
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural and thermodynamic analysis of triple conducting ceramic materials BaCo0.4Fe0.4Zr0.2−XYXO3−δ†
Jack H.
Duffy‡
 a,
Nancy
Birkner‡
a,
Chiyoung
Kim
b,
Ryan
Jacobs
b,
Dane
Morgan
b,
Shivani
Sharma
c,
Scott T.
Misture
c,
Erik M.
Kelder
d,
Harry W.
Abernathy
e and
Kyle S.
Brinkman
*ae
a,
Nancy
Birkner‡
a,
Chiyoung
Kim
b,
Ryan
Jacobs
b,
Dane
Morgan
b,
Shivani
Sharma
c,
Scott T.
Misture
c,
Erik M.
Kelder
d,
Harry W.
Abernathy
e and
Kyle S.
Brinkman
*ae
aDepartment of Materials Science and Engineering, Clemson University, Clemson, South Carolina 29634, USA. E-mail: ksbrink@clemson.edu
bDepartment of Materials Science and Engineering, University of Wisconsin–Madison, Madison, Wisconsin 54706, USA
cKazuo Inamori School of Engineering, Alfred University, Alfred, New York 14802, USA
dDepartment of Radiation Science & Technology, Delft University of Technology, 2629 JB Delft, The Netherlands
eNational Energy Technology Laboratory, United States Department of Energy, Morgantown, West Virginia 26507, USA
First published on 5th March 2025
Abstract
Triple ionic–electronic conductors, capable of concurrent conduction of protons, oxygen ions, and electrons, are promising cathodes in ceramic fuel cells. Though thoroughly studied, extensive evaluation and explanation of their transport phenomena are crucial for guiding future research. In this work, the structure, composition, and formation enthalpies of BaCo0.4Fe0.4Zr0.2−XYXO3−δ (BCFZYX, X = 0, 0.05, 0.1, 0.15, 0.2) are correlated with its trends in defect mobility. Three compositions of BCFZYX, X = 0, 0.1, 0.2, are measured under X-ray diffraction and neutron powder diffraction to reveal a common cubic perovskite structure with increasing oxygen vacancy concentration upon increasing yttrium concentration. Formation enthalpies obtained by high-temperature calorimetry reveal a general destabilization of this perovskite structure with yttrium substitution, with deviations at the endmember compositions. Further analysis suggests that high yttrium substitution causes yttrium–vacancy pairs to be more energetically favorable than randomized oxygen vacancies. This phenomenon, along with changes in oxygen vacancy concentration, helps explain observed trends in oxygen-ion and proton mobility in the compositional suite as well as criteria for the selection of high performing, durable protonic ceramic fuel cell materials.
1. Introduction
The design of efficient materials for energy conversion and storage applications is a critical challenge facing modern materials science.1–3 Perovskite ceramic materials, including mixed ionic–electronic conductors, have shown great potential for use in gas separation membranes, fuel cells, and catalysis.4–8 Among these, triple ionic–electronic conductors (TIECs), known to concurrently transport protons, oxide ions, and electrons under humid oxidizing conditions, are especially interesting due to their high ionic transport at low-to-intermediate temperatures.Among relevant TIECs, BaCo0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY0.1) and its closely related derivatives are among the most widely studied in the field.9 It is known that BCFZY0.1 can incorporate a wide range of metal ion dopants10,11 and substitutions,12–16 as well as withstand broad changes in metal-ion and oxygen non-stoichiometry.17,18 Our recent work has focused on changing the dopant levels of Zr and Y in the BaCo0.4Fe0.4Zr0.2−XYXO3−δ (BCFZYX) compositional series to understand the dopant effects on ionic and electronic mobility and surface kinetics of these materials.19,20 Shin, et al. have performed a similar study focusing on varying the levels of multivalent dopants Co and Fe to understand similar phenomena.21 Focusing on incremental changes in dopant levels helps reveal the properties which produce high-performance cathode materials in oxide-based fuel cells.
The properties of TIEC materials, including defect mobility and surface exchange reactions, can be significantly affected by oxygen vacancies22–28 and structural changes.29–32 Understanding these effects is crucial for the design and optimization of new materials with improved properties. Neutron powder diffraction (NPD) is a powerful tool to understand the structure of these materials. NPD has an additional benefit of probing low atomic weight elements, like lithium and oxygen, unlike traditional X-ray diffraction (XRD). Because of this resolution, the absolute oxygen vacancy concentration can be measured, which is important for TIEC materials with multivalent cations such as Co and Fe, as the vacancy concentration cannot be calculated without knowing the valence state.33,34 Combined with relative probing of vacancy concentration using methods like thermogravimetric analysis (TGA), the nominal composition of these materials can be elucidated throughout its working temperature ranges.
A method used to correlate the structure, defect chemistry, and mobility properties of solid oxide materials is the measurement of enthalpy of formation from the oxides by high temperature oxide melt solution calorimetry.35 Previous efforts have correlated interesting property changes with respect to composition in perovskites,36 fluorites,37 and other-structured oxide ceramics.38,39 A general trend is emerging which suggests defect mobility and thermodynamic stability are negatively correlated up to a certain level of substitution, after which structural changes inhibit the further improvement of mobility throughout these complex systems. Indeed, this work supports this trend.
In this study, we present the trends in structure, oxygen vacancy concentration, and enthalpy of formation from the oxides with respect to yttrium concentration. NPD revealed the increase in oxygen vacancy concentration with increases in aliovalent doping. Enthalpy of formation from the oxides was measured using high temperature oxide melt solution calorimetry. These thermodynamic data give new insights to the defect chemistry of BCFZYX; changes in trends in the enthalpy of formation from the oxides in this BCFZYX perovskite series suggest causes for the optimum concentration for surface and bulk-level defect mobility, which, among the compositions studied, occurs at 10% B-site doping Y3+ for Zr4+.
2. Methods
2.1 Material synthesis
BaCo0.4Fe0.4Zr0.2−XYXO3−δ (BCFZYX) materials with 0 ≤ X ≤ 0.2 were synthesized using a wet chemistry sol–gel method.40 Stoichiometric amounts of precursor nitrates were dissolved in distilled water. Citric acid and EDTA were then added to the solution in a ratio of citric acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EDTA:metal of 1.5:1.5:1 as chelating agents. While stirring, ammonium hydroxide was added to the solution until the solution achieved a pH of 9. The solution was heated at 80 °C until a gel formed and was subsequently placed in an oven at 150 °C for 36 h. The resultant complex was fired at 1000 °C for 10 h to remove organic residues and calcine the perovskite phase.
:EDTA:metal of 1.5:1.5:1 as chelating agents. While stirring, ammonium hydroxide was added to the solution until the solution achieved a pH of 9. The solution was heated at 80 °C until a gel formed and was subsequently placed in an oven at 150 °C for 36 h. The resultant complex was fired at 1000 °C for 10 h to remove organic residues and calcine the perovskite phase.
To ensure full calcination of the perovskite phase, membranes of each composition were synthesized by pressing the powder at 160 MPa for two minutes into 15 mm diameter pellets. Pellets were covered with their own calcined powder to reduce the potential for Ba loss during sintering. All samples were sintered at 1275 °C for 8 h in air to form dense membranes. These membranes were subsequently crushed for future measurements.
2.2 Structural characterization
The phase assemblages of all the samples were measured using powder XRD (Hitachi SmartLab Powder Diffractometer) equipped with Cu Kα radiation. NPD was also used to characterize BaCo0.4Fe0.4Zr0.2O3−δ (BCFZ), BaCo0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY0.1), and BaCo0.4Fe0.4Y0.2O3−δ (BCFY) at ambient and high temperature using the PEARL diffractometer available at Delft University of Technology.41 The constant-wavelength diffractometer employs a Ge monochromator and wavelength of λ = 1.667 Å was used. Samples were measured at RT and at 800 K. Samples consisted of sintered pellets that were crushed and ground, loaded in a cylindrical vanadium sample can (50 mm length, 6 mm inner diameter) and sealed under air. The NPD patterns were recorded over a 2θ range from 11° to 158°. Each diffraction pattern was collected for about 3 hours. Rietveld refinements of XRD and NPD data were performed using the software JANA 2006.422.3 Thermogravimetry
Thermogravimetric analysis (TGA) was performed using a PerkinElmer TGA8000. Samples were measured to determine the oxygen loss characteristics of each sample at high temperatures. Approximately 20 mg of powdered, sintered pellets were utilized for the measurement. Samples were loaded and the temperature was increased to 200 °C at a heating rate of 50 °C min−1 and held for 30 minutes to remove adsorbed water from the surface of the material. Next, the temperature was increased to 950 °C at a heating rate of 10 °C min−1. This measurement was performed in both air and nitrogen as the purge gas to quantify the weight loss in oxidizing and reducing conditions.2.4 Quantification of chemical composition
The metals content of each sample was measured using ICP-MS (Thermo X Series II). Approximately 10 mg of each BCFZYX powder was dissolved in 10 mL of 2% HNO3 to give solutions containing 10–500 ng mL−1 of each element. Calibration standards were prepared in 2% HNO3 by diluting single-element plasma standards. The ICP-MS analysis yielded a limit of detection (LOD) of 0.066, 0.027, 0.376, 0.064, and 0.012 ng mL−1 for 137Ba, 59Co, 56Fe, 90Zr, and 89Y, respectively. Measured concentrations of each ion in the BCFZYX solutions were always one to three orders of magnitude larger than the LODs. Each dissolution and each analysis was repeated in triplicate, except for BCFZ which was repeated in duplicate, which yielded standard deviations of the calculated molar ratios of less than 1%. Oxygen content in the samples is estimated from thermogravimetric analysis and Rietveld analysis of the BCFZYX powders measured via NPD.2.5 High-temperature oxide melt solution calorimetry
Formation enthalpy is a fundamental thermodynamic property which may be used as a gauge of phase stability. Calorimetry is the only direct means to access the formation enthalpy from the oxides and the standard heats of formation from the elements, which it does through the application of appropriate thermochemical cycles. In this method, the total heat effect of drop solution (ΔHds) includes the heat of solution (ΔHsln) and heat content (ΔHT) of the sample. The drop solution enthalpy for each sample was measured using an AlexSys 1000 (Setaram®) superbill twin Tian-Calvet high temperature solution calorimeter at 700 °C. Hand-pressed sample pellets (nominally 4–6 mg) were dropped from room temperature into molten sodium molybdate (3Na2O–4MoO3) (20 g) solvent. Oxygen was flushed through the glassware assembly at 40 mL min−1 and bubbled through the solvent at 4–5 mL min−1. Flushing ensures constant vapor pressure above the solvent, while bubbling aids dissolution. Experiments were replicated for each sample to achieve statistical relevance (typically not less than 8 measurements) which produced the mean enthalpy of drop solution (ΔHds). The reported mean measurement uncertainties were computed as two standard deviations. Calibration was done against the known heat content of trace metal grade alpha-alumina (Sigma Aldrich, 99.999%). The calorimetry and calibration methods have been described previously.43–462.6 Computational methods
To calculate the enthalpy of formation from oxides of BCFZYX materials, spin-polarized DFT calculations were conducted using the Vienna Ab Initio Simulation Package (VASP).47 The calculations employed the generalized gradient approximation (GGA) for the exchange–correlation functional, incorporating a Hubbard U correction within the PBE framework.48–50 The U values in PBE + U method were consistent as those from the Materials Project:51U = 5.3 eV for Fe and 3.32 eV for Co. Core electrons were described using projector augmented wave (PAW) pseudopotentials.52 The calculations were performed with an energy cutoff of 500 eV. A 4 × 4 × 1 Monkhorst–Pack53k-point mesh in the Brillouin zone was used for a 2 × 2 × 10 supercell of the primitive perovskite unit, containing a total of 200 atoms with 4 different cation configurations in order to cancel out the cation arrangement effects. This somewhat unusual supercell was used so that we could exactly match the desired oxygen stoichiometries. For the binary oxides, Monkhorst–Pack k-point mesh of 8 × 8 × 8, 8 × 8 × 8, 5 × 5 × 5, 5 × 5 × 5 and 3 × 3 × 3 were used for BaO (space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), CoO (space group F
m), CoO (space group F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m), Fe2O3 (space group Rc), ZrO2 (space group P21/c) and Y2O3 (space group Ia) respectively.
3m), Fe2O3 (space group Rc), ZrO2 (space group P21/c) and Y2O3 (space group Ia) respectively.
The 200-atom supercell was also used to calculate the binding energy of an oxygen vacancy and a Y atom by calculating the difference between two cells, one with Y–vacancy close (nearest neighbors), Y–vacancy intermediate and one with Y–vacancy far (separated as widely as possible in the cell, here 4.63 Å). The coordinates for the three calculations have been included in the ESI† as POSCAR_Y_Vac_Close, POSCAR_Y_Vac_Intermediate and POSCAR_Y_Vac_Far and the location of the vacancy (removed oxygen) being studied are included with the label “Vac” (this line must be removed to run these files in VASP). The binding energy of a 40-atom unit cell and a 320-atom supercell, with the same Y–vacancy close and Y–vacancy far distances as in DFT calculations, was additionally calculated using molecular dynamics (MD) simulations with Interatomic Potentials (M3Gnet).54 The cohesive energy of metal oxide compositions under air conditions (Co3O4 with space group Rm, Fe2O3 with space group Rc, and Y2O3 with space group Ia) was calculated using data extracted from the Materials Project.51
3. Results
3.1 Structural analysis
Fig. 1 shows the Rietveld refinement of the room temperature XRD patterns, where all the peaks in each pattern are indexed using the cubic Pmm space group, which is consistent with previous reports.12,40,55,56 The refined lattice parameters are shown in the respective figures, demonstrating that the Y3+ substitution for Zr4+ caused a lattice expansion of ∼0.003 nm (about 2.4% expansion by volume) as the yttrium content is increased to X = 0.2. This increase is expected due to the larger ionic radius of Y3+ compared to Zr4+, and the magnitude of expansion is reasonably consistent with our and others' previous syntheses of BCFZYX.12,19,20,40,57 The goodness of fit parameters for BCFZ, BCFZY and BCFY are 1.2, 1.1 and 1.1, respectively, which shows the high quality of fit for all the samples.
 | ||
| Fig. 1 Rietveld refined powder X-ray diffraction patterns of (a) BaCo0.4Fe0.4Zr0.2O3−δ (BCFZ), (b) BaCo0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY), and (c) BaCo0.4Fe0.4Y0.2O3−δ (BCFY). | ||
Powder neutron diffraction measurements were performed on all three compositions at 27 °C and 500 °C. Fig. 2a–c shows the NPD patterns for BCFZ, BCFZY and BCFY. As confirmed from the room temperature XRD, the crystal system for all compositions is cubic with only slight variation in lattice parameters. Therefore, one can expect to see similar peak patterns at all compositions in the absence of additional structural or magnetic ordering.
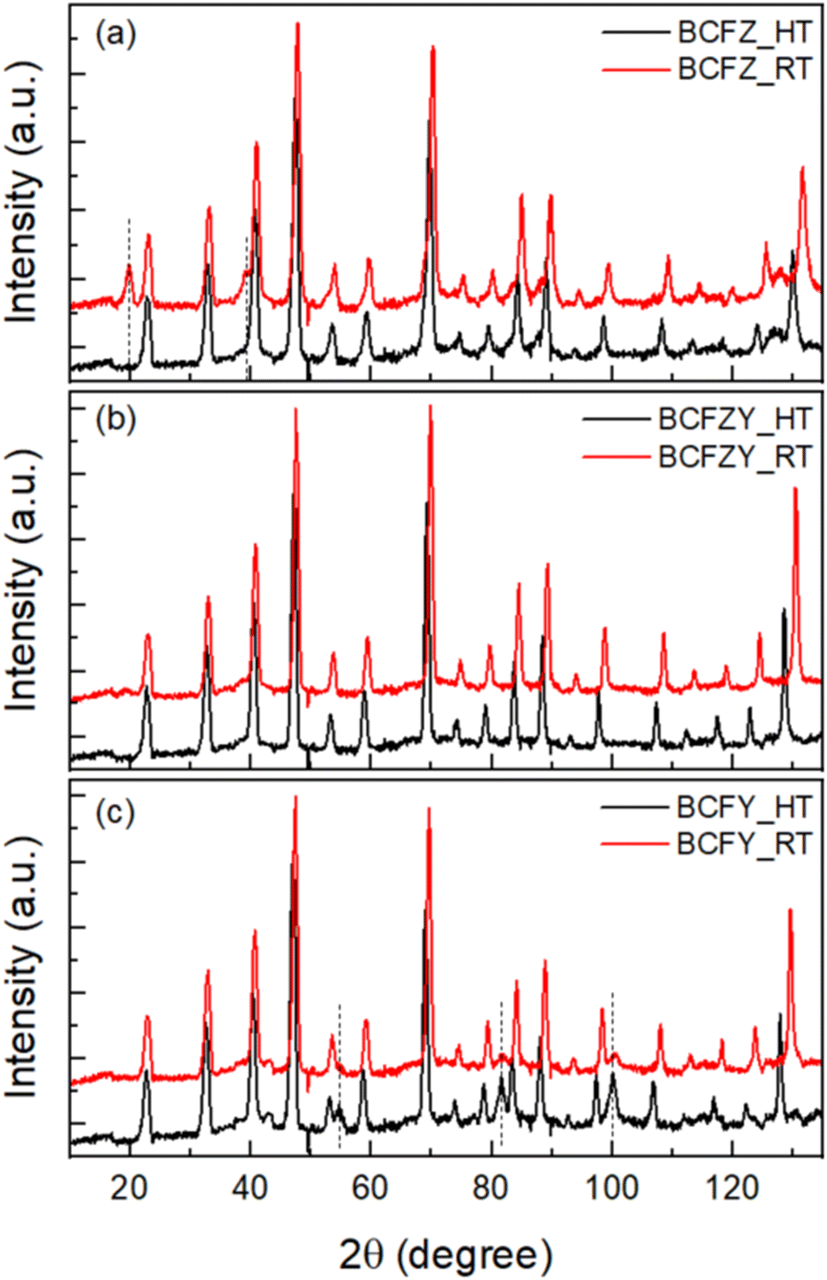 | ||
| Fig. 2 Neutron powder diffraction patterns for (a) BaCo0.4Fe0.4Zr0.2O3−δ (BCFZ), (b) BaCo0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY) and (c) BaCo0.4Fe0.4Y0.2O3−δ (BCFY) recorded at 27 and 500 °C. | ||
However, while comparing the NPD for different samples as a function of temperature, several interesting features emerge. First, for BCFZ, as shown in Fig. 2a, two extra peaks at lower 2θ values of ∼20° and ∼40° can be seen, unlike the other two samples at room temperature. Since these additional peaks appear only at lower 2θ angle, and because the XRD data do not demonstrate peaks at the same d-spacings, they are associated with long range magnetic ordering of Co/Fe spins. Further, magnetic ordering is confirmed when these peaks disappear at high temperatures. Simple observation suggests that the high temperature data belongs to the paramagnetic phase of BCFZ where Co/Fe spins are randomly orientated. The dashed line at 2θ values at ∼20° and ∼40° in Fig. 2a highlights the absence of magnetic peaks in the high temperature data. The presence of magnetic field was confirmed in BCFZ using Mössbauer spectroscopy. As shown in ESI Fig. S1,† BCFZ was the only observed composition with magnetic hyperfine splitting, while the other BCFZYX compositions exhibited only quadrupole splitting. On close observation of the NPD patterns, it is clear that the same magnetic ordering peaks are present, though just above background, in BCFZY0.1 but are absent in BCFY. This phenomenon is not observed in the Mössbauer spectroscopy, although it is possible that iron does not play a role in the material's magnetism as yttrium is doped into the structure, and that Co is the primary magnetic ion. The presence of magnetic peaks in the Zr containing samples, unlike in BCFY, indicates that the onset of magnetic ordering is linked to Zr doping in the studied compounds and the ordering becomes stronger with increasing Zr content.
Another prominent feature of the NPD data is the presence of 3 additional peaks with increased intensity at elevated temperature (500 °C) for the BCFY sample. The FWHM of the peaks is quite broad and the peaks cannot be indexed using any high temperature phase reported for this family of structures. Therefore, we attribute these to interference due to the beam clipping some component of the furnace, creating additional diffraction peaks.
The high temperature NPD of each composition was subjected to Rietveld fitting using cubic symmetry to extract the structural parameters in the paramagnetic region. For refining the NPD data of BCFY, we have excluded several broad peaks that appeared at this temperature at higher 2θ angle. Fig. 3 shows the fitted NPD patterns measured at 500 °C. In the crystal structure, the Ba ions reside at the 1a (0, 0, 0) position while the Co/Fe/Zr/Y ions are located at the 1b (0.5, 0.5, 0.5). The oxygen ion is situated at the 3c (0.5, 0.5, 0) position. In addition to refining the peak shapes, background, lattice parameters and other global parameters, the data enabled refinement of the oxygen site occupancy as well as the displacement parameters for the transition metal ions and oxygen. Table 1 presents the occupancies and displacement parameter values for each ion for the data measured at 500 °C. Since Co, Fe, Zr and Y are all randomly distributed on the 1b site, the refinement included a restraint keeping the displacement parameters for Co, Fe and Zr/Y the same. The refined compositions are BaCo0.4Fe0.4Zr0.2O2.32, BaCo0.4Fe0.4Zr0.1Y0.1O2.28 and BaCo0.4Fe0.4Y0.2O2.27.
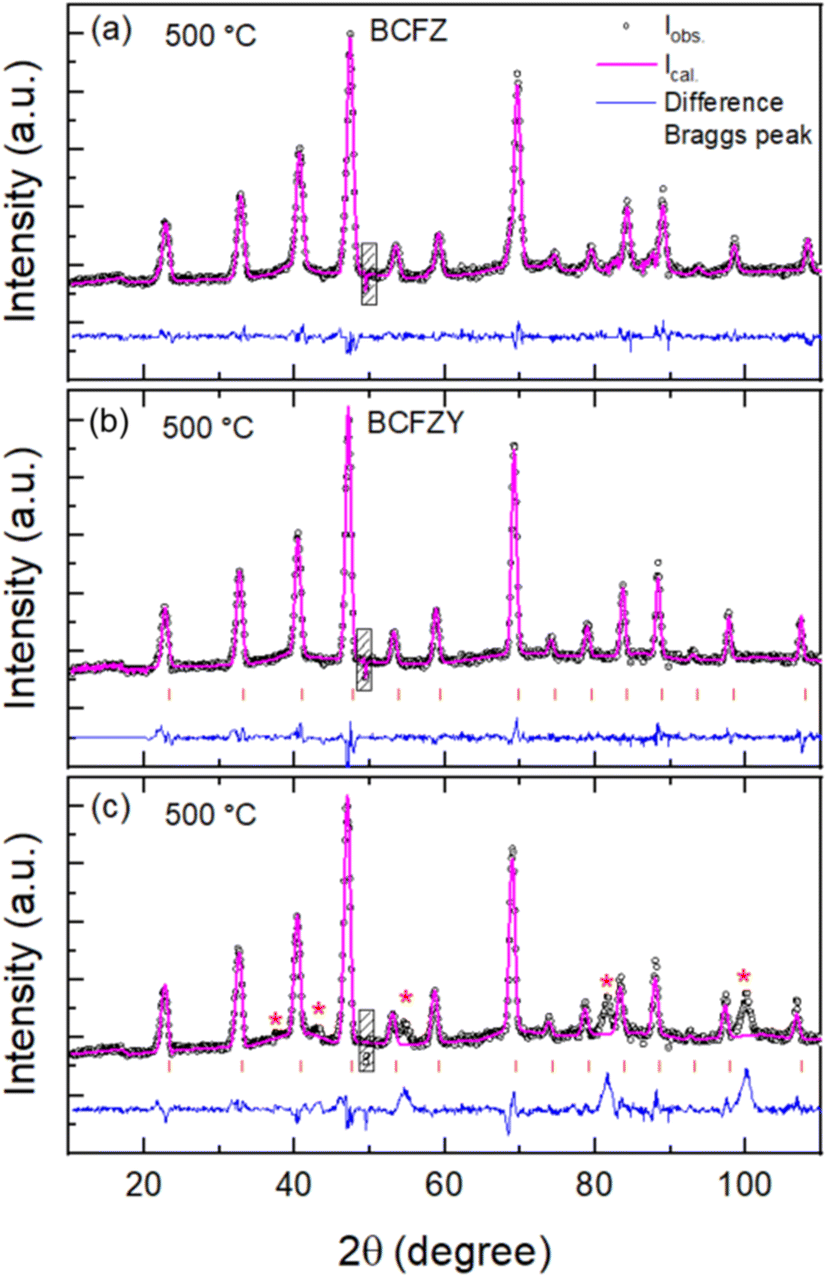 | ||
| Fig. 3 Rietveld refinements of high temperature (500 °C) NPD patterns for (a) BaCo0.4Fe0.4Zr0.2O3−δ (BCFZ), (b) BaCo0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY) and (c) BaCo0.4Fe0.4Y0.2O3−δ (BCFY) using cubic symmetry. The hatched regions show the excluded regions from the refinement. | ||
m using NPD data
| Composition | BaCo0.4Fe0.4Zr0.2O3−δRwp = 5.04 and Rp = 3.96 | BaCo0.4Fe0.4Zr0.1Y0.1O3−δRwp = 5.17 and Rp = 3.94 | BaCo0.4Fe0.4Y0.2O3−δRwp = 5.72 and Rp = 4.46 | |||
|---|---|---|---|---|---|---|
| Atom | Occupancy | Uiso | Occupancy | Uiso | Occupancy | Uiso |
| Ba | 1 | 0.0355(13) | 1 | 0.0313(12) | 1 | 0.0386(13) |
| Co/Fe/Zr/Y | 0.4/0.4/0.2/0 | 0.0467(13) | 0.4/0.4/0.1/0.1 | 0.0452(12) | 0.4/0.4/0/0.2 | 0.0539(13) |
| O | 0.773(7) | 0.0437(11) | 0.760(7) | 0.0479(11) | 0.756(7) | 0.0580(13) |
The NPD data collected at room temperature has also been fitted to extract the temperature variation of the lattice parameters, shown in Fig. 4. The NPD pattern of BCFZ was fitted in the triclinic setting (P-1) to include the satellite reflections coming from magnetic ordering using the incommensurate propagation vector k = (0.295, 0.5, 0). With the available data quality, the complete refinement of the magnetic structure was not possible, and future studies would benefit from neutron diffraction studies of single crystals to enable determination of the spin structure for the given incommensurate propagation vector. Table 2 shows the values of the displacement parameters at room temperature.
 | ||
| Fig. 4 Room temperature NPD patterns for (a) BaCo0.4Fe0.4Zr0.2O3−δ (BCFZ) (b) BaCo0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY) and (c) BaCo0.4Fe0.4Y0.2O3−δ (BCFY). The NPD pattern of BCFZ is fitted using triclinic setting (P-1) to include magnetic reflections whereas the data of BCFZY and BCFY was fitted using cubic symmetry. The hatched regions show the excluded regions from the refinement. | ||
| Composition | BaCo0.4Fe0.4Zr0.2O3−δRwp = 5.05 and Rp = 4.06 | BaCo0.4Fe0.4Zr0.1Y0.1O3−δRwp = 5.40 and Rp = 4.20 | BaCo0.4Fe0.4Y0.2O3−δRwp = 5.72 and Rp = 4.46 | |||
|---|---|---|---|---|---|---|
| Atom | Occupancy | Uiso | Occupancy | Uiso | Occupancy | Uiso |
| Ba | 1 | 0.0279(14) | 1 | 1.5459(11) | 1 | 0.0210(12) |
| Co/Fe/Zr/Y | 0.4/0.4/0.2/0 | 0.0412(14) | 0.4/0.4/0.1/0.1 | 2.5556(12) | 0.4/0.4 | 0.0342(13) |
| O | 0.792(9) | 0.0361(12) | 0.782(7) | 2.713(2) | 0.766(8) | 0.0379(13) |
The oxygen occupancies were also refined. While comparing the values of oxygen occupancies, one can see a clear increase in oxygen vacancy content with increasing temperature. Table 3 compares the room temperature values of δ with high temperature values. A small increase in δ values at elevated temperature is consistent with the TGA results as discussed later.
| Sample | δ (27 °C) | δ (500 °C) |
|---|---|---|
| BaCo0.4Fe0.4Zr0.2O3−δ | 0.624(9) | 0.681(7) |
| BaCo0.4Fe0.4Zr0.1Y0.1O3−δ | 0.654(7) | 0.720(7) |
| BaCo0.4Fe0.4Y0.2O3−δ | 0.702(8) | 0.732(7) |
Temperature dependence of the lattice parameters and oxygen vacancies as a function of temperature along with the thermal expansion coefficient values for each sample is shown in Fig. 5a–c. The lattice expands on heating as evident from significant increase in lattice parameters values. As mentioned above, the oxygen vacancies increase slightly in each sample at 500 °C as compared to room temperature data. The thermal expansion coefficient shows a maximum for the equally co-doped sample BCFZY0.1.
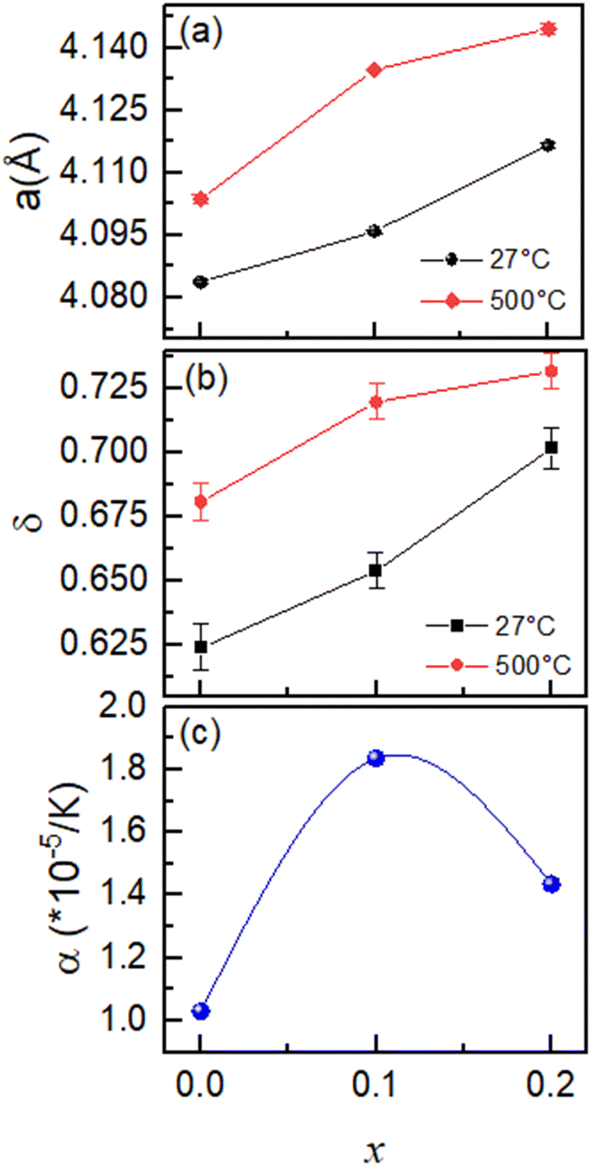 | ||
| Fig. 5 Temperature dependence of (a) lattice parameters, (b) oxygen vacancies and (c) thermal expansion coefficient as a function of X for BaCo0.4Fe0.4Zr0.2−XYXO3−δ. | ||
Using the oxygen vacancy concentration refined from room temperature NPD as an initial starting value, the vacancy concentration at the operating temperature (500 °C) is estimated using thermogravimetric analysis. The values from TGA are then compared to the refined vacancy concentration from NPD at 500 °C. Fig. 6a displays mass as a function of temperature for BCFZ, BCFZY, and BCFY. A small mass loss occurred during the drying process, followed by mass increase from 300 to 450 °C (approx.), and then continuous mass loss above 450 °C. Mass gain over the intermediate temperature range suggests oxidation of the multivalent metal cations due to oxygen uptake, as observed in other ferrite and cobaltite materials.58–60 On comparison with TGA in inert conditions (pure N2) as shown in ESI Fig. S2,† mass gain is not observed, supporting this hypothesis of oxidation. Subsequent mass loss indicates thermal reduction as a result of metal cations releasing their bound oxygen from the structure. In terms of mass loss, little difference is evident in the TGA profiles for these samples, however oxygen vacancy formation appears greater in the presence of Y-substitution, particularly in BCFZY0.1. The mass loss is used to estimate oxygen vacancy concentration (δTGA) using eqn (1).12,61
 | (1) |
 | ||
| Fig. 6 Thermogravimetric analysis of three BCFZYX materials, showing (a) weight loss as a function of temperature, and (b) oxygen vacancy concentration calculated from weight loss, with initial vacancy concentration calculated from neutron powder diffraction. | ||
| δ i,NPD 27 °C | δ NPD 500 °C | δ TGA 500 °C | ΔNPD (δNPD,500 − δNPD,27) | ΔTGA (δTGA,500 − δNPD,27) | (ΔNPD − ΔTGA)/ΔNPD | |
|---|---|---|---|---|---|---|
| BCFZ | 0.624(9) | 0.681(7) | 0.647(5) | 0.056(8) | 0.022(8) | 60% |
| BCFZY0.1 | 0.654(7) | 0.720(7) | 0.669(3) | 0.066(0) | 0.014(6) | 78% |
| BCFY | 0.702(8) | 0.732(7) | 0.727(2) | 0.029(9) | 0.024(4) | 18% |
3.2 Calorimetry
Five BCFZYX compositions were measured with high temperature oxide melt solution calorimetry in molten sodium molybdate at 700 °C under continuous oxygen flow. The samples fully dissolved in the sodium molybdate melt within 35 to 40 minutes as evidenced by the heat flow peaks returning to baseline without baseline shifts, tailing, or other anomalies. Table 5 presents two thermochemical cycles, which compute the formation enthalpy from the oxides (ΔHf,ox) as well as the standard formation enthalpy from the elements (ΔHel) for each sample. The former uses the measured mean drop solution enthalpies of the samples and the binary oxides (BaO, CoO, Co3O4, FeO, Fe2O3, ZrO2, and Y2O3) and the latter from the elements. Table 6 summarizes the mean drop solution enthalpies (ΔHds) of the five BCFZYX compositions.| Reactions | ΔH (kJ mol−1) |
|---|---|
| a Notations: s = solid and sln = solution. Measurement mean uncertainty was calculated as a two-standard deviation. The content of Co and Fe in BaCo0.4Fe0.4Zr0.2−XYXO3−δ are merely representative of the BCFZYX target. | |
| (1) BaCo0.4Fe0.4Zr(0.2−X)YXO(3−δ)(s,25°C) → BaO(sln,700°C) + aCoO(sln,700°C) + (⅓)bCo3O4(sln,700°C) + nFeO(sln,700°C) + (½)mFe2O3(sln,700°C) + (0.2 − X)ZrO2(sln,700°C) + (½)XY2O3(sln,700°C) + 0.25O2(g,700°C) | ΔH1 = ΔHds (BCFZYX) |
| (2) BaO(s,25°C) → BaO(sln,700°C) | ΔH2 = ΔHds (BaO) = −185.30 ± 3.12 (ref. 62) |
| (3) CoO(s,25°C) → CoO(sln,700°C) | ΔH3 = ΔHds (CoO) = 15.66 ± 0.59 (ref. 63) |
| (4) Co3O4(s,25°C) → Co3O4(sln,700°C) | ΔH4 = ΔHds (Co3O4) = 271.05 ± 3.72 (ref. 64) |
| (5) FeO(s,25°C) → FeO(sln,700°C) | ΔH5 = ΔHds (FeO) = −85.21 ± 0.92 (ref. 65) |
| (6) Fe2O3(s,25°C) → Fe2O3(sln,700°C) | ΔH6 = ΔHds (Fe2O3) = 95.63 ± 0.50 (ref. 65) |
| (7) ZrO2(s,25°C) → ZrO2(sln,700°C) | ΔH7 = ΔHds (ZrO2) = 19.5 ± 0.9 (ref. 66) |
| (8) Y2O3(s,25°C) → Y2O3(sln,700°C) | ΔH8 = ΔHds (Y2O3) = −120.07 ± 2.5 (ref. 67) |
|
|
| Calculated ΔH f,ox (25 °C) of BCFZY X from binary oxides: | |
| BaO(s) + aCoO(s) + (⅓)bCo3O4(s) + nFeO(s) + (½)mFe2O3(s) + (0.2 − X)ZrO2(s) + (½)XY2O3(s) + 0.25O2(g) → BaCo0.4Fe0.4Zr(0.2−X)YXO(3−δ)(s) | |
| ΔH9 = ΔHf,ox = ΔH2 + aΔH3 + (⅓)bΔH4 + nΔH5 + (½)mΔH6 + (0.2 − X)ΔH7 + (½)XΔH8 − ΔH1 | |
|
|
| Calculated ΔH f,el (25 °C) from the elements and the ΔH f,ox of BCFZY X | |
| Ba(s) + aCo(II)(s) + (⅓)bCo(II,III) + nFe(II)(s) + (½)mFe(III)(s) + (0.2 − X)Zr(s) + XY(s) + 1.5O2(g) → BaCo0.4Fe0.4Zr(0.2−X)YXO(3−δ)(s) | |
| ΔHf,el = ΔH9 + ΔHf,el(BaO) + aΔHf,el(CoO) + (⅓)bΔHf,el(Co3O4) + nΔHf,el(FeO) + (½)mΔHf,el(Fe2O3) + (0.2 − X)ΔHf,el(ZrO2) + (½)XΔHf,el(Y2O3) | |
| Target stoichiometry | ΔHds (kJ mol−1)a | ΔHf,oxb (kJ mol−1) | ΔHf,elc (kJ mol−1) |
|---|---|---|---|
| a The measurement mean of multiple experiments, as shown in parentheses, yields the enthalpy of drop solution (ΔHds). The reported uncertainty is the two-standard deviation of the measurement average. For the enthalpies of formation from oxides and elements, uncertainties are the propagated errors computed as the square root of the sum of the squares. b ΔHf,ox = enthalpy of formation from the binary oxides. c ΔHf,el = enthalpy of formation from the elements. Ancillary data used in the thermochemical cycles to compute the standard heats of formation are from Robie and Hemingway.68 Thermochemical values for a comparative BZY sample are from the research group of Alexandra Navrotsky.62 | |||
| BaCo0.4Fe0.4Zr0.2O3−δ | −50.57 ± 2.08 (7) | −95.60 ± 5.57 | −1072.90 ± 10.95 |
| BaCo0.4Fe0.4Zr0.15Y0.05O3−δ | −60.27 ± 1.08 (6) | −99.63 ± 5.28 | −1069.68 ± 10.81 |
| BaCo0.4Fe0.4Zr0.1Y0.1O3−δ | −70.19 ± 1.66 (6) | −96.94 ± 5.43 | −1033.27 ± 10.88 |
| BaCo0.4Fe0.4Zr0.05Y0.15O3−δ | −82.43 ± 1.47 (8) | −92.92 ± 5.37 | −1011.43 ± 10.85 |
| BaCo0.4Fe0.4Y0.2O3−δ | −90.05 ± 1.64 (8) | −96.96 ± 5.42 | −1002.25 ± 10.88 |
| BaZr0.80Y0.20O2.90 (Navrotsky 2014) | −85.53 ± 3.95 (8) | −92.89 ± 5.7 | −1634.94 ± 10.63 |
Using the oxygen nonstoichiometry, the average valence state for Co and Fe can be elucidated in each measured sample. Extra steps are taken to decouple each valence for Co and Fe. Mössbauer spectroscopy (Table S1†) was used to calculate the ratio of binary oxides in Fe. The ratio of binary oxides in Co was estimated using a charge balance based on the valence states of all other metal cations with the oxygen concentration elucidated from neutron powder diffraction. In intermediate compositions BCFZY0.05 and BCFZY0.15, the oxygen vacancy concentration is estimated using a linear interpolation. To characterize and correlate thermodynamic and transport phenomena for these complex oxides, accurate elemental composition is paramount. Using the above methods, the reported compositions are the best estimates possible for the actual composition of each sample. Compositions used for these thermodynamic analyses are given in Table S2.† For the following analyses, only binary oxides of Co and Fe with valence (II) and (III) are considered, as the binary oxides with valence (IV) are not stable and have not been characterized by thermochemical methods.
As presented in Table 6 and Fig. 7a, the enthalpy of formation from oxides and elements are both strongly exothermic and are thus favorable with respect to their binary oxides. The standard formation enthalpy from the elements decreases in stability tracking inversely with the Y content increase and directly with the decrease in Zr. The stability of these compositions is less favorable with simple mixing of the elements. They are all significantly less thermodynamically stable in their standard formation from the elements that the BZY (ΔHf,el = −1634.94 ± 10.63 kJ mol−1) that is not substituted with the transition metals, Co and Fe. The increasingly negative drop solution enthalpies indicate dissolution tendency and track directly with the increasing Y content. Although not a hard and fast rule, typically increased favorability of sample dissolution corresponds to decreased stability of the phase, and in this case would produce an increasingly endothermic trend in the enthalpy of formation from the oxides for perovskite samples with increasing Y-substitution. The expected trend holds for the three intermediate phases, BCFZY0.05, BCFZY0.1, and BCFZY0.15. However, the endpoint compositions BCFZ (ΔHds = −50.57 ± 2.08 kJ mol−1 and ΔHf,ox = −95.60 ± 5.57 kJ mol−1) and BCFY (ΔHds = −90.05 ± 1.64 kJ mol−1 and ΔHf,ox = −96.96 ± 5.42 kJ mol−1) break the expected trend for the mixing of binary oxides to form these two samples and, in spite of the large 40 kJ mol−1 difference in drop solution enthalpy, the ΔHf,ox remains relatively constant.
 | ||
| Fig. 7 (a) Enthalpies of drop solution (left axis, left-facing arrow) as measured in sodium molybdate at 700 °C and formation enthalpies from the oxides at 25 °C (right axis, right-facing solid arrow) and (b) comparison of empirical enthalpies of formation from the oxides with DFT calculations, as a function of X yttrium content in BCFZY solid solutions. Reported uncertainty values were calculated as two standard deviations from the mean for both experimental and DFT plotted data. | ||
Comparison between the empirical and the DFT-calculated enthalpy of formation from the oxides is shown in Fig. 7b. All DFT-calculated values are within 15% of the experimentally determined values, indicating good agreement between the model and experimental data. The DFT data show many similarities to the experiments in the first four compositions. Specifically, DFT also predicts a slight decrease in ΔHf,ox from BCFZ (ΔHf,ox,DFT = −112.14 ± 4.46 kJ mol−1) to BCFZY0.05 (ΔHf,ox,DFT = −113.69 ± 1.46 kJ mol−1), followed by a general increase to BCFZY0.15 (ΔHf,ox,DFT = −105.53 ± 1.02 kJ mol−1). This significant increase in ΔHf,ox from BCFZY0.05 to BCFZY0.15 closely matches between the empirical data and predicted DFT calculations. However, DFT calculations appear to underestimate the ΔHf,ox in the final BCFY composition. Overall, the BCFZYX compositions are relatively close to previously reported values for yttrium-doped barium zirconate in both drop solution enthalpy and enthalpy of formation from the oxides.52
4. Discussion
Neutron diffraction revealed that this compositional suite of materials has high oxygen vacancy concentration, especially compared to other ionic conductors such as BaZr1−XYXO3−δ and Ce1−XGdXO2−δ.62,69,70 Previous neutron diffraction studies on a similar mixed ionic–electronic material Ba0.5Sr0.5Co0.8Fe0.2O3−δ showed similar levels of initial oxygen non-stoichiometry of δ ≈ 0.6.34 From a structural symmetry standpoint, Ba2+ is a large cation, meaning that larger B-site cations are necessary for structural stability/symmetry of the cubic perovskite system. As redox state in Fe/Co is reduced, the ionic radius is increased given the same spin state.57 It is therefore structurally favorable for the lattice to contain reduced redox states of Fe/Co, which are closer to the ionic radius of Zr4+, as suggested in this work. In recent studies in this system, specifically on BCFZY0.1, iodometry has suggested a wide range of oxygen vacancy concentrations from δ = 0.23 to δ = 0.5,12,17,18 lower than the value reported here, leaving wide uncertainty of the actual vacancy concentration of this composition. Other work on BaCoO3−δ and BaCo0.5Fe0.5O3−δ have shown that high vacancy concentrations (δ = 0.78 and δ = 0.84, respectively) are necessary for cubic perovskite symmetry, supporting this work;71–73 at lower vacancy concentrations with more oxidized metal cation redox states, the structural symmetry is reduced to hexagonal. Contrary to this work, these compounds were synthesized in reducing conditions, though the compositions were shown to be stable in air until 220 °C through TGA measurements.71 It is possible that the larger and univalent Zr4+ and Y3+ cations in the BCFZYX system help stabilize the lattice across a wide range of temperatures. These works also present an interesting question on the upper-bound of vacancy concentration for compositions synthesized in air.Although it is known that small changes in dopant concentration have notable effects on transport properties in BCFZYX, the underlying reasons for the trends in transport phenomena have not been clearly defined in literature. Neutron diffraction revealed that the three compositions are structurally quite similar; all compositions exhibit cubic perovskite symmetry with only small changes in lattice parameter across the compositional range. The substitution of yttrium for zirconium increases the lattice parameter, which increases ionic hopping distance and therefore can decrease ionic conductivity. This phenomenon is observed in oxide ion conductivity (σO2−) given in Table 7. The same trend is not observed in proton conductivity (σH+); it is more likely that localized atomic changes caused by Y for Zr doping are the main cause for the optimal proton conductivity observed in BCFZY0.1, as discussed later. Additionally, neutron diffraction showed increasing oxygen vacancy concentration with increasing yttrium doping. Increased vacancy content likely increases the available sites for surface exchange, increasing oxygen surface exchange (kO,chem)and hydroxyl surface exchange (kOH,chem)as noted in Table 7. The increased vacancy concentration also decreases the available B–O bonds in the system, thus decreasing the electronic conductivity (σe−).
| Composition | σ e− (S cm−1) | σ H+ (S cm−1) | σ O2− (S cm−1) | k O,chem (cm s−1) | k OH,chem (cm s−1) |
|---|---|---|---|---|---|
| BCFZ | 2.63 | 8.48 × 10−4 | 7.56 × 10−2 | 7.3 × 10−4 | 8.62 × 10−5 |
| BCFZY0.1 | 1.71 | 3.23 × 10−3 | 3.56 × 10−2 | 2.8 × 10−3 | 2.26 × 10−4 |
| BCFY | 1.23 | 2.7 × 10−3 | 1.41 × 10−2 | 4.6 × 10−3 | 3.91 × 10−4 |
Thermodynamic destabilization of the perovskite phase was expected with aliovalent substitution of Y3+ for Zr4+ in this series. This trend was expected in part due to the endothermic effects of increasing number of oxygen vacancies as well as local lattice strain caused by the larger size of Y3+ compared to Zr4+ and Co/Fe multivalent ions. In Fig. 7, the experimental enthalpy of formation from the oxides exhibits this trend in the three intermediate compositions. However, the endpoint compositions BCFZ and BCFY show interesting deviations from the expected trend. Some of the deviations can be explained by the larger changes in barium concentration at the endmembers. However, when only accounting for the barium concentration change, the roughly linear trend still cannot be resolved compared to the three intermediate concentrations. BCFZ shows greater endothermicity than expected. Observations from literature suggest difficulties for this composition to achieve cubic symmetry, including lower symmetry structures and secondary phases.19,74,75 This difficulty may provide some explanation for why BCFZ is more endothermic than expected.
In BCFY, the greater exothermicity from expected may be the result of vacancy-dopant association, where the yttrium atom becomes associated with the vacancy it forms, and therefore yttrium–vacancy pairs become more energetically stable than randomized vacancies. This phenomenon has also been hypothesized by high temperature oxide melt solution calorimetry in other fluorite and perovskite structures.76–79 Vacancy association is typically correlated with decreases in conductivity of mobile ions due to the trapping of vacancies, reducing the number of available mobile defects for kinetic transport.80,81 Vacancy association can especially be supported by the previously measured proton conductivity of the BCFZYX series, where BCFZY0.1 (X = 0.1) exhibited the highest proton conductivity followed by a decrease with increased yttrium doping as shown in Table 7 and Fig. S3.19† Yttrium–vacancy association is also noted in BZY, where proton conductivity reaches a relative maximum at X = 0.2 and decreases once vacancy association (suggested from high temperature solution calorimetry) is present at X = 0.3 and greater.62,70 Like in BZY,82 the vacancy-dopant association in BCFY outweighs the increased propensity for proton uptake from yttrium incorporation19 and leads to its decreased proton conductivity. A similar explanation may also apply to the increased exothermicity of BCFZY0.05 against BCFZ, where some local vacancy-dopant association is occurring with the introduction of Y3+. However, in this case, the propensity for proton uptake from Y3+ incorporation outweighs the local association effect, so proton conductivity increases.
Vacancy association was investigated using computational approaches. Density functional theory (DFT) calculations of an O vacancy next to and farther from Y in a 200-atom unit cell of BaCo0.4Fe0.4Y0.2O3 show a binding energy of −2.22 eV, a very strong repulsion. This result contradicts expectations from electrostatics, where Y and oxygen vacancies in the perovskite lattice should attract due to the negative charge of Y and the positive charge of the oxygen vacancies. The result is also inconsistent with the size effects expected from the larger Y atoms. The ionic radii for B-site cations in the perovskite system are Y3+ (0.90 Å), Co4+ (0.53 Å) and Fe4+ (0.58 Å). The comparably larger ionic radii of Y3+ compared to Co4+ and Fe4+ would be expected to attract oxygen vacancies to reduce the strain in the system, effectively helping the larger Y3+ fit by creating space nearby. However, the binding energy is consistent with the stronger metal–oxygen bond of Y compared to Co or Fe. Specifically, we can estimate the bond strengths by dividing the cohesive energy of each phase by the number of metal–oxygen bonds, which gives: Y2O3 cohesive energy/bond = −19.53 eV/12 bonds = −1.68 eV/bond; Co3O4 cohesive energy/bond = −9.03 eV/18 bonds = −0.50 eV/bond, and Fe2O3 cohesive energy/bond = −8.54 eV/12 bonds = −0.71 eV/bond, as calculated using DFT (see Methods). We therefore believe that it is the strong yttrium–oxygen bond compared to the other metals that attracts the oxygen, or equivalently, repels the vacancy in this estimation.
To ensure a robust calculation of the binding energy of yttrium–vacancy pair, an additional binding energy calculation was performed for a configuration where the distance of Y and the oxygen vacancy was intermediate between the near and far case. This calculation yielded a binding energy of −1.91 eV, consistent with the repulsion decreasing as the vacancy moves farther away from nearby yttrium. Additionally, the M3Gnet universal machine learning potential was used to evaluate the binding energy of the yttrium–vacancy pair. Oxygen vacancies adjacent to and farther from yttrium in a 40-atom and 320-atom unit cell of BaCo0.4Fe0.4Y0.2O3 showed the same binding energy of −0.89 eV. This result is potentially quite approximate as the potential was not fit to vacancy binding data but is qualitatively consistent with the DFT in that it shows strong repulsion between the yttrium and the oxygen vacancy and no strong dependence on the size of the cell used in the study. These DFT and M3Gnet calculations are not consistent with the attractive yttrium–vacancy binding that has been posited in the explanations above. It is possible that some other effect underlies the above thermodynamic and kinetic observations, or that the complex magnetic, electron localization, redox, and correlated electron effects that may be occurring in the BaCo0.4Fe0.4Y0.2O3 are poorly enough captured by the PBE DFT and M3Gnet that they get the wrong sign for the yttrium–vacancy binding. Overall, although opposite in potential, a strong association is suggested with yttrium in both the computational work (Y–O bond) and the experimental work (Y–Vac bond), each of which could decrease mobile species in the lattice. More work is needed in this area to develop a robust and consistent understanding.
In terms of oxygen transport, BCFZYX shows a clear tradeoff between bulk oxygen conductivity and surface exchange19,20 as shown in Table 7 and Fig. S4.† The increase in oxygen surface exchange can be explained by the increased vacancy concentration with increasing yttrium concentration, allowing for more sites for oxygen incorporation. For bulk transport, we can see the reduced oxygen diffusion kinetics with increasing yttrium concentration by considering the oxygen ionic diffusion coefficient, DO2−. Using the previously determined oxygen ionic conductivity,20σO2−, and the Nernst–Einstein equation,83,84DO2− can be calculated:
 | (2) |
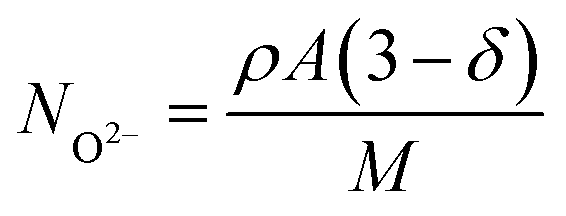 | (3) |
 | (4) |
 | ||
| Fig. 8 Estimated oxygen ionic self-diffusivity as a function of temperature from 550–700 °C, using structural data and previously determined oxygen ion conductivity measurements. | ||
Based on both the observed thermodynamic and structural phenomena, the observed trends in oxygen mobility are supported by the increased hopping distance due to the increases in lattice parameter from BCFZ to BCFY and the hypothesis of dopant vacancy association, leading to increased vacancy immobilization around the Y ion.62,80,81,88 This estimation is also supported by the assertion that increased vacancy content decreases the ionic mobility.89 The trend observed with the ionic diffusion coefficient aligns reasonably well with the previously measured chemical diffusion coefficient, DO,chem, except at low temperatures, as shown in Fig. S4.† The low temperature measurements of the chemical diffusion for BCFZ may be hindered by the low surface exchange coefficient at low temperatures. If BCFZ was surface limited in this measurement, this could potentially skew the bulk diffusivity to be lower than expected.20
5. Conclusions
This work analyzed trends in the enthalpy of formation from the oxides as a function of oxygen vacancy and structure effects through the application of high temperature solution calorimetry, XRD, TGA, and neutron powder diffraction. The results were then correlated with previously measured proton and oxygen kinetics in the BCFZYX perovskite ceramic compositional suite. All compositions exhibit a cubic perovskite structure which shows increasing oxygen vacancy concentration with increasing yttrium substitution. It is hypothesized through the thermochemical data that the increase in yttrium for zirconium substitution induces dopant-vacancy association, correlating with a decrease in defect mobility in the fully yttrium-doped BCFY.Data availability
The data supporting this article have been included as part of the ESI† with additional data available upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by the Department of Energy, National Energy Technology Laboratory an agency of the United States Government, through an appointment administered by the Oak Ridge Institute for Science and Education. Neither the United States Government nor any agency thereof, nor any of its employees, nor the support contractor, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof. The authors would like to acknowledge Boris Merkies at TU Delft for his work on the Mössbauer spectroscopy measurements. JHD was supported in part by an International Research Experiences for Students program through National Science Foundation Award #1952528. KSB was supported in part by an appointment to the NETL Research Participation Program, sponsored by the U.S. Department of Energy and administered by the Oak Ridge Institute for Science and Education.References
- E. Fabbri, D. Pergolesi and E. Traversa, Chem. Soc. Rev., 2010, 39, 4355–4369 RSC.
- J. Irvine, J. L. M. Rupp, G. Liu, X. Xu, S. Haile, X. Qian, A. Snyder, R. Freer, D. Ekren, S. Skinner, O. Celikbilek, S. Chen, S. Tao, T. H. Shin, R. O'Hayre, J. Huang, C. Duan, M. Papac, S. Li, V. Celorrio, A. Russell, B. Hayden, H. Nolan, X. Huang, G. Wang, I. Metcalfe, D. Neagu and S. G. Martín, J. Phys., 2021, 3, 031502 CAS.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- J. Kim, S. Sengodan, S. Kim, O. Kwon, Y. Bu and G. Kim, Renew. Sustain. Energy Rev., 2019, 109, 606–618 CrossRef CAS.
- R. Merkle, M. F. Hoedl, G. Raimondi, R. Zohourian and J. Maier, Annu. Rev. Mater. Res., 2021, 51, 461–493 CrossRef CAS.
- U. Balachandran, B. Ma, P. S. Maiya, R. L. Mieville, J. T. Dusek, J. J. Picciolo, J. Guan, S. E. Dorris and M. Liu, Solid State Ionics, 1998, 108, 363–370 CrossRef CAS.
- J. Sunarso, S. S. Hashim, N. Zhu and W. Zhou, Prog. Energy Combust. Sci., 2017, 61, 57–77 CrossRef.
- N. Kochetova, I. Animitsa, D. Medvedev, A. Demin and P. Tsiakaras, RSC Adv., 2016, 6, 73222–73268 RSC.
- L. R. Tarutina, M. A. Gordeeva, D. E. Matkin, M. T. Akopian, G. N. Starostin, A. V. Kasyanova, A. P. Tarutin, N. A. Danilov, I. A. Starostina, D. A. Medvedev and Z. Shao, Chem. Eng. J., 2024, 490, 151615 CrossRef CAS.
- M. Liang, F. He, C. Zhou, Y. Chen, R. Ran, G. Yang, W. Zhou and Z. Shao, Chem. Eng. J., 2021, 420, 127717 CrossRef CAS.
- M. Liang, Y. Song, D. Liu, L. Xu, M. Xu, G. Yang, W. Wang, W. Zhou, R. Ran and Z. Shao, Appl. Catal., B, 2022, 318, 121868 CrossRef CAS.
- M. S. Alam, I. Kagomiya and K. Kakimoto, Ceram. Int., 2023, 49, 11368–11377 CrossRef CAS.
- H. Lv, Z. Jin, R. Peng, W. Liu and Z. Gong, Ceram. Int., 2019, 45, 23948–23953 CrossRef.
- Y. Xu, F. Hu, Y. Guo, J. Zhang, Y. Huang, W. Zhou, J. Sun, B. He and L. Zhao, Sep. Purif. Technol., 2022, 297, 121482 CrossRef CAS.
- J. Ma, F. Zhu, Y. Pan, H. Zhang, K. Xu, Y. Wang and Y. Chen, Sep. Purif. Technol., 2022, 288, 120657 CrossRef CAS.
- Z. Xu, J. Yu and W. Wang, Membranes, 2022, 12, 831 CrossRef CAS PubMed.
- R. Ren, Z. Wang, C. Xu, W. Sun, J. Qiao, D. W. Rooney and K. Sun, J. Mater. Chem. A, 2019, 7, 18365–18372 RSC.
- X. Kuai, G. Yang, Y. Chen, H. Sun, J. Dai, Y. Song, R. Ran, W. Wang, W. Zhou and Z. Shao, Adv. Energy Mater., 2019, 9, 1902384 CrossRef CAS.
- J. H. Duffy, H. W. Abernathy and K. S. Brinkman, J. Mater. Chem. A, 2023, 11, 8929–8938 RSC.
- J. H. Duffy, Y. Meng, H. W. Abernathy and K. S. Brinkman, Membranes, 2021, 11, 766 CrossRef CAS PubMed.
- Y. Shin, Y. Kim, M. Sanders, S. P. Harvey, M. Walker and R. O'Hayre, J. Mater. Chem. A, 2022, 10, 24839–24853 RSC.
- D. Poetzsch, R. Merkle and J. Maier, Adv. Funct. Mater., 2015, 25, 1542–1557 Search PubMed.
- H. Takahashi, I. Oikawa and H. Takamura, J. Phys. Chem. C, 2018, 122, 6501–6507 CrossRef CAS.
- X. Wang, W. Li, C. Zhou, M. Xu, Z. Hu, C.-W. Pao, W. Zhou and Z. Shao, ACS Appl. Mater. Interfaces, 2023, 15, 1339–1347 CrossRef CAS.
- R. Ren, Z. Wang, X. Meng, X. Wang, C. Xu, J. Qiao, W. Sun and K. Sun, ACS Appl. Energy Mater., 2020, 3, 4914–4922 CrossRef CAS.
- L. Zhang, B. Pang, S. Hu, Z. Cao, P. Zhang, X. Zhu and W. Yang, Int. J. Hydrogen Energy, 2023, 48, 10180–10190 CrossRef CAS.
- F. Feng, W. Yang, Q. Zheng, J. Milewski, L. Zhao, C. Wang and Y. Guo, Energy Fuels, 2021, 35, 8953–8960 CrossRef CAS.
- M. A. Gomez, Z. Lin, S. Lin, Y. Tian, A. van Bokkelen and M. Valerio, J. Phys. Chem. C, 2020, 124, 27954–27964 CrossRef.
- M. Coduri, M. Karlsson and L. Malavasi, J. Mater. Chem. A, 2022, 10, 5082–5110 RSC.
- M. Mogensen, D. Lybye, N. Bonanos, P. Hendriksen and F. Poulsen, Solid State Ionics, 2004, 174, 279–286 CrossRef CAS.
- J. Ding, J. Balachandran, X. Sang, W. Guo, J. S. Anchell, G. M. Veith, C. A. Bridges, Y. Cheng, C. M. Rouleau, J. D. Poplawsky, N. Bassiri-Gharb, R. R. Unocic and P. Ganesh, Chem. Mater., 2018, 30, 4919–4925 CrossRef CAS.
- J. F. Basbus, M. D. Arce, F. R. Napolitano, H. E. Troiani, J. A. Alonso, M. E. Saleta, M. A. González, G. J. Cuello, M. T. Fernández-Díaz, M. P. Sainz, N. Bonanos, C. E. Jimenez, L. Giebeler, S. J. A. Figueroa, A. Caneiro, A. C. Serquis and L. V. Mogni, ACS Appl. Energy Mater., 2020, 3, 2881–2892 CrossRef CAS.
- S. McIntosh, J. F. Vente, W. G. Haije, D. H. A. Blank and H. J. M. Bouwmeester, Solid State Ionics, 2006, 177, 833–842 CrossRef CAS.
- S. McIntosh, J. F. Vente, W. G. Haije, D. H. A. Blank and H. J. M. Bouwmeester, Chem. Mater., 2006, 18, 2187–2193 CrossRef CAS.
- A. Navrotsky, AIP Conf. Proc., 2000, 535, 288–296 CrossRef CAS.
- J. Cheng, A. Navrotsky, X. D. Zhou and H. U. Anderson, J. Mater. Res., 2005, 20, 191–200 CrossRef CAS.
- H. J. Avila-Paredes, T. Shvareva, W. Chen, A. Navrotsky and S. Kim, Phys. Chem. Chem. Phys., 2009, 11, 8580–8585 RSC.
- M. Zhao, Y. Xu, L. Shuller-Nickles, J. Amoroso, A. I. Frenkel, Y. Li, W. Gong, K. Lilova, A. Navrotsky and K. S. Brinkman, J. Am. Ceram. Soc., 2019, 102, 4314–4324 CrossRef CAS.
- H. Xu, L. Wu, J. Zhu and A. Navrotsky, J. Nucl. Mater., 2015, 459, 70–76 CrossRef CAS.
- C. Duan, J. Tong, M. Shang, S. Nikodemski, M. Sanders, S. Ricote, A. Almansoori and R. O'Hayre, Science, 2015, 349, 1321–1326 CrossRef CAS.
- L. Van Eijck, L. D. Cussen, G. J. Sykora, E. M. Schooneveld, N. J. Rhodes, A. A. Van Well and C. Pappas, J. Appl. Crystallogr., 2016, 49, 1398–1401 CrossRef CAS.
- V. Petříček, M. Dušek and L. Palatinus, Z. Kristallogr. - Cryst. Mater., 2014, 229, 345–352 CrossRef.
- A. Navrotsky, MRS Bull., 1997, 22, 35–41 CrossRef CAS.
- A. Navrotsky, Engineering, 2019, 5, 366–371 CrossRef CAS.
- A. Navrotsky, Phys. Chem. Miner., 1997, 24, 222–241 CrossRef CAS.
- A. Navrotsky, J. Am. Ceram. Soc., 2014, 97, 3349–3359 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. Hubbard, Proc. R. Soc. London, Ser. A, 1963, 276, 238–257 Search PubMed.
- V. I. Anisimov, F. Aryasetiawan and A. I. Lichtenstein, J. Phys.: Condens. Matter, 1997, 9, 767–808 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- C. Chen and S. P. Ong, Nat. Comput. Sci., 2022, 2, 718–728 CrossRef.
- M. Shang, J. Tong and R. O'Hayre, RSC Adv., 2013, 3, 15769 RSC.
- L. Zhang, J. Shan and Q. Wang, J. Alloys Compd., 2019, 771, 221–227 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- F. Dong, Y. Chen, D. Chen and Z. Shao, ACS Appl. Mater. Interfaces, 2014, 6, 11180–11189 CrossRef CAS.
- V. Cascos, J. A. Alonso and M. T. Fernández-Díaz, Materials, 2016, 9, 1–12 Search PubMed.
- A. Aguadero, J. A. Alonso, D. Pérez-Coll, C. De La Calle, M. T. Fernández-Díaz and J. B. Goodenough, Chem. Mater., 2010, 22, 789–798 CrossRef CAS.
- J. I. Lee, K. Y. Park, H. Park, H. Bae, M. Saqib, K. Park, J. S. Shin, M. Jo, J. Kim, S. J. Song, E. D. Wachsman and J. Y. Park, J. Power Sources, 2021, 510, 230409 CrossRef CAS.
- M. D. Gonçalves, P. S. Maram, R. Muccillo and A. Navrotsky, J. Mater. Chem. A, 2014, 2, 17840–17847 RSC.
- L. Wang, A. Navrotsky, R. Stevens, B. F. Woodfield and J. Boerio-Goates, J. Chem. Thermodyn., 2003, 35, 1151–1159 CrossRef CAS.
- N. H. Perry, T. O. Mason, C. Ma, A. Navrotsky, Y. Shi, J. S. Bettinger, M. F. Toney, T. R. Paudel, S. Lany and A. Zunger, J. Solid State Chem., 2012, 190, 143–149 CrossRef CAS.
- K. I. Lilova, F. Xu, K. M. Rosso, C. I. Pearce, S. Kamali and A. Navrotsky, Am. Mineral., 2012, 97, 164–175 CrossRef CAS.
- A. V. Radha, O. Bomati-Miguel, S. V. Ushakov, A. Navrotsky and P. Tartaj, J. Am. Ceram. Soc., 2009, 92, 133–140 CrossRef CAS.
- P. Zhang, A. Navrotsky, B. Guo, I. Kennedy, A. N. Clark, C. Lesher and Q. Liu, J. Phys. Chem. C, 2008, 112, 932–938 CrossRef CAS.
- R. A. Robie and B. S. Hemingway, Thermodynamic Properties of Minerals and Related Substances at 298.15 K and 1 Bar (105 pascals) Pressure and at Higher Temperatures, 1995 Search PubMed.
- T. S. Zhang, J. Ma, H. Cheng and S. H. Chan, Mater. Res. Bull., 2006, 41, 563–568 CrossRef CAS.
- D. Han and T. Uda, J. Mater. Chem. A, 2018, 6, 18571–18582 RSC.
- O. Mentré, M. Iorgulescu, M. Huvé, H. Kabbour, N. Renaut, S. Daviero-Minaud, S. Colis and P. Roussel, Dalton Trans., 2015, 44, 10728–10737 RSC.
- A. Diatta, C. V. Colin, R. Viennois, M. Beaudhuin, J. Haines, P. Hermet, A. van der Lee, L. Konczewicz, P. Armand and J. Rouquette, J. Am. Chem. Soc., 2024, 146, 15027–15035 CrossRef CAS PubMed.
- S. W. Strauss, I. Fankuchen and R. Ward, J. Am. Chem. Soc., 1951, 73, 5084–5086 CrossRef CAS.
- K. Efimov, O. Czuprat and A. Feldhoff, J. Solid State Chem., 2011, 184, 1085–1089 CrossRef CAS.
- O. Czuprat, M. Arnold, S. Schirrmeister, T. Schiestel and J. Caro, J. Membr. Sci., 2010, 364, 132–137 CrossRef CAS.
- J. Cheng and A. Navrotsky, J. Solid State Chem., 2004, 177, 126–133 CrossRef CAS.
- W. Chen and A. Navrotsky, J. Mater. Res., 2006, 21, 3242–3251 CrossRef CAS.
- M. Aizenshtein, T. Y. Shvareva and A. Navrotsky, J. Am. Ceram. Soc., 2010, 93, 4142–4147 CrossRef CAS.
- S. Buyukkilic, T. Shvareva and A. Navrotsky, Solid State Ionics, 2012, 227, 17–22 CrossRef CAS.
- M. S. Khan, M. S. Islam and D. R. Bates, J. Phys. Chem. B, 1998, 102, 3099–3104 CrossRef CAS.
- K. Huang, R. S. Tichy and J. B. Goodenough, J. Am. Ceram. Soc., 1998, 81, 2565–2575 CrossRef CAS.
- E. Fabbri, D. Pergolesi, S. Licoccia and E. Traversa, Solid State Ionics, 2010, 181, 1043–1051 CrossRef CAS.
- S. Kotsantonis and J. A. Kilner, Solid State Ionics, 2011, 192, 168–171 CrossRef CAS.
- P. J. Gellings and H. J. M. Bouwmeester, The CRC Handbook of Solid State Electrochemistry, CRC Press, 1997 Search PubMed.
- S. Y. Jeon, M. B. Choi, H. N. Im, J. H. Hwang and S. J. Song, J. Phys. Chem. Solids, 2012, 73, 656–660 CrossRef CAS.
- L. Wang, R. Merkle and J. Maier, J. Electrochem. Soc., 2010, 157, B1802 CrossRef CAS.
- L. Wang, R. Merkle, J. Maier, T. Acartürk and U. Starke, Appl. Phys. Lett., 2009, 94, 1–4 Search PubMed.
- S. Imashuku, T. Uda, Y. Nose, G. Taniguchi, Y. Ito and Y. Awakura, J. Electrochem. Soc., 2009, 156, B1 CrossRef CAS.
- Z. Li, X. Mao, D. Feng, M. Li, X. Xu, Y. Luo, L. Zhuang, R. Lin, T. Zhu, F. Liang, Z. Huang, D. Liu, Z. Yan, A. Du, Z. Shao and Z. Zhu, Nat. Commun., 2024, 15, 9318 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta06417h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |