
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exsolved Cu–ZnO interfaces for methanol production from CO2 at atmospheric pressure†
Sanaz
Soodi‡
a,
Eleonora
Calì‡
b,
Shailza
Saini‡
a,
William S.
Skinner
 c,
Alex Martinez
Martin
a,
Gwilherm
Kerherve
c,
Marco Pietro
Mezzapesa
b,
Samir
Bensaid
b,
David J.
Payne
cd,
Melis S.
Duyar
a and
Kalliopi
Kousi
*a
c,
Alex Martinez
Martin
a,
Gwilherm
Kerherve
c,
Marco Pietro
Mezzapesa
b,
Samir
Bensaid
b,
David J.
Payne
cd,
Melis S.
Duyar
a and
Kalliopi
Kousi
*a
aSchool of Chemistry and Chemical Engineering, University of Surrey, Guildford, GU2 7XH, UK. E-mail: k.kousi@surrey.ac.uk
bDepartment of Applied Science and Technology, Politecnico di Torino, Corso Duca degli Abruzzi, 24, Turin 10129, Italy
cDepartment of Materials, Imperial College London, Exhibition Road, London SW7 2AZ, UK
dNEOM Education, Research, and Innovation Foundation, Al Khuraybah, Tabuk 49643-9136, Saudi Arabia
First published on 17th October 2024
Abstract
An intimate Cu–ZnO interface that allows for the conversion of CO2 to methanol even at atmospheric pressures was produced using the exsolution method. Detailed characterization and operando IR spectroscopy were employed to deconvolute the materials' properties that allow for this to happen.
 Kalliopi Kousi | Dr Kalliopi Kousi is a Lecturer at the School of Chemistry and Chemical Engineering at the University of Surrey. Her research background is in Heterogeneous Catalysis and Materials Synthesis and Characterisation. She obtained her degree in Chemistry from the University of Patras, Greece in 2011, where she also got her MRes in 2013 and PhD in 2016. Her current research focuses around nanoengineering of materials for the conversion of greenhouse gases and the production of clean energy. She is also very interested in Science Communication and has been recognised for her work in the EDI space. |
Introduction
The UK and EU, along with the rest of the world, have pledged net-zero greenhouse gas emissions by 2050, significantly cutting back on emissions by 2030.1 There is a wide range of technologies that can help us achieve decarbonisation of our energy supply but in addition to this, we need to capture and utilise the CO2 still being emitted. The use of hydrogen, coupled with the conversion of that excess CO2, could result in a ∼90% drop in CO2 emissions associated with the synthesis of chemical feedstocks as compared to current industry practice.2 Methanol is already acknowledged as a viable bridge to low or net zero emission fuels and chemicals,3 as it can be used in pure form or as a blend, in internal combustion engines or in direct methanol fuel cells.4 It is a clean energy resource used to fuel cars, trucks, ships, with the UK spending roughly £150 M to import it yearly.5 Beyond that, traditional application of methanol in plastics, building blocks, paints, car parts, and construction materials are still of great interest.Industrially, methanol is currently produced at relatively high temperatures and pressures (200–300 °C, 50–100 bar) over a Cu/Zn/Al2O3 catalyst using syngas produced through the steam reformation of natural gas.6 CO2 hydrogenation to methanol has large-scale application prospects using existing infrastructure and while the conventional industrial methanol process uses syngas that contains CO2, the majority of the carbon feedstock consists of CO.7 There are a lot of catalysts that have been studied with various compositions, presenting a range of active metal sites such as Cu, Ni and Pt interfaced with oxides of Ga, In or Zn.8–13 Although scale up of a CO2 hydrogenation process has been carried out using these industrial Cu/Zn/Al2O3 catalysts,14 switching from a CO to CO2 feed presents major kinetic and stability challenges for the materials used.15 Specifically, due to the production of formate as intermediate of CO2 hydrogenation, Cu catalysts demonstrate a “poisoning” effect from high formate coverages.16,17 High surface coverages of water which occur during CO2 hydrogenation also lead to deactivation of Cu catalysts.18 To improve kinetics of CO2 hydrogenation, catalyst synthesis strategies adopting weakening the binding energy of the formate19 intermediate on the surface or promoting its conversion to methanol can be adopted. Various active sites have been reported for hydrogenation of CO2 including metallic Cu, oxidized Cu, Zn decorated Cu, and isolated Cu–Zn interfaces14,20–26 while for the commercial catalyst, there is not a consensus regarding the active interface, and both CuZn bimetallic sites27,28 and ZnO/Cu29 interfaces have been proposed as active sites. Creating highly active Cu–Zn interfaces that remain stable under CO2 hydrogenation conditions (i.e. rapid generation of water) and can overcome limitations of Cu–Zn catalysts for CO2 hydrogenation, are needed to make this process economically favourable.
Exsolution has attracted a lot of interest the last few years as a method to produce stable and highly active catalysts.30,31 As a disassembly method where nanoparticles are born from within the material, exsolution provides us the opportunity to create systems where the active sites (nanoparticles) have increased interactions with their supports as they are crystallographically aligned with and partly embedded in them.32 This leads to materials that possess high stability and increased activity as compared to their traditionally prepared analogues and there have already even been some efforts to exsolve copper from different matrixes for different applications.33–35
Herein, for the first time, we take advantage of the unique features of the exsolution method to create an intimate relationship between exsolved copper particles and ultra-high dispersed ZnO. We designed a material capable of exsolving Cu surrounded by a pool of Zn and create a powerful interface that allows us to produce methanol from CO2 at even atmospheric pressures.
Results and discussion
Materials with increased Cu–ZnO interactions and low metal loadings capable of producing methanol at atmospheric pressure
To design a material that would exsolve Cu particles but would also be stable enough to be used in such a challenging application we needed to incorporate an element on the B-site that would keep the material's structure largely intact, hence the choice of Ti. However, the need of also having a labile structure that would promote exsolution led us to the use of A-site and oxygen deficiency.31 We also decided to use Ca on the A-site as this is also known to facilitate cation mobility and hence enhance exsolution.35,36 The A-site deficiency is providing the materials with a thermodynamically stable structure after exsolution, which has the promise of withstanding high temperatures and pressures due to this and the embedded nature of the exsolved particles formed.37 Lastly, due to the oxidation states and coordination environments as well as size of Cu and Zn we designed a material that would be able to incorporate both on the B-site of the perovskite. Taking into consideration the above factors we designed and prepared a La0.5Ca0.4Cu0.1Zn0.1Ti0.8O3−δ perovskite (Fig. 1a). As observed by the stoichiometry, the material does indeed contain a minimal amount of Cu (∼3.5 wt%) as compared to the normally ∼50 wt% that is found in traditionally prepared (deposition, precipitation) materials.38,39 This is because materials prepared by exsolution have demonstrated enhanced activity for various reactions due to the enhanced interface of the nanoparticles with their support.40 | ||
| Fig. 1 Synthesizing a system with low substitution of metal and increased Cu–ZnO interactions. (a) Crystal structure of the new material. (b) Microstructure of the as-synthesised catalyst. (c) Room temperature XRD pattern. (d) Rietveld refinement analysis corresponding to a fresh sample shown in (c). | ||
The perovskite was originally synthesised with a solid-state method to ensure control over stoichiometry and was sintered at 1000 °C for 12 h. Fig. 1b demonstrates the microstructure of the as-synthesised catalyst that appears to have a small grain size, a result of the low sintering temperature. The material was indeed of high purity as indicated by X-ray diffraction and Rietveld refinement (Fig. 1c and d) seemingly adopting an orthorhombic crystal structure. The Cu and Zn dopants seem to have been fully incorporated in the structure apparent when compared to the undoped sample (Fig. S1a†) (La0.4Ca0.4TiO3).
After a 10 h reduction at 500 °C, the microstructure of the catalyst remains intact as seen in Fig. 2a, keeping its grain size. X-ray diffraction showed that the sample retained overall its crystallographic properties (Fig. 2b). Two extra peaks appear with the main one at 43.2° (Fig. 2c) and are assigned to metallic Cu particles. Using the Scherrer equation (Fig. S1b†) but also Scanning and Transmission electron microscopies we identify that the metallic copper phase did indeed exsolve in the form of hundreds (∼25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 particles per μm2) of small ∼2–6 nm sized particles (Fig. 2d–f and S2†). We did not pinpoint the presence of any other phase exsolving with any of the analyses conducted, hence we believe that we have created a sample where metallic Cu particles sit within a pool of atomically dispersed Zn ions. This is expected to produce a highly intimate interface between the two phases leading to the production of a highly active new catalyst. The overall calculated amount of Cu exsolved41 seems to be approximately 10% (∼0.32 wt%) (see calculations in ESI†) of the originally doped copper. This value was further corroborated by the refinement of the XRD pattern of the exsolved catalyst. Rietveld refinement showed a 0.3% metallic Cu with high accuracy (Rwp = 5.18) (Fig. S1c†).
000 particles per μm2) of small ∼2–6 nm sized particles (Fig. 2d–f and S2†). We did not pinpoint the presence of any other phase exsolving with any of the analyses conducted, hence we believe that we have created a sample where metallic Cu particles sit within a pool of atomically dispersed Zn ions. This is expected to produce a highly intimate interface between the two phases leading to the production of a highly active new catalyst. The overall calculated amount of Cu exsolved41 seems to be approximately 10% (∼0.32 wt%) (see calculations in ESI†) of the originally doped copper. This value was further corroborated by the refinement of the XRD pattern of the exsolved catalyst. Rietveld refinement showed a 0.3% metallic Cu with high accuracy (Rwp = 5.18) (Fig. S1c†).
 | ||
| Fig. 2 Activating the exsolved Cu–ZnO interface. (a) Microstructure of the reduced sample. (b) Room temperature XRD pattern for the as sintered and the reduced sample. (c) Close up of the Cu peak. (d) Nanostructure of the reduced sample via TEM. (e) Nanostructure of the reduced sample via SEM. (f) Particle size distribution obtained via ImageJ analysis on the SEM images. | ||
Nevertheless, the newly prepared samples were tested for their activity and selectivity to methanol in the CO2 hydrogenation reaction at ambient pressure (Fig. 3a and b) at the 200–350 °C temperature range. Our results show that the newly designed material is capable of converting CO2 to methanol starting at 200 °C. Methanol production keeps increasing throughout the temperature range tested as can be seen in Fig. 3e which is interesting as published literature and theory (Fig. S3†) suggests that methanol production would peak at ∼250–280 °C and then decrease while CO would increase with temperature.42,43 In contrast, here we see an increase in CO production, accompanied by an increase in methanol as well. The TOF values, i.e., the no. of CO2 molecules hydrogenated per Cu site per sec (s−1) increased significantly from 0.4 × 10−3 at 200 °C to 3.2 × 10−3 at 350 °C (Fig. 3c). This value is ∼4 times higher than reported value 0.4 × 10−3 (measured at high p, 3 MPa) for Cu–Zn–Zr catalysts with copper ∼45 wt%.44 This suggests that our catalyst design is of great interest potentially due to the strained nature of the copper sites or the enhanced interaction of Cu with the support i.e. ZnO phase.44 And while the main product is indeed CO, methanol formation at atmospheric pressure is remarkable. Almost the entirety of the published literature that reports methanol production from CO2 hydrogenation, is mainly under high-pressures (∼30–50 bars).45 In addition,46 the apparent activation energy (Ea) of CO2 hydrogenation was also determined (Fig. 3d) and compared to Ea values reported for other catalysts (from 65–86 kJ mol−1) in the literature17,43 (typically measured at high pressure, 2 MPa) (see Table 1). The Ea for our material was found to be ∼36 kJ mol−1, which is significantly lower than the Ea values reported for other catalysts containing much higher copper contents (>50 wt%)43 and also much lower than its impregnated equivalent (104 kJ mol−1) (Fig. S8c†) (Table 1). Our results indicate that although only a small percentage of the doped copper phase was actually exsolved, indicating that there is still a lot of room for improvement in the material design to be able to drive more copper to the surface of the support, methanol production is possible at atmospheric pressure while using merely 0.3% wt of metal loading, probably owing to the unique interface of the Cu with the Zn containing support. The Cu–Zn interface is known to be crucial for achieving CO2 conversion to methanol and the production of methanol at ambient pressure is an indication of the successful interface formation upon exsolution, which complements our characterisation data. Materials characterisation after testing using SEM, TEM, XRD and XPS showed that the micro and nanostructure of the catalyst are intact (Fig. 3f–g and S7†). Specifically, comparison of the Cu 2p and Zn 2p core levels of the exsolved sample measured before catalytic testing (Fig. 4b and d) with those measured afterwards (Fig. S7†) does not indicate any change in chemical state of Cu or Zn, suggesting that the surface is stable under reaction conditions. STEM-EDX elemental maps acquired on several areas of the CuZn exsolved sample after catalytic testing showing dispersed Cu NPs of the same size as the ones found for the reduced sample while XRD showed no additional phases being generated after testing.
 | ||
| Fig. 3 Producing methanol at atmospheric pressure. (a) Schematic showcasing the use of exsolution in CO2 to methanol process. (b) Catalytic performance, conditions (P = 1 atm; H2/CO2 = 3; GHSV = 7500 mL g cat−1 h−1) – grey points represent the support tested under the same conditions. (c) Turn over frequency as the function of temperature. (d) Arrhenius plot of TOF of CO2 conversion versus inverse temperature. (e) Catalytic stability La0.5Ca0.4Cu0.1Zn0.1Ti0.8O3−δ catalyst under the identical conditions mentioned above. (f) SEM and (g) TEM images after testing. | ||
| Material | Activation energy (kJ mol−1)/TOF (s−1) | P (bar) | MeOH yield (mmol gcat−1 h−1) | Byproducts | Ref. |
|---|---|---|---|---|---|
|
a Authors of the study conducted CO2 hydrogenation at H2:CO2 ratio different of 3:1.
|
|||||
| Ru/In2O3 (Ru wt% = 2) | E a not provided/1.77 | 1 | 0.12 | CO | 47 |
| Au–Ni–Ga (Ni wt% = 49) | E a not provided/3 | 10 | 1 | CO | 48 |
| Au/CeO2 (Au wt% = 1) | E a not provided/1.67 × 10−3 | 1 | 0.15 | CO, CH4 | 49 |
| Au/ZrO2 (Au wt% = 1) | E a not provided/2.3 × 10−2 | 5 | 0.66 | CO | 50 |
| NiGa alloy (ni wt% = 68) | E a not provided/4.8 × 10−4 | 10 | 0.29 | CO | 51 |
| NiO–Ga2O3/m-SiO2/Al2O3 fibers (Ni/Ga wt% = 7.5) | E a not provided/9 × 10−4 | 1 | 0.60 | CH4, CO | 52 |
| Cu–Al/ZnO rods (Cu wt% = 15) | 45.9/4.7 × 10−4 | 1 | 1.9 | CO | 53 |
| Cu/ZnO/Al2O3–La2O3 support (Cu wt% = 54) | E a not provided/5.21 × 10−5 | 1 | 0.60 | CO | 54 |
| Cu/ZrO2 (Cu wt% = 5) | E a not provided/4.5 × 10−2 | 1 | 0.43 | CO | 55 |
| Cu/ZnO (Cu wt% = 5) | E a not provided/7.57 × 10−4 | 1 | 0.21 | CO | 56 |
| Pd–Cu–Zn (Cu wt% = 71) | E a not provided/8.5 × 10−6 | 1 | 0.29 | CO | 57 |
| Cu/Zn/Al2O3 Commercial | E a not provided/Cu wt% not provided | 1 | 0.15 | CO | 58 |
| Cu/ZnO (Cu wt% = 15) | 75/7.8 × 10−5 | 7 | 0.11 | CO | 59 |
| CuZn–LCT (Cu wt% = 0.3) | 36/3.2 × 10−3 | 1 | 0.8 | CO | This work |
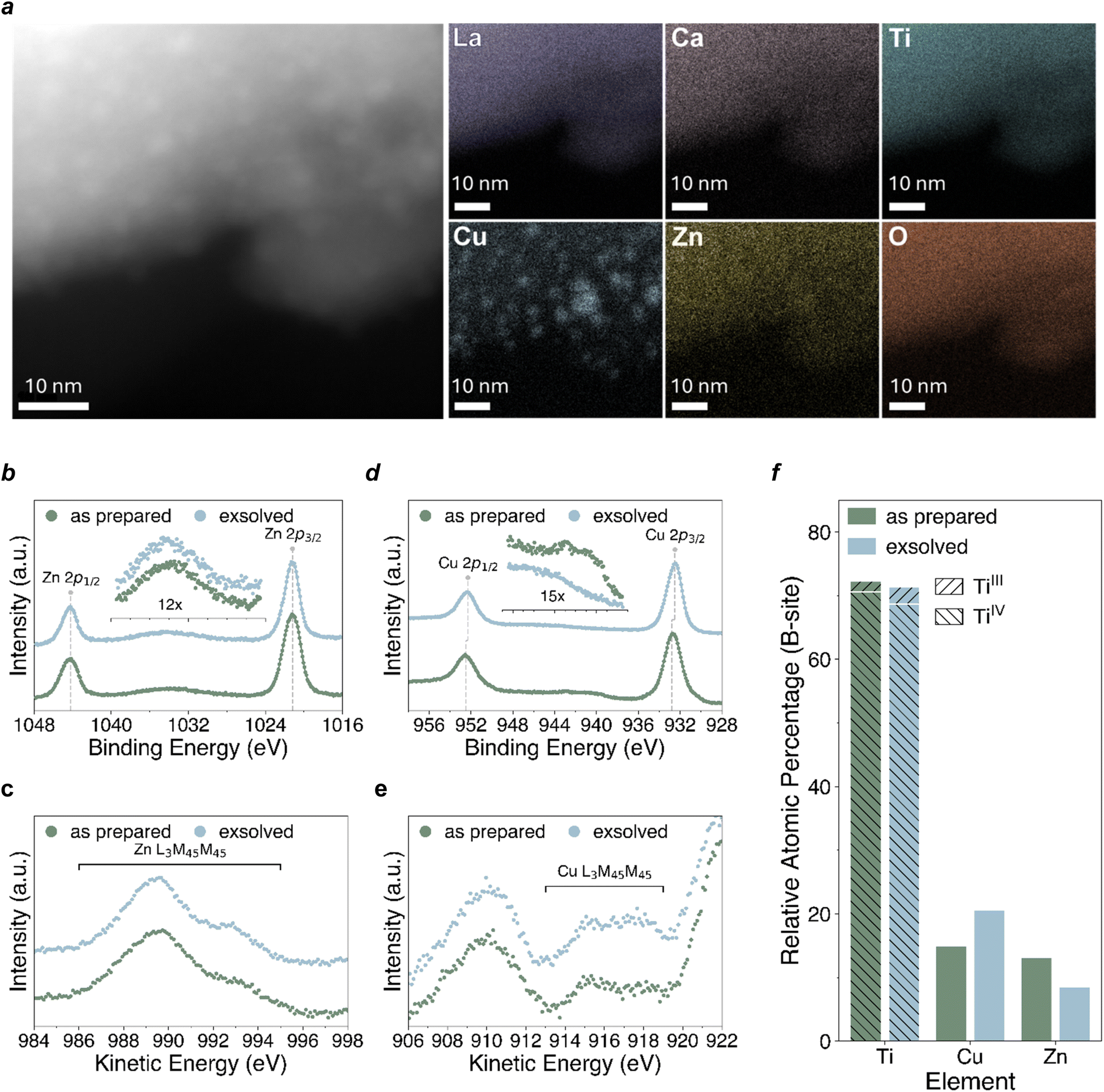 | ||
| Fig. 4 Deconvoluting the Cu–ZnO relationship. (a) STEM-EDX elemental maps of La, Ca, Ti, Cu, Zn and O acquired on grains of the reduced sample. (b) Zn 2p levels. (c) Zn L3M4,5M4,5. (d) Cu 2p core levels. (e) Cu L3M4,5M4,5 spectra. (f) Relative atomic percentage of the B-site components. | ||
Deconvoluting the Cu–ZnO relationship in the exsolved catalysts
In order to better characterise the Cu–ZnO interface and identify the material advancement presented in the exsolved system we employ transmission electron microscopy (Fig. 4a) to look at the surface of the novel material prepared here. STEM-EDX characterization indeed revealed the presence of nanometer-sized nanoparticles, evenly distributed on the surface of the analysed grains, in line with the main characteristic of exsolution. Elemental maps acquired over several regions confirmed the Cu nature of these exsolved nanoparticles, with the other elements composing the novel perovskite material found evenly distributed in the sample. The size distribution of the exsolved nanoparticles was measured to be 2–6 nm, with interparticle distances along the same scale and an intimate Zn distribution possibly surrounding the Cu particles, as partly visible form the EDX elemental distribution map.To get a deeper understanding of the relationship between copper (Cu) and the support material, X-ray photoelectron spectroscopy (XPS) was employed to measure the sample before and after exsolution. Examination of the XPS data revealed that for both the as-prepared and the exsolved sample the oxidation state and coordination environment of the A-site is essentially unchanged throughout, indicative of A-site stability throughout the redox process (the A-site core level spectra (La 3d, Ca 2p) can be found in the ESI (Fig. S4†), alongside full survey scans along with a table, Table 1, with the measured energies). The signals of La3+, Ca2+, Ti4+ and O2− are characteristic of the ones usually reported for a lanthanum calcium titanate. The La 3d and Ca 2p core levels remain essentially unchanged following reduction. Similarly, the Ti 2p core level also remains stable following reduction, aside from the slight change in the relative amounts of Ti3+ and Ti4+ (Fig. S4†), which could be linked to an increase in the concentration of oxygen vacancies upon reduction. No considerable changes are observed in the O 1s core level following reduction.
The measured Zn 2p (Fig. 4b) and Zn L3M45M45 (Fig. 4c) spectra of the as prepared and exsolved samples suggest a predominance of Zn(II). The Zn 2p3/2 peak position of the sample before and after reduction (1021.0 eV) matches closely that of ZnO in previous XPS measurements carried out by Biesinger et al.60 The Zn L3M45M45 Auger signal at 989.7 eV has the characteristic shape of Zn(II), but its position is 1.5 eV higher than has previously been observed,61 corresponding to a modified Auger parameter (2p3/2, L3M45M45) of 2011.7, which is 1.5 eV higher than that previously calculated for ZnO.60,61 This could be a result of the difference in chemical environment between Zn(II) as a dopant in the lanthanum calcium titanate perovskite lattice and Zn(II) in ZnO. The considerable similarity between the Zn XPS and Auger spectra of the as prepared and exsolved samples suggests that reduction does not have an observable effect on the chemical state of Zn. In contrast, the measured Cu 2p (Fig. 4d) and Cu L3M45M45 (Fig. 4e) spectra of the as prepared and exsolved samples suggest that all Cu(II) present in the as prepared sample is reduced to either Cu(I) or Cu metal following reduction, owing to the disappearance of the characteristic Cu(II) satellite previously observed for CuO.60,61 The Cu 2p3/2 peak position of the sample before reduction (932.8 eV) is approximately 1 eV lower than that typically measured for CuO, lying between the measured binding energies of Cu2O and CuO.60–62 Despite considerable overlap with the Ti 2s and Zn Auger peaks, it is still possible to identify the point of highest intensity in the Cu L3M45M45 Auger region, which lies at approximately 915.2 eV. This is 1.3 eV lower than that previously observed for Cu2O, and 2.6 eV lower than that observed for CuO.61 We expect that this mismatch between the measured Cu 2p3/2 and Cu L3M45M45 energies of the as-prepared sample and literature values for Cu2O and CuO could be explained by either (1) a single chemical state of Cu(II) in the lanthanum calcium titanate perovskite lattice that is distinctly different to that of both Cu2O and CuO but still presents the characteristic Cu(II) satellite, or (2) a mixture of Cu(I) and Cu(II) states that manifests as a signal lying somewhere between those observed for Cu2O and CuO. Following reduction, the disappearance of the characteristic Cu(II) satellite feature is accompanied by a 0.3 eV downwards shift in the Cu 2p3/2 peak position to 932.5 eV, and the apparent emergence of a higher kinetic energy feature in the Cu L3M45M45 Auger region at approximately 918 eV. While it is difficult to be certain of the initial chemical state of Cu in the as prepared sample, we see evidence of reduction in the exsolved sample to either Cu(I) or Cu metal.
The variation in the relative atomic percentage of the different B-site components is plotted in Fig. 4f. In comparing the as-prepared and exsolved samples, a notable decrease in Zn content and corresponding increase in Cu content are observed following reduction, while the overall Ti content remains relatively unchanged. This suggests that some form of exchange may have taken place between Cu ions in the bulk and Zn ions in the near-surface region, with Cu ions preferentially migrating towards the surface which alters their ratio after reduction. Additionally, despite the overall proportion of Ti remaining relatively unchanged, an increase in the relative proportion of Ti3+ to Ti4+ is observed following reduction. This may be linked to an increase in the concentration of positively charged oxygen vacancy defects upon reduction, which is compensated by reduction of Ti4+ to Ti3+.
Mechanistic insights into the process
To investigate the mechanism behind the observed catalytic results, operando FTIR spectroscopy was performed on the powder sample pressed into a thin disk for high-temperature gas analysis. A 75% H2:25% CO2 reagent mixture was introduced in the gas cell over a temperature range between 180–350 °C (7 bar, 15 mL min−1) and outlet signal measured by mass spectrometry (Fig. S5†). Fig. 5 shows the main results obtained while studying the reaction in real time. To investigate intermediate species evolution, the region where formates and carbonates are expected was analysed (∼1300–1650 cm−1). Temperature increase resulted in an increase of formate species present between 1560–1600 cm−1.63 coupled with a decrease of carbonate and carboxylate species associated to TiOx-based materials (1610–1630 cm−1),64 as visible in Fig. 5a. This is in line with the already proposed CO2 hydrogenation mechanism, which suggests an initial formation of carbonates at the interface between the support and the metal,64 Cu in our case. In the literature, this was followed by reduction of the formed carbonates to formates, with consequent CO evolution, and a rapid formate decomposition to surface hydroxyl groups and CO, with production of water.64 Similarly, here, peaks associated with the presence of formates on Ti4+, Cu0, and ZnO can be seen at the 1575–1587 cm−1 region (Fig. 5b), that would suggest that indeed the mechanism involved in the CO2 hydrogenation to methanol occurs via carbonate–carboxylate consumption and formate evolution over temperature. The support does play an important role in this mechanism, as it has been proposed that CO2 can be adsorbed over titania surfaces rich in oxygen vacancies, with consequent oxidation of Ti3+ to Ti4+, causing CO2 activation through electron transfer occurring from the Ti in the reduced form to the adsorbed CO2. To quantitatively verify the CO2 adsorption properties of our system, CO2 TPD tests were carried out and the results are reported in the ESI (ESI, Table 2 and Fig. S9†). Three main peaks were identified for our system when running the test up to 500 °C, specifically at ∼139 °C, ∼232 °C and ∼350–360 °C. The first peak can be attributed to CO2 desorption from weak basic sites on titanate structures, as reported in other works,65–67 whereas the higher T peaks have been attributed to the presence of medium-strength basic sites from TiO2 and Cu/TiO2 structures reduced in the same temperature range as our system,67,68 and La-based oxides,54 respectively, with the latter also suggested to increase the adsorption capacity of titanate-based samples.65 Moreover, the identification of a small amount of Ti3+ measured through XPS on the reduced sample after exsolution seems to corroborate this hypothesis, highlighting the role of the high oxygen vacancy concentration in these novel samples for the investigated reaction. A final interesting feature is the broad band observed increasing at ∼1500 cm−1 when the sample was tested at 300 °C and 350 °C (Fig. 5c), which has been assigned to the ZnO contribution in the CO2 hydrogenation reaction. Specifically, the intensity of such broad band might be indicative of the amount of defect ionized VO+ sites during catalytic reaction, and therefore contributing to the conversion of CO2 to methanol.69 This band is already present after N2 pretreatment in our case which is not surprising, since the analysed sample had undergone extensive reduction to induce exsolution (100% H2, 500 °C, 10 h), hence also inducing formation of a high extent of VO. Under operando FTIR conditions, photoionization induced by the measurement conditions would cause partial thermal ionization of such mono-ionized oxygen vacancies to VO2+, thought responsible for the presence of this broad band. The increase in intensity observed in our case under reaction mixture at 300 and 350 °C compared to what was seen for the exsolved sample pre-testing might suggest formation of VO+ donor-type defects by CO2 during methanol synthesis. The intensity of such band has recently been related to the existence of the metal-oxide (Cu–ZnO) interaction, with higher catalytic activity connected to a higher intensity of the band. This could imply that Znδ+ sites in partially reduced ZnO under reaction conditions act as CO2 adsorption sites and reaction intermediates, while the metallic Cu sites interact with H2 atoms, highlighting the synergy of the active sites present in our exsolved system69 as also previously reported in various studies.70,71 Finally, some methane was also detected by MS during the operando test. Previous works reported production of methane on pure TiO2 (011) faceted surfaces through deoxygenation of methoxy groups from Ti cations-VO vicinity. In our case, a high concentration of oxygen vacancies is produced after nanoparticle exsolution, which could also explain the amount of methane observed.72
 | ||
| Fig. 5 Mechanistic insights into the process. Operando FTIR spectra for the exsolved catalyst showing the evolution of the reaction intermediates in the (a) 1630–1560 cm−1 region, (b and c) 1700–1200 cm−1 region highlighting the different species evolved during the operando CO2 hydrogenation reaction experiment (75% H2:25% CO2, 7 bar, 15 mL min−1, NTP). | ||
Post testing analysis verified the presence of nanoscale Cu nanoparticles still present on the support, surrounded by a pool of ZnO (Fig. S6†) which demonstrates the stability of the materials presented here even after submitting them to harsh reaction environments.
Conclusions
There is a wide range of technologies that can help us achieve decarbonisation of our energy supply and achieve our net-zero pledges but in addition to this, we need to utilise the CO2 still being emitted. CO2 hydrogenation to methanol, a versatile chemical and fuel, can be performed using existing infrastructure but so far material challenges have hindered its wide application. We have designed and prepared a material with Cu exsolved nanoparticles within a pool of ZnO. The material is able to produce methanol starting at 200 °C and atmospheric pressure with only 0.3 wt% of Cu on its surface. We use atmospheric pressure methanol synthesis activity to evaluate the catalytic implications and the success of our strategy to form Cu–ZnO exsolved interfaces. X-ray photoelectron spectroscopy, transition electron microscopy and operando infrared spectroscopy indicate that the intimate relationship between the particles and support could be responsible for this. Lastly, although, the material concept demonstrated here requires optimisation in order to drive more Cu sites on the surface of the support, and hence make it even more active, it represents an important advancement in the field of CO2 hydrogenation reactions. This is also because there is a growing interest in the field of catalysis on non-equilibrium processes, such as forced periodic oscillation73 or catalytic resonance theory to overcome Sabatier curve rate limitations as well as thermodynamic equilibrium limitations at milder conditions,74 as well as on dynamic integrated CO2 capture and conversion processes where steady-state thermodynamic conversion limitations no longer apply.75 Hence designing catalysts with low-pressure activity for classically high-pressure reactions such as methanol synthesis holds value beyond the realm of steady-state operation.Data availability
The research data for this article can be accessed at DOI: https://doi.org/10.15126/surreydata.901036.Author contributions
Sanaz Soodi data curation; formal analysis; investigation; methodology; writing – original draft; Eleonora Calì data curation; formal analysis; investigation; methodology; writing – original draft; Shailza Saini data curation; formal analysis; investigation; methodology; writing – original draft; William S. Skinner data curation; formal analysis; writing – original draft; Alex Martinez Martin formal analysis; investigation; Gwilherm Kerherve formal analysis; investigation; methodology; Marco Pietro Mezzapesa data curation; formal analysis; Samir Bensaid methodology, funding acquisition; resources; writing – review & editing David J. Payne methodology, funding acquisition; resources; writing – review & editing, Melis S. Duyar methodology, supervision, writing – review & editing Kalliopi Kousi conceptualization; funding acquisition; methodology; project administration; resources; supervision; writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The research leading to these results was funded by the School of Chemistry and Chemical Engineering (Chemical and Process Engineering), by the Royal Society RGS\R2\222062, and the Engineering and Physical Science Research Council EP/Y015487/1. E. C. gratefully acknowledges support from the European Union HORIZON-MSCA-2021-PF-01 Marie-Curie Project: 101063146 (MEXCAT).References
- United Nations, Net Zero Coalition, https://www.un.org/en/climatechange/net-zero-coalition, accessed April 27, 2024 Search PubMed.
- D. Wang, J. Li, W. Meng, Z. Liao, S. Yang, X. Hong, H. Zhou, Y. Yang and G. Li, J. Cleaner Prod., 2023, 412, 137388 CrossRef CAS.
- M. Lashgari and S. Soodi, RSC Adv., 2020, 10, 15072–15078 RSC.
- Q. Shi, Y. He, X. Bai, M. Wang, D. A. Cullen, M. Lucero, X. Zhao, K. L. More, H. Zhou, Z. Feng, Y. Liu and G. Wu, Energy Environ. Sci., 2020, 13, 3544–3555 RSC.
- UK, https://www.statista.com/statistics/482190/methanol-import-value-in-the-united-kingdom-uk/, accessed April 27, 2024.
- J. Kotowicz, M. Brzęczek, A. Walewska and K. Szykowska, Energies, 2022, 15, 1480 CrossRef CAS.
- J. Zuo, K. Chen, J. Zheng, L. Ye and Y. Yuan, J. CO2 Util., 2021, 52, 101699 CrossRef CAS.
- Y. Wang, Y. Liu, L. Tan, X. Lin, Y. Fang, X. F. Lu, Y. Hou, G. Zhang and S. Wang, J. Mater. Chem. A, 2023, 11, 26804–26811 RSC.
- A. Gallo, J. L. Snider, D. Sokaras, D. Nordlund, T. Kroll, H. Ogasawara, L. Kovarik, M. S. Duyar and T. F. Jaramillo, Appl. Catal., B, 2020, 267, 118369 CrossRef CAS.
- S. K. Sharma, B. Paul, R. S. Pal, P. Bhanja, A. Banerjee, C. Samanta and R. Bal, ACS Appl. Mater. Interfaces, 2021, 13, 28201–28213 CrossRef CAS PubMed.
- Z. Shi, M. Pan, X. Wei and D. Wu, Int. J. Energy Res., 2022, 46, 1285–1298 CrossRef CAS.
- O. Martin, A. J. Martín, C. Mondelli, S. Mitchell, T. F. Segawa, R. Hauert, C. Drouilly, D. Curulla-Ferré and J. Pérez-Ramírez, Angew. Chem., 2016, 128, 6369–6373 CrossRef.
- T. Pinheiro Araújo, C. Mondelli, M. Agrachev, T. Zou, P. O. Willi, K. M. Engel, R. N. Grass, W. J. Stark, O. V. Safonova, G. Jeschke, S. Mitchell and J. Pérez-Ramírez, Nat. Commun., 2022, 13, 5610 CrossRef PubMed.
- H. Zhang, J. Chen, X. Han, Y. Pan, Z. Hao, S. Tang, X. Zi, Z. Zhang, P. Gao, M. Li, J. Lv and X. Ma, Ind. Eng. Chem. Res., 2024, 63, 6210–6221 CrossRef CAS.
- J. Zhong, X. Yang, Z. Wu, B. Liang, Y. Huang and T. Zhang, Chem. Soc. Rev., 2020, 49, 1385–1413 RSC.
- S. Kuld, M. Thorhauge, H. Falsig, C. F. Elkjær, S. Helveg, I. Chorkendorff and J. Sehested, Science, 2016, 352, 969–974 CrossRef CAS PubMed.
- F. Studt, M. Behrens, E. L. Kunkes, N. Thomas, S. Zander, A. Tarasov, J. Schumann, E. Frei, J. B. Varley, F. Abild-Pedersen, J. K. Nørskov and R. Schlögl, ChemCatChem, 2015, 7, 1105–1111 CrossRef CAS.
- A. Beck, M. A. Newton, L. G. A. van de Water and J. A. van Bokhoven, Chem. Rev., 2024, 124, 4543–4678 CrossRef CAS PubMed.
- M. S. Duyar, C. Tsai, J. L. Snider, J. A. Singh, A. Gallo, J. S. Yoo, A. J. Medford, F. Abild-Pedersen, F. Studt, J. Kibsgaard, S. F. Bent, J. K. Nørskov and T. F. Jaramillo, Angew. Chem., Int. Ed., 2018, 57, 15045–15050 CrossRef CAS PubMed.
- R. Dalebout, L. Barberis, G. Totarella, S. J. Turner, C. La Fontaine, F. M. F. de Groot, X. Carrier, A. M. J. van der Eerden, F. Meirer and P. E. de Jongh, ACS Catal., 2022, 12, 6628–6639 CrossRef CAS PubMed.
- D. Allam, S. Bennici, L. Limousy and S. Hocine, C. R. Chim., 2019, 22, 227–237 CrossRef CAS.
- C. Wu, L. Lin, J. Liu, J. Zhang, F. Zhang, T. Zhou, N. Rui, S. Yao, Y. Deng, F. Yang, W. Xu, J. Luo, Y. Zhao, B. Yan, X.-D. Wen, J. A. Rodriguez and D. Ma, Nat. Commun., 2020, 11, 5767 CrossRef CAS PubMed.
- J. S. Hayward, The Effects of Secondary Oxides on Copper-Based Catalysts for Green Methanol Synthesis, ChemCatChem, 2017, 9, 1655–1662 CrossRef CAS.
- Z. Zhong, U. Etim and Y. Song, Front. Energy Res., 2020, 8 DOI:10.3389/fenrg.2020.545431.
- Z. Zhang, X. Chen, J. Kang, Z. Yu, J. Tian, Z. Gong, A. Jia, R. You, K. Qian, S. He, B. Teng, Y. Cui, Y. Wang, W. Zhang and W. Huang, Nat. Commun., 2021, 12, 4331 CrossRef CAS PubMed.
- M. Lashgari, S. Soodi and P. Zeinalkhani, J. CO2 Util., 2017, 18, 89–97 CrossRef CAS.
- K. Stangeland, H. Li and Z. Yu, Energy, Ecol. Environ., 2020, 5, 272–285 CrossRef.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 CrossRef CAS PubMed.
- C. Tang, K. Kousi, D. Neagu and I. S. Metcalfe, Chem.–Eur. J., 2021, 27, 6666–6675 CrossRef CAS PubMed.
- K. Kousi, C. Tang, I. S. Metcalfe and D. Neagu, Small, 2021, 17, 2006479 CrossRef CAS PubMed.
- D. Neagu, J. T. S. Irvine, J. Wang, B. Yildiz, A. K. Opitz, J. Fleig, Y. Wang, J. Liu, L. Shen, F. Ciucci, B. A. Rosen, Y. Xiao, K. Xie, G. Yang, Z. Shao, Y. Zhang, J. Reinke, T. A. Schmauss, S. A. Barnett, R. Maring, V. Kyriakou, U. Mushtaq, M. N. Tsampas, Y. Kim, R. O'Hayre, A. J. Carrillo, T. Ruh, L. Lindenthal, F. Schrenk, C. Rameshan, E. I. Papaioannou, K. Kousi, I. S. Metcalfe, X. Xu and G. Liu, J. Phys.: Energy, 2023, 5, 031501 CAS.
- S. Jo, H. G. Jeong, Y. H. Kim, D. Neagu and J. Myung, Appl. Catal., B, 2021, 285, 119828 CrossRef CAS.
- L. Fu, J. Zhou, J. Yang, Z. Lian, J. Wang, Y. Cheng and K. Wu, Appl. Surf. Sci., 2020, 511, 145525 CrossRef CAS.
- M. Wang, E. I. Papaioannou, I. S. Metcalfe, A. Naden, C. D. Savaniu and J. T. S. Irvine, Adv. Funct. Mater., 2023, 33, 2302102 CrossRef CAS.
- J. G. Lee, J.-H. Myung, A. B. Naden, O. S. Jeon, Y. G. Shul and J. T. S. Irvine, Adv. Energy Mater., 2020, 10, 2070044 CrossRef CAS.
- D. Neagu, G. Tsekouras, D. N. Miller, H. Ménard and J. T. S. Irvine, Nat. Chem., 2013, 5, 916–923 CrossRef CAS PubMed.
- D. Kordus, J. Jelic, M. Lopez Luna, N. J. Divins, J. Timoshenko, S. W. Chee, C. Rettenmaier, J. Kröhnert, S. Kühl, A. Trunschke, R. Schlögl, F. Studt and B. Roldan Cuenya, J. Am. Chem. Soc., 2023, 145, 3016–3030 CrossRef CAS.
- M. Kourtelesis, K. Kousi and D. I. Kondarides, Catalysts, 2020, 10, 183 CrossRef CAS.
- M. A. Naeem, P. M. Abdala, A. Armutlulu, S. M. Kim, A. Fedorov and C. R. Müller, ACS Catal., 2020, 10, 1923–1937 CrossRef CAS.
- D. Neagu, E. I. Papaioannou, W. K. W. Ramli, D. N. Miller, B. J. Murdoch, H. Ménard, A. Umar, A. J. Barlow, P. J. Cumpson, J. T. S. Irvine and I. S. Metcalfe, Nat. Commun., 2017, 8, 1855 CrossRef PubMed.
- M. Ren, Y. Zhang, X. Wang and H. Qiu, Catalysts, 2022, 12, 403 CrossRef CAS.
- R. Ye, L. Ma, J. Mao, X. Wang, X. Hong, A. Gallo, Y. Ma, W. Luo, B. Wang, R. Zhang, M. S. Duyar, Z. Jiang and J. Liu, Nat. Commun., 2024, 15, 2159 CrossRef CAS PubMed.
- G. Bonura, M. Cordaro, C. Cannilla, F. Arena and F. Frusteri, Appl. Catal., B, 2014, 152–153, 152–161 CrossRef CAS.
- X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS PubMed.
- A. Prašnikar, A. Pavlišič, F. Ruiz-Zepeda, J. Kovač and B. Likozar, Ind. Eng. Chem. Res., 2019, 58, 13021–13029 CrossRef.
- B. Deng, H. Song, Q. Wang, J. Hong, S. Song, Y. Zhang, K. Peng, H. Zhang, T. Kako and J. Ye, Appl. Catal., B, 2023, 327, 122471 CrossRef CAS.
- M. S. Duyar, A. Gallo, J. L. Snider and T. F. Jaramillo, J. CO2 Util., 2020, 39, 101151 CrossRef CAS.
- A. Vourros, I. Garagounis, V. Kyriakou, S. A. C. Carabineiro, F. J. Maldonado-Hódar, G. E. Marnellos and M. Konsolakis, J. CO2 Util., 2017, 19, 247–256 CrossRef CAS.
- Y. Hartadi, D. Widmann and R. J. Behm, ChemSusChem, 2015, 8, 456–465 CrossRef CAS PubMed.
- L. F. Rasteiro, M. A. L. S. Rossi, J. M. Assaf and E. M. Assaf, Catal. Today, 2021, 381, 261–271 CrossRef CAS.
- P. Chen, G. Zhao, Y. Liu and Y. Lu, Appl. Catal., A, 2018, 562, 234–240 CrossRef CAS.
- S. Du, W. Tang, X. Lu, S. Wang, Y. Guo and P.-X. Gao, Adv. Mater. Interfaces, 2018, 5, 1700730 CrossRef.
- M. Kourtelesis, K. Kousi and D. I. Kondarides, Catalysts, 2020, 10, 183 CrossRef CAS.
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 CrossRef CAS.
- J. Díez-Ramírez, F. Dorado, A. R. de la Osa, J. L. Valverde and P. Sánchez, Ind. Eng. Chem. Res., 2017, 56, 1979–1987 CrossRef.
- J. Díez-Ramírez, J. A. Díaz, P. Sánchez and F. Dorado, J. CO2 Util., 2017, 22, 71–80 CrossRef.
- Y.-N. Yang, C.-W. Huang, V.-H. Nguyen and J. C.-S. Wu, Catal. Commun., 2022, 162, 106373 CrossRef CAS.
- A. Karelovic and P. Ruiz, Catal. Sci. Technol., 2015, 5, 869–881 RSC.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. St. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- G. Deroubaix and P. Marcus, Surf. Interface Anal., 1992, 18, 39–46 CrossRef CAS.
- S. W. Goh, A. N. Buckley, R. N. Lamb, R. A. Rosenberg and D. Moran, Geochim. Cosmochim. Acta, 2006, 70, 2210–2228 CrossRef CAS.
- G. Bonura, S. Todaro, L. Frusteri, I. Majchrzak-Kucęba, D. Wawrzyńczak, Z. Pászti, E. Tálas, A. Tompos, L. Ferenc, H. Solt, C. Cannilla and F. Frusteri, Appl. Catal., B, 2021, 294, 120255 CrossRef CAS.
- L. F. Bobadilla, J. L. Santos, S. Ivanova, J. A. Odriozola and A. Urakawa, ACS Catal., 2018, 8, 7455–7467 CrossRef CAS.
- Z. Wang, L. Huang, B. Su, J. Xu, Z. Ding and S. Wang, Chem.–Eur. J., 2020, 26, 517–523 CrossRef CAS PubMed.
- Y. Ma, J. Liu, M. Chu, J. Yue, Y. Cui and G. Xu, Catal. Lett., 2020, 150, 1418–1426 CrossRef CAS.
- H. Deng, X. Zhu, Z. Chen, K. Zhao and G. Cheng, Carbon Lett., 2022, 32, 1671–1680 CrossRef.
- C. Zhang, L. Wang, U. J. Etim, Y. Song, O. M. Gazit and Z. Zhong, J. Catal., 2022, 413, 284–296 CrossRef CAS.
- S. M. Fehr, K. Nguyen and I. Krossing, ChemCatChem, 2022, 14, e202101500 CrossRef CAS.
- X. Liu, J. Luo, H. Wang, L. Huang, S. Wang, S. Li, Z. Sun, F. Sun, Z. Jiang, S. Wei, W.-X. Li and J. Lu, Angew. Chem., Int. Ed., 2022, 61, e202202330 CrossRef CAS PubMed.
- X. Liu, H. Wang and J. Lu, J. Catal., 2024, 436, 115561 CrossRef CAS.
- M. Manzoli, A. Chiorino and F. Boccuzzi, Appl. Catal., B, 2005, 57, 201–209 CrossRef CAS.
- J. Leipold, C. Seidel, D. Nikolic, A. Seidel-Morgenstern and A. Kienle, Comput. Chem. Eng., 2023, 175, 108285 CrossRef CAS.
- M. A. Ardagh, T. Birol, Q. Zhang, O. A. Abdelrahman and P. J. Dauenhauer, Catal. Sci. Technol., 2019, 9, 5058–5076 RSC.
- L.-P. Merkouri, A. I. Paksoy, T. Ramirez Reina and M. S. Duyar, ACS Catal., 2023, 13, 7230–7242 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta05812g |
| ‡ All authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |