
Synergistic influence of multivalent Ruδ+ on a CeOx nanocatalyst for self-powered efficient electrochemical water splitting†
Papri
Mondal
 and
Sujoy
Baitalik
*
and
Sujoy
Baitalik
*
Department of Chemistry, Inorganic Chemistry Section, Jadavpur University, Kolkata 700032, India. E-mail: sujoy.baitalik@jadavpuruniversity.in; sbaitalik@hotmail.com; Fax: +91 33 24137121; Tel: +91 33 24572032 Tel: +91 033 2414 6666
First published on 25th November 2024
Abstract
In spite of the rapid development of portable water-splitting devices based on rechargeable metal–air batteries, there is a scarcity of efficient multifunctional electrocatalysts (ECs) for oxygen reduction, oxygen evolution, and hydrogen evolution reactions (ORR/OER/HER). Herein, a multicomponent–multivalent coupling strategy involving surface-interface engineering is adopted for the development of an efficient trifunctional (TF) EC through the integration of dominant CeOx (CO) with a small fraction of Ru0 (R0) and RuOx (RO). Under tuned metal valency-composition, the designed multi-interfacial CO/R0/RO nanocomposite (NComp) shows enhanced ORR (E1/2 of 0.936 V), OER [overpotential (η10) of 166 mV at 10 mA cm−2], and HER (η10 of 58 mV) activity. Moreover, the drastically enhanced activity of CO/R0/RO is achieved with a small cell voltage of as low as 1.49 V, required to accomplish overall water splitting (OWS) at 10 mA cm−2. In addition, a large peak power density of 376.4 mW cm−2 and a low charge–discharge voltage gap of 0.247 V are observed for zinc–air batteries (ZABs) with admirable cycling stability (over 2000 h/12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles). In addition to ZABs, CO/R0/RO NComp is employed to mimic the functionality of Li–air batteries (LABs). For practical utility, an integrated device consisting of a symmetric two-electrode water splitting electrolyzer powered by two series-connected ZABs is realized using CO/R/RO as a single catalyst, which efficiently drives OWS and effectively produces H2 and O2 with production rates of 399.3 and 199.6 μL min−1, respectively. Moreover, the observed high faradaic efficiencies of 98.9% for the HER and 98.4% for the OER illustrate the effective energy conversion and high efficiency of this self-powered system. Thus, this work presents a feasible strategy of surface engineering via alteration of surface electronic states and concurrent fabrication of value-added highly efficient TF ECs, paving the way for their widespread adoption in energy-related devices.
000 cycles). In addition to ZABs, CO/R0/RO NComp is employed to mimic the functionality of Li–air batteries (LABs). For practical utility, an integrated device consisting of a symmetric two-electrode water splitting electrolyzer powered by two series-connected ZABs is realized using CO/R/RO as a single catalyst, which efficiently drives OWS and effectively produces H2 and O2 with production rates of 399.3 and 199.6 μL min−1, respectively. Moreover, the observed high faradaic efficiencies of 98.9% for the HER and 98.4% for the OER illustrate the effective energy conversion and high efficiency of this self-powered system. Thus, this work presents a feasible strategy of surface engineering via alteration of surface electronic states and concurrent fabrication of value-added highly efficient TF ECs, paving the way for their widespread adoption in energy-related devices.
Introduction
With regard to the ever-growing global energy demand and environmental pollution, rechargeable metal–air batteries and electrocatalytic overall water splitting (OWS) systems have attracted immense attention as green and renewable energy sources due to their high efficiency, sustainability, and environmental friendliness.1–9 These electrochemical systems principally rely on three half-reactions, viz. ORR, OER, and HER. In particular, the ORR and OER are two key reactions for rechargeable metal–air batteries10–12 while the OER and HER are core processes for OWS.13–17 Usually, Pt-based materials are the universal choice for the HER and ORR18–20 while Ru/Ir-based oxides represent the state-of-the-art catalysts for the OER.21–23 But the large-scale utilization of platinum metals is mainly hindered by their scarcity, high cost, and limited multifunctional catalytic activities.24–26 Moreover, the sluggish kinetics, large overpotential (η), and requirement of unreasonable quantities of precious metals for these multielectron reactions are the major bottlenecks for their effective utilization in multifunctional electrocatalysis.27–32 Therefore, fabrication of cost-effective and highly efficient TF ECs is highly desirable.33–36 Recently, research on developing cost-effective TF ECs has primarily focused on two important aspects, viz. either decreasing the noble metal (NM) content or designing NM-free catalysts.A wide variety of NM-free transition metal-based (such as Fe, Co, Ni, Cu and Zn) oxides17,37 hydroxides,38 and chalcogenides,39,40 to name a few, have already been developed as ECs.41–44 But in terms of both catalytic activity and durability, catalysts based on NM are found to be superior to their NM-free counterparts, especially when used as TF ECs for the ORR/OER/HER simultaneously. To this end, substantial efforts have been devoted to minimizing the NM content in the catalyst as much as possible without compromising their catalytic activity. Ru-based catalysts are now drawing attention as a probable substitute for Rh/Ir/Pt, primarily because of their much lower cost. Additionally, Ru is less corrosive but exhibits Pt-like hydrogen adsorption properties. Hence, our objective is to design Ru-based ECs that can efficiently catalyze the ORR, OER and HER. Profitably, the Ru–*H interaction is strong during the HER, while the adsorption of *OH on Ru is very weak for the OER. This unbalanced adsorption capability of the reaction intermediates leads to sluggish reaction kinetics. In addition, pure Ru-based materials possess unsatisfactory ORR performance due to their improper electronic structure. Thus, appropriate electronic structural modification of Ru is required for its efficient utilization as multifunctional ECs.45–48 Compositing Ru with other non-precious metal oxides could be an effective way which not only adjusts the required properties but also reduces the cost of the resulting catalyst. Such fabrication provides a large heterogeneous interface, which in turn facilitates efficient charge transport. Under this scenario, we decided to composite Ru with CeOx (CO) because of its facile and reversible inter-conversion between Ce3+ and Ce4+ states.49–53 In addition, CO possesses some other interesting features including more oxygen vacancy defects, faster oxygen ion conductivity, a moderate band gap, multiple valence states, and high oxygen utilization ability.
The present work aims to fabricate a low-cost, easily synthesizable multicomponent–multivalent (MV) smart electrocatalytic system with the merit of exhibiting superior TF activity. Herein, our primary emphasis is to develop suitable interface structures and increase the active site density, which in turn can boost the catalytic efficiency, selectivity, and stability during the ORR, OER, and HER. To this end, we have designed a CO-based nanocomposite (NComp) and thoroughly studied the effect of MV Ruδ+-loading on their electrocatalytic performances. The initial introduction of Ru0 (R0) over CO leads to improved TF activity of the CO/R0 NComp and this improved activity is further boosted substantially upon loading of an ultralow amount of RuOx (RO). Detailed electrochemical investigations indicate that the catalytic performance of the designed catalysts strongly relies on the oxidation states of Ce and Ru metals. The coexistence of Ce3+ and Ce4+ together with Ru0, Ru3+ and Ru4+ improves the adsorption of reactive intermediates during the ORR/OER/HER and owing to these unique merits, the optimized CO/R0/RO shows excellent TF activities for the ORR (0.936 V of E1/2), OER (η10 = 166 mV), and HER (η10 = 58 mV) which are far superior to those of CO, CO/R0, CO/RO counterparts as well as most of the recently reported advanced TF ECs. The symmetrically assembled two-electrode OWS electrolyzer delivers a current density of 10 mA cm−2 at a cell voltage of only 1.49 V and exhibits excellent durability even at reasonably large current density. Moreover, when CO/R0/RO is used as the air electrode EC, the as-constructed ZAB exhibits a large peak power density of 376.4 mW cm−2 with a low charge–discharge voltage gap of 0.247 V over 2000 h/12000 cycles. In addition to ZABs, the CO/R0/RO NComp is employed to mimic the functionality of LABs. Based on its excellent TF features toward the ORR, OER and HER, a self-driven water splitting system powered by two series connected ZABs is constructed, employing CO/R0/RO as the sole catalyst which successfully shows the formation of O2 and H2 bubbles at the respective electrodes, achieving a high H2 evolution rate of 399.3 μL min−1. Hence, this work offers a feasible strategy for rationally integrating various active sites into a single EC to enhance its multifunctional efficiency in renewable energy applications.
Results and discussion
Synthesis and characterization
Scheme 1 depicts a three-step synthetic process of R0 and RO anchored CO, the details of which are illustrated in the ESI (Section S1.2–S1.5†). Additionally, CO/RO, Ru0 (R0), RuOx (RO), and R0/RO are prepared for the sake of activity comparison (see the ESI†). The introduction of R0 onto CO is performed through a deposition–precipitation method. It is probably a physical adsorption phenomenon which involves weak interactions where the respective components (CO, R0, and RO) adhere to each other without forming strong chemical bonds. Herein, the pH point of zero charge (pHpzc) of the synthesized CO nanorods is measured to be 6.7, which indicates that the surface of CO has net electrical neutrality at pH 6.7 (Fig. S1a†). Below pH 6.7, the net surface charge is expected to be positive, while the pH value above 6.7 corresponds to the negative surface. Thus, according to the measured pHpzc value of CO (pHpzc = 6.7), it is expected that during the synthesis of CO/R0 (pH of reaction medium: ∼7.2) and CO/R0/RO (pH of reaction medium: ∼9), initially the Ru species from Ru(NO)(NO3)3 should randomly adsorb on the CO surface owing to the negative charges present on its surface (as pH > pHpzc). Then further reaction takes place for the subsequent development of CO/R0 and CO/R0/RO (Section S1.3 and S1.4).† The approximate molar ratio of CO, R0 and RO in the catalyst is 1:0.0011:0.000011.
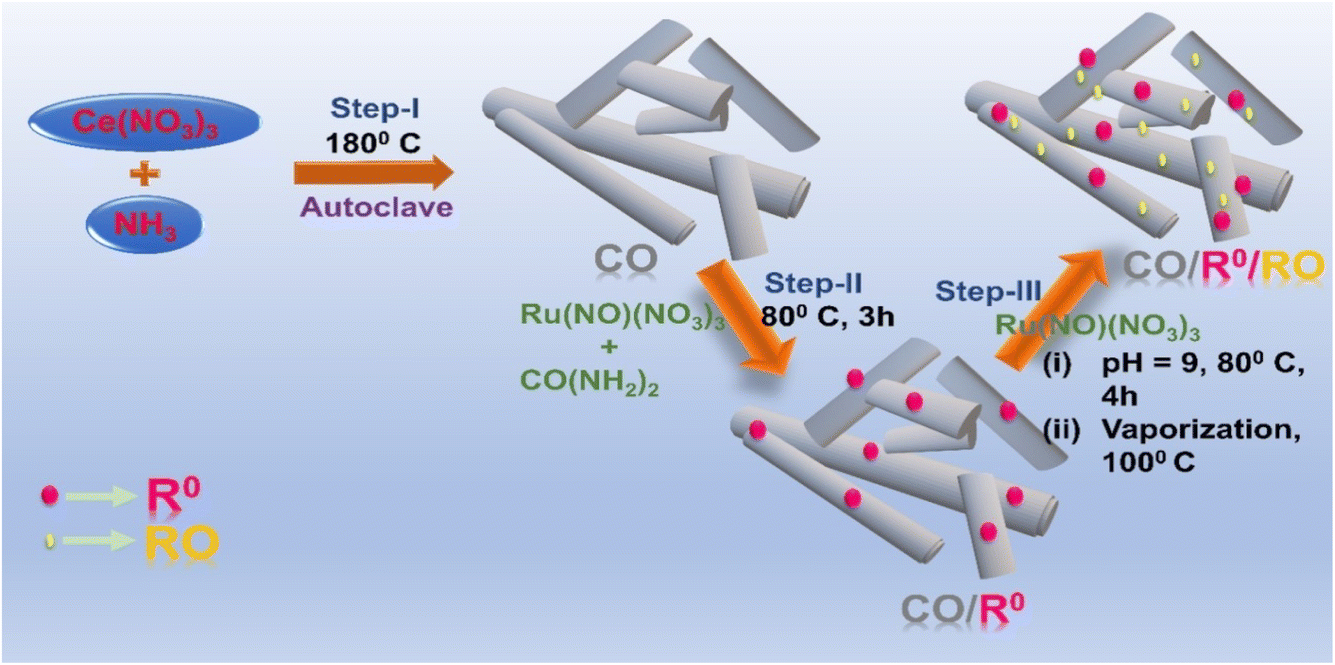 | ||
| Scheme 1 An illustration of the synthetic scheme of CO (Step-I), CO/R0 (Step-II), and CO/R0/RO (Step-III). | ||
The X-ray diffraction (XRD) technique is used to study the effect of Ce/O and Ru/O ratios on the crystalline phase of the respective materials as well as their compositions. The well indexed diffraction pattern shown in Fig. 1a, S1b and S2 (ESI†) confirms good crystallization and effectual fabrication of CeO2 with R0 and RuO2, the detailed discussion of which is presented in Section S2.2 of the ESI.† Further characterization of the chemical compositions and chemical valences of the materials is conducted through X-ray photoelectron spectroscopy (XPS) analyses. High-resolution Ce 3d XPS spectra for each of CO, CO/R0, CO/RO and CO/R0/RO (Fig. 1b and S4a, ESI†) exhibit two multiplets, denoted as v and u, corresponding to the spin–orbit coupling of Ce 3d5/2 and Ce 3d3/2, respectively. Herein, each spin–orbit component of Ce 3d derived from Ce3+ oxides refers to two u0/v0 and u′/v′ features, while each component of Ce4+ oxides is dominated by three distinct features, viz. u/v, u′′/v′′, and u′′′/v′′′ (Table S1, ESI†).54–57 Thus, the presence of characteristic and well-defined peaks in the Ce 3d spectrum suggests the coexistence of both Ce3+ and Ce4+ in CO, CO/R0, CO/RO, and CO/R0/RO. Two distinct peaks at 283.4 and 279.0 eV in the Ru 3d XPS spectrum, assignable to the Ru 3d3/2 and 3d5/2 states of metallic Ru, respectively, confirm the effective formation of R0 over CO (denoted CO/R0) (Fig. 1(c(i))). The Ru 3d XPS signal shown in Fig. S4b (ESI†) can be divided into two spin–orbit doublets and two shakeup satellites. Two strong peaks at 284.9 and 280.8 eV can be designated as 3d3/2 and 3d5/2 for Ru4+, while two satellite peaks at 284.2 and 279.8 eV correspond to 3d3/2 and 3d5/2 states of Ru3+, indicating the formation of mixed valence RuOx (RO) over CO (denoted CO/RO).58,59 Finally, the deconvolution of the Ru 3d XPS signal in Fig. 1(c(ii)) leads to two additional shakeup satellites at 283.5 and 279.0 eV, corresponding to the 3d3/2 and 3d5/2 states of R0, which in turn confirm the formation of CO/R0/RO. It is worth noting that a small C 1s peak at 284.8 eV is observed in all cases for binding energy calibration. Additionally, the presence of mixed valence Ruδ+ (δ = 0, 3, and 4) species in CO/R0/RO is confirmed by Ru 3p binding energy peaks (3p1/2: 486.4 eV, 483.3 eV, and 481.6 eV; 3p3/2: 464.1 eV, 459.8 eV, and 457.2 eV) as shown in Fig. S5 (ESI†).60 Thus, the XPS results confirm the presence of both Ru3+ and Ru4+ along with Ru0 which in turn is the testimony for the successful formation of CO/R0/RO. Further confirmation of the existence of mixed-valence states for both Ce (Ce3+ and Ce4+) and Ru (Ru0, Ru3+ and Ru4+) has been done via X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectroscopic techniques [Fig. 1e–i].61,62 The O1s XPS spectra of the materials are also thoroughly examined (Fig. 1d and S4c, ESI).† The overlapping broad peaks in O1s spectra of CO/R0 (Fig. 1(d(ii))), CO/RO (Fig. S4c, ESI†), and CO/R0/RO (Fig. 1(d(iii))) can be deconvoluted into three distinct components, viz. lattice oxygen (Ol), oxygen of adventitious surface –OH groups (OOH), and oxygen of surface chemisorbed H2O groups (Oc). On the other hand, the O1s spectra of CO (Fig. 1(d(i))) possess only two peaks that can be assigned to Ol and OOH. Herein, the surface-active oxygen (Oa) is defined as the sum of OOH and Oc (Oa = OOH + Oc). The intense binding energy peak at ∼529.1 eV corresponds to Ol, while the peak in range of 530.9–531.2 eV is attributed to OOH sites for all the four materials. An additional peak is also observed within the range of 532.4–533.1 eV in CO/R0, CO/RO, and CO/R0/RO which can be attributed to Oc at surface defects or to adsorbed H2O.63–67
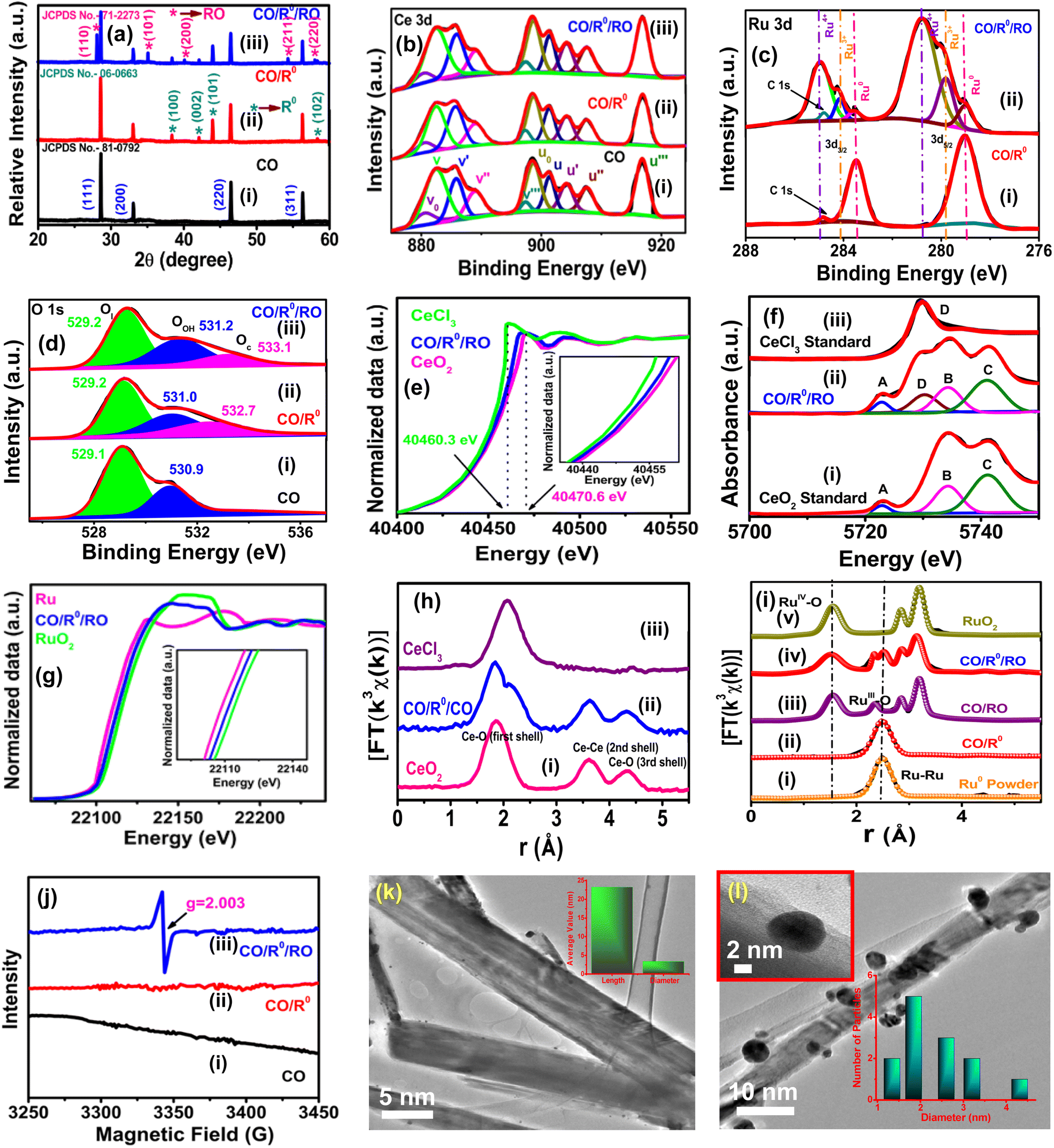 | ||
| Fig. 1 (a) XRD pattern of CO, CO/R0, and CO/R0/RO; XPS spectra of Ce 3d (b), Ru 3d (c), and O 1s (d) of the materials; (e) Ce K-edge XANES spectra of CO/R0/RO together with CeCl3 and CeO2 as references; the inset shows partially enlarged spectra; (f) Ce L3 XANES spectra of CO/R0/RO along with CeCl3 and CeO2 standards; (g) Ru K-edge XANES spectra of CO/R0/RO and references (Ru0 powder and RuO2) (the inset shows partially enlarged XANES spectra); (h) Fourier transform (FT) (magnitude) Ce K-edge k3-weighted EXAFS spectra of CO/R0/RO together with CeCl3 and CeO2 as the references in R space; (i) k3-weighted Ru K-edge FT-EXAFS (magnitude) spectra of CO/R0, CO/RO, CO/R0/RO, and references (Ru0 powder and RuO2) in R space; (j) EPR spectra of CO, CO/R0, and CO/R0/RO; TEM images of CO (k, the inset shows the particle size distribution) and CO/R0/RO (l, insets show the magnified TEM image and particle size distribution). | ||
To determine the relative surface-active oxygen content, the peak areas of the Oa and Ol components are integrated, and the corresponding results of Oa/(Oa + Ol) are presented in Table S2 (ESI).† Compared to CO/R0 and CO/RO, the CO/R0/RO NComp possesses a higher amount of activated surface oxygen (46.62%) which could be responsible for its enhanced catalytic activity. The overall analyses, thus unambiguously confirm the successful formation of Ruδ+-fabricated CO and also corroborate the coexistence of both Ce and Ru with their variable oxidation states [Ce (+3 and +4) and Ru (0, +3, and +4)] in CO/R0/RO which is expected to be highly beneficial for effective catalytic reactions.
The electronic structure and local coordination environment of Ce and Ru atoms in the materials are further analyzed by X-ray absorption spectroscopy (XAS). Fig. 1e displays the Ce K-edge XANES profile of CO/R0/RO together with CeCl3 and CeO2 standards. The edge absorption energy of Ce in CO/R0/RO is found to lie in between CeCl3 and CeO2 which is indicative of the coexistence of both Ce3+ and Ce4+. In addition, the absorption edge is observed to be slightly inclined towards CeO2, implying the slight predominance of Ce4+ over Ce3+. The average valence state of Ce in CO/R0/RO is estimated to be around +3.7 by using the linear relationship between the valence state and absorption energy [Fig. S6a (ESI)].† Additionally, Ce L3 absorption edge XANES measurements are also carried out to identify the oxidation states of Ce. Herein, the linear combination fitting (LCF) protocol is adopted for processing the Ce L3 XANES data. In the LCF method, the X-ray absorption spectrum is modelled by least-squares fitting via a linear combination of known species to fit an unknown spectrum. Subsequently, we measured the Ce L3 XANES spectra of CO/R0/RO and reference materials, viz. CeCl3 and CeO2. CeO2 displays three peaks (denoted as A, B, and C) at 5722.8, 5734.3 and 5741.0 eV that correspond to Ce4+ [Fig. 1f(i)], while CeCl3 exhibits only a single strong peak at 5729.8 eV, assignable to Ce3+ [Fig. 1f(iii)]. Upon close scrutiny, it is clearly evident that the XANES spectrum of CO/R0/RO [Fig. 1f(ii)] possesses distinctive features of both Ce3+ and Ce4+. Thus, by employing the LCF method through the use of Athena software,68,69 the coexistence of both Ce3+ and Ce4+ is confirmed in CO/R0/RO. The Ru K-edge XANES spectra of CO/R0/RO along with Ru0 powder and commercial RuO2 reference compounds are also acquired to know the electronic structural characteristics and coordination environment as well as the valence states of Ru in the NComp (Fig. 1g). The absorption threshold of CO/R0/RO is found to lie in between Ru0 powder and RuO2, indicating the presence of multiple oxidation states of Ru in between 0 and 4+, which is well consistent with the XPS results.
EXAFS spectra are also acquired to analyze the electronic structure, in particular the existence of different oxidation states of Ce and Ru, in the CO/R0/RO NComp. EXAFS spectra of CeO2 [Fig. 1h(i)] and CeCl3 [Fig. 1h(iii)] are also recorded for the sake of comparison. Ce K-edge EXAFS oscillations in k space (k3-weighting) for CO/R0/RO are provided in Fig. S6b (ESI).† In Fig. 1h(ii), the CO/R0/RO NComp displays broad and overlapping shells within 1–3 Å and two successive shells with their shell maxima at ∼3.62 and ∼4.30 Å. CeO2 exhibits three prominent peaks at ∼1.85, ∼3.62, and ∼4.30 Å, corresponding to Ce–O (1st shell), Ce–Ce (2nd shell), and Ce–O (3rd shell) (denoted Ce–O1) contributions, respectively. CeCl3, on the other hand, displays a single shell at 2.06 Å due to Ce–Cl. Upon comparison, the overlapping peaks within 1–3 Å in CO/R0/RO are attributed to both Ce4+ and Ce3+, while the peaks at ∼3.62 and ∼4.30 Å correspond to Ce–Ce (2nd shell), and Ce–O (3rd shell), respectively. To obtain quantitative structural parameters around Ce atoms, a least-squares curve fitting for Ce–O, Ce–Ce, and Ce–O1 paths is conducted. The IFEFFIT package is employed herein to analyze the EXAFS data via a theoretical model generated by the FEFF 8.4 code. The corresponding fitting results are displayed in Fig. S6c (ESI†), while the best fit EXAFS data of CO/R0/RO are presented in Table S3 (ESI).†
Fig. S7 (ESI†) shows the Ru K-edge EXAFS oscillations in k space (k3-weighting) for CO/R0/RO. The FT Ru k-edge EXAFS spectra of CO/R0/RO along with Ru0, RuO2, CO/R0, and CO/RO are presented in Fig. 1i. The sole peak at 2.45 Å for metallic Ru0 powder is ascribed to Ru–Ru interactions [Fig. 1i(i)], while the peaks at 1.53, 2.82, and 3.18 Å correspond to Ru–O/Ru interactions in RuO2 [Fig. 1i(v)]. The peak at 1.53 Å is associated with the scattering path for Ru–O with its nearest neighbour O atom, while the peaks at 2.82 and 3.18 Å arise from Ru–Ru scattering paths in the second and the third shells, respectively. An EXAFS fitting analysis is also conducted to reveal the differences in the local coordination environment of Ru species in CO/R0/RO and RuO2 (Table S4, ESI†). In Fig. 1i(iv), CO/R0/RO displays lower peak intensity for the first coordination shell (Ru–O) compared to RuO2, which could be due to a lower coordination number and/or higher structural disorder in the local coordination environment (Fig. S8, ESI†). Fitting of the first coordination shells reveal that CO/R0/RO possesses low coordination numbers, viz. 5.5 ± 0.2, which is indicative of the presence of the oxygen vacancies (OV) in the NComp. The mean Ru–O bond lengths are found to be longer in CO/R0/RO (1.99 ± 0.01 Å) compared to RuO2 (two Ru–O bonds with a length of 1.94 ± 0.01 Å and four Ru–O bonds with a length of 1.99 ± 0.01 Å). The slightly reduced oxidation state of Ru in CO/R0/RO, indicated by X-ray absorption near edge structure analyses, is also explained by the lower coordination numbers and longer bond lengths of Ru with neighbouring O atoms. The structural disorder of a given path in the material could be accessed using the Debye–Waller factor (σ2). The σ2 value of 0.005 ± 0.001 for CO/R0/RO is found to be much higher than that of RuO2 (0.001 ± 0.002) (Table S4, ESI†). Thus, the structural disorder is significantly higher in CO/R0/RO than in RuO2. This disorder facilitates charge transportation by shortening the charge-transfer paths, which in turn improves the electrocatalytic performance.
The EPR spectra of CO/R0/RO along with CO and CO/R0 are also acquired to investigate the contribution of oxygen vacancies in the water splitting reaction. CO/R0/RO displays a characteristic EPR signal at g = 2.003 due to oxygen vacancies [Fig. 1j(iii)]. By contrast, CO [Fig. 1j(i)] and CO/R0 [Fig. 1j(ii)] do not exhibit any such signal which indicates that when CO, R0, and RO form the heterointerface, additional defects are introduced due to lattice distortion.
The field emission scanning electron microscopy (FESEM) image indicates an overall nanorod morphology for both CO (Fig. S9a, ESI†) and CO/R0/RO, with additional adherence of some stacked small particles in the latter case [Fig. S9b (ESI)†]. The transmission electron microscopy (TEM) image again confirms the rod-like structure of CO with an average length and diameter of ∼23.28 nm and ∼3.38 nm, respectively (Fig. 1k). Upon incorporation of R0 and RO, CO/R0/RO does not show any such morphological changes; only some R0 and RO particles are found to be firmly grown on CO, thereby representing good consistency with the SEM images (Fig. 1l) [see Section S2.3 (ESI†) for detailed discussions]. The energy dispersive X-ray spectra (EDX) shown in Fig. S13(a–d) (ESI†) indicate a homogeneous distribution of elements (Ce and O in CO and Ce, Ru, and O in CO/R0, CO/RO, and CO/R0/RO) in the materials (Table S5, ESI†).
Electrocatalytic performances toward Zn–air batteries (ZABs)
Moreover, CO/R0/RO delivers a cathodic peak at 0.921 V which is much higher than those of CO/RO (0.869 V), CO/R0 (0.837 V), and CO (0.747 V), demonstrating that CO/R0/RO stands out as the best among the three ORR ECs. The ORR activity is then assessed by linear sweep voltammetry (LSV) on a rotating disk electrode (RDE) with a rotating speed of 1600 rpm (Fig. 2a and S15a, ESI†). Interestingly, the limiting current densities of CO/R0/RO (Fig. 2a–d) remain constant in the potential window of 0.2 to 0.7 V while for other catalysts, it shows a gradual decreasing trend (Fig. 2a and S15a, ESI†). This difference in the trend is mainly due to the nature of the catalyst and the reaction conditions. As the reaction conditions remain almost similar for all the catalysts, the difference in limiting current density observed in LSV is mainly due to the nature of the catalyst. The constancy in the limiting current density for CO/R0/RO indicates that the reaction is kinetically controlled. Herein, the activity of the CO/R0/RO NComp is high enough that the current density reaches a plateau and remains stable as the overpotential (η) increases. Moreover, it also reveals that the transport of reactants to the surface or products away from the surface is not a limiting factor. The overall result illustrates that CO/R0/RO, an efficient catalyst with a large number of active sites, can sustain a high current density without significant losses. Thus, in CO/R0/RO the mass transport (or diffusion) effects are minimal, leading to a stable limiting current. In contrast, the decreasing trend in limiting current density of CO, CO/R0, and CO/RO (Fig. 2a and S15a, ESI†) can be attributed to mass transport limitations or catalyst deactivation. For these catalysts, as η increases, the rate of electrochemical reaction might exceed the rate at which reactants are delivered to the catalyst surface or products are removed. This results in a drop in current density as the reaction becomes diffusion-limited.
 | ||
| Fig. 2 ORR performances of corresponding electrodes: (a) LSV (1600 rpm) of the ECs, (b) bar diagram representing Eonset, E1/2, and Eoverpotential, (c) Tafel plots, (d) LSV of CO/R0/RO at different rotational speeds, (e) corresponding K–L plots, (f) LSV-RRDE curves, (g) graphical representation of the electron transfer number (n) and peroxide yield at 1600 rpm rotational speed, (h) chronoamperometric durability tests at 0.936 V with 1600 rpm rotational speed, and (i) MeOH tolerance test (green arrow indicates the addition of 18 mL of MeOH into 72 mL of 0.1 M KOH after ≈2000 s). | ||
The highest ORR performance is observed for CO/R0/RO with a more positive Eonset of 1.056 V, a large half-wave potential (E1/2) of 0.936 V, and a high diffusion-limited current density (jL) of −13.7 mA cm−2 (at 0.2 V), which confirms its superiority over CO/RO, CO/R0, CO, and even commercial Pt/C (Fig. 2b and Table S6, ESI†) as well as other counterparts (Fig. S15c, ESI†). Interestingly, CO/R0/RO also possesses the smallest Tafel slope of 53.2 mV dec−1 compared with the other three (Fig. 2c and S15b, ESI†), suggesting its faster and favourable reaction kinetics. For an in-depth understanding of the ORR kinetics of CO/R0/RO, LSVs are recorded upon varying the rotation speeds within the domain of 400–2000 rpm (Fig. 2d) and it is observed that limiting current density enhances with increasing rotational speed. To analyze the reaction pathway and to calculate the number of electron transfers (n) during the ORR, the Koutecky–Levich (K–L) equation is employed for CO/R0/RO. Fig. 2e depicts good linearity and strong coincidence in the K–L plot within the potential window of 0.2–0.7 V, indicating first order reaction kinetics along with a similar number of electron transfers per O2. The calculated n value is found to be ∼4 for CO/R0/RO, revealing that the 4e− transfer pathway is energetically favourable for O2 reduction. The result matches well with the rotating ring-disk electrode (RRDE) measurements wherein CO/R0/RO shows a high value of disk current (Fig. 2f). Alternatively, the Pt-ring current related to H2O2 oxidation is found to be negligible, signifying that the OH− species are the key products during the ORR. This illustration is strongly supported by the calculated n value of ∼4 and negligible HO2− formation (∼0.82%, estimated by using eqn S3, S4 and Table S7, ESI†) for CO/R0/RO in the potential range from 0.2 to 0.7 V, indicating its efficient and high ORR selectivity (Fig. 2g).
To assess the stability of CO/R0/RO, an accelerated durability test is performed. Fig. S14c (ESI†) shows the outstanding long-term durability of CO/R0/RO with negligible degradation of E1/2 and limiting current density even after 300000 cycles. This cycling number displays the overwhelming superiority of our EC over most of the previously reported cycling numbers.4 The ORR stability is further assessed by chronoamperometric measurement for 40 h, wherein CO/R0/RO possesses enhanced durability with 99.7% retention of its original value (Fig. 2h). As evident from Fig. 2i, CO/R0/RO exhibits an enhanced tolerance toward MeOH poisoning where the performance of CO/R0/RO remains almost unchanged even after incorporation of 3.0 M MeOH (20 vol%) into O2 saturated 0.1 M KOH at 2000s (Fig. S14d, ESI†). Thus, the outcomes of the above experiments unequivocally confirm that CO/R0/RO possesses efficient activity, enhanced durability, and high selectivity toward the ORR in alkaline electrolyte (Table S13, ESI†).
During the ORR, O2 adsorption, facile dissociation of O–O bonds and moderate binding with oxygenated intermediates on the catalyst surface are vital factors for an efficient ORR EC. Herein, the reversible redox couples of Ce and Ru in various oxidation states facilitate the activation of O2, subsequently accelerating the ORR kinetics. Moreover, the strong oxophilicity of Ru favours the preferential adsorption of O2, promote ORR processes. In addition, the large heterointerface in the NComp facilitates O2 adsorption and *OH desorption processes. The favourable work function values of CO, R0, and RO also promote electron transfer from R0 to neighbouring CO and RO in the CO/R0/RO NComp. The said electron transfer creates d-band vacancies in R0 with a concomitant increase in the d-band centre which in turn favours O–O bond splitting and facilitates the adsorption/desorption of intermediates during the ORR.70,71
000 cycling tests (Fig. S17, ESI†). In addition, this excellent durability of CO/R0/RO is further confirmed by a 40 h chronopotentiometric test, recorded at a constant current density of 10 mA cm−2 (Fig. 3c). Detailed investigations on the OER activities of the studied materials will also be discussed later in the electrochemical water splitting section. The bifunctional (BF) electrocatalytic activity of our fabricated ECs (Fig. 3d) is evaluated through the calculation of the potential difference between the OER and ORR; i.e., ΔE = Ej10 − E1/2, where Ej10 is the OER potential at a current density of 10 mA cm−2 and E1/2 is the ORR half-wave potential. As expected, CO/R0/RO shows the smallest ΔE of 0.460 V (Table S6, ESI†) which is not only superior to CO/RO (0.506 V), CO/R0 (0.566 V), and CO (0.728 V), but also better than the commercial Pt/C + RuO2 couple (0.729 V) as well as other recently reported advanced BF ECs (Table S16, ESI†).
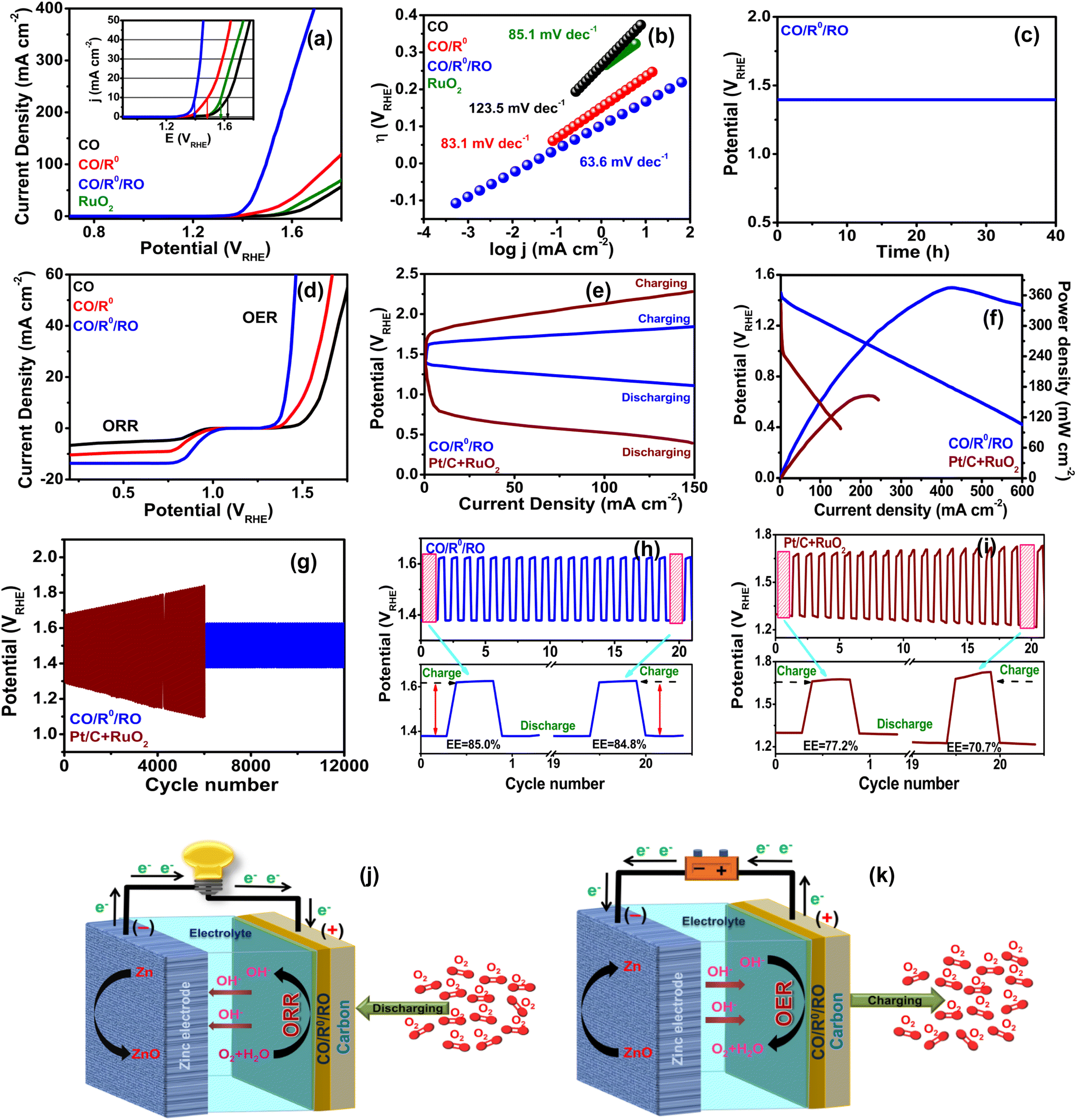 | ||
| Fig. 3 (a) OER polarization curves (inset: LSV plots up to a current density of 50 mA cm−2), (b) OER Tafel plots, (c) chronopotentiometric measurements at a current density of 10 mA cm−2, (d) ORR/OER BF LSV curves, (e) discharge/charge polarization curves of the RZAB using CO/R0/RO and Pt/C + RuO2 as air electrodes, (f) discharge voltage curve and corresponding power density plots, (g) discharge/charge cycling curves of CO/R0/RO and Pt/C + RuO2 assembled RZABs at a current density of 1 mA cm−2. Long-term cycling performance of CO/R0/RO (h) and Pt/C + RuO2 (i) up to 20 cycles. Schematic presentation of the RZAB under discharging (j), and charging (k) conditions. | ||
The superior BF performance and durability of the CO/R0/RO NComp inspired us to investigate its practical application in RZABs using CO/R0/RO as the catalyst for air cathodes [Fig. 3j and k]. The CO/R0/RO-based ZAB offers a stable open circuit potential of 1.54 V over the time span of over 9 h (Fig. S18, ESI†). The voltage difference between discharge–charge polarization curves of the CO/R0/RO-based battery is also found to be less (Fig. 3e). Additionally, large current densities of 125.3 and 246.7 mA cm−2 are attained at 1.2 and 1.0 V, respectively (Fig. 3f). Besides, a CO/R0/RO-based battery delivers a higher peak power density of 376.4 mW cm−2 at 420.1 mA cm−2 compared to a Pt/C + RuO2 based battery (164.7 mW cm−2 at 219.2 mA cm−2) (Fig. 3f). The efficiency and cyclic stability of the battery, assessed by executing continuous galvanostatic discharging (5 min)–charging (5 min) cycling tests at 1 mA cm−2, indicate an ultralong lifespan of 12000 cycles (corresponding to 2000 h of operation) without any noticeable increase in voltage gaps (Fig. 3g). In particular, CO/R0/RO exhibits initial charge and discharge potentials of 1.620 V and 1.378 V (ΔE = 0.242 V), respectively with an energy efficiency (EE) of ∼85.1% (EE = discharge end voltage divided by charge end voltage) (Fig. 3h). Even after completing the 20th cycle, these two voltages are found to be almost unaltered [1.625 and 1.378 V (ΔE = 0.247 V)] with a negligible loss of EE (0.2%), revealing the excellent stability of CO/R0/RO. On the other hand, the Pt/C + RuO2 assembled device exhibits poor cycle life with a large ΔE of 0.506 V (Fig. 3i) after the 20th cycle stability test and its performance deteriorates with time. Thus, the above results conclude the superiority of our CO/R0/RO EC over most of the reported cathode ECs (Table S17, ESI†). Therefore, CO/R0/RO shows promise for achieving a high-performance RZAB.
Apart from ZABs, LABs are also designed with the CO/R0/RO NComp. The galvanostatic discharge/charge performance of the CO/R0/RO cathode is tested in the voltage range between 2.5 V and 3.3 V, under an O2 atmosphere. In Fig. 4a, the battery catalyzed by CO/R0/RO is tested at 200 and 1000 mA g−1 with a limited capacity of 1200 mA h g−1. Evidently, CO/R0/RO efficiently reduces the charge overpotential for the first cycle (charge voltage is 3.12 V at 200 mA g−1 and 3.21 V at 1000 mA g−1) which further confirms the excellent OER activity of CO/R0/RO for LABs. Even at 1000 mA g−1, the battery catalyzed by CO/R0/RO shows an extremely low overpotential (the voltage gap at half capacity) of 0.43 V. Fig. 4b demonstrates the discharge/charge load curves after the 1st, 20th, 50th, 70th, and 100th cycles at a current density of 1000 mA g−1. The voltage difference between the discharge and charge plateau in the first cycle is 0.44 V due to the distinguished electrocatalytic ORR and OER activity. The lower charge/discharge overpotential improves the energy efficiency (EE) of the battery. In particular, CO/R0/RO (Fig. 4b) exhibits an initial charge potential of 3.21 V and a discharge potential of 2.76 V with an EE of ∼85.9%. The discharge/charge overpotential slightly increased during cycling. After the 100th cycle, the charge voltage increases by 29.9 mV and the discharge voltage decreases by 23 mV with a 1.4% loss of EE, revealing the good stability and superior BF catalytic activity of CO/R0/RO toward the ORR and OER. The high specific capacity and reduced voltage gap are due to the appropriate design of the CO/R0/RO NComp. In addition, the LAB with the CO/R0/RO electrode sustains ∼400 cycles with the capacity maintained at 1200 mA h g−1 (Fig. 4c) which surpasses most of the reported oxide-based batteries.72 Noticeably, the discharge cut-off potential of the CO/R0/RO electrode remains above 2.5 V, and the charge terminal voltage is below 3.5 V during the entire catalytic cycle (Fig. 4c). Thus, the CO/R0/RO-based battery exhibits superior cycling ability, maintaining the lowest charge terminal voltage. It is interesting to note that compared to LABs, ZABs exhibit a low charge–discharge voltage gap while maintaining a long operational life with negligible loss of EE as indicated in Table 1. Moreover, RZABs are becoming more attractive due their low cost, environmental friendliness, high theoretical energy density, and inherent safety. Thus, our CO/R0/RO based ZAB may be considered as a better alternative to LABs for the advancement of sustainable battery systems.
 | ||
| Fig. 4 (a) First discharge/charge curves at 200 mA g−1 and 1000 mA g−1 of CO/R0/RO with a capacity of 1200 mA h g−1, (b) discharge/charge curves of CO/R0/RO at 1000 mA g−1 under various cycles, and (c) cyclic performance of LABs with the CO/R0/RO-based cathode at a current density of 1000 mA g−1. | ||
| Battery | Initial charge discharge voltage gap (V) | EE (initial) (%) | Charge discharge voltage gap after 100th cycle (V) | EE (after 100th cycle) (%) | Loss of EE after 100th cycle (%) |
|---|---|---|---|---|---|
| LAB | 0.44 | 85.9 | 0.503 | 84.5 | 1.4% |
| ZAB | 0.242 | 85.1 | 0.248 | 84.7 | 0.4% |
Electrocatalytic performances toward overall water splitting (OWS)
 | ||
| Fig. 5 (a) LSV curves of the HER (inset: plots up to a current density of −50 mA cm−2), (b) plot denoting the Eonset and η10 (inset: corresponding bar diagram), (c) mass activity plots (inset: bar diagram representing mass activity at η = 155 and 90 mV), (d) HER Tafel plots (inset: plot for Tafel slopes), and (e) plot representing the comparison of η10 and η20 (left) and exchange current densities (right). | ||
| Stability | ||||||||
|---|---|---|---|---|---|---|---|---|
| ECs | E onset (mVRHE) | η j = 10 mA cm−2 (mV) | j HER (mA cm−2) at η = 155 mV | TOF (s−1) | Mass activity (A g−1) at 155 mV | Δηj = 10 mA cm−2 (mV) | Tafel slope (mV dec−1) (before) | Tafel slope (mV dec−1) (after) |
| CO | 94 | 172 | 7.4 | 3.59 × 10−3 | 67.1 | +26 | 75.2 | 78.4 |
| CO/R0 | 61 | 103 | 22.7 | 3.96 × 10−2 | 205.6 | +18 | 47.4 | 49.7 |
| CO/RO | 45 | 78 | 40.0 | 1.01 × 10−1 | 365.0 | +6 | 44.3 | 45.9 |
| CO/R0/RO | 18 | 58 | 50.0 | 1.54 × 10−1 | 450.0 | 0 | 34.5 | 34.1 |
| Pt/C | 53 | 89 | 30.4 | 7.28 × 10−2 | 278.7 | +12 | 47.8 | 51.9 |
Additionally, CO/R0/RO exhibits the smallest Tafel slope of 34.5 mV dec−1 among others, demonstrating its faster and favourable HER kinetics with splendid electron transfer (Fig. 5d and S19c, ESI†). The Tafel slope of CO/R0/RO suggests that the HER occurs through the Volmer–Heyrovsky mechanism (eqn S25 and S26, ESI†) wherein the Heyrovsky reaction is the rate determining step, the details of which are presented in the ESI (Section S5, ESI†) (Fig. 5d and S19c, ESI†). To gain deeper insight into the HER rate and the number of active sites, the exchange current density (j0) is also assessed. CO/R0/RO exhibits a ∼1.7-fold enhancement of j0 (0.34 mA cm−2) compared with CO (0.20 mA cm−2) which again highlights its significantly enhanced efficiency (Fig. 5e).
The alkaline HER involves initial water dissociation followed by associative desorption of H2. The latter step becomes more facile in the majority of reported ECs, but the overall HER process is restricted because of the energy-demanding H–OH bond cleavage. Thus, boosting the initial water dissociation has now become a great challenge for researchers. To this end, we are motivated to design multicomponent and MV NComp systems encompassing two or more metal oxides that can modify their electrochemical properties via enhancement of electroactive sites and enabling faster charge and mass transportation. Due to the excellent H2O adsorption capacity of R0 (0.85 eV) and RuO2 (0.64 eV), the R0/RuO2 on the CO/R0/RO NComp surface displays higher M–H2O binding energy (0.60 eV) than Pt/C (0.48 eV), which improves H2O adsorption and accelerates the Volmer step (Fig. S20a, ESI†).73 In the subsequent step, H2O dissociates into H and OH, and the easier the water dissociates, the more protons can be supplied and the faster the reaction rate will be. In Fig. S20b (ESI†), the H2O dissociation on the surface of the NComp is much easier than on the commercial Pt/C surface as RuO2 shows the lowest H2O dissociation energy. Additionally, the moderate H and OH binding energies at the R0/RuO2 interface in the NComp lead to efficient hydrogen evolution and rapid regeneration of active sites. In the case of CO/R0, the water dissociation step is restricted due to the higher H2O dissociation energy of R0 whereas CO/R0/RO demonstrates an outstanding H2O adsorption–dissociation capability, and appropriate binding energies for H and OH, showing its superiority as a potential HER catalyst. Thus, the synergistic influence of CO, R0 and RO in the CO/R0/RO NComp promotes the adsorption of OH− on the MV metal sites which in turn accelerates the water dissociation process. It is of interest to note that the judicious fabrication of the material allows the development of a Schottky barrier type heterojunction that could accelerate the catalytic performance of CO/R0/RO (Scheme 2).
 | ||
| Scheme 2 Schematic representation of the band energy diagram when R0 and RO are brought into contact with CO [(a) before contact, and (b) after contact]. | ||
The slope of the Mott–Schottky plot in Fig. S22 (ESI†) indicates that CO is an n-type semiconductor with a flat band potential (Efb) of −0.44 V which can be ascribed to its conduction band minimum (ECB). Subsequently, using band gap energy, obtained from the Tauc plot (Fig. S21b, ESI†), the valence band potential (EVB) of CO is calculated to be 2.56 V (Table S8, ESI†). By contrast, RO is a metallic oxide with its electronic conductivity being half of R0. Thus, the contact among CO, R0, and RO leads to the formation of a Schottky barrier at their interfaces. Based on this, an illustration of the plausible band diagram of CO/R0/RO is represented in Scheme 2a. As the work function (Φ) of RO (6.2 eV)74 is higher than that of CO (5.34 eV),75 the electrons are transferred from CO to RO until their Fermi levels (EF) get aligned under thermal equilibrium (Scheme 2b). As a consequence, upward band bending of CO is observed which may be highly beneficial for charge separation. Subsequently, a space-charge region is developed, thereby facilitating the formation of an electric field from CO to RO (Scheme 2b). This will lead to the formation of abundant active sites at the contact area and numerous electrons from the bias could be consumed, thereby improving the HER activity.
The long-term HER durability is further evaluated by chronopotentiometric measurements at a static current density of 10 mA cm−2 (Fig. 6a and S23b, ESI†). CO/R0/RO exhibits a negligible change in η even after 30 h of continuous HER testing, as confirmed by their LSV curves (Fig. 6b and S24a–d, ESI†). In contrast, other ECs experience poor durability with a significant loss of η (Table 2 and Fig. 6c), probably due to the peel-off of the ECs. Additionally, no obvious attenuation of η could be observed in the LSV curves after 300000 cycles of stability tests (Fig. 6d). The superior activity and accelerated durability of CO/R0/RO is also confirmed from its decreased Tafel slope (34.1 mV dec−1) (Fig. 6e and S25, ESI†). In addition, after the stability test, a slight decrease in charge transfer resistance (Rct) in electrochemical impedance spectroscopy (EIS) strongly suggests the efficient functionality of CO/R0/RO (Fig. S26a, ESI†). The structural and compositional stability of CO/R0/RO upon completion of the catalytic cycles is again confirmed by XPS (Fig. S27, ESI†), XRD (Fig. S28, ESI†), and TEM analyses (Fig. S29a, ESI†). Lastly, to scrutinize the HER stability of our synthesized ECs, a graphical comparison (Fig. 6f) has also been made which further confirms the superior durability and excellent catalytic activity of CO/R0/RO over others (see Section S7, ESI† for detailed discussion).
 | ||
| Fig. 6 (a) Chronopotentiometric tests for 30 h at 10 mA cm−2, (b) HER polarization curves of CO/R0/RO after the 30 h stability test (inset: LSV plots up to a current density of −50 mA cm−2), (c) change in η10 of the ECs before and after the 30 h stability test, (d) polarization curves of CO/R0/RO before and after the 300000th cycle stability test (inset: LSV plots up to a current density of −50 mA cm−2), (e) HER Tafel plots (inset: corresponding Tafel slopes), and (f) graphical representation of η10 before and after the stability test. | ||
| Stability | ||||||||
|---|---|---|---|---|---|---|---|---|
| ECs | E onset (VRHE) | η j = 10 mA cm−2 (mV) | j OER (mA cm−2) at η = 320 mV | TOF (s−1) | Mass activity (A g−1) at 1.43 V | Δηj = 10 mA cm−2 (mV) | Tafel slope (mV dec−1) (before) | Tafel slope (mV dec−1) (after) |
| CO | 1.52 | 357 | 6.6 | 3.38 × 10−3 | 9.0 | +28 | 119.3 | 136.2 |
| CO/R0 | 1.39 | 240 | 30.1 | 1.71 × 10−2 | 47.1 | +10 | 82.6 | 83.3 |
| CO/RO | 1.36 | 180 | 94.5 | 7.48 × 10−2 | 171.0 | +8 | 71.9 | 72.0 |
| CO/R0/RO | 1.34 | 166 | 177.1 | 1.12 × 10−1 | 251.8 | 0 | 63.4 | 58.8 |
| RuO2 | 1.51 | 340 | 6.7 | 1.72 × 10−3 | 8.8 | +25 | 86.5 | 91.2 |
 | ||
| Fig. 7 (a) LSV plots of the OER (inset: plot up to a current density of 50 mA cm−2), (b) current density vs. η plots (inset: plot up to a current density of 50 mA cm−2), (c) plot representing the Eonset and η10 (inset: corresponding bar diagram), (d) mass activity plots (inset: bar diagram representing mass activity at η = 200 and 160 mV), (e) corresponding activity enhancement of the materials relative to RuO2, and (f) OER Tafel plots (inset: Tafel slope of the materials). | ||
Interestingly, CO/R0/RO exhibits a far lower Eonset of 1.34 V compared to CO/RO (1.36 V), CO/R0 (1.39 V), and CO (1.52 V) which is merely 170 mV behind the commercial RuO2 (Table 3), revealing its advanced OER performance. Moreover, the oxygen evolution current of CO/R0/RO is observed to be significantly improved compared to that of the others, reaching 177.1 mA cm−2 at a η of 320 mV (Table 3) which again demonstrates its enhanced conductivity and faster electron transport. The OER current densities of CO/R0/RO are found to increase almost linearly at two selected potentials of 1.46 and 1.43 V with a similar mass loading of 0.11 mg cm−2 (Fig. 7d and S30b, ESI†) which reveals that the highly active MV surface structure is advantageous for the exposure of abundant reactive sites and enabling excellent charge-ion transport toward the deeper layers of the EC. Specifically, a moderately small η (200 mV) is required for CO/R0/RO to achieve a current density of about 251.8 A g−1, which is much lower than those of CO/RO, CO/R0, CO, and even commercial RuO2 (216, 318, 414, and 413 mV), respectively (Fig. 7d). In particular, CO/R0/RO demonstrates a significant improvement in activity over RuO2 by factors of about 6.1, 25.8, and 74.2 times at η values of 120, 170, and 220 mV, respectively (Fig. 7e). The remarkable OER performance of CO/R0/RO is also reflected in its smaller Tafel slope of 63.4 mV dec−1 compared to the other ECs (Fig. 7f and Table 3), suggesting its more facile and faster OER kinetics through efficient electron–mass transport. Fig. 7f and S30c (ESI†) depict a reasonable synchronous decrease in the Tafel slope with a gradual increase in the mixed-valence coexistence of Ruδ+ on passing across the CO → CO/R0 → CO/RO series and the minimum slope is achieved in CO/R0/RO where the number of variable oxidation states (Ce3+, Ce4+, Ru0, Ru3+, and Ru4+) is maximum. The Tafel slope for CO/R0/RO is close to 60 mV dec—1, indicating that the initial electron transfer process is the rate-determining step in the OER (see eqn S20, ESI†) and the overall oxidation reaction follows the first order kinetics. Thus, this result clearly establishes that the OER activity of CO is increased upon R0 loading and a sequential increase in the variable oxidation states of Ru further enhances the OER efficiency of CO/R0/RO.
Detailed electrochemical desorption experiments were conducted to understand the adsorption characteristics of the oxygenated intermediates on the surface of the ECs. During the negative potential sweeps, the ECs display desorption of oxygenated intermediates within the potential window of 0.75–0.64 V after 5 min of anodic polarization at 1.45 V. Fig. 8a clearly indicates a gradual negative shift in Eonset upon the successive changes in the electrode materials across the series (CO → CO/R0 → CO/RO → CO/R0/RO). Among others, CO/R0/RO displays a much slower increase in desorption current density along with a decreased Eonset, indicating the sluggish stripping characteristics of adsorbates from its surface, thereby improving the adsorption of oxygenated intermediates on the EC surface. The observed results highlight the importance of the MV states in the OER activity of CO/R0/RO.
 | ||
Fig. 8 (a) LSV curves corresponding to the electrochemical desorption behaviour, (b) 30 h chronopotentiometric stability tests at 10 mA cm−2, (c) LSV of CO/R0/RO after 30 h of OER stability test (inset: plots up to a current density of 50 mA cm−2), (d) η10 of the ECs before and after the 30 h stability test, (e) LSV curves of CO/R0/RO before and after the 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) 000th cycle of the stability test (inset: plots up to a current density of 50 mA cm−2), (f) chronopotentiometric stability of CO/R0/RO up to 25 h under different chopping current densities, (g) OER Tafel plots after the stability test (inset: corresponding Tafel slopes), (h) changes in Tafel slopes of the ECs before and after 30 h of stability test, and (i) graphical representation of η10 before and after the stability test. 000th cycle of the stability test (inset: plots up to a current density of 50 mA cm−2), (f) chronopotentiometric stability of CO/R0/RO up to 25 h under different chopping current densities, (g) OER Tafel plots after the stability test (inset: corresponding Tafel slopes), (h) changes in Tafel slopes of the ECs before and after 30 h of stability test, and (i) graphical representation of η10 before and after the stability test. | ||
Generally, the metal oxide-based OER functions via the adsorbate evolution mechanism (AEM)17,76,77 involving four concerted proton coupled electron transfer (PCET) pathways78–80 (Fig. S31a, ESI).† There is an additional mechanism called non-concerted PCET, which is basically different from the conventional scheme, wherein the produced O2 may originate from the lattice oxygen oxidation of some highly covalent active metal oxides. The OER of metal oxides involving four concerted PCET pathways exhibits pH-independent activity on the RHE scale, whereas the extremely active metal oxides usually display pH-dependent performance on a similar scale indicating the participation of non-concerted PCET during the OER. Fig. S32a (ESI†) depicts pH-independent OER activity of our CO/R0/RO NComp which confirms the involvement of concerted PCET steps wherein the metals with their varying oxidation states act as the active centres for the OER. The spectroscopic evidence supporting the OER catalytic cycle in Fig. S31a (ESI†) is provided by an in situ XPS study (Fig. S33, ESI†). During the in situ OER measurement, the Ru 3d XPS spectrum (Fig. S33, ESI†) of CO/R0/RO is found to exhibit two additional peaks at around 281.2 eV and 286.2 eV (corresponding to 3d5/2 and 3d3/2 of Ru5+, respectively) as compared to the Ru 3d spectrum of CO/R0/RO before the OER measurement. Thus, the deconvolution of the Ru 3d in situ XPS spectrum proves the existence of Ru0, Ru3+, Ru4+, and Ru5+ in CO/R0/RO. This result may conclude that the OER functions via the AEM involving four concerted PCET pathways. Thus, during the OER, Ru3+ and Ru4+ act as active sites for O2 evolution and transform to Ru4+ and Ru5+, respectively, during the catalytic cycle and finally revert back to respective Ru3+ and Ru4+ states after the completion of the reaction cycle. Moreover, the enhancement of Ce–O and Ru–O bond covalence with the increase in metal oxidation states in CO/R0/RO leads to the involvement of both surface oxygen and active metal sites for promoting the OER kinetics [Fig. S31(b and d), ESI†].
A moderate metal–adsorbate bond formed through the adsorption of oxygenated intermediates (e.g., *OH) on the metal centres is a crucial factor for efficient OER which gradually becomes stronger with increasing metal oxidation states. In the early stage of the OER, some O2 may originate from the decomposition of surface oxides and hence, unquestionably, CO with Ce3+ and Ce4+ facilitates O2 evolution to some extent. After R0 loading, the generated heterointerface between CO and R0 accelerates the OER kinetics probably because of increased electrochemical conductivity, charge–mass transport, and electrochemically active surface area. Additionally, Ruδ+ species in their 3+ and 4+ oxidation states together with Ce3+ and Ce4+ in CO/RO, enhance the adsorption of oxygenated anions, facilitating the formation of Ce–O and Ru–O bonds which in turn increases the O2 evolution efficiency. Interestingly, our designed CO/R0/RO NComp exhibits substantially enhanced OER performance which may be attributed to the more active heterointerfaces among CO, R0 and RO, high active site density and the mesoporous surface.
To check the long-term durability, chronopotentiometric analysis is also conducted on the ECs under investigation (Fig. 8b and S34, ESI†). An increase in η for each of CO, CO/R0, CO/RO and even RuO2 is observed whereas almost no change in η is observed in the case of CO/R0/RO even after 30 h of stability test. The durability results are further confirmed by respective polarization curves [Fig. 8c and S35(a–d), ESI†] wherein the floating bar diagram represents that the ηt=0 h for CO, CO/R0, CO/RO and RuO2 increased from 0.357 to 0.385 V, 0.24 to 0.25 V, 0.18 to 0.188 V, and 0.34 to 0.365 V, respectively after 30 h (ηt = 30 h) under a constant current density of 10 mA cm−2 (Fig. 8d). In sharp contrast, both ηt = 0 h and ηt = 30 h of CO/R0/RO remain unaltered under similar conditions, demonstrating its robustness and efficacy in practical applications. The durability of CO/R0/RO was again assessed using the LSV plot of CO/R0/RO for up to 300000 cycles wherein no detectable change was observed (Fig. 8e). Moreover, chronopotentiometric measurement carried out for 25 h under both ascending and descending current density (10, 20, 30, 20, 10 mA cm−2) also confirmed the supremacy of the NComp (Fig. 8f). The reduced value of the Tafel slope from 63.4 to 58.8 mV dec−1 for CO/R0/RO in contrast to the increase observed for the others is the testimony of the superiority of the present NComp (Table 3, Fig. 8g, h and S36, ESI†). In addition, after the long-term stability test, the structure and composition of CO/R0/RO remains unaltered as demonstrated by XPS (Fig. S27, ESI†) and XRD (Fig. S28, ESI†) analyses. HRTEM analysis also concludes the retention of the morphology of the composite (Fig. S29b, ESI†). The EIS plots of the CO/R0/RO NComp after the stability test further bear testimony to its enhanced performance (Fig. S26b, ESI†). Finally, the linear stability plot offers strong indication of the outstanding stability and durability of our superior CO/R0/RO EC (Fig. 8i).
Taking advantage of the excellent BF OER/HER properties (Fig. 9a), we evaluated the OWS capability of our CO/R0/RO NComp using a two-electrode configuration electrolyzer. CO/R0/RO affords a small cell voltage of only 1.49 V during OWS [Fig. 9b and S41, ESI†] to achieve a current density of 10 mA cm−2 which is far lower (0.037 V) than that of Pt/C‖RuO2 (1.527 V) and comparable to those of some of the reported efficient ECs (Table S19, ESI†). Moreover, under a low applied potential of 1.49 V, 10 mA cm−2 current density is maintained even after 30 h of chronoamperometric stability testing (Fig. 9c). The long-term durability of the EC at a large current density is demonstrated under 1.7 V applied potential where the CO/R0/RO NComp shows exceptional stability with only a little increase in current density after 60 h of continuous testing (Fig. 9d). This result is further verified using the LSV curve (Fig. 9e) after 60 h of stability testing wherein the η10 gets shifted negatively about 3 mV compared to its initial state, demonstrating the supremacy and excellent durability of our device. Additionally, the electrolyzer remains steady enough to support the long-term chronopotentiometric test (30 h) of electrolysis with no obvious change in the potential of 1.49 V at 10 mA cm−2 current density (Fig. 9f).
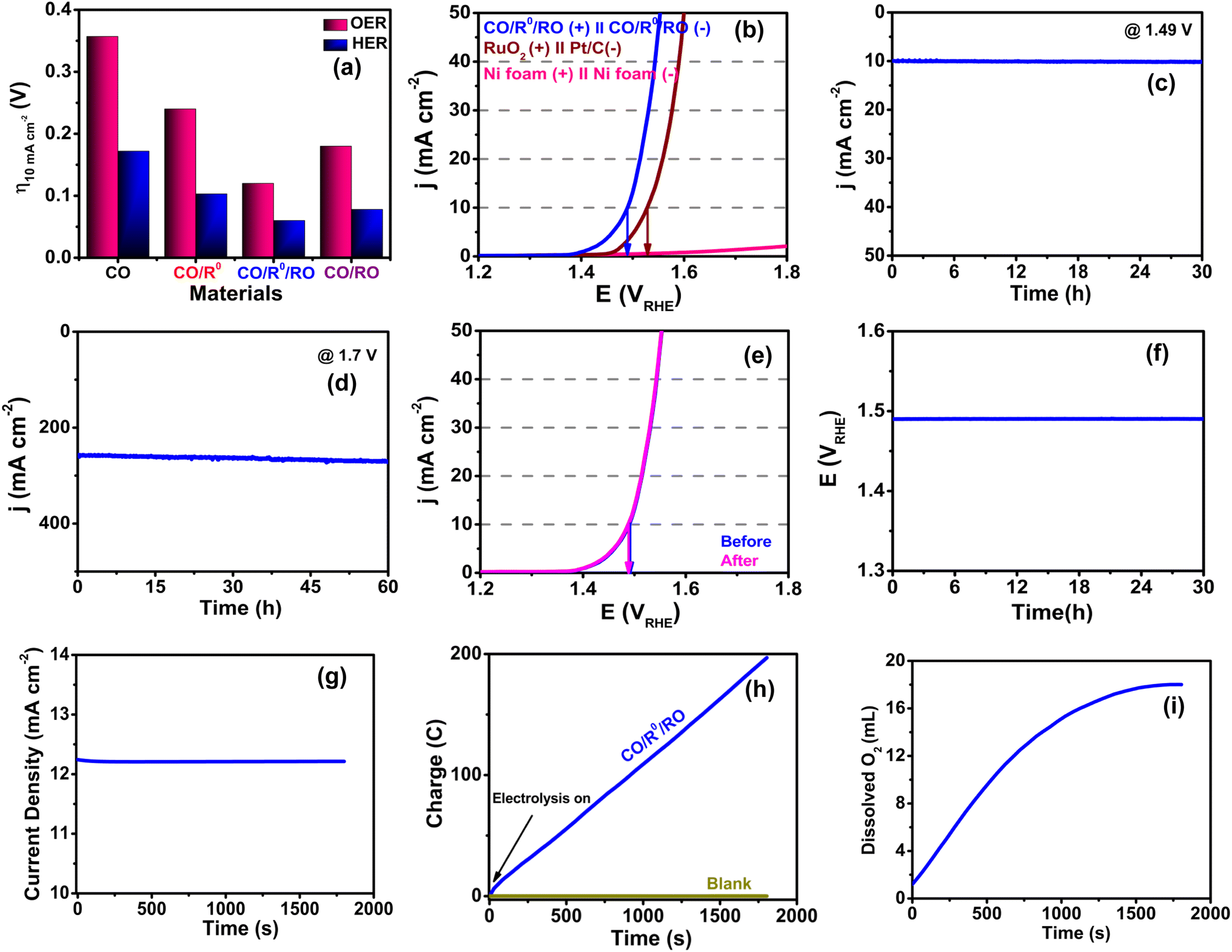 | ||
| Fig. 9 (a) η10 for both the OER and HER, (b) polarization curves of a self-assembled water splitting electrolyzer, (c) chronoamperometric measurement of CO/R0/RO at 1.49 V applied potential, (d) chronoamperometric stability test of CO/R0/RO at 1.7 V for 60 h. (e) LSV plot of CO/R0/RO before and after 60 h of stability test, (f) chronopotentiometric stability measurements of a CO/R0/RO-based electrolyzer at 10 mA cm−2; (g) controlled-potential electrolysis (CPE) of CO/R0/RO in 1.0 M KOH (pH ∼11.7), (h) charge vs. time plot during CPE, and (i) the subsequent O2 evolution plot. | ||
Anin situ EPR experiment was also executed to investigate the contribution of oxygen vacancies in the water splitting reaction. During the experiment under open-circuit voltage, a typical paramagnetic signal was found at g = 2.003 which confirms the presence of oxygen vacancies in the NComp (Fig. S42a, ESI†). When the cell voltage is increased to 1.5 V, the signal intensity corresponding to oxygen vacancies remarkably diminishes, indicating that some of the oxygen vacancies act as active sites for water splitting. It is worth mentioning that the signal intensity of oxygen vacancies remains almost unaltered upon a systematic increase in voltage up to 2.7 V, indicating that no new oxygen vacancies are generated during the OER process even at high potentials (Fig. S42b, ESI).† Thus, the lattice oxygen of the EC does not take part in the oxidation process even at high potential which in turn indicates the structural stability of the NComp.
The oxygen evolution during water splitting is confirmed by controlled potential electrolysis (CPE) (Fig. 9g) at 1.32 V where CO/R0/RO exhibits the highest catalytic current density of ∼12.2 mA cm−2 which remains constant throughout the electrolysis (30 min) and the concentration of dissolved O2 in the solution dramatically increases from 1.2 mL to 18 mL (Fig. 9i). Moreover, the faradaic efficiency for the O2 evolution is observed to lie above 98.2% (Fig. 9h and Table S20, ESI†) and this admirable efficiency strongly depends on the choice of metal, fabrication of composite materials, and regulation of the metal oxidation state. The long-term structural and compositional stability of CO/R0/RO upon completion of water electrolysis is again confirmed by XAFS analysis (Fig. S43, ESI†). The surface morphology of the EC is found to be mostly preserved with good structural ordering after the stability test (Fig. S44, ESI†). Thus, the splendid high catalytic activity together with excellent long-term stability makes the CO/R0/RO NComp a prospective candidate for future energy applications.
:1 ratio which tallies well with the theoretical value of 2:1. Taking into consideration the molar ratio of the generated gas and Zn consumed, the faradaic efficiency for the HER and OER is calculated to be 98.9% for H2 and 98.4% for O2 at a current density of 50 mA (Fig. S47).†
In addition, the rate of gas evolution is measured by plotting the amount of gas evolved against time (Fig. S47, ESI†) and the rate of H2 and O2 evolution is found to be 399.3 and 199.6 μL min−1, respectively. Hence, the ratio of H2/O2 production is estimated to be 2, which is the desired stoichiometry for OWS. In addition, the CO/R0/RO-based ZABs are capable of illuminating a LED for more than 10 h without any significant change in brightness (Fig. S48, ESI).†
The outcomes of extensive investigations on ORR/OER/HER strongly indicate that the TF activity of CO improves upon R0 loading and the subsequent incorporation of RO onto CO/R0 leads to the dramatic enhancement of catalytic performance of CO/R0/RO. The enhanced TF performance of our designed CO/R0/RO is probably because of the following factors. (1) Mesoporous surface: the larger BET surface area of CO/R0/RO (Fig. S37 and Table S9, ESI†) enables more interactions between the catalyst and electrolyte, eventually contributing to its increased electrochemically active surface area (ECSA) (Tables S10–S12, Fig. S15d, S23a and S32b, ESI†). Moreover, the mesoporous surface provides a large number of accessible active sites which facilitate the faster charge–mass transport and ultimately enhances the accessibility of the electrolyte ions onto the outer surface of the EC. (2) Charge transfer resistance (Rct): The smallest Rct of CO/R0/RO [Rct: CO > CO/R0 > CO/RO > CO/R0/RO, measured from EIS (Fig. S38 and Tables S10–S12, ESI†)] offers rapid charge transfer kinetics between CO/R0/RO and the substrate, facilitating electrochemical reactions, which validates the key role of MV metal in enhancing ORR/OER/HER performances. In addition, the greater double layer capacitance (Cdl) (Table S10–S12, ESI†) of CO/R0/RO contributes to its largest effective ECSA, offering abundant active sites for boosting its TF activity. (3) Roughness factor (Rf): the substantial improvement in surface roughness of CO/R0/RO [Rf: 2711.2 (ORR), 10281.7 (OER), and 8943.6 (HER)] compared to others (Tables S10–S12, ESI†) efficiently facilitates ion and gas transport, leading to the highest TF activity. (4) Turn over frequency (TOF): Tables 2 and 3 show the highest TOF of CO/R0/RO during both the HER and OER which is about 43- and 33-fold greater than that of CO, respectively, and this result strongly contributes to the notable electrocatalytic performance of CO/R0/RO. Moreover, these maximum TOF values probably offer a solid clue about the much-exposed active sites and superior conductivity of the NComp. (5) Synergistic effect: the drastically enhanced catalytic activity of CO/R0/RO may also be ascribed to the strong synergistic effect of CO, R0, and RO, substantially modifying its surface electronic properties.
Furthermore, MV Ceδ+ (δ = +3, +4) and Ruδ+ (δ = 0, +3, +4) species are more advantageous for CO/R0/RO as they (i) offer abundant active sites that can energetically adsorb the oxygenated intermediates and enable solid state ion-electron transport, (ii) open up opportunities for creating a strong electronic interaction and enhance the synergy between Ceδ+ and Ruδ+, (iii) act as a conducting bridge that suitably balances the binding of oxygenated intermediates and alters the exchange kinetics of intermediates, (iv) induce Ce3+/Ru3+ to smoothly adopt +4 oxidation states during water oxidation through the AEM, (v) provide an active heterointerface that facilitates tandem electron transfer, (vi) modulate the physicochemical and electrochemical properties through effective polarization of the oxygen coordination environment, and (vii) facilitate smooth multi-electron processes, which in turn are responsible for the outstanding catalytic performance of the EC. Thus, in the presence of MV metal sites, multicomponent CO/R0/RO becomes a highly efficient TF catalyst which is far superior to other synthesized ECs and well ahead of recently reported state-of-the-art materials (Tables S13–S19, ESI†).
Conclusions
In summary, we report a multicomponent–multivalent-assisted strategy involving surface–interface engineering for the development of robust, low-cost, and highly efficient trifunctional electrocatalysts. Herein, the novel multivalent nanostructure design not only substantially modulates the surface electronic structure but also abundantly exposes the active sites, maintaining its catalytic stability. The resulting assembly, CO/R0/RO exhibits significantly enhanced trifunctional activity [E1/2 (ORR): 0.936 V, η10 (OER): 166 mV, η10 (HER): 58 mV] and excellent durability which is much superior to those of CO/RO and CO/R0 counterparts. Impressively, CO/R0/RO-based ZABs display a large power density of 376.4 mW cm−2 and excellent long-term durability (over 2000 h, 12000 cycles). Moreover, the CO/R0/RO-assembled overall water splitting device delivers an ultra-small cell voltage of only 1.49 V at 10 mA cm−2, maintaining its activity for more than 60 h. Finally, the excellent trifunctionality of CO/R0/RO is exploited to operate a self-powered water-splitting device which delivers a fast H2 generation rate (399.3 μL min−1) together with a high faradaic efficiency of 98.9% (HER). Thus, this work opens a new avenue toward the fabrication of an advanced material for achieving multifunctionality and maximizing the energy efficiency in renewable energy applications.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
P. Mondal conducted all experimental work, analyzed the data and prepared the draft manuscript. S. Baitalik supervised and validated this project. The final version of the manuscript was written by S. Baitalik.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
P. M. appreciatively acknowledges DST-SERB (file no. PDF/2022/000517) for the fellowship and research funding. S. Baitalik is thankful to SERB [grant no. CRG/2020/001233] and CSIR [grant no. 01(2945)/18/EMR-II], New Delhi, for funding.References
- L. Zhang, Y. Zhu, Z. Nie, Z. Li, Y. Ye, L. Li, J. Hong, Z. Bi, Y. Zhou and G. Hu, ACS Nano, 2021, 15, 13399–13414 CrossRef CAS PubMed.
- F. Yang, J. H. Xie, X. Q. Liu, G. Z. Wang and X. H. Lu, Small, 2021, 17, 2007085 CrossRef CAS.
- J. Y. Zhu, Q. Xue, Y. Y. Xue, Y. Ding, F. M. Li, P. J. Jin, P. Chen and Y. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 14064–14070 CrossRef CAS PubMed.
- C. Zhou, S. Zhao, H. Meng, Y. Han, Q. Jiang, B. Wang, X. Shi, W. Zhang, L. Zhang and R. Zhang, Nano Lett., 2021, 21, 9633–9641 CrossRef CAS PubMed.
- W. Zhang, J. Zhao, J. Zhang, X. Chen, X. Zhang and F. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 10299–10306 CrossRef CAS.
- K. Tang, L. Chen, Y. Xiong, L. Zhang and M. Wu, ACS Appl. Nano Mater., 2023, 6, 11553–11560 CrossRef CAS.
- P. Da, Y. Zheng, Y. Hu, Z. Wu, H. Zhao, Y. Wei, L. Guo, J. Wang, Y. Wei, S. Xi, C.-H. Yan and P. Xi, Angew. Chem., Int. Ed., 2023, 62(1–8), e202301802 CAS.
- W. Shen, Y. Zheng, Y. Hu, J. Jin, Y. Hou, N. Zhang, L. An, P. Xi and C.-H. Yan, J. Am. Chem. Soc., 2024, 146, 5324–5332 CrossRef CAS PubMed.
- Y. Wei, Y. Hu, P. Da, Z. Weng, P. Xi and C.-H. Yan, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(1–9), e2312224120 CrossRef CAS.
- T. Tang, W.-J. Jiang, X.-Z. Liu, J. Deng, S. Niu, B. Wang, S.-F. Jin, Q. Zhang, L. Gu, J.-S. Hu and L.-J. Wan, J. Am. Chem. Soc., 2020, 142, 7116–7127 CrossRef CAS.
- J. Yu, B.-Q. Li, C.-X. Zhao, J.-N. Liu and Q. Zhang, Adv. Mater., 2020, 32, 1908488 CrossRef CAS.
- P. Mondal, U. K. Ghorui, J. Satra, S. Mardanya, D. N. Srivastava, G. R. Bhadu and B. Adhikary, ACS Appl. Nano Mater., 2020, 3, 3876–3891 CrossRef CAS.
- T. Zhu, J. Han, T. Sun, J. Chen, S. Wang, S. Ren, X. Pi, J. Xu and K. Chen, ACS Appl. Mater. Interfaces, 2023, 15, 8200–8207 CrossRef CAS.
- P. Zhu, Y. Shen, L. Dai, Q. Yu, Z. M. Zhang and C. An, ACS Appl. Mater. Interfaces, 2022, 14, 1452–1459 CrossRef CAS PubMed.
- Y. Li, X. Wei, L. Chen and J. Shi, Angew. Chem., Int. Ed., 2021, 60, 19550–19571 CrossRef CAS PubMed.
- H. Li, W. Ma, X. Ma, M. Guo and G. Li, ACS Sustainable Chem. Eng., 2022, 10, 16214–16224 CrossRef.
- P. Mondal, J. Satra, D. N. Srivastava, G. R. Bhadu and B. Adhikary, ACS Catal., 2021, 11, 3687–3703 CrossRef CAS.
- H. A. Gasteiger and N. M. Markovic, Science, 2009, 324, 48–49 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- K. L. Zhou, Z. Wang, C. B. Han, X. Ke, C. Wang, Y. Jin, Q. Zhang, J. Liu, H. Wang and H. Yan, Nat. Commun., 2021, 12, 3783 CrossRef CAS.
- Y. Pi, Q. Shao, P. Wang, J. Guo and X. Huang, Adv. Funct. Mater., 2017, 27(1–8), 1700886 CrossRef.
- C. Spöri, P. Briois, H. N. Nong, T. Reier, A. Billard, S. Kühl, D. Teschner and P. Strasser, ACS Catal., 2019, 9, 6653–6663 CrossRef.
- S. Hao, M. Liu, J. Pan, X. Liu, X. Tan, N. Xu, Y. He, L. Lei and X. Zhang, Nat. Commun., 2020, 11, 5368 CrossRef CAS PubMed.
- M. A. Ahsan, A. R. P. Santiago, Y. Hong, N. Zhang, M. Cano, E. Rodriguez-Castellon, L. Echegoyen, S. T. Sreenivasan and J. C. Noveron, J. Am. Chem. Soc., 2020, 142, 14688–14701 CrossRef CAS PubMed.
- Y. Gao, Z. C. Xiao, D. B. Kong, R. Iqbal, Q. H. Yang and L. J. Zhi, Nano Energy, 2019, 64, 103879 CrossRef CAS.
- H. T. Liu, J. Y. Guan, S. X. Yang, Y. H. Yu, R. Shao, Z. P. Zhang, M. L. Dou, F. Wang and Q. Xu, Adv. Mater., 2020, 32, 2003649 CrossRef CAS PubMed.
- Y. Li, F. M. Li, X. Y. Meng, X. R. Wu, S. N. Li and Y. Chen, Nano Energy, 2018, 54, 238–250 CrossRef CAS.
- P. Arunkumar, S. Gayathri and J. H. Han, ChemSusChem, 2021, 14, 1921–1935 CrossRef CAS.
- T. L. L. Doan, D. T. Tran, D. C. Nguyen, D. H. Kim, N. H. Kim and J. H. Lee, Adv. Funct. Mater., 2021, 31, 2007822 CrossRef CAS.
- Z. Ji, J. Liu, Y. Deng, S. Zhang, Z. Zhang, P. Du, Y. Zhao and X. Lu, J. Mater. Chem. A, 2020, 8, 14680–14689 RSC.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- (a) S. Dresp, F. Dionigi, M. Klingenhof and P. Strasser, ACS Energy Lett., 2019, 4, 933–942 CrossRef CAS; (b) Z. Fang, L. Peng, H. Lv, Y. Zhu, C. Yan, S. Wang, P. Kalyani, X. Wu and G. Yu, ACS Nano, 2017, 11, 9550–9557 CrossRef CAS PubMed.
- H. Wang, Z. Chen, D. Wu, M. Cao, F. Sun, H. Zhang, H. You, W. Zhuang and R. Cao, J. Am. Chem. Soc., 2021, 143, 4639–4645 CrossRef CAS.
- J. Dai, D. Zhao, W. Sun, X. Zhu, L. J. Ma, Z. Wu, C. Yang, Z. Cui, L. Li and S. Chen, ACS Catal., 2019, 9, 10761–10772 CrossRef CAS.
- G. Li, K. Zheng, W. Li, Y. He and C. Xu, ACS Appl. Mater. Interfaces, 2020, 12, 51437–51447 CrossRef CAS PubMed.
- H. P. Guo, B. Y. Ruan, W. B. Luo, J. Deng, J. Z. Wang, H. K. Liu and S. X. Dou, ACS Catal., 2018, 8, 9686–9696 CrossRef CAS.
- Y. Li, F. M. Li, X. Y. Meng, S. N. Li, J. H. Zeng and Y. Chen, ACS Catal., 2018, 8, 1913–1920 CrossRef CAS.
- (a) X. Zhang, A. N. Marianov, Y. Jiang, C. Cazorla and D. Chu, ACS Appl. Nano Mater., 2020, 3, 887–895 CrossRef CAS; (b) G. Shi, C. Yu, Z. Fan, J. Li and M. Yuan, ACS Appl. Mater. Interfaces, 2019, 11, 2662–2669 CrossRef CAS.
- H. Li, X. Wu, P. Wang, S. Song, M. He, C. Li, W. Wang, Z. Fang, X. Yuan, W. Song and Z. Li, ACS Sustainable Chem. Eng., 2022, 10, 13112–13124 CrossRef CAS.
- D. Rathore, S. Ghosh, J. Chowdhury and S. Pande, ACS Appl. Nano Mater., 2023, 6, 3095–3110 CrossRef CAS.
- (a) Y. Wu, X. Tao, Y. Qing, H. Xu, F. Yang, S. Luo, C. Tian, M. Liu and X. Lu, Adv. Mater., 2019, 31(1–9), 1900178 CrossRef PubMed; (b) C. Ray, S. C. Lee, B. Jin, A. Kundu, J. H. Park and S. C. Jun, ACS Sustainable Chem. Eng., 2018, 6, 6146–6156 CrossRef CAS.
- (a) Y. Shi, F. Yang and L. Tao, ACS Appl. Mater. Interfaces, 2023, 15, 28055–28063 CrossRef CAS PubMed; (b) E. T. Nguyen, I. A. Bertini, A. J. Ritz, R. A. Lazenby, K. Mao, J. R. McBride, A. V. Mattia, J. E. Kuszynski, S. F. Wenzel, S. D. Bennett and G. F. Strouse, Inorg. Chem., 2022, 61, 13836–13845 CrossRef CAS PubMed.
- (a) N. Yao, P. Li, Z. Zhou, Y. Zhao, G. Cheng, S. Chen and W. Luo, Adv. Energy Mater., 2019, 9(1–8), 1902449 CrossRef CAS; (b) M. A. R. Anjum, M. H. Lee and J. S. Lee, ACS Catal., 2018, 8, 8296–8305 CrossRef CAS.
- (a) J. K. He, M. C. Wang, W. B. Wang, R. Miao, W. Zhong, S. Y. Chen, S. Poges, T. Jafari, W. Q. Song, J. C. Liu and S. L. Suib, ACS Appl. Mater. Interfaces, 2017, 9, 42676–42687 CrossRef CAS PubMed; (b) N. Pradhan, J. Phys. Chem. C, 2017, 121, 18973–18974 CrossRef CAS.
- M. Chen, Z. H. Fan, L. H. Ai and J. Jiang, Appl. Surf. Sci., 2021, 564, 150478 CrossRef CAS.
- B. B. Yang, J. Y. Xu, D. Bin, J. Wang, J. Z. Zhao, Y. X. Liu, B. X. Li, X. N. Fang, Y. Liu, L. Qiao, L. F. Liu and B. H. Liu, Appl. Catal., B, 2021, 283, 119583 CrossRef CAS.
- M. X. Li, H. Y. Wang, W. D. Zhu, W. M. Li, C. Wang and X. F. Lu, Adv. Sci., 2020, 7, 1901833 CrossRef CAS PubMed.
- G. Lin, Y. Wang, J. Hong, K. Suenaga, L. Liu, L. Y. Chang, C. W. Pao, T. Zhang, W. Zhao, F. Huang, M. Yang, Y. Y. Sun and J. Wang, ChemSusChem, 2020, 13, 2739–2744 CrossRef CAS PubMed.
- X. Wu, Y. Yang, T. Zhang, B. Wang, H. Xu, X. Yan and Y. Tang, ACS Appl. Mater. Interfaces, 2019, 11, 39841–39847 CrossRef CAS PubMed.
- H. Wang, H. Liu, T. Feng, L. Wang, W. Yuan, Q. Huang and Y. Guo, Dalton Trans., 2022, 51, 675–784 RSC.
- H. Xu, J. Cao, C. Shan, B. Wang, P. Xi, W. Liu and Y. Tang, Angew. Chem., Int. Ed., 2018, 57, 8654–8658 CrossRef CAS PubMed.
- J. H. Kim, K. Shin, K. Kawashima, D. H. Youn, J. Lin, T. E. Hong, Y. Liu, B. R. Wygant, J. Wang, G. A. Henkelman and C. B. Mullins, ACS Catal., 2018, 8, 4257–4265 CrossRef CAS.
- N. K. Shrestha, S. A. Patil, J. H. Seok, A. S. Salunke, S. Cho, A. I. Inamdar, Y. Park, S. U. Lee, H. Kim and H. Im, Mater. Today Phys., 2023, 38, 101252 CrossRef CAS.
- S. Luo, T.-D. Nguyen-Phan, A. C. Johnston-Peck, L. Barrio, S. Sallis, D. A. Arena, S. Kundu, W. Xu, L. F. J. Piper, E. A. Stach, D. E. Polyansky, E. Fujita, J. Rodriguez and S. D. Senanayake, J. Phys. Chem. C, 2015, 119, 2669–2679 CrossRef CAS.
- A. I. Martín-Perales, D. Rodríguez-Padron, A. G. Coleto, C. Len, G. d. Miguel, M. J. Muñoz-Batista and R. Luque, Ind. Eng. Chem. Res., 2020, 59, 17085–17093 CrossRef.
- T. Mori, K. Tong, S. Yamamoto, S. Chauhan, T. Kobayashi, N. Isaka, G. Auchterlonie, R. Wepf, A. Suzuki, S. Ito and F. Ye, ACS Omega, 2022, 7, 25822–25836 CrossRef CAS PubMed.
- K. Fugane, T. Mori, P. Yan, T. Masuda, S. Yamamoto, F. Ye, H. Yoshikawa, G. Auchterlonie and J. Drennan, ACS Appl. Mater. Interfaces, 2015, 7, 2698–2707 CrossRef CAS.
- (a) J. Cored, A. García-Ortiz, S. Iborra, M. J. Climent, L. Liu, C. H. Chuang, T. S. Chan, C. Escudero, P. Concepcion and A. Corma, J. Am. Chem. Soc., 2019, 141, 19304–19311 CrossRef CAS PubMed; (b) Z. Liu, X. Yang, G. Hu and L. Feng, ACS Sustainable Chem. Eng., 2020, 8, 9136–9144 CrossRef CAS.
- X. Wang, G. Lan, H. Liu, Y. Zhu and Y. Li, Catal. Sci. Technol., 2018, 8, 6143–6149 RSC.
- H. Wang, X. Li, Q. Ruan and J. Tang, Nanoscale, 2020, 12, 12329–12335 RSC.
- M. Newville, Rev. Mineral. Geochem., 2014, 78, 33–74 CrossRef CAS.
- S. D. Kelly, D. Hesterberg and B. Ravel, Methods of Soil Analysis Part 5-Mineralogical Methods, 2008, vol. 5, pp. 387–463 Search PubMed.
- Y. Lin, L. Yu, L. Tang, F. Song, R. Schlögl and S. Heumann, ACS Catal., 2022, 12, 5345–5355 CrossRef CAS.
- D. Ding, K. Shen, X. Chen, H. Chen, J. Chen, T. Fan, R. Wu and Y. Li, ACS Catal., 2018, 8, 7879–7888 CrossRef CAS.
- T. Zhu, J. Huang, B. Huang, N. Zhang, S. Liu, Q. Yao, S.-C. Haw, Y.-C. Chang, C.-W. Pao, J.-M. Chen, Q. Shao, Z. Hu, Y. Ma and X. Huang, Adv. Energy Mater., 2020, 10(1–10), 2002860 CrossRef CAS.
- S. Asim, M. S. Javed, J. Khan, M. Khalid, S. S. A. Shah, M. Idrees, M. Imran, M. Usman, S. Hussain, I. Ahmad and T. S. AlGarni, Electrochim. Acta, 2021, 378(1–12), 138139 CrossRef CAS.
- H. Idriss, Surf. Sci., 2021, 712(1–6), 121894 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 96–100 CrossRef CAS PubMed.
- L. Liu, Q. Wei, X. Yu and Y. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 34068–34076 CrossRef CAS PubMed.
- Y. Qiu, Y. Rao, Y. Zheng, H. Hu, W. Zhang and X. Guo, InfoMat, 2022, 4(1–13), e12326 CrossRef CAS.
- J. Li, H. Zhou, Z. Jian, H. Li, X. Guan, Y. Xing, S. Zhang and H. Xu, ACS Sustainable Chem. Eng., 2021, 9, 5334–5344 CrossRef CAS.
- W. Zhang, J. Zhao, J. Zhang, X. Chen, X. Zhang and F. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 10299–10306 CrossRef CAS PubMed.
- Y. He, D. Langsdorf, L. Li and H. Over, J. Phys. Chem. C, 2015, 119, 2692–2702 CrossRef CAS.
- K. Zhao, J. Qi, H. Yin, Z. Wang, S. Zhao, X. Ma, J. Wan, C. Lin, Y. Gao, R. YYu and Z. Tang, J. Mater. Chem. A, 2015, 3, 20465–20470 RSC.
- M. Kim, J. Park, M. Kang, J. Y. Kim and S. W. Lee, ACS Cent. Sci., 2020, 6, 880–891 CrossRef CAS.
- M. Rana, S. Mondal, L. Sahoo, K. Chatterjee, P. E. Karthik and U. K. Gautam, ACS Appl. Mater. Interfaces, 2018, 10, 33737–33767 CrossRef CAS.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2016, 7, 11053 CrossRef CAS PubMed.
- R. P. Forslund, W. G. Hardin, X. Rong, A. M. Abakumov, D. Filimonov, C. T. Alexander, J. T. Mefford, H. Iyer, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2018, 9, 3150 CrossRef PubMed.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y. L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta04989f |
| This journal is © The Royal Society of Chemistry 2025 |