
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnvironmental degradation and durability of bulk 3D-printed parts from biodegradable polyester blends of PBS, PLA, and PHB in seawater†
Alisa
Sabalina
 *a,
Sergejs
Gaidukovs
*a,
Oskars
Platnieks
a,
Olesja
Starkova
ab,
Gerda
Gaidukova
a,
Liga
Orlova
c and
Maksims
Jurinovs
a
*a,
Sergejs
Gaidukovs
*a,
Oskars
Platnieks
a,
Olesja
Starkova
ab,
Gerda
Gaidukova
a,
Liga
Orlova
c and
Maksims
Jurinovs
a
aInstitute of Chemistry and Chemical Technology, Faculty of Natural Sciences and Technology, Riga Technical University, P. Valdena 3, Riga, LV-1048, Latvia. E-mail: alisa.sabalina@rtu.lv; sergejs.gaidukovs@rtu.lv
bInstitute for Mechanics of Materials, Faculty of Science and Technology, University of Latvia, Jelgavas 3, Riga, LV-1004, Latvia
cInstitute of Materials and Surface Engineering, Faculty of Natural Sciences and Technology, Riga Technical University, P. Valdena 3, Riga, LV-1048, Latvia
First published on 18th July 2025
Abstract
The environmental degradation of biodegradable polyester parts prepared via fused filament fabrication (FFF) from poly(butylene succinate) (PBS)/poly(lactic acid) (PLA) and PBS/poly(hydroxybutyrate) (PHB) blends (5/5 and 7/3 w/w) was systematically studied in static artificial seawater over six months. In contrast to typical thin-film degradation studies, bulk specimens provide realistic insights into the degradation behavior of thicker polymer products encountered in practical marine applications. 3D-printed dumbbell specimens fabricated with concentric and rectilinear infill patterns were investigated to tackle this issue and respond to emerging additive manufacturing trends. Changes in mechanical performance were significant, with the PBS/PHB (5/5) blend showing a pronounced 3.3-fold reduction in ultimate strength and a 2.5-fold reduction in elastic modulus. A three-stage sorption model was applied, quantifying water diffusion, hydrolytic degradation, and leaching of polymer components. Morphological examinations using scanning electron microscopy and energy-dispersive X-ray spectroscopy revealed crystalline salt deposits forming preferentially at interlayer interfaces, contributing to accelerated structural deterioration. Differential scanning calorimetry further showed shifts in crystallization temperature and crystallinity, underscoring alterations in polymer structure due to degradation. These results demonstrate that bulk part dimensions and 3D printing parameters critically influence degradation pathways, emphasizing the necessity of bulk-scale studies to predict real-world degradation behavior in marine environments accurately.
Sustainability spotlightThis research systematically investigates the environmental durability and degradation behavior of bulk-scale 3D-printed parts fabricated from biodegradable polyester blends (PBS/PLA and PBS/PHB) via fused filament fabrication (FFF) exposed to artificial seawater, directly addressing sustainability concerns related to conventional non-biodegradable polymers widely used in additive manufacturing. Moving beyond typical thin-film experiments to bulk-scale printed specimens, the study accurately captures complex, real-world degradation phenomena relevant to marine applications. Results revealed significant mechanical performance deterioration, particularly pronounced in PBS/PHB printed parts, due to hydrolytic degradation processes accelerated by printing-induced interlayer defects and subsequent crystalline salt deposition. A quantitative three-stage sorption model provided deeper insights into the individual contributions of water diffusion, polymer hydrolysis, and leaching mechanisms, essential for reliably predicting long-term environmental performance of bio-based polyesters. |
1 Introduction
The rapid growth of fused filament fabrication (FFF), a popular additive manufacturing (AM) technology known for its simplicity, affordability, and versatility, raises increasing concerns regarding the environmental impacts of the predominantly non-biodegradable polymers commonly used.1–3 The expanding use of 3D printing generates considerable plastic waste, highlighting an urgent need for sustainable and environmentally friendly alternatives.4 Biodegradable polyesters such as poly(lactic acid) (PLA), poly(hydroxybutyrate) (PHB), and poly(butylene succinate) (PBS) offer promising solutions. However, each polymer exhibits limitations: PLA's brittleness and moisture sensitivity,5–7 and PHB's brittleness, and challenging processing.8,9 Also, some existing studies have highlighted the slow marine degradation rates of PBS and PLA.9–11 Blending these polymers leverages their complementary properties, potentially improving mechanical durability and performance.12,13 Yet, understanding their long-term behavior under marine conditions remains limited. Typically, biodegradable polyesters are expected to degrade through hydrolytic mechanisms, with mechanical properties diminishing as polymer chains undergo cleavage and low-molecular-weight components leach out.14 Clarifying the degradation behaviour of these blended polymers in marine conditions is crucial to advancing their application in conventional processing and additive manufacturing.15PLA is a fully bio-based aliphatic thermoplastic polyester that can be used to enhance the resilience of polymer blends.16,17 The widespread adoption of PLA in FFF is attributed to its advantageous properties, such as exceptional processing capabilities, tensile strength, elastic modulus, and barrier properties.18,19 However, PLA's utility is somewhat constrained by its inherent brittleness and low heat resistance.20 Because PLA hydrolyses slowly under natural conditions, manufacturers usually certify it only for industrial composting, even though the wider public regards the polymer as generally biodegradable.21,22 This sensitivity adversely affects several critical parameters, such as molecular weight, glass transition temperature (Tg), melting temperature (Tm), and crystallinity (χc).23 Polyhydroxyalkanoates (PHA) are a class of aliphatic polyesters synthesized by microorganisms during fermentation.24,25 Among this group, PHB is notable for its biodegradability in marine environments, while its thermo-mechanical properties are relatively similar to PLA.26 PBS was developed as a biodegradable alternative to conventional plastics such as polyethylene (PE) and polypropylene (PP). Characterized by its high ductility and relatively low melting point, PBS is an attractive material for enhancing blend properties.27,28
Deroiné et al. reported that PLA's molecular weight does not change significantly after six months of immersion in seawater.29 Similarly, Bagheri et al. conducted an experiment where PLA films were submerged in artificial seawater at ambient temperature and exposed to fluorescent light for a year.30 Their findings indicated no notable weight loss in the PLA films. Sawpan et al. observed that neat PLA's crystallization increased by 50% due to accelerated weathering.31 Sashiwa et al. emphasized PHB's superior biodegradability in seawater compared to other polymers.32 They found that PHB exhibits higher water absorption rates than PLA, resulting in significant mass loss after 56 days, whereas PLA's mass loss remained unchanged after three weeks of water immersion. Limited research suggests that PBS exhibits poor degradation capabilities in marine environments.33–35 As a ductile polymer, PBS can enhance blends' toughness and elongation properties, addressing the limitations of brittle, glassy polymers like PHA or PHB.36,37 Wang et al. demonstrated that PLA/PBS blends improve the ductility of PLA.38 Righetti et al. concluded that adding PBS or PBSA to PHB enhances the flexibility and stiffness of the blends.39 Additionally, it has been reported that amorphous polymers tend to absorb more water than semicrystalline polymers like PBS and PHB.40–42
Reports on the durability and long-term performance of biopolyesters remain scarce, with the literature frequently noting that aliphatic polyesters are particularly susceptible to hydrolysis. Brzeska et al. investigated the chemical and enzymatic hydrolysis of polyurethane/PLA blends, observing that the presence of PLA accelerated the degradation of the blend in both hydrolytic and enzymatic environments.43 Zubir et al. examined the effects of additives on water absorption and plasticization in PLA/polyhydroxybutyrate-valerate (PHBV) blends.44 In contrast, some studies have focused on assessing the mechanical properties of 3D-printed products, conducting experiments to validate their potential applications.45–47 To the author's knowledge, only a few studies have been conducted on the durability of 3D-printed parts made from biopolyesters and their blends.48
Another significant limitation is that most biodegradable polyester degradation studies are conducted on thin polymer films, which fail to fully capture practical application scenarios due to their constrained thickness and simplified geometry. Bulk-scale parts offer more realistic scenarios, but their long-term degradation behaviors remain poorly understood. This study addresses this critical gap by investigating the environmental degradation of bulk-scale parts produced via fused filament fabrication (FFF) from PBS/PLA and PBS/PHB blends. A quantitative three-stage sorption model is applied to describe the complex water uptake behavior, clearly distinguishing the contributions of diffusion, hydrolysis, and leaching processes. The research examines explicitly how polymer blend composition and infill orientation influence long-term mechanical performance, water absorption, degradation, and morphological changes in artificial seawater. Results provide essential insights to better predict the practical environmental performance of biodegradable polyester blends used in marine applications.
2 Experimental section
2.1. Materials
Polybutylene succinate (PBS) is produced by PTT MCC Biochem Company Limited with grade FZ71PB® and trademark BioPBS™. It is made from succinic acid and is 50% bio-based and compostable, and its suggested applications include injection molding, lamination, and film blowing. PBS's melt flow index is 22 g/10 min (2.16 kg at 190 °C), density is 1.26 g cm−3, and inherent melting point is 115 °C. PLA, produced by NatureWorks LLC with trademark Ingeo™ and grade 6201D, is 100% bio-based, and it is suggested to apply it for the preparation of fibers. PLA's melt flow index is 15–30 g/10 min (210 °C, ASTM D1238), density is 1.24 g cm−3, and melting temperature is 170 °C. Poly-β-hydroxybutyrate (PHB) is produced by PHAradox with grade ENMAT Y3000P. It is made from 3-hydroxybutanoic acid from Cupriavidus necator fermentation of D-glucose and is 100% bio-based, and its suggested applications include extrusion, film blowing, thermoforming, and injection molding. PHB's melt flow index is 10–25 g/10 min (2.16 kg at 190 °C), density is 1.25 g cm−3, and melting temperature is 175–180 °C. Carbodilite® HMV-15CA (CDI) was granted from Nisshinbo Chemical Inc. and used as a thermal processing additive. Sodium chloride (NaCl), sodium sulfate (Na2SO4), sodium bicarbonate (NaHCO3), potassium bromide (KBr), boric acid (H3BO3), and sodium fluoride (NaF) were purchased from Merck KGaA (Darmstadt, Germany).2.2. Polymer blends and 3D printing filament processing
Biodegradable binary blends from PBS/PHB and PBS/PLA were prepared at a ratio of 7/3 and 5/5. CDI acted as chain extender to control molecular weight and viscosity. The PBS/PHB and PBS/PLA blends are defined in Table 1. The authors' previous study fully presents the information on melt blending.49 Briefly, according to the manufacturing recommendations, PBS, PHB, and PLA polymer granules were dried in a vacuum furnace (60 °C, 8 hours). The polymer blend compositions were compounded in twin-screw extruder Thermo Electron PRISM TSE 16 TC at barrel temperatures 190 °C (feeding zone), 185 °C, 180 °C, 175 °C, and 175 °C (die) and screw speed 30 rpm. Then, they were pelletized with Thermo Electron PRISM VARICUT 1 to a length of 2 mm and stored in sealed plastic bags. Pellets were used to produce filaments of a diameter 1.75 ± 0.05 mm by a 3Devo filament extruder with temperature distribution of 185 °C, 180 °C, 175 °C, and 175 °C and a screw rotation speed of 30 rpm.| Sample | PBS, wt% | PHB, wt% | PLA, wt% | CDI, wt% |
|---|---|---|---|---|
| S5B5 | 49.9 | 49.9 | — | 0.2 |
| S7B3 | 69.9 | 29.9 | — | 0.2 |
| S5L5 | 49.9 | — | 49.9 | 0.2 |
| S7L3 | 69.9 | — | 29.9 | 0.2 |
2.3. 3D modeling and printing
3D models were designed using CAD software (SolidWorks). The samples, according to ISO 527-20212 standards, were printed using Mass Portal Pharaoh ED Desktop 3D Printer with a nozzle diameter of 0.40 mm, printing speed of 40 mm s−1, extrusion multiplier of 1.25 ± 0.1, a printing line width of 0.50 mm and a layer height of 0.20 mm. The 3D printing process was as follows: firstly, the desired CAD model was created and saved in STL format. The 3D model was then sliced using the Simplify3D program with the desired printing properties, such as layer orientation. Then, the created G-code was loaded into the 3D printer, and the model was printed using the desired material. The samples were printed at 185 °C with the printing bed temperature set to 20 °C. Dumbbell-shaped samples with dimensions of 80 mm × 11 mm × 0.6 mm (Fig. 1) were printed with two types of infill directions (along the length of sample – vertical (V) and perpendicular – horizontal (H)). The infill type of horizontal prints was set to rectilinear, while it was set to concentric for vertical prints. The internal and external infill angle offsets were the same for vertical and horizontal prints and equal to 90°.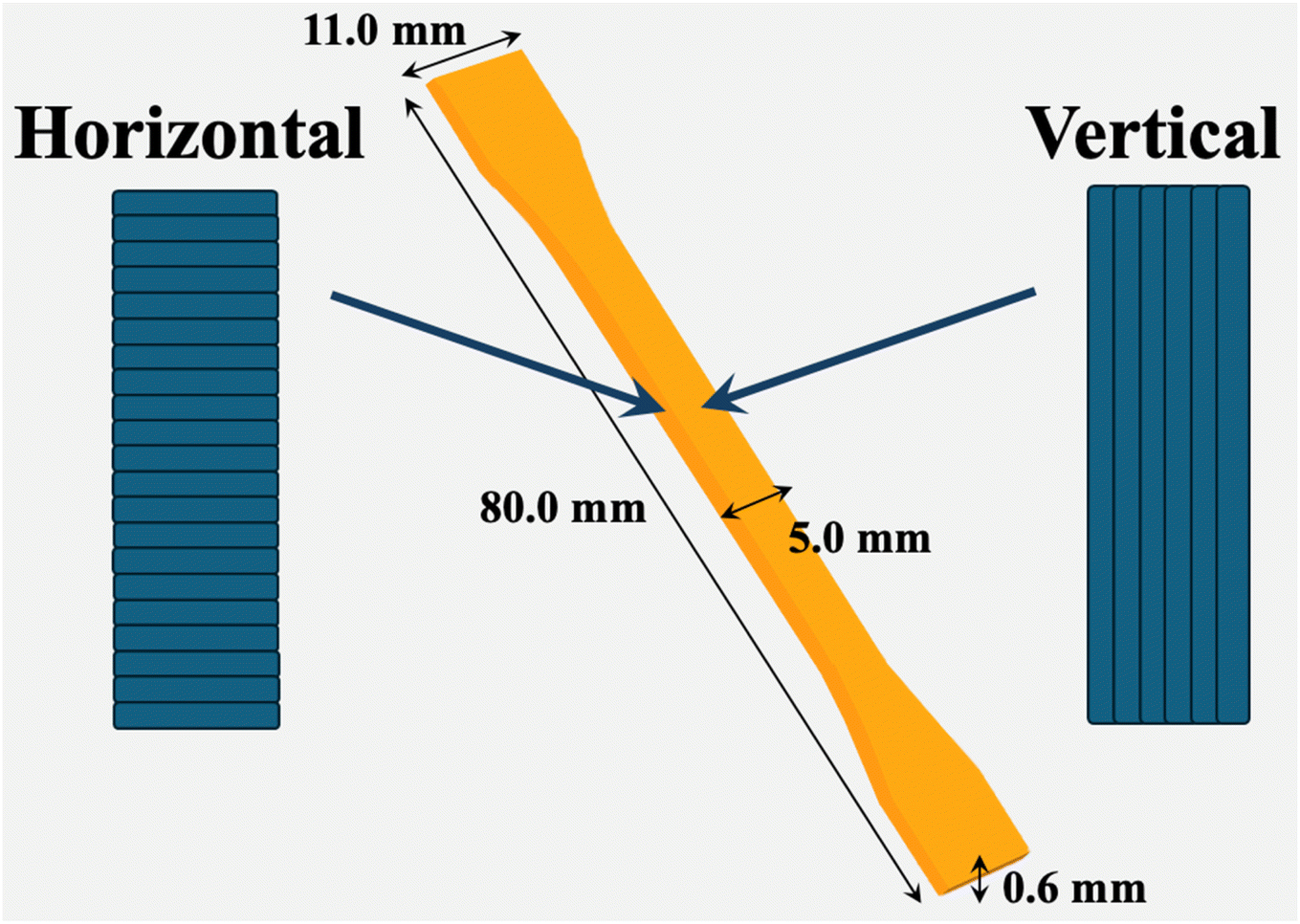 | ||
| Fig. 1 Model of 3D printed dumbbell shape sample. | ||
Some of the non-blended polymers did not produce printable filaments; therefore, compression molding was chosen to provide reference values for the blends and their mechanical properties and to assess the impact of 3D printing. Films with a thickness of 0.45 mm were prepared with the compression molding technique using Carver CH 4386 and used for tensile and absorption measurements. The temperature for hot plates was set at 185 °C. The samples were preheated for 2 min, compressed for 2 min, and cooled for 3 min between steel plates of 30 kg of thermal conductive mass.
2.4. Testing methods
The hydrodensitometry method was used to determine the density of samples. Measurements were done using Sartorius KB BA 100 electronic scales equipped with a Sartorius YDK 01 hydrostatic density measurement kit in air and ethanol. Ten replicates of each sample were used for measurements. The density of the blends was calculated using the following eqn (1):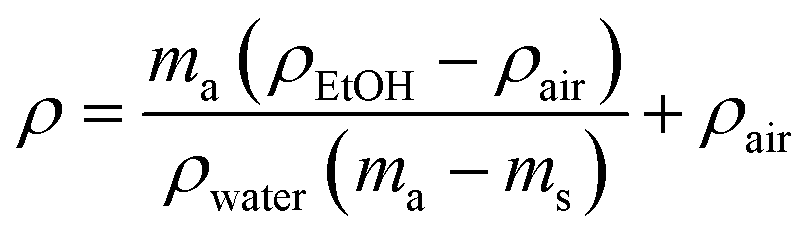 | (1) |
Field emission scanning electron microscope (FESEM) FEI Nova NanoSEM650 (Eindhoven, The Netherlands) equipped with an energy dispersive X-ray (EDX) analysis detector EDAX™ (Pleasanton, CA, USA) was used to perform measurements. Fractured sample cross-section surfaces were prepared through immersion in liquid nitrogen and were mounted on electrically conductive double-sided carbon tape for imaging. An acceleration voltage of 10 kV was used for image generation.
Water absorption was monitored via gravimetric measurements. To simulate the marine environment, samples were immersed in artificial seawater (AW) at ambient temperature (T = 21 ± 2 °C). Artificial seawater was prepared using the NaCl 24.53 g L−1, Na2SO4 4.09 g L−1, NaHCO3 0.20 g L−1, KBr 0.10 g L−1, H3BO3 0.03 g L−1, and NaF 0.03 g L−1 salts. Weight changes of samples were measured using analytical scales Precisa XT220A ± 0.0001 g, and the relative weight change (w, %) was determined by the following eqn (2):50–52
 | (2) |
Spectra were recorded using a Fourier Transform Infrared Spectrometer (FTIR) Nicolet 6700 (Thermo Scientific, Waltham, Germany) with an attuned total reflectance sampling method (FTIR-ATR). Spectra of 3D printed samples were collected at a resolution of 4 cm−1 in the region of 4000–600 cm−1, with sixteen runs for every sample transformed into the average spectrum with a measurement error below 1%.
Thermogravimetric analysis (TGA) was performed on a Mettler Toledo TG50 instrument, according to the ASTM D3850 standard. The measurements were made in an air environment at 10 °C min−1 from 25 °C to 600 °C. Materials thermal stability was evaluated from the weight loss heating curves.
Differential Scanning Calorimetry (DSC) was performed using a DSC-1 (Mettler Toledo) analyzer. The measurements were made under a nitrogen atmosphere, from 0 °C to 220 °C. The heating and cooling rate was 10 °C min−1, and the average weight of samples was approximately 10 mg. The crystallinity of the PBS and PHB components in the PBS/PHB and PBS/PLA blends was calculated from their melting peak using eqn (3):51,53
 | (3) |
 | (4) |
Tensile tests were carried out on a Tinius Olsen model 25ST (Horsham, PA, USA) universal testing machine with a 5 kN load cell. The test methodology was under ISO 527. The step-wise tensile tests determined Young's 0.2–0.5% modulus with a 1 mm min−1 crosshead speed. Then, the crosshead speed was increased to 5 mm min−1 (3–100%) and 15 mm min−1 (>100%).58,59 The 3D-printed dumbbell specimen models are visible in Fig. 1. Three replicate samples were tested for each composition, printing direction, and immersion/aging time (two weeks, one month, and six months). Aged samples were tested within five minutes after their removal from the artificial seawater. PBS, PHB, and PLA films used for tensile tests were cut into rectangle strips of 10 mm in width and about 60 mm long. The gauge length between the grips was 40 mm.
3 Results and discussions
3.1. Morphology
Firstly, the structural characteristics of the printed specimens after removal from a simulated artificial seawater environment were examined using SEM. In comparison, the morphology of PBS/PHB and PBS/PLA samples before and after aging is summarized in Fig. 2. The unique characteristics of FFF products include increased voids between layers and a rougher surface texture. These defects can be attributed to PBS's high melt-flow rate, which introduces defects during polymer melt deposition, allowing the lines to separate more easily.49 This finding highlights the critical influence of initial surface finish and print quality. Specifically, over-extrusion was also found to cover junction sites between layers, locations susceptible to stress concentrations, thereby facilitating crack formation (Fig. 2). Similar results were also reported in the literature, where increased air void content and void size in the printed structure led to the reduction of the strength of interlayers.60–62 | ||
| Fig. 2 SEM images of the surface morphology of PBS/PHB and PBS/PLA 3D-printed reference samples before aging (solid line) and samples after six months of aging in artificial seawater (dashed line). | ||
Water infiltrates through gaps and cracks, leading to the material's swelling and initiating hydrolytic degradation of the material. Similar observations are reported, considering that the hydrolytic degradation of PLA 3D-printed samples occurs primarily within the bulk rather than on the material surface.63,64 Our previous research has also highlighted that vertically printed samples exhibit line defects and cavities due to gaps between printing lines.49 Conversely, horizontally printed samples displayed signs of over-extrusion on their surfaces. After six months of water exposure, vertically printed specimens reveal noticeable voids and gaps between adjacent extrusion lines.49 Moreover, vertically printed parts of PBS-rich blends become slightly visually rougher over immersion time due to possible surface erosion.65 In contrast, among horizontal prints, only samples printed from PBS/PLA blends showed noticeable surface structure changes. However, it was a static seawater test, meaning the samples were not exposed to flowing water or physical abrasion conditions that typically drive surface-intensive erosion and formation of microplastics.66
Micrographs in Fig. 3 illustrate the cross-sectional morphology of PBS/PLA and PBS/PHB samples after a six months immersion period. PBS/PHB dumbbells exhibited rougher cross-sectional surfaces, while S5B5 vertical and horizontal samples displayed regions of clear phase separation between the polymers. In general, the images reveal that the sample cross-sections appear homogeneous, consistent with the density values measured before and after aging in artificial seawater, as shown in Table S1.†
 | ||
| Fig. 3 SEM images of the cross-sectional morphology of 3D-printed PBS/PHB and PBS/PLA samples, produced by liquid nitrogen fracture, after aging in artificial seawater for six months. S7L3 reveals salt crystals deposited on its surface after six months' immersion in artificial seawater. The EDX table shows the normalized elemental compositions of these crystals at analysis sites spot 1, spot 2, and spot 3. | ||
Blends have two distinct morphologies, which originate from blend ratios. A 7/3 ratio yields typical matrix/droplet morphology, while a 5/5 ratio yields a co-continuous phase structure. Samples S7B3 and S7L3 showed some pull-out voids due to the dispersed particle morphology of the blends. Furthermore, at higher magnification, Fig. 3 highlights the growth of inorganic salt crystals at the layer junctions, appearing as numerous small, regularly shaped crystallites.
Energy-dispersive X-ray spectroscopy (EDX) measurements (Fig. 3) confirmed the formation of regularly shaped crystalline inclusions across the respective areas. EDX analysis revealed that the inclusions primarily consist of K and Cl, with the normalized atomic percentages closely matching those of KCl salt crystals. Other detected elements, such as Na, Br, and Ca, were present in minor quantities. These findings align well with the chemical composition of the artificial seawater used in the study. It has been reported that chemical elements in seawater can promote the degradation of macromolecular chains, specifically through hydrolytic scission of polymer chains. The hydrolysis process can directly and negatively impact the thermal and mechanical properties of polymer materials.67 Several external factors, including chemical reactivity, salt concentration, pH, solvent and polymer molecular mobility, water diffusion rate, and other conditions influence the hydrolysis process. Studies have shown that the hydrolysis of polyesters occurs more rapidly in alkaline solutions compared to acidic ones, leading to an accelerated degradation rate.68,69 Consequently, the hydrolytic scission of biopolyester chains is strongly amplified in the presence of dissolved inorganic salts in aquatic environments. Table S2† shows that the immersion solution remained alkaline, with a pH range of 8.0–8.5, throughout the six months experiment. In 3D-printed samples, structural defects such as air voids and cracks – arising from differences in printing direction and infill patterns – can markedly increase the rate of hydrolytic degradation.
3.2. Water absorption
The weight gain curves of the 3D-printed neat polymer samples aged in artificial seawater are shown in Fig. 4. During the initial stages of water absorption, PBS, PLA, and PHB samples exhibited similar behavior, characterized by a rapid increase in weight followed by stabilization. All samples reached apparent saturation within two weeks (approximately ) of immersion in seawater. The water absorption (or weight gain) of the samples is driven by the diffusion mechanism, referred to as Fickian diffusion, which is described by the following eqn (5):51,70,71
) of immersion in seawater. The water absorption (or weight gain) of the samples is driven by the diffusion mechanism, referred to as Fickian diffusion, which is described by the following eqn (5):51,70,71 | (5) |
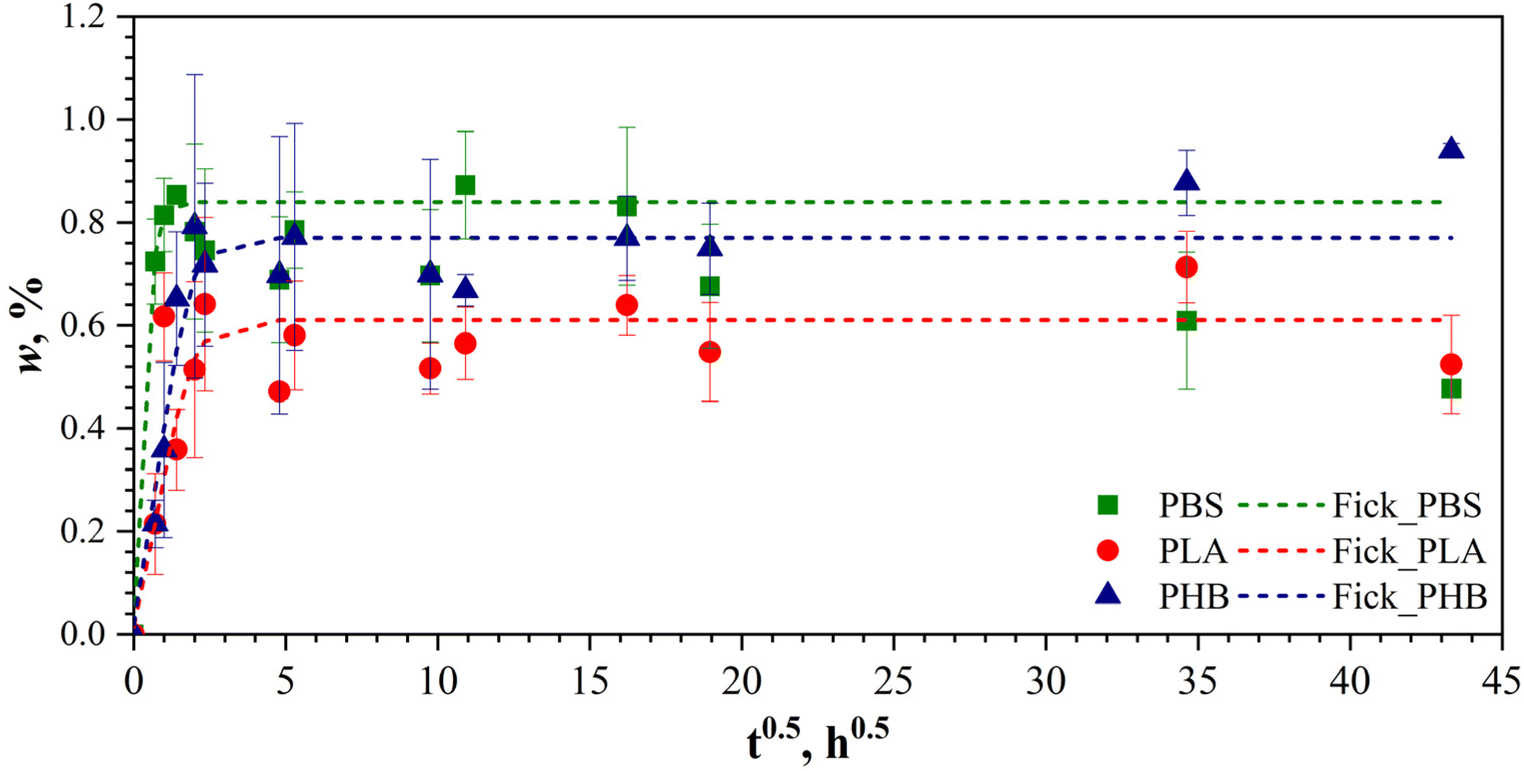 | ||
| Fig. 4 Weight gain curves of neat PBS, PHB, and PLA samples immersed in artificial seawater are presented. Lines are approximations by eqn (6). | ||
The corresponding values for the apparent Fickian water equilibrium wd∞ were determined to be 0.83% ± 0.15%, 0.77% ± 0.08%, and 0.6% ± 0.09% for PBS, PHB, and PLA, respectively. The diffusion coefficients of neat polymers were defined by fitting the weight gain curves (Fig. 4), and their values are D = 0.072 mm2 h−1, 0.009 mm2 h−1, and 0.015 mm2 h−1 for PBS, PHB, and PLA, respectively. However, with prolonged exposure to seawater, deviations from the equilibrium state became evident in the PHB and PBS samples. Notably, opposite trends were observed for the neat polymers PBS and PHB: a decrease in weight for PBS and an increase in weight for PHB.
It is known that water absorption in biopolyesters such as PBS and PHB can lead to a polymer chain hydrolysis process, which may be accompanied by the leaching of low-molecular-weight degradation products or remnants of hydrolysis, along with additional water absorption.72,73 The declining trend in the weight gain curve of PBS suggests that leaching mechanisms are predominant, while the continuous increase in weight of PHB samples can be attributed to additional hydrolysis-driven water absorption. The observed water absorption behaviour and the obtained values of weight gain for PBS, PLA, and PHB are consistent with data reported in other studies. The reported average sorption capacity value for PBS was about 1%,7,74,75 and 1–5%.76 PLA demonstrated close to Fickian water absorption behaviour with wd∞ = 0.6%, which also agrees well with literature data.51,77,78
The weight gain curves of vertically printed samples of various blends are compared in Fig. 5, with the sorption characteristics provided in Table 2. Notably, no significant differences in water sorption capacity were observed between the 5/5 and 7/3 pairs of specimens. The maximum weight gain wmax for S5B5 and S5L5 samples is within the range of 1.95 ± 0.55%, while S7B3 and S7L3 samples absorbed a higher amount of water, with an approximate wmax value of 2.20 ± 0.80%. These wmax values for the printed blend samples are up to two to three times higher than those predicted by the rule of mixture (RoM). In the first approximation, it can be assumed that the equilibrium (Fickian) water content in binary blends is additively contributed by each component:51
| wd∞(blend) = wd∞(polymer 1) × φ(polymer 1) + wd∞(polymer 2) × φ(polymer 2) | (6) |
| wd∞(S7B3) = wd∞(PBS) × 0.7 + wd∞(PHB) × 0.3 | (7) |
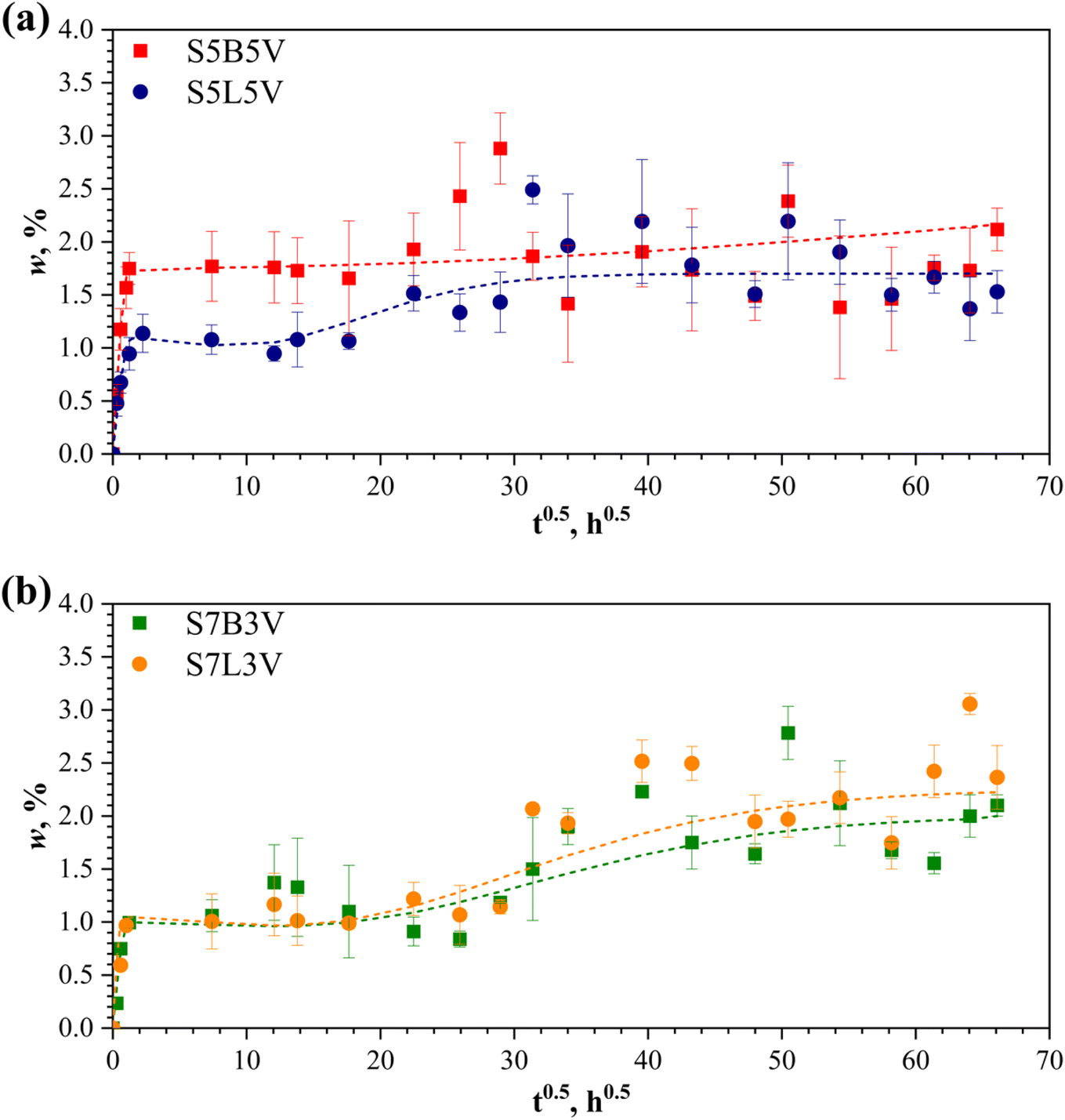 | ||
| Fig. 5 Weight gain curves of vertically printed blend samples (a) S5B5 and S5L5, and (b) S7B3 and S7L3. Lines are approximations by eqn (8)–(10). | ||
| Sample | w max, % |
|
I | II | III | |||
|---|---|---|---|---|---|---|---|---|
| D, mm2 h−1 | w ∞d, % | k h, h−1 | w ∞h, % | k l, h−1 | w ∞l, % | |||
| a Calculated by the rule of mixture (RoM). b Similar for vertical and horizontal orientations. | ||||||||
| S5B5V | 2.12 ± 0.20 | 0.77 | 0.20 | 1.75 | 0.0005 | 0.37 | — | — |
| S5B5H | 1.22 ± 0.30 | 0.77b | 0.20 | 0.60 | 0.001 | 0.73 | — | — |
| S7B3V | 1.82 ± 0.07 | 0.82 | 0.20 | 1.00 | 0.002 | 1.25 | 0.004 | 0.50 |
| S7B3H | 1.33 ± 0.24 | 0.82b | 0.20 | 1.00 | 0.003 | 0.60 | 0.009 | 0.27 |
| S5L5V | 1.53 ± 0.15 | 0.73 | 0.30 | 1.00 | 0.003 | 1.20 | 0.004 | 0.48 |
| S5L5H | 1.62 ± 0.21 | 0.73b | 0.30 | 1.00 | 0.001 | 1.20 | 0.004 | 0.48 |
| S7L3V | 2.34 ± 0.25 | 0.80 | 0.25 | 1.00 | 0.001 | 2.00 | 0.005 | 0.76 |
| S7L3H | 1.88 ± 0.07 | 0.80b | 0.25 | 1.00 | 0.002 | 1.73 | 0.006 | 0.90 |
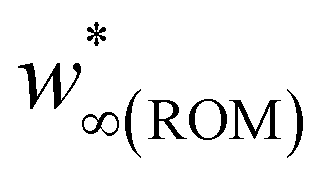
The differences between wmax and wd∞(blend) can be attributed to the 3D printing features and the inevitable formation of voids in the structure of the samples due to the layer-by-layer deposition of filaments. Defects in the blend structure, the filaments themselves, and those introduced during the printing process contribute to additional water absorption. It is also important to note that the higher water absorption of the 7/3 samples, compared to their 5/5 counterparts, cannot be solely explained by the higher contribution of PBS and its greater water absorption capacity (Fig. 4). The 7/3 samples exhibited poorer processability and printability, leading to more structural defects, as shown by SEM images and, consequently, a higher void content in the samples.
The weight gain curves of vertical and horizontal prints of 5/5 blends are shown in Fig. 6. The water absorption of S5B5 samples shows differences in properties, i.e., vertical samples exhibit slightly higher water absorption than horizontally printed samples. This difference can be explained by the higher number of voids and pores observed in the vertical samples' specific printing conditions (layering). At the same time, these effects do not appear in S5L5 samples due to their better processability compared to S5B5 blend samples. Similar behaviour was observed for S7B3 and S7L3 (Fig. S1†).
 | ||
| Fig. 6 Weight gain curves of vertically and horizontally printed specimens of (a) S5B5 and (b) S5L5. Lines are approximations by eqn (8)–(10). | ||
Water absorption by biodegradable polymers, such as PBS, PHB, and PLA, is a complex process involving different mechanisms and is accompanied by the degradation of materials.79,80 When immersed in water, especially salt water, irreversible (chemical) biopolyester degradation can occur due to hydrolytic, enzymatic, and oxidative degradation.43,81 Simultaneously, the physical effects caused by the polymer/water interaction, such as plasticization and polymer swelling, are usually reversible.52,82 Considering the complexity and interconnection of all involved processes, it is usually hard to divide the contribution of each involved mechanism and model the water absorption–biodegradation phenomena in biopolyester. In this study, we consider a simplified approach to model anomalous water absorption by biopolyester blends and assume the additive contribution of several processes.
The total changes in the weight of the samples are related to three processes: water absorption by the diffusion mechanism, additional water absorption caused by hydrolytic decomposition of the polymer and leaching of decomposition products. Then, the mathematical representation of the proposed three-stage model of weight change w(t) can be written as follows:71
| w(t) = wd(t) + wh(t) − wl(t) | (8) |
The diffusion component is given by eqn (5). The hydrolysis-driven water absorption and leaching of biopolyester degradation products are assumed to be the first-order kinetic processes. Then, wh is given by the following eqn (9):71
| wh(t) = wh∞(1 − e−kht) | (9) |
| wl(t) = wl∞(1 − e−klt) | (10) |
The three-stage model eqn (5) and (8)–(10) was applied to model weight changes of the produced biopolyester blends when immersed in artificial seawater. The data approximation procedure consisted of several steps. First, the diffusion constants wd∞ and D were estimated from the start regions of the w vs. curves. Then, wh∞ and kh and wl∞ and kl were adjusted to get the best fit of the curves. Note, the wl(t) component was considered only for compositions demonstrating s-shape weight change kinetics. Polymer blends that did not lose weight when immersed in water were modeled using a two-stage model (diffusion plus additional water absorption due to hydrolysis).
curves. Then, wh∞ and kh and wl∞ and kl were adjusted to get the best fit of the curves. Note, the wl(t) component was considered only for compositions demonstrating s-shape weight change kinetics. Polymer blends that did not lose weight when immersed in water were modeled using a two-stage model (diffusion plus additional water absorption due to hydrolysis).
The approximation results of the three-stage water absorption model are shown in Fig. 5 and 6. The model parameters are summarized in Table 2. All prints reached apparent saturation at  (two weeks). However, after that, non-Fickian sorption was observed. All specimens, except S5B5, showed a similar three-stage sorption behaviour: weight gain curves have a typical s-shape with apparent secondary equilibrium after about two months, followed by steady growth. S5B5 weight gain increases steadily after primary Fickian saturation is reached without secondary equilibrium occurring. As seen in Fig. 5, S5B5 samples showed slightly higher water absorption than S5L5, while the water sorption capacity variations were insignificant for PBS/PHB and PBS/PLA 7/3 compositions. This can be related to the higher hydrophilicity of PHB compared to PLA; however, for 7/3 samples, these differences were mitigated by the main PBS contribution. Changing the printing orientation to the horizontal S5B5 demonstrated some print direction-based properties in sorption behaviour and wmax values for S5B5V and S5B5H samples differ by a factor of two (Table 2 and Fig. 6). At the same time, no significant changes in the weight gain curves of S5L5V and S5L5H specimens were observed. According to the data from Table 2, the diffusivity of 3D-printed binary blend specimens is higher than that of neat polymers. Also, the difference in sorption parameters related to the first stage (Fickian) sorption is insignificant for specimens with different compositions and orientations. Parameters (kh, w∞h, kl, w∞l) related to degradation processes (hydrolysis and leaching) shifted with changing blend composition and printing orientation.83,84 Structural imperfections (e.g., 7/3 vertical prints) result in additional water ingress followed by more extensive degradation processes.
(two weeks). However, after that, non-Fickian sorption was observed. All specimens, except S5B5, showed a similar three-stage sorption behaviour: weight gain curves have a typical s-shape with apparent secondary equilibrium after about two months, followed by steady growth. S5B5 weight gain increases steadily after primary Fickian saturation is reached without secondary equilibrium occurring. As seen in Fig. 5, S5B5 samples showed slightly higher water absorption than S5L5, while the water sorption capacity variations were insignificant for PBS/PHB and PBS/PLA 7/3 compositions. This can be related to the higher hydrophilicity of PHB compared to PLA; however, for 7/3 samples, these differences were mitigated by the main PBS contribution. Changing the printing orientation to the horizontal S5B5 demonstrated some print direction-based properties in sorption behaviour and wmax values for S5B5V and S5B5H samples differ by a factor of two (Table 2 and Fig. 6). At the same time, no significant changes in the weight gain curves of S5L5V and S5L5H specimens were observed. According to the data from Table 2, the diffusivity of 3D-printed binary blend specimens is higher than that of neat polymers. Also, the difference in sorption parameters related to the first stage (Fickian) sorption is insignificant for specimens with different compositions and orientations. Parameters (kh, w∞h, kl, w∞l) related to degradation processes (hydrolysis and leaching) shifted with changing blend composition and printing orientation.83,84 Structural imperfections (e.g., 7/3 vertical prints) result in additional water ingress followed by more extensive degradation processes.
In addition, to assess how well a model explains and predicts future outcomes based on the data, it is fitted to the coefficient of determination (R2) and calculated using the following eqn (11):
 | (11) |
The R2 values for S5B5H, S5L5V, S5L5H, and S7L3V were 80%, while for S5B5H it is 74%. S5B5V demonstrated the lowest R2 value, which was only 58%. While the model did not achieve a perfect fit, it demonstrated satisfactory agreement between the experimental and calculated data. The poorer fit observed for the S5B5V sample may be attributed to the complex interrelated contributions of water diffusion and hydrolysis-driven leaching mechanisms and the presence of micro-voids resulting from specific printing quality. Also, the S5B5V samples are characterized by relatively high void content, which is reflected with wmax and w∞(ROM) values in Table 2.
3.3. FTIR spectroscopy
Fig. 7 shows the FTIR spectra of S5B5 and S5L5 before and after 6 months of sorption in artificial seawater. The PBS/PHB blends are characterized by the asymmetric and symmetric C–H stretching at 3020–2840 cm−1, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of aliphatic ester at 1715 cm−1, and 1200–1000 cm−1 C–O–C and C–O stretching region.72,85,86 PBS/PLA show the asymmetric and symmetric stretching vibrations of C–H bonds at 3030–2790 cm−1, the CO peaks at around 1712 cm−1, bending vibrations of C–H bonds at 1470–1300 cm−1, and C–O–C and C–O stretching vibration at 1250–1000 cm−1.87–89
O stretching of aliphatic ester at 1715 cm−1, and 1200–1000 cm−1 C–O–C and C–O stretching region.72,85,86 PBS/PLA show the asymmetric and symmetric stretching vibrations of C–H bonds at 3030–2790 cm−1, the CO peaks at around 1712 cm−1, bending vibrations of C–H bonds at 1470–1300 cm−1, and C–O–C and C–O stretching vibration at 1250–1000 cm−1.87–89
 | ||
| Fig. 7 FTIR spectra of S5B5 and S5L5 blends: (a) full spectra, (b) CO stretching vibrations, and (c) C–O–C stretching vibrations before and after immersion in artificial seawater. | ||
FTIR analysis revealed only minor spectral changes in the biopolyester blends after six months of water exposure. For PBS/PHB and PBS/PLA, a slight decrease was recorded in the ester-related band at 1250–1000 cm−1, accompanied by a modest reduction in the carbonyl stretching peaks at 1712 and 1719 cm−1 (PBS/PHB) and 1712 cm−1 (PBS/PLA). In sample S5L5, small shifts appeared for the C–O–C stretching band (to 1155 cm−1) and the aliphatic-ester CO band (to 1712 cm−1). These subtle intensity losses and shifts may reflect limited chain scission via hydrolysis, but overall, they indicate slow degradation. Comparable spectra were obtained for the S7B3 and S7L3 prints (Fig. S2†).
Hydrolytic degradation of PBS/PHB and PBS/PLA blends has previously been linked to a drop in molecular weight and to changes in crystalline–amorphous balance.90,91 Aldehyde and carboxyl end-groups produced during degradation can alter the relative area of the carbonyl band associated with the free-amorphous phase of PBS.66,92,93 Because the present FTIR data show only marginal variations, DSC was performed on reference and aged samples to obtain a clearer picture of crystalline-structure evolution; those results are discussed in the next section.
3.4. Thermal analysis
DSC was employed to monitor changes in the thermal properties of neat and blended polymers resulting from prolonged seawater aging. Fig. 8 illustrates DSC's first heating, cooling, and second heating scans, while thermal characteristics, i.e., melting temperature and degree of crystallinity of the first heating scan for blends before aging and after one and six months of exposure, are visualized in Fig. 9. Precise values from DSC curves are summarized in Tables S3–S5.† | ||
| Fig. 8 DSC curves: (a) first heating, (b) cooling, and (c) second heating scans. | ||
 | ||
| Fig. 9 Melting temperature (dashed lines) and degree of crystallinity (solid lines) extracted from first-run DSC heating scans for neat polymers and for PBS/PLA and PBS/PHB blends. | ||
Fig. 8(a) shows typical peaks observed for neat polymers: the PHB curve contains a melting peak at 180 °C, while the PBS curve shows a melting peak centered at 115 °C with a shoulder shifted towards lower temperatures. It is typical of PBS to have imperfectly crystallized lamellae undergo melting and subsequent recrystallization into more perfect lamellae during heating.35 PBS second melting curve also has a small exothermic recrystallization peak (Fig. 8(c)), which also appears in some blends. PLA scan reveals glass transition at around 60 °C, cold crystallization at 100 °C, and a melting peak at 173 °C. At the same time, PHB demonstrated a melting peak at 180 °C. Focusing on unaged PBS/PHB blends, crystallinity (χm) of PBS in blends significantly decreases compared to neat PBS, dropping from 51.2% in neat PBS to 34.8% in S5B5 and 30.8% in S7B3 at 0 months (Table S3†). This indicates that PHB hinders the crystallization process of PBS.94 PHB in blends maintained similar crystallinity levels to neat PHB but exhibited lower melting temperatures.
In general, the crystalline phase acts as a barrier to water diffusion, thereby reducing overall water molecules permeability,95 while degradation predominantly occurs in the more accessible amorphous regions.96 Our data support this assumption and indicate that crystallinity does not significantly change after 6 months of aging compared to the unaged samples. In addition, changes observed in polymer blends can deviate from expected performance in single polymer systems, and this is attributed to the aspects of developed blend morphology, which introduce additional interfaces that can disrupt or promote secondary crystallization.97,98 After immersion, PHB crystallinity in the S5B5 blend was 56.5%, 60.7%, and 58.5% for 0, 1, and 6 months of immersion, respectively. There is substantial evidence from the literature demonstrating that PHAs degrade in seawater.99–101 PLA in the PBS/PLA blends followed the same pattern. As reported by other authors, the increase in crystallinity could be explained by the erosion of some of the amorphous parts of PLA that resulted in a loss of molecular fragments.102–104 The degree of crystallinity in PBS increased steadily with immersion time. Similarly to PLA and PHB, semicrystalline PBS's amorphous regions are more susceptible to hydrolytic degradation.105,106
Cooling curves (Fig. 8(b)) and corresponding data (Table S4†) highlight significant changes in the crystallization process of PBS, particularly in PBS/PLA blends. The crystallization exotherm of PBS shifted to lower temperatures, indicating that nucleation has become less efficient. Either because existing nucleating surfaces were partially masked by the second polymer or because hydrolytic fragmentation disrupted chain alignment and formation of imperfect crystals.105 Consequently, PBS requires greater undercooling before nuclei can form and crystal growth can proceed, so its crystallization temperature drops.97,98
Second heating (Fig. 8(c)) can examine and compare aged and unaged samples by removing pre-history. In the case of second heating (Table S5†), a similar observation is observed. Table S5† shows that the crystallinity of PLA in the S5L5 and S7L3 blends increased to 19.6% and 17.3% from 13.1% and 7.9%, respectively, after six months of immersion in artificial seawater. Similarly, PHB in PBS/PHB blends exhibited some increase in crystallinity, although the change was less pronounced than for PLA. In contrast, PBS showed only a slight increase in crystallinity within PBS/PLA blends, while in PBS/PHB blends, the presence of PHB appeared to suppress PBS crystallization more significantly. These results align with previously discussed hydrolytic degradation mechanisms.
As shown in Table S5,† the glass transition temperature (Tg) of PLA in the blends showed a decrease after immersion in artificial seawater. A decrease in Tg during aging aligns with observed hydrolytic degradation, indicated some plasticization and is supported by the literature.104,105 It has also been reported that the Tg of PLA in PLA/PBS blends changes non-linearly with the polymer composition.106
Fig. S3† presents the TGA thermograms along with their corresponding first derivative curves. The TGA curves display comparable profiles for both the reference and aged samples. The derivative curves indicate a two-stage degradation process for all compositions, corresponding to each polymer's component's expected primary degradation peaks. Similarly, no significant differences were observed in the derivative curves. These TGA results suggest hydrolysis is relatively slow.
3.5. Tensile tests
The effect of the printing parameters and water influence on the 3D-printed samples' mechanical characteristics was measured. Table 3 demonstrates the average values of the elastic modulus (E), ultimate strength (σu), and ultimate strain (εu) of neat PBS, PLA, and PHB films. Hereinafter for specimens exhibiting necking and yielding, the yield strength and yield strain are considered as ultimate characteristics σu and εu, respectively. PLA and PHB are brittle compared to PBS, which shows ductile failure behaviour. After three months of water sorption, PLA and PHB show a 1.2-fold decrease in ultimate strength, but σu value for PBS did not change and remains within the scatter of data and is equal to 31.33 ± 0.89 MPa. PLA and PBS show a decrease in ultimate strain by 18%, whereas εu of PHB decreases by 45%. The E values of PLA and PBS were in the data scattering range, while the modulus of PHB slightly increased.| Sample | Reference | 3 months in artificial seawater | ||||
|---|---|---|---|---|---|---|
| E, GPa | σ u, MPa | ε u, % | E, GPa | σ u, MPa | ε u, % | |
| PBS | 0.51 ± 0.02 | 28.19 ± 0.55 | 18.11 ± 0.95 | 0.57 ± 0.04 | 31.33 ± 0.89 | 14.84 ± 1.90 |
| PLA | 1.78 ± 0.09 | 62.56 ± 5.91 | 4.45 ± 0.27 | 2.04 ± 0.13 | 53.33 ± 8.90 | 3.670 ± 0.57 |
| PHB | 1.89 ± 0.18 | 42.87 ± 3.67 | 3.55 ± 0.46 | 2.66 ± 0.20 | 37.54 ± 1.95 | 1.97 ± 0.20 |
Fig. 10 shows stress–strain curves of vertical (V) and horizontal (H) PBS/PHB and PBS/PLA blends before and after immersion in artificial seawater. The effects of aging on the mechanical response were evaluated for 3D-printed blend samples immersed in artificial seawater for 6 months. A bar chart comparing various tensile properties, including error bars, is presented in Fig. 11. The data in Table 3 indicate that these mechanical properties are clearly limited by the polymer matrix and significantly affected by selected printing parameters. Notably, S5L5V exceeds the ultimate strength of pure PBS, showing around 35.32 ± 2.15. At the same time, S5B5V shows only about 23.10 ± 0.85 MPa. Fig. 10(a) illustrates the mechanical changes observed in S7L3 samples over the immersion period (two weeks, one month, and six months). In contrast, the aging effects on vertically and horizontally printed S5B5, S7B3, and S5L5 samples are presented in Fig. S5–S7,† respectively. As seen in Fig. 10(a), S7L3V and S7L3H elongation at break dropped to 18.5- and 1.8-fold, respectively, after six months aging. At the same time, ultimate strength values decreased by 2.2- and 3.1-fold for S7L3V and S7L3H, respectively.
 | ||
| Fig. 10 Stress–strain diagrams for (a) vertical (V) and horizontal (H) S7L3 prints for two weeks, one month, and six months aging in seawater; (b) S5B5 and S5L5 vertical and horizontal prints; (c) S5B5, S5L5, and S7B3, S7L3 vertical samples in referenced and after aging in artificial seawater. | ||
 | ||
| Fig. 11 (a) and (b) Elastic moduli (E), (c) and (d) ultimate strength (su), (e) and (f) ultimate strain (eu) for PBS/PHB and PBS/PLA vertical and horizontal prints in the reference state and aged for one month and six months. | ||
PBS/PHB and PBS/PLA print demonstrated the same tendency in the vertical and the horizontal orientation (Fig. 10(b)). Both reference and aged vertical prints achieved significantly higher values than horizontal ones. The vertical prints exhibit necking formation, which was not observed for the horizontal prints due to their lower contact surfaces between extruded filaments.107 Additionally, the orientation of vertical prints aligns with the strain orientation, while the orientation of horizontal samples depends on the created interface and print line overlays.49
In addition, stress–strain curves for vertical PBS/PHB and PBS/PLA samples are compared in Fig. 10(c). PBS/PLA prints demonstrate the highest ultimate strength and ultimate strain values, but the lowest elastic modulus compared to PBS/PHB.108 Tao et al. reviewed defect formation in FFF 3D printing, highlighting inherent shortcomings, such as void formation, poor interlayer adhesion, and anisotropic mechanical behavior, which can significantly reduce the mechanical performance of printed parts.109 Moreover, these defects are closely linked to the selected processing parameters. This is particularly relevant to selected print orientations (vertical or horizontal). The literature indicates that compared to the single-layer bulk, spherulites at the layer weld interface show enhanced birefringence, indicating greater structural perfection.109 However, the bulk region hosts a larger number of spherulites, suggesting higher nucleation but reduced individual spherulite quality. These findings suggest that layer weld interfaces, due to their localized structural characteristics, commonly serve as preferential sites for stress concentration and subsequent mechanical failure.
Tensile tests were also performed on samples aged for two weeks, but changes in mechanical characteristics were in the data scatter range and were therefore not shown. After one month of immersion in seawater, the mechanical properties showed minimal change, with the ultimate values falling within the data scatter of the unaged prints. In DSC, it was observed that crystallinity increased after one month of immersion, but mechanical properties did not increase. This could be related to slight hydrolytic degradation and leaching compensating for the crystallinity increase of the immersed samples. Likewise, inherent defects from the 3D printing process and blend morphology likely play a role in determining mechanical performance. After six months of aging, a notable decrease in properties is observed, but PBS/PHB, both vertical and horizontal, blends retain slightly higher E values than PBS/PLA blends. The most extraordinary drop, 2.5-fold in E values, was for S5B5, which was attributed to the higher degradation rate of PHB.110
Vertical and horizontal PBS/PLA prints reach the highest ultimate strength values and show this trend during 6 months of aging. After six months of water sorption, a remarkable drop in σu values were observed for S7B3V and S5B5H samples, about 2.1- and 3.3-fold, respectively. This may be attributed to the lower compatibility with PBS and the higher degradation rate of PHB. During the printing process of thermoplastic polymers, layer welding is dominated by an amorphous region.109,111 As was previously discussed, the amorphous phase is most sensitive to hydrolytic degradation. In this regard, the observed significant decrease in elastic modulus and ultimate strength can be attributed to hydrolytic degradation, complemented by the structural design induced by 3D printing.
Strain values after six months of immersion indicate a total loss of ductility in the blends, except in cases where the material was already brittle. Ultimate strain (Fig. 11) also declines in nearly every blend composition, pointing to the incipient embrittlement of polymer material.110 It can be observed that vertical prints have more significant decreases in mechanical properties than horizontal prints. This is attributed to the higher water content of the vertical samples.48
4 Conclusion
This study examined the influence of FFF printing orientation and blend composition on the hydrolytic aging behavior of PLA-, PHB-, and PBS-based binary polymer blends in artificial seawater. A key focus was placed on how printing parameters, particularly orientation, correlate with changes in structure and performance over time. All samples showed rapid initial water uptake, reaching their primary Fickian sorption plateau within two weeks, with equilibrium contents between 1.2 and 2.3 wt%. Non-Fickian sorption kinetics followed, including additional uptake from hydrolytic decomposition and mass loss from leaching. Higher water uptake was observed in 7/3 blends, which contained more structural voids.After six months of immersion, DSC results showed that the degree of crystallinity increased, particularly for PLA and PHB components. Tensile testing showed that mechanical properties remained close to unaged values after one month. However, after six months, ultimate strength and elastic modulus declined significantly by up to 3.3-fold and 2.5-fold, respectively, with PHB-containing blends showing greater reductions. No increase in ultimate strain was observed, indicating that plasticization did not occur. Instead, material embrittlement was evident, particularly in vertically printed samples, which also exhibited higher water uptake. Vertical prints consistently deteriorated more than horizontal prints, which is attributed to their higher porosity and greater water uptake.
SEM and EDX analyses revealed the formation of salt crystals at interlayer interfaces, confirming localized hydrolytic degradation pathways facilitated by printing-induced structural imperfections. FTIR and TGA results showed only minor spectral changes, indicating slow chain scission throughout the six months exposure.
The data clearly point towards the observed hydrolytic degradation of polymers. Still, the overall breakdown of bulk parts remained slow under static marine conditions. This suggests that, if environmental plastic pollution is the primary concern, such materials, especially in their solid printed form, may persist significantly longer than expected. Future perspectives should include microbial activity and mechanical abrasion studies to represent real-world marine environments and predict polymer blend durability.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Alisa Sabalina: data curation; formal analysis; investigation; validation; visualization; writing – original draft. Sergejs Gaidukovs: conceptualization, methodology; resources; supervision; writing – review and editing. Oskars Platnieks: formal analysis; writing – original draft; writing – review and editing. Olesja Starkova: data curation; formal analysis; writing – original draft; writing – review and editing. Gerda Gaidukova: resources, validation. Liga Orlova: investigation. Maksims Jurinovs: investigation; writing – review and editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This activity/work has been supported by the EU Recovery and Resilience Facility within the Project No. 5.2.1.1.i.0/2/24/I/CFLA/003 “Implementation of consolidation and management changes at Riga Technical University, Liepaja University, Rezekne Academy of Technology, Latvian Maritime Academy and Liepaja Maritime College for the progress towards excellence in higher education, science and innovation” academic career doctoral grant (ID 1027).References
- A. Dey, M. M. Rahman, N. Yodo and D. Grewell, Mater. Today Commun., 2023, 34, 105316 CrossRef CAS.
- T. Yao, H. Ouyang, S. Dai, Z. Deng and K. Zhang, Compos. Struct., 2021, 268, 113970 CrossRef CAS.
- F. Pallottino, L. Hakola, C. Costa, F. Antonucci, S. Figorilli, A. Seisto and P. Menesatti, Food Bioprocess Technol., 2016, 9, 725–733 CrossRef.
- J. G. Rosenboom, R. Langer and G. Traverso, Nat. Rev. Mater., 2022, 7, 117–137 CrossRef PubMed.
- R. Pantani, F. De Santis, F. Auriemma, C. De Rosa and R. Di Girolamo, Polymer, 2016, 99, 130–139 CrossRef CAS.
- J. V. Ecker, A. Haider, I. Burzic, A. Huber, G. Eder and S. Hild, Rapid Prototyp. J., 2019, 25, 672–678 CrossRef.
- D. M. Nieto, M. Alonso-García, M. A. Pardo-Vicente and L. Rodríguez-Parada, Polymers, 2021, 13, 1036 CrossRef CAS PubMed.
- W. Kanabenja, K. Passarapark, T. Subchokpool, N. Nawaaukkaratharnant, A. J. Román, T. A. Osswald, C. Aumnate and P. Potiyaraj, Addit. Manuf., 2022, 59, 103130 CAS.
- T. Narancic, S. Verstichel, S. R. Chaganti, L. Morales-Gamez, S. T. Kenny, B. De Wilde, R. B. Padamati and K. E. O'Connor, Environ. Sci. Technol., 2018, 52, 10441–10452 CrossRef CAS PubMed.
- P. Shaiju, B. B. Dorian, R. Senthamaraikannan and R. B. Padamati, Molecules, 2020, 25, 5766 CrossRef CAS PubMed.
- M. He, Y. I. Hsu and H. Uyama, J. Hazard. Mater., 2024, 474, 134819 CrossRef CAS PubMed.
- M. M. Hedrick, F. Wu, A. K. Mohanty and M. Misra, RSC Adv., 2020, 10, 44624–44632 RSC.
- J. Y. Boey, L. Mohamad, Y. S. Khok, G. S. Tay and S. Baidurah, Polymers, 2021, 13, 1544 CrossRef CAS PubMed.
- T. Glaskova-Kuzmina, O. Starkova, S. Gaidukovs, O. Platnieks and G. Gaidukova, Polymers, 2021, 13, 1–27 Search PubMed.
- D. Fico, D. Rizzo, R. Casciaro and C. E. Corcione, Polymers, 2022, 14, 465 CrossRef CAS PubMed.
- J. Muller, C. González-Martínez and A. Chiralt, Materials, 2017, 10, 1–22 Search PubMed.
- Z. Qi, B. Wang, C. Sun, M. Yang, X. Chen, D. Zheng, W. Yao, Y. Chen, R. Cheng and Y. Zhang, Int. J. Mol. Sci., 2022, 23, 6746 CrossRef CAS PubMed.
- M. S. Andrés, R. Chércoles, E. Navarro, J. M. de la Roja, J. Gorostiza, M. Higueras and E. Blanch, J. Cult. Herit., 2023, 59, 181–189 CrossRef.
- A. Dey, I. N. R. Eagle and N. Yodo, J. Manuf. Mater. Process., 2021, 5, 69 CAS.
- Q. Ou-Yang, B. Guo and J. Xu, ACS Omega, 2018, 3, 14309–14317 CrossRef PubMed.
- S. J. Royer, F. Greco, M. Kogler and D. D. Deheyn, PLoS One, 2023, 18, e0284681 CrossRef CAS PubMed.
- Z. Terzopoulou and D. N. Bikiaris, Mater. Lett., 2024, 362, 136174 CrossRef CAS.
- N. Tripathi, M. Misra and A. K. Mohanty, ACS Eng. Au, 2021, 1, 7–38 CrossRef CAS.
- M. Mehrpouya, H. Vahabi, M. Barletta, P. Laheurte and V. Langlois, Mater. Sci. Eng., C, 2021, 127, 112216 CrossRef CAS PubMed.
- B. McAdam, M. B. Fournet, P. McDonald and M. Mojicevic, Polymers, 2020, 12, 1–20 CrossRef PubMed.
- E. Markl, Nov. Tech. Nutr. Food Sci., 2018, 2, 206–209 Search PubMed.
- J. Xu, P. H. Manepalli, L. Zhu, S. Narayan-Sarathy and S. Alavi, J. Polym. Res., 2019, 26, 188 CrossRef.
- S. A. Rafiqah, A. Khalina, A. S. Harmaen, I. A. Tawakkal, K. Zaman, M. Asim, M. N. Nurrazi and C. H. Lee, Polymers, 2021, 13, 1–28 CrossRef PubMed.
- M. Deroiné, A. Le Duigou, Y. M. Corre, P. Y. Le Gac, P. Davies, G. César and S. Bruzaud, Polym. Degrad. Stab., 2014, 108, 319–329 CrossRef.
- A. R. Bagheri, C. Laforsch, A. Greiner and S. Agarwal, Glob. Chall., 2017, 1, 1700048 CrossRef PubMed.
- M. A. Sawpan, M. R. Islam, M. D. H. Beg and K. Pickering, J. Polym. Environ., 2019, 27, 942–955 CrossRef.
- H. Sashiwa, R. Fukuda, T. Okura, S. Sato and A. Nakayama, Mar. Drugs, 2018, 16, 1–11 CrossRef PubMed.
- P. Shaiju, B. Dorian, R. Senthamaraikannan and R. B. Padamati, Molecules, 2020, 25, 5766 CrossRef CAS PubMed.
- A. Nakayama, N. Yamano and N. Kawasaki, Polym. Degrad. Stab., 2019, 166, 290–299 CrossRef CAS.
- O. Platnieks, S. Gaidukovs, V. K. Thakur, A. Barkane and S. Beluns, Eur. Polym. J., 2021, 161, 110855 CrossRef CAS.
- E. Hassan, Y. Wei, H. Jiao and Y. Muhuo, J. Fiber Bioeng. Inf., 2013, 6, 85–94 CrossRef.
- X. Zhao, D. Zhang, S. Yu, H. Zhou and S. Peng, E-Polymers, 2021, 21, 793–810 CrossRef CAS.
- X. Wang, M. Jiang, Z. Zhou, J. Gou and D. Hui, Composites, Part B, 2017, 110, 442–458 CrossRef CAS.
- M. C. Righetti, P. Cinelli, L. Aliotta, E. Bianchi, F. Tricoli, M. Seggiani and A. Lazzeri, Polym. Int., 2022, 71, 47–56 CrossRef CAS.
- R. Pantani, G. Gorrasi, G. Vigliotta, M. Murariu and P. Dubois, Eur. Polym. J., 2013, 49, 3471–3482 CrossRef CAS.
- E. L. De Paula, V. Mano and F. V. Pereira, Polym. Degrad. Stab., 2011, 96, 1631–1638 CrossRef.
- F. Luzi, E. Fortunati, A. Jiménez, D. Puglia, D. Pezzolla, G. Gigliotti, J. M. Kenny, A. Chiralt and L. Torre, Ind. Crops Prod., 2016, 93, 276–289 CrossRef CAS.
- J. Brzeska, A. Heimowska, W. Sikorska, L. Jasińska-Walc, M. Kowalczuk and M. Rutkowska, Int. J. Polym. Sci., 2015, 2015, 795985 Search PubMed.
- N. H. M. Zubir, S. T. Sam, N. N. Zulkepli and M. F. Omar, Polym. Bull., 2018, 75, 61–76 CrossRef CAS.
- H. Tanabi, Int. Adv. Res. Eng. J., 2021, 5, 188–193 CrossRef.
- J. Kingman and M. K. Dymond, Chem. Data Collect., 2022, 40, 100884 CrossRef CAS.
- S. Shabana, R. V. Nikhil Santosh, J. Sarojini, K. Arun Vikram and V. V. K. Lakshmi, Int. J. Eng. Adv. Technol., 2019, 8, 2351–2356 Search PubMed.
- S. Souissi, W. Bennour, R. Khammassi and A. Elloumi, J. Elastomers Plast., 2022, 55, 184–200 CrossRef.
- A. Sabalina, S. Gaidukovs, M. Jurinovs, L. Grase and O. Platnieks, J. Appl. Polym. Sci., 2023, 140, e54031 CrossRef CAS.
- S. Beluns, O. Platnieks, S. Gaidukovs, O. A. Starkova, A. Sabalina, L. Orlova, V. K. Thakur and G. Gaidukova, Int. J. Mol. Sci., 2021, 22, 12939 CrossRef CAS PubMed.
- O. A. Starkova, O. Platnieks, A. Sabalina and S. Gaidukovs, Polymers, 2022, 14, 221 CrossRef CAS PubMed.
- D. Gibhardt, A. E. Krauklis, A. Doblies, A. Gagani, A. Sabalina, O. Starkova and B. Fiedler, Polym. Test., 2023, 118, 107901 CrossRef CAS.
- S. Baidurah, Polymers, 2022, 14, 4928 CrossRef CAS PubMed.
- V. C. Agbakoba, N. Webb, E. Jegede, R. Phillips, S. P. Hlangothi and M. J. John, Macromol. Mater. Eng., 2023, 309, 2300276 CrossRef.
- A. Gowman, T. Wang, A. Rodriguez-Uribe, A. K. Mohanty and M. Misra, ACS Omega, 2018, 3, 15205–15216 CrossRef CAS PubMed.
- C. Zhang, Q. Lan, T. Zhai, S. Nie, J. Luo and W. Yan, Polymers, 2018, 10, 1181 CrossRef PubMed.
- C. H. Chan, C. Kummerlöwe and H. W. Kammer, Macromol. Chem. Phys., 2004, 205, 664–675 CrossRef CAS.
- C. Phillips, M. Kortschot and F. Azhari, Addit. Manuf., 2022, 58, 103050 Search PubMed.
- P. Striemann, D. Huelsbusch, M. Niedermeier and F. Walther, Addit. Manuf., 2021, 46, 102095 CAS.
- B. Panda, S. C. Paul, N. A. N. Mohamed, Y. W. D. Tay and M. J. Tan, Measurement, 2018, 113, 108–116 CrossRef.
- T. T. Le, S. A. Austin, S. Lim, R. A. Buswell, A. G. F. Gibb and T. Thorpe, Mater. Struct., 2012, 45, 1221–1232 CrossRef CAS.
- J. Van Der Putten, G. De Schutter and K. Van Tittelboom, in RILEM Bookseries, Springer Netherlands, 2019, vol. 19, pp. 234–244 Search PubMed.
- S. Bergaliyeva, D. L. Sales, F. J. Delgado, S. Bolegenova and S. I. Molina, Polymers, 2022, 14, 1–12 CrossRef PubMed.
- G. Gorrasi and R. Pantani, Adv. Polym. Sci., 2018, 279, 119–151 CrossRef CAS.
- J. Kim, S. Park, S. Jung, H. Yun, K. Choi, G. Heo, H. J. Jin, S. Park and H. W. Kwak, Polym. Degrad. Stab., 2023, 216, 110490 CrossRef CAS.
- N. Koca, C. Aversa and M. Barletta, J. Appl. Polym. Sci., 2023, 140, e54659 CrossRef CAS.
- R. R. A. Silva, C. S. Marques, T. R. Arruda, S. C. Teixeira and T. V. de Oliveira, Macromol, 2023, 3, 371–399 CAS.
- Q. Qin, Y. Yang, C. Yang, L. Zhang, H. Yin, F. Yu and J. Ma, Sci. Total Environ., 2022, 842, 156775 CrossRef CAS PubMed.
- X. F. Wei, M. Bohlén, C. Lindblad, M. Hedenqvist and A. Hakonen, Water Res., 2021, 198, 117123 CrossRef CAS PubMed.
- J. W. Westwater and H. G. Drickamer, J. Am. Chem. Soc., 1957, 79, 1267–1268 CrossRef.
- O. Starkova, S. Gaidukovs, O. Platnieks, A. Barkane, K. Garkusina, E. Palitis and L. Grase, Polym. Degrad. Stab., 2021, 191, 109670 CrossRef CAS.
- A. Kovalcik, S. Obruca, M. Kalina, M. Machovsky, V. Enev, M. Jakesova, M. Sobkova and I. Marova, Materials, 2020, 13, 1–21 CrossRef PubMed.
- S. Rogovina, L. Zhorina, A. Yakhina, A. Shapagin, A. Iordanskii and A. Berlin, Polymers, 2023, 15, 645 CrossRef CAS PubMed.
- M. A. S. Mazlan, Z. Zakaria, M. A. A. Saidi, A. Hassan and C. J. Xin, J. Chem. Inf. Model., 2020, 21, 1–9 Search PubMed.
- A. D. Banjo, V. Agrawal, M. L. Auad and A. D. N. Celestine, Compos., Part C: Open Access, 2022, 7, 100243 CAS.
- C. S. Wu, D. Y. Wu and S. S. Wang, Polym. Bull., 2021, 78, 4817–4834 CrossRef CAS.
- N. Petchwattana, J. Sanetuntikul, P. Sriromreun and B. Narupai, J. Bionic Eng., 2017, 14, 781–790 CrossRef.
- R. Cosquer, S. Pruvost and F. Gouanvé, Membranes, 2021, 11, 1–25 CrossRef PubMed.
- L. G. Engler, N. C. Farias, J. S. Crespo, N. M. Gately, I. Major, R. Pezzoli and D. M. Devine, Polymers, 2023, 15, 2874 CrossRef PubMed.
- R. P. Brannigan and A. P. Dove, Biomater. Sci., 2017, 5, 9–21 RSC.
- B. S. Ndazi and S. Karlsson, eXPRESS Polym. Lett., 2011, 5, 119–131 CrossRef CAS.
- H. Yu, C. Zhu, L. Yao, Y. Ma, Y. Ni, S. Li, H. Li, Y. Liu and Y. Wang, Mathematics, 2023, 11, 1160 CrossRef.
- X. Yin, Y. Liu, Y. Miao and G. Xian, Polymers, 2019, 11, 505 CrossRef PubMed.
- M. Wrosch, G. Xian and V. M. Karbhari, J. Appl. Polym. Sci., 2008, 107, 3654–3662 CrossRef CAS.
- M. Zarei and S. Karbasi, J. Porous Mater., 2018, 25, 259–272 CrossRef CAS.
- D. C. Lingegowda, J. K. Kumar, A. G. D. Prasad, M. Zarei and S. Gopal, Rom. J. Biophys., 2013, 22, 137–143 Search PubMed.
- P. Chaiwutthinan, V. Pimpan, S. Chuayjuljit and T. Leejarkpai, J. Polym. Environ., 2015, 23, 114–125 CrossRef CAS.
- M. Rasheed, M. Jawaid, B. Parveez, A. H. Bhat and S. Alamery, Polymers, 2021, 13, 1–15 Search PubMed.
- X. Hu, T. Su, P. Li and Z. Wang, Polym. Bull., 2018, 75, 533–546 CrossRef CAS.
- V. Berthé, L. Ferry, J. C. Bénézet and A. Bergeret, Polym. Degrad. Stab., 2010, 95, 262–269 CrossRef.
- Y. J. Phua, W. S. Chow and Z. A. M. Ishak, Polym. Degrad. Stab., 2011, 96, 1194–1203 CrossRef CAS.
- Y. V. Tertyshnaya, M. V. Podzorova, I. A. Varyan, V. V. Tcherdyntsev, M. Y. Zadorozhnyy and E. V. Medvedeva, Polymers, 2023, 15, 1029 CrossRef CAS PubMed.
- E. Sasimowski, Ł. Majewski and M. Grochowicz, Materials, 2021, 14, 1–35 Search PubMed.
- S. F. Yao, X. T. Chen and H. M. Ye, J. Phys. Chem. B, 2017, 121, 9476–9485 CrossRef CAS PubMed.
- K. Zhang, A. K. Mohanty and M. Misra, ACS Appl. Mater. Interfaces, 2012, 4, 3091–3101 CrossRef CAS PubMed.
- S. Zeman and L. Kubík, Tech. Sci., 2007, 10, 33–34 Search PubMed.
- B. Kost, M. Basko, M. Bednarek, M. Socka, B. Kopka, G. Łapienis, T. Biela, P. Kubisa and M. Brzeziński, Prog. Polym. Sci., 2022, 130, 101556 CrossRef CAS.
- A. Sabalina, O. Platnieks, G. Gaidukova, A. Aunins, T. V. Eiduks and S. Gaidukovs, RSC Adv., 2025, 15, 501–512 RSC.
- A. Sabalina, S. Gaidukovs, A. Aunins, A. Gromova, G. Gaidukova, L. Orlova and O. Platnieks, Polymers, 2024, 16, 2288 CrossRef CAS PubMed.
- T. Read, C. M. Chan, C. Chaléat, B. Laycock, S. Pratt and P. Lant, Sci. Total Environ., 2024, 931, 172771 CrossRef CAS PubMed.
- C. Kato, A. Honma, S. Sato, T. Okura, R. Fukuda and Y. Nogi, High Press. Res., 2019, 39, 248–257 CrossRef CAS.
- T. G. Volova, A. N. Boyandin, A. D. Vasiliev, V. A. Karpov, S. V. Prudnikova, O. V. Mishukova, U. A. Boyarskikh, M. L. Filipenko, V. P. Rudnev, B. Bá Xuân, V. V. Dũng and I. I. Gitelson, Polym. Degrad. Stab., 2010, 95, 2350–2359 CrossRef CAS.
- J. Vazquez-Armendariz, R. Tejeda-Alejandre, A. Rodriguez-Garcia, Y. I. Vega-Cantu, C. Mendoza-Buenrostro and C. A. Rodriguez, Materials, 2020, 13, 1–17 CrossRef PubMed.
- C. X. F. Lam, S. H. Teoh and D. W. Hutmacher, Polym. Int., 2007, 56, 718–728 CrossRef CAS.
- G. Gorrasi and R. Pantani, Polym. Degrad. Stab., 2013, 98, 1006–1014 CrossRef CAS.
- K. Cho, J. Lee and K. Kwon, J. Appl. Polym. Sci., 2001, 79, 1025–1033 CrossRef CAS.
- I. Lyyra, N. Sandberg, V. S. Parihar, M. Hannula, H. Huhtala, J. Hyttinen, J. Massera and M. Kellomäki, Mater. Today Commun., 2023, 37, 107242 CrossRef CAS.
- S. Souissi, W. Bennour, R. Khammassi and A. Elloumi, J. Elastomers Plast., 2023, 55, 184–200 CrossRef CAS.
- Y. Tao, F. Kong, Z. Li, J. Zhang, X. Zhao, Q. Yin, D. Xing and P. Li, J. Mater. Res. Technol., 2021, 15, 4860–4879 CrossRef CAS.
- A. J. dos Santos, L. V. O. D. Valentina, A. A. H. Schulz and M. A. T. Duarte, Ing. Cienc., 2017, 13, 269–298 CAS.
- D. Vaes, M. Coppens, B. Goderis, W. Zoetelief and P. Van Puyvelde, Polymers, 2021, 13, 2677 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5su00275c |
| This journal is © The Royal Society of Chemistry 2025 |