysis detecting the concentration of the ECs and allowing for the calculation of the degradation percentage (details in ESI, Section 5‡). In detail, the degradation percentage was calculated as a ratio between the detected contaminant concentration after the photodegradation cycle and the initial spiked solution concentration. Control samples of each contaminant without photocatalysts were treated under the same conditions to evaluate the potential effect of interaction between radiation and the water-soluble contaminant molecule. The possible adsorption of the three contaminants by alginate beads has been excluded in previously reported experiments.70Fig. 7 shows the EC degradation percentage as a function of irradiation time. RhB, BPA and OFLOX showed negligible degradation without photocatalysts, proving the photostability of these ECs under the experimental conditions used. | ||
| Fig. 6 (a) Chemical structure of the selected emerging contaminants (ECs), (b) photoreactor used in degradation experiments with LED off and (c) LED on. (d) Alg–BT aerogels (Alg–BTO2-T2 in the image) were suspended in contaminant spiked water (RhB case study in the image) and irradiated with blue visible light at room temperature. | ||
 | ||
| Fig. 7 % Degradation of (a) RhB, (b) BPA, and (c) OFLOX obtained by Alg–BT aerogels (V = 3 mL, CIN = 4 mg L−1, 5 mg of photocatalysts). | ||
The results obtained from the five photocatalysts (Alg–BT-T2, Alg–BTO-T2, Alg–BTO2-T2, Alg–T2(BTO2)3, and Alg–BTO2-Tz2) showed a common trend for all the tested ECs. Alg–BTO2-Tz2 reached a photodegradation percentage higher than 99% within 2 h of contact time for RhB and OFLOX and 3 h for BPA, making it the fastest photocatalyst among those tested. Alg–BTO2-T2 and Alg–T2(BTO2)3 showed comparable performance towards RhB and OFLOX, achieving complete degradation in 4 h and 3 h of contact time, respectively. For BPA, Alg–T2(BTO2)3 showed higher degradation capacity, reaching 85% in 5 h, compared to Alg–BTO2-T2 which showed a slightly lower degradation percentage (65%). The best performances in the degradation of the three ECs tested, were achieved by using Alg–BTO2-Tz2 and Alg–BTO2-T2 with the trend that closely matches the amount of ROS generated by the Alg–BT aerogels, reported in Fig. 5c. The ability to reuse photocatalytic materials is fundamental for reducing waste generation and process costs. With the aim of assessing the potential of recycling the photocatalysts for multiple reuses, the stability of the best performing Alg–BTs aerogels (Alg–BTO2-T2, Alg–T2(BTO2)3, and Alg–BTO2-Tz2) was investigated (Fig. 8a).
 | ||
| Fig. 8 (a) Sketch of the photodegradation (irradiated at λ = 460 nm) and reuse process (aerogels washed with deionized water). (b) Recycling of Alg–BTO2-T2, Alg–T2(BTO2)3 and Alg–BTO2-Tz2 aerogels on RhB showed as a case study (BPA/OFLOX, in Fig. S9, ESI‡). | ||
Photodegradation of RhB, BPA and OFLOX was performed for five consecutive runs. At the end of each cycle, the floating aerogels were quickly recovered from the solution by using a small colander and reused for the next run after washing with deionized water.
Interestingly, after five consecutive runs, no significant change was observed in the degradation efficiency of RhB for any of the BT photocatalysts, with degradation percentages ranging from 89% to 96%. A similar trend was observed for the other pollutants studied: specifically, OFLOX was completely degraded (98–99%), and BPA degradation ranged from 59% to 65% for Alg–BTO2-T2, from 79% to 85% for Alg–T2(BTO2)3, and from 93% to 96% for Alg–BTO2-Tz2 (Fig. S9, ESI‡).
Ecotoxicity experiments
Water solutions of OFLOX were analysed with the bioluminescent bacterium A. fischeri to assess the possible intrinsic acute ecotoxicity and, in the case of irradiated solutions, the ecotoxicity of degradation by-products. The bioluminescent bacterium A. fischeri, was exposed for 5, 15 and 30 minutes to solutions in accordance with the standardised method and the experimental details are reported in Section 6, ESI.‡62The effects are considered toxic if the inhibition of bioluminescence is >20% (toxicity threshold) compared to the light emission of the bacterium under optimal conditions (in saline solution, 20 g per L NaCl).84 The effects are reported as average values % of bioluminescence inhibition (±standard error). The 30-min toxicity results are shown in Fig. 9; results for all exposure times (5, 15 and 30 min) are reported in Table S4 in ESI.‡ All solutions showed no toxic effects at any of the exposure times on A. fischeri. Indeed, bioassays on the OFLOX antibiotic and its irradiated solutions showed values below the ecotoxicity threshold (<20%), with bioluminescence inhibition values ranging between 0% and 11.64%.
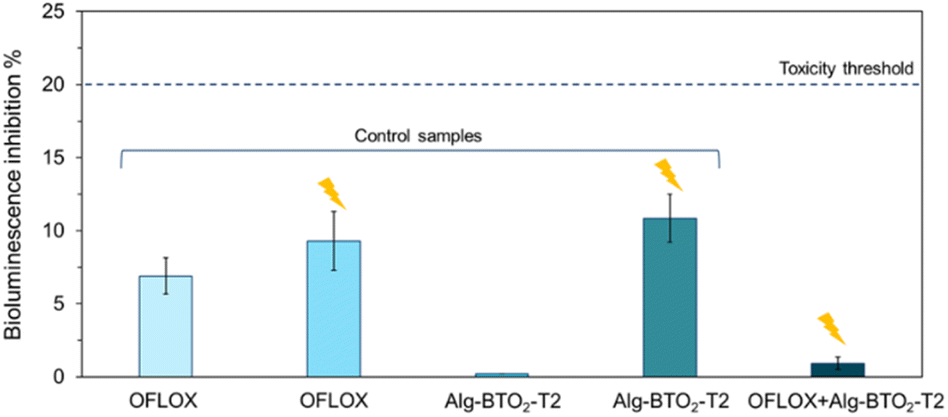 | ||
| Fig. 9 A. fischeri bioluminescence inhibition% at 30 min induced by solutions containing (from left to right): OFLOX kept in the absence of light, OFLOX irradiated, Alg–BTO2 in the absence of light, Alg–BTO2 irradiated, and OFLOX irradiated in the presence of Alg–BTO2-T2 aerogels. The flash highlights the irradiated samples. | ||
Consequently, the irradiated antibiotic with and without the photocatalyst displayed no ecotoxicity. In particular, OFLOX in water displays a bioluminescence inhibition of 6.91% ± 1.23%, and the antibiotic irradiated in the presence of the photocatalyst (OFLOX + Alg–BTO2-T2 + UV) displayed even lower inhibition (0.93% ± 0.42%). This means that the photodegradative by-products were not toxic, showing even lower inhibition compared to the starting OFLOX solution. In addition to OFLOX solutions, the test was also performed on ultrapure water left in contact with the Alg–BTO2-T2 aerogel in the absence of light. In this case, the solution proved to be non-toxic and no bioluminescence inhibition was observed. This confirms that the BT molecules are stable and are not released into water, once encapsulated in the alginate matrices. Finally, Alg–BTO2-T2 aerogels in water were irradiated in the absence of OFLOX as a control, and the resulting solution (Alg–BTO2-T2 + UV) was analysed under the same conditions. The solution was not toxic being below the 20% threshold but displayed the highest bioluminescence inhibition (10.86% ± 1.65%) compared to the other samples. Probably, in the absence of contaminants to be degraded, the radicals formed during irradiation cause oxidative stress to A. fischeri.
Conclusions
A family of V-shaped oligothiophenes, derived from a benzo[b]thiophene (BT) core and S-oxide or S,S-dioxide moieties, was synthesized by the Stille cross-coupling reaction, with yields in the range of 67–83%. The optical properties of BT molecules were studied by spectroscopy and DFT calculations. Upon blue-light irradiation, the targeted BT molecules demonstrated the ability to generate ROS following both type I and type II mechanisms. BT molecules were encapsulated in an alginate matrix by ionotropic gelation–freeze drying, resulting in porous aerogels that were dispersed in water and tested in the photodegradation of three emerging contaminants, i.e., RhB, BPA and OFLOX. Among the synthesized alginate–BT aerogels, Alg–BTO2-Tz2 proved to be the best performing photocatalyst, achieving complete degradation of all the three ECs within 2 h, followed by Alg–BTO2-T2 and Alg–T2(BTO2)3, which still showed high efficiency (complete degradation in less than 5 h). This trend perfectly reproduces the amount of ROS generated by the Alg–BT aerogels and the obtained results highlight the important role of the S,S-dioxide moiety in the generation of ROS. The stability of the aerogels was evaluated by performing five photodegradation cycles with the same photocatalysts, showing no noticeable change in degradation efficiency, proving the reusability of the photocatalysts. Furthermore, an ecotoxicological bioassay using Aliivibrio fischeri was performed on the OFLOX antibiotic and its photodegradation by-products. Both solutions showed no toxicity (values below the threshold: inhibition of bioluminescence < 20%), and the photodegradation by-products displayed less bioluminescence inhibition in the bacterium, lower than the initial OFLOX solution. This work demonstrates the possibility of exploiting oligothiophenes as photocatalysts and sheds some light on the structure–properties relationships in S-oxide and S,S-dioxide based materials, opening new perspectives for water treatment and the degradation of persistent emerging contaminants.Data availability
Detailed description of materials synthesis and characterization, theoretical calculations, light-induced reactive oxygen species quantification, photodegradation experiments, high performance liquid chromatography analyses, and ecotoxicity experiments.Author contributions
Andrea Trifoglio: methodology, investigation, data curation, editing. Francesca Tunioli: methodology, investigation, data curation, editing. Laura Favaretto: methodology, investigation. Massimo Zambianchi: methodology, investigation. Cristian Bettini: methodology, investigation. Ilse Manet: methodology, investigation, formal analysis. Livia Mariani: methodology, investigation. Anna Barra Caracciolo: methodology, investigation. Paola Grenni: methodology, investigation. Manuele Di Sante: investigation, methodology, formal analysis. Matteo Di Giosia: investigation, methodology, formal analysis. Tainah Dorina Marforio: investigation, methodology, formal analysis. Edoardo Jun Mattioli: investigation, methodology, formal analysis. Matteo Calvaresi: investigation conceptualization. Manuela Melucci: conceptualization, validation, writing – original draft.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge the support for this work by the projects Life-Remembrance, ENV/IT/001001 Life Resource and Environment LIFE20 ‘Give plastic wastes from the production of hollow-fiber membranes a second life’, project PNRR MUR project ECS_00000033_ECOSISTER, and ‘Le materie prime del futuro da fonti non-critiche, residuali e rinnovabili’ (FUTURAW, FOE 2022). Edoardo Jun Mattioli was supported by Fondazione Umberto Veronesi.Notes and references
- F. Xiao, B. Deng, D. Dionysiou, T. Karanfil, K. O'Shea, P. Roccaro, Z. J. Xiong and D. Zhao, Nat. Water, 2023, 1, 1004–1015 CrossRef.
- B. Rathi, P. Kumar and D.-V. Vo, Sci. Total Environ., 2021, 797, 149134 CrossRef CAS PubMed.
- X. Tong, S. Mohapatra, J. Zhang, N. H. Tran, L. You, Y. He and K. Y.-H. Gin, Water Res., 2022, 217, 118418 CrossRef CAS.
- O. S. Arvaniti and A. S. Stasinakis, Sci. Total Environ., 2015, 524–525, 81–92 CrossRef CAS.
- https://www.epa.gov/sdwa/and-polyfluoroalkyl-substances-pfas .
- P. Thamarai, R. Kamalesh, A. Saravanan, P. Swaminaathan and V. C. Deivayanai, Environ. Nanotechnol., Monit. Manage., 2024, 21, 100913 CAS.
- H. Azzahra, A. F. Nisaa and M. A. Mardyanto, IOP Conf. Ser. Earth Environ. Sci., 2024, 1307, 012013 CrossRef.
- D. Yadav, S. Karki and P. G. Ingole, J. Environ. Chem. Eng., 2022, 10, 108109 CrossRef CAS.
- M. M. Zubair, H. Saleem and S. J. Zaidi, Desalination, 2023, 564, 116705 CrossRef CAS.
- S. Feijoo, X. Yu, M. Kamali, L. Appels and R. Dewil, Rev. Environ. Sci. Bio/Technol., 2023, 22, 205–248 CrossRef CAS.
- J. Yuan, E. Passeport and R. Hofmann, Water Res., 2022, 210, 118026 CrossRef CAS.
- E. Menya, J. Jjagwe, H. M. Kalibbala, H. Storz and P. W. Olupot, Chem. Eng. Res. Des., 2023, 192, 412–440 CrossRef CAS.
- A. Katsigiannis, C. Noutsopoulos, J. Mantziaras and M. Gioldasi, Chem. Eng. J., 2015, 280, 49–57 CrossRef CAS.
- N. Singh, G. Nagpal and S. Agrawal, Environ. Technol. Innovation, 2018, 11, 187–240 CrossRef.
- M. N. Soliman, F. Z. Guen, S. A. Ahmed, H. Saleem, M. J. Khalil and S. J. Zaidi, Process Saf. Environ. Prot., 2021, 147, 589–608 CrossRef CAS.
- D. Van Thuan, H. L. Ngo, H. P. Thi and T. T. H. Chu, Environ. Res., 2023, 229, 116000 CrossRef CAS PubMed.
- H. C. Yap, Y. L. Pang, S. Lim, A. Z. Abdullah, H. C. Ong and C. H. Wu, Int. J. Environ. Sci. Technol., 2019, 16, 601–628 CrossRef CAS.
- H. Kumari, S. Sonia, R. Ranga, S. Chahal, S. Devi, S. Sharma, S. Kumar, P. Kumar, S. Kumar, A. Kumar and R. Parmar, Water, Air, Soil Pollut., 2023, 234, 349 CrossRef CAS PubMed.
- S. Peiris, H. B. de Silva, K. N. Ranasinghe, S. V. Bandara and I. R. Perera, J. Chin. Chem. Soc., 2021, 68, 738–769 CrossRef CAS.
- S. Murgolo, S. Franz, H. Arab, M. Bestetti, E. Falletta and G. Mascolo, Water Res., 2019, 164, 114920 CrossRef CAS.
- M. N. Chong, B. Jin, C. W. K. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS.
- S.-Y. Lee and S.-J. Park, J. Ind. Eng. Chem., 2013, 19, 1761–1769 CrossRef CAS.
- V. Prashanth, P. Jayasree, P. Rajput and N. Remya, in Advanced Oxidation Processes for Effluent Treatment Plants, ed. M. P. Shah, Elsevier, 2021, pp. 69–85, DOI:10.1016/B978-0-12-821011-6.00004-9.
- S. Ramu, I. Kainthla, L. Chandrappa, J. M. Shivanna, B. Kumaran and R. G. Balakrishna, Environ. Sci. Pollut. Res. Int., 2024, 31, 167–190 CrossRef CAS.
- X. Li, K. Kawai, M. Fujitsuka and Y. Osakada, Surf. Interfaces, 2021, 25, 101249 CrossRef CAS.
- Y. Feng, X. Su, Y. Chen, Y. Liu, X. Zhao, C. Lu, Y. Ma, G. Lu and M. Ma, Mater. Res. Bull., 2023, 162, 112207 CrossRef CAS.
- C. Santhosh, V. Velmurugan, G. Jacob, S. K. Jeong, A. N. Grace and A. Bhatnagar, Chem. Eng. J., 2016, 306, 1116–1137 CrossRef CAS.
- A. Feliczak-Guzik, Materials, 2023, 16, 193 CrossRef CAS.
- Q. Wang, Q. Gao, A. M. Al-Enizi, A. Nafady and S. Ma, Inorg. Chem. Front., 2020, 7, 300–339 RSC.
- L. Gan, M. T. Nord, J. M. Lessard, N. Q. Tufts, A. Chidambaram, M. E. Light, H. Huang, E. Solano, J. Fraile, F. Suárez-García, C. Viñas, F. Teixidor, K. C. Stylianou and J. G. Planas, J. Am. Chem. Soc., 2023, 145, 13730–13741 CrossRef CAS.
- H. Jia, D. Ma, S. Zhong, L. Li, L. Li, L. Xu and B. Li, Chem. Eng. J., 2019, 368, 165–174 CrossRef CAS.
- S. Silvestri, A. R. Fajardo and B. A. Iglesias, Environ. Chem. Lett., 2022, 20, 731–771 CrossRef CAS.
- K. S. Min, R. Manivannan and Y.-A. Son, Dyes Pigm., 2019, 162, 8–17 CrossRef CAS.
- M. Moosavifar, S. M. Heidari, L. Fathyunes, M. Ranjbar, Y. Wang and H. Arandiyan, J. Inorg. Organomet. Polym. Mater., 2020, 30, 1621–1628 CrossRef CAS.
- H. Ozawa, S. Kusaba, M. Matsunaga and M.-a. Haga, Langmuir, 2018, 34, 2952–2958 CrossRef CAS PubMed.
- L.-S. Wan, J. Wu and Z.-K. Xu, Macromol. Rapid Commun., 2006, 27, 1533–1538 CrossRef CAS.
- Z.-L. Ma, G.-F. Huang, D.-S. Xu, M.-G. Xia, W.-Q. Huang and Y. Tian, Mater. Lett., 2013, 108, 37–40 CrossRef CAS.
- D. M. Mafukidze and T. Nyokong, J. Photochem. Photobiol., A, 2021, 409, 113142 CrossRef CAS.
- W.-j. Sun, J. Li, G.-p. Yao, F.-x. Zhang and J.-L. Wang, Appl. Surf. Sci., 2011, 258, 940–945 CrossRef CAS.
- R. Rahimi, S. Zargari, A. Ghaffarinejad and A. Morsali, Environ. Prog. Sustainable Energy, 2016, 35, 642–652 CrossRef CAS.
- A. Gogoi, P. Mazumder, V. K. Tyagi, G. G. Tushara Chaminda, A. K. An and M. Kumar, Groundw. Sustain. Dev., 2018, 6, 169–180 CrossRef.
- A. Tkaczyk, K. Mitrowska and A. Posyniak, Sci. Total Environ., 2020, 717, 137222 CrossRef CAS PubMed.
- T. L. Yusuf, B. O. Orimolade, D. Masekela, B. Mamba and N. Mabuba, RSC Adv., 2022, 12, 26176–26191 RSC.
- J. Sharma, S. Sharma, U. Bhatt and V. Soni, J. Hazard. Mater. Lett., 2022, 3, 100069 CrossRef CAS.
- Y. Qian, X. Jia, T. Ding, M. Yang, B. Yang and J. Li, Sci. Total Environ., 2021, 758, 143606 CrossRef CAS.
- L. E. Lesser, A. Mora, C. Moreau, J. Mahlknecht, A. Hernández-Antonio, A. I. Ramírez and H. Barrios-Piña, Chemosphere, 2018, 198, 510–521 CrossRef CAS PubMed.
- P. Kovalakova, L. Cizmas, T. J. McDonald, B. Marsalek, M. Feng and V. K. Sharma, Chemosphere, 2020, 251, 126351 CrossRef CAS.
- G. Barbarella, L. Favaretto, A. Zanelli, G. Gigli, M. Mazzeo, M. Anni and A. Bongini, Adv. Funct. Mater., 2005, 15, 664–670 CrossRef CAS.
- G. Barbarella, L. Favaretto, G. Sotgiu, M. Zambianchi, V. Fattori, M. Cocchi, F. Cacialli, G. Gigli and R. Cingolani, Adv. Mater., 1999, 11, 1375–1379 CrossRef CAS.
- C. Santato, L. Favaretto, M. Melucci, A. Zanelli, M. Gazzano, M. Monari, D. Isik, D. Banville, S. Bertolazzi, S. Loranger and F. Cicoira, J. Mater. Chem., 2010, 20, 669–676 RSC.
- R. Peng, Y. Luo, Q. Cui, J. Wang and L. Li, ACS Appl. Bio Mater., 2020, 3, 1305–1311 CrossRef CAS PubMed.
- A. Cantelli, M. Malferrari, A. Soldà, G. Simonetti, S. Forni, E. Toscanella, E. J. Mattioli, F. Zerbetto, A. Zanelli, M. Di Giosia, M. Zangoli, G. Barbarella, S. Rapino, F. Di Maria and M. Calvaresi, JACS Au, 2021, 1, 925–935 CrossRef CAS PubMed.
- M. Piacenza, M. Zambianchi, G. Barbarella, G. Gigli and F. Della Sala, Phys. Chem. Chem. Phys., 2008, 10, 5363–5373 RSC.
- R. A. Raus, W. M. F. W. Nawawi and R. R. Nasaruddin, Asian J. Pharm. Sci., 2021, 16, 280–306 CrossRef.
- Y. Sun, T. Zhou, W. Li, F. Yu and J. Ma, Chemosphere, 2020, 241, 125110 CrossRef CAS.
- D. Eluk, O. Nagel, A. Gagneten, U. Reno and R. Althaus, Water Environ. Res., 2021, 93, 2914–2930 CrossRef CAS PubMed.
- T.-D. Nguyen, T. Itayama, R. Ramaraj, N. Iwami, K. Shimizu, T.-S. Dao, T.-L. Pham and H. Maseda, Environ. Pollut., 2021, 291, 118095 CrossRef CAS PubMed.
- T.-D. Nguyen, T. Itayama, N. Iwami, K. Shimizu, T.-S. Dao, T. L. Pham, V. Q. Tran and H. Maseda, Drug Chem. Toxicol., 2023, 47(5), 662–673 CrossRef.
- S. Jouanneau, M.-J. Durand-Thouand and G. Thouand, Environ. Sci. Pollut. Res., 2016, 23, 4340–4345 CrossRef CAS.
- S. Parvez, C. Venkataraman and S. Mukherji, Environ. Int., 2006, 32, 265–268 CrossRef CAS.
- M. Abbas, M. Adil, S. Ehtisham-ul-Haque, B. Munir, M. Yameen, A. Ghaffar, G. A. Shar, M. Asif Tahir and M. Iqbal, Sci. Total Environ., 2018, 626, 1295–1309 CrossRef CAS.
- L. Mariani, P. Grenni, A. Barra Caracciolo, E. Donati, J. Rauseo, L. Rolando and L. Patrolecco, Ecotoxicology, 2020, 29, 815–824 CrossRef CAS.
- H. Shimotani, S. Ikeda, N. Asao, Y. Yamamoto, K. Tanigakiab and T. Jin, Chem. Commun., 2016, 52, 4926–4929 RSC.
- J. Hassan, L. Lavenot, C. Gozzi and M. Lemaire, Tetrahedron Lett., 1999, 40, 857–858 CrossRef CAS.
- M. J. T. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian16, Wallingford, CT, 2016 Search PubMed.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- S. Canola, L. Mardegan, G. Bergamini, M. Villa, A. Acocella, M. Zangoli, L. Ravotto, S. A. Vinogradov, F. Di Maria and P. Ceroni, Photochem. Photobiol. Sci., 2019, 18, 2180–2190 CrossRef CAS PubMed.
- B. Wang, Y. Wan, Y. Zheng, X. Lee, T. Liu, Z. Yu, J. Huang, Y. S. Ok, J. Chen and B. Gao, Crit. Rev. Environ. Sci. Technol., 2019, 49, 318–356 CrossRef CAS.
- F. Tunioli, S. Khaliha, S. Mantovani, A. Bianchi, A. Kovtun, Z. Xia, M. S. S. Bafqi, B. S. Okan, T. D. Marforio, M. Calvaresi, V. Palermo, M. L. Navacchia and M. Melucci, J. Environ. Chem. Eng., 2023, 11, 109566 CrossRef CAS.
- E. Turrini, L. Ulfo, P. E. Costantini, R. Saporetti, M. Di Giosia, M. Nigro, A. Petrosino, L. Pappagallo, A. Kaltenbrunner, A. Cantelli, V. Pellicioni, E. Catanzaro, C. Fimognari, M. Calvaresi and A. Danielli, Cell. Mol. Life Sci., 2024, 81, 144 CrossRef CAS.
- A. Petrosino, R. Saporetti, F. Starinieri, E. Sarti, L. Ulfo, L. Boselli, A. Cantelli, A. Morini, S. K. Zadran and G. Zuccheri, iScience, 2023, 26, 108032 CrossRef CAS.
- B. Bortot, M. Apollonio, G. Baj, L. Andolfi, L. Zupin, S. Crovella, M. di Giosia, A. Cantelli, R. Saporetti, L. Ulfo, A. Petrosino, G. Di Lorenzo, F. Romano, G. Ricci, M. Mongiat, A. Danielli, M. Calvaresi and S. Biffi, Free Radical Biol. Med., 2022, 179, 242–251 CrossRef CAS.
- L. Ulfo, A. Cantelli, A. Petrosino, P. E. Costantini, M. Nigro, F. Starinieri, E. Turrini, S. K. Zadran, G. Zuccheri and R. Saporetti, Nanoscale, 2022, 14, 632–641 RSC.
- M. Di Sante, A. Kaltenbrunner, M. Lombardo, A. Danielli, P. E. Costantini, M. Di Giosia and M. Calvaresi, Pharmaceuticals, 2023, 16, 1329 CrossRef CAS.
- A. Marconi, G. Giugliano, M. Di Giosia, T. D. Marforio, M. Trivini, E. Turrini, C. Fimognari, F. Zerbetto, E. J. Mattioli and M. Calvaresi, Pharmaceutics, 2023, 15, 919 CrossRef CAS PubMed.
- G. Greco, L. Ulfo, E. Turrini, A. Marconi, P. E. Costantini, T. D. Marforio, E. J. Mattioli, M. Di Giosia, A. Danielli and C. Fimognari, Cells, 2023, 12, 392 CrossRef CAS.
- E. J. Mattioli, L. Ulfo, A. Marconi, V. Pellicioni, P. E. Costantini, T. D. Marforio, M. Di Giosia, A. Danielli, C. Fimognari and E. Turrini, Biomolecules, 2022, 13, 68 CrossRef PubMed.
- C. Alvarez-Lopez, D. Cavazos-Elizondo, B. Heyne, I. E. Kochevar and A. Aguirre-Soto, Photochem. Photobiol., 2023, 99, 580–592 CrossRef CAS.
- H. Ding, H. Yu, Y. Dong, R. Tian, G. Huang, D. A. Boothman, B. D. Sumer and J. Gao, J. Controlled Release, 2011, 156, 276–280 CrossRef CAS PubMed.
- A. Cantelli, F. Piro, P. Pecchini, M. Di Giosia, A. Danielli and M. Calvaresi, J. Photochem. Photobiol., B, 2020, 206, 111852 CrossRef CAS PubMed.
- P. Sanchez-Cruz, F. Dejesus-Andino and A. E. Alegria, J. Photochem. Photobiol., A, 2012, 236, 54–60 CrossRef CAS.
- K. Plaetzer, B. Krammer, J. Berlanda, F. Berr and T. Kiesslich, Lasers Med. Sci., 2009, 24, 259–268 CrossRef CAS PubMed.
- G. Persoone, B. Marsalek, I. Blinova, A. Törökne, D. Zarina, L. Manusadzianas, G. Nalecz-Jawecki, L. Tofan, N. Stepanova, L. Tothova and B. Kolar, EnTox, 2003, 18, 395–402 CAS.
Footnotes |
| † In memory of Giovanna Barbarella. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta06689h |
| § These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |