
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCircular plastic economy for sustainable development: current advances and future perspectives
Muhammad
Anwar
 *,
Maria E.
Konnova
and
Sarim
Dastgir†
*
*,
Maria E.
Konnova
and
Sarim
Dastgir†
*
Qatar Environment and Energy Research Institute (QEERI), Hamad Bin Khalifa University, Qatar Foundation, PO Box. 34110, Doha, Qatar. E-mail: muanwar@hbku.edu.qa
First published on 16th June 2025
Abstract
More than 8 billion tonnes of plastic have been produced globally since 1950, with almost 80% of the plastic generated annually turning into waste. This plastic waste represents a significant environmental challenge and reflects a major economic loss. Catalytic methods capable of transforming plastic waste into valuable chemicals and fuels offer the opportunity to turn plastic pollution into a viable resource, promoting a circular plastic economy that is crucial for achieving sustainability in energy sectors. This review examines the latest research advancements in catalytic processes for recycling plastic waste into chemicals and fuels. These technologies are emerging as potential solutions in the search for a sustainable circular plastic economy and energy markets, offering alternatives that incineration and mechanical recycling have largely failed to deliver. Various catalytic processes are comprehensively accessed, including pyrolysis, hydrocracking, chemolysis, hydrogenolysis, photocatalysis, electrocatalysis, biocatalysis, and metathesis, which efficiently convert plastic waste into valuable chemical building blocks, fuels, and other high-value products. These technologies not only address the environmental issues associated with plastic pollution but also contribute to resource recovery and energy sustainability with potential to produce low-carbon fuels, chemicals and building blocks to enhance plastic circularity. Moreover, this review addresses the current challenges and future research directions essential for accelerating the transition towards sustainable circular plastic economy. It offers a comprehensive evaluation of catalytic recycling technologies, including pyrolysis, hydrocracking, chemical depolymerisation, and metathesis, with a focus on mitigating Scope 3 Emissions and fostering sustainable energy solutions. The objective is to promote the advancement of catalytic technologies, recognizing the potential of catalysis to enhance economic efficiency and capitalize on the conversion of plastic waste into high value chemical feedstocks and energy. The review highlights recent developments in catalytic processes, including catalysts, plastic feedstocks, reaction parameters, and their impact on product distribution and yield. While the gasification method is briefly mentioned, this review does not cover thermosetting plastics, physical recycling, or non-catalytic processes such as thermal recycling, mechanical recycling, or incineration.
 Muhammad Anwar | Muhammad Anwar obtained his DPhil. in Chemistry from the University of Oxford, United Kingdom, under the supervision of Professor Mark Moloney in 2006. His doctoral research focused on the development of synthetic methodologies for nitrogen heterocycles and the total synthesis of bioactive natural products. He subsequently joined the group of Professor Sir Jack Baldwin, FRS, as a Postdoctoral Research Fellow (2006–2009), and later continued at the University of Oxford as a Senior Postdoctoral Researcher in Professor Moloney's group (2009–2013). Dr Anwar has acquired diverse experience in sustainable catalysis, biomimetic synthesis, structure-based drug design and development, target- and diversity-oriented synthesis, and polymer chemistry while working for Roche (F. Hoffmann-La Roche Ltd), Pfizer (UK), and Tocris Bioscience prior to joining the Qatar Environment and Energy Research Institute (QEERI) at Hamad Bin Khalifa University, Qatar Foundation, as a Scientist in 2016. His current research interests include reaction engineering, development of novel catalytic processes and their applications in energy and environment, CO2 valorisation, polymerisation, polymer recycling, sustainable fuels, and materials for chemical and biomedical applications. |
 Maria E. Konnova | Maria E. Konnova earned her Diploma (2011) in Physical Chemistry from the Samara National Research University named after Academician S. P. Korolev (Russia). In 2023, she received her PhD degree in Chemistry from the Samara State Technical University (Russia) under the supervision of Prof. Sergey P. Verevkin. She is currently a research associate at Hamad Bin Khalifa University in Qatar Energy and Environment Research Institute (QEERI). Her research focuses on chemical methods to recycle/upcycle waste plastics, the study of catalysis and renewable energy. |
 Sarim Dastgir | Sarim Dastgir studied chemistry at the University of Oxford, UK, where he completed his DPhil. (PhD) thesis in 2005 in the research group of Malcolm L. H. Green, FRS. He then joined the Research Group of Professor Chao-Jun Li at McGill (2006–2007) and later the Research Group of Professor Gino Lavoie at York University (2007–2012), as a postdoctoral research fellow. Before joining Green Global Group of Companies as partner and member of board, Sarim worked as a Senior Scientist and Program Director at Qatar Energy and Environment Research Institute (QEERI), Hamad Bin Khalifa University (HBKU), a part of Qatar Foundation. At Green Global Group, Sarim guides strategic growth, enhances market competitiveness, and oversees physical commodity trading, engineering and emerging technology portfolios. He has extensive project management & business background and effectively merges strategy with execution by leveraging experience from R&D to Startups and industrial deployment in a forward-thinking and collaborative approach. Sarim has strong focus on sustainable development, life cycle assessments (LCAs), quality & environmental management systems, regulatory compliance, net zero, risk management and business continuity planning for industry 5.0 transition. Sarim's career highlights includes brown & green field technologies for carbon management (CCUS), hydrogen economy, gas processing and treatments, waste management, polymer recycling, olefin polymerisation, oligomerisation, engineering plastics, sustainable fuels and base oils and materials for chemical and biochemical applications along with his strong focus on circular economy, management & cost recovery systems, certification, accreditation, compliance & regulatory overview, audits, risk management, HT/HO data management, data analytics & software controls. He is also a strong influencer & negotiator with expertise that includes creative problem-solving, intellectual property, technology development & transfer, product development, manufacturing, commercialisation, and merger & acquisitions across diverse sectors. |
Sustainability spotlightA circular plastic economy is essential in tackling the global plastic waste crisis through sustainable and scalable solutions. It investigates how advanced catalytic and chemical recycling technologies can create new value from plastic waste while reducing reliance on fossil resources. By analysing recent advancements in chemical recycling and catalysis-driven approaches, the article outlines methods for converting plastic waste into valuable materials. It highlights innovations aimed at lowering lifecycle emissions, minimising environmental impacts, and promoting circularity in production systems. The technological insights shared directly contribute to the achievement of several UN Sustainable Development Goals, including SDG 9 (Industry, Innovation, and Infrastructure), SDG 12 (Responsible Consumption and Production), and SDG 13 (Climate Action), by enabling a resilient shift toward low-carbon, resource-efficient, and innovation-led plastic systems. |
1. Introduction
Plastic has become a ubiquitous part of human life and plays a significant role in the improvement of human living standards through its versatile applications across various industries.1 Plastics are used for manufacturing a wide range of products, and are crucial to innovation in many industrial segments including automotive, aviation, electronic, construction, healthcare, packaging etc. The mass production of plastic started in the 1950s, since then the worldwide production of plastic materials has reached more than nine billion metric tonnes (Fig. 1). With the massive increase in the world's population, the plastic demand is continuously increasing due to its various advantages such as good insulating properties, light weight, durability, comparatively low cost, erosion resistance, ease of processing, mouldability, etc.2–5 In 2023, the annual plastic production was 413.8 million tonnes and is projected to rise to 590 million tonnes by 2050.6–8 In 2015, fossil fuel-based plastic production contributed 1.7 Gt CO2 equivalent (CO2e) in greenhouse gas (GHG) emissions over their life cycle. If current trends continue, both plastic production and associated GHG emissions are expected to nearly quadruple by 2050.9 | ||
| Fig. 1 Global annual plastic production. Source: Statista Research, 2024. | ||
Plastic demand mainly involves high-density polyethylene (HDPE), low density polyethylene (LDPE), polyethylene terephthalate (PET), polypropylene (PP), polystyrene (PS), polyurethane (PU), and polyvinyl chloride (PVC). HDPE is used in the manufacture of storage boxes, bottles, pipes, cable insulation, toys, etc. LDPE is applied in agricultural films, packaging, reusable bags, and computer components. PET is used in beverage bottles, food containers, and films. PVC is used to manufacture plumbing pipes, tiles, cables for insulation, garden hoses, automotive upholstery. PP is used in packaging, automotive parts, bank notes, and microwave containers. PS serves in insulation, food packaging, electrical equipment, eyeglass frames, etc.Fig. 2 represents the global demand and use of these polymers across segments.10
 | ||
| Fig. 2 Global plastic demand by application in 2021 (left); global plastic distribution by polymer type in 2023 (right). Data are based on Plastics-the Facts 2022 and 2024 reports. | ||
The continuous increase in plastic production has led to increased plastic waste accumulation, with almost 60% of the global plastic ending up in the environment as plastic waste. Plastic waste is categorised as industrial and municipal waste. Industrial plastic waste, being more homogeneous and less contaminated, is easier to recycle into lower grade products. In contrast, municipal plastic waste is heterogeneous and often contaminated, comprising HDPE, LDPE, PET, PP, PS, and PVC. Packaging, single use plastics, dominates plastic consumption and accounts for over 50% of global plastic waste.11–13 Different polymers reach the waste status at varying rates, as shown in Fig. 3.14 Plastic packaging has a short life span and typically becomes waste within 6 months of its production, leading to a significant economic loss of ∼100 billion dollars annually, in addition to disposal and environmental costs.15 About ∼11% (by mass) of the total metropolitan solid waste (MSW) stream is made up of synthetic polymers but occupy an uneven volume in landfills.16 Most of these polymer materials degrade slowly in landfills, and stay there for an indefinite period. The collection of municipal solid waste can cost hundreds of dollars per metric tonne, though disposal cost varies by region.17 In addition, cleaning up of synthetic polymers that skip collection can be very costly. For instance, about 500 million dollars is spent annually to remove litter, mostly plastics, from the west coast of US only.18
 | ||
| Fig. 3 Production volume and waste generation in million tonnes (MT per year) from major plastics in 2019. The values are taken from ref. 14. | ||
Scope emissions encompass greenhouse gases emitted across the entire value chain. In particular, Scope 1 emissions are direct emissions from an organization's controlled processes. In plastic manufacturing, a major source of carbon emissions is the extraction of raw materials, such as petroleum hydrocarbons from fossil fuels. Another significant contributor is the conversion of raw materials into polymer resins and final plastic products. Scope 1 emissions, being directly controlled by organisations, are the most manageable. Scope 2 emissions, however, arise indirectly from purchased energy, with energy-intensive processes like injection and extrusion moulding amplifying their impact. To mitigate these emissions, organisations can implement energy-efficient practices or transition to low-carbon and renewable energy sources.
Scope 3 emissions refer to indirect emissions occurring from the entire value chain of an industry or organization, accounting for majority of the footprint. These emissions originate from activities beyond direct control but resulting from operations, including supply chain activities, distribution, product usage, and waste disposal. Scope 3 emissions are extremely difficult to track and are potentially the largest contributor to an organisation's total emissions.
According to the OECD (Organisation for Economic Co-operation and Development) report, plastics account for 3.4% of global greenhouse gas emissions, with 90% resulting from their production and fossil fuel-based conversion.19 Similarly, end-of-life processes, such as recycling, incineration or disposal, generate downstream Scope 3 emissions. Incineration has the highest carbon emissions, releasing two tonnes of CO2 by incinerating one tonne of plastic.20,21 However, upstream Scope 3 emissions can be significantly reduced by integrating recycled plastics into manufacturing. Recycling can reduce the emissions from incineration by 40–50%.22 Organisations can mitigate various embedded emissions at the extraction and processing stages by reusing and reprocessing waste into recycled inputs instead of relying on virgin raw materials. Circular economy strategies provide potential benefits to reduce and decarbonise both upstream and downstream Scope 3 emissions by:
• Reducing waste during manufacturing and designing products to ensure their reuse or recyclability.
• Using recycled materials to create products leading to less use of virgin materials, thereby reducing emissions from extraction and processing.
• Implementing downstream solutions alongside upstream solutions to reduce emissions throughout the entire value chain.
Despite numerous reviews on plastic recycling technologies, most of them focus either on specific polymer classes or on individual catalytic approaches, such as pyrolysis or chemolysis. While these studies offer valuable depth, they often lack a holistic approach that considers the interconnected technical and environmental factors shaping the broader plastic circularity landscape. This review addresses that gap by encompassing a broad spectrum of catalytic strategies, ranging from thermal catalysis (e.g., pyrolysis, hydrocracking) to precision depolymerisation methods (e.g., chemolysis, metathesis), as well as emerging low-temperature processes (e.g., photocatalysis, electrocatalysis, biocatalysis). It provides a comprehensive analysis of how these diverse catalytic technologies can synergistically contribute to the advancement of a circular plastic economy. A distinctive aspect of this review is its emphasis on the potential of catalytic recycling in reducing Scope 3 emissions, an often-overlooked yet crucial metric in life-cycle assessments and sustainability reporting. By highlighting catalytic pathways that enable decentralised, low-carbon valorisation of plastic waste, this review contributes to the expanding discourse on climate accountability within the chemical and energy sectors.
2. Plastic pollution: a global environmental concern
Plastic pollution causes serious risks to human health, ecosystems, and environment. In 2015, the incineration of plastic packaging wastes emitted about 16 million tonnes of CO2 equivalents. In 2019, only 9% plastic waste was recycled, 19% incinerated, 50% went to landfills, and 22% evaded waste management systems and ended up in open dumps or natural environments, or burned in open areas.19 Moreover, the Covid-19 led to significant increase in single-use plastic waste, with Wuhan generating 240 metric tonnes of medical plastic waste which is nearly 6 times compared to pre-pandemic levels.23 Similarly, Singapore produced an additional 1400 tonnes waste from packaging and food delivery during the eight-weeks period of lockdown.24 Although recycling rates are expected to increase to 17% by 2060, landfilling (50%) and incineration (18%) will remain dominant, leading to continued waste accumulation.19 Landfilling is an unsustainable solution as plastic waste dominates a large volume of landfill sites and can remain there for many years, or even for decades, due to their high resistance to degradation against physical, chemical, and biological actions.25,26 Continuous disposal of plastic waste into landfills creates serious environmental risks, including pollution and greenhouse gas emissions (GHG).27,28 On the other hand, incineration generates harmful emissions with limited energy recovery. Therefore, severe pollution or disasters are caused both by incineration and landfilling. The net material value recovery is either none or minimum at large scale. The interaction of plastic with groundwater and toxic materials in landfill sites can lead to production of harmful leachate, eventually contributing to environmental degradation and deterioration of surrounding land. Therefore, landfilling the plastic waste is the most undesirable waste management strategy. The environmental damage caused by plastic pollution is often considered more severe than carbon footprints.29,30The Ellen MacArthur Foundation reported the loss of about 1/3rd of global plastic packaging waste into the environment.15 Accumulation of plastic waste in oceans has become a major environmental challenge.31 The Great Pacific Garbage Patch (GPGP) now spans approximately 1.6 million km2, i.e., three times the area of Spain, comprising 99.9% plastic debris. In 2016, the foundation reported that plastic mass in oceans would become equal to the fish that live in them by 2050, if current plastic management trends persist (Fig. 4).15,32 Even conservative studies have estimated that 8 MMT of plastic materials end up in oceans annually.13 Plastic waste now reaches even the most remote locations, from the Virgin Islands to the ocean's deepest depths.33 Plastic waste in the north Pacific Ocean has increased by 100 fold during the last 40 years. The OECD's 2022 report estimates that 30 million tonnes of plastic waste have already accumulated in oceans and seas, with additional 109 million tonnes in rivers.19 Environmental pollution by plastic is undoubtedly alarming, according to UNCTAD 2022 and UNEP 2022 reports, out of 369 million tonnes of plastic waste generated annually, about 11 million metric tonnes ends up in the ocean.34
 | ||
| Fig. 4 Plastic pollution in oceans and reservoirs. Figure partially generated using iStock AI Image Generator. | ||
Plastic waste fragments over time in oceans, eventually forming microscopic particles (∼20 μm diameter) which eventually enter aquatic animals. Large particles could harm ocean species via entanglement, resulting a significant loss to aquatic habitat.35,36 Animals can get entangled in plastic waste, which can lead to injury and even death. Plastic pollution can also affect the eating habits and reproduction of marine life, contributing to reduction in their population. According to United Nations, almost 1 million marine mammals and seabirds are killed annually by plastic waste.37 If current trends continue unchecked, it is projected that by 2100, the total mass of microplastics in the marine environment may increase by 50 times, compared to 4.9 × 105 in 2010.38 The composition of microplastics is mainly represented by the following polymers: PE, PET, PP, PS, PVC, PA (polyamide), nylon, and polyvinyl alcohol (PVA).39 Microplastics are mixtures of small particles, ranging from 1 micron to 5 mm size, and are categorised into two types: primary and secondary.40,41 Primary microplastics are produced for a specific industrial or household use, such as exfoliants in face scrubs, toothpastes, and secondary microplastics are generated through the breakdown of large plastic materials under the influence of UV radiation or mechanical wear and tear. Microplastics enter the environment primarily through industrial and domestic wastewater.41 However, the most likely and large-scale process of microplastic formation results from the fragmentation of improperly disposed plastics, such as abrasion of larger fragments of plastic on beaches. In water, secondary microplastics degrade slowly due to low temperatures and ultraviolet radiation, eventually breaking down into microscopic particles. These are easier to swallow by marine organisms, which increases the vulnerability to toxic leaching, desorption, and adsorption. In addition, microplastics can be transferred to various ecosystems through animal migration or water currents, affecting even remote areas.41,42 Recent studies show that a variety of marine organisms, including mussels, fish, shrimp, oysters, oarfish, worms and even whales, swallow these particles.43 According to studies, ingestion of these particles causes severe health effects, including pathological stress, false saturation, reproductive complications, blocking of enzyme production, decreased growth rate, and oxidative stress.44 While whales and some large marine animals may pass microplastics without any harm due to lack of enzymatic pathways, these microplastics adsorb toxic chemicals such as organochlorine pesticides and polycyclic aromatic hydrocarbons, transferring toxins into the food chain.42 Thus, even if some organisms avoid direct contact with microplastics they still face exposure through other organisms containing these particles. Current methods for removing microplastics from water include flotation, enzymatic degradation, photocatalytic degradation, membrane separation, and coagulation deposition.45
Properties of plastics are often improved with additives such as plasticisers for flexibility, stabilisers for thermal and UV, flame retardants, and dyes. However, some of these additives can potentially be transmitted into the blood stream and animal tissues via ingestion, potentially damaging blood cells and tissues.46 Among the most concerning additives are bisphenol A, brominated flame retardants, phthalate plasticisers, and antimicrobial agents. BPA and phthalates are often found in products including computers, cosmetics, food packaging, floor coverings, medical devices, perfumes, and toys. Due to their volatile nature, they contaminate the aquatic environment, air, and dust.47 These chemicals have shown to affect reproduction and deteriorate the development of crustaceans and amphibians.48
Microplastics are not confined to oceans but also contaminate the soil and atmosphere. Landfill sites, which contain the highest concentration of microplastics, are a major source of soil contamination. These microplastics can infiltrate soil through wind, dust, rain, and erosion, where microorganisms and enzymes break them down into even smaller fragments than those found in the aquatic environment.41 Microplastics with a higher density remain in the ground, moving to deeper layers and transferring pollution to groundwater, and potentially transmitting to plants and the food chain. Microplastics with a lower density remain on the surface and can be carried by wind and water because of erosion. Hence, microplastics can have an impact not only on marine life but also get into the food chain of terrestrial animals and plants. Plastics can change the pH of the soil, for example, polylactic acid and low-density polyethylene increase the pH, whereas high-density polyethylene lowers it. Such changes in the soil certainly have an impact on organisms living in it.49 Microplastics can disrupt the growth, development and ecological processes of plants. However, some studies have suggested beneficial effects of microplastics such as a decrease in volume density, increased aeration, and enhanced root growth.50
3. World energy demand and sustainable chemical industry
The world population is steadily increasing, projected to exceed 9.7 billion in 2050 and potentially reaching 10.9 billion by 2100.51 Life expectancy is also expected to rise from the current 67 years to 75 years in 2050. This demographic shift will drive a surge in global energy demand, which, under a “business as usual” could triple from 13.1 million tonnes (MT) of oil equivalents in 2015 to 35 MT in 2050, potentially reaching unsustainable levels.51 Industries will remain the dominant drivers of this demand, with economic growth and urbanisation accelerating consumption, particularly in resource intensive sectors. However, this increasing energy demand will represent unique challenges for the energy sector, which must balance increasing energy needs with environmental concerns. To mitigate climate change, greenhouse gas emissions must be reduced to half from current levels, requiring a dramatic reduction in fossil fuel dependence and a shift towards sustainable energy solutions.Significant economic growth is anticipated over the next few decades, with the world economy projected to expand to 280 trillion US dollars in 2050 from 75 trillion dollars in 2010, and this in turn would produce an enormous growth prospect for the chemical industry (Fig. 5).52 The size of chemical industry could potentially increase to US$ 18.7 trillion. Rising life expectancy and economic growth will increase chemical demand strongly across various sectors, from basic and specialty chemicals to pharmaceuticals. However, there will be enormous pressure to reduce greenhouse gas emission and fossil fuel consumption to circumvent the environmental challenges. Consequently, energy industries in future will be under pressure to substitute their naphtha and oil demands for more sustainable feedstocks.53,54
 | ||
| Fig. 5 Status of the world in 2050 – BAU scenario. The values are taken from ref. 52. Figure partially generated using iStock AI Image Generator. | ||
By 2050, the current industrial practices and BAU scenarios suggest that two and a half earth resources would be required to support the basic demand, leading to an unsustainable future.48 For a sustainable economy, a corrective path is vital with the development of a circular economy by improved industrial practices. To address these challenges, innovation and increased efficiency would play a key role for a sustainable society. The circular economy provides a structured framework to address these challenges by minimising waste and maximising resource utilisation. One potential approach involves reusing waste plastics to make feedstock chemicals, offering a viable solution to support circularity and resource efficiency.
The recent global stocktake by the United Nations Framework Convention on Climate Change (UNFCCC) underscores the alarming gap in current climate mitigation efforts. Projections reveal that the remaining global carbon budget to limit warming to 1.5 °C has reduced to 275 gigatonnes of CO2 which is equivalent to less than seven years of emissions at current rates. Fossil-based transportation fuels contribute approximately 20% of global CO2 emissions, requiring a rapid transition to low and zero carbon alternatives. Initiatives such as the EU's ReFuelEU Aviation program signify this shift directing a gradual increase in the adoption of sustainable aviation fuels (SAFs) and synthetic fuels. By 2035, a minimum of 20% of aviation fuel supplied at EU airports must comprise SAFs, a target representing a 900% rise from 2025 levels. This transition is crucial in decarbonising the aviation sector and mitigating its environmental footprint.
Concurrently, the global waste crisis presents another huge challenge, with over 2 billion tonnes of solid waste generated annually. Plastic waste, which constitutes about 12% of global solid waste, predominantly ends up in landfills, incinerators, or is poorly managed, further worsening environmental degradation. The circular plastic economy has the potential to play a crucial role in addressing the world energy demand and developing a sustainable chemical industry by transforming waste into a resource, thus reducing the dependence on virgin fossil fuels. By recycling and reprocessing plastics, the circular economy significantly cuts down on the energy-intensive extraction and processing of raw materials, leading to energy conservation and reduction in greenhouse gas emissions. This shift also drives innovation in the chemical industry, promoting the development of sustainable processes and materials that are more energy-efficient and ecofriendly. Advanced recycling technologies such as chemical recycling enables the conversion of plastic waste into valuable chemicals and fuels, contributing to more resilient and diversified energy supply. By enabling the recycling of a broader range of plastics, these innovations support the sustainable energy landscape by contributing to resource efficiency, reducing greenhouse gas emissions, conserving energy, and fostering circular economy. Overall, the circular plastic economy plays a vital role in shaping a sustainable chemical industry that meets global energy demands and minimises environmental impact. Continuous advancements in recycling technologies, particularly chemical recycling, can convert plastic waste into monomers, chemical building blocks, high-quality raw materials, or even fuels, contributing to the development of sustainable energy markets.
The growing urgency to address plastic waste has reinforced the need for effective recycling to reduce the environmental impact. Plastic-to-energy projects are gaining attraction in the energy industry as awareness about the severe environmental harm caused by single use plastics grows. Energy recovery from plastic waste, along with other forms of domestic, industrial, and agricultural waste, is now recognised as an environmentally benign strategy to produce renewable energy.55 Sustainable solutions that harness the energy potential of plastic waste while mitigating its environmental harm are imperative. Beyond recycling, the search for clean, low-cost energy resources is vital amid growing population growth and rising demand for plastics and energy. Generally, plastic waste-to-energy conversion processes transform plastics into electricity, heat, or alternative fuels. These technologies, which are well established in Europe, can produce electricity, heat, fertilisers, and biofuels from plastic waste, offering a good feedstock for energy recovery.56
There are different ways to valorise plastic waste, including chemical recycling to feedstocks and energy. These methods harness the chemical energy stored in the hydrocarbon structure of plastics to produce chemical feedstock and fuels, however, currently not all of them are economically viable. Continued innovation in technology and processes will be critical to enhance efficiency, scalability, and cost-effectiveness in plastic to energy conversion.
4. Plastic circular economy
Synthetic polymers are typically derived from non-renewable resources, with petrochemicals used in plastic production accounting for 14% of world's total oil demand and 8% of gas demand.57 These figures are anticipated to rise, making petrochemicals the world's biggest driver of oil demand, surpassing sectors such as aviation, shipping, and trucks. From the energy security and economic perspective, the large scale at which synthetic polymers are made from petroleum resources can be concerning due to high energy security and resource depletion, as most plastics are still produced using fossil-based feedstocks. A major portion of plastic waste ends up with an uncertain fate, either lost during processing or escapes collection systems, leading to severe environmental consequences (Fig. 6). The limitations of linear plastic value chain have intensified interest in the circular economy.58 Plastic recycling processes are considered necessary alternatives that offer economic and environmentally friendly solutions for plastic waste management. Transitioning to a circular and climate neutral economy will require substantial investment and innovation across the plastic value chain. This shift includes developing new business models focused on reuse, producing more recycled plastics, and creating alternative feedstocks that reduce dependency on fossil fuels. The circular economy presents an alternate and more sustainable model to linear economy which follows the “Take–Make–Waste” model by using valuable resources for longer periods and reintegrating materials into production cycles. Consequently, a circular economy reduces the demand for primary raw materials, enforces waste prevention, enhances recycling, and potentially increases resource efficiency. It is aimed at ‘designing out’ waste and prioritising reuse and recycling of resources. The industrial evolution has been dominated by a linear model of production over the past few centuries through technology advancements, resulting in unprecedented prosperity to our society.59–61 The “New Plastics Economy” must be a circular economy that can eliminate waste, maximise value, and use plastic efficiently. A hierarchical approach has been proposed for dealing with plastic wastes, according to the following options in the order of priority.62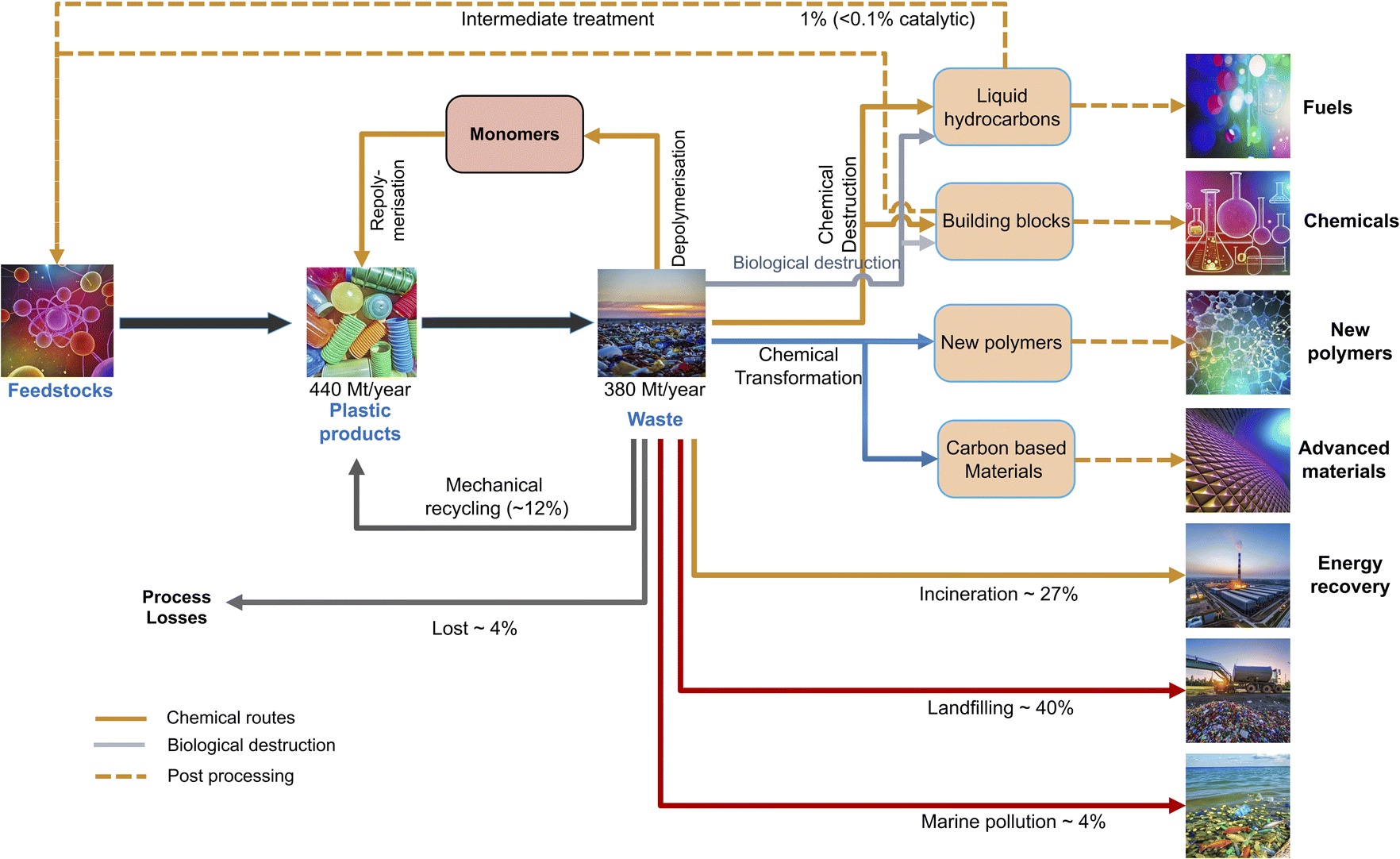 | ||
| Fig. 6 Current pathways for plastic waste management. The percentages represent the global average values in 2017 based on ref. 63. Figure partially generated using iStock AI Image Generator. | ||
(a) Reduction: reducing the number of materials employed in the manufacture of many plastic products, such as food packaging.
(b) Reuse: widely applied in the packaging sector, reuse strategies extend the lifespan of plastic items by using for the same or similar purpose multiple times.
(c) Recycling: it allows the recovery of valuable materials from plastic products.
(d) Energy recovery: it is aimed at taking advantage of the high heat value of plastics (20 MJ kg−1 for PET, ∼40–44 MJ kg−1 for polyolefins), being similar to that of petroleum-derived fuels (45 MJ kg−1 for crude oil) and superior to that of coal.64 Through incineration, plastic waste can produce electricity, or partially replace fossil fuels in specific applications like cement kilns. However, the significant concern of air pollution from directly burning plastic wastes leads to a strong opposition in many countries. An alternative is the conversion of plastic residues by thermochemical methods into hydrocarbon mixtures for subsequent transformation into carbon neutral ultra-low sulphur fuels.
(e) Controlled disposal: plastic wastes are accumulated in landfills, where they occupy significant space. This option is the least preferred one within the hierarchical approach as it does not afford any recovery of material or energy, leading to a loss of natural resources.
For sustainable industrial development, recycling raw materials into useful chemicals is utmost important both from economic and environmental perspectives. Plastic waste recycling can be applied to address the exponential growth of plastic waste, which is threatening the United Nation's Sustainable Development Goals.65 Efficient recycling of plastic waste on a global scale could save up to 3.5 billion barrels of oil annually, resulting in an estimated economic benefit of US$176 billion.66 Furthermore, chemical recycling has the potential to drive the shift towards a circular economy, enabling achieving closed-loop recycling of materials and supporting long-term sustainability.67
4.1 Plastic waste recycling
Recycling plastic waste is gaining increasing attention as a solution to environmental challenges and the transition towards a sustainable economy. Advanced chemical recycling technologies can help to curb the plastic pollution while generating valuable chemical feedstocks and fuels (Fig. 6). In terms of economy, 95% of plastic packaging material is lost every year after a first use cycle, with an estimated value of $80–120 billion.65 Recycling one tonne of plastic can save 130 million kJ of energy, highlighting its efficiency in resource conservation.65 Recycling processes for plastics can be classified into primary, secondary, tertiary, and quaternary processes, as defined by ISO15270:2008 and ASTM standards D5033 and D7209.684.1.3.1 Thermolysis. Thermolysis involves the chain scission of plastics using heat to produce low-molecular weight compounds and monomers.74 Thermolysis is divided into three methods:
(a) Pyrolysis.
(b) Hydrocracking.
(c) Gasification.
Thermolysis of plastic waste allows conversion of heavy and long chain polymeric molecules into valuable hydrocarbon rich petrochemical-based feedstock such as gas, oil, and char at high temperature. The thermolysis processes differ in operating conditions as well as product composition. Pyrolysis and hydrocracking processes lead to formation of oil and gas, whereas gasification produces only gas (CO, CO2 and H2). The pyrolysis process is conducted under an inert atmosphere, hence dioxins are not produced unlike gasification, and it requires a low reaction temperature that reduces operational cost.75,76
4.1.3.2 Chemolysis. Chemolysis is a resource recovery method where waste plastic is depolymerised or chemically treated to recover monomers (Fig. 7).69 Chemical recycling is the only method that aligns with sustainable development principles because it regenerates original raw materials.77 Nylon, polylactide, polyamide, polycarbonate, polyester, polyethylene terephthalate, and polyurethane wastes are recycled by this process, with PET and PU being the most commonly recycled. Chemolysis can be further categorised into hydrolysis, glycolysis, methanolysis, and aminolysis. Plastic sorting is the main challenge for this method as the operating conditions are plastic specific, hence, its application is limited.78
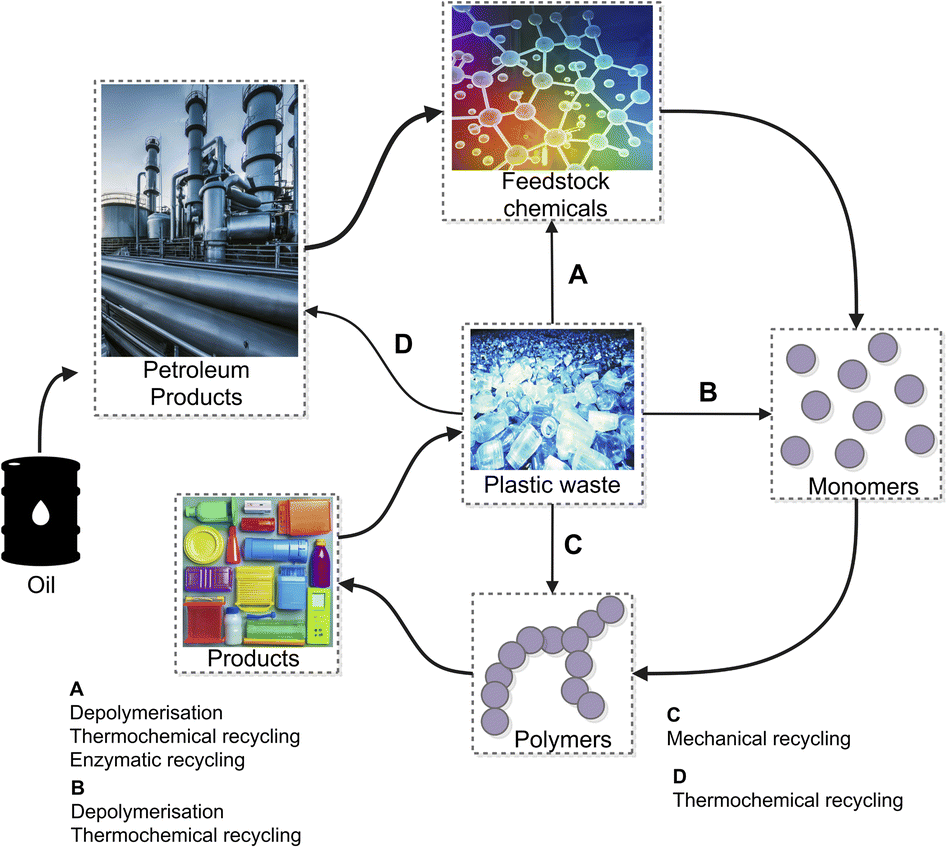 | ||
| Fig. 7 Plastic recycling technologies. Figure partially generated using iStock AI Image Generator. | ||
4.2 Landfilling
A major portion of plastic waste is disposed of in landfills, where it undergoes slow degradation (Fig. 6). Under environmental conditions, plastic materials experience substantial changes in their chemical structure, leading to loss of some of their properties. Polymeric characteristics such as molecular weight, functional groups, crystallinity, and the nature of additives present in polymers significantly influence their degradation.Degradation of plastic involves a series of processes that break down plastics into lower molecular weight oligomers, and by-products such as CO2, methane, or water.80 Plastic is degraded by four pathways: biodegradation by enzymes, hydrolytic degradation, photodegradation, and thermooxidative degradation.81 Natural plastic degradation typically starts with photodegradation, which triggers thermooxidative degradation. Microbial degradation of plastics is environmentally benign, requires less energy and leads to complete degradation, making it a popular option. However, most plastic waste is non-biodegradable, limiting the effectiveness of microbial degradation. Hence, various advanced pre-treatment methods are employed to enhance their biodegradability. These pre-treatment methods include mechanochemical degradation, photodegradation, and thermal degradation.29 Different recycling technologies have different entry points into a plastic circular economy and produce variable size circular loops subject to feedstocks and process efficiencies (Fig. 7).
5. Pyrolysis
Pyrolysis is a thermochemical process that involves heating plastic waste under an inert atmosphere at temperatures from 300 to 800 °C to break the polymer into small molecules such as hydrocarbons and monomers. Pyrolysis is a well-defined technology which was developed at the large scale for the valorisation of plastic wastes several decades ago, although its market adoption has remained quite limited until now. While gasification technology is focused on the production of syngas, pyrolysis is an alternative thermochemical route where the targeted product is a liquid. The liquid product can be further used for either energy or as a pool of valuable hydrocarbon feedstock. One of the advantages is the flexibility for varying the operating conditions to achieve products in the desired yield. In addition, from the environmental perspective, it prevents dioxin emissions as well as reduces CO and CO2 emissions.82,83 The gaseous product yield can vary from minimal values to about 90 wt%, with compositions comprising light hydrocarbons (ethane, ethene, propane, butane, etc.), CO, CO2 and even Cl (from PVC wastes). Thermal pyrolysis of polyolefinic wastes, such as PE and PP, may produce waxes. Such waxes comprise linear, branches, saturated and unsaturated hydrocarbons, in proportions depending on the feedstock properties and pyrolysis conditions. The long-chain hydrocarbons (>C20) are typically solid at room temperature. Nevertheless, waxes from polyethylene can be targeted products with interesting applications, including hot liquid fuels, electrical insulations, inks, greases, lubricants, etc., as well as co-feeding into cracking units in refineries for producing fuels and petrochemicals.62,84,85 In an opposite trend to gas production, waxes are favoured at lower temperatures. High yields (85–94 wt%) can be achieved from PE and PP cracking at temperatures in the range of 450–525 °C.86,87 The liquid oil fraction is usually maximised during the plastic pyrolysis, as it serves as a precursor for fuels and platform mixture for valuable chemicals. As pyrolysis at moderate temperature occurs through a random radical scission mechanism, the obtained oil presents a complex mixture of compounds. Only in the case of polystyrene and poly(methyl methacrylate), a real depolymerisation occurs where the oil can be enriched in their corresponding monomers (styrene and methyl acrylate).88,89 Furthermore, from an economic perspective, pyrolysis has a lower net operating cost and annual capital investment compared to incineration and plasma arc gasification, making it a more advantageous option.Catalytic pyrolysis is based on two steps: the first stage is pyrolysis where radicals are formed as intermediates, and the second stage involves catalytic cracking where carbocations are formed as intermediates over an acid catalyst. Use of an acid catalyst often leads to more efficient operation, i.e., with lower temperature and better yield of C2–C4 olefins.90 Plastic pyrolysis provides a better and environmentally benign option, as it not only facilitates the disposal of plastic waste but also allows for energy recovery.
5.1 Suitable plastics for catalytic pyrolysis
Pyrolysis and hydrocracking are the most suitable recycling techniques for heterogeneous plastic waste such as municipal plastic waste (MPW). While most of the plastic waste in daily life is suitable for pyrolysis, the process is economically feasible only when conducted on a large scale and when there is abundant supply of MPW.91 | ||
| Fig. 8 Composition of MPW by weight. | ||
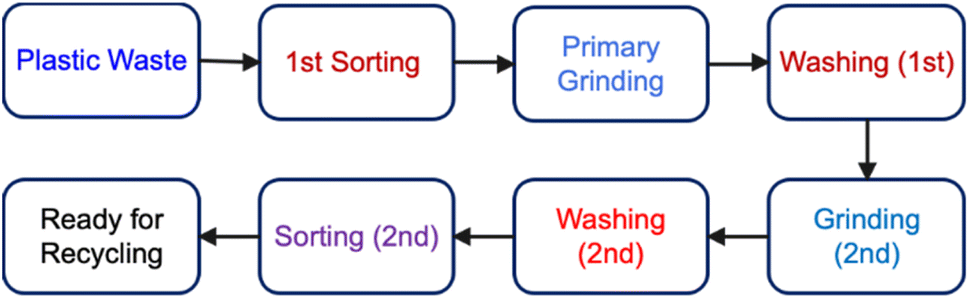 | ||
| Fig. 9 Pretreatment scheme for plastic waste. | ||
5.1.3.1 Plastic waste sorting. Plastic waste sorting, due to its complex composition, is an important step in a Material Recovery Facility (MRF) to ensure high quality materials for recycling.94 In an MRF, plastic waste is subjected to manual and automatic sorting processes including waste screening, air, ballistic, magnetic, and sensor-based separations in order to generate plastic waste as pure as possible (Fig. 9).92 Foreign materials are removed by gravity-based methods using air flow or water streams, whereas metals can also be eliminated through magnetic attraction or magnetic repulsion.95 With these advanced sorting techniques, the resultant materials can reach purity levels of up to 97% or higher.94
5.1.3.1.1 Manual sorting. Manual sorting is highly effective; however, it can be costly. Operators are trained to separate items such as milk bottles (HDPE), soda bottles (PET), garden furniture, cable sleeves (HDPE), and window profile (PVC), among others.92
5.1.3.1.2 Sorting by gravity. Some plastics can be sorted by gravity based on their density differences. For instance, polyolefins (PP and PE) having densities of ∼0.9 g mL−1 can be separated from denser plastics such as PET and PVC, which have densities around 1.4 g mL−1. The effectiveness of gravity can be further improved by applying electrostatic or magnetic fields, although this method's success is highly sensitive to ultimate contamination levels in the waste.95 Usually, this technique is employed to separate shredded waste using water as a floatation agent. In a water bath, PP and PE, which have densities below 1 g cm−3, will float, whereas other plastics such as PVC, PET, PS, and ABS will sink.96
5.1.3.1.3 Automatic sorting. Various plastics are sorted on a conveyor belt through identification of plastic types using infrared detectors such as near infrared (NIR) or short wave infrared (SWIR), and then directing into appropriate categories using air jet or actuator.95 NIR technology can efficiently remove PLA (polylactic acid) bioplastics and carbon board from mixed packaging by irradiating unsorted plastic with near infrared light of different wavelengths.96 FT-NIR is widely regarded as a highly efficient and high-speed identification method for sorting waste plastic into categories such as HDPE and PET. In addition to NIR detectors, X-ray fluorescence detectors can be used to identify heavy elements, such as Br and Cl, enabling the sorting of more challenging plastic combinations, like HDPE/LDPE and PET/PLA, which are difficult to distinguish with conventional NIR.96
These advanced sorting techniques lead to fractions that contain mostly HDPE, films, PP, PET, and mixed plastics. These methods improve efficiency and plastic recovery rates.97 In addition, logistics costs get reduced by enabling the transport of the entire waste stream, rather than different sorted fractions, making centralised post-sorting more efficient and cost-effective compared to household-level pre-sorting. Therefore, post-sorting of plastic waste has the potential to become a widely adopted global solution.
5.1.3.1.4 Electrostatic sorting. Electrostatic separation is an effective method to separate binary mixtures such as ABS/PC, PET/PVC, and PP/PE. In this process, plastic flakes collide with charging units, causing one type of plastic to acquire positive charge and the other either gains negative charge or remains neutral at the surface. The particles with different charges are then separated due to their varying deflection in an electric field.96 This technology enables separation of different materials based on their electrical properties and offers low cost and high efficiency separation without concerns for secondary pollution.98
5.1.3.1.5 Froth floatation. Froth floatation is used to separate plastics with similar densities. The process relies on the principle in which hydrophobic particles adhere to air bubbles and float to the surface for recovery, whereas the hydrophilic particles remain in the liquid phase. This method is particularly used to separate mixed polymers with densities higher than water, generally binary mixtures such as PC, PET, PS, PVC, or POM (polyoxymethylene).95
Tracer-based sorting technologies are also being developed by embedding fluorescent pigments into plastic substrates or sleeve, which are detectable under UV light only at sorting facilities.99 Digital watermarks, integrated into packaging design, are also used in another technology, and cameras can detect these watermarks on high-speed sorting lines. A watermark contains information about the product and assists in precise waste sorting. One prominent project, HolyGrail 2.0, aims to prove the technical and economic viability of using digital watermarks for more efficient sorting of packaging waste.100 This initiative has been joined by 160 plus EU companies. Robotic sorting is another emerging technology that uses artificial intelligence to enhance the efficiency of waste management.101 In this process, AI-powered cameras and robotic arms work together to identify and separate different types of plastics as they move along conveyor belts.
5.1.3.2 Plastic grinding. Plastic grinding is a crucial step in the entire recycling process (Fig. 9). After sorting (if applicable and desired), plastic waste on the conveyor belts is directed to a crude shredder, which reduces the plastic material into initial-sized particles through a grinding process.
5.1.3.3 Plastic washing. The sorted plastic waste often requires washing for removing general dirt, label residues, and other impurities, which is particularly crucial for mechanical recycling and may also be imperative for chemical recycling.
The washing process typically involves the use of cold or hot water, along with caustic agents or detergents.92 This cleaning step is generally combined with the sorting chain, often following shredding. This type of washing requires specialised equipment along with drying systems and wastewater treating facilities. For example, odorous components are partially removed with caustic wash, whereas detergents or organic solvents are used to remove apolar components.102 Alternate dry-cleaning methods are being explored to reduce the cost and water consumption associated with traditional wet cleaning.103 These dry cleaning methods have been shown to achieve results comparable to those of conventional caustic washing.
5.1.3.4 Feeding system. Some reactors such as fluidised bed technology require the feed to be uniformly sized to maintain consistent thermodynamic conditions. Various feeding systems have been investigated to address this requirement, with screw feeders currently used in several pyrolysis processes. However, screw feeders can present several challenges. For instance, high temperatures and pressure variations at the reactor's inlet often result in severe blockages. Additionally, many of the preferred pyrolysis feeds are highly cohesive particulate solids, which frequently cause clogging in the feeding lines. If the feed contains plastic films, they can become tangled around the screw and form a melt around the heated screw, further complicating the feeding process.
5.2 Pyrolysis principle
Thermochemical recycling includes processes such as pyrolysis, gasification, depolymerisation, and upcycling. This method offers a viable alternate approach to traditional plastic waste management methods along with addressing the transition towards a plastic circular economy. Thermochemical recycling processes avoid rigorous sorting methods and operate under flexible conditions with minimum environmental impact.104Fig. 10 illustrates the basic flow of the pyrolysis process. Pyrolysis is a tertiary recycling where plastics are transformed into liquid oil, gaseous fuels, and carbonised char at high temperature in the absence of oxygen.105 Various pyrolysis studies have been conducted at temperatures of 300–900 °C, however 450–550 °C is the optimal temperature range for pyrolysis of plastic waste.104 Additionally, pyrolysis of waste plastic has been studied under various reaction conditions, such as heating rates of 4 °C min−1 and 10 °C min−1, with varying retention times ranging between 40 and 70 minutes.106,107 Catalytic pyrolysis is a crucial process for converting plastic waste into valuable products. With catalytic pyrolysis, high quality liquid oil can be produced at low temperatures and process times compared to thermal pyrolysis. These important parameters can significantly decrease energy consumption while improving the efficiency and selectivity of the pyrolysis process.108 High-quality catalytic pyrolysis liquids are typically associated with low oxygen contents. There is a significant advantage in using catalysts in fast pyrolysis due to their ability to directly produce high quality hydrocarbons from biomass or liquids, which can be utilised in subsequent processes. The catalyst either reduces the activation energy of reactions or alters the reaction mechanism to follow a more efficient pathway, making the process more effective. | ||
| Fig. 10 Basic flow diagram of the pyrolysis process. | ||
Catalytic pyrolysis can be classified into in situ and ex situ processes. In the in situ process, both the catalyst and polymer materials are combined directly in the pyrolysis reactor, where the upgradation occurs. In contrast, the ex situ process involves placing the catalyst in a separate catalyst bed to upgrade the pyrolysis vapours. These two approaches lead to different compositions of the pyrolysis products. Numerous catalysts such as metal oxides, FCC catalysts, silica–alumina, metal incorporated alumina, single atom catalysts, zeolites, SBA, and mesoporous MCM-41 have been used for pyrolysis of various polymers. The efficiency of these catalysts depends upon their properties such as specific surface area, crystallite size, Lewis versus Brønsted acidity, basic sites, pore size distribution, etc., which influence the composition and yield of the pyrolysis oil.109
5.3 Pyrolysis mechanism
Polymers undergo multiple initiation reactions that occur simultaneously during thermal degradation. These reactions differ depending on whether the polymer is a thermoplastic or thermosetting plastic.• Chain-end scission: the degradation process is initiated at the gas–liquid interfaces, where the polymer chain breaks at its ends and successively generates monomer units as shown in the following reactions:110,111
 | (1) |
 | (2) |
• Randon-chain scission: in this process, the polymer chain breaks at random points, leading to the formation of low molecular weights without release of monomers as shown in eqn (3).111
 | (3) |
• Cross-linking: upon heating thermoset plastic to a high temperature, cross-linking occurs via rearrangement of carbon chains.111,112 In catalytic degradation, the catalyst under heat treatment leads to breakage of the C–C bonds of the polymer chain.
• The activation energy in catalytic degradation decreases because of the catalyst effect compared to thermal degradation. In catalytic pyrolysis, initially carbenium ions are formed. This occurs in two ways: if the catalyst behaves as a Lewis acid, it removes a hydride ion from the polymer chain, however, when the catalyst behaves as a Brønsted acid, it adds a proton to the polymer chain.72
The degradation of PE in catalytic pyrolysis proceeds through various steps as follows.
Initiation: initiation occurs at a weaker area of the polymer chain, where proton addition converts an olefinic link to a carbenium ion (eqn (4)).72 Hydride ion abstraction leads to a carbenium ion as in eqn (5).
–CH2CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2CH2– + HX → –CH2CH2CH+CH2–CH2CH2– + X− CHCH2CH2– + HX → –CH2CH2CH+CH2–CH2CH2– + X− | (4) |
| –CH2CH2CH2CH2CH2– + R+ → –CH2CH2CH+CH2CH2– + RH | (5) |
Then β-scission occurs to further break the chains (eqn (6)).
| –CH2CH2CH+CH2–CH2CH2– → –CH2CH2CHCH2 + +CH2CH2– | (6) |
Depropagation: in this step, the molecular weight of the polymer reduces by reactions on acid sites, leading to the formation of oligomer fractions, which further break down into gases and oils through β-scission.
Isomerisation: double-bond isomerisation takes place due to the rearrangement of hydrogen or carbon ions (eqn (7)).
 | (7) |
Aromatisation: some of the carbenium ions undergo a cyclisation reaction. Abstraction of a hydride ion leads to olefinic carbenium ion formation that can attack the double bonds and initiate cyclisation leading to aromatic compound formation (eqn (8)).
| R1+ + R2HCCHCH2CH2CH2CH2CH3 ↔ R1H + R2HCCHCH2CH2CH2+CHCH3 | (8) |
5.4 Reactors for pyrolysis
The reactor choice is critical for plastic waste pyrolysis, and its design plays a significant role in achieving the desired products. Typically, the following reaction parameters can impact the efficiency of reactors to obtain higher selectivity for the desired products:• Plastic type and its particle size.
• Catalyst type and amount.
• Mixing efficiency of reactants and catalyst.
• Reaction temperature.
• Heat transfer rate.
• Residence time.
Fig. 11 presents the configurations of different reactor types used by researchers for the thermochemical conversion of waste plastics, which are discussed in this section.
 | ||
| Fig. 11 Various reactor configurations for pyrolysis. | ||
Fixed bed reactors have been widely utilised by numerous researchers for plastic pyrolysis.114 Various reactor configurations have been explored,115 including single-stage and two-stage fixed bed systems.114 For instance, Renzini et al. utilised a fixed bed reactor for catalytic upgradation of pyrolysis gases derived from polyethylene (PE) using a Y-zeolite catalyst, and achieved liquid products with 85 wt% yield.116,117 However, without using a catalyst, 95 wt% liquid was obtained at 500 °C, with minimal or no coke formation and minor gas yield. Moreover, the yield of the gaseous product and liquid product was found to increase and decrease, respectively, upon increasing the temperature. Al-Salem et al. employed a fixed bed reactor for recovering wax by pyrolyzing virgin plastics including LDPE, HDPE, and plastic solid waste (PSW).118 The pyrolysis of LDPE gave the highest wax yield (64.5 wt%) at 500 °C, and HDPE produced 32 wt% under the same conditions; however, pyrolysis of PSW at 700 °C produced less wax (9.25 wt%). Similarly, pyrolysis of HDPE at 500 °C in a fixed bed reactor, utilising nitrogen as an inert carrier gas gave the oil yield of 95 wt%, with minimum gas production in the absence of a catalyst,119 whereas the use of the zeolite Y catalyst resulted in a slightly lower oil yield of 85 wt%.
Many researchers prefer to use fluidized bed reactors rather than fixed bed reactors for catalytic cracking of plastics due to their superior mixing and heat transfer capabilities, which provide better access and contact between reagents and the catalyst. Typically, pyrolysis of plastics in fluidised bed reactors is performed at temperatures in the range of 290–850 °C. Sharratt et al. successfully conducted pyrolysis of HDPE in a specially designed fluidised bed reactor using ZSM-5 catalyst.123 Luo et al. employed a fluidised bed reactor to study the pyrolysis of PP and HDPE catalysed by silica–alumina, and achieved a high liquid yield from PP at 500 °C.124 Lin et al. utilised a fluidised bed reactor for PP pyrolysis with various catalysts for the production of valuable hydrocarbons.125 Mastellone et al.126,127 evaluated the influence of temperature and residence time on the pyrolysis of polyethylene in a fluidised bed reactor; Mastral et al.128 performed HDPE pyrolysis using HZSM-5 catalyst at 350 °C and 500 °C, yielding high amounts of gas contents.
Elordi et al. investigated a continuous pyrolysis of HDPE in CSBR at 500 °C using different zeolite catalysts, with HZSM-5 exhibiting high selectivity towards light olefins (58 wt%). In contrast, Hβ and HY resulted in higher yield of non-aromatic C5–C11 hydrocarbons (∼45 wt%).132 In a similar study, pyrolysis of HDPE using the spent catalyst resulted in complete conversion leading to 50 wt% of gasoline (C5–C11) and 28 wt% of C2–C4 olefin fractions.133 Authors reported that the reactor efficiently enhanced the melting of the polymer and its coating onto the catalyst. Similarly, Elordi and co-workers conducted the pyrolysis of HDPE using CSBR at a temperature of 500–700 °C.134 At 500 °C, waxes (C21+) were obtained in a high yield reaching 67 wt%, but with the temperature rising to 700 °C the product distribution shifted towards gases and gasoline fractions (C5–C11), with a yield of 39 wt% and 33 wt%, respectively. Artetxe et al. used CSBR for the pyrolysis of HDPE with HZSM-5 catalysts of varying acidity to selectively produce olefins.135 The study achieved a maximum yield of light olefins (58 wt%) using a highly acidic catalyst with a SiO2/Al2O3 ratio of 30. However, complex design and various technical challenges, including catalyst entrainment and feeding, and product collection pose difficulties in the operation of this reactor.130 Moreover, this reactor usually has high operation cost due to the need for multiple pumps to maintain adequate pressure for the spouting behaviour.75,121
The microwave-induced pyrolysis of HDPE pellets and toothpaste packaging was investigated by Ludlow-Palafox carrying out degradation experiments at temperatures ranging from 500 to 700 °C.143 The results obtained from the microwave reactor were comparable to those achieved from conventional reactors. Various researchers studying pyrolysis in microwave reactors have concentrated on its application with various types of wastes, including plastic waste and sewage sludge,144,145 scrap tyres,146 heavier hydrocarbon liquids,147 and papers.148 Undri et al. studied the pyrolysis of HDPE and PP in a microwave reactor using two types of microwave absorbents to maintain the reactor temperature, and achieved a high liquid yield (83.9 wt%) from HDPE, whereas the yield from PP was 74.7 wt%.149 Khaghanikavkani et al. used a rotating microwave reactor for plastic pyrolysis to evaluate the influence of various reaction parameters on product yield and composition, aiming for potential industrial applications.150 They concluded that microwave heating provides superior and uniform heat distribution without any changes in the product compared to conventional methods. The type of material used for pyrolysis was found to influence the efficiency of microwave-assisted pyrolysis,151 and pyrolysis outcome was also influenced by the sample density.152 Recent studies have demonstrated co-pyrolysis of PE and PS with rice straw and sugarcane bagasse using HZSM-5 under microwave heating.153 PE and PS furnished significantly higher amounts of liquid oil, with 82 wt% and 98 wt% yields, respectively, whereas straw and bagasse gave oil yields of 26 wt% and 29 wt%, respectively.
Microwave technology offers various advantages over the conventional pyrolysis process, including increased production rate, rapid heating, efficient energy transfer, lower production cost, process flexibility, equipment portability, and the avoidance of undesired oxygen containing hydrocarbons. Moreover, preheating is also not required in microwave pyrolysis.154 For example, a study by Ding et al. on the pyrolysis of LDPE using a microwave reactor at 1.6 MPa and 425 °C revealed that the yield of liquid oil could reach as high as 89.5 wt%.155 However, there is insufficient data on the dielectric properties of treated waste streams which is the primary limitation of this technology for large scale implementation.121 This limitation exists because few investigations have been performed on microwave-assisted pyrolysis.149,156 In conclusion, our review suggests that microwave-assisted pyrolysis can be effectively employed for energy recovery from plastic waste. However, further studies are necessary to understand the characteristics of pyrolysis for plastic waste materials using microwave technology and to fully assess its potential for large-scale implementation.
Each reactor type and design has its advantages and disadvantages, and selecting the appropriate reactor type for pyrolysis can improve the quality and yield of the required products. A comparison of different reactors is summarised in Table 1.
| Reactor type | Working mechanism | Advantages | Disadvantages | References |
|---|---|---|---|---|
| Batch reactor | • Operates as a closed system based on thermodynamic principles | • Best reactor for thermal pyrolysis | • Batch to batch product variation | 159 and 160 |
| • Simple in design and operation | • Unsuitable for large scale pyrolysis production | |||
| • Heat transfer is carried out by the bed material | • Preferable for char production | • Unsuitable for catalytic pyrolysis | ||
| • High labour and energy cost per batch especially in large scale | ||||
| Semi-batch reactor | • Reactants can be fed into the reactor at intervals during pyrolysis | • Better control over the reaction rate and selectivity | • High labour and energy cost per batch | 121, 161 and 162 |
| • Variable performance | ||||
| • Suitable for small scale operation | ||||
| • Simultaneous addition of reactants and extraction of products is possible | • Suitable for production of high yield of oil | • Unsuitable for catalytic pyrolysis | ||
| • Production rate is limited by inefficient heat and mass distribution | ||||
| Fixed bed reactor | • Catalyst is arranged in a fixed bed and shaped into pellets | • Simple construction and easy operation | • Unsuitable for feedstocks with irregular sizes and shapes | 121, 160, 163 and 164 |
| • Both thermal and catalytic processes are possible | ||||
| • Temperature control difficult | ||||
| • Economical operation | • The catalyst's surface area available for reaction is significantly restricted | |||
| • Longer residence time required for high carbon conversion | • Process results in low output of both liquid and gaseous products | |||
| Fluidized bed reactor | • Feedstock and bed material are combined through fluidisation | • Uniform particle mixing | • Complex design and operation | 121, 165 and 166 |
| • Uniform temperature distribution | • High capital cost especially for small scale operation | |||
| • Continual operation | • Melt plastic attaches to the surface of the bed particles | |||
| • Mixing occurs due to the velocity of the fluidized medium | • Significant versatility on gas residence times | • Separation of fine catalyst particles from the exhaust gas is challenging | ||
| • Provides larger accessibility to the surface area of the catalyst | ||||
| • Suitable for scale-up operation | • Corrosions of pipes and blockage of feeding system | |||
| • Maintenance costs are moderate | ||||
| Conical spouted bed reactor | • An alternate to fluidised bed reactor and suitable for flash pyrolysis and continuous feed operations | • Capable of accommodating large and irregular particle sizes, and densities | • Encounters technical difficulties including catalyst entrainment, feeding issues, and challenges in collecting the final products | 121, 129, 134 and 160 |
| • Provides good mixing | ||||
| • Minor defluidization | • Complex design requiring many pumps | |||
| • Have low attrition rate and bed segregation | ||||
| • Design allows high heat transfer between phases | • High operation cost | |||
| Rotary kiln reactor | • Cylinder with a certain slope rotates at a certain speed | • High yields of pyrolysis oil | • Complex design | 105, 164 and 167 |
| • Slow reaction process | ||||
| • Uses centrifugal forces for mixing the sand and moving feed upward | • Provides good mixing | • Substantial char formation | ||
| • Low capital cost | • Low heating rate | |||
| Screw kiln reactor (auger reactor) | • Continuous feed to a screw (auger) which is typically filled with sand | • Easy control over the product | • Uneven heating of processed material with hot spots near hot surfaces | 139 and 168 |
| • Capable of producing a wide variation of products | • Poor heat exchange | |||
| • Output ranges from wax to crude-like oil to diesel-quality oil | ||||
| • Offers high flexibility for treating municipal waste plastics (MWPs) of varying shapes and sizes | • Poor scaling up potential | |||
| Microwave reactor | Transfer of energy through atomic or molecular interactions using a microwave | • Efficient heat transfer | • Scaling up is questionable | 121, 131, 140 and 169 |
| • Short residence time | ||||
| • Low by-product formation | • Inadequate mixing | |||
| • Higher production speed and product selectivity | ||||
| • Suitable for co-pyrolysis applications | • Strong reliance on the dielectric properties of the waste material | |||
| Electromagnetic induction reactor | • Electromagnetic induction directly heats materials | • Lower energy consumption | High operational costs | 158 |
| • Quick and effective heating | ||||
| • The process can be precisely controlled by adjusting the frequency and power of the current | • Consistent temperature distribution | |||
| • Offers precise control over temperature | ||||
| • Delivers high efficiency |
5.5 Advances in catalysis for pyrolysis
Thermal pyrolysis is performed at high temperature for decomposing waste plastic under an inert atmosphere. Polyolefins derived plastics usually undergo random-chain scission process, resulting in various products such as n-paraffins and α-olefins, which often require further treatment to improve their quality.170,171 With a radical-chain transfer mechanism, thermal cracking is completed. However, thermal degradation also leads to the formation of more core/waxes that can block the apparatus due to the low heat transfer rate and high viscosity.172Some key drawbacks of thermal pyrolysis include high-temperature requirement, broad product distribution, and longer residence time. Furthermore, at elevated temperatures (up to 900 °C), the products formed from random scission of plastics are challenging to separate, while thermal cracking at low temperature leads to the formation of waxy oil fractions. However, due to the poor quality of products, thermal breakdown is not considered a cost-effective process.75 In contrast, pyrolysis assisted by a catalyst considerably reduces the optimal temperature required for this process by reducing the activation energy. Therefore, catalytic pyrolysis offers several advantages over thermal pyrolysis, including low energy consumption, a more narrow distribution of hydrocarbon products, and improved product selectivity (Fig. 12).93 Catalysts provide numerous benefits including reduced reaction times, lower activation energy to break C–C bonds, and their capability to produce lower molecular weight products and reduce reaction temperatures.173,174
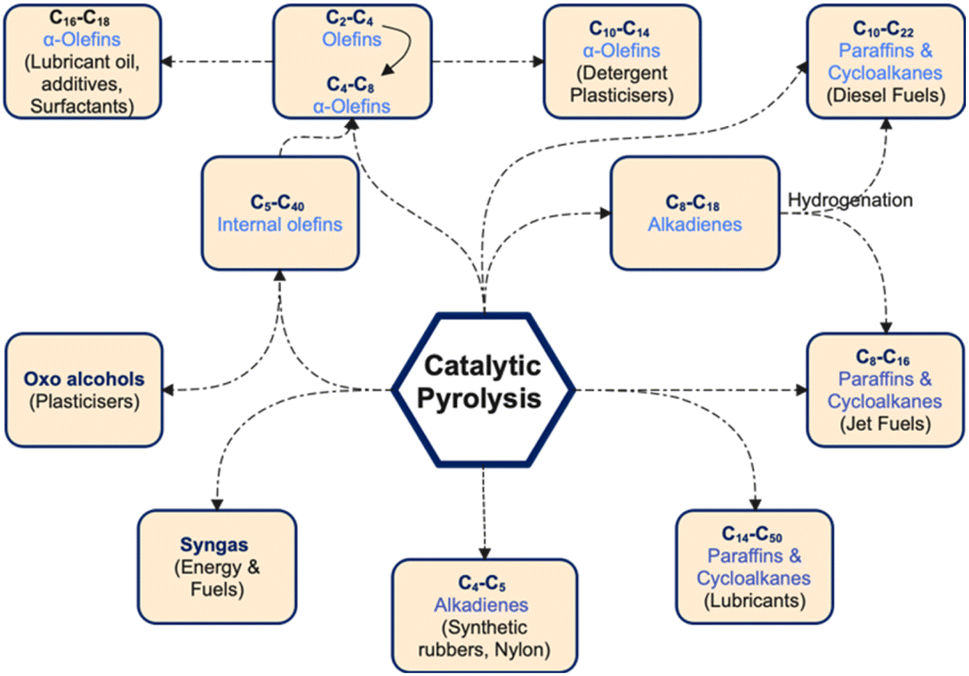 | ||
| Fig. 12 Production of value-added products from pyrolysis of plastic waste. | ||
Catalytic pyrolysis of common plastics, including HDPE, LDPE, PP, PS, and PET, is usually carried out with three types of catalysts that include FCC catalysts, silica–alumina, and zeolites.
5.5.1.1 Silica–alumina catalysts. Silica–alumina is an amorphous acidic catalyst characterised by medium–strong tuneable acidity. In this material, the Lewis acid sites act as electron acceptors, and Brønsted acid sites contain the ionisable hydrogen atom. These catalysts, composed of silicon dioxide (SiO2) and aluminium oxide (Al2O3), are particularly valued for their ability to facilitate various reactions such as cracking, hydrocracking, and isomerization. The unique combination of silica and alumina provides a high surface area, strong acidity, and thermal stability, making these catalysts effective in breaking down complex hydrocarbons into simpler molecules. The acid ratio of silica–alumina catalysts has a strong influence on plastic pyrolysis and is determined by the silica to alumina molar ratio. These catalysts have higher acidity when their silica to alumina ratio is high which is in contrast to zeolites.178
The synthesis of silica–alumina catalysts typically involves a combination of sol–gel and precipitation methods. These catalysts can be synthesised from fly ash via an activation process that involves treatment of fly ash with sodium hydroxide, followed by the aging method. Catalysts exhibited good efficiency similar to commercially available silica–alumina catalysts.75,91 Uddin et al. evaluated the effect of Si–Al catalysts with different SiO2/Al2O3 ratios, specifically 83.3/16.7 and 21.1/78.9, which resulted in liquid yields of 68 wt% and 77 wt%, respectively.179 The study revealed that the yields and product distributions could be controlled by changing the SiO2/Al2O3 ratio. The optimal SiO2/Al2O3 ratio for pyrolysis of HDPE was found to be 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, achieving a liquid fuel in 87% yield, with significantly improved calorific values.178,180
:3, achieving a liquid fuel in 87% yield, with significantly improved calorific values.178,180
Achyut et al. used kaolin and silica–alumina catalysts to investigate the catalytic pyrolysis of polypropylene at temperatures between 400 and 500 °C in a semi-batch reactor.181 Silica–alumina showed better performance with less residence time than kaolin catalyst, and gave liquid oil in 91% yield versus 89.5% with kaolin, which is attributed to the higher acidity of the silica–alumina catalyst. The resultant oil can potentially be used as engine fuel after undergoing additional processing.121 In 2020, Gopinath et al. investigated the pyrolysis of used LDPE polymer (ULDP) in a semi-batch reactor using silica–alumina as the catalyst and nitrogen as the fluidizing gas at 500 °C, and obtained a liquid in 93.5 wt% yield along with gas and char in 5.4 wt% and 1.1 wt% yields, respectively.182 The authors also studied the efficiency of plastic oil blends (20%, 40%, 60%, 80%, and 100%) in a direct injection (DI) diesel engine without any modification. The efficiency of ULDP20 was found to be higher than that of other blends, leading them to conclude that ULDP20 could be used as a fuel for diesel engines.
Recently, the silica–alumina catalyst was used to convert polyethylene into aromatic compounds at temperatures of ≤280 °C.183 The authors compared the activity of the silica–alumina catalyst with Pt(1 wt%)/Al2O3 and found comparable product yields under similar conditions. The acid site density of the silica–alumina catalyst strongly influenced the product selectivity. Particularly, the increase in Brønsted acid density increased polyaromatic products as well as the extent of polymer deconstruction. Additionally, the catalytic activity improved with each recycling, achieving soluble product yields of up to 83%. The acid sites on the catalyst were identified as key factors in initiating both depolymerisation and aromatisation reactions.
5.5.1.2 Zeolite catalysts. Zeolites are crystalline solid structures composed of aluminosilicates with a three-dimensional framework where oxygen atoms are tetrahedrally connected.184 Zeolites are also known as molecular sieves. Natural zeolites are comprised of numerous earth metals including Ca, Fe, K, Mg, and Na.185 These structured minerals have a surface area of 4.3 m2 g−1, pore size of 18.7 Å, and volume of 0.02 cm3 g−1. There are >250 zeolite frameworks identified and accepted by the Structure Commission of International Zeolite Association with 40 known naturally occurring zeolites.186 The ratio of SiO2/Al2O3 in the zeolite structure determines their reactivity and effectiveness, which in turn influence the conversion rates and quality and type of pyrolysis products from plastics.187 The acidity of zeolites has been demonstrated to favour the conversion of aliphatic hydrocarbons to aromatics and cyclic compounds.188 Various zeolite catalysts, such as HUSY, HMOR, SAHA, and HZSM-5, have been investigated for the catalytic pyrolysis of HDPE. Among these, HZSM-5 demonstrated the highest activity, achieving the highest conversion in 30 min achieving 93.23 wt% gaseous products with the lowest residue of 4.53 wt% compared to other catalysts.187 The typical activity was HZSM-5 > HUSY ≈ HMOR > SAHA. However, using HZSM-5 resulted in a product comprising mostly gases, which is considered its major drawback.91 When HUSY and HZSM-5 were used for the pyrolysis of polyethylene at 550 °C with a catalyst to plastic feed ratio of 10 wt%, different product selectivities were observed with these catalysts.178 The use of HUSY and HZSM-5 catalysts for the pyrolysis of PP led to comparable results, however the liquid product yield was reduced when the polymer to catalyst ratio was 40 wt%.125 Seo et al. also studied the effect of HZSM-5 catalyst on the pyrolysis of HDPE at 450 °C with 20 wt% catalyst to polymer ratio, achieving liquid and gas products in 35 wt% and 63.5 wt%, respectively.189 However, using HZSM-5 catalyst for pyrolysis at 550 °C led to liquid hydrocarbons in 4.4 wt% yield and gaseous products in 86.1 wt% yield.190
Ding et al. studied the pyrolysis of LDPE using HY zeolite catalyst and evaluated the influence of the catalyst to plastic ratio on reactivity and product distribution.155 The optimised catalyst to polymer ratio was found to be 1:10, yielding 56.54 wt% of oil with significant increase in high octane compounds, mainly aromatics and isomerised hydrocarbons. An increase in catalyst to plastic ratio from 0 to 1:15 resulted in significant increase in the oil yield, from 51.56 wt% to 60.01 wt%. But, further increasing the catalyst to polymer ratio to 1:5 led to a decrease in oil yield, whereas gas yield increased.155 Ajibola et al. studied the pyrolysis of LDPE waste using zeolite Y, and reported similar results with 53.3 wt% liquid yield comprising 56 wt% of gasoline fractions.191 Milato et al. explored the co-pyrolysis of oil sludge with various polyolefins, such as HDPE, and PP, using Y zeolite catalysts, and evaluated the effect of acidic and textural properties to obtain paraffin-rich pyrolysis liquids.192 The study demonstrated a direct correlation between the gas fraction and catalyst's acid strength. The catalyst with higher acidity resulted in a higher gas fraction compared to thermal degradation alone.
Some researchers have studied the use of natural zeolites for pyrolysis of waste plastic and found them to be promising candidates. However, optimising the temperature is essential to achieve the desired selectivity from these catalysts. Natural zeolites have been demonstrated to increase the oil formation during pyrolysis of waste LDPE by increasing the temperature from 300 °C to 550 °C. However, additional increase in temperature resulted in higher gas yield.193 Erawati et al. reported a similar effect of temperature variation (410–440 °C) on natural zeolite for pyrolysis of plastics (HDPE &PP).194 The liquid yield was increased with temperature, reaching a maximum yield of 68.42 wt% at 440 °C. Moreover, the highest yield (87.31 wt%) was achieved by using a plastic to catalyst ratio of 67.33 wt%, and the resulting product was deemed suitable for use as diesel fuel. Similar studies were carried out by Hendrawati et al. for pyrolysis of HDPE and PP using a natural zeolite catalyst, producing liquid hydrocarbons in 69.69% and 65.60% yields, respectively, after 8 hours at 450 °C.195 The product distribution of liquid hydrocarbons from PP pyrolysis was C5–C12, C17–C20, and C13–C16 in 74.16%, 22.32%, and 3.52% ratio, respectively, whereas the liquid hydrocarbons from HDPE pyrolysis were C13–C16, C5–C12, C17–C20, and >C20 in 40.39%, 30.36%, 24.69%, and 4.56% ratio, respectively. Fig. 13 illustrates the influence of catalyst properties and distribution of main products.
 | ||
| Fig. 13 Main products from the catalytic pyrolysis of polyolefins on different acid catalysts. Adapted with permission from ref. 84. Copyright 2017, Elsevier. | ||
Sivagami et al. used various commercial and synthesised ZSM-5 zeolite catalysts to study the pyrolysis of mixed plastic waste.196 The authors tested various types of plastic wastes, including single and multilayer materials. Biaxial oriented polypropylene (BOPP)-based plastic waste was found to give a higher oil yield compared to PET-based plastic waste. The synthesised ZSM-5 zeolite catalyst provided 70 wt% oil, 16 wt% gas, and 14 wt% char for LDPE plastic.194 The synthesised ZSM-5 catalyst with strong acidic properties resulted in increased cracking and isomerisation that enhanced the breakup of larger molecules resulting in more oil yield. Co-catalysts have also been used with zeolites to improve product quality. Ding et al. demonstrated that adding NiO as a co-catalyst with HY for pyrolysis of LDPE increased the production of high-octane number compounds while decreasing the coke formation compared to HY catalysts alone.155 Use of NiO with HY zeolite in a 1:10 ratio furnished oil in 51 wt% yield, the oil product contained gasoline with the highest-octane number and 46.61 wt% aromatic content. The inclusion of NiO also increased the gas yield, primarily containing H2 and C1–C3 hydrocarbons. Co-catalysis is considered beneficial not only for improving product quality but also for reducing catalyst costs, as the total required quantity of co-catalysis (0.15 g NiO, 1.5 g HY) is significantly lower than using HY alone (3 g).155
Zeolites are suitable catalysts for plastic catalytic pyrolysis due to their reactivity and selectivity. The liquid product achieved from the pyrolysis of polystyrene using zeolite catalysts can be used for energy generation after removing acids, contaminants and other residues, and can be upgraded for blending with diesel.197 However, zeolites are less favourable catalysts for achieving higher liquid yield due to their microporous structure, which often results in a substantial fraction of gaseous products. Therefore, there is an increased interest to synthesise composite zeolites that can produce higher liquid products with good reactivity. Zhou et al. investigated the activity of ZSM-5 zeolite catalysts coated on SiC foam for pyrolysis of polyethylene in a continuous process.198 The structured ZSM-5 catalyst demonstrated high selectivity for gasoline-range aromatic hydrocarbons and their stability performance was almost 37 times better than that of conventional ZSM-5 catalysts. The improved stability was ascribed to more efficient mass transport, facilitated by the reduced diffusion pathway provided by zeolite coating and the irregular silicon carbide structure.
Due to the high viscosity and bulky nature of plastics, catalysts must be designed to ensure good accessibility to the acid sites. In this sense, nanocrystalline and hierarchical zeolites (having bimodal micro and mesoporosity) have shown better activity than conventional zeolites.199,200 This is because polyolefins are often unable to access the micropores of HZSM-5 zeolite, whereas the mesopores of materials like Al-MCM-41 are totally accessible.199,201 Furthermore, the acidic properties of these zeolites when combined with appropriate pore sizes are capable of significantly increasing the selectivity towards monoaromatics, such as BTX, which are valuable marketable products.82 The most important limitation to be addressed in zeolites is their deactivation by coke deposition, which tends to occur more rapidly in highly acidic and large pore-containing materials. However, this phenomenon can also be reduced by designing a catalyst with a proper combination of acidic and porous properties. When different zeolites are compared under analogous catalytic pyrolysis conditions, ZSM-5 zeolite is almost always concluded to be the most efficient in terms of oil yield, aromatics production, and reduced coke formation.82
Colantonio et al. investigated the performance of HUSY and HZSM5 zeolite catalysts for the pyrolysis of packaging plastic waste. Both catalysts were found to exhibit good cracking performance, leading to reduction in the heavy oil content while increasing the light hydrocarbon contents and preventing the formation of wax.202 HUSY was very effective in increasing the yield of monoaromatics, especially benzene and ethylbenzene. In contrast, HZSM5 was more effective for gas production due to small pores and strong acid sites. Liu et al. investigated three catalysts, ZSM-5, HY and MCM-41 for cracking of LDPE to compare their performance.203 MCM-41 was found to increase the oil yield to 78.4% at 650 °C, indicating the reduced secondary cracking of intermediate components. This high selectivity was attributed to MCM-41's relatively high BET surface area and moderate acidity. Meanwhile, ZSM-5 and HY produced high amount of gas products at yields of 61.4% and 67.1%, respectively. ZSM-5 produced an oil with an aromatic yield of 65.9% at 500 °C. Recently, Wong et al. used HZSM-5 (MFI-type) catalyst for pyrolysis of polyolefins (HDPE, LDPE and PP) in a fixed bed reactor using induction heating, with a reaction time of 30 min204 The catalyst significantly increased both the gas yields (70.6–73.9 wt%) and liquid yields (24.0–27.2 wt%) compared to the thermal process. The gas products were mainly comprised of C3 compounds, followed by C4 fractions, irrespective of the type of plastic. The liquid oil was rich in aromatics, particularly C7–C10 aromatics comprising ethylbenzene, naphthalene, toluene, xylene, and various alkylbenzene isomers.
5.5.1.3 Fluid catalytic cracking catalysts. Fluid catalytic cracking (FCC) is one of the largest catalytic conversion technologies in petroleum industries.205,206 FCC catalysts are comprised of zeolite crystals, mainly the zeolite-Y, as the main active component, some non-zeolite silica–alumina material for producing a meso- and microporous matrix, and a binder to bind the system components.184 Significant advancements have been made to enhance the efficiency of these catalysts to improve accessibility, hydrothermal stability, cracking activity, and coke selectivity by creating microporosity in the zeolite crystals or by manipulation of aluminium's extra-framework.205 FCC catalysts, previously used in petroleum refineries, are called spent FCC catalysts and contain different contaminants. However, these spent catalysts can be used for pyrolysis of plastics. Lee et al. utilised a spent FCC catalyst to evaluate its effectiveness for pyrolysis of HDPE.83 They found that the catalyst increased the liquid oil yield from 75.5 wt% to 79.7 wt% at a pyrolysis temperature of 430 °C, while the gas yield slightly decreased compared to thermal degradation. Additionally, there was significant reduction in the formation of residue content. In another study, the effect of catalyst to polymer (HDPE) ratio (from 10 wt% to 60 wt%) was investigated at 450 °C. The study revealed that although the catalyst had a minimal effect on the yield of non-condensable products, the yield of gaseous products increased with a higher catalyst to polymer ratio.161 However, the coke formation was found to increase, most likely due to more aromatisation and dehydrogenation on the catalyst surface, which promoted coke formation.
Various comparative studies have been conducted on pyrolysis of waste plastics to evaluate the reactivity and product distribution between FCC and zeolite catalysts. Onwudili et al. investigated the catalytic pyrolysis of mixed plastic waste (PE, HDPE, LDPE, PP, PS, PET) using FCC, ZSM-5, and zeolite Y catalysts.207 They reported a higher oil yield (76 wt%) with zeolite Y catalyst compared to FCC catalyst (71.5 wt%). While the properties of the obtained liquid oil with both catalysts were suitable for fuel, the oil obtained with the spent FCC catalyst had a higher aromatic content (36.2 wt%) than that from the zeolite Y catalyst (33.7 wt%). Aisien et al. studied the use of FCC catalyst for pyrolysis of PP and achieved an oil yield of 77.6 wt%, which was notably higher than the yield (71.5 wt%) reported by Onwudili et al. using the same catalyst.208 The high activity was attributed to the difference in feedstock, as PP tends to produce more oil than mixed plastic feed. Moreover, liquid oil was found to contain a range of hydrocarbons (C4–C17), including olefins (44.6%), paraffins (30.83%), naphthalene (19.44%), and aromatics (5.13%), with fuel properties comparable to those of gasoline and diesel. The same authors recently studied the pyrolysis of waste LDPE using FCC catalyst at temperatures in the range of 350 °C to 550 °C, with a catalyst to plastic ratio of 0.10–0.25.209 At a ratio of 0.2 and temperature of 550 °C, the process yielded 92.7 wt% liquid oil, along with gas and char at 6.1 wt%, and 1.2 wt%, respectively. The obtained liquid consisted of C7–C29 hydrocarbons. Orozco et al. evaluated the effect of oxygen co-feeding on the deactivation of an equilibrium FCC catalyst during pyrolysis of HDPE and observed a great impact on the catalyst performance.210 The distribution of pyrolysis products moved towards lighter compounds, with significant increase in light olefins. Moreover, there was a significant reduction in catalyst deactivation rate under oxidative conditions.
Huiyan et al. explored the efficiency of spent FCC catalyst for co-pyrolysis of plastics (PE, PP, and PS) with pine sawdust to evaluate how different feedstocks affect product distribution, especially aromatics and olefins.211 They observed a synergetic effect between these feedstocks.211 The optimum blend ratio of the catalyst and PE/pine sawdust was found to be 4:1, producing the best petrochemical carbon yield of 71%, which was better than that achieved from individual feedstocks. A notable reduction was observed in the total carbon yield of petrochemicals by increasing the PE ratio, however there was a significant reduction in coke and char yields. The authors concluded that the effective H/C ratio increased with the addition of plastic to biomass in catalytic pyrolysis, which enhanced the conversion of biomass to aromatic compounds and olefins. The co-pyrolysis of PS/biomass produced the highest aromatic yield of 47%, while olefin yield was relatively lower at 11.4%.
Some researchers have investigated the use of FCC catalyst with silica and alumina for plastic pyrolysis, reporting a higher oil yield and better selectivity than with FCC catalyst alone. Streiff et al. conducted the pyrolysis of mixed plastic using a mixture of FCC (75%) and silica (25%), obtaining liquid oil in 72.7 wt% yield that contained gasoline, kerosine, diesel, polycyclic aromatics, dicyclic aromatics, and monocyclic aromatic fractions in 35.78 wt%, 26.87 wt%, 16.07 wt%, 11.8 wt%, 11.2 wt%, and 8.1 wt%, respectively.212 Wong et al. used FAU-type FCC catalyst with distinct Brønsted acidity for the pyrolysis of HDPE, LDPE and PP using induction heating.204 The gas and liquid yields significantly increased to 62.4–75.2 wt% and 0–35.9 wt%, respectively. The liquid products were rich in alkanes and alkenes, in the range of C9–C40, and coke formation was observed at 1.32–1.70 wt%.
FCC catalysts have been emerging as some of the most widely used catalysts for plastic pyrolysis.213 Now a days, Fluid Catalytic Cracking Units (FCCUs) are recognised as the main unit for production of gasoline in refineries,214 suggesting that FCC technology has a significant potential for large scale pyrolysis of plastic wastes in the near future.
5.5.1.4 Bifunctional catalysts. Zeolite catalysts can be modified to increase their activity and selectivity by impregnating active metals to make them bi-functional catalysts. Akubo et al. used Y-zeolite catalysts impregnated with various transition metals such as Co, Fe, Ga, Mo, Ni, and Ru at 1 wt% and 5 wt% loadings to evaluate the effect of these promotors on the composition of aromatic fuel.215 The addition of these promotors produced oils with an increased aromatic content, predominantly consisting of single-ring aromatic hydrocarbons. However, the presence of these promotors resulted in substantial increase in coke formation on impregnated catalysts. Steriff et al. used ECAT (equilibrated FCC) impregnated with 4 wt% Ga for pyrolysis of plastic (PE 80%, PS 20%) in a semi-batch reactor at 425 °C.216 They also used another catalyst prepared from the combination of 4 wt% Ga loaded over ZSM-5 in combination with ECAT. The bifunctional catalyst ECAT with 4% Ga gave oil in 85 wt% yield, with 42.1 wt% gasoline having a Research Octane Number (RON) of 80 and Motor Octane Number (MON) of 77.5. However, the second catalyst, synthesised by the addition of HZSM-5, produced oil in a relatively low yield (73.7 wt%), but the produced oil was of higher quality with 47.5 wt% gasoline having an RON of 84.7 and MON of 81.4.216 In another study, Wang et al. investigated the effect of Zn loaded ZSM-5 catalyst for pyrolysis of PE to prepare aromatics in a high pressure reactor.217 Authors evaluated the influence of Zn loading, temperature, and pressure on the yield of aromatic hydrocarbons. Their finding revealed a significant enhancement in the selectivity of aromatics using 3 wt% at 420 °C, with the monocyclic aromatics content rising to 90% in the liquid product.
Recently, Nandakumar et al. investigated the co-pyrolysis of biomass and HDPE using HZSM-5 catalyst modified with Mn, Ni, and Zn.218 Their findings demonstrated that a 1% Zn-modified HZSM-5 catalyst gave oil with relatively high aromatic contents (25.12%), whereas 5% Ni and 5% Zn-modified HZSM-5 catalysts produced oil with 16.22 wt% and 15.76 wt% aromatics, respectively. In contrast, 5% Mn-HZSM-5 resulted in increased production of light olefins (C2–C4), accounting for 15.84 wt% of pyrolysis gases, followed by 1% Mn (13.97 wt%) and 10% Ni (13.61 wt%). Yousefi et al. explored the effect of iron (Fe) and boron (B) promotors (10 wt%) on ZSM-5 catalyst during the pyrolysis of polypropylene (PP) to produce petrochemicals.219 They found that boron incorporation resulted in a mesoporous structure, and iron incorporation reduced the active sites of the catalyst. This modification produced a high-quality oil with a substantial aromatic content of 76.4 wt%, along with iso-paraffins and olefins in 7.1 w% and 14.2 wt%, respectively.
One of the major limitations with zeolites is their small pore size, which prevents bulky polymer compounds from accessing the catalyst, limiting the conversion to smaller molecules. In order to address this issue, Musavi loaded MIL-53 (Cu) onto the surface of the zeolite and used it for pyrolysis of plastic waste.220,221 The resulting liquid fuel, determined by using Response Surface Methodology, was categorised into gasoline, jet fuel, and diesel.
5.5.1.5 Composite catalysts. Researchers have extensively studied the use of zeolites as catalysts for cracking waste plastic to achieve high conversion rates. However, the microporous structure of traditional zeolites often leads to products with a high gas yield. Therefore, there is growing interest in micro–mesoporous composite zeolites because they possess both micropore and mesopore characteristics. Typically, the zeolite structure is modified by inducing mesopores to synthesise composite catalysts with the goal of enhancing liquid yields from the pyrolysis of plastic waste.
Ratnasari et al. used different ratios of MCM-41 and ZSM-5 catalysts for the pyrolysis of HDPE at 500 °C and reported the reduced formation of aliphatic hydrocarbons by increasing the ratio of ZSM-5 zeolitic catalyst.213 By using a 1:1 mixture of MCM-41 and ZSM-5, and a catalyst to plastic ratio of 1:2, they achieved a remarkable oil yield of 97.72 wt%, with 95.85 wt% aromatic contents in the gasoline range hydrocarbons. The authors also used MCM-41 and ZSM-5 alone to compare the efficiency with the composite MCM-41/ZSM-5 catalyst. ZSM-5 catalyst resulted in 15% of aliphatic hydrocarbons, with 80% of hydrocarbons in the gasoline range (nC8–nC12), whereas MCM-41 catalyst produced 70% of aliphatic hydrocarbons, with 45% hydrocarbons in the gasoline range (nC8–nC12). Yunhao and coworkers in 2020 synthesised hierarchical HZSM-5 zeolites by steam-assisted crystallisation using silica sol as the silica source to evaluate their performance in the catalytic cracking of polyethylene.222 The composite catalysts contained both micropores and mesopores. The HZSM-5 (30)-34 zeolite catalyst, containing a Si/TPA+ optimal ratio of 34 and Si/Al ratio of 30, exhibited the highest number of micropores and a large surface area of mesoporous Brønsted active sites. The authors also synthesised a conventional microporous HZSM-5 catalyst to compare its performance with HZSM-5 (30)-34. When these catalysts were used for cracking of LDPE, the hierarchical catalyst HZSM-5(30)-34 outperformed the conventional catalyst. The enhanced performance of hierarchical zeolites was attributed to the better accessibility of Brønsted acid sites, which lowered the cracking temperature of LDPE.
The fine balance of mesoporous content, which provides large surface area and accessibility, and acidic contents, which are responsible for catalytic reactivity, determines the performance of composite catalysts. De Souza et al. synthesised composite catalysts of ZSM-35/MCM-41 with different compositions of ZSM-35 and MCM-41 and investigated their performance for the pyrolysis of PVC.223 A catalyst with a 3:1 ratio of ZSM-35 to MCM-41 exhibited the highest conversion rate (91%) and significantly reduced the degradation temperature of PVC. This was attributed to the high surface area provided by MCM-41 and strong acidity of ZSM-35. Similarly, Li et al. utilised an HZSM-5/MCM-41 catalyst for co-pyrolysis of plastic waste films and rice husk, investigating the effects of temperature and synergistic interaction between the two feedstocks.224 The micro–mesoporous composite catalyst demonstrated exceptional catalytic performance, achieving a maximum hydrocarbon yield of 71.1% at 600 °C using a feedstock ratio of 1:1.5 (rice husk to waste plastic film).
5.5.1.6 Activated carbon catalysts. In recent years, activated carbon has emerged as a cost-effective catalyst for converting waste plastics into hydrocarbons suitable for jet fuel. Zhang et al. demonstrated the effectiveness of an activated carbon (CAC2) catalyst for pyrolysis of LDPE at 500 °C, obtaining oil in 70.4 wt% yield, with 93% of hydrocarbons falling within the jet fuel range (C8–C16).225 The jet fuel range hydrocarbons were comprised of 71.8% alkanes and 28.2% aromatics (Fig. 14). Typically, 60–80% of components in aviation jet fuel are C8–C16 alkanes, and their selectivity is influenced by reaction temperature, with increase in reaction temperature resulting in a higher fraction of these alkanes in liquid products.
 | ||
| Fig. 14 Product yields and distribution of liquid components from LDPE waste pyrolysis, over CAC2 and CAC5 catalysts. | ||
In a different approach to producing hydrogen and liquid oil rich in jet fuel components from LDPE, Huo et al. utilised corncob derived activated carbon (BAC) and MgO as a catalyst for pyrolysis at 500 °C.226 Using a 1:1 ratio of BAC and MgO, the process yielded 72.0% liquid and 23.9% gas products. Remarkably, 98.7% of the liquid output comprised hydrocarbons in the jet fuel range, with 65.3% being C8–C16 hydrocarbons and 33.4% composed of aromatic hydrocarbons larger than C16. Moreover, C17–C23 alkanes had a selectivity of 1.3% without any additional products. In another study, biomass-derived activated carbon was used for pyrolysis of LDPE at 500 °C for obtaining jet fuel range hydrocarbons.227 The authors reported a substantial liquid yield of 75.3 wt%, with 93.1% of hydrocarbons within the jet fuel range, consisting primarily of C8–C16 alkanes, aromatics below C16, and C17–C23 alkanes. Furthermore, only low coke yield (1.3 wt%) was produced by the catalyst.
Bifunctional activated carbon catalysts have been used for plastic waste pyrolysis to produce jet fuel oil.228 In one study, pyrolysing plastic at 500 °C with 10% Fe/AC catalyst yielded a maximum oil of 53.67%. Increasing the temperature from 450 °C to 600 °C further enhanced the aromatic selectivity due to dehydroxylation of phenol. The Fe/AC catalyst outperformed standard activated carbon (AC) by producing more aromatics, especially mono aromatics, attributed to the formation of new acid sites. Ali et al. employed graphite as the catalyst for pyrolysing waste plastics to produce jet fuel.229 A strong correlation was observed between the temperature and product composition when waste plastic was heated at 350–450 °C in an inert atmosphere. At higher pyrolysis temperatures, the composition of pyrolysis oil shifted from alkanes to diesel range alkanes, eventually producing jet fuel range alkanes. Duan et al. used chestnut derived activated carbon as the catalyst for pyrolyzing waste LDPE.230 The carbonisation temperature was found to drastically influence the acidity and surface area of the catalyst. The resultant oil contained 100% components in the jet fuel range. The optimum aromatic selectivity of 95.88% was attained at carbonization and catalytic temperatures of 850 °C and 550 °C, respectively. Activated carbon catalysts have also been used for pyrolysis of polystyrene (PS) in a microwave reactor.231 Pyrolysis was performed at different temperatures (340–580 °C) and microwave power levels (450–850 W) to evaluate their influence on the yield and components of oils for aviation fuel production. An impressive oil yield of 98.78 wt% was achieved at 460 °C with 650 W microwave power. The generated oil was comprised of monocyclic and polycyclic aromatic hydrocarbons, along with cycloalkenes (C8–C16).
Some researchers have explored biochar as the catalyst for pyrolysis of plastics, although its catalytic reactivity and oil yield have not been very promising. Li et al. employed biochar, synthesised from wood chips, to catalyse the pyrolysis of HDPE and LDPE, yielding 5.8 wt% of liquid and 20 wt% gas at 500 °C.232 These findings indicate that biochar catalyst is less active, producing lower yields of products from plastic conversion when compared to similar studies under the same reaction conditions. Wang et al. synthesised a biochar catalyst from corn stover and Douglas fir, which were then used for pyrolysis of LDPE and real plastic waste.233 The corn stover-derived biochar gave liquid and gas products in 40 wt% and 60 wt% yields, respectively. The liquid product contained C8–C16 aliphatic (60%), mono-aromatic (20%), and C17–C23 aliphatic hydrocarbons (20%), while the gas was comprised of 60–80 vol% H2. Biochar derived from Douglas fir favoured CH4 in contrast to corn stover derived biochar that generated more H2 gas.
5.5.1.7 Other types of catalysts. Researchers have developed various other catalysts for catalytic cracking of plastic waste to enhance pyrolysis performance. These include clay (calcium bentonite, halloysite clay, Fe, Ti, Zr-pillared, etc.),109 BaCO3,234 bimetallic catalysts,109 Al–Zn composite,235 Al-MCM-41,213 Fe and Mo, Fe and Ni catalysts,236 kaolin,237 and CuCO3.238 In 2019, Panda and Singh used kaolin and a SiO2/Al2O3 catalyst at temperatures ranging from 400 to 500 °C to produce liquid fuel oil.181 The SiO2/Al2O3 catalyst demonstrated superior performance to the kaolin catalyst in this experiment.181
Montmorillonite, hectorite, and beidellite have been used for polymer cracking for decades due to their high selectivity for liquid products and low coke deposition formation on catalysts, resulting from their trivial acidity. Recently, pillared clays (PILCs) have gained attention as catalysts. These feature interlayered structures that allow the incorporation of various metal oxides to enhance their textural properties and acidity.239 While these catalysts have demonstrated outstanding performance in the pyrolysis of single polymers such as HDPE and PP,240 their efficiency was later optimised for more complex feedstocks containing multiple types of plastics. Li et al. investigated the application of various metal-loaded pillared clays, including Fe-PILC, Ti-PILC, and Zr-PILC, to catalyse the pyrolysis of mixed plastic waste, rather than single polymer applications.241 For comparison, authors also used Al-PILC (pillared clay). The Fe-PILC catalyst produced an oil product in 79.3% yield, with a high diesel fraction (80.5%) and a gas product rich in H2 (47.7 vol%), owing to its optimal acidity and nano-architecture. In another study, they developed Co/Ni pillared montmorillonites for catalytic pyrolysis of post-consumer film waste, and reported 80.2% liquid yield, with significant selectivity for C10–C13 range hydrocarbons (43.5%), and 42.0 vol% of H2.242 Wang et al. used Co and Ni supported vermiculite as a catalyst support for the pyrolysis of plastic waste and achieved decent selectivity for C3, the main gaseous hydrocarbon of the pyrolysis product, and substantial diesel fraction in the oil product.243 The improved reactivity and selectivity for condensable products were attributed to the pillared structure, even distribution of acidic sites, and high surface area. Iron-pillared bentonite clays, due to their mesoporosity, high surface area, and thermal stability, have shown superior performance over HZSM-5 in the co-pyrolysis of HDPE and petroleum residue, yielding 63% more light linear hydrocarbons (C10–C23) with a composition similar to conventional diesel.244
A tandem pyrolysis–catalytic upgradation methodology was used by Li et al. to convert plastic waste to kerosene-range fuels by using transition-metal modified Si-pillared vermiculite (SPV) catalysts, specifically incorporating Co, Fe, and Ni.245 Among these, Co–Fe/SPV demonstrated excellent thermal stability and good catalytic efficiency for selective production of kerosene-range oils during tandem pyrolysis and catalytic upgradation process. The product distribution from post-consumer plastic using various SPV catalysts, as illustrated in Fig. 15, showed that the silicon pillared vermiculite significantly enhanced the thermal stability of catalysts. Meanwhile, the transition metals helped in fine-tuning the acidity and porosity of the catalysts. In particular, the Co–Fe/Si-pillared vermiculite gave the highest yield of oil product (75.7 wt%), with excellent selectivity towards valuable hydrocarbons, 7.5% alkanes and 27.8% aromatics.
 | ||
| Fig. 15 Product distribution in the pyrolysis of grocery bags with various metal modified Si-pillared vermiculite catalysts. | ||
Mesoporous MCM-22 has garnered significant attention in acid catalysed reactions such as benzene alkylation, glycerol dehydration, and cracking.246 However, there are few studies on its use as a catalyst for polymer pyrolysis, although catalytic systems for polymer cracking with high product selectivity for liquid products over mesoporous silica have been established.247 However, reports showed that similar processes still use mesoporous catalysts at temperatures of 400–500 °C.248 Other mesoporous silicas with pore sizes ranging from 20 to 300 Å, larger surface areas, and large pore diameters have also been investigated. Particularly, SBA-15 has been extensively studied in various acid catalysed chemical reactions. Furthermore, its acidity can be tuned by adding Lewis acids into the framework such as Al ions.249 The cracking of polypropylene with aluminium exchanged MCM-41 and SBA-15 catalysts resulted in further degradation of heavier molecules to lower hydrocarbons (<C14) due to the presence of these catalysts.247,250 In recent studies, Xu et al. used MCM-41 for the pyrolysis of LDPE under N2 and CO2 environments, and obtained olefins with selectivity up to 44.66% under a nitrogen atmosphere.251 The olefin selectivity was further increased to 60.39% under the CO2 atmosphere.251 Furthermore, there was 14.66% increase of C5–C12 olefin selectivity under the CO2 atmosphere by increasing the catalyst ratio to 5:4 (LDPE:MCM-41). Zn/SBA-15 has been identified as an efficient catalyst to break down HDPE pyrolysis wax into small chain olefins.252 The authors also investigated other SBA-15 catalysts impregnated with metals such as Co, Fe, Mn, Cs, and Cu but the Zn/SBA-15 catalyst exhibited much higher activity compared to these catalysts or pure SBA-15. The tandem catalysis was further applied for catalytic cracking of different polyolefin wastes such as HDPE, LDPE, and PP, leading to the production of high-quality naphtha. The process achieved C5–C12 hydrocarbon selectivity ranging from 50% to 71%. Moreover, the use of a real plastic mixture produced high quality naphtha in 60.39% yield, with 57.16% selectivity for C5–C12. The authors concluded that the reaction proceeds through two catalytic zones, the cracking occurs in the first zone via free radicals, whereas hydrogenation takes place in the second zone.252 The probable mechanism of HDPE pyrolysis through tandem catalysis is illustrated in Fig. 16. Other mesoporous catalysts, such as Fe and Mo containing catalysts, have demonstrated the ability to enhance hydrogen contents in the gas phase during steam cracking of heavy oil.253 Moreover, Fe catalysts have shown to increase the H:C ratio in liquid products while reducing the viscosity of the resulting liquid product. Fe coated SBA-15 (Fe-SBA-15) has been reported to exhibit good catalytic activity for PP cracking, with significant increase in the liquid yield from 61% to 73–77% along with reduction in gas yield from 31% to 24–21%, compared to thermal cracking.254
 | ||
| Fig. 16 Reaction mechanism for catalytic reforming of HDPE pyrolysis volatiles by a tandem catalysis. Reproduced with permission from ref. 252. Copyright 2021, Elsevier. | ||
Most of the reported results are derived from experiments using pure plastics, such as PE and PP, without considering the additives and contaminants commonly found in real plastic waste. However, when real plastic wastes have been used as feedstocks, differences in results are observed. For instance, a real plastic film waste consisting of LDPE and EVA polymers from Almeria (Spain) greenhouse was used as feedstock in the catalytic cracking over different acidic solids including nanocrystalline ZSM-5, aluminium/MCM-41, and aluminium/SBA-15.255 Among these catalysts, only nanocrystalline ZSM-5 zeolite maintained a high activity and selectivity for C1–C5 hydrocarbons, achieving 60 wt%. In contrast, the mesoporous solids (aluminium/SBA-15 and aluminium/MCM-41) showed outstanding activity for cracking of pure LDPE, but were found inactive for real plastic waste, possibly due to their mild acidic properties. Ma et al. used Fe and Ni impregnated MCM-41 and ZSM-5 catalysts for pyrolysis of polystyrene, resulting in oil yields of 63.2 wt% and 61.2 wt%, respectively. However, higher yields were achieved with Fe-MCM-41 (65.9 wt%) and Ni-MCM-41 (65.3 wt%), with these oils containing a greater concentration of single-ring aromatics compared to the ZSM catalysts.256Table 2 contains additional examples of catalysts used for processing plastics, along with process conditions and product yields.
| Catalyst | Feedstock | Reaction conditions | Product and conversion | Reference |
|---|---|---|---|---|
| a Cat – catalyst; MPW – municipal plastic waste; ILs – ionic liquids; DEP – deep eutectic solvent. | ||||
| Co/SPV | Polyethylene grocery bags | Two-stage fixed-bed reactor Cat to feedstock ratio = 1:2.5 300 °C (10 min), 490 °C (30 min) |
74.7% – liquid; 18.33% – gas; 6.97% – char and coke | 245 |
| Co–Ni/SPV | 71.9% – liquid; 22.3% – gas; 6.8% – char and coke | |||
| Co–Fe/SPV | 75.7% – liquid; 18.62% – gas; 5.68% – char and coke | |||
| Fe–Ni/SPV | 71.0% – liquid; 21.8% – gas; 7.2% – char and coke | |||
| CuBr–NiO | LDPE | Cat = 0.1 wt% 700 °C | 56.5% carbon nanomaterials (CNMs) | 257 |
| Al-MCM-41 | PE/PP-mixture | Cat to feedstock ratio = 0.03:1 N2 atmosphere, 2 stage process: T1 = 300 °C (5 min), T2 = 500 °C (30 min) |
64.9% – liquid and wax; 34.8% – gas; 0.3% – solid residue | 258 |
| ZSM | 41.5% – liquid and wax; 57.7% – gas; 0.8% – solid residue | |||
| Highly uniform nanocrystalline ZSM-5 | 61.0% – liquid and wax; 38.9% – gas; 0.1% – solid residue | |||
| Al-SBA-15 | 89.1% – liquid and wax; 10.9% – gas | |||
| Kanemite-derived folded silica (KFS-16B) | 64.9% – liquid and wax; 34.8% – gas; 0.3% – solid residue | |||
| β-Zeolite | 45.9% – liquid and wax; 54.0% – gas; 0.1% – solid residue | |||
| Coconut shell activated carbon | HDPE | Cat to feedstock ratio = 1:1 microwave heating: 400 °C, 45 min |
99.22% – liquid and gas (methane gas is 10.2%; 54.09% – cycloparaffin and n-paraffin) | 259 |
| 0.78% – residue | ||||
| Activated carbon | PS, PP | Cat to feedstock ratio = 1:5 microwave pyrolysis: 900 W, 20 min |
84.3% – liquid; 15.7% – gas | 260 |
| Mordenite | Polyolefin mix + PS | Cat to feedstock ratio = 1:5 fluidized bed bath reactor; 360 °C, 15 min |
36.8% – liquid; 57.7% – gas; 5.5% – residue coke | 261 |
| MCM-41 | 66.4% – liquid; 26.5% – gas; 7.1% – residue coke | |||
| HUSY | 56.6% – liquid; 34.6% – gas; 8.8% – residue coke | |||
| HZCM-5 | 30.3% – liquid; 65.8% – gas; 3.6% – residue coke | |||
| HZSM-5 | PP, HDPE, LDPE | Cat to feedstock ratio = 1:5 ultra-fast heating, the heating rate is estimated to be 50–85°C min−1; 500–700 °C, 10 min |
24.0–27.2% – liquid; 70.6–73.9% – gas; 0% – wax | 204 |
| FCC | 0–35.9% – liquid; 62.4–75.2% – gas; 0–25.4% – wax | |||
| FCC used | HDPE | Cat to feedstock ratio = 6:1 fluidized bed reactor; 450 °C |
34.1% – liquid; 41.6% – gas; 24.3% – residue coke | 262 |
| FCC | PE | Fluidized bed reactor; PE = 1 kg h−1; Cat = 1–2 kg h−1, 450–515 °C, 10–12 s | 37.1–38.3% – liquid; 48.3–51.8% – gas; 9.2–14.1% – residue coke; 0.5–0.7% – char | 263 |
| Si–Al | PE | Catalyst = 0.1 wt%, fixed bed reactor; 430 °C, 30 min | 61.2% – liquid; 37.3% – gas; 1.5% – residue coke | 264 |
| MgO | LDPE | Cat to feedstock ratio = 1:5 microwave-assisted pyrolysis: ex situ; T1 = 500 °C, T2 = 450 °C, 20 min |
52.8–67.1% – liquid; 46.3–30.3% – gas; 1.5% – residue coke (36% – aromatic compounds C5–C12; 38% – alkenes C5–C12; 11% – alkanes) | 265 |
| Ni/CuO | PS | Catalyst = 5 wt%, 390 °C, 10–90 min | 93.5% – liquid (21.29% olefins, 68.9% aromatics, 2.2% oxygenates) | 266 |
| Kaolin clay | PP | Catalyst = 10 wt%, 600 °C, 1 hour | 22% – liquid (25–35% alkenes, 2–7% n-alkanes, 30–40% aromatics, 5–10% cycloparaffins) | 237 |
| Fe/Al oxide-pillared clay | PE | Cat to PE ratio = 1:1, 300 °C, 3 hours |
60% – liquid (100% – aliphatic) | 240 |
| Ni/ZSM-5 | MPW (84% LDPE, HDPE, 15% PP) | Cat to feedstock ratio = 1:3.3 pyrolysis: 500 °C; gasification: 800 °C, 1 hour |
7.6–11.8% – liquid; 73.8–81.4% – gas; 10.4–15.8% – residue coke | 267 |
| Ce/Ni/ZSM-5 | ||||
| Ce and La had very little effect on the product yields, but significantly affected the product distribution | ||||
| Ni/La/ZSM-5 | ||||
| Ce/La/ZSM-5 | ||||
| — | HDPE | Batch reactor; 390–430 °C, P = 2.4 bar; N2 atmosphere, 1–2.5 hours | 72.7–85% – liquid | |
| — | PP | 650 °C 30 min | 67.5–98.5% – liquid | |
| ZSM | Polyethylene film waste | Catalyst = 10 wt% batch reactor; 500 °C, 2 hours | 56.12% – liquid; 39.64% – gas; 10.27% – char | 268 |
| FCC | 23.92% – liquid; 61.65% – gas; 14.42% – char | |||
| HUSY-5.1 | 37.33% – liquid; 57.25% – gas; 5.42% – char | |||
| HUSY-5.1-Ru (1%) | 29.06% – liquid; 66.99% – gas; 3.95% – char | |||
| HUSY-5.1-Ni (1%) | 24.98% – liquid; 68.72% – gas; 6.29% – char | |||
| HUSY-30 | 39.06% – liquid; 53.08% – gas; 7.87% – char | |||
| SiO2 | 57.70.85% – liquid; 36.53% – gas; 5.77% – char | |||
| Sulphated ZrO2 | PET | Catalyst = 10 wt% cylindrical horizontal furnace: 450–600 °C, 20 s | 48.0% – liquid; 35.0% – gas; 17.0% – solid residue (27–32% – yield of benzoic acid) | 269 |
| Kaolin | PP waste | Cat to feedstock ratio = 1:2 500 °C, 30 min |
80.75% – liquid; 17.55% – gas; 1.7% – char | 270 |
| Fe/Al2O3 | PP | Cat to feedstock ratio = 1:2 T1 = 500 °C, T2 = 800 °C, 30 min |
18.1% – liquid (80.6 wt% C8–C16); 41.7% – gas (58.7 vol% H2, 32.4 vol% CH4); 30.2% – solid | 271 |
| HDPE | 20% – liquid (81.4 wt% C8–C16); 43.4% – gas; 36.9% – solid | |||
| LDPE | 20.7% – liquid (80.2 wt% C8–C16); 41.5% – gas; 35.9% – solid | |||
| Ni/ZSM-5 | PE | Cat to feedstock ratio = 1:2 T1 = 500 °C, T2 = 800 °C, 40 min |
22.1% – liquid; 47.2% – gas (19.1 mmol per g H2); 30.7% – solid (66 wt% filamentous carbon) | 272 |
| Ni/ZSM-5-M | 6.4% – liquid; 48.7% – gas (26.3 mmol per g H2); 44.9% – solid (66 wt% filamentous carbon) | |||
Fly ash, with its rich composition of Al2O3, SiO2, and Fe2O3 (typically 80–90 wt%) has merged as an effective catalyst for pyrolysis applications.273 Gaurh et al. demonstrated the catalytic potential of fly ash in the pyrolysis of PE at 700 °C and obtained oil containing twice the amount of valuable aromatic contents (22.12%) compared to the noncatalytic process.273 When this catalyst was used for pyrolysis of LDPE at 500 °C, the produced oil was lighter in colour compared to the oil obtained via thermal pyrolysis.274 Incinerated fly ash (IFA) has also been used as a catalyst for the pyrolysis of plastic waste such as polyolefin detergent bottles and plastic bags, leading to improved conversion efficiency and liquid yields.275 Particularly, the conversion of virgin HDPE pellets using IFA was significantly improved from 46.7% to 92.8%, whereas the liquid yield increased to 81.6% from 35.1%. Recently, the pyrolysis of waste LDPE plastic was investigated using a fuel oil fly ash as a catalyst across various different temperatures (450–600 °C) and catalyst loadings (5–20%).276 The resulting oil contained both aliphatic and aromatic hydrocarbons, and has the potential for use in transportation fuel and industrial applications.
Valášková et al. investigated the use of vermiculites from Brazil and Palabora for depolymerisation of polystyrene in order to evaluate their catalytic effects.277 Thermal depolymerisation of polystyrene produced styrene monomer (SM) in 54.5 mass% yield, and volatile oligomers in 32.1 mass%. However, using VerS and VerP as catalysts changed the product distribution, with SM yields of 50.7 mass% and 53.6 mass%, respectively, and oligomer yields of 37 mass% and 33.3 mass%, respectively. This shift in product distribution was attributed to the formation and deposition of carbon on catalysts. The variable yields were likely due to the intrinsic properties of the natural vermiculites.
5.5.1.8 Supercritical water pyrolysis. Sub-critical (sub-CW) and supercritical water (SCW) are used in oil cracking and plastic processing. This method has also been used in plastic processing due to their dual role as solvents and catalysts. This method is advantageous because the supercritical fluid mitigates coke formation and reduces the possibility of subsequent condensation.278
In a study by Bai et al., co-pyrolysis of polyethylene and residual oil was conducted in sub-CW and SCW at 420 °C, using water density in the range of 0.10–0.30 g cm−3.279 As the water density increases, the phase structure of the system changes from a liquid to a gaseous/solid state, eventually stabilising into a two-phase liquid/solid state. The main product of pyrolysis of polyethylene under these conditions is paraffin with a high hydrogen content. When the water density is higher, the favourable two-phase structure of the liquid/solid component enhances the interaction among aromatic radicals formed from pyrolysis of PE, which produced residual oil and paraffins. This interaction provides a link between the pyrolysis chains of oil and polymer. Subsequently, aromatic radicals can undergo dealkylation, thus the formation of components contributing to coke formation is effectively suppressed. In another investigation, the degradation of PE in supercritical water had several advantages over conventional thermal cracking, including shorter reaction times, enhanced oil yields, and reduced coke formation.280 The process achieved an oil yield exceeding 90 wt% with a water-to-PE ratio of 6 at 460 °C and reaction time of 1 min. The water-to-PE ratio significantly influenced the yield as well as the composition of oil products.
Fang et al. reported that the “polyethylene + water” system remains heterogeneous under various conditions.281 However, the reaction products are rapidly distributed in the supercritical phase, along with a substantial change in the volume of polyethylene. Hence, it is possible to control the phase behaviour of the “PE + SCW” system and then distribute the PE conversion product to SCW. Therefore, the SCW can influence the reaction rate, equilibrium state, and main reaction pathway, due to significant changes in the properties of the solvent. Zhao et al. explored the liquification of the LDPE and PP mixture into oil using supercritical water (scH2O), and achieved a high conversion rate of 99.75% and oil yield of 90.7 wt%, without the use of a catalyst or hydrogen.282 The supercritical water co-liquefaction of this polymer mixture improved the oil yield compared to using a single polymer. Production of cyclic hydrocarbons was increased, whereas generation of paraffins was suppressed. TGA data revealed that the process favoured the production of diesel and lubricant oils, with a reduction in gasoline fractions.
Supercritical water (SCW) processes effectively address issues related to low thermal conductivity of polymers and can tolerate common additives such as stabilisers. SCW processes have been demonstrated to produce high-value products after careful optimisation and operation.283 Depending on the polymer's chemical structure, monomers can be recovered for producing new plastics, make valuable chemical building blocks, or produce fuels. Increasing the temperature and residence time in supercritical water (SCW) processes for plastic can enhance the reaction rate, conversion yield, as well as the production of gas products.283,284 Conversely, increasing the pressure typically has a minimal impact on reaction rates. In these processes, water serves as both an oxidising and solubilising agent, dissolving constituents released during the breakdown of polymer chains. However, increasing the plastic to water ratio decreases the dispersion of macromolecular free radical fragments, leading to reduced reaction rates. In general, supercritical fluid technology is an encouraging process for liquification of mixed plastic waste into oils with a high conversion rate.
Pyrolysis of plastic waste typically produces a mixture of liquid hydrocarbons, including gasoline range fractions (C6–C12), kerosene (C10–C16) and diesel range fractions (C13–C18) and waxes (C19+). These liquid hydrocarbons consist of various functional groups including aromatics, naphthenes, olefins, and paraffins.191
Among pyrolysis products, the most demanding product is gasoline due to its extensive applications and significant economic value. As a result, much of the research in this field is concentrated on increasing the yield of gasoline-range oil with high aromatic content by changing the various reaction parameters, including the type of catalyst, temperature, catalyst to plastic ratio, metal loading on the catalyst, and reaction time.155 Low-carbon naphtha has also emerged as a valuable product of pyrolysis, particularly because it can potentially be used as feedstock to produce new plastics. Researchers are increasingly focusing on optimising the conditions for generating low-carbon naphtha to promote sustainable plastic production and support circular economy initiatives.285,286
5.5.2.1 Pressure. The pyrolysis of plastic waste has been typically performed at ambient pressure, as higher pressure with higher reaction temperature has been found to increase gas yield in the final product.121 Therefore, the influence of pressure on pyrolysis remains unexplored and requires further detailed investigation. However, some researchers have conducted pyrolysis at low pressure, either in an inert diluent or under vacuum, which have been found to favour the formation of primary products. In contrast, high-pressure conditions often lead to more complex liquid fractions.287 As pressure increases, the boiling point of pyrolytic products rises, causing heavier hydrocarbons to undergo further pyrolysis instead of vaporising at the operating temperature.287 Lopez et al. investigated the pyrolysis of waste tires under vacuum conditions (25 kPa and 50 kPa) with a continuous feeding system at 500 °C, achieving excellent yield of diesel like hydrocarbons, similar to those produced under elevated pressure.288 The use of vacuum pressure increased the liquid product yield compared to atmospheric pressure. Moreover, the BET surface areas of the formed carbon blacks improved due to reduced pore blockage, positively influencing the porous structure. The introduction of vacuum also helped to devolatilise and diffuse volatiles within the particles. Moreover, the isoprene yield was increased by performing pyrolysis under vacuum but the yield of limonene was lower. On the other hand, increasing pressure led to greater formation of gaseous products and a high yield of low molecular weight liquid hydrocarbons.289
5.5.2.2 Reaction time. Residence time, which refers to the average time a particle resides in a reactor during degradation, is an important factor in determining the product distribution. A prolonged residence time generally enhances the conversion of primary pyrolysis products into lower molecular weight hydrocarbons. However, beyond certain temperature limit, further changes in residence time have a minimal impact on product distributions. An investigation on pyrolysis of HDPE concluded that longer residence times at 600 °C resulted in higher yield of liquid products, with 2.6 wt% aromatics obtained at 5.6 seconds compared to 0.8 wt% at 1.4 seconds.290 However, when the temperature exceeded 685 °C, residence time had little impact on oil and liquid yields. Kulas et al. recently evaluated the influence of residence time on plastic feed (HDPE) at 600 °C in a waste plastic pyrolysis pilot plant.291 Their findings showed that extending the residence time from 1 to 4.5 seconds significantly changed the product distribution, with 11 wt% increase in light oil fractions (C5–C10), 9 wt% reduction in wax (C20–C30) formation, and a 4 wt% rise in gaseous products.
5.5.2.3 Temperature. Temperature plays a pivotal role in regulating and optimising the catalytic pyrolysis of waste plastics to achieve the desired yield and quality of oil and gas products.292 The thermal degradation behaviour of plastics can be measured using a thermal gravimetric analyser (TGA). TGA analysis of PET indicates that its degradation occurs at temperatures ranging from 350 °C to 500 °C.293 In contrast, the complete thermal degradation of HDPE takes place between 404 °C and 539 °C.294 Moreover, the thermal degradation rate also depends on the heating rate, with the maximum degradation temperatures of HDPE and LDPE reported at 465 °C and 460 °C, respectively.295 Bagri et al. investigated the catalytic pyrolysis of LDPE using activated carbon and silica gel, finding the optimal degradation temperature as 450 °C.119 In another study, the effect of temperature on the degradation of LDPE was evaluated using activated carbon as a catalyst, identifying 500 °C as the optimum temperature for obtaining maximum oil yield (71.5 wt%).225 Increase in temperature from 500 °C to 571 °C increased gas fractions but oil yield was reduced to 54.0 wt%. However, decrease in temperature from 450 °C to 430 °C resulted in the formation of waxes.225
The thermal degradation temperature range for polypropylene (PP) is between 300 °C and 500 °C.296 Comparative studies on the degradation of HDPE and PP degradation have shown that PP undergoes thermal degradation at a lower temperature than HDPE, with a maximum degradation temperature of 447 °C for PP compared to 467 °C for HDPE.294 The thermal degradation temperature for polystyrene (PS) ranges from 350 °C to 500 °C. The maximum oil yield (97.0 wt%) from catalytic pyrolysis of PS was achieved at 425 °C using a batch reactor.297 Further increase in temperature to 581 °C resulted in the reduction of oil yield whereas the gas yield was increased.91 Dement'ev et al. performed the pyrolysis of PS waste at 450–500 °C in hydrocarbon media over zeolite catalysts.298 At an optimum temperature of 500 °C, the conversion was almost 100% yielding ethylbenzene, benzene, and toluene at 80%, 12.7%, and 6.3%, respectively. Polyvinyl chloride (PVC) shows a different thermal degradation behaviour. The initial weight loss involving dehydrochlorination occurs at around 200–360 °C, and weight loss of dechlorinated-PVC occurs at around 360–550 °C.299 The weight loss is insignificant by raising the temperature up to 800 °C. Hence, the thermal degradation range for PVC is approximately 200–520 °C. The optimal temperature for maximising liquid oil yield from PVC was determined to be 550 °C, however, increasing the temperature to 600 °C led to reduced oil yield.300
Onwudili et al. investigated the effect of catalyst bed temperature in a two stage fixed bed reactor on product distribution during pyrolysis of mixed plastic waste.207 The study, conducted at temperatures of 500 °C and 600 °C, revealed that a higher catalyst bed temperature of 600 °C significantly increased the gas yield, particularly in the hydrocarbon range of C2–C4. Moreover, zeolite Y gave the highest yield of hydrogen gas compared to other catalysts like HZSM-5 and FCC. The temperature increase also led to higher content of low molecular weight aromatics in the oil product, indicating the quality improvement of gasoline type fuel.207 Similarly, Yao et al. reported that increasing the temperature during pyrolysis affects the composition of gas yield, specifically leading to higher concentrations of CH4 and H2 gases with reduction in C2+ gases. The study found that the highest levels of H2 (25.14 mmol per gpp) and CH4 (33.12 vol%) were achieved at a temperature of 800 °C.236
In another investigation, the effect of variation in catalysis temperature (in the range of 350–500 °C) was evaluated on product distribution using HY catalyst with LDPE in 1:10 ratio.155 As the catalysis temperature increased from 350 °C to 450 °C, the oil yield gradually decreased from 66.11 wt% to 56.54 wt%, decreasing further to 38.16 wt% at 500 °C (Fig. 17A). In contrast, the gas yield rose significantly, nearly doubling from 32.21 wt% as the temperature increased to 500 °C. Huo et al. investigated the temperature's effect on yield and product distribution during LDPE pyrolysis using activated carbon with MgO as the catalyst.226 The liquid product yield was found to increase from 60.9 wt% at 450 °C to 72.0 wt% at 500 °C, but further temperature elevation to 600 °C reduced the liquid yield to 68.5 wt% due to secondary thermal cracking.
 | ||
| Fig. 17 (A) Effects of catalysis temperature on product yield and distribution during LDPE pyrolysis. (B) Composition of liquid fuel from HDPE pyrolysis with different modified kaolin catalysts. (C) Distribution of aromatics in the liquid fraction of PE and PP catalytic pyrolysis. (D) Liquid, gas, solid and coke yield with different catalysts during pyrolysis of mixed plastics. Reproduced with permission from ref. 155 (A), ref. 301 (B), ref. 302 (C), ref. 303 (D). Copyrights 2019 Elsevier, 2023 Elsevier, 2018 Elsevier, 2022 Elsevier, respectively. | ||
5.5.2.4 Catalyst type. The type of catalyst significantly influences the distribution of products in pyrolysis. Ajibola et al. employed a zeolite Y catalyst, synthesised from kaolin deposits, for pyrolysis of LDPE waste. The resulting liquid fuel primarily consisted of alkenes and aromatics within the hydrocarbon range of C8–C29, with 56 wt% of gasoline fraction (C6–C12), 26 wt% of diesel and kerosene fraction (C13–C18), 10 wt% as fuel oil (C19–C23), and 6 wt% as residual fuel (>C24).191 Akubo et al. also used zeolite Y catalyst for pyrolysis of HDPE and achieved oil formation with 80 wt% aromatic content, in contrast to the oil from non-catalytic pyrolysis, which was composed entirely of aliphatic components.215 Moreover, loading metals such as Co, Ni, Mo, Ga, Fe, and Ru onto Y-zeolite reduced the overall oil yield but significantly increased the aromatic contents in the resulting product. For example, 1 wt% Ga-Y-zeolite, 1 wt% Fe-Y-zeolite, and 1 wt% Ru-Y-zeolite produced oils with aromatic contents of 93.2%, 93.5%, and 94.4%, respectively. Increasing the metal loading from 1% to 5% Y-zeolite didn't lead to further increase in aromatic hydrocarbon content. Aromatic hydrocarbons produced with metal incorporated Y-zeolite consisted of 97–99% of mono and bicyclic aromatic compounds including ethylbenzene, toluene, xylene, naphthalene, and alkylated naphthalene. Similarly, Onwudili et al. also used acidic Y-zeolite for pyrolysis of waste plastic and reported the highest yields of aromatic compounds in the oil product at 500 °C and 600 °C.207 Y-zeolite with a Si/Al ratio of 80
:1 produced a significantly higher oil yield of 68 wt% compared to Y-zeolite with a Si/Al ratio of 50:1, which yielded only 53 wt%. Moreover, 90% of aromatic components were comprised of benzene and toluene using the strong acidic Y-zeolite catalyst. This indicates that valuable aromatic hydrocarbons can be efficiently obtained in high yields from mixed plastic waste by using acidic Y-zeolite catalyst.
Zeolitic catalysts such as HUSY and HZSM-5 are highly acidic catalysts and have been extensively tested in catalytic pyrolysis of polymers, showing very promising results. Colantonio et al. studied pyrolysis of plastic packaging waste using HUSY and HZSM-5 catalysts, achieving an oil yield of 74.1 wt% with 22.5 wt% aromatics in the hydrocarbon range of C5–C9.202 The aromatic fraction produced with the HUSY catalyst primarily consisted of monoaromatics (benzene, toluene, and ethyl benzene). Furthermore, pyrolysis using HZSM-5 catalyst resulted in a high gas yield and significantly reduced the char formation. No obvious difference in gas composition was observed between HUSY and HZSM-5 pyrolysis reactions. However, HZSM-5 tends to favour the formation of gas due to its small pores and strong acidic sites, these properties also favour the formation of styrene in the resulting oil.
When natural kaolin was used as a catalyst for pyrolysis of polypropylene (PP), it produced condensate oil containing 90–97% of hydrocarbons in the C6–C20 range, which are key components of gasoline, diesel, and naphtha.237 The acid modification of kaolin with HCl significantly reduced the condensate oil yield and increased the C6–C11 component of hydrocarbons. This change is attributed to the stronger acid sites of HCl-modified kaolin that promoted the further cracking of diesel components into gasoline range hydrocarbons. Furthermore, acid modification increased the aromatic content of the condensate oil through aromatisation and Diels–Alder reaction of alkanes and alkenes. As a result, there was a reduction in straight chain alkenes and cyclo-paraffins, in contrast to natural kaolin, which produced higher aliphatics, straight alkenes, and cycloparaffins, and less aromatics in the condensate oil.237 Similar studies were performed by Mohan et al. for the pyrolysis of waste HDPE using acid and alkaline activated kaolin.301 The resulting oil from pyrolysis with acid and alkaline modified kaolin clay catalysts was rich in hydrocarbons, particularly in the range of C10–C25, predominantly consisting of alkanes and olefins. Among the various modified kaolin catalysts, HNO3-treated kaolin (KC(HNO3)) gave the highest oil yield (∼79%) compared to KC(HCl), KC(CH3COOH), KC(H3PO4) and KC(NaOH). KC(HNO3) resulted in oil yield with the highest percentage of alkanes (74.7%) compared to oils with 56% alkanes obtained by using KC(H3PO4) and KC(NaOH) catalysts (Fig. 17B).301
Pyrolysis of LDPE using activated carbon as the catalyst produced a fuel that, with the addition of cyclohexane, was found suitable for direct use as jet fuel. In addition, the liquid oil produced using different activated carbon catalysts was found to contain 97–100% of diesel range hydrocarbons.225 Dai et al. investigated the pyrolysis of polyolefins towards low-carbon naphtha, which holds potential as a feedstock for producing new plastics.285 They compared the effect of Al2O3 pillared montmorillonite clay and ZSM-5 catalysts on the selectivity of product oil. The liquid obtained with Al2O3 pillared montmorillonite clay contained C5–C12 alkanes in 60.3% yield, whereas ZSM-5 produced liquid with high aromatic contents (46%) and olefins (35%). In another investigation, the same authors employed a relay catalysis method and used Al2O3 followed by HZSM-5 catalyst, resulting in a liquid with 100% selectivity for monoaromatics and C5–C12 alkanes/olefins.286 The oil obtained from pyrolysis of LDPE using relay catalysis contained monoaromatics, C5–C12 alkanes/olefins, and naphtha, making it a suitable feedstock for producing new plastics. Li et al. used Ni–Co/Ni pillared montmorillonite catalysts for pyrolysis of waste plastics and reported that Ni–Co pillared montmorillonite catalysts gave a high liquid product yield of 80.2% with 43.5% selectivity for C10–C13 range hydrocarbons.242 Sivagami et al. studied the pyrolysis of polyolefin based plastic waste using different zeolite catalysts in a bench scale pyrolysis reactor.304 The synthesised ZSM-5 zeolite catalyst gave the highest oil yield of 70%, with 16% gas and 14% char from the pyrolysis of LDPE plastic compared to other catalysts (mordenite and g-alumina). The total petroleum hydrocarbon (TPH) distribution in LDPE pyrolysis oil obtained by using ZSM-5 catalyst was comprised of C1–C10 (28.8%), C11–C20 (42.6%) and C21–C30 (28.6%). In contrast, use of g-alumina furnished oil with hydrocarbon fractions C1–C10, C11–C20, and C21–C30 in 17.4%, 50.2%, and 22.3% yields, respectively.
5.5.2.5 Metal loading. Increasing the metal loading on a catalyst can significantly impact the composition of the product fuel. Studies have shown that raising the metal content from 1 wt% to 5 wt% enhances the hydrogen content in the gas yield, while simultaneously reducing the concentration of C1–C4 hydrocarbons.215 For example, pyrolysis of LDPE using Y-zeolite with 1% Ni loading afforded an oil yield of 36 wt% along with a gas yield of 36%. The gas composition included H2, CH4, C2 hydrocarbons, C3 hydrocarbons and C4 hydrocarbons in 52 vol%, 18 vol%, 11 vol%, 16 vol% and 3 vol%, respectively. Increasing the metal loading to 5% Ni on Y-zeolite increased the oil yield to 43 wt% and hydrogen content to 66 vol%, while significantly reducing the amounts of C1–C4 hydrocarbon contents. The order for hydrogen yield with 5 wt% metal incorporation was found to be Co > Ni > Mo > Ga > Fe > Ru.
5.5.2.6 Plastic feed. The type of plastic significantly influences both the yield and selectivity of pyrolysis products. In a pilot-scale study using bentonite clay as a catalyst, pyrolysis of polystyrene (PS) produced oil composed of 95% aromatic hydrocarbons.305 Catalytic pyrolysis of PP, LDPE, and HDPE produced C10–C13 fraction in 16.49 wt%, 22.28 wt%, and 21.43 wt%, respectively. However, only oil fractions produced from PP have aromatic content, comprising 1.44 wt%. Use of USY zeolite catalysts for pyrolysis of PE and PP led to the production of C8–C11 hydrocarbons in both cases.302 However, the oil produced from PE had a higher aromatic content (8.7 wt%) compared to the oil from PP (5.7 wt%). This difference is attributed to steric hindrance, which affects the transition state during the cyclisation and aromatisation processes. The steric hindrance is more pronounced in branched molecules like PP, thereby reducing the formation of aromatic compounds. Fig. 17C shows the distribution of aromatic fractions in the liquid oil.
Reaction temperature plays a pivotal role and impacts coke deposition on catalysts during pyrolysis. For example, during the pyrolysis of PP, carbon deposition on a bimetallic catalyst (FeNi2) increased significantly with temperature, rising from 6 wt% at 600 °C to 29 wt% at 800 °C.236 However, the impact of temperature on coke formation changes depending on the type of catalyst in use. In contrasting studies, Zhang et al. reported the different behaviour of coke formation with temperature increase, and found that during the pyrolysis of LDPE, less than 5% coke was formed on an activated carbon (AC) catalyst at higher temperature of 500 °C, compared to more coke formation at a temperature of 430 °C.225 The amount of coke deposited on activated carbon catalysts (CACs) varied between 2.5 wt% and 5.4 wt% during LDPE pyrolysis with different catalysts. Comparable results were reported for catalytic cracking of LDPE using ZSM-11 and beta zeolite catalysts.116 Marco and colleagues studied the effect of various catalysts (HY, CaO, MgO, and HZSM-5) on coke deposition during the pyrolysis of mixed plastic waste. Coke formation was not observed during pyrolysis without using a catalyst, however coke was formed for all catalysts in the range of 2.4–6.3% (Fig. 17D).303
Several researchers have reported that using co-catalysts can help reduce coke formation. Ding et al. found that incorporating NiO as a co-catalyst with HY in catalytic pyrolysis of LDPE not only improved the production of high-octane number compounds in the liquid product but also significantly reduced the coke yield compared to using HY catalysts alone.155 The addition of NiO enhanced the primary degradation of larger molecular fragments into smaller radicals, thereby reducing the likelihood of coke formation caused by large molecules. Research has also demonstrated that the ratio of catalyst to plastic influences coke formation. Varying the catalyst-to-plastic blending ratios (from 10 wt% to 60 wt%) affects coke formation. Increasing the catalyst-to-polymer ratio led to higher coke production during LDPE pyrolysis using an FCC catalyst.91 Additionally, metal loading on bi-functional catalysts influences both gas composition and coke deposition. Akubo et al. studied different metal loadings (1 wt% and 5 wt%) on zeolite Y for catalytic pyrolysis of PS and found that higher metal loading resulted in greater coke deposition on the catalyst. Among the catalysts tested, Co-Y zeolite catalyst exhibited the highest level of coke formation.215
Regeneration of deactivated catalyst during plastic pyrolysis is crucial for maintaining catalyst efficiency and prolonging its lifespan. This regeneration process typically involves the removal of coke deposits from the catalyst surface, which blocks active sites and consequently reduces catalytic performance. Regeneration is typically achieved by heating the coked catalyst to high temperatures, around 600 °C for zeolites, to remove the accumulated coke.216 Advanced techniques such as gasification has been used by some researchers, where the coke is converted into gas, and the energy released is utilised to further support the pyrolysis process. This method not only restores the catalyst's activity but also contributes to overall energy efficiency of the system. Effective catalyst regeneration ensures consistent performance in plastic pyrolysis, enabling the sustainable recycling of plastic wastes into valuable products. Colantonio et al. developed a method for regenerating coked catalysts through gasification in a catalyst regenerator.202 In their study, the catalyst was applied in a fluidized bed reactor for pyrolysis of mixed plastics. The authors reported that energy generated from the combustion of coke and other byproducts in the catalyst regenerator could be used to enhance the pyrolysis process when the regenerated catalyst is reintroduced into the reactor.
5.6 Commercial technologies
Catalytic pyrolysis of plastics has been implemented at a semi-commercial scale and there are even commercial plants operational in the market. Tukker et al. reported the development and deployment of a low temperature pyrolysis plant for polyolefin waste at a semi-commercial scale according to the Fuji process.307 The unit processed 500 tonnes of plastic waste per year using ZSM-5 as catalysts at 400 °C, yielding a pyrolysis oil fraction of 8 wt%. Recently, Plastic Energy has developed and implemented a technology of thermal processing, namely thermal anaerobic conversion (TAC), at a pilot scale.308 The TAC process is already being used on a large scale at two pilot plants in Spain. Pre-treated plastic (up to 20 tonnes per day) is thermally decomposed in a reactor at a temperature of 320–425 °C using a catalyst. The plastic is converted to naphtha and diesel fuel.Quantafuel company (Norway) uses a pyrolysis process to convert plastic waste into oil, which is fed to a line to remove impurities, including ashes and chlorine, and the resultant oil is finally converted into high quality fuel in a two-stage catalytic process.308,309 Agrob Eko company operates one of the largest plastic pyrolysis plant in Zabrze, Poland, and processes 10k tonnes of plastic waste per year. It is based on Smuda technology, where nickel silicate and iron silicate are used as a catalyst.138,310 The Reentech process (Korea) is a highly efficient process for recycling plastic waste (polyolefins and polystyrene) into useful products such as kerosene, gasoline, and diesel fuel. The plastic thermally decomposes in the presence of a catalyst for dehalogenation. The resultant melt is then transferred to a fluid catalytic cracking unit, where it decomposes in the presence of the aluminum silicate catalyst. This process enables the recovery of up to 75% fuel oil, which is fractionated to obtain the final products.308 One of the most widely used technologies for plastic waste recycling is the PYROPLEQ process, which is used in industries in Austria, Italy, Germany, Korea, and Switzerland. This process was actively used during 1978 to 1996 and involved the pyrolysis of plastic waste at temperatures of 450–500 °C in a rotating drum and the combustion of the resulting gas at 1200 °C. PSW is usually used as the raw material for this process. Agilex processes mixed plastic–polystyrene waste to produce synthetic oil and has a capacity of 1–50 tonnes per day.310 Royco Beijing (China) and Mogami-Kyoko (Japan) companies also use pyrolysis technology for processing plastics with a capacity of 6 kilo tonnes per year and 3 tonnes per day, respectively.311 In the case of the Royco Beijing process, PE, PP, PS, and waste oils are used as raw materials to produce oil with 87% yield. The pyrolysis products of PE and PP in the Mogami-Kyoko process are 79% oil and 12% gas. The Akzo Nobel process is a method of recycling PVC-containing waste. This is a fast pyrolysis process with a capacity of 30 kg h−1 using a circulating fluidized bed system, followed by waste incineration. Crushed mixed waste with a high content of PVC is used as the starting material and processed at 700–900 °C. The main products of the process are HCl, CO, H2, and CH4, along with various other hydrocarbons and fly ash, depending on the feedstock composition.311
Other commercial plants include Plastic2Oil in the USA, which processes 1.8 tonnes per year of plastic waste (HDPE-LDPE-PP-their combinations) to produce fuels. Henan Doing plant in China processes mixed plastics by low temperature catalytic pyrolysis to produce liquid fuel for diesel engines. The plant operates at 50 tonnes per day capacity. Recycling Technologies in UK transforms 7000 tonnes of mixed plastic waste annually into chemical feedstock using its RT7000 technology, called Plaxx®.312
6. Hydrocracking
Hydrocracking is a catalytic refining process generally used for upgradation of heavier fractions obtained from crude oil distillation or residues.313 Thus, this process is used for converting high molecular weight fractions into lower molecular weight compounds by breaking C–C bonds in the presence of H2, with concurrent or sequential hydrogenation of the unsaturated molecules produced during the reaction. It is also referred to as the hydrogenation process. Hydrocracking process is typically performed in a batch stirred reactor or closed tubing bomb reactor. The resulting products from the hydrocracking process usually consist of a liquid fraction comprising asphaltenes and pre-asphaltenes extracted using THF, an oil fraction composed of a low molecular weight fraction obtained by solvent extraction, and a gas fraction.174,314Hydrocracking is one of the most effective methods to convert plastic wastes into high quality liquid fuels. It has several advantages over pyrolysis, and produces highly quality liquid fuel, saturated liquid fuel, without requiring an additional hydrogenation step.315 This process operates at lower temperatures,91 leads to reduced amount of olefins in the product and less coke formation that extends the catalyst life,316 and offers more selectivity in producing gasoline range hydrocarbons (C5–C12).317 Hydrocracking process produces less amount of aromatics compared to pyrolysis.29 Moreover, it significantly removes heteroatoms like bromine, chlorine, and fluorine, which are commonly present in plastic waste.318 Dioxin and other toxic products are not produced in hydrocracking. Hydrocracking operating conditions for a defined catalyst depend on various factors such as feedstock nature, and the desired product composition. While considering catalyst requirements for hydrocracking, it is essential to account that the main reactions include cracking and isomerisation (acidic support like amorphous silica–alumina or a zeolite), and hydrogenation (metal active centres). Typical metals for catalysts include noble metals (Pd, Pt) or non-noble metal sulphides from Co, Ni, Mo, and W. Process configuration also plays a crucial role, and a design engineer must choose from options such as using one or multiple catalysts, operating in one or two stages, and selecting between once thorough or recycle mode, depending upon the required outcome and operating conditions.
According to Munir et al.,314 research publications in the plastic hydrocracking area have decreased in recent years, which reveals a loss of interest on application of this technology to waste valorisation. This situation may be explained in terms of the challenges to develop a competitive process and catalysts able to deal with a wide variety of polymers and contaminants typically present in real world plastic waste. These contaminants include non-plastic materials (fibres, cellulose, biomass, etc.), organic (halogen, nitrogen, sulphur, etc.) and inorganic (fillers) additives. Hydrocracking results are well known to vary with the polymer type, and the key influencing variables for a fixed polymer mixture include the composition, catalyst type, hydrogen pressure, and temperature. Typical operating conditions are 300–450 °C, 2–15 MPa H2 pressure and bi-functional catalysts that consist of an acidic support with a metal supported on it.314 The products obtained from hydrocracking are highly saturated liquid hydrocarbons.
Plastics suitable for pyrolysis are also ideal for hydrocracking, such as PE (HDPE, LDPE), PP, and PS. These plastics are commonly found in municipal plastic waste (MPW) and are well-suited for conversion through the hydrocracking process. PET can also be recycled by hydrocracking; however, its thermal degradation generates less quantity of oil. PVC can also be used in a two-stage liquefaction hydrocracking method, where the first stage involves the dechlorination of PVC materials.314
6.1 Advances in catalysis for hydrocracking
From a conceptual point of view, hydrocracking is a promising technology to convert polymeric waste into high quality liquids, which can be useful for added value applications like solvents or monomers for the petrochemical industry or transportation fuels.92 Liquid quality is the main advantage of hydrocracking over thermal or catalytic cracking where unsaturated compounds are produced in high concentration as a consequence of C–C bond scission during the cracking of polymer macromolecules. However, a significant amount of energy is required for both pyrolysis and hydrothermal cracking, and both processes have limited selectivity for products. These features hinder their broader commercial applications. The development of more efficient catalysts and catalytic processes could help to increase depolymerisation and degradation reactions, thereby lowering the operating temperatures and potentially expanding the commercial viability of these technologies.Hydrocracking of a polymer usually takes places at moderate temperatures, typically between 300 °C and 450 °C, and comparatively high hydrogen pressure (2–15 MPa) in the presence of a bifunctional catalyst.319 From an energy point of view, cracking and hydrogenation are complementary reactions being endothermic and exothermic, respectively, hence saves energy.320 High partial pressure of hydrogen helps to suppress undesirable coking or repolymerisation during the process.321 Although thermal hydrocracking can occur without a catalyst, the presence of a catalyst stimulates hydrogen addition. A suitable catalyst for hydrocracking of plastics should have both cracking and hydrogenation–dehydrogenation functions. Typically, a hydrocracking catalyst comprises an acidic support impregnated with a metal. The acidic support facilitates cracking and isomerisation, whereas the metal provides the hydrogenation function.314,322,323 Acidic supports can include amorphous oxides such as silica–alumina or strong acids like sulphated zirconia,324 crystalline zeolites such as beta, USY, HY, HZSM-5,325,326 mesoporous aluminosilicates such as Al-SBA-15 and Al-MCM-48. Active metals may include noble metals such as Pt and Pd known for their hydrogenation/dehydrogenation capability,318,327 or non-noble metals from group VI-A (Sn or Mo)328 or VIII-A (Ni or Co)329 of the periodic table.
A mechanism involving carbenium ion intermediates has been reported for PE and PP hydrocracking.330,331 In the case of PP, the presence of strong acid sites leads to the formation of tertiary carbenium ions which are subsequently cracked to generate olefins (Fig. 18). These olefins are then hydrogenated at the metal centres of the catalyst, whereas the remaining smaller carbenium ions undergo metathesis with hydrogen, resulting in the formation of lower molecular weight paraffin hydrocarbons (Fig. 18). The resulting species can undergo further cleavage, producing lower branched alkanes. In contrast, weakly acidic materials have been proposed to follow the free radical mechanism.332
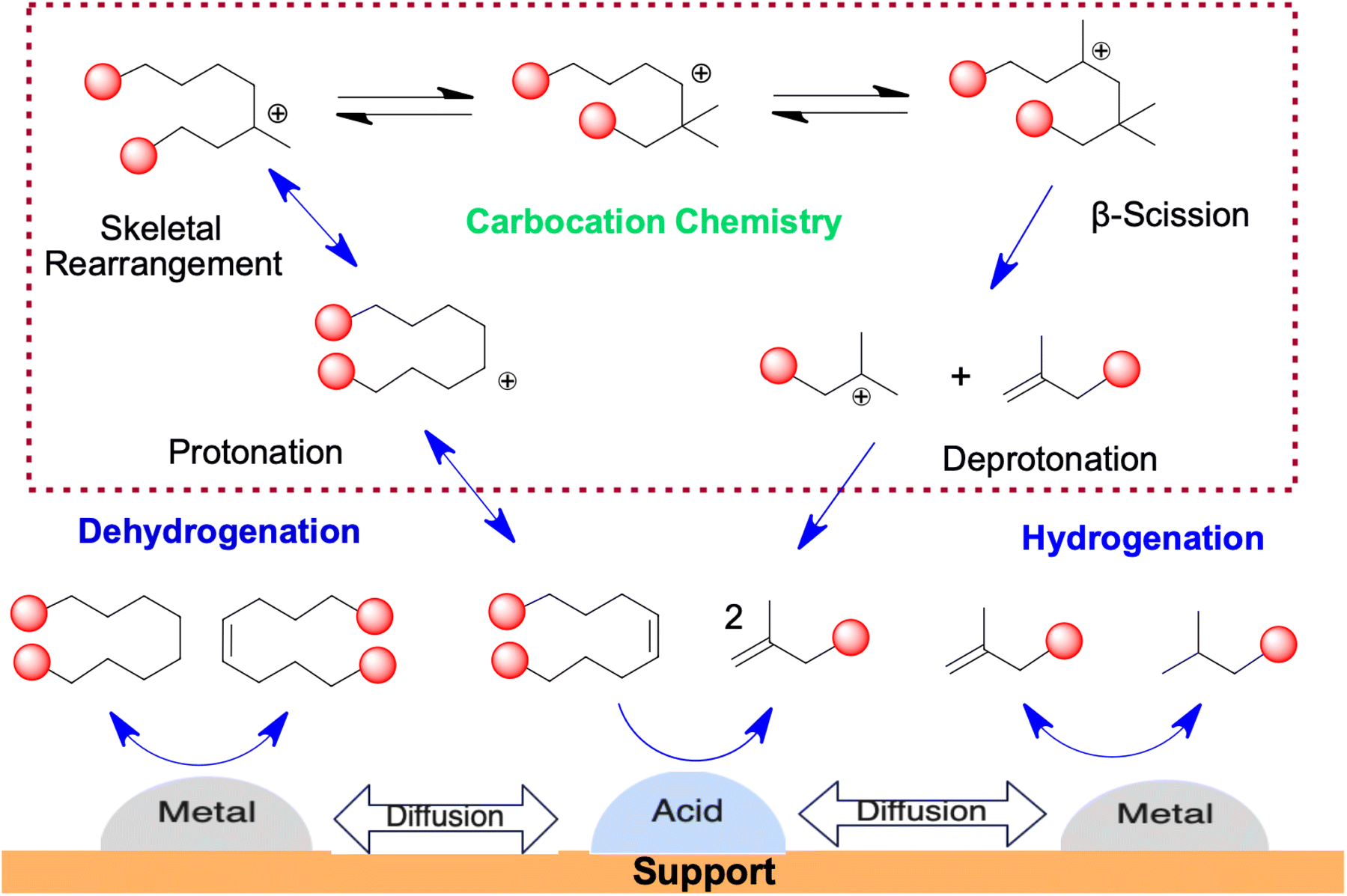 | ||
| Fig. 18 Mechanism of PP hydrocracking, reproduced with permission from ref. 331. Copyright 2023, Wiley-VCH GmbH. | ||
MCM-48 is a mesoporous silica that belongs to the M41S family and has a cubic pore structure.333 Its three-dimensional structure offers higher accessibility compared to the one-dimensional hexagonal pore structure of MCM-41.334 However, due to its weak acidic nature, MCM-48 alone does not exhibit strong activity in the hydrocracking of plastics. The cracking activity of the MCM-48 mesoporous catalyst can be enhanced by impregnating with aluminium (Al). Liu et al. investigated the effect of aluminium and platinum (Pt) loading on MCM-48 and observed that aluminium impregnation increased the catalyst's acidity due to a decrease in SiO2/Al2O3 ratio.335 Additionally, loading platinum onto MCM-48 significantly enhanced its hydrogenation capability that changed the product distribution to a large degree. This is due to platinum's ability to efficiently split molecular hydrogen into chemically active atomic hydrogen, which then becomes available for subsequent chemical reactions. According to Balandin's multiplet theory, which studies the parameters of the crystal lattice of metals, metals with a specific electronic structure can be catalysts for hydrogenation and dehydrogenation. Such metals include Ni, Co, Cu, Ru, Ir, Pd, Pt, Rh, Os and Re. The Al-MCM-48 catalyst containing Pt produced a substantially higher amount of jet fuel range hydrocarbons (C9–C15) from hydrocracking of PE compared to the Al-MCM-48 catalyst without platinum.335 The yield of C9–C15 hydrocarbons was significantly increased to 85.9% with 1 wt% Pt loading. Munir and Usman investigated the effect of aluminium impregnation on the SBA-16 mesoporous catalyst and found a significant increase in catalyst activity for hydrocracking of mixed plastics, achieving conversion yields and selectivity comparable to the USY catalyst.336 Although the addition of aluminium increases the catalyst activity of the mesoporous catalyst, their performance has not been extensively compared with highly acidic catalysts such as HZSM-5 and beta. Consequently, there is increasing interest in developing mesoporous structures within highly acidic zeolites to improve their effectiveness in hydrocracking of plastics.
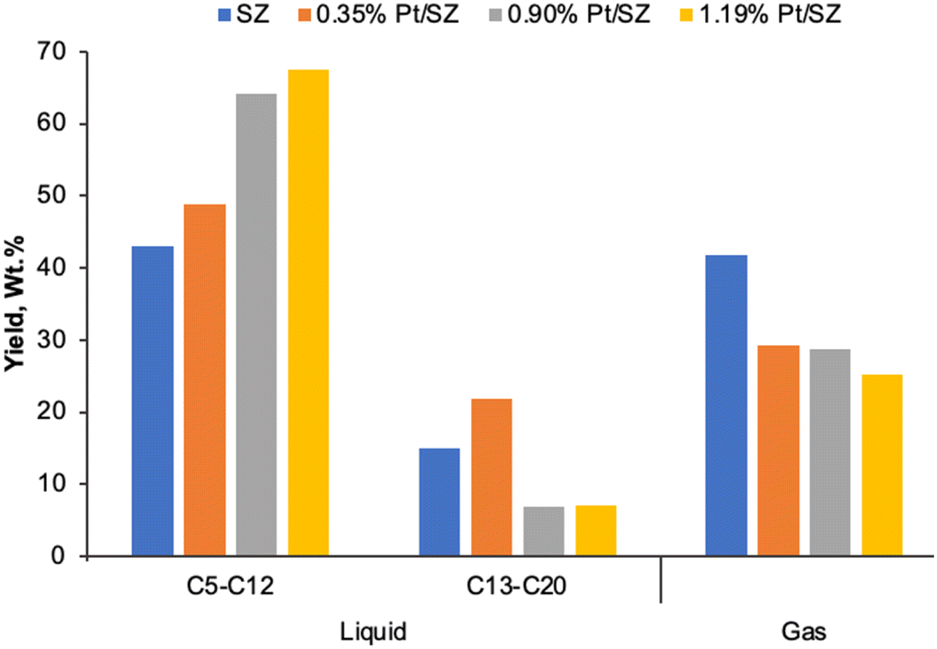 | ||
| Fig. 19 Hydrocarbon product distribution from LDPE hydrocracking with sulphated zirconia catalysts at 250 °C, H2 20 mL min−1, 3–4 h. | ||
Hydrocracking of LDPE was also studied by Amin et al. using Ni incorporated sulphated zirconia (Ni–SZ), and its catalytic activity was compared with ZrO2 and SZ.342 The total acidity and surface area of SZ catalysts were reported to increase considerably with the incorporation of nickel.343 The use of SZ and Ni–SZ led to increased liquid yield, as well as substantial reduction in coke formation and increase of gasoline fraction within the liquid product. Cr/sulphated zirconia (Cr/SZ) also exhibited good activity and selectivity in the hydrocracking of LDPE, and produced higher liquid and gasoline yields of 40.99 wt% and 93.42 wt%, respectively.324 All three catalysts (Cr1/SZ, Cr2/SZ, and Cr3/SZ) exhibited greater selectivity for the gasoline content (C5–C12) than the diesel range hydrocarbons (C13–C20). Among these, the Cr2/SZ catalyst showed the highest selectivity for the gasoline fraction, reaching 93.42 wt%.
Metal impregnated sulphated zirconia has demonstrated promising results in terms of selectivity towards gasoline and overall liquid product yield. However, its long-term stability for these reactions remains uncertain due to the potential loss of sulphur over time.314 To enhance its suitability for hydrocracking applications, it will be necessary to develop novel catalyst formulations incorporating ZrO2 that can maintain higher stability under severe reducing conditions.
The hydrocracking of plastics often produces a large fraction of gaseous components. To improve the selectivity towards the liquid fraction, some investigators have carried out the desilication and dealumination of zeolites to change their acidic properties and introduce microporosity. Desilication and dealumination of zeolites are carried out by removing alumina and silica from the zeolite framework and by modifying the SiO2/Al2O3 ratio. The desilication and dealumination process increases the surface area of zeolite support, thereby improving its catalytic performance in hydrocracking reactions. Marcos et al. used desilicated Pt-HY and dealuminated Pt-HBeta zeolites for hydrocracking of PS and reported that dealumination of beta zeolite increased its selectivity towards the gasoline fraction, which contained high contents of paraffins and iso-paraffins.353 However, desilication of Pt-HY gave a gasoline fraction with a higher naphthenic content.
Zeolite catalysts demonstrate outstanding catalytic activity in the hydrocracking of plastic, however they yield products rich in gaseous contents due to their microporous nature. Among various zeolites, ZSM-5 gave the highest gas yield followed by HBeta, HY, and USY. However, ZSM-5 has excellent thermal stability and was found to be the most active in these reactions. Therefore, to enhance selectivity toward liquid products, hierarchical zeolites are being developed.354–356 These hierarchical zeolites combine the traditional micropores with mesopores, introducing additional secondary porosity into the zeolite structure. This combination improves the diffusion of reactants and products in the catalyst, leading to better selectivity for liquid products during hydrocracking.
Jumah et al. found that platinum incorporated beta zeolite was a highly effective catalyst for hydrocracking of post-consumer plastic mixtures (LDPE, HDPE, PP, and PS), achieving high conversion and low coke formation compared to the Pt-USY catalyst.345 However, the Pt-USY catalyst produced a higher yield of liquid hydrocarbons (C5–C20) and heavier fractions due to its more open structure, while Pt-beta zeolite resulted in a higher gas fraction. Both catalysts exhibited a high iso/normal alkane ratio.357 The ultrastable Y-zeolite, also known as USY, is produced through a steaming process at high temperatures above 500 °C. This steaming process also creates mesopores within the zeolite structure, which enhances its selectivity towards liquid products, particularly naphtha, compared to other zeolites. As a result, USY zeolites are more effective in producing a higher yield of liquid hydrocarbons and narrower range of alkanes during the hydrocracking of plastics.
The concentration of active metal on bifunctional catalysts significantly influences the product distribution in the hydrocracking of plastic. Studies have shown that increasing the Ni loading on sulfated zirconia (SZ) from 0.5 wt% to 1.5 wt% enhanced hydrogenation activity, leading to higher yield of more saturated compounds such as n-paraffins, iso-paraffins, and naphthene from hydrocracking of LDPE. Additionally, higher metal loading increased the selectivity towards gasoline-range hydrocarbons (C5–C12).342 Since nickel (Ni) has lower hydro-/dehydrogenation activity compared to platinum (Pt), a higher amount of nickel is required to achieve a similar catalytic performance. Recently, Tedstone et al. synthesised sulfided nickel catalysts by impregnating different supports with a single-source precursor (SSP) “Ni(S2COC2H5)2” and investigated these catalysts for the hydrocracking of mixed polyolefin waste.358 These SSP-derived catalytic materials provided higher conversion to liquid products at 330 °C and 20 bar H2 pressure compared to previous benchmark catalysts synthesised through the wet impregnation method (Fig. 20). The SSP-derived 5wt%Ni@beta catalyst demonstrated over 95 wt% conversion of mixed polyolefin plastic into liquid products, significantly outperforming the 39.8 wt% conversion achieved with 5 wt% Ni@beta catalyst. Increasing the Ni loading from 1 wt% to 5 wt% substantially improved the conversion (>95 wt%) while maintaining selectivity. The 5%Ni@beta(SSP) catalyst produced saturated C4, C5, and C6 hydrocarbons in yields of 37.3 wt%, 21.6 wt% and 12.8 wt%, respectively, from pure LDPE.
 | ||
| Fig. 20 Hydrocracking of mixed plastic with platinum and nickel zeolite catalysts at 330 °C, 60 min time, 20 bar H2 pressure. | ||
6.2 Effect of operating parameters
The key parameters of a hydrocracking catalyst involve its hydrogenation capabilities (metallic function), cracking, and isomerisation abilities (related to the acidity of the support). Common catalyst supports include amorphous oxides such as silica–alumina, zeolites like ZSM-5, and strong solid acids such as sulphated zirconia, often in combination with other materials. For the metallic function, both non-noble metals (like cobalt, nickel, molybdenum, and tungsten) and noble metals (like Pd and Pt) are commonly used. Most studies have been focused on pure polymeric materials, mainly HDPE, LDPE, PP and their mixtures according to reported investigations.332,359,360 However, when dealing with real plastic waste as a hydrocracking feedstock, the presence of contaminants, such as metals and halogens, must be considered due to their potential to accelerate catalyst deactivation. To address these challenges and develop cost-effective catalysts suitable for processing waste plastics with impurities, various catalysts, both with and without metal loading, have been studied for hydrocracking of PP.361 Supported activated carbons have shown promising results, suggesting that this system holds potential to be an exceptional catalyst for the liquefaction of waste plastics due to its outstanding performance. Several studies performed during 1990s reported notable advancements in hydrocracking plastic waste, emphasising the potential of activated carbon-based catalysts in this application.362,363Although studies on the effect of hydrogen pressure are limited, it is generally observed that increasing hydrogen pressure improves the quality of liquid product, helps to remove impurities from plastic waste, and reduces coke formation. Higher pressure tends to enhance conversion rates and liquid yield, but only to a certain level. According to Munir et al., the optimum pressure range for hydrocracking is 2.0–6.0 MPa and the optimum temperature range is 370–400 °C.314 The ease of degradation and properties of liquid fraction obtained from the hydrocracking process depend on the type of polymer being processed. For example, the liquid obtained from polyolefins like PP or PE has a higher content of alkanes than the liquid obtained from PS that is typically richer in alkyl benzenes and phenyl alkanes.
Product distribution is different for different types of polymers, the products from HDPE and LDPE hydrocracking are mainly comprised of gases, mostly C3 and C4 hydrocarbons in 36–56 wt% yield. However, the hydrocracking of post-consumer PP results in a liquid product rich in branched alkanes.345,367 For polystyrene (PS), the products of hydrocracking are primarily aromatic compounds, with 60–80% consisting of benzene and ethylbenzene and around 5% being gas. When hydrocracking is performed under mild reaction conditions with reduced H2 pressure then PS results in a gas stream with a higher surplus of hydrogen compared to the product gas streams from PE and PP.345 This is due to insufficient hydrogenation of aromatic rings in PS under these conditions. Munir et al. reported a similar behaviour in the hydrocracking of HDPE, which resulted in a low liquid yield (52 wt%) and a higher gas yield compared to the 65.7 wt% liquid yield obtained from a plastic mixture (PP, LDPE, HDPE, PS).350 The higher liquid yield from mixed plastics is attributed to the presence of PS and PP, which produce more liquid than HDPE. However, the reactivity of both virgin HDPE and mixed plastic appeared to be similar when subjected to the same temperature conditions. Moreover, the liquid fraction obtained from HDPE hydrocracking contained lower gasoline content (58 wt%) and higher heavy hydrocarbons than from waste plastic mixture, which had a gasoline content of 70.5 wt%.350
Real waste plastic mixture contains additives including heteroatoms, trace metals, printed inks, organic and inorganic residues, which make it more complex for hydrocracking compared to virgin polymers as these additives can deactivate the catalysts used in the process.368,369 Munir et al. used composite beta catalysts for hydrocracking of virgin and post-consumer plastic waste and found lower conversion of post-consumer plastic mixture than the virgin plastic mixture.350 This effect can probably be explained by an increase in the number of active sites on the surface of the catalyst as a result of calcination, which led to the decomposition of coke deposits accumulated in these areas. In addition, partial sintering of the catalyst particles may have occurred, which led to a decrease in the average pore size. However, both types of feeds gave a similar quantity of liquid yield, with the waste plastic yielding 64 wt% of gasoline fractions compared to 68 wt% from the virgin plastic mixture. Jumah et al. reached a similar conclusion using 1% Pt loaded zeolite beta for hydrocracking of model mixtures consisting of LDPE (34%), PP (33%), HDPE (24%), and PS (10%).345 They achieved 72 wt% conversion of virgin plastic mixture compared to 66 wt% from post-consumer plastic mixture.
The functionality of active metals is important for achieving the desired product distribution. However, even a highly functional active metal may not deliver good selectivity when used with weakly acidic supports such as alumina and silica. These supports are not ideal for plastic hydrocracking and exhibit low cracking activity. Even when palladium is impregnated on these supports, the resulting liquid tends to consist of very heavy hydrocarbons.370 But, palladium loading on zeolite beta has been shown to increase catalytic efficiency in the hydrocracking of LDPE, resulting in higher gas yields (C2–C4), while the liquid yield (C5, C6+) remained below 50%. The Pd-beta catalyst also resulted in a higher yield of liquid branched isomers (Ciso/Cn = 0.65) and higher paraffin shares (66%) in the C2–C4 fraction, and lower olefins due to increased hydroisomerisation. Moreover, Pd-beta produced a higher aromatic content (5.5%) in the C6–C9 hydrocarbon fraction than the beta catalyst.370
Platinum metal has strong hydrogenation/dehydrogenation ability, making it highly effective when impregnated on highly acidic supports such as zeolite beta and USY, creating excellent bifunctional catalysts for hydrocracking of plastics. Jumah et al. used Pt incorporated zeolite beta for hydrocracking of plastic mixtures and found that the liquid product contained no olefins due to efficient hydrogen transfer. Furthermore, the product showed a high ratio of iso-butane (i-C4) to normal butane (n-C4) due to hydroisomerisation.345 Mesoporous catalysts such as MCM48, SBA15, SBA16, and microporous catalysts such as USY give different product distributions in hydrocracking of plastics. Composites of USY and SBA-16 have shown improved conversion and better liquid yields compared to USY alone catalyst.336 The USY catalyst produced the highest yield of gasoline (67 wt%) and light diesel (23 wt%) compared to the composite of USY and Al-SBA-16 with a residence time of 60 min. The micropores in USY promote a higher yield of liquids in gasoline (C5–C12) and light diesel (C13–C18) fractions, whereas the mesopores in Al-SBA-16 enhance the production of heavier hydrocarbons (C19+) at 27.6 wt%, and result in a comparatively lower gasoline fraction (51.54 wt%). However, composite catalysts produced gasoline, light diesel, and heavy diesel fractions in 65.58 wt%, 17.93 wt% and 16.48 wt% yields, respectively.336
Coke deposition on the catalyst reduces its activity during the hydrocracking process by blocking the active sites. The activity of coked catalysts has been found to decrease significantly. Munir et al. studied the use of a spent catalyst for hydrocracking a mixed plastic feed and achieved a lower conversion (73 wt%) compared to 97 wt% with a fresh composite zeolite beta catalyst.350 However, there was no significant reduction in the quantity of liquid produced, though the liquid became less selective towards gasoline and produced more heavier fractions. Catalyst activity can be restored by regeneration of the coked catalyst.371 Zeolite catalysts, for example, can be completely regenerated at room temperature with low energy consumption. When Munir et al. used the regenerated catalyst for hydrocracking of mixed plastic waste, its catalyst activity was improved and resulted in similar conversion and liquid yield as obtained using the fresh catalyst. Furthermore, the regenerated catalyst enhanced selectivity towards gasoline (C5–C12) by breaking down heavier hydrocarbons (C19+) into lighter fractions.350 In another study, using a regenerated nickel loaded zeolite catalyst led to an increased conversion (95.9 wt%) compared to 91.6 wt% with the spent catalyst (Fig. 21).372 In addition, the regenerated catalysts also increased the selectivity towards liquid products and decreased the selectivity of gaseous products (Fig. 21).372 However, the regenerated catalyst demonstrated increased selectivity toward gasoline-range fuels, achieving 85.5 wt%, while selectivity towards diesel-range fuels reduced to 14.5 wt%.
 | ||
| Fig. 21 Conversion and selectivity of liquid fractions from hydrocracking of surgical face masks over a 5% Ni-ST-HY catalyst at 325 °C and 10 bar H2. | ||
Similarly, increasing chromium loading on sulphated zirconia (SZ) has been shown to increase the selectivity for the gasoline fraction (C5–C12) while reducing the diesel fraction (C13–C20).373 Using 1 wt% Cr on sulphated zirconia produced a C5–C12 hydrocarbon fraction in 93.42 wt% and C13–C20 hydrocarbon fraction in 6.58 wt% yield, compared to 91.63 wt% (C5–C12) and 8.37 wt% (C13–C20) with 0.5 wt% Cr/SZ in the hydrocracking of LDPE plastic waste (Fig. 22). However, gasoline selectivity was slightly reduced by further increasing the chromium loading to 1.5 wt%. In addition to metal loading, the catalyst to plastic ratio has a significant impact on product distribution, and an optimised catalyst to feed ratio is essential to achieve the desired product. Zhang et al. studied the effect of various catalyst to feed ratios for the LDPE hydrocracking process, and found that increase in catalyst loading enhanced the hydrogenation reaction, and consequently increased the aliphatic and cyclic alkane contents and decreased aromatic hydrocarbons.374 A catalyst to plastic ratio of 0.2 resulted in 10.7% selectivity for aliphatic alkanes and 80% for cycloalkanes, compared to 7.4% aliphatic alkanes and 24% cycloalkanes at a ratio of 0.05. The higher catalyst loading provides additional active sites for hydrogenation and hydrocracking reactions, leading to increased yields of aliphatic and cyclic alkanes with significant reduction in aromatic hydrocarbons.
 | ||
| Fig. 22 Chemical reactions involved in gasification of plastic waste. | ||
Zhang et al. investigated the influence of temperature on product distribution in the hydrogenation step (150–200 °C) during microwave assisted hydrocracking of LDPE using Raney Ni-4200 catalyst.374 Their findings indicated that higher temperature increased the hydrogenation rate of aromatics. Moreover, although the content of aliphatic alkanes remained relatively constant, aromatic and hydroaromatic hydrocarbons decreased at 200 °C. In addition, there was significant impact on the carbon selectivity of specific alkanes with the increase in reaction temperature. Similarly, Hauli et al. evaluated the temperature effect on product distribution in the hydrocracking of LDPE-based plastic waste using Cr2/SZ (1% Cr on SZ).373 They demonstrated a significant increase in gas yield, from 59 wt% to 87.65 wt%, by increasing the temperature from 250 °C to 350 °C, while the liquid yield reduced from 40.99 wt% to 11.43 wt%.
Catalyst is also an important factor to consider during the optimisation of hydrocracking temperature as highly acidic catalysts such as zeolites yield higher conversion even at lower process temperatures, whereas less acidic catalysts such as mesoporous catalysts require higher temperature for significant conversion. Studies have shown that catalytic activity is more prominent at lower hydrocracking temperatures. In a study by Munir et al., it was reported that after an optimised temperature, all catalysts lead to similar conversion including thermal hydrocracking without a catalyst.332 The optimal temperature for hydrocracking of mixed plastics using composite and mesoporous catalysts was found to be 400 °C. Moreover, product distribution is also affected by temperature changes with different catalysts. With the USY composite catalyst, increase in temperature from 375 °C to 425 °C resulted in a higher yield of lighter fractions having the highest gasoline hydrocarbons (69.5 wt%) at 425 °C compared to 375 °C and 400 °C. However, under the same reaction conditions using Al-SBA-16 mesoporous catalyst, increasing the temperature furnished low gasoline yield and high C19+ hydrocarbons.336 Qiu et al. explored the effect of temperature on the hydrocracking of polyolefins using a noble-metal free bifunctional catalyst, MoSx-Hbeta.375 They reported a significant increase in non-solid yield (NSY), rising from 13.5% to 89.2% as the temperature increased from 180 °C to 250 °C. All commonly used aliphatic polyolefins were found to convert into liquid products in high yields of 85.7–97.2% at 250 °C compared to 56.0–81.8% yield at 200 °C, with C5–C12 alkanes being the major liquid products.
For catalysts such as USY, Al-SBA16, and their composite catalysts, increase in reaction time for hydrocracking of mixed plastic can enhance both the conversion and liquid yield. However, the most prominent improvement in catalyst activity and liquid yield occurs when reaction duration was increased from 0.5 hour to 1 hour. However, further increase in reaction time to 1.5 hours didn't have a significant effect on reactivity and liquid quantity.336 Hence, when these catalysts are used for hydrocracking, the optimised reaction time is found to be in the range of 1–1.5 hours. Further increase in residence time is not recommended for these reaction systems.336 Zhang et al. also investigated the impact of varying the residence time from 1 to 4 hours on product distribution during catalytic microwave degradation of LDPE using 20 wt% ZSM-5 catalyst. Prolonging the reaction time significantly enhanced cycloalkane formation, increasing from 26.47% at 1 hour to 56.28% at 2 hours, and reaching 75.99% after 4 hours, attributable to enhanced hydro-addition activity. The authors identified the optimum reaction time of 2 hours as further increase in reaction time was found to initiate the carbon loss reactions that consequently reduced aromatic and hydro-aromatic hydrocarbons, and produced higher cycloalkanes, making further increase in reaction time unnecessary.374 Although prolonged residence time enhances the hydrogenation, hydrocracking, and hydroisomerisation reactions, changes in product distribution becomes minimal after a certain residence time, so this parameter must be carefully optimised (Table 3).
| Feedstock | Catalyst | Reaction conditions | Yield (wt%) | Product distribution | Reference |
|---|---|---|---|---|---|
| LDPE | Beta | Cat to feedstock ratio = 1:4, 250 °C, 1 hour |
28.6% gas, 16.4% liquid, 55.0% char | 15.9% (C5–C12), 0.5% (C13) | 347 |
| W/beta | 26.9% gas, 18.6% liquid, 54.5% char | 18.1% (C5–C12), 0.5% (C13) | |||
| Pt/beta | 33.2% gas, 55.5% liquid, 11.3% char | 54.9% (C5–C12), 0.5% (C13) | |||
| Pt/W/beta | 30.3% gas, 63.7% liquid, 6.0% char | 63.6% (C5–C12), 0.1% (C13) | |||
| LDPE | Pt/WO3/ZrO2 + HZSM5 | Cat to feedstock ratio = 1:20, 250 °C, 2 hours |
70% gas, 29% liquid, 13% solid | 29% (C5–C12), 1% (C13) | 351 |
| Pt/WO3/ZrO2 + HMOR | 21% gas, 54% liquid, 20% solid | 73% (C5–C12), 7% (C13) | |||
| Pt/WO3/ZrO2 + HBEA | 21% gas, 79% liquid | 79% (C5–C12) | |||
| Pt/WO3/ZrO2 + HY(30) | 9% gas, 83% liquid, 6% solid | 81% (C5–C12), 2% (C13) | |||
| Mixed plastic (HDPE, LDPE, PS, PP) | Pt-USY | Cat to feedstock ratio = 1:10, 330 °C, 55 min |
27% gas, 61% liquid, 12% solid | 27% (C5–C12) | 345 |
| Pt-beta | 50% gas, 45% liquid, 5% solid | 19% (C5–C12) | |||
| LDPE | Pt/MCM-48 | Cat to feedstock ratio = 1:4, 300 °C, 4 hours |
6.2% (C1–C8), 0.9% (C9–C15) | 335 | |
| Pt/Al/MCM-48 | 8.9% (C1–C8), 85.9% (C9–C15) | ||||
| Pt-HY | 80.7% (C1–C8), 18.9% (C9–C15) | ||||
| Mixed plastic (HDPE, LDPE, PS) | Beta | Cat to feedstock ratio = 1:20, 400 °C, 1 hour |
33.5% gas, 59.1% liquid, 3.6% solid | 68% (C5–C12), 18.6% (C13–C18), 11% (C19+) | 350 |
| HDPE | Beta (composite) | 24.5% gas, 65.7% liquid, 3.8% solid | 70% (C5–C12), 21.9% (C13+) | ||
| Waste plastic (HDPE, LDPE, PS) | Beta (composite) | 32% gas, 52% liquid | 58% (C5–C12), 24% (C13+) | ||
| HDPE | Beta (composite) | 18% gas, 68% liquid | 64% (C5–C12), 22% (C13+) | ||
| LDPE | ZrO2 | Cat to feedstock ratio = 1:100, 300 °C, 1 hour |
38.1% gas, 17% liquid | 79.9% (C5–C12), 15.9% (C13–C22) | 324 |
| SZ | 32.5% gas, 24.2% liquid | 82.3% (C5–C12), 16.2% (C13–C22) | |||
| Cr/SZ | 23.6% gas, 40.2% liquid | 89.9% (C5–C12), 9.1% (C13–C22) | |||
| Waste LDPE | Nano ZrO2 | Catalyst = 1 wt% 300 °C, 1 hour | 92.6% gas, 7.1% liquid, 0.3 char | 4.8% (C5–C12), 2.3% (C13–C22) | 376 |
| Nano ZrO2–SO4 | 75.2% gas, 23.8% liquid, 1% char | 18.1% (C5–C12), 5.7% (C13–C22) | |||
| Pt-nano ZrO2–SO4 | 44.9% gas, 54.8% liquid, 0.3% char | 37.1% (C5–C12), 17.7% (C13–C22) | |||
| Nano ZrO2 | Catalysts = 1 wt% 350 °C, 1 hour | 98.1% gas, 1.3% liquid, 0.6% char | 1.0% (C5–C12), 0.3% (C13–C22) | ||
| Nano ZrO2–SO4 | 91.1% gas, 8.0% liquid, 0.9% char | 6.1% (C5–C12), 1.9% (C13–C22) | |||
| Pt-nano ZrO2–SO4 | 83.7% gas, 15.9% liquid, 0.4% char | 12.0% (C5–C12), 3.9% (C13–C22) | |||
| LDPE | ZrO2 | Catalyst = 1 wt% 300 °C, 1 hour | 83.7% gas, 15.9% liquid, 0.4% char | 342 | |
| SZ | 83.7% gas, 15.9% liquid, 0.4% char | ||||
| 1.5Ni–SZ | 83.7% gas, 15.9% liquid, 0.4% char | 66.3% (C5–C12), 30.44% (C13+) | |||
| 1Ni–SZ | 83.7% gas, 15.9% liquid, 0.4% char | 62.9% (C5–C12), 30.4% (C13+) | |||
| 0.5Ni–SZ | 83.7% gas, 15.9% liquid, 0.4% char | 61.2% (C5–C12), 30.4% (C13+) | |||
| LDPE | ZrO2 | Cat to feedstock ratio = 1:100, 250 °C, 1 hour |
50.1% gas, 49.6% liquid, 0.1% char | 33.5% (C5–C12), 16.2% (C13–C22) | 341 |
| SZ | 41.7% gas, 57.9% liquid, 0.4% char | 42.6% (C5–C12), 15.4% (C13–C22) | |||
| 1.5Pt–SZ | 25.4% gas, 74.0% liquid, 0.23% char | 67.5% (C5–C12), 6.5% (C13–C22) | |||
| Mixed plastic (HDPE, LDPE, PS) | UC1.2 (USY composite) | Catalyst = 5 wt% 400 °C, 1 hour | 14% gas, 40% liquid | 65.6% (C5–C12), 17.9% (C13–C22) | 336 |
| USY | 10% gas, 33% liquid | 67.3% (C5–C12), 22.6% (C13–C22) | |||
| Al-SBA16 | 9% gas, 29% liquid | 51.5% (C5–C12), 20.8% (C13–C22) | |||
| Mixed plastic (HDPE, LDPE, PS) | Beta | Catalyst = 5 wt% 400 °C, 1 hour | 33.5% gas, 59.1% liquid, 3.6% char | 68% (C5–C12), 18.6% (C13–C22) | 350 |
| Beta composite | 24.5% gas, 65.7% liquid, 3.7% char | 70% (C5–C12), 21.8% (C13–C22) | |||
| PP | NiMo/Al2O3 | Cat to feedstock ratio = 1:10, 450 °C, 20 bar pressure |
13.8% gas, 86.0% liquid, 0.22% char | 34.5% (C5–C12), 65.6% (C13–C22) | 377 |
| Pt/Al2O3 | 15.6% gas, 84.0% liquid, 0.4% char | 49.9% (C5–C12), 50.1% (C13–C22) | |||
| PP | 5% Ni-ST-HY | Cat to feedstock ratio = 1:10, 325 °C, 2 hours |
14.5% gas, 59.1% liquid | 92.0% (C5–C12), 8.0% (C13–C22) | 372 |
| 5% Ni-ST-HY-regenerated | 27.8% gas, 59.1% liquid | 85.5% (C5–C12), 14.5% (C13–C22) | |||
| Mixed plastic (HDPE, LDPE, PS) | Al-SBA15 | Cat to feedstock ratio = 1:20, 400 °C, 1 hour |
9% gas, 34% liquid | 332 | |
| MZ-15 (composite) | 10% gas, 33% liquid | ||||
| Al-SBA16 | 8% gas, 22% liquid | ||||
| MZ-16 (composite) | 12% gas, 39% liquid | ||||
| PS | Pt-HY | PS (5 wt%) dissolved in decahydronaphthalene | 86.7% gas | 353 | |
| Catalyst = 2.36 g L−1 400 °C, 40 min | |||||
| Pt-beta | 72.9% gas | ||||
| Pt-HY (desilicated) | 84.6% gas | ||||
| Pt-beta (dealuminated) | 77.2% gas |
7. Gasification
Gasification is a thermochemical process that transforms carbon-based materials into a gaseous fuel, known as syngas, a versatile fuel comprising largely H2 and CO. This complex process is carried out by heating the feedstock to high temperatures (1100–1600 °C) under the atmosphere of oxygen, air, carbon dioxide, or steam. Gasification involves multiple steps such as drying, pyrolysis, cracking, reforming, oxidation, and reduction reactions. The significance of these steps in gasification performance is influenced by the properties of the feedstock and the process operating conditions. Gasification holds significant potential as a pivotal technology for generating low-carbon synthetic biofuels and advancing plastic circularity.The valorisation of plastic waste through gasification has been explored using various gasification methods to produce syngas with different compositions for various applications. However, research on plastic gasification remains in its early stages, with a limited number of studies conducted in this area.378 When plastic is the feedstock then the drying stage has minimal impact on the overall gasification process, as plastics generally contain much less moisture compared to other types of feedstocks. However, pyrolysis is a critical stage in the gasification of plastics, mainly due to the distinctive physical and chemical characteristics of plastic materials. The pyrolysis stage usually occurs at high heating rates, and involves a series of complex endothermic reactions that produce volatile compounds and char. Therefore, when the feedstock consists of only plastic waste such as PE, PET, PS, PP, PVC, etc. then their high volatile content can be almost entirely converted into vapours with minimal residual char formation.378 Therefore, the composition of volatiles during the gasification of plastics will be determined by temperature and the degradation mechanism, which typically involves cracking and random scission of the plastics. For example, at high temperature, polyolefins can break down into light olefins,379 whereas PET and PS can also lead to the formation of monoaromatic and polyaromatic species that could contribute to primary tars.111 These volatiles and tars then undergo further reforming, gasification, and oxidation reactions, ultimately leading to syngas production. The overall gasification process of plastic waste consists of several simultaneous chemical reactions, which are summarised in Fig. 22.
Initially, during the heating phase, the solid polymeric chains undergo a random scission and break down into monomeric units. Then, a series of exothermic reactions occur in an oxygen-limited environment, leading to partial or complete oxidation reactions to achieve the temperatures required for gasification. The addition of steam into the gasifier plays a pivotal role in H2 production through a sequence of endothermic gasification and reforming reactions. The water–gas shift (WGS) reaction, a key exothermic equilibrium process, controls the H2/CO in the gasifier but becomes less favoured at high gasification temperatures. Consequently, the ratio of gasifying agents governs the overall thermodynamics in the gasifier, balancing the preceding exothermic and endothermic reactions. Moreover, though CO2 produced from oxidation reactions is less reactive than steam, initially, it still acts as a gasifying agent through dry reforming and Boudouard reactions.
During gasification of plastics, the formation of tars and their progress in the gasifier are influenced by the plastic composition, in addition to operation conditions. Polystyrene and polyethylene terephthalate are the only plastics that can produce primary aromatic tars during gasification. However, gasification of polyolefins may result in the formation of secondary and tertiary aromatic tars due to polymerisation of light olefins, which serve as tar precursors.378 The most likely mechanism for tar formation from these light olefins is the Hydrogen Abstraction–Acetylene Addition (HACA) mechanism.380 The mechanism involves a cyclic sequence in which a hydrogen atom is first abstracted from an aromatic hydrocarbon, forming a reactive radical site. Subsequently, an acetylene molecule (C2H2) adds to this radical centre, generating a vinyl-type radical or an intermediate PAH (polycyclic aromatic hydrocarbon) precursor. This is followed by internal rearrangement, ring closure, and dehydrogenation steps, which facilitate the formation of additional aromatic rings. This derives the growth of PAHs, leading to the formation of larger and more complex aromatic structures typical of tar. Therefore, the higher yield of tar during plastic gasification is directly related to the increased formation of light olefins compared to biomass gasification.381 The final application of syngas from plastic waste gasification will determine the gasifier type and operational parameters. For syngas used in energy production through engines and turbines, the tar content must be below 10 mg Nm−3, whereas for syngas used in synthesis processes, the tar contents need to be even lower.382 Tar characteristics, especially its dew point, are key factors contributing to operational challenges such as deposition in heat exchangers and other equipment.383 Recent advancements in pre-conversion technologies have significantly expanded the potential of gasification. These innovations enable the conversion of low-value feedstocks including mixed municipal unsorted plastic waste into syngas which can substitute virgin hydrocarbons to produce fuels, electricity, chemicals, and plastics (Fig. 23). Pre-conversion processes such as plastic liquification can transform unsorted plastic waste, unsuitable for other recycling methods, into intermediate feedstocks suitable for gasification. This approach, integrating plastic liquification and gasification, has the potential to play an important role in assisting to close the plastic circularity loop by producing high-value circular plastics, thereby addressing both economic and environmental objectives (Fig. 23).
 | ||
| Fig. 23 Gasification of plastic waste to produce syngas for conversion into lower-carbon and more circular products. | ||
7.1 Effect of operating parameters
The composition of syngas produced during gasification is affected by different process parameters, including the composition of gasifying agent, temperature, pressure, reactor type, and the characteristics of the gasifying process using plastic waste as feedstock. The main challenge in plastic waste gasification, irrespective of the gasifying agent, is effectively managing tar content in the resulting syngas.384–387 However, the tar content is reduced if air, oxygen or CO2 is used as the gasifying agent instead of steam. Lopez et al. conducted a review of air gasification of plastic wastes, including PE, PP, and mixed plastics, in fluidised bed reactors at bench and pilot scales, and concluded that the gasification process operating at 800–950 °C produced syngas in the range of 2.5 to 6 m3 kg−1.378 The heating value of syngas (3–12 MJ Nm−3) was found to depend largely on the composition of plastic wastes, and the equivalence ratio (0.2–0.35). The equivalence ratio refers to the ratio of air-to-fuel in the gasifier to the stochiometric air-to-fuel ratio needed for total consumption. Tar can be eliminated by different approaches, which are categorised as the primary method (within the gasifier) and secondary method (outside the gasifier). In the primary gasification method, both dolomite and olivine have been used as catalysts, with dolomite showing better tar removal efficiency, whereas olivine exhibited superior mechanical performance. For secondary methods, activated carbon and Ni-loaded activated carbon have been tested as tar-cracking agents, demonstrating good tar removal capabilities and leading to higher H2 and CO concentrations.388,389 Consequently, air gasification presents a promising alternative for energy applications, particularly for electricity generation in engines and turbines.In contrast, plastic steam gasification produces a hydrogen rich syngas, which is a feedstock for production of methanol and dimethyl ether (DME). Moreover, the absence of nitrogen in the process results in a syngas with heating value that can exceed 1.5 MJ Nm−3, making it also suitable for energy applications. Temperature is the key factor in this process, enabling the reforming and cracking of hydrocarbon tars, which increases gas and hydrogen production. Despite the high quality of syngas produced, the steam gasification of plastics faces significant challenges, including higher tar content in the syngas and endothermic nature of the process, which complicates large-scale implementation.390 Although many technologies deployed for laboratory scale investigations for gasification of plastic waste were tested at standard atmospheric pressure, this parameter is not as important as previous ones.391,392 High pressure gasification produces syngas rich in methane and hydrocarbon due to the methanation reaction. One of the main advantages of working at elevated pressures at the industrial scale is the direct utilisation of high-pressure syngas for follow-up applications, such as power generations in gas turbines. In general, plastic waste steam gasification remains underexplored and less developed, and it is not yet as mature or well-established as the air gasification approach.
7.2 Reactors for gasification
Various configurations are used for gasifiers that include the fixed bed, fluidised bed, spouted bed, entrained bed, and plasma reactor. The advantages and shortcomings for both fixed and fluidised bed gasifiers have been reported in the literature.3897.3 Commercial processes
The selection of a suitable process for a specific use depends on various factors, including the scale, feedstock compositions, moisture contents, type and quality of the required product.103,391The Waste Gas Technology UK Limited process is widely used and involves drying and mechanical pre-treating of various types of wastes such as plastics, municipal solid waste, rubbers, and wood, followed by sorting out incombustibles and granulation. The gasification is carried out in a cylindrical reactor at 700–900 °C and produces a high heating value gas. Another well-known commercial technology is the Texaco gasification process, which was tested on a large scale with mixed plastic waste at a pilot plant (10 tonnes per day) in California, USA. This process includes a liquification stage and an entrained bed gasification stage. During the liquefaction stage, plastic waste undergoes mild thermal cracking, breaking down into synthetic oil and a mixture of condensable and non-condensable gases. The non-condensable gases are typically recycled as fuel to sustain the liquefaction process, whereas synthetic oil and condensed gases are fed into an entrained gasifier. In the gasification stage, oxygen and steam are introduced at temperatures ranging between 1200 °C and 1500 °C. After several cleaning steps, clean, and dry syngas is produced, primarily consisting of CO and H2, with small amount of CO2, CH4, water vapours, and trace inert gases. Ebara has developed various gasification technologies to process wastes including plastic waste, such as an internally circulating fluidised-bed gasification system (ICFG), and pressurised twin internally circulating fluidised-bed gasification (PTIFG).414 The PTIFG process can gasify plastic waste into syngas, with the ash recovered as molten slag, which can be reused in cement industry and other applications. An example of large-scale plastic waste gasification is Showa Denko K. K. (SDK), which employs a pressurised two-stage gasification furnace. This system processes 195 tons per day of hydrogen gas from non-recyclable plastic waste.
Plasma gasification, a more recent technology compared to other gasification methods, is particularly suitable for processing municipal solid waste and non-recycled plastics (NRP).415 In this technology, a plasma arc powered by electricity generates extremely high temperatures to initiate and enhance gasification reactions. Within the plasma gasifier, feedstocks are broken down into their fundamental elements, allowing even hazardous wastes to be transformed into valuable syngas.392 As a result, plasma gasification is considered an environmentally friendly technology.
A key advantage of gasification is its flexibility to valorise mixtures of different plastic waste types along with other feedstocks, including biomass or municipal solid waste. The differences observed in the co-gasification of mixed feedstocks is limited to gas composition in the resulting product and the yield of by-products, making the process adaptable. This flexibility, combined with the well-established coal gasification technologies, has driven research into plastic co-gasification, with plastic content reaching up to 55% in combination with coal and biomass. Germany has developed various gasification technologies for waste gasification such as British Gas-Lurgi (BGL) fixed-bed gasification, GSP entrained-flow gasification, etc.416 The BGL gasifier operates at 2.5 MPa, with a feed capacity of 27–32 tonnes per hour.
Currently, waste plastics are primarily gasified within municipal solid waste (MSW) stream by companies like Enerkem. However, there appear to be few developers focusing on the gasification of isolated plastics. In 2018, Tsiamis and Castaldi investigated the effects of incorporating increasing percentages (up to 50%) of non-recycled plastics into a biomass feedstock at a gasification pilot plant for ethanol production.417 They concluded that increasing the plastic contents in the feedstock significantly enhanced the gasification process, leading to 42% increase in methanol production, and 28% improvement in thermal efficiency. Moreover, by increasing the energy input by 2%, the generation of syngas increased by nearly 80%.
Gasification is a mature technology with 686 gasification plants globally operating in 272 large facilities.418 Their syngas production capacity is about 200 GW. In most gasification plants, coal is the primary feedstock, followed by petroleum heavy residues. These feedstocks provide a platform to produce chemicals such as NH3, MeOH, DME, liquid fuels, and gaseous fuels including hydrogen and synthetic natural gas. Current trends in the gasification market show a shift towards larger-scale projects, primarily located in Asia and the Middle East. However, there is also growing interest in smaller, modular gasifiers, which offer flexibility in feedstock and can process materials like biomass, municipal solid waste, non-recycled plastics, and used tires. To date, the application of gasification technology for waste conversion has primary been limited to directly utilising syngas for fuelling a boiler or methanol production.378 However, there has been growing attention from the scientific community and the chemical industry on waste gasification, including non-recycled plastics (NRP).378 The gasification of NRP aligns with the principles of sustainability and circular economy, especially when the syngas is used to produce chemicals that can be transformed into second-life products.417 Plasma gasification has also gained traction, with commercial scale plants operating in countries like Japan, Canada, UK and India. Many plasma conversion plants are being developed worldwide by companies including GeoPlasma, Plasco Energy Group, and StarTech, particularly in regions where landfill disposal costs are high and the renewable electricity generated from gasification facilities has a high value.
Gasification has been recognised as a sustainable technology that can convert plastic waste into syngas for various applications, including steel production. In Europe, plastic waste is currently used as a reducing agent in blast furnaces replacing coke, coal or natural gas to produce syngas. Voestalpine in Austria operates the world's largest plant to inject plastic waste into blast furnaces, utilising 220000 tonnes of plastic waste annually since 2007, significantly reducing the need for heavy oil input.419 However, there are still challenges that need to be addressed when processing plastic waste in the gasifier. These include heterogeneity of the feedstock, difficulties in feeding highly viscous melted plastics that can cause operational issues, and the formation of corrosive compounds such as HF, HCl, and HBr when processing plastics such as PTFE, PVC, ABS, etc.
8. Chemical depolymerisation
The engineered plastics show outstanding thermal and mechanical properties. The global market value of these polymer materials reached US$ 121.9 billion in 2023, and their demand is projected to increase from 10.26 million metric tonnes in 2020 to 15.26 million metric tonnes by 2026, with a compound annual growth rate (CAGR) of 7.03%.420 Polycarbonate (PC) and polyethylene terephthalate (PET) are the two mostly used engineering plastic globally. Polycarbonate materials are usually not recycled by established mechanical recycling procedures mainly due to post-use material processing complexities. Consequently, PC waste usually ends up in landfills after use.421 In the case of PET, mechanical recycling methodologies have been implemented, however polymer chains suffer from breakdowns due to residual moisture and contaminants present in the waste during melt processing. As a result, polymer molecular weight significantly reduces and accordingly strength and melt viscosity are reduced.422 Therefore, overall utilisation and common applications of these methodologies are not large, and much of the used PET material eventually ends up in landfills contributing several billion pounds of polymer waste annually.Chemical depolymerisation, also referred to as chemolysis, converts plastic wastes either into initial monomers that can be repolymerised to make original high quality polymers, or new building block molecules with potential use for producing chemicals or other polymer materials.423 The major challenge facing chemolysis is the diverse range of chemical structures corresponding to plastic waste streams, each requiring different treatment methods, making it a technically complex process, particularly when the plastic wastes are mixed. On the other hand, plastics usually contain additives or plasticisers, further complicating their recyclability. Therefore, there is no single solution for the degradation of all plastic types, eventually specific strategies are required to recycle each polymer family or plastic waste stream. Instead, specific strategies must be developed to recycle each polymer family or plastic waste stream. Chemolysis is typically a preferred method to recycle condensation polymers such as PET, PLA, nylon, and PC, with the exception of PU, which is an addition polymer.423–425 In contrast to hydroconversion of polymers, solvolysis process utilises a solvent phase to depolymerise condensation polymers such as amides, carbonates, polyesters, etc. Various solvents such as amines, ethanol, glycol, methanol, and water have been employed to depolymerise post-consumer plastics. Scheme 1 shows the typical methods for solvolysis of PET. Glycolysis and hydrolysis, after pyrolysis and hydrocracking, are the third most applicable recycling routes, which focus on oxygen-containing plastics such as PET and PUR feedstocks.
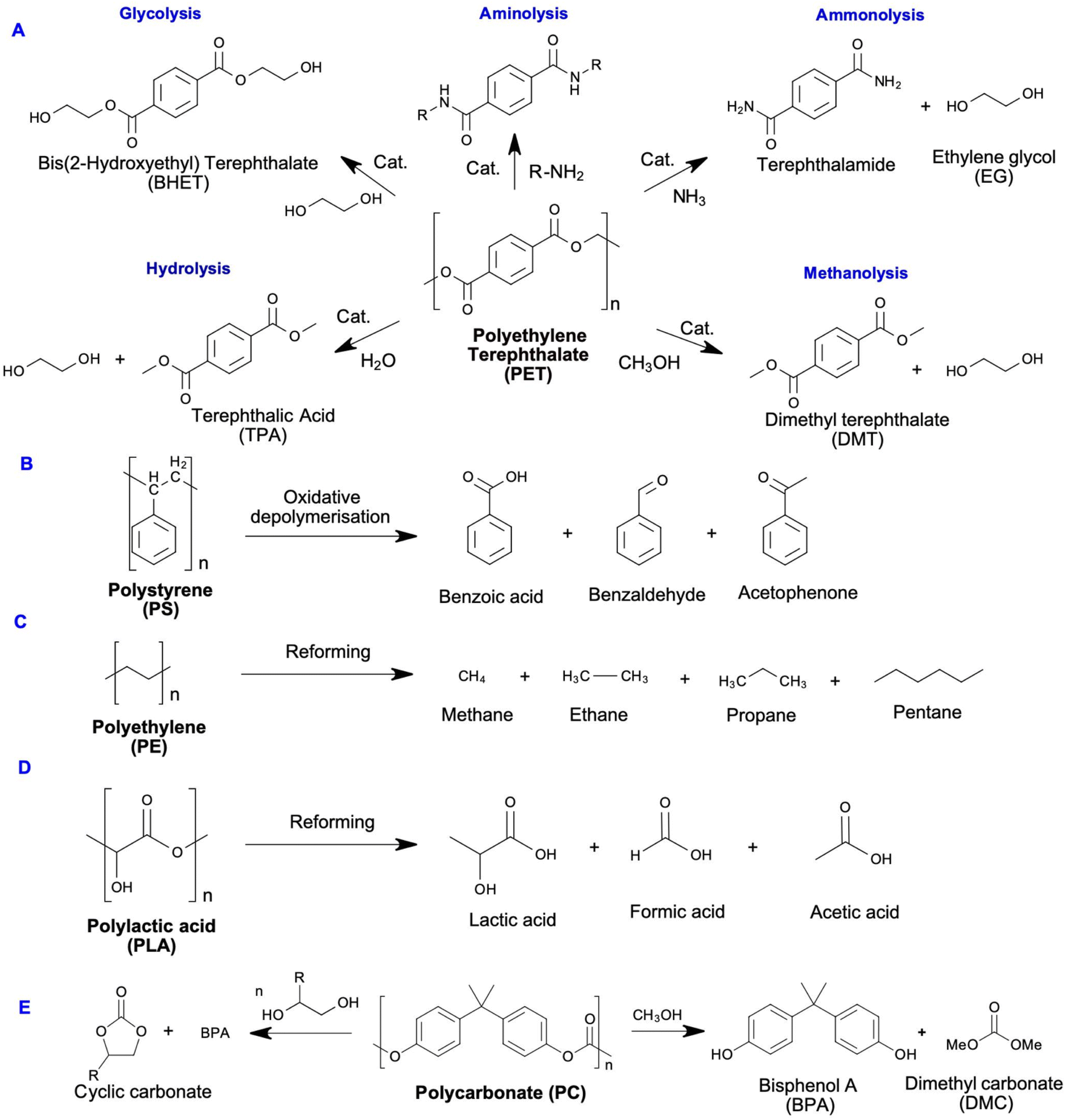 | ||
| Scheme 1 Routes for chemical recycling (A–E) of different plastics and their possible products. | ||
Polyesters are the most easily chemically processed type of polymers since the ester bonds of the polymer chains of these substances easily react with various nucleophilic reagents. As a result of such reactions, a high yield of useful products is obtained. Theoretically, all polyesters can undergo such a recycling process. However, in practice this is implemented only with PET. This is due to the simplicity of its collection and the large number of used bottles, fibres and photographic films made of PET. Also, chemical processing allows the removal of any pollutants from PET, even if they are attached to its polymer chain. The demand for secondary PET is also the most well-established compared to other large-volume polymers.
8.1 Glycolysis
Glycolysis is mostly carried out using heterogeneous solid catalysts, which typically comprise metal oxides and supported metal nanoparticles, designed to overcome the challenges of catalyst recovery commonly encountered with homogeneous catalysts. Glycolysis is predominantly applied to PET due to the economically appealing market of recycled PET. However, PET's accessibility to catalytic sites can be hindered if the conditions do not permit its proper fluidification. Most research in this area has concentrated on breaking down PET into the bis(2-hydroxyethyl) terephthalate (BHET) monomer, which can be utilised to produce new PET (Scheme 1). PET glycolysis consists of PET transesterification with ethylene glycol at high temperature (180–240 °C), which results in its depolymerisation to produce BHET. The glycolysis reaction proceeds slowly in the absence of a catalyst and it requires 32 kcal mol−1 activation energy, which is almost double that of the catalysed glycolysis reaction.426 An exception occurs when the process is performed at high temperature and under pressure, where PET exhibits resistance to glycolytic decomposition. The process of PET degradation by glycolysis involves three stages that lead to oligomers, then dimers, and finally monomers. During the reaction, glycol penetrates the polymer, causing it to swell and increase the diffusion rate. This swelling facilitates the glycol's attack on ester bonds in the polymer chain, breaking down PET into smaller fragments.424 Higher EG/PET ratios (≥5:1) favour the monomer formation (BHET), whereas low EG/PET ratios (<4:1) favour higher molecular weight products such as oligomers (dimer and trimer).424 In glycolytic depolymerisation, the macromolecules of the polymer are degraded by glycols (polyalcohols), in the presence of transesterification catalysts.427–429 Baliga and Wong concluded that in the process of glycolysis, ethylene glycol (EG) acts both as a reagent and as a catalyst.430 Depolymerisation of PET occurs through a transesterification reaction between its ester and alcohol. The catalyst is essential in this process, as it enhances the electrophilicity of carbonyl, facilitating the nucleophilic attack from the diol (Scheme 2). The glycolysis reaction is specifically suited for polymers that contain heteroatoms in their backbone.
 | ||
| Scheme 2 Mechanism of transesterification via diol insertion promoted by cationic centres. | ||
Various investigations have been directed to understand the degradation process of PET into BHET (Scheme 2). Kinetic studies reveal that the reaction exhibits 1st order kinetics with respect to PET and 2nd order concerning EG.424 But, this model doesn't account for the commonly observed induction period, which is thought to result from chain scission-induced recrystallisation and the subsequent reorganisation of polymer chains during nucleation and growth phases.431 Esquer et al. suggested a pathway where the depolymerisation rate decreases as the reaction progresses.432 In this mechanism, the interaction between the metal and carbonyl at the ester site enhances nucleophilic attack by EG on the carbonyl group, leading to the breakdown of polymers into oligomers (Scheme 2). These oligomers continue to react similarly to form dimers, eventually reaching an equilibrium with the BHET monomer (Scheme 2).
Homogeneous catalysts such as ionic liquids and metal acetates that include acetates of Zn, Pb, Mg, and Co, demonstrate high efficiency in the process of PET glycolysis. However, these catalysts have few disadvantages, including slow reaction rates and the difficulty of catalyst separation as most catalysts dissolve in ethylene glycol. This requires additional processing in a chemical reaction. Furthermore, zinc salts do not accelerate glycolysis reaction at temperatures beyond 245 °C and cannot be recycled or reused. Additionally, zinc salts can cause side reactions that reduce the purity of products, limiting their practical application.439
Stable and highly active organometallic catalysts including sodium/potassium sulphate, titanium phosphate, zinc or lead acetates have been widely used for depolymerisation processes. Esquer et al. investigated the depolymerisation of polyurethane (PU) and PET using phosphine/phosphite ligands and [Ni(COD)2] as the catalytic precursor (Fig. 24).432 Their results showed that PET degradation improved significantly when bidentate phosphine ligands were used with [Ni(COD)2] and CoCl2. Wang et al. carried out a series of experiments using the sodium titanium tris(glycolate) catalyst, which showed significantly higher efficiency compared to traditional catalysts such as sodium carbonate, tetrabutyl titanate and zinc acetate, due to the simultaneous action of two catalytic mechanisms.440 At a temperature of 190 °C, the yield of BHET was 76.5%, better than using the zinc acetate catalyst which gave a yield of 69.3%, under the same conditions. In addition, the sodium titanium tris(glycolate) allowed the process to be carried out at significantly lower concentrations of the catalyst without loss of efficiency.
 | ||
| Fig. 24 Degradation of PU by transesterification. (a) Flexible foam, (b) rigid foam, (c) reactions involved in PU degradation. Reprinted with permission from ref. 432. Copyright 2019, Elsevier. | ||
Excellent results on PET glycolysis were achieved using transition-metal-substituted polyoxometallates (POM) as catalysts. Five catalysts, K6SiW11MO39(H2O) (M = Zn, Mn, Co, Cu, Ni), were successfully employed for PET glycolysis under moderate conditions.441 The catalytic performance followed the order SiW11Ni < SiW11Cu < SiW11Co < SiW11Mn < SiW11Zn. Among these, SiW11Zn showed the highest catalytic performance, achieving 100% PET conversion and 84% BHET yield with a catalyst/PET molar ratio of 0.13% and PET/EG weight ratio of 1:4. In a parallel study, Fang et al. synthesised a Finke-type POM catalyst, K10[M4(H2O)2(PW9O34)2]-H2O incorporating different metals. The Zn-substituted POM catalyst, K10[Zn4(H2O)2(PW9O34)2]-H2O, demonstrated outstanding catalytic activity at 240 °C, achieving a 92.8% yield of BHET in 8 minutes.442 Wang et al. used a pseudo-homogeneous system based on a carbon nitride colloid for PET glycolysis.443 This catalyst achieved complete conversion of PET with 80.3% BHET yield, reducing the reaction time by 2–8 times compared to traditional metal-based catalysts.
Glycolysis is among the most widely investigated processes for recycling polyurethane (PUR) and involves heating PUR waste with glycols in the presence of catalysts for facilitating transesterification reactions. Various catalysts such as NaOH, KOH, CH3COONa, CH3COOK, Zn(CH3COO)2, metal octoates, amines, etc. have been reported for PUR glycolysis.444–447 The process is typically performed at temperatures of 160–250 °C and ambient pressure for 2–10 hours, leading to polyol formation in 70–95% yield. However, process parameters need to be optimised carefully due to the diverse chemical structures in polyurethanes and the presence of composite materials in PUR-based products.444 Diethylene glycol (DEG) is the most widely used agent for cleaving PUR, although glycerol has recently emerged as a more favourable alternative.448 The efficient degradation of PUR foams by glycolysis is more challenging due to their low density and specific chemical properties, often requiring high catalyst loadings and large reactor volumes, reducing their economic demand. However, when a dual phase glycolysis method was applied using DEG and stannous octoate as the catalyst at 189 °C, over 80% polyol was recovered from flexible PUR foam in 50 minutes.446 Recently, Esquer and García used metal chlorides for depolymerisation of flexible foams with EG and achieved outstanding performance.432 Among the metal chloride tested, the highest yield was achieved with FeCl3 followed by CoCl2. The proposed mechanism, shown in Fig. 24, involves PUR degradation (k1) and subsequent release of polyols (k2). For rigid foams, the degradation rate (k1) is higher than the polyol release rate (k2) due to their highly cross-linked molecular structure, whereas for flexible foams k2 > k1, resulting in a higher yield of polyols (>80%).
Poly(bisphenol A carbonate) (BPA-PC) is a widely utilised thermoplastic experiencing rapid market growth, hence the need for effective end-of-life management strategies becomes more critical. In addition to pyrolytic methods, chemical recycling approaches that cleave the carbonate bond via alcoholysis, aminolysis, and hydrolysis not only offer a viable route for regenerating bisphenol A (BPA), enabling its reuse in the production of virgin polycarbonate (BPA-PC), but also facilitate the synthesis of value-added chemicals, enhancing the sustainability of PC recycling. Quaranta et al. studied the glycolysis of PC using DBU catalyst, which effectively and selectively depolymerised PC with 1,2-propanediol or glycerol, yielding BPA and the analogous cyclic carbonates.449 The reactions were performed under solventless conditions and in THF, which enhanced polymer dissolution. Glycolysis of PC with 1,2-propanediol in THF resulted in BPA and cyclic carbonate in 95% and 94% yields, respectively. The catalyst was effectively regenerated in the form of a BPA/DBU adduct, enabling reuse with sustained efficiency. Dove et al. demonstrated a selective and efficient chemical depolymerisation of PC, PLA, and PET using cost-effective metal salt/organobase dual catalysts.450 By optimising catalyst selection and reaction parameters, selective and sequential depolymerisation of polymer mixtures was achieved. At 180 °C, DMAP exhibited superior catalytic activity for PET glycolysis, achieving 94% conversion to BHET, while imidazole reached only 17%. PLA was fully depolymerised to 2-hydroxyethyl lactate in 2 hours at 120 °C using DMAP. For PC glycolysis at 180 °C, imidazole outperformed DMAP, yielding a higher depolymerisation conversion (96% vs. 89%) and a greater BPA-to-BPA-SP ratio (66% vs. 47%). PLA was found to completely depolymerise at 150 °C and 180 °C when both Lewis acids and bases were used. For PET, dual-catalyst systems incorporating DMAP showed greater activity than imidazole-based systems. The study identified three optimal reaction systems for a selective, sequential depolymerisation process: (a) MgCl2 at 150 °C for PLA depolymerisation, (b) MgCl2/imidazole at 150 °C for PC, and (c) Zn(OAc)2/DMAP at 180 °C for complete PET depolymerisation.
Olazabal et al. developed a novel low-temperature depolymerisation method for BPA- PC, achieving efficient conversion into trimethylene carbonate (TMC).451 The process relies on a solvent that enhances catalyst–polymer interaction and favours TMC cyclisation over polymerisation into poly(trimethylene carbonate) (PTMC). Using imidazole and 1-methylimidazole as the catalyst and solvent, respectively, BPA-PC was completely depolymerised at 50 °C in 3 hours, yielding 81% TMC. As sustainable TMC synthesis remains inefficient due to its tendency to ring opening, this approach offers a promising alternative. The method was also extended to other nucleophiles, showcasing its potential for producing carbonyl-containing cyclic molecules from plastic waste.
In addition to ionic liquids, metal chlorides and protic ionic salts also show exceptional catalytic activity. In particular, 1,5-diazabicyclo[4,3,0]non-5-ene (DBN), DBU, and TBD successfully degraded PET entirely at 190 °C within 10 min.435 Interestingly, a combination of TBD and methanesulfonic acid (MSA) in equal molar amounts achieved complete depolymerisation of PET in 2 h, producing BHET in 91% yield.456 Yue et al. used [Bmim]ZnCl3 ionic liquid for glycolysis of PET and achieved complete conversion with 84% selectivity for BHET using 0.16 wt% catalyst loading.457
Deep eutectic solvents (DESs) have garnered significant interest due to various advantages such as low toxicity, cost-effectiveness, simple preparation, and their comparable features to ionic liquids. These solvents have been successfully demonstrated for chemolysis of PET. Wang and co-workers used a DES system comprising [n(urea)/n(ZnCl2)] for glycolysis of PET at 170 °C and atmospheric pressure, and achieved 100% PET conversion with 83% BHET selectivity in 30 minutes.458 Similarly, Liu et al. utilised a 1,3-dimethylurea based deep eutectic solvent with 5 wt% Zn(OAc)2 in PET glycolysis and converted 100% of PET in 20 min at 190 °C with 82% selectivity for BHET.459 The high selectivity was attributed to the synergy between 1,3-dimethyl urea and Zn(OAc)2, demonstrating the potential of DESs for efficient PET recycling. In another study, Wang et al. have used 1,3-dimethylimidazolium-2-carboxylate and achieved complete depolymerisation in less than 1 hour at 180 °C, with a BHET yield of up to 60%.460
Metals containing ionic liquids have emerged as highly effective and environmentally friendly catalysts. These ionic liquids offer a great advantage of not emitting toxic materials into the environment, and have been used as catalysts for depolymerisation of various plastic wastes such as polyesters, PET, polycarbonates, nylon, etc.456,461,462 However, while acidic ionic liquids tend to degrade at temperatures above 180 °C, basic ionic liquids, despite their complex and costly synthesis, effectively accelerate the glycolysis depolymerisation of PET.424 Liu et al. carried out alcoholysis of waste PLA to produce lactate esters and achieved methyl lactate in 91% yield using a DBU based ionic liquid.463 They also investigated polyester depolymerisation, catalysed by imidazole-anion-derived ionic liquids ([HDBU][Im] ILs), via alcoholysis to recover the corresponding monomers.464 The authors investigated the detailed catalytic behaviour and catalyst recyclability for alcoholysis of polyester under mild conditions and compared the activity with the reported catalysts, the results highlighted the superior efficiency of [HDBU][Im] for polyester alcoholysis.
Al-Sabagh et al. conducted glycolysis of waste PET using bentonite impregnated with [Bmim-Fe] [(OAc)3], achieving 100% conversion and 44% BHET yield.465 Wang et al. recently synthesised composites of DES@ZIF-8, where different metal salts and acetamide were used to synthesise DES, and investigated their catalytic efficiency for glycolysis of PET.466 Under optimised conditions, PET was converted completely into BHET with 83.2% yield at 195 °C for 25 min. Thus, ionic liquids could be a reasonable choice of catalysts for depolymerisation reactions under mild and benign conditions. From a practical perspective, despite increased research using ionic liquids for recycling of polymeric waste, industrial scale adaptation remains rare. This is mainly due to the high costs of processes such as dry conditions, inert atmosphere, and expensive reagents. Wang et al. used CoFe2O4 modified with ionic liquids for glycolysis of PET, resulting in 100% conversion with 95.8% BHET yield.467 Wang et al. evaluated the synergistic effect of protic ionic liquids for glycolysis at 180 °C using a PIL-Zn(OAc)2 composite catalyst, and achieved complete conversion of PET in 20 min, furnishing BHET in 91.25% yield, which was significantly higher compared to that of either ionic liquids or metal salts alone,468 as ionic liquids remove a hydrogen atom from ethylene glycol's hydroxyl group, increasing its nucleophilicity. Simultaneously, Zn2+ ions interact with the carbonyl of the ester group in PET, enhancing the electrophilicity of the CO bond.469 The presence of Zn+2 can improve the nucleophilicity of ethylene glycol on the carbonyl group in the PET polyester. This highlights the potential of combining ionic liquids and metal salts for more efficient and high yield depolymerisation processes.
Microwave heating has also been investigated for the glycolysis of PET using different catalysts with some advantages over conventional heating. Generally, it has been observed that catalytic reaction rates are enhanced with simultaneous decrease in activation energy.470 Recently, Sirohi et al. evaluated the alcoholysis of PET waste using ZnCl2 as the catalyst and 1-decanol as the alcohol.471 The resulting oligomers were then used as the plasticiser in polynitrile and nitrile/PVC rubber blends.
Kim and co-workers used superparamagnetic g-Fe2O3 nanoparticles for PET depolymerisation, achieving 90% BHET yield in 1 h at 300 °C.474 Moreover, the catalyst was recycled 10 times without significantly affecting the BHET yield. Additionally, two dimensional layered iron nanosheets have recently been introduced for PET glycolysis.475 The ultrathin FeIII and FeII/FeIII layered double hydroxide (LDH) nanosheets have a hexagonal structure, and completely converted PET with 100% BHET yield in 30 minutes at 200 °C. The FeIII nanosheet catalyst showed excellent recyclability and gave 96.7% BHET yield after 5 cycles. Al-Sabagh et al. developed a sustainable Fe3O4-boosted multiwalled carbon nanotube (MWCNT) catalyst for PET glycolysis and obtained 100% BHET yield in 2 hours at 190 °C.476 The outstanding performance was attributed to the synergistic effects by magnetite and MWCNTs. In another study, Lima et al. used titanate nanotubes (TNTs) for glycolysis of virgin PET and obtained 84% BHET yield in 2 h, which is slightly better compared to the commercial Zn(OAc)2 catalyst.477
Graphene oxide has been used as a support to prepare nanocatalyst materials for PET glycolysis. Park et al. prepared a graphene oxide–manganese oxide (GO–Mn3O4) nanocomposite and obtained 96.7% BHET yield using this catalyst at 300 °C.478 To reduce the temperature requirement and optimise BHET yield, Jin et al. incorporated MnO2 on holey graphene oxide (HGO) nanosheets via the oxidative etching method.479 This catalyst completely converted PET to BHET in 100% yield in 10 min at 200 °C. The presence of abundant active sites, resulting from the interaction of the large surface area of GO with MnO2, was claimed to enhance the overall catalyst activity. Hexagonal boron nitride (h-BN) has also been used as a support for glycolysis of PET. Kim et al. synthesised metal nanoparticles deposited hexagonal boron nitride (hBN) nanohybrid catalysts for PET glycolysis.480 Metal nanoparticles containing Pd, Pt, Ag, and RuO2 were investigated and it was found that Pd/hBN catalysed glycolysis achieved 98.4% PET conversion in 30 min at 100 °C, and produced BHET in 92.1% yield. The glycolysis of PET has also been investigated using various other transition metals. For example, the use of Fe2O3 nanoparticles encapsulated in the hexagonal boron nitride nanosheets (h-BNNS) achieved BHET in 100% yield after 5 hours at 200 °C.481 This exceptional activity was attributed to the unique polarity of boron nitride bonds and hexagonal nanostructures, which enhanced the catalyst efficiency.
Fehér et al. tested various organocatalysts, silica gel functionalised with organic bases including trialkylguanidine (Si-GUA), dialkylthiourea (Si-THU), trialkylamine (Si-TEA), and triazabicyclodecene (Si-TBD) for glycolysis of PET.482 Trialkylamine functionalised silica exhibited the highest thermal stability and decent catalytic activity, whereas triazabicyclodecene functionalised silica furnished the highest activity. Similarly, both trialkylamine and triazabicyclodecene functionalised silicas produced high cumulative yields of 89% and 88%, respectively.482 One of the latest advancements in catalytic PET glycolysis involves the use of zeolite as the support material.483 Doping the zeolite support with an optimal amount of ZnO enhances its activity compared to other SBA-15 supported metal oxide catalysts (Fig. 25). SBA-15 offers high acid strength, high surface area, and significant pore volume, which facilitates the dispersion of the active catalyst component and the adsorption of reactants. Increasing the zinc content from 1% to 5% increased PET conversion as well as the yield of BHET, however, catalyst efficiency started to decline after further increase in zinc concentration (Fig. 25). Consequently, the 5% ZnO/SBA-15 catalyst efficiently facilitated PET glycolysis, achieving a BHET yield of 91% at 197 °C. The catalyst exhibited good stability and recyclability for PET degradation. Notably, this catalytic method produced colourless BHET, which is more valuable for industrial applications. Additionally, bimetallic zeolites present an emerging alternative f or catalytic PET glycolysis.
 | ||
| Fig. 25 PET glycolysis with SBA-15 supported with different metal oxide catalysts (left). PET conversion with different ZnO loadings (right). Adapted with permission from ref. 483. Copyright 2022, Elsevier. | ||
 | ||
| Fig. 26 Sluggish kinetics of BHET formation from PET glycolysis. Adapted with permission from ref. 472. Copyright 2013, Elsevier. | ||
Metal oxides are the most investigated catalysts due to their Lewis acid cationic sites, tuneable properties, and wider availability, making them highly effective in depolymerisation reactions. The catalytic sites in mixed metal oxides (MMOs) could be increased by modifications in the electronic structure of active metals, which increases the interaction between the catalyst and substrate, hence the rate of the reaction increases.484 Moreover, their acid–base properties can be modified by changing the molar ratio of individual metal precursors.485 BHET is the most widely reported product of PET glycolysis using metal oxides as catalysts. Layered double hydroxide (LDH) catalysts have been used for PET glycolysis due to their cost effectiveness and benign synthesis.486 Chen and co-workers pioneered the use of Mg–Al mixed oxides, synthesised from hydrotalcite calcination, for PET glycolysis.487 These oxides showed higher catalytic activity. In particular, the mixed oxide with an Mg/Al molar ratio of 3, calcined at 500 °C, exhibited the highest activity for PET glycolysis, affording BHET in 81.3% yield. The high catalyst performance was due to the moderate basic sites of the catalyst. Guo et al. synthesised nanosized Mg–Al double oxides sintered on Fe3O4 microparticles, offering a cheap and sustainable catalyst for PET glycolysis.488 The synthesised hierarchically structured Mg–Al–O@Fe3O4 catalyst demonstrated high activity and afforded 80% yield of BHET due to its large active surface area. However, the catalyst efficiency decreased after two cycles because of active site blockage, although it can be regenerated by heat treatment.
Zn–Al hydrotalcites have been shown to be effective catalysts for PET glycolysis.489 The basicity and surface area of Zn–Al hydrotalcite mixed oxides were significantly influenced by the calcination temperature.490 Al3+ addition decreased the basicity of the catalyst, however increasing the calcination temperature increased the basicity. The Zn–Al hydrotalcite catalyst calcined at 500 °C (ZnAl-3 catalyst) gave a higher PET conversion (92%) and BHET yield (79%) compared to other tested catalysts. In a separate study, Eshaq et al. added Mg2+ cation to modify its acid–base, resulting in the (Mg–Zn)–Al LDH catalyst that completely converted PET at 196 °C, producing BHET in 75% yield.491 Sulphated metal oxides have shown good catalytic activity in PET glycolysis. Zhu et al. synthesised a range of sulphated solid acid catalysts, such as S/Ti, S/Zn, and S/Zn–Ti, by varying calcination temperatures from 200 °C to 600 °C.492 Among these, the sulphated S/Zn–Ti mixed oxide was hypothesised to develop more moderate acidic sites than the single metal oxide. Particularly, the S/Zn–Ti catalyst calcined at 300 °C demonstrated the highest efficiency, achieving complete PET conversion and yielding 72% BHET. This improved performance was attributed to its optimal acidity and high surface area.
The application of clays usually depends upon the special properties of clay particles, particularly their chemical properties, which include active internal and external surfaces. Kaolinite, a natural, cost-effective, and environmentally benign material, has been widely used as an additive, adsorbent, coating, and filler. In addition, kaolinite has been used as a catalyst to promote organic reactions including petroleum cracking or depolymerisation. Clay catalysts have been commonly used for hydrolysis of cellulosic polymers and lignin depolymerisation.493–495 Jeya et al. investigated the depolymerisation of post-consumer PET via glycolysis using Al3+, Fe3+ and Zn2+ containing kaolin clays as catalysts.496 The clay catalysts containing 0.5–5.0 wt% of metals including Al3+, Fe3+ and Zn2+ were synthesised using the wet impregnation method, and their effectiveness in catalysing the glycolytic depolymerisation of post-consumer beverage bottles using EG was evaluated. BHET was obtained as a major product, with its yield significantly enhanced as the metal ion content in the catalysts increased from 0.5 to 5.0 wt%. Al and Zn-kaolin catalysts with 5 wt% metal ion loading afforded much higher yield of BHET, up to 85%, compared to Fe-kaolin catalyst under optimised conditions.496
8.2 Aminolysis
The aminolysis process can be performed under atmospheric pressure and at significantly lower temperatures (<200 °C) compared to glycolysis and hydrolysis. This method has been extensively investigated for polyurethane (PUR) recycling. The basicity of the reaction medium is a key factor influencing the decomposition rate of PUR chains, and varies depending on the solvents used, such as alkanolamines, amines, and ammonia. The effective depolymerisation catalysts are KOH and NaOH, when combined with alkanolamines, enabling complete depolymerisation of PUR foams. The aminolytic routes for PUR depolymerisation are detailed in the review by Bhandari et al.497Ghorbantabar et al. carried out a catalyst-free aminolysis of PET using excess monoethanolamine at 160 °C, achieving bis(2-hydroxyethylene) terephthalamide (BHETA) in 85% yield.498 Fukushima et al. used the triazabicyclodecene (TBD) catalyst for aminolysis of post-consumer PET to produce various crystalline terephthalamides.499 These materials include additives and chemical building blocks for advanced material applications. The aminolysis reactions were performed under milder conditions (110–120 °C) due to the thermodynamic favourability of aminolysis compared to alcoholysis. The bifunctional nature of TBD played a critical role in aminolysis to produce terephthalamides in good yields. The aminolysis products of PET have been recently investigated to be used for asphalt modifications.500 In recent investigation, researchers carried out nucleophilic aromatic substitution polymerisations on terephthalamide monomers to produce poly(arylether sulfone-amide)s (PAESA), which are thermoplastic materials.501 The terephthalamide was incorporated at 10–30 mol% and the resulting PAESA materials showed strong thermal characteristics, including a maximum Tg of 215 °C due to hydrogen bonding. Furthermore, these polymers were found to have high mechanical strength, with tensile strengths of 40–60 MPa and ductility between 6 and 11%. Tawfik and Eskander studied the depolymerisation of waste PET bottles through aminolysis, utilising ethanolamine in the presence of a dibutyl tin oxide catalyst.502 Ethanolamine was particularly effective in this process because it has an amino group and a hydroxyl group, which increases its ability to cleave ester bonds in polyester. This resulted in the formation of BHETA, which can serve as valuable feedstock to produce polyurethanes.
Holmes carried out the aminolysis of PET fibres using n-butyl amine, resulting in the cleavage of the polymer chain.503 The process left one end of the chain with an amide group, while the other had a hydroxyl group. This cleavage led to changes in PET's properties, including reduction in molecular weight and tensile strength. Consequently, the crystallinity of fibres increased, leading to the formation of surface cracks. Recently, Syeda et al. investigated the glycolysis of PET with amino alcohols (AmOH's) at 180 °C under atmospheric pressure, and produced oligo ethylene terephthalate in high yields (98%).504 These oligo ethylene terephthalates were subsequently converted back to PET. Amino alcohols were found to exhibit greater catalytic reactivity than tertiary amines. These results highlighted the industrial potential of amino alcohols as effective organic catalysts, capable of not only producing oligomers in large yields but also generating BHETA.
In addition to conventional heating methods, microwave heating presents an excellent alternative for aminolysis of PET wastes, offering efficient processing without sacrificing product yield. As is well known, microwave irradiation has significant advantages over conventional heating, including fast, non-contact and selective heating, which reduces reaction time and improves product conversion. This technique relies on in-core heating, where microwaves interact directly with the molecules of the reaction mixture, causing polar molecules to rotate and generate heat via dipolar polarization. The efficiency of this energy conversion is characterized by the dielectric loss tangent.505 Pingale and Shukla used microwave irradiation to supply thermal energy for the aminolysis of PET with ethanolamine, mediated by sodium salts as catalysts, and produced BHETA in excellent yield (>90%) in just 4 minutes.506Scheme 3 illustrates the proposed mechanism in which the sodium ion from the catalyst forms a complex with the –CO of the ester. This facilitates the attack of ethanolamine's NH group on the –CO bond of PET, thereby initiating BHETA formation. While ethanolamine possesses two nucleophilic centres, amine being more nucleophilic than hydroxyl attacks the ester linkage of PET. Different sodium salts showed similar performance, with the reaction largely driven by the PET/ethanolamine ratio. In another study, using Na2CO3 as the catalyst in hydrazine monohydrate solvent resulted in the production of terephthalic dihydrazide in 84% yield. The catalyst significantly reduced the reaction duration from 24 h to 3 h.507 Bäckström et al. recycled PET flacks by microwave assisted aminolysis under catalyst free conditions and produced terephthalamide in 61 wt%, 91 wt%, 82 wt%, and 64 wt% yields using allylamine, ethanolamine, furfurylamine, and hexylamine as solvents, respectively.508 Terephthalamide (TA) is a promising constituent for plastic films or plasticisers for polylactic acid, and exhibits superior strain tolerance than virgin polylactic acid. The authors further transformed the resulting diallylterephthalamide through the thiol–ene reaction to produce a resin suitable for film fabrication (Scheme 4).508
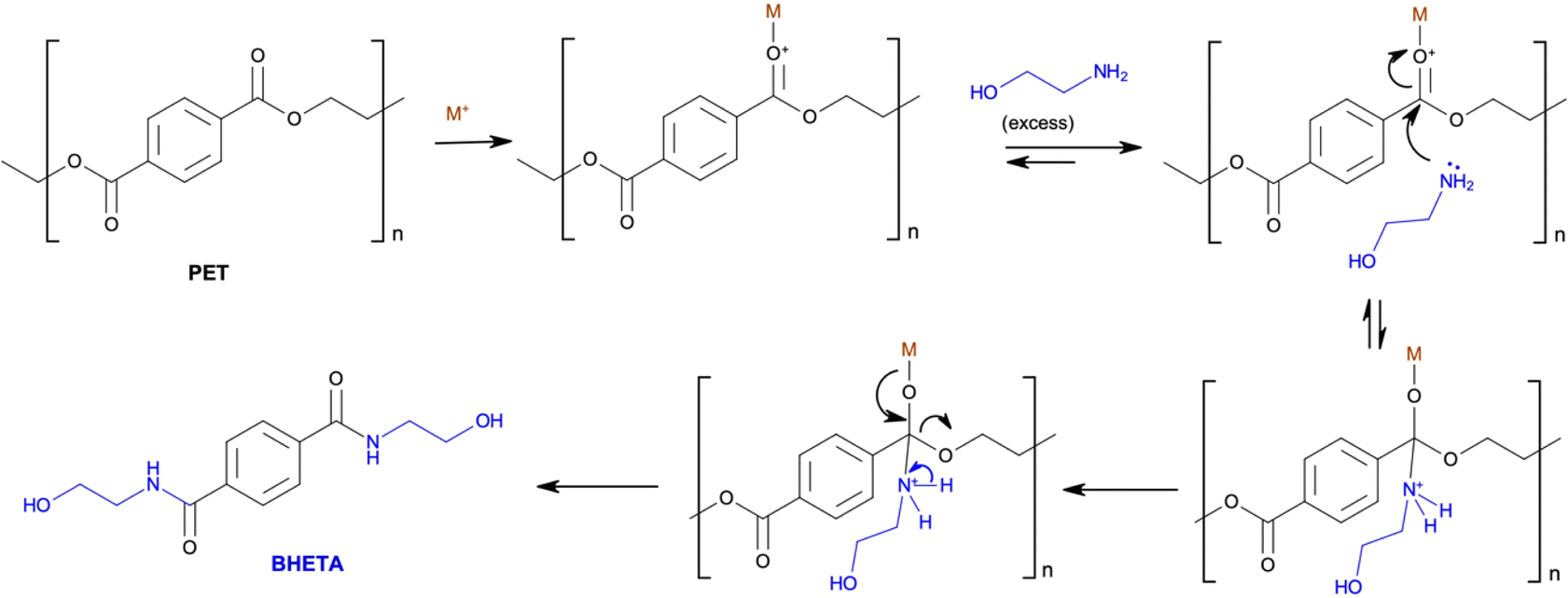 | ||
| Scheme 3 Mechanism of PET aminolysis catalysed by a metal containing catalyst. | ||
 | ||
| Scheme 4 PET recycling for the fabrication of plastic films. Reproduced from ref. 508. | ||
When ammonia serves as solvent, the process is known as ammonolysis and can be carried out at relatively low temperatures. Du Pont has commercialised a phosphate-catalysed process for ammonolysis of PUR carpets, though harsher temperature and pressure conditions are required. Despite the numerous beneficial properties of ammonolysis products, the incorporation of nitrogen groups in the resulting material complicates its integration into PET manufacturing processes that rely on BHET polymerisation. This requires additional conversions, reducing its economic attractiveness. Therefore, this process is primarily valuable for producing terephthalamides for specialised applications such as Kevlar production.
PET can undergo ammonolysis with ammonia to produce terephthaldiamide (TPHA) and EG as primary products at 70–180 °C under low pressure, typically with catalysts such as zinc acetate. The intermediate products of ammonolysis include 1,4-bis(amino-methyl)cyclohexane, 1,4-bis(amino-ethyl) cyclohexane, terephthalonitrile, and p-xylylenediamine.424 TPHA can be converted to terephthalyl dinitrile, which can be further hydrogenated to p-xylenediamine (bis-(aminomethyl)benzene) or 1,4-diaminodimethylcyclohexane. p-Xylenediamine can be used in the production of heat-resistant polymers, as an epoxy resin hardener, corrosion inhibitor, etc. Kenneth et al. developed a low-pressure process for ammonolysis of PET using NH3 in an EG environment with Zn(CH3CO2)2 catalyst at a concentration of 0.05 wt%.509 The process was carried out at 70 °C, producing terephthalamide with a yield of about 87%. Gupta and Bhandari studied the chemical depolymerisation of PET bottles via aminolysis and ammonolysis, yielding diamides of TPA.510 The conventional analytical pathways to determine the aminolytic degradation of PET waste were investigated by Ghorbantabar and co-workers.498 Bäckström et al. synthesised a series of terephthalamides by the aminolysis of PET using microwave heating under catalyst-free conditions.508 Demarteau et al. used organocatalysts, such as a mixture of triazabicyclodecene and methanesulfonic acid (TBD:MSA), and triazabicyclodecene and benzoic acid (TBD:BA), for aminolytic upcycling of PET waste using various amino-alcohols, and subsequently used these alcohols to produce poly(ester-amide)s.511 Recently, Gabrielli et al. used different β-hydroxy amines for aminolysis of PET water bottles using biocompatible catalysts, such as CH3COONa, at 180 °C with microwave heating under solvent-free conditions, and obtained terephthalamide diol monomers in a high yield of up to 98%.512 The terephthalamide diol monomers were later used to synthesise poly(urethane acrylates).
BPA-PC can be recycled by aminolysis to produce BPA monomer that can be used as feedstock to make new polymers. Singh et al. developed an efficient method to convert waste polycarbonate into urea derivatives through reaction with primary amines under catalyst and solvent-free conditions at 80 °C.513 This process enabled the direct extraction of carbonyl groups from polycarbonate, yielding functionalised urea compounds and BPA from waste sources such as CDs and DVDs. The authors also optimised the process for maximum polymer-to-urea conversion while avoiding chromatographic purification. In another study, Quaranta et al. valorised PC waste by converting it into BPA and polyureas (PUs) through a non-isocyanate route that involved the reaction of BPA-PC with diamines at a temperature of 120 °C in THF, yielding BPA (83–95%) and polyureas (78–99%).514 Basic diamines (e.g., 1,6-diaminohexane, 4,7,10-trioxa-1,13-tridecanediamine, m-xylylenediamine, p-xylylenediamine) facilitated the reaction without catalysts, while less reactive aromatic diamines (e.g., 4,4′-diaminodiphenylmethane; 2,4-diaminotoluene) required base catalysts such as NaOH or DBU. The process proceeds via a carbamation step, forming urethane intermediates that are further converted into polyureas and additional BPA.
Haung et al. demonstrated the feasibility of recycling PC waste into high-performance thermoplastic polyurethanes (TPUs) via selective aminolysis.515 The process generated monomer mixtures incorporating newly introduced flexible ether linkages, which, without prior purification, can be directly re-polymerised into TPUs using isocyanate reagents. The enhanced reactivity of terminal phenolic hydroxyl groups enabled the formation of high molecular weight TPUs (Mw: 3000 to 158000 g mol−1). By incorporating low-melting polyether-type polyols and optimising reaction conditions, the TPUs exhibited distinct phase separation morphologies (10–25 nm domain sizes). Wang et al. recently developed a ZnX2-catalysed amino-alcoholysis method to upcycle PC waste into BPA monomers and high-value chiral 2-oxazolidinones under mild conditions.516 This method also enabled the sequential depolymerisation of PC and PET mixed plastics with excellent yields and selectivity. Various zinc catalysts, including Zn(OAc)2, ZnEt2, Zn(HMDS)2, and ZnCl2, exhibited high activity, with Zn(HMDS)2 being the most effective, though ZnCl2 was identified as more cost-efficient for industrial applications. The approach effectively recovers BPA while utilising the carbonyl groups in BPA-PC to generate valuable chiral chemicals. Furthermore, PC plastics were successfully processed using this method.
8.3 Hydrolysis
Hydrolysis has been moderately investigated for depolymerisation of PET (Fig. 27). In this process, the ester bonds in PET are hydrolysed by water, operating in acidic, alkaline, or neutral condition media at elevated temperatures and pressures, breaking the polymer chains to monomers such as TPA and EG.517 Neutral hydrolysis typically utilises water or steam at temperatures between 200 and 300 °C in the presence of catalysts like alkali metal acetates. This environmentally benign process doesn't cause pollution, however, impurities in PET waste remain in the product, which is a major drawback. Hydrolysis proceeds slowly compared to glycolysis and methanolysis as water acts as a weaker nucleophile compared to ethylene glycol and methanol. Acid hydrolysis is commonly carried out using conc. H2SO4, HNO3, or H3PO4.518 Sulfuric acid can lead to complete depolymerisation of PET into monomers. However, this requires highly concentrated sulfuric acid (>80% by weight). Even a slight reduction in acid concentration can slow down the reaction by more than 50%.518 Acid hydrolysis can take place at low temperatures and pressure (25–90 °C and 1 atm), but reactor corrosion as well as purification of decomposition products from sulfuric acid and inorganic salts are major challenges.519 Sulfonic acid catalysts have shown strong potential for PET hydrolysis. Yang et al. reported a 96.2% yield of terephthalic acid (TPA) using p-toluenesulfonic acid (PTSA) at 150 °C within 90 minutes, maintaining consistent performance over five cycles.520 Other sulfonic acids, including 2-naphthalenesulfonic acid (2-NSA), and 1,5-naphthalenedisulfonic acid (1,5-NDSA) also yielded over 90% TPA under similar thermal conditions.521 However, the required reaction times varied significantly, with PTSA, NSA and 1,5-NDSA requiring 3, 3, and 8 hours, respectively for comparable yields. Mishra et al. performed an in-depth study on the kinetics and thermodynamics of nitric acid catalysed PET hydrolysis.522 However, severe corrosion and formation of a large amount of inorganic salts remain significant drawbacks in acid hydrolysis. To address these challenges, Ügdüler et al. performed alkaline hydrolysis of pure PET using 5 wt% NaOH in an ethanol:water mixture (60:40 vol%) at 80 °C and obtained 95 wt% yield of TPA, which was separated by filtration.523 During alkaline hydrolysis of PET in NaOH solution, PET was completely depolymerised at 200–250 °C under high pressure for a longer reaction duration, forming a solution that contained the sodium salt of TPA. Then, by subsequent acidification, TPA is precipitated as a solid. Hydrolysis is the only known method for depolymerisation of PET, allowing the production of pure TPA and EG, which can be used for producing new PET. However, this method has significant drawbacks as it requires high operating pressures (1.4–2 MN m−2), temperatures (200–250 °C) and longer reaction times (3–5 hours or more). In addition, TPA purification is quite expensive, which makes this method less attractive for some industries, especially for those producing food-grade recycled PET.424 Depolymerisation of waste PET by hydrolysis has been reviewed briefly in the literature, i.e. M. Han in 2019 compared hydrolysis and methanolysis from a process perspective, including industrial feasibility studies524 and Pohjakallio et al. in 2020 provided a brief description of the technical aspects of hydrolysis.525
 | ||
| Fig. 27 (A–D) Depolymerisation of PET with phase transfer catalysts (PTCs). (A) (TOMAB is the highly studied PTC with microwave heating.) Adapted with permission from ref. 526. Copyright 2010, John Wiley & Sons. (B) Influence of reaction temperature on the evolution of the TPA yield. Adapted with permission from ref. 527. Copyright 2014, Elsevier. (C) Mechanism of PET hydrolysis, adapted with permission from ref. 528. Copyright 2012, Elsevier. (D) Lamellar mechanism, adapted with permission from ref. 527; Copyright 2014, Elsevier. (E) PUR hydrolysis to diamine and 1,4-butanediol. (F) Depolymerisation yield of aliphatic (H-PU and I-PU) and aromatic (M-PU) PUR, adapted with permission from ref. 529. Copyright 2017, John Wiley & Sons. | ||
Hirota et al. investigated the depolymerisation of PET by hydrolysis within the pH range of 3.0–10.5 at 80 °C, and obtained TPA and EG in high yield at pH 10.5.530 The hydrolysis of PET waste and methyl benzoate under alkaline conditions at 190–200 °C resulted in TPA and benzoic acid formation with 87–95% and 84–89% yields, respectively.531 Yoshioka et al. explored the effect of NaOH concentration on the distribution of products, including EG, TPA, CO2 and oxalic acid.532 The oxalic acid was formed as a result of base-catalysed oxidation and exhibited first-order dependence on OH− concentration. Zhang et al. recently developed a binuclear zinc catalyst for intramolecular PET hydrolysis.533,534 Their kinetic studies and DFT calculation revealed that substituents on the binuclear zinc complex strongly influenced the rate limiting step. Substituents that enhanced the electron density at catalytic centres significantly accelerated the hydrolysis. Notably, the catalyst incorporating n-pentyl groups showed a remarkable specific activity of 778 ± 40 gPET h−1 gcat−1 in 0.1 M NaOH, which is 23 times more efficient compared to traditional alkaline hydrolysis approaches.533
Recently, a new metal-free catalyst with multiple hydrogen bonds has demonstrated exceptional activity in the hydrolysis of PET.535 This catalyst provides almost complete depolymerisation of PET (≥99%) under relatively mild conditions (100 °C, pH ≈ 8). Mechanical studies have shown that hydrogen bonds play a crucial role in stabilizing the transition state, and both the number and strength of these interactions are key factors in the effectiveness of the catalyst. This approach offers a sustainable and efficient alternative to PET processing, potentially reducing dependence on metal-based catalysts and providing milder reaction conditions. Cations containing two N–H groups replaced the oxyanion hole, while anions containing carboxyl groups activated nucleophilic water molecules, assuming the role of a traditional catalytic triad. Based on this structural feature, [TBDH]+ (with two N–H groups) was chosen as the cation and [HCO3]− as the anion to facilitate the hydrolysis of PET under mild conditions.
Alkaline hydrolysis of PET has also been carried out using phase transfer catalysts including tetrabutylammonium bromide, tetrabutylammonium iodide, trioctylammonium bromide,536 and ultrasonic assisted radiation.537 More insightful work on depolymerisation of waste PET focusing on the catalyst and mechanistic understanding was carried out using phase transfer catalysts (PTCs) under alkaline conditions (Fig. 27).526,528 Quaternary ammonium salts including trioctylmethylammonium bromide (TOMAB) and hexadecyltrimethylammonium bromide (HDTMAB) were investigated as phase transfer catalysts. Good performance was achieved with appropriately sized alkyl groups, which facilitated polymer solvation and avoided steric hindrance. Kinetic studies concluded that TOMAB catalyst reduced the activation energy from 99 to 83 kJ mol−1, supporting the hypothesis that phase transfer catalysts worked with an interfacial mechanism instead of an extraction mechanism, thereby enabling OH− ions to interact with PET chains more effectively.538 Applying microwave irradiation with TOMAB catalyst resulted in shortening the reaction time to less than 30 minutes for almost complete depolymerisation of PET (97%) (Fig. 27A).526 By following a similar approach, Khalaf and Hasan jointly applied tetraethylammonium iodide catalyst and microwave irradiation to completely convert PET into TPA in 60 min.528 They suggested a similar interfacial mechanism to that proposed by Glatzer et al.538 above (Fig. 27C), where the metal carbanion (MR) is formed at the interface between organic and aqueous phases, initiating the reaction. A phase transfer catalyst then extracts MR into the organic phase, where it transforms into a highly reactive intermediate (RQ). This intermediate is subsequently attacked by the OH− anion, significantly accelerating the depolymerisation process. Barredo et al. conducted PET depolymerisation at temperatures of 80–100 °C and atmospheric pressure using tributylhexadecylphosphonium bromide (TBHDPB) as a quaternary salt catalyst.539 Optimal results were achieved after 4 hours at 100 °C, with a PET particle size of 1–1.4 mm, a catalyst-to-PET ratio of 0.2, and stirring rate of 525 rpm. Under these conditions, PET conversion of 99.9% and a TPA yield of 93.5% were obtained, demonstrating an effective balance between conversion efficiency and product yield.
Quaternary ammonium polytungstophosphates as dual phase transfer catalysts have been used to depolymerise PET (Fig. 27B).527 Kinetic studies found an activation energy of 68 kJ mol−1 for PET hydrolysis at temperatures of 115–145 °C and proposed a chain-end scission mechanism (Fig. 27D), where depolymerisation occurred on the external surface and PET flakes were lamellarly depolymerised. However, the authors ignored the autocatalytic effect during hydrolysis, attributed to protons from the carboxylic group of the depolymerisation products.538 For such an autocatalytic model, a reaction order of 0.5 was proposed for carboxylic acid.540 Based on these results, ionic liquid catalysts have shown high performance for PET depolymerisation under mild operation conditions. However, the high cost of this process along with concerns for the separation and recyclability is a key factor in hindering their applicability for large scale processes.
De Paula et al. demonstrated hydrolytic depolymerisation of PET and the bioderived based polyester poly(ethylene 2,5-furandicarboxylate) (PEF) using eutectic solvents under alkaline conditions.541 The approach effectively broke down the polymers into their respective monomers, TPA and 2,5-furandicarboxylic acid (FDCA). Using a design of experimental methodology, the process was optimised to achieve monomer recovery yields exceeding 90% for TPA and 80% for FDCA under mild conditions, with reaction temperatures below 150 °C and durations under five hours. Structural analyses confirmed the high chemical purity of the recovered monomers. Furthermore, the eutectic solvent exhibited excellent recyclability, with minimal loss of activity over successive cycles, supporting waste minimisation.
The efficient depolymerisation of PET by alkaline hydrolysis at low temperatures has been successfully demonstrated to produce high-quality TPA.542 A key discovery was the significant effect of pH regulation during acidification, where maintaining a pH of 2 resulted in an impressive TPA yield of 99.6%. The required amount of HCl for acidification was directly proportional to the amount of NaOH used in alkaline hydrolysis, where higher concentrations of NaOH increased PET conversion but increased overall reagent costs. A conversion rate of up to 90% was achieved using 23 g of PET waste with a 30:70 ratio of water and ethanol, 2.2 molar ratio of NaOH to PET, 10% NaOH concentration, and a 30-minute reaction time. Optimal depolymerisation was achieved using PET granules in a water to ethanol ratio of 30:70, with 10 wt% NaOH in 100 mL of solvent, at 82 °C for 60 minutes. Wu and co-workers reported the hydrolysis of PLA using diphenyl phosphate (DPP) as an organocatalyst under solvent-free conditions, without the need for high pressure or an inert atmosphere.543 The process yielded oligomeric products with low moisture content within 1.5 hours. The resulting oligolactic acid (OLA) oligomers can be directly used to synthesise lactide (LA) followed by conversion to PLA through the established OLA–LA–PLA industrial cycle, enabling closed-loop recycling of PLA. DPP demonstrated remarkable catalytic durability, maintaining consistent product quality over ten consecutive reaction cycles. By increasing the water content during hydrolysis, the process also produced lactic acid solution with preserved stereochemistry and a purity comparable to commercial-grade lactic acid.
Azeem et al. developed a highly efficient two-step microwave-assisted method for PET depolymerisation, beginning with a microwave pre-treatment using glycerol as a green reagent, enhancing PET's susceptibility to degradation.544 Optimisation via Box–Behnken design identified the ideal conditions, 12 mL glycerol and 182 W microwave irradiation for 3 minutes, achieving 11% weight loss, increased carbonyl index (up to 4.22), and 33% crystallinity. Subsequent microwave-assisted hydrolysis using sodium bicarbonate and EG achieved 99.9% PET conversion within 3 minutes, yielding 79.1% TPA, 17.6% monohydroxyethyl terephthalate (MHET), and 1.8% BHET. The recovered TPA, with 95% purity and favourable physicochemical properties, was confirmed suitable for repolymerisation into virgin PET. Benninga and colleagues demonstrated a highly efficient microwave-assisted depolymerisation method for poly(p-phenylene terephthalamide) (PPTA), commercially available as Kevlar or Twaron.545 The process used alkaline hydrolysis in a microwave reactor, operating at 240–260 °C for durations between 1 and 15 minutes. Optimal depolymerisation occurred at 260 °C after 15 minutes, achieving a conversion rate of 96%. The resulting monomers, TPA and p-phenylenediamine, were isolated with purities exceeding 99% via sequential extraction and precipitation. This study represents the fastest reported depolymerisation of PPTA under mild conditions and offers a promising route towards a circular life cycle for high-performance aramid polymers.
Pereira et al. investigated the hydrolysis of PET in pure water over a temperature range of 190–400 °C and pressures between 1 and 35 MPa, resulting in the formation of TPA.546 They used various physical states of water including saturated vapour, superheated vapour, saturated liquid, and supercritical fluid to treat both solid and molten PET. Among these, hydrolysis of molten PET in saturated liquid water produced the highest TPA yields. Notably, rapid heating of the reaction mixture at a rate of 5–10 °C s−1 enabled high TPA yields in 1 minute, significantly outperforming the traditional 30-minute isothermal approach.
Onwucha et al. investigated the neutral hydrolysis of PET, highlighting its advantages in reducing catalyst use, simplifying product purification, and minimising waste generation.547 Their findings showed that extended reaction times and high PET-to-water ratio significantly improved the selectivity, yield, and purity of TPA. In particular, the TPA yield increased from approximately 86% to 98% as the hydrolysis duration was extended from 6 to 24 hours, confirming the effectiveness of catalyst-free hydrolysis under optimised conditions.
Regarding PUR recycling, Motokucho et al. investigated the depolymerisation of aliphatic and aromatic polyurethanes using pressurised CO2 in water, where in situ produced carbonic acid acted as a catalyst (Fig. 27E).529 Various polyurethanes, such as poly(1,4-tetramethylene 1,6-hexamethylene dicarbamate), poly(methylene bis-(1,4-phenylene)hexamethylene dicarbamate), etc. were successfully depolymerised into their respective diamines and 1,4-butane diol in high yields after 24 h (Fig. 27F). The urethane linkages in aliphatic structures were selectively cleaved more efficiently, possibly due the higher hydrophilicity around the urethane linkages in the terminal moieties of these polymer. The mechanism involves the carbonic acid attacking the carbonyl group, followed by hydrolysis to cleave the urethane group.548
Polylactide (PLA) is a renewable polyester produced by condensation polymerisation of lactic acid monomers. However, it is generally more expensive than petroleum-based polymers due to the costly fermentation and purification processes involved in lactic acid production.549 Biodegradation of PLA can take place in duration of 12 to 52 weeks.550 Consequently, there is increasing research interest for efficient recycling of polylactide. Piemonte and Gironi performed hydrolysis of PLA at 180 °C using PLA-to-water in a mass ratio of 2:1, obtaining lactic acid in 95% yield within 2 h.551 Hirao et al. used microwave radiation for hydrolysis of PLA with a PLA-to-water ratio of 3:1 and compared the yield and reaction duration with conventional heating. With microwave-assisted heating, 45 wt% yield of lactic acid was obtained in 2 h compared to the same yield produced in over 13 h by conventional heating.552 Liu et al. conducted hydrolysis of PLA using [Bmim][OAc] ionic liquid at 130 °C, achieving 93.93% conversion and 76.08% yield of calcium lactate.553
Hydrolysis of PLA under acidic and basic conditions follows alternate pathways for depolymerisation (Scheme 5). Under acidic conditions, the protonation of the terminal OH group of PLA promotes hydrolysis, leading to direct conversion to lactic acid by a chain-end scission mechanism.554 The degradation rate under acidic conditions depends on the polymer chain length due to the increased hydrophilic nature of the chain end. However, under basic conditions the back-biting reactions lead to a random chain scission to produce lactam, which on subsequent hydrolysis is converted to lactic acid (Scheme 5).555
 | ||
| Scheme 5 Acid and base catalysed mechanism of PLA hydrolysis. | ||
Song et al. explored the use of various ionic liquids, acting simultaneously as solvents and catalysts, for the hydrolysis of PLA.553 [Bmim][OAc] achieved the highest conversion to lactic acid, with yields up to 94% at 130 °C in 2 h. The ionic liquid facilitated the partial solubilisation of PLA, and the ions assisted the hydrolysis mechanism. The calcium lactate product was isolated by adding calcium carbonate, yielding up to 76%. Remarkably, the ionic liquid maintained its performance over seven cycles. However, from an economic perspective, complete hydrolysis of PLA is deemed impractical due to the significant energy costs involved in removing water, coupled with the racemisation of lactic acid. Additionally, alcoholysis of PLA lacks stereospecificity.556 Despite the challenges, NatureWorks has pioneered an efficient hydrolysis process to convert off-grade PLA resin to lactic acid.557 Since 2004, more than 17 million pounds of PLA resin have been hydrolysed with this process.
Polycarbonate has been shown to undergo rapid hydrolysis under high pressure, high temperature steam (300 °C), producing bisphenol A (BPA) in 5 min, with a maximum yield of 80%.558 PC was completely decomposed in high pressure steam at 300 °C, but it didn't decompose in the liquid water phase even after 50 min. The excellent yield was attributed to its remarkable stability in high pressure steam. This process also proved to be cost-effective, as the elevated pressure significantly reduced the amount of water needed for PC degradation. Quaranta et al. explored the use of Lewis acids, specifically M(O3SCF3)3 (M = La, Yb, Sc) triflate salts, to catalyse the hydrolysis of PC, offering a promising route for its chemical recycling.559 In THF, these salts effectively depolymerised PC into BPA and CO2. Among them, La(O3SCF3)3 exhibited the highest BPA yield and selectivity (97%), minimising the undesired conversion of BPA into and 4-isopropylphenol, phenol, and 4-isopropenylphenol. In 2021, they used natural clay as the catalyst for depolymerisation of poly-(bisphenol A carbonate) wastes.560 The authors investigated the natural clinochlore clay for the hydrolysis reaction of poly-(bisphenol A carbonate) in THF solvent at 200 °C. The clinochlore efficiently promoted the depolymerisation (up to 99%, after 6 h) of PC by H2O and afforded bisphenol A monomer with nearly 99% selectivity.
Sun et al. investigated the catalytic degradation of PC using copper sulfide nanoparticles (CuS NPs) in DMF and DMSO solvents.561 In DMF solvent, PC underwent hydrolytic depolymerisation, producing BPA with ∼80% yield. Conversely, degradation in DMSO resulted in the formation of oligomeric or branched/crosslinked PC (B/X PC) due to a radical-assisted chain cleavage, with the balance between chain scission and branching/crosslinking being dependent on temperature and solution heterogeneity. Higher temperatures promote both chain oligomerisation and branching/crosslinking by accelerating radical generation, whereas heterogeneous conditions primarily facilitate branching/crosslinking due to the strong chain entanglement. These findings highlight the critical role of solvents in polymer degradation pathways and offer insights into solvent selection for plastic recycling. Rubio Arias et al. investigated the depolymerisation of PC and PET in both individual and mixed streams using a KOH in MeOH hydrolysis process.562 This method enabled the selective breakdown of mixed PET/PC streams and the efficient one-step separation of their monomers with high energy efficiency. The activation energies for PC and PET depolymerisation were determined to be 68.6 and 131.4 kJ mol−1, respectively. Complete depolymerisation of randomly mixed streams was achieved within 2 minutes at 120 °C using 30 mL of solution per gram of polymer. The process yielded BPA and PTA with purities of 98% and 97%, respectively, without secondary reactions. Furthermore, the method proved effective for simultaneous depolymerisation of mixed plastic waste streams.
The use of ionic liquids in polycarbonate waste recycling via hydrolysis has demonstrated significant potential.464 Researchers reported the synthesis of a series of DBU based ionic liquids and conducted PC hydrolysis without the need of metals and solvents. Under optimised conditions, the process achieved complete PC conversion and 97% yield of bisphenol A at 140 °C in 3 h.462
8.4 Methanolysis
Methanolysis depolymerisation reaction is mostly applicable to polymers containing heteroatoms in their backbone. Studies on different polymers have been reported in the literature for depolymerisation via methanolysis such as PET, polylactides, poly (bisphenol A carbonate), polycarbonate, etc.563–566 Methanolysis consists of trans-esterification using methanol at 180–280 °C and pressure of 20–40 bar accelerated by catalysts to produce corresponding monomers such as dimethyl terephthalate (DMT) and EG in the case of PET.524 The process has been mostly applied to PET recycling. The obtained building blocks may be used to make virgin quality PET. Methanolysis progresses in the presence of typical transesterification catalysts (e.g., acetates of Co, Mg, and mostly Zn), while the reaction can be achieved by heating and melting PET and allowing it to react with methanol in a subsequent step or providing directly superheated methanol vapours that work as a heat transfer and chemolysis agent. Catalyst deactivation is a must at the end of the reaction for avoiding transesterification of DMT with EG. The crude DMT upon separation from the mixture is subjected to vacuum distillation or centrifugation followed by crystallisation for purification. Although methanolysis is less economically viable than glycolysis, it offers the significant advantage of effectively processing low-quality feedstock due to its superior tolerance to contamination. However, when it is used for post-consumer PET, then the cost of separation and purification from the mixture becomes higher than that associated with the depolymerisation step.567 DuPont, Eastman, and Hoechst have used this process at large for many years in the past, but it was discarded due to high corrosiveness and high cost related to separation and refining of the reaction mixture.424Depolymerisation of PET through methanolysis occurs in two phases: initially solvolysis produces smaller oligomers which on catalytic action leads to DMT and EG formation. While investigating PET methanolysis by Kurokawa et al. in the presence of aluminium triisopropoxide catalyst, the addition of 20 vol% toluene resulted in increase of DMT yield from 64% to 88%.568 The authors concluded that the increased solubility of PET accelerated the rate determining oligomer formation, resulting in a higher yield of monomers. Vanlaldinpuia and co-workers reported the successful decomposition of PET bottles by methanolysis using bamboo leaf ash as the catalyst.569 The process was conducted in an autoclave at 200 °C for two hours, leading to the formation of DMT and EG with respective yields of 78% and 76%. Bamboo leaf ash has excellent thermal stability, mesoporous structure, biocompatibility, cost-effectiveness, ease of preparation, and recyclability, which makes it an attractive and more environmentally friendly alternative to solid catalysts for PET depolymerisation.
McKeown and co-workers have developed an organocatalyst, [NMe4]+[OCO2Me]−, using dimethyl carbonate and tetramethylammonium hydroxide for degradation of various polymers.570 They proposed a three-step decomposition mechanism: (1) formation of active sites in situ due to the decomposition of carbonate anions with the release of methanol and CO2, (2) substitution of carbonate anions with alkoxides upon addition of alcohol, and (3) transesterification facilitated by alkoxides, activating the carbonyl group using methyl hydrogen atoms acting as Brønsted acids. This method provides successful depolymerisation of PLA, PCL, PC and PET. Recently, Li et al. established a straightforward and highly effective method for the simultaneous upcycling of BPA-PC and PET into a high-performance, transparent engineering plastic known as poly(aryl ether ketone) (PAR).571 Under relatively mild conditions, diverse post-consumer BPA-PC and PET materials, including textiles, were methanolysed using a [TBDH]Ac ionic liquid catalyst, yielding recycled bisphenol A (r-BPA) and dimethyl terephthalate (r-DMT) monomers with purities of 98% and 99%, respectively. Notably, this process eliminates the need for additional purification steps typically required in conventional recycling routes to produce monomers suitable for re-polymerisation, thus streamlining the overall recycling workflow.
The methanolysis method can be coupled with the polymer manufacturing process, offering a major advantage as the recycled DMT has the same quality as that of virgin DMT.572 The advantages of methanolysis also include the simplicity of DMT purification and the ease of extraction of ethylene glycol and methanol. Moreover, methanolysis has good resistance to pollutants, which allows the processing of even low-quality raw materials.569 However, the reaction products are initially composed of a complex mixture including DMT, glycol, derivatives of phthalate, and other alcohols, making the separation process costly and lengthy. This limitation restricts the broader use of methanolysis, making the hydrolysis and glycolysis methods the preferred options. The major drawback is related to the new industrial trends to produce PET from terephthalic acid and BHET based products instead of DMT as the raw material.573 Consequently, DMT needs to be hydrolysed into TPA and EG, which are the required monomers for the re-polymerisation to PET. Nonetheless, the process scheme involves the use of additional processing units, adversely impacting the economic and environmental aspects of the methanolysis process. Because the production of PET is increasingly dependent on terephthalic acid compared to DMT, interest in the DMT route has declined in recent decades. However, an indirect route to obtain DMT by methanolysis of PET, followed by its hydrolysis towards terephthalic acid, can address the expensive purification of BHET typically derived from PET glycolysis.568
Methanolysis has also been reported for polylactide (PLA) recycling.563,565,566,574,575 PLA can be depolymerised into alkyl lactate through alcoholysis. Even et al. depolymerised PLA to methyl lactate, feedstock for new poly(lactide) products, by using methanol as the depolymerisation reagent and Zn salt as the catalyst.576 In another study, 4-dimethylaminopyridine (DMAP) catalyst was used for depolymerisation of PLA under microwave irradiation to yield >99% methyl lactate in a duration of 10–20 min.506 Alberti et al. in 2020 also reported the use of bismuth subsalicylate catalyst to depolymerise PLA to methyl lactate with an excellent yield of >99% and turnover frequencies of 13800 h−1 under microwave irradiation.565 Bismuth subsalicylate catalyst was also found to be recyclable for depolymerisation. Pham et al. used potassium carbonate as a low energy catalyst for depolymerisation of PET to dimethyl terephthalate (DMT).577 2-Hydroxyethyl methyl terephthalate and monomethyl terephthalate are the other reported products, however, the selectivity for DMT was claimed to be increased by controlling the moisture content.
If long-chain primary alcohols are used instead of MeOH, then plasticisers for PVC can be obtained. For instance, when 2-ethylhexanol was used in the process of alcoholisation of PET bottles, the plasticiser dioctyl terephthalate was obtained with high yield, and its quality was not inferior to commercial plasticisers.578,579 Ethanolysis has been carried out at temperatures of 180 °C to 300 °C in the liquid phase, both without a catalyst and using titanium isopropoxide, zinc acetate, manganese or copper as catalysts. If titanium isopropoxide is used, the process does not depend on the moisture content of the starting alcohol, which allows the use of industrial alcohol without additional purification to decompose the polymer.580
Methanolysis is a widely used method for recovering pure BPA and dimethyl carbonate (DMC) monomers from polycarbonate polymers (Scheme 1E). However, PC is not soluble in methanol, methanolysis typically requires high temperatures and pressures, along with significant quantities of conc. acids or bases as catalysts. These catalysts present several challenges, such as their inability to be reused, equipment corrosion, complex post-reaction workup, and environmental concerns. While the supercritical methanolysis method can address some of these issues, its application is limited due to the severe reaction conditions required. Studies have shown that polycarbonate can be fully decomposed into its BPA monomer under high pressure and high temperature steam (300 °C).558 However, the low stability of BPA under these conditions results in a relatively low yield. To improve PC recycling, a reactive environment that maintains BPA while maximising PC reactivity is necessary.
Ionic liquids such as succinimide-based ionic liquids (SIILs) have been investigated for selective conversion of PC into BPA via methanolysis.581 The authors evaluated the catalytic behaviour of SIILs in depolymerisation reactions, focusing on the impact of ionic liquid structures, reaction conditions, catalyst recyclability, and the interaction between the catalyst and methanol. The ionic liquid [HDBU][Suc], consisting of DBU cation and succinimide anion, demonstrated higher performance with complete depolymerisation of PC and producing BPA in 96% yield at 70 °C in 2 h. Additionally, [HDBU][Suc] was easily recyclable and exhibited reasonable reusability, making it a promising green catalyst for PC depolymerisation. A potential mechanism for [HDBU][Suc]-catalysed methanolysis of PC was also proposed. Potassium fluoride was also reported as a catalyst for depolymerisation of PC to products containing building block monomers bisphenol A and dimethyl carbonate.564 Imidazolium-based ionic liquids such as [Bmim][Cl] and [Bmim][Ac] were investigated for the methanolysis of BPA-PC.582,583 Both catalysts achieved complete depolymerisation within 3 hours with high yields (>95%). The excellent catalytic efficiency of [Bmim][Ac] under milder conditions was due to its better solubility in BPA-PC. Liu et al. investigated the use of Lewis-basic ionic liquids to catalyse the alcoholysis of PC.584 They discovered that 1,8-diazabicyclo[5,4,0]undec-7-enelactate [HDBU][Lac], a thermally stable ionic salt, could catalyse PC methanolysis with 100% conversion and 99% yield for BPA and DMC at 120 °C. When other alcohols including ethanol, propanol, and butanol were used under similar conditions, the solvolysis rate reduced as the carbon chain length (C number) of alcohol increased.584 Tanaka et al. recently reported a low-temperature and efficient depolymerisation process for recycling polyester fibres via methanolysis, using dimethyl carbonate as a trapping agent for ethylene glycol.585 Polyester fibres from textile products were depolymerised at 50 °C to afford DMT in high yield (>90%) in 2 h. Using this approach, real polyester textiles blended with other fibres and coloured polyester textiles were depolymerised to produce DMT selectively in high yields. D'Anna et al. developed an optimised methanolysis process for polycarbonate, yielding dimethyl carbonate and bisphenol A (BPA) using cholinium-based ionic liquids with eco-friendly anions.586 The process achieved high conversion and BPA yields under milder conditions than previously reported, aligning with green chemistry principles. The most effective catalyst demonstrated excellent reusability without significant performance loss. Furthermore, the methodology was effectively implemented for the recycling of post-consumer polycarbonate waste. In another approach, PC and PLA were upcycled into BPA and methyl lactate (ML) with high efficiency, using SBA-15 functionalised basic ionic liquids under solvent-free conditions.587 Among the developed catalysts, SBA-15-Pr-MIM-OH showed the highest basicity (0.98 mmol g−1) and demonstrated outstanding catalytic efficiency, achieving complete depolymerisation of PC and PLA at 120 °C within 1 hour and 4 hours, respectively, with monomer yields exceeding 98%. Optimised reaction conditions ensured maximum catalytic efficiency and product selectivity. The integrated “one-pot” methodology was successfully applied to mixed PC/PLA waste, achieving full conversion and excellent monomer recovery, demonstrating the potential of this approach for sustainable plastic recycling.
Huang et al. synthesised imidazolium-based deep eutectic solvents (DESs) and demonstrated their efficiency in catalysing BPA-PC methanolysis.588 The DES [EmimOH]Cl-2Urea exhibited excellent catalytic activity and reusability, achieving nearly complete PC conversion with 98–99% BPA yield under optimised conditions (DES:PC mass ratio of 0.1:1, CH3OH:PC molar ratio of 5:1, 120 °C, 2 h). Kinetic studies confirmed a pseudo-first-order reaction, determining an activation energy of 133.59 kJ mol−1. The catalyst showed good thermal stability and was reusable for five cycles without significant loss of efficiency. This study provides valuable insights for designing of efficient DESs for polymer degradation and transesterification reactions. Despite the advantages of metal-based catalysts, they present various drawbacks including difficulty in their separation from the crude product, poor selectivity, and high economic and environmental cost. Hence, the use of green solvents, particularly ionic liquids or their combinations with organocatalysts, has increased in recent studies.
Quaranta et al. used organocatalysts, such as DBU, DABCO and DMAP, to catalyse alcoholysis of BPA-PC under solvent-free conditions, enabling selective depolymerisation to BPA and organic carbonates under mild conditions (22–100 °C).589 Using MeOH/PC at a molar ratio of 4.6 and DBU concentration of 10 mg mLMeOH−1, PC was selectively converted into BPA and DMC with a quantitative yield in 30 min. DBU exhibited superior catalytic activity compared to DABCO and DMAP. Ethanolysis led to selective production of BPA and diethyl carbonate (DEC), although at a slower rate than methanolysis. In mixed MeOH/EtOH systems, the process generated BPA along with DMC, DEC, and methyl ethyl carbonate (MEC), achieving a 60% MEC yield in a single step. Additionally, DBU was successfully recycled multiple times without any loss of catalytic activity. In a study by Do et al. the use of 1,5,7-triazabicyclo [4.4.0]-dec-5-ene (TBD) as a catalyst for the methanolysis of BPA-PC resulted in high yields (>96%) of BPA and DMC at 75 °C.590 In addition, depolymerization of polycarbonate in 2-methyltetrahydrofuran (2-Me-THF) with the addition of small diols enabled the formation of five-membered cyclic carbonates with good yields (89–97% for carbonates and 93–99% for BPA).
In a recent study by Parida et al., methanolysis of PC using minimal NaOH (≤0.05 wt%) was demonstrated as an effective approach, achieving high BPA yields (93–94%) at moderate temperatures (125–75 °C).591 Optimised conditions minimised BPA degradation and simplified purification. Recovery from end-of-life PC roof panels using 0.004 wt% NaOH reached 93% in 30 minutes at 175 °C, while end-of-life PC/ABS required additional processing due to additive contamination, ultimately yielding ∼94% BPA. The process showed a lower environmental impact factor compared to existing methods. Krisbiantoro et al. used sodium aluminate (NaAlO2) as a solid base catalyst for PC depolymerisation via methanolysis, using THF solvent.592 The catalyst exhibited high activity, achieving 98.1% PC conversion with 96.8% BPA yield at 60 °C within 2 h. THF was identified as the optimal solvent due to its polarity similarity to PC, facilitating dissolution. Mechanistic studies indicated a methoxide pathway, and NaAlO2 showed excellent reusability over four cycles with minimal deactivation. The reaction exhibited a low activation energy (75.1 kJ mol−1), the lowest recorded for solid catalysts in PC methanolysis. Recently, Xu et al. developed polymeric carbon nitride nanosheets (PCNS) using thermal polymerisation followed by thermal exfoliation.593 The catalyst demonstrated broad applicability and high efficiency in the methanolysis of PC, PET, and PLA. The catalyst exhibited high activity, achieving 87% BPA yield from PC after six hours at 130 °C. PET and PLA were converted to dimethyl terephthalate (DMT) and methyl lactate (ML) with yields of 85% and 98%, respectively.
Methanolysis depolymerisation has been reported with enhanced efficiency when supported by microwave heating, leading to an excellent rate of depolymerisation with short reaction time and high turnover frequencies. Excellent results have been reported for depolymerisation of PLA in the presence of microwave heating to produce monomers in >99% yield.506 Similarly, depolymerisation of poly(bisphenol A carbonate) proceeds with an accelerated rate under microwave conditions to produce BPA and DMC monomers, both of which can be repurposed and used as monomers to produce new poly(bisphenol A carbonate), creating a closed loop recycling process.564 Ikenaga et al. depolymerised PC via methanolysis using pressurised microwave heating without a catalyst, and obtained BPA in 94% yield at 90 °C and pressure of 3 MPa in 3 hours.566 Similarly, PET was depolymerised by Han et al. via a methanolysis reaction.524 Hofmann et al. depolymerised end-of-life PET via methanolysis using Zn(OAc)2 catalyst and obtained dimethyl terephthalate (DMT) and ethylene glycol.594 Use of microwave heating resulted in excellent yield and selectivity. The authors also applied this catalytic system for depolymerisation of a combination of PET, poly(bisphenol A carbonate), and poly(lactide) (PLA) producing corresponding monomers in excellent yield.594 The potential of obtained monomers for repolymerisation was also investigated to reproduce PET successfully.
Moreover, some hybrid methods including glycolysis–hydrolysis, glycolysis–methanolysis, and methanolysis–hydrolysis have been developed to take benefit of the advantages of each individual process.62,424 Other less conventional methods such as acetolysis and biodegradation have also been used for recycling of plastic waste. Acetolysis, an exchange reaction between carboxylic acid and ester, has been applied for processing of PET waste.595 Among carboxylic acids, acetic acid turned out to be more effective for PET depolymerisation due to its strongest acidity. Studies have shown that PET bottle flakes can be completely depolymerised at 280 °C via acetolysis in two hours, yielding 95.8% TPA with purity over 99.7% and 95.3% EGDA (ethylene glycol diacetate) with purity over 98.0%. Another interesting way of PET recycling is the process of biodegradation by enzymes. The process is typically carried out at temperatures between 30 °C and 63 °C, over a period of 3 to 14 days at atmospheric pressure.596 The enzymes used are produced from various microorganisms including Micromonospora viridis, Thermobifida fusca, cutinase, lipase, etc. The enzymes primarily target the ester bond of PET. The process of biodegradation by microorganisms as described by Muller is a heterogeneous process. At the beginning of the process a biochemical reaction occurs on the polymer surface (PET does not dissolve in water and has a high molecular weight).597 After partial degradation, intermediate water-soluble compounds are formed, which further participate in cellular metabolism. Finally, monomers (N,N′-bis(2-hydroxy)ethyl terephthalate, terephthalic acid), ethylene glycol, carbon dioxide, water, methane, and other products are obtained at the end of the process. Degradation by enzymatic methods is discussed in Section 13.
Ionic liquids such as [Bmim][BF4] can effectively catalyse ethanolysis of PET under supercritical conditions, which can reduce the PET decomposition time from 6 hours to 45 minutes with 98% conversion of PET.455 Supercritical methanol can be used to depolymerise PET, requiring temperatures of 270–300 °C and pressures in the range of 0.1–15 MPa. High-molecular-weight PET depolymerises more rapidly in this process compared to PET with a lower molecular weight.519
8.5 Hydrogenolysis
Depolymerisation of polymers by hydrogenolysis reaction is carried out in the presence of molecular hydrogen and a catalyst. Typically, hydrogenolysis involves breaking of C–C bonds followed by hydrogenation on a catalyst, and is often facilitated by mono-functional metal catalysts.598 This process involves low energy as there is almost no need of extraction and it has less environmental impact, hence it is considered as a promising approach to achieve the circular plastic economy compared to traditional thermal and catalytic cracking technologies. Both homogeneous and heterogeneous catalysts have been used in the hydrogenolysis process for depolymerisation of different polymers such as PET, polyester, and polycarbonates.598,599The research groups of Milstein and Robertson were pioneers in the depolymerisation of polyesters into alcohol monomers via hydrogenolysis using homogeneous catalysts.600,601 They successfully employed ruthenium-N,N,P-pincer complexes (ruthenium(II) PNN) to achieve high yields of monomers through catalytic breakdown of polymers. Polyesters were depolymerised into diols, whereas polycarbonates underwent hydrogenolysis to produce glycols and methanol. Interestingly, polyesters containing two CH2 units between ester groups were found to depolymerise into carboxylic acids instead of diols when treated with the ruthenium(II) PNN complex.600 Using this strategy, post-consumer PET was completely depolymerised at 160 °C under 54 bar H2 within 24 h, with the process effectively tolerating impurities such as additives and pigments. Fuentes et al. developed advanced ruthenium-N,N,P-pincer complexes to enhance the hydrogenolysis of PET.602 However, the practical application of this method is limited by high catalyst loading and prolonged reaction durations, making it challenging for consumer products and engineered polymers. One ruthenium catalyst from the same family of complexes produced TPA in 53% yield via hydrogenolysis of PET in THF/anisole at 110 °C in 48 h.602 However, the resulting ethylene glycol deactivated the catalyst. Feghali and Cantat explored a similar strategy to depolymerise polymeric materials such as polycarbonates, polyethers, and polyesters using hydrosilane as the reductant in the presence of organocatalysts [B(C6F5)3] or [Ph3C+,B(C6F5)4−].603
Westhaues et al. studied the hydrogenolytic depolymerisation of PLA, PC and PET wastes using a ruthenium catalyst ([Ru(triphos-xyl)methylallyl]NTf2) with bis(trifluoromethanesulfonyl) imide (HNTf2) as a co-catalyst (Fig. 28A–C).599 Hydrogenolysis of PC resulted in the production of 1,6-hexanediol only, and PLA was completely converted to 1,2-propanediol. However, PET hydrogenolysis proved more challenging, only 42% PET was converted, with 64% selectivity for 1,4-benzene dimethanol and EG (Fig. 28B). Fig. 28C shows the selective hydrogenolysis of PLA and PET polymers using ([Ru(triphos-xyl)methylallyl]NTf2) catalyst. The acid-activated catalyst also facilitated the successive formation of ether by-products, which decreased the product selectivity, an effect particularly pronounced in the hydrogenolysis of polybutylene terephthalate (PBT). Though PBT was completely depolymerised, only 22% of the products were monomeric diols including 1,4-butanediol, with the remainder being ether by-products (Fig. 28B). The use of the ruthenium complex, [Ru(triphos-xyl)tmm], along with HNTf2 as a co-catalyst quantitatively depolymerised various PET materials into TPA and EG at 140 °C and 100 bar H2 pressure in 16 h, and remarkably tolerated impurities (Fig. 28).599,604 Kumar et al. used a ruthenium pincer catalyst for hydrogenative depolymerisation of extensively used nylons and polyamides in DMSO at 150 °C and 70 bar H2 pressure.605 The same catalyst was used to hydrogenate polyurethane, resulting in the production of diamine, diol, and methanol.
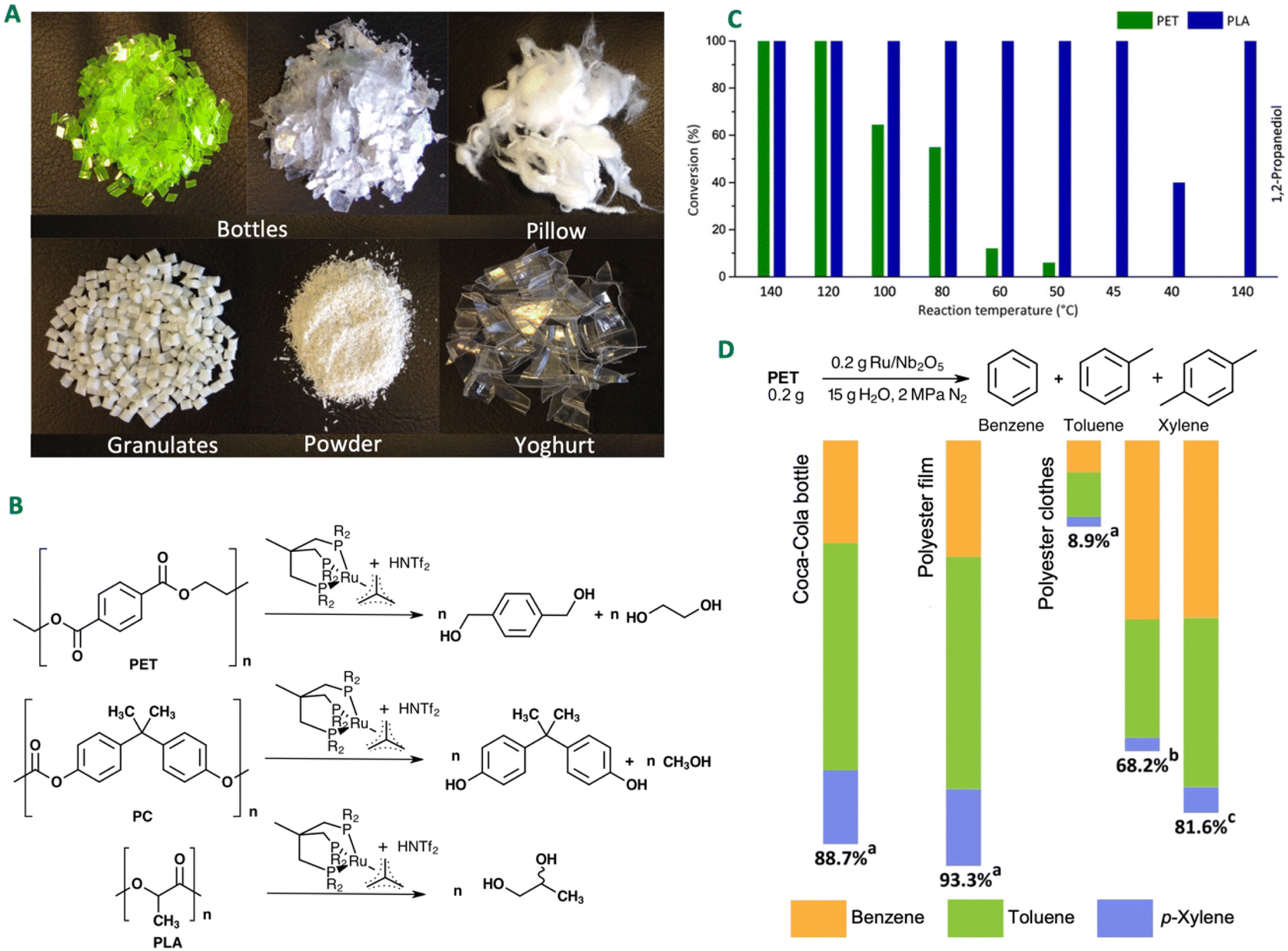 | ||
| Fig. 28 Depolymerisation of plastic waste feedstocks by catalytic hydrogenolysis. (A–C) PET, PC and PLA at 1400 °C with 190 bar H2 pressure. Adapted with permission from ref. 599. Copyright 2018, American Association for the Advancement of Science. (D) Hydrogen-free PET conversion over Ru/Nb2O5: (a) 220 °C, 12 h; (b) 280 °C, 12 h; (c) 280 °C, 16 h. Adapted with permission from ref. 598. Copyright 2021, John Wiley & Sons. | ||
Single site carbon-supported molybdenum-dioxo catalysts (MoO2/C) have been used for selective depolymerisation of PET to TPA and ethylene via hydrogenolysis.606 This solvent-free depolymerisation was carried out at 260 °C with 1 bar H2 pressure, and PET was completely deconstructed to TPA with 87% yield. This catalyst showed high depolymerisation efficiency for both pure and waste PET due to the active Mo sites that enabled selective activation and cleavage of PET's ester groups. Yan et al. carried out PET depolymerisation by hydrodeoxygenation to recover arenes using a titanium supported cobalt catalyst (Co/TiO2).607 Initial studies were focused on a pure TPA monomer, leading to the formation of 75% xylene and 9% toluene at 340 °C and 30 bar H2 pressure in 4 h. Under the same conditions, depolymerisation of PET afforded ca. 79% yield of toluene and xylene after 24 h. However, catalyst stability was not satisfactory, and catalytic activity was reduced from 90 mol% to 35 mol% after 2 cycles due to Co leaching and degradation of the support. Wu et al. developed a bimetallic catalyst CoMo@NC derived from Mo@ZIF-CoZn at 900 °C.608 Mo@ZIF-CoZn was pyrolysed at high temperatures into a N-doped graphitic matrix where metals were anchored as Co nanoparticles and Mo nanoclusters, respectively. The synthesised bimetallic catalyst showed excellent activity in PET hydrogenolysis, affording TA in 91% yield at 260 °C and 1 atm H2 pressure. The synergic catalysis between cobalt and molybdenum sites within the catalyst promoted hydrogenolysis under mild conditions. This catalyst exhibited stable activity in six cycles. Jing et al. reported the upgradation of aromatic plastic waste to simple arenes through hydrogenolysis using a multifunctional Ru/Nb2O5 catalyst.609 This catalyst selectively cleaved C–O and C–C bonds within aromatic plastics, producing arenes in high yield (75–85%). In addition to converting single-component aromatic plastics, this catalytic system also allowed the simultaneous conversion of mixed aromatic plastics into arenes with high selectivity.
Lu and co-workers carried out the hydrogen-free conversion of PET to BTX by leveraging the inherent hydrogen within the ethylene glycol segment, using Ru/Nb2O5 and Ru/NiAl2O4 catalysts via simultaneous hydrogenolysis and decarboxylation reactions (Fig. 28D).598 The Ru/Nb2O5 catalyst showed superior hydrogenolysis and inferior decarboxylation performance compared to the Ru/NiAl2O4 catalyst, leading to different selectivities of alkyl aromatics in BTX. The overall process occurred in three sequential stages: hydrolysis, reforming, and C–O/C–C bond cleavage, with the latter identified as the rate determining step that involved parallel hydrogenolysis and decarboxylation pathways. This H2-free system was also tested to convert different common real PET plastic feedstocks into BTX (Fig. 28D).598 This depolymerisation strategy can be taken as a new possible solution in the circular economy of PET. Yiang et al. reported a bimetallic RuFe catalyst (Ru-280/Fe–N–C-800) for hydrodeoxygenation (HDO) of PET plastics and DMT (dimethyl terephthalate), achieving yields of para-xylene at 82.6% and 88.9%, respectively.610 The catalyst selectively cleaved C–O bonds without hydrogenating aromatic rings or breaking C–C bonds to afford BTX in 96% yield from PET plastics, and 93.8% BTX yield from DMT in 90 min.
Recently, Leshkov investigated the hydrogenolytic depolymerisation of commercial and LDPE waste using carbon supported Ru nanoparticles at 200 °C without solvents.611,612 Initial studies were focused on degradation of octadecane using various catalysts such as Ru/Al2O3, Ru/C, Ru/CeO2, Ru/SiO2, and Ru/TiO2. However, the Ru/C catalyst gave the highest reactivity using octadecane as a model template. This methodology was then extended to LDPE with a Mw of ∼4000 Da, targeting both commercial and post-consumer LDPE. Hydrogenolysis using Ru/C catalyst (wt%) at 200 °C with 20 bar H2 resulted in 45% yield of liquid hydrocarbons, with C1–C6 hydrocarbons being present in the residue. Increasing the temperature to 250 °C resulted in stoichiometric production of CH4. Hydrogenolysis of LDPE waste at 200 °C generated C7–C45 alkanes, highlighting the catalyst's ability for valorising plastic waste.
Román-Leshkov and colleagues later applied this method to degrade PP and mixed polyolefin waste, and achieved isoalkanes (C5–C32) under moderate conditions (200–250 °C, 20–50 bar H2).612 Under the same conditions, depolymerisation of high-molecular-weight PP (∼340000 Da) resulted in liquid and gas products. In another study, CeO2-supported ruthenium catalyst successfully degraded LDPE into liquid hydrocarbons (C5–C21) and wax (C22–C45) at 202 °C and 2 MPa H2 in excellent yield (92%).613 Similarly, HDPE and PP were degraded to valuable chemicals in high yield (83–90%) using this method.
8.6 Oxidative depolymerisation
Oxidative depolymerisation involves the breakdown of macromolecules by the action of oxygen and requires the presence of an oxidative agent. Oxidative routes have been investigated to deconstruct polyolefins. Catalysts capable of promoting oxidation reactions can significantly increase the rate of such depolymerisation reactions. The oxidative depolymerisation of PPO [poly(2,6-dimethyl-1,4-phenylene oxide)], an engineering plastic, was first reported by Saito et al. in 2003 in 2,6-dimethylphenol solvent.614 In this work, the CuCl/pyridine catalyst resulted in depolymerisation of PPO to yield oligomeric products. Shimoyama and Nakajima recently developed a more advanced oxidative depolymerisation method to recycle PPO, producing 2,6-dimethylbenzoquionone (26DMBQ) as the only aromatic product. Using nitronium ions (NO2+), produced from a mixture of silicotungstic acid and nitrate salts at 120 °C, as a mild oxidant, they achieved 66% yield of the 26DMBQ product (Scheme 6).615 Mechanistic investigation indicated that NO2+ oxidised PPO. This was followed by the interaction of NO2+ with H2O, which cleaved the C–O bonds, leading to 26DMBQ formation (Scheme 6B). Increasing the temperature led to a gradual increase of 26DMBQ yield, however the yield of AA reached the maximum at 80 °C and then reduced (Scheme 6C). The PPO conversion was about 80% after 48 h, and the reaction was saturated with 26DMBQ at 64 h without changing after additional reaction time (Scheme 6D). The authors also used 26DMBQ in a polymerisation reaction with a dianiline to produce a polyimide. | ||
| Scheme 6 (A) Oxidative depolymerisation of PPO at 160 °C, 64 h. (B) Possible mechanism for PPO depolymerisation. (C) Effect of temperature on product yields. (D) Product yield with time: 26DMBQ (red), AA (blue) and FA (green). Adapted with permission from ref. 615. Copyright 2023, John Wiley & Sons. | ||
Potassium permanganate (KMnO4) solution has been used by Kim et al. as an oxidising agent to decompose the cross-linked epoxy resin of carbon fibre reinforced plastic (CFRPs) for recovering carbon fibres.616 The mechanical properties of the recovered material were found to be comparable to those of virgin fibres. The oxidative depolymerisation strategy has been effectively applied to depolymerise lignin, a natural polymeric material. This method is widely used on industrial scale in paper industry.617–619 The lignin from wood pulp could be efficiently and selectively depolymerised and removed under mild reaction conditions.620 In another application, Sadaka et al. used oxidation to depolymerise ground waste tyres using periodic acid as the oxidising agent.621 The process involved the epoxidation of polyisoprene's double bonds, followed by the cleavage of resulting oxirane structures. By precisely controlling the amount of periodic acid, which acted as both an oxidant and catalyst, the researchers successfully synthesised low molecular weight telechelic polymers with aldehyde end groups. Luo et al. developed a method for acid-mediated oxidative decomposition of PS in solution.622 To facilitate activation by benzyl hydrogen, they introduced nitric acid as an inexpensive acid along with an oxidiser. A mixture of 20% nitric acid and PS was heated to 180 °C in an autoclave in an oil bath. After 3 hours, 90 mol% of PS was successfully converted to benzoic acid. Although the exact mechanism remains poorly understood, previous studies suggest that NO2 can catalyse the oxidation of alkanes through the formation of NOx compounds and superoxides. Probably, nitric acid promotes the formation of benzyl radicals, while O2 serves as the final oxidant for cleavage of C–C bonds, forming benzoic acid. Additionally, the inclusion of radical scavengers such as 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) suppressed the formation of benzoic acid, confirming that decomposition occurs by a radical chain mechanism.
A microwave assisted process was reported by Bäckström et al. for oxidative depolymerisation of LDPE into valuable chemicals.623 In this method, they used nitric acid as an oxidant, and after 1 hour of microwave heating at 180 °C, the process produced predominantly water soluble dicarboxylic acids such as adipic acid, succinic acid, acetic acid, and propionic acid. The process was also applied to depolymerise LDPE freezer bags, successfully achieving dicarboxylic acids in 71% yield. The same group later reported a microwave-assisted process to convert HDPE waste to well-defined chemicals that include adipic, glutaric, and succinic acids. These acids were then used to synthesise plasticisers for polylactide (PLA) films.624
A novel method of solvent-free mechanocatalytic cracking of polyethylene (PE) has been developed using the heterogeneous Fenton process.625 This method involves the accidental oxidation of carbon atoms in the polymer chain, which contributes to its activation and subsequent fragmentation into oxygenates with a lower molecular weight. The main reaction pathway involves hydrolysis of ester intermediates, while decarbonylation and decarboxylation contribute to the formation of small amounts of alkanes and CO2/CO. The resulting oligomers can be integrated into existing oil refining processes for further processing into valuable products.
Recently, CeO2 has been used as a catalyst for converting PE waste into oxygen-containing compounds that can serve as suitable substrates for biological processing.626 This process employs a heterogeneous catalyst, which improves product separation and minimises the risk of metal contamination in subsequent biological stages. The process also utilises O2 as an oxidising agent. The oxidised PE derivatives served as the only carbon source for cultivating Cupriavidus necator H16, facilitating their microbial conversion into the biodegradable polymer poly(3-hydroxybutyrate) (PHB). This integrated process achieved impressive PHB yields, 0.22 g PHB per g of PE powder, 0.21 g g−1 for LDPE bags and 0.28 g g−1 for HDPE bottles. Wang et al. developed a low-temperature, hydrogen-free catalytic process for oxidising PE into valuable aliphatic dicarboxylic acids using Ru/TiO2 catalyst.627 Under mild conditions (160 °C, 1.5 MPa air, 24 h), LDPE achieved 95% conversion with 85% yield of liquid products, primarily low molecular weight dicarboxylic acids. The method demonstrated high efficiency across various PE feedstocks and produced minimal volatile by-products. NMR and IR analyses confirmed that dicarboxylic acids dominated the product mixture, with only trace amounts of esters. Notably, the recycled catalyst outperformed the fresh one, attributed to increased hydrophobicity and enhanced interaction with molten PE due to surface accumulation of organic residues. Zhang et al. carried out the direct oxidative conversion of PE into high-value saturated dicarboxylic acids, achieving a carbon yield of 85.9%.628 Long-chain dicarboxylic acids (C10–C20) accounted for 58.9% of the yield, facilitated by MCM-41 molecular sieves doped with cobalt, without the use of solvents or precious metal catalysts. By adjusting the cobalt content in MCM-41 through nanocomposite modification, it is possible to control the distribution of dicarboxylic acids from short-chain (C4–C10) to long-chain (C10–C20) products. Chang et al. developed an innovative low-temperature oxidative cracking strategy to upcycle polyolefin waste into recyclable elastomers.629 HDPE was oxidatively cleaved at 110 °C to produce carboxyl-terminated oligomers with tunable molecular weights (Mn 1500–5500 Da) and broad dispersities (Đ 2.91–3.33). These crystalline oligomers were directly esterified with oxidised cis-polybutadiene containing epoxy groups to form a dynamically cross-linked elastomer. The resulting material exhibited excellent mechanical properties, including a tensile strength of 16.4 MPa, elongation at break of 600%, and toughness of 46.0 MJ m−3, outperforming commercial polyolefin elastomers. The presence of dynamic ester bonds enables recyclability, offering a streamlined chemical upcycling route for polyolefins without the need for complex product separation.
Liu et al. developed a temperature-gradient pyrolysis technique to convert PE and PP into waxes with controlled chain lengths, achieving an 80% yield.630 Using a custom reactor with distinct hot and cold zones, the process prevents complete breakdown into small molecules, enabling precise control over depolymerisation. The resulting waxes were subsequently oxidised using a Mn(II) stearate catalyst to produce long chain organic acids and small amount of esters, which were further converted into high-value surfactants. This approach eliminates the need for expensive catalysts and harsh conditions, offering a cost-effective and scalable solution for plastic waste recycling.
Organocatalysts such as N-hydroxyphthalimide (NHPI) and its derivatives have been shown to catalyse the oxidative upcycling of PS to carboxylic acids without the need for metal catalysts.631 In a system comprising PS, an organocatalyst (0.01 mmol), nitric acid, and glacial acetic acid, heating the reaction mixture at 120 °C in air for 24 hours resulted in a mixture primarily composed of benzoic acid and 4-nitrobenzoic acid. Among the catalysts evaluated, 4-F-NHPI and N,N′,N′′-trihydroxyisocyanuric acid (THICA) showed the highest efficiencies, producing approximately 30 mol% benzoic acid and 10 mol% 4-nitrobenzoic acid relative to styrene units. Minor byproducts included formic acid and partially oxidised oligomers, highlighting the potential of metal-free organocatalysis for PS valorisation. A sustainable oxidative upcycling method was developed to convert PS waste into benzoic acid using a NiO/TiO2 catalyst in an aqueous medium, with air as the oxidant.632 Operating at 200 °C and 1 MPa for 18 hours, the process achieved a carbon yield of up to 51.1% and was successfully applied to real-world PS waste. Mechanistic studies identified superoxide radicals (˙O2−) as the key reactive species, initiating C–H bond activation in the aliphatic segments of PS, followed by depolymerisation through successive CC bond cleavage. This pathway generates progressively smaller oxygenated fragments, ultimately forming benzoic acid. The approach offers a green and efficient route for PS valorisation.
Chen et al. carried out the catalytic oxidation of PET, producing TPA and glycolic acid (GA) instead of ethylene glycol.633 Using an Au/NiO catalyst enriched with oxygen vacancies, they achieved exceptional yields of 99% for TPA and 87.6% for GA. This approach demonstrates a promising strategy to convert PET waste into valuable chemical raw materials. Yu et al. developed an oxidative upcycling strategy to convert waste PET into aniline and TPA using Ru/TiO2 as the catalyst and nitrobenzene as the hydrogen acceptor.634 The process resulted in complete PET conversion, yielding 94% aniline and 99% TPA at 220 °C in 10 hours. This high performance is driven by strong Ru–TiO2 interactions that facilitate electron transfer and accelerate the dehydrogenation of EG unit, the rate-limiting step. Additionally, controlled hydrogen release enhances selectivity towards aniline, while the use of an aqueous medium allows for energy-efficient product separation. Life cycle and techno-economic assessments highlight the method's environmental and industrial viability, projecting significant energy savings, reduced carbon footprint, and an annual profit of $36.13 million for processing 100k tonnes of PET waste. This approach offers a selective, efficient, and scalable solution for PET upcycling into high-value chemicals.
Sullivan et al. integrated chemical and biological catalysis to convert mixed plastic waste into valuable compounds.635 A catalytic system comprising Co(II), Mn(II) and N-hydroxyphthalimide was used to oxidise HDPE, PS, and PET, individually and in mixed streams, under 8 bar O2 with an additional 72 bar of N2 introduced for safety. Oxidation of HDPE produced a broad range of dicarboxylic acids (C4–C22) in ∼34 mol% yield relative to polymer carbon, while PS yielded over 60 mol% benzoic acid. The oxidised plastic waste was further bioconverted by Pseudomonas putida strains into β-ketoadipate, a precursor for polyamide synthesis. Strain AW164 also produced polyhydroxyalkanoates incorporating 3-hydroxydodecanoic and 3-hydroxydecanoic acid monomers. Recently, dicarboxylic acids obtained by PE oxidation were further transformed by Aspergillus nidulans into metabolites with potential pharmaceutical applications.636
8.7 Reductive depolymerisation
Reductive depolymerisation involves breaking down polymer chains into their monomeric or oligomeric constituents under reductive conditions. This efficient methodology holds significant potential for addressing the global plastic waste challenge by transforming plastic waste into valuable chemicals over catalysts such as boron, iridium, and ruthenium in the presence of reducing agents like H2 or silanes. Unlike oxidative depolymerisation, which involves the use of oxidizing agents, reductive depolymerisation employs hydrogen or hydrogen-donating compounds to break the polymer bonds. This method is effective in transforming waste plastic into chemicals such as diols, tetrahydrofuran, p-xylene, etc. However, one significant drawback is the high cost of the catalyst. For plastic recycling processes to be sustainably scaled for industrial applications, the catalysts need to be highly active, affordable, and stable.599,637 This methodology allows for the selective depolymerisation of waste plastics into functional chemicals including alcohols and phenols at ambient temperature. A major advantage of reductive depolymerisation is its selectivity and ability to tolerate additives commonly found in waste plastics, allowing it to selectively depolymerise mixtures of polymers.425During catalytic hydrogenation, PET is converted into 1,4-cyclohexanedicarboxylate (PECHD), which offers superior biodegradability and is thermally stable compared to BHET, owing to the presence of an aliphatic ring.638 Typically, PECHD is synthesised by hydrogenating dimethyl terephthalate followed by polymerisation. However, this process is hindered by harsh conditions, the high cost associated with noble metal catalysts, and challenging separation of the intermediate “1,4-dimethyl cyclohexanedicarboxylate” from the reaction mixture, making it highly expensive. Therefore, Tan and coworkers developed a sustainable process for PECHD polyester by selective hydrogenation of PET in a single step at 50 °C and H2 pressure of 6.89 MPa.639 By this method, PECHD was produced in 98% yield with 100% PET conversion using a Vulcan supported Rh–Pt bimetallic catalyst and HFIP solvent. The remarkable catalytic activity was ascribed to rhodium's exceptional ability of strong adsorption to the aromatic ring, along with platinum's ability to enhance hydrogen spillover. The same research group further explored the Rh–Pt bimetallic system supported on SBA-15 (Rh2.5Pt2.5/SBA-15) for PET hydrogenation, highlighting the synergistic role of Rh and Pt, which promoted PET hydrogenation in an aqueous medium under moderate conditions.640
Monsigny et al. investigated the depolymerisation of oxygenated plastics under moderate conditions using a pincer complex [Ir(POCOP)H(THF)][B(C6F5)4] (POCOP = 1,3-(tBu2PO)2C6H3) as a catalyst, with hydrosilanes as reducing agents.425 The iridium catalyst, with 0.3–1 mol% loading, selectively produced either silyl ethers or alkanes, based on the reaction temperature. Moreover, this method successfully depolymerised actual plastic waste, including PET bottles and PLA derived from 3D printers, without interference from dyes or other plastic additives present in the plastic waste. The Fernandes group has developed a highly effective and selective method for reductive depolymerisation of different polymers, such as PET, PLA, polycaprolactone (PCL), etc. They used an inexpensive and air-stable dioxomolybdenum catalyst, MoO2Cl2(H2O)2, along with silanes as reducing agents.641 This process converted plastic waste into valuable chemicals and fuels such as 1,6-hexanediol, propane, and xylene. The catalyst showed excellent stability, maintaining high activity over eight cycles during the reductive depolymerisation of PCL. Moreover, the PMHS/MoO2Cl2(H2O)2 system proved highly effective in the gram scale production of propane from depolymerisation of PLA, and demonstrated efficiency in selectively reducing mixed plastic waste, such as PCL, PLA, and PET.641
Fernandes' group used a commercial and stable zinc catalyst (Zn(OAc)2·2H2O) for reductive depolymerisation of plastic waste and successfully converted PBT, PCL, PET, and PLA waste into valuable compounds including THF, p-xylene, 1,6-hexanediol, and 1,2-propanediol with realistic yields.599,637 Zinc catalyst was proved to be effective in 7 catalytic cycles and selectively reduced PCL with good yields. Moreover, this catalyst was efficient for depolymerisation of mixed polyester wastes, such as PCL + PBT and PCL + PET, using (EtO)2MeSiH as the reducing agent in THF, resulting in 1,6-hexanediol with excellent yields. Marie et al. studied the homogeneous reductive depolymerisation of polyesters and polycarbonates using hydroboranes, catalysed by lanthanum(III) tris(amide).642 This process efficiently converted the polymers into valuable alcohols under moderate conditions, using 1 mol% La[N(SiMe3)2]3 in combination with pinacolborane (HBpin). The reaction demonstrated high selectivity for producing alcohols and diols upon hydrolysis. They later developed an efficient zirconium based catalytic system, Cp2Zr(H)Cl combined with dimethylmethylsilane reductant, for hydrosilylation of esters into alcoholic products.643 Using this catalytic system, the authors successfully depolymerised a number of polyesters, including PCL, PLA, and PET wastes, to their corresponding monomeric diols in high yields.
In metal supported catalysts, the particle size, dispersion, and content of the active metal play a significant role in achieving high product yield.
8.8 Product distribution in depolymerisation
Product distribution depends on the polymer type and the reaction conditions being used for depolymerisation (Table 4). PET, most widely recycled plastic all over the world, has ester functionalities that can be broken down by a range of different reagents such as alcohols, amines, ammonia, glycols, hydrogen, and water. Therefore, PET can undergo a variety of chemical recycling processes, including alcoholysis, aminolysis, ammonolysis, glycolysis, hydrolysis, and others. Depolymerisation products for PET include monomers, dimers, trimers, and oligomers, such as BHET, dimethyl terephthalate (DMT), terephthalic acid (TPA), glycols, chemicals, gases, hydrocarbon liquids, etc. PET also leads to BTX formation by hydrogenolysis. These products can be purified by distillation, drying, crystallisation, or further chemical reactions, depending on their intended use. The reaction conditions for depolymerisation depend on the catalysts used and process applied for depolymerisation.| Feedstock | Catalyst | Reaction conditions | Product | Reference |
|---|---|---|---|---|
| a BPA: bisphenol A; DES: beep eutectic solvent; DMAP: 4-dimethylaminopyridine; DMC: dimethyl carbonate; DMT: dimethyl terephthalate; EG: ethylene glycol; BHET: bis(2-hydroxyethyl) terephthalate; BHETA: bis(2-hydroxyethyl) terephthalamide; sC: supercritical; PPO: poly(2,6-dimethyl-1,4-phenylene oxide); PEHD: 1,4-cyclohexanedicarboxylate; RT: room temperature; TPA: terephthalic acid. | ||||
| PET | Co3O4, 1 wt% | 260 °C, 60 min, EG | 63% BHET | 472 |
| Mn3O4, 1 wt% | 74% BHET | |||
| ZnMn2O4, 1 wt% | 92.2% BHET | |||
| PET | FeIII nanosheet, 0.01 wt% | 200 °C, 30 min, EG | 100% BHET | 475 |
| PET | MWCNTs, 5 wt% | 190 °C, 120 min, EG | 78% BHET | 476 |
| Fe2O3-MWCNTs, 5 wt% | 100% BHET | |||
| PET | MnO2/HGO, 0.01 wt% | 200 °C, 10 min, EG | 100% BHET | 479 |
| PET | Pd/h-BN, 1 wt% | 100 °C, 30 min, EG | 92.1% BHET | 480 |
| PET | ZnAl hydrotalcite (Zn/Al molar ratio: 1), 1 wt% | 196 °C, 85 min, EG | 65.6% BHET | 490 |
| ZnAl hydrotalcite (Zn/Al molar ratio: 2), 1 wt% | 74.8% BHET | |||
| ZnAl hydrotalcite (Zn/Al molar ratio: 3), 1 wt% | 76.4% BHET | |||
| PET | ZnO/SBA-15, 1 wt% | 197 °C, 60 min, EG | 91% BHET | 483 |
| PET | 70% ZnCl2/H2O, 0.5% mass ratio | 180 °C, 8 h | 98.31% TPA | 651 |
| PET | Zinc acetate, 0.26 wt% | 196 °C, 2 h, EG | 70% BHET | 652 |
| PET | Fe3O4 NPs@h-BNNS, 0.67 wt% | 200 °C, 300 min, EG | 100% BHET | 481 |
| PET | [(CH3)3N(C16H33)]3PW12O40, 1 wt% | 160 °C, 3 h, water | 93% BHET | 653 |
| PET | K6SiW11ZnO39(H2O), 0.13 wt% | 185 °C, 30 min, EG | 84% BHET | 441 |
| PET | K10[Zn4(H2O)2(PW9O34)2]-H2O, 2 wt% | 240 °C, 8 min, EG | 92% BHET | 442 |
| PET | Na12[WZn3(H2O)2(ZnW9O34)2], 0.5 wt% | 190 °C, 40 min, EG | 84.5% BHET | 654 |
| PET | (Cu(OAc)2-[Bmim] [OAc]), 33.3 wt% | 190 °C, 3 h, EG | 59.3% BHET | 655 |
| (Zn OAc)2-[Bmim] [OAc], 33.3 wt% | 45.6% BHET | |||
| PET | DES: [n(urea)/n(ZnCl2)], 5 wt% | 170 °C, EG | 83% BHET | 458 |
| PET | TBD: 1,5,7-triazabicyclo[4.4.0]dec-5-ene, 3.6 wt% | 150 °C, 60 min, dibenzylamine | 87% N,N′-dibenzylterephthalamide | 499 |
| 110 °C, 60 min, ethylene diamine | 89% N,N′-bis(2-aminoethyl)terephthalamide | |||
| 120 °C, 120 min, ethanolamine | 93% BHETA | |||
| PET | NaOAc, 1 wt% | 180 °C, MW, 60 min, ethanolamine | 98% BHETA | 512 |
| PET | [(CH3)3N(C16H33)]3[PW12O40], 1 wt% | 145 °C, 14 MPa, 3 h, H2O (acidic) | >90% TPA | 527 |
| PET | SO42−/TiO2 (solid super-acid catalyst), 10 wt% | 160 °C, 15 MPa, 12 h, H2O, sCCO2 | 99.19% TPA | 656 |
| PET | Tributylhexadecylphosphonium bromide, 0.07 mol L−1 | 90 °C, 8.1 MPa, 12 h, H2O, NaOH | TPA | 657 |
| PET waste | Bamboo leaf ash, 22.2 wt% | 190 °C, 3.5 h, EG | 83% BHET | 427 |
| PET | Deep eutectic solvent@ZIF-8 composite, 0.4 wt% | 195 °C, 25 min, EG | 83.2% BHET | 466 |
| Polyurethane | Stannous octoate, 0.9–2.3 wt% | 189 °C, 45 min | Polyol | 446 |
| LDPE | Ru/CeO2, 2.94 wt% | 240 °C, 6 MPa H2, 8 h | 9.7% gas (C1–C4); 83% liquid (C5–C21); 6.5% wax (C22–C45) | 613 |
| Ru/ZrO2, 2.94 wt% | 240 °C, 6 MPa H2, 4 h | 11% gas (C1–C4); 61% liquid (C5–C21); 12% wax (C22–C45) | ||
| Pt/H-USY, 2.94 wt% | 260 °C, 6 MPa H2, 42 h | 18% gas (C1–C4); 82% liquid (C5–C21); <0.1% wax (C22–C45) | ||
| LDPE | HNO3, 0.1 g mL−1 | 180 °C, 1 h, MW | 71% dicarboxylic acid | 623 |
| PPO | CuCl/pyridine = 1:100, PPO = 0.2 unit mol per L toluene solution |
2,6-Dimethylphenol, RT, 1–70 min | Oligomeric products | 614 |
| Natural rubber | Periodic acid in THF, 5 mol L−1 | Up to 50 °C, up to 120 min, THF solvent | Carbonyl telechelic oligomers | 621 |
| PET | Zn(OAC)2, 1 mol% | MeOH (46.2 equiv.), 20 min | 98% DMT and 96% EG | 594 |
| Microwave heating, 160 °C | ||||
| PET | K2CO3, up to 1 mol% | 25 °C, MeOH, 24 h | 93.1% DMT | 577 |
| Polylactide | DMAP, 5 mol% | 180 °C, MeOH (23.1 equiv.), MW, 20 min | 99% methyl lactate | 563 |
| Poly(bisphenol A carbonate) | KF, 2.5 mol% | MeOH (46.3 equiv.), MW, 180 °C, 10 min | 99% BPA and 79% DMC | 564 |
| Multilayer & coloured PET waste | NaOH, 5–15 wt% | 80 °C, EtOH:H2O (60:40 vol%), 20 min |
95% TA & EG | 523 |
| Poly(bisphenol A carbonate) | Chlorite, 70 wt% vs. PC | 200 °C, THF, 6 h | 98% BPA | 560 |
| PET | Rh2.5Pt2.5/Vulcan XC-72-polyol, 1 wt% | 50 °C, H2 (6.89 MPa), 60 min | 98% PECHD | 639 |
| PET | Rh2.5Pt2.5/SBA-15, 20 wt% | 90 °C, H2 (1000 psi), 80 min | 100% PECHD | 640 |
| PET | C/MoO2, 5 mol% | 260 °C, H2 (1 atm), neat | 87% TPA | 606 |
| PET waste | 260 °C, H2 (1 atm), neat | 86% TPA | ||
| PET | 260 °C, Ar (1 atm), neat | 58% TPA | ||
| PBT | Zn(OAc)2·2H2O, 10 mol% | PhSiH3, 160 °C, C6H5Cl, 4 days | 67% p-xylene | 637 |
| 70% THF | ||||
| PBT | (EtO)2MeSiH, 160 °C, C6H5Cl, 4 days | 15% p-xylene | ||
| PCL | (EtO)2MeSiH, 65 °C, THF, 24 h | 98% 1,6-hexanediol | ||
| PET | (EtO)2MeSiH, 160 °C, C6H5Cl, 4 days | 30% p-xylene and 55% 1,4-benzenedimethanol | ||
| PLA | PhSiH3, 110 °C, toluene, 48 h | 65% 1,2-propanediol | ||
| PLA | (EtO)2MeSiH, 110 °C, toluene, 48 h | 60% 1,2-propanediol | ||
Polycarbonates (PC) can be transformed into monomers such as bisphenol A (BPA) via aminolysis, glycolysis, hydrolysis, and methanolysis. The hydrogenolysis route of PC depolymerisation leads to hexane diols. Similarly, polyesters can be converted to diols via hydrogenolysis, and polyesters with two CH2 units between ester groups can be depolymerised to carboxylic acids. Depolymerisation of polylactide via methanolysis and hydrogenolysis leads to the production of methyl lactate and 1,2-propanediol, respectively. Hydrolysis of PLA results in lactic acid formation. In the case of LDPE, oxidative depolymerisation yields water-soluble acids such as succinic acid, adipic acid, acetic acid, propionic acid, etc. under microwave heating. Polystyrene produces benzoic acid, benzaldehyde, and acetophenone upon oxidative depolymerisation. Polyurethane on depolymerisation by glycolysis leads to mainly polyols, and by hydrogenolysis results in various products such as diol and diamines. These processes offer multiple routes for recovering valuable chemicals and monomers from plastic wastes, contributing to a more sustainable strategy to plastic recycling.
8.9 Commercial processes
Several companies are investing in chemical depolymerisation of plastic wastes, especially PET waste, to develop innovative technologies to enhance recycling efficiency.644 Axens, JEPLAN, and IFPEN developed and licensed an innovative PET recycling process, called Rewind® PET, to recycle various PET wastes, and started their Rewind® PET semi-industrial unit in Kitakyushu, Japan. The plant operates at a production capacity of 1000 tonnes per annum to produce BHET, which can easily be polymerised again in existing (or new) polymerisation plants.645Ioniqa Technologies has developed ionic liquids and a unique separation process for coloured PET using glycolysis to convert polyester into BHET.646 In 2019, Ioniqa Technologies has built its first PET plastic up-recycling plant in The Netherlands, with a capacity to convert 10000 tonnes per year of PET waste into high grade ‘virgin’ raw materials (BHET), for creating new high end PET through polymerisation. Gr3n, a Swiss company, has developed a DEMETO (Depolymerisation by Microwave Technology) process, which focuses on PET waste depolymerisation. The process only requires water and energy to hydrolyse PET, operating in continuous mode and using a microwave technology to produce virgin grade EG and PTA, with significantly reduced reaction time from 180 to 10 minutes.647 LOOP industries has developed a PET depolymerisation process that operates with zero energy input, in which PET is chemically depolymerised to form TPA as a salt containing less than 3 wt% of impurities and EG.648 This process was patented in 2017, which involves simultaneous mixing of PET waste with a halogenated non-polar solvent (3–5 vol%) and a solution of a linear alcohol, C1–C4 (95–97 vol%), along with hydroxide.649 The non-polar solvent to alcohol ratio is from 1:10 to 1:50 v/v. The method operates at atmospheric pressure without external heat, and involves continuous admixing for a sufficient duration, typically less than 5 hours, to depolymerise at least a portion of the PET.
GARBO S.r.l. has developed a recycling process called “ChemPET”, capable of using a heterogeneous mixture of PET wastes, including dyed PET.650 The process is based on glycolysis, in which PET reacts with EG and it is transformed into BHET after various purification steps, including removal of solids, salts, colour, other polymers, and contaminants. Gabro has implemented this technology at medium scale, 55–65 tonnes per day of BHET production.
9. Metathesis
Metathesis is a bimolecular reaction in which bonds are exchanged between two reacting chemical molecules. In metathesis, two hydrocarbons (alkanes, alkenes, or alkynes) are transformed into two new hydrocarbons by exchanging carbon–carbon single, double or triple bonds assisted by a metal catalyst. Alkane metathesis is an innovative catalytic methodology that converts two alkane compounds into both lower and higher homologues.One of the key advantages of metathesis reactions is their reversibility, which allows the reaction to achieve equilibrium rapidly, sometimes in seconds, using an appropriate catalyst system.585 Various categories of metathesis have been reported such as olefin metathesis, ring opening metathesis, ring closing metathesis, alkane metathesis, enyne metathesis, and cross-metathesis. Metathesis processes use catalysts only to chemically repurpose plastics by deconstruction as compared to hydroconversion and solvent conversion methods. Metathesis processes are specifically applicable to polyolefins and have the potential to upcycle these materials into more valuable chemicals, such as fuels, instead of recycling them back into monomers.
The chemical inertness of C–H and C–C bonds in alkanes poses a significant challenge for their chemical transformation, as these bonds are relatively unreactive. Similarly, the degradation of polyolefins through low energy methods is a daunting challenge due to the inherent chemical inertness where atoms are attached through these unreactive bonds.658 Alkane metathesis introduced a novel concept of alkane reactivity, enabling the transformation of alkanes into lower and higher molecular weight hydrocarbons. Alkane metathesis becomes feasible through a tandem catalytic mechanism that circumvents the direct cleavage of inert C–C and C–H bonds by first dehydrogenating alkanes to form reactive alkenes. These alkenes undergo olefin metathesis via metal–carbene complexes, followed by hydrogenation to regenerate alkanes. Transition metals facilitate bond activation through oxidative addition, σ-bond metathesis, and agostic interactions, which lower activation energy barriers. Catalyst design, including bifunctional systems and tailored supports, further enhances reactivity and selectivity, enabling the transformation of saturated hydrocarbons under milder, energetically accessible conditions. The metathesis reaction was reported by Basset et al. in 2010 on silica-supported tantalum hydride.659 The reaction involved the rearrangement of saturated linear and branched chains into larger saturated molecules. For example, n-propane was successfully converted into a range of paraffins such as C1, C2, C4, C5, etc., under relatively mild conditions. This development was a breakthrough considering the inert sp3 C–H or C–C bonds. Basset's group developed highly efficient single-site heterogeneous tantalum and tungsten–alkylidene complexes with siloxy ligands for alkane metathesis.659 The Chevron company in 1970s achieved butane metathesis using Pt/Al2O3 at 400 °C.659 Since then numerous catalysts have been developed for alkane metathesis.659–663
Cross-metathesis is also a powerful methodology where two different olefins with similar reactivities can react to produce a dynamic mixture of metathesis products in equilibrium. In Alkane Cross-Metathesis (ACM), two different alkanes can be upgraded to more valuable products in the presence of a catalyst.664 The process is cost-effective compared to other processes as it proceeds in a single step with a single catalyst. Alkane cross-metathesis can lead to the production of saturated hydrocarbons via dehydrogenation of alkanes followed by olefin metathesis. Basset's research group studied ACM between light and heavy paraffins (n-propane and n-decane) using silica-supported catalysts, i.e., [(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiO)W(CH3)2(H)3], and reported products with distribution from C4 to C19 saturated paraffinic compounds.664 Moreover, alkane metathesis reactions, which involve an alkane dehydrogenation reaction followed by an olefin metathesis reaction, have also been studied by various research groups.659,665,666 The in situ generated ruthenium hydride species from Grubbs catalysts have been reported for dehydrogenation of alcohols coupled with cross-metathesis reactions.666 In an investigation of the selectivity of Grubbs' first generation catalyst for dehydrogenative alcohol coupling with alkane metathesis, the introduction of tricyclohexylphosphine improved the efficiency of the Hoveyda–Grubbs 2nd generation catalyst (HG2) towards the selective synthesis of ester derivatives. By using allyl alcohol as both self and cross-metathesis substrates, HG2 catalysed the quantitative formation of γ-butyrolactone and long-chain esters.666 Various catalysts have been reported for cross-alkane metathesis reactions.659,664,667,668
SiO)W(CH3)2(H)3], and reported products with distribution from C4 to C19 saturated paraffinic compounds.664 Moreover, alkane metathesis reactions, which involve an alkane dehydrogenation reaction followed by an olefin metathesis reaction, have also been studied by various research groups.659,665,666 The in situ generated ruthenium hydride species from Grubbs catalysts have been reported for dehydrogenation of alcohols coupled with cross-metathesis reactions.666 In an investigation of the selectivity of Grubbs' first generation catalyst for dehydrogenative alcohol coupling with alkane metathesis, the introduction of tricyclohexylphosphine improved the efficiency of the Hoveyda–Grubbs 2nd generation catalyst (HG2) towards the selective synthesis of ester derivatives. By using allyl alcohol as both self and cross-metathesis substrates, HG2 catalysed the quantitative formation of γ-butyrolactone and long-chain esters.666 Various catalysts have been reported for cross-alkane metathesis reactions.659,664,667,668
The degradation and conversion of polymer waste into useful chemicals such as fuel is an important area of research due to the large volume of polymeric materials produced all over the world. With other chemical recycling methods, metathesis reactions are also being studied for breaking down polymers into useful fuel. The metathesis reaction can efficiently degrade polymeric chains by converting them into small alkane molecules. In the literature, significant work has focused on the depolymerisation of polyolefins (e.g., polyethylene, polypropylene) and rubbers using metathesis to produce hydrocarbons and monomers.669–671
9.1 Polyolefin degradation via metathesis reactions
Almost two-thirds of world's plastics consist of polyolefins, mainly polyethylene (PE) and isotactic polypropylene (i-PP), with an annual market value of about $200 billion globally.672 However, their degradation through low energy methods is a daunting challenge due to the inherent chemical inertness where atoms are attached through C–C and C–H bonds.658 The immiscible PE and i-PP are usually recycled together by melt processing to low value brittle materials, due to the high cost associated with separating them from each other.673 Polyethylene, which is synthesised by polymerisation of ethylene, produces various types of materials such as LDPE, LLDPE, HDPE, UHDPE and other cross-linked types, which complicates recycling efforts.62 PE recycling by metathesis has been mostly studied as an alternative to pyrolysis.Hydroconversion of polyolefins can yield products comprising linear and branched alkanes, as well as aromatics. In contrast, cross-alkane metathesis (CAM) has been shown to selectively produce linear alkanes without the need for hydrogen. In this methodology, polyolefins and short alkanes first dehydrogenate to produce olefins. The resulting unsaturated species then participate in cross-metathesis, generating two new olefins. Subsequent hydrogenation of resulting olefins leads to the formation of saturated alkanes, completing the transformation process (Fig. 29B).674–676 Jia et al. in 2016 developed a mild cross-alkane metathesis process to selectively degrade polyethylenes into liquid fuels and waxes (Fig. 29).674 Iridium based pincer type alkane dehydrogenation catalysts, such as (t-BuPOCOP)IrH2 and [(MeOiPrPOCOP)Ir(C2H4)] supported on g-Al2O3, were used for dehydrogenation to eliminate hydrogen from PE, and Re2O7/g-Al2O3 was used for olefin metathesis which assisted the breakdown of PE (Fig. 29A).674,677 By using low-value alkanes as co-reactants in cross-metathesis, numerous polyethylene materials of a wide range of molecular weights could be transformed into valuable oils and low molecular weight waxes as major and minor products, respectively. The formation of alkenes or aromatic products was not evident, and the process showed high selectivity for linear alkanes.
 | ||
| Fig. 29 Alkane metathesis and cross alkane metathesis. (A) Catalyst used for PE depolymerisation. (B) PE degradation pathway through catalytic CAM with n-hexane. (C–E) The distribution of degradation products after 4 days at 175 °C. Adapted from ref. 674. | ||
Particularly, HDPE with a Mw of 3350 and PDI of 1.6 underwent cross-metathesis with excess n-hexane at 150 °C under an inert atmosphere to produce a liquid n-alkane oil in 56 wt% yield, with almost half of the fractions in the diesel range (Fig. 29C).674 There was a substantial decrease in the molecular weight of wax products compared to the original PE (Mw = 680). Olefin metathesis must take place at internal CC double bonds, formed during the dehydrogenation of polyolefins, to achieve significant molecular weight (MW) reduction. The alumina supported (t-BuPOCOP)Ir catalyst was found to form internal CC bonds and the oil yield was increased to 98%. This catalyst system successfully degraded a wide range of polyolefins (with MW range: thousands to million and PDI up to 13) except LDPE, which produced wax products with MW < 1000 and PDI of 1.3 (Fig. 29D and E). The cascade catalytic systems efficiently degraded various waste polyethylene products, including bags, bottles, and films, converting them into high value liquid fuels and waxes. The product distribution can be finely tuned by modifying catalyst structures and reaction times to produce liquid fuels. The system was also found to be efficient in the presence of polymer stabilisers and plasticisers such as zinc stearate and polyphenol present in commercial polyolefins. The recycling of iridium and ruthenium catalysts supported on alumina exhibited reduced activity for both olefin metathesis and alkane dehydrogenation.
Ellis et al. used SnPt/g-Al2O3 and Re2O7/g-Al2O3 catalysts for depolymerisation of PE via olefin mediated CAM with n-pentane.671 SnPt/g-Al2O3 catalyst was synthesised with 1.7% (g g−1) Sn and 0.8% (g g−1) Pt loading through sequential deposition, whereas Re2O7/g-Al2O3 catalyst with 8% (g g−1) loading (Re basis) was synthesised using 75–80% perrhenic acid. This catalyst system generated a broad spectrum of n-alkanes from a model linear C20 alkane as well as linear PE feedstock. The PE substrate resulted in 73% reduction in molecular weight at 200 °C in 15 h, achieving 99 wt% yield of liquid alkanes.671 Zhang et al. used platinum supported on g-Al2O3 for converting PE (Mn = 1.85 g mol−1) into liquid linear dialkylbenzenes via a tandem catalytic hydrogenolysis/aromatisation in the absence of external H2 at 250–330 °C.678 Overall, hydrogenolysis of low molecular weight PE produced organic soluble hydrocarbons, insoluble hydrocarbons, and gaseous products in 75 wt%, 12 wt%, and 9 wt% yields, respectively. The reaction mechanism suggests that H2 is produced in situ during depolymerisation, which is subsequently used in simultaneous reactions for hydrogenating aromatic rings and hydrogenolysing the PE substrate. This mechanism also involves ring closure, resulting in the formation of alkylated aromatics and cyclic hydrocarbons.
9.2 Rubber degradation via metathesis reactions
Rubber degradation via metathesis reactions is a chemical process that involves breaking down rubber polymers into smaller, more useful molecules through the action of catalysts. This method leverages the principles of olefin metathesis, a reaction in which CC bonds of olefins are cleaved and reformed in different arrangements, effectively “shuffling” the molecular components.
The alkane metathesis methods have been widely investigated for depolymerisation of polydienes. Partial depolymerisation of 1,4-polybutadiene (Mw = 1800–500,000 g mol−1) was accomplished via a tandem ring-opening ring-closing metathesis method using ruthenium homogeneous catalysts.669 Macrocyclic oligo(butadiene) compounds (C16–C44) were produced in high yield (up to 90%) from 1,4-polybutadiene. The type of ligand structure in the ruthenium catalyst strongly influenced the distribution of cyclic compounds. Particularly, first-generation ruthenium catalysts favoured the selective formation of C16–C44 oligomers, whereas 2nd generation ruthenium catalysts containing N-heterocyclic carbene ligands favoured the formation of undesired t,t,t-cyclododecatriene (CDT). When 1st generation Ru complexes containing tricyclohexylphosphine ligands were used, the process resulted in highly selective (up to 98%) mixtures of C16 to C44 macrocyclic oligobutadienes with moderate conversions (59–88%).669 Michel et al. carried out depolymerisation of commercial polydienes and polybutadienes via cross-metathesis (CM) reactions, employing a ruthenium catalyst in combination with an acyclic bis(trialkoxysilyl) chain-transfer agent. They selectively depolymerised polybutadienes and poly(butadiene-co-isoprene) differing in their end functionalisation, producing low viscosity hydrocarbon α,ω-bis(trialkoxysilyl) telechelic (co)polydienes with exceptional chemoselectivity. The process demonstrated notable catalytic efficiency, with turnover numbers reaching up to 24000, though this figure was non-optimised.679 A controlled degradation process to depolymerise poly(cis-1,4-isoprene) was reported by Mouawia et al. via olefin metathesis using trihexyl-(tetradecyl)phosphonium chloride and N,N-dioctylimidazolium bromide with 2nd generation Grubbs catalyst.680 They successfully produced acetoxy telechelic polyisoprenic oligomers in high yield (99%). Furthermore, the methodology was successfully applied to waste tire depolymerisation, producing telechelic oligomers with high efficiency.680 These oligomers are key intermediates for synthesis of block copolymer compatibilisers and thermoplastic elastomers.
Olefin cross-metathesis has been utilised for degradation of carboxylated nitrile butadiene rubber to carboxyl content, using acrylonitrile as the chain transfer agent.681 The reaction was performed at 60 °C using 2% acrylonitrile and 0.2% catalyst under an inert atmosphere. The weight-average molecular weight of the degraded polymer was significantly reduced to 0.98 × 104. Herman et al. developed a novel approach to depolymerise cross-linked polybutadiene via alkene metathesis. They used HeatMet, Ru catalyst, and 2nd generation Hoveyda–Grubbs catalyst for metathesis depolymerisation of PB and compared their https://www.google.com/search?sca_esv=5037f467212c212c%26rlz=1C1GCEB_enIN1116IN1116%26q=activity%26spell=1%26sa=X%26ved=2ahUKEwjhzN-8yf-MAxX_yzgGHY5LLFkQkeECKAB6BAgJEAE.670 HeatMet demonstrated excellent stability and minimal activity towards PB under standard atmospheric conditions. However, when the catalyst was used in low concentrations (0.004–0.024 mol%), at 100 °C the cross-linked PB network rapidly depolymerised to oil. This in situ depolymerisation using latent catalysis provides an efficient pathway for recycling cross-linked PB waste, offering a potential solution for polymer waste management.
Hu et al. in 2021 studied the metathesis degradation and functionalisation of natural rubber (NR) using Grubbs 2nd generation catalyst, incorporating both asymmetric and symmetric olefins as chain transfer agents (CTAs).682 They used different CTAs such as 1-hexene, 1-decene, 1-dodecene, 1-octene, 4,4′-dibromo-trans-stilbene, and trans-stilbene to produce natural rubber based telechelic oligomers. Oligomers with low Mn were formed when terminal olefins with longer chain length were introduced. However, the incorporation of symmetrical stilbene derivatives, exhibiting significant phenyl steric hindrance, led to the generation of oligomers with slightly increased molecular weights (Mn above 10 kg mol−1). Recently, Burelo et al. successfully depolymerised industrial-grade polybutadiene and poly(styrene–butadiene–styrene) via metathesis, achieving excellent yields (>94%) with the 2nd generation Grubbs–Hoveyda catalyst, using 10-undecen-1-ol as the chain transfer agent. This approach led to synthesis of hydroxyl-terminated polybutadiene (HTPB).683 The molar ratio of rubber to chain transfer agent and the catalyst loading were critical factors in determining the molecular weight and tailoring the characteristics of resulting oligomers from PB degradation. The catalyst loadings of [CC]/Ru = 5000:1 were found to be optimum, producing oligomers with Mn in the range of 583–6580 g mol−1.
9.3 Product distribution via metathesis
Degradation of various polymers has been reported via cross-alkane metathesis reactions leading to the formation of products ranging from C2 to C40 depending upon the type and molecular weight of polymers. For example, Jia et al. depolymerised various types of polyolefins with different molecular weights to produce oils in the range of C3 to C42.674 They also investigated degradation of different commercial wastes, and found that distribution of degradation products was strongly influenced by dehydrogenation catalysts and reaction time. Their strategy uses a system with two catalysts: one for alkane dehydrogenation and the other for olefin metathesis. At the first stage, an iridium-based dehydrogenation catalyst removes hydrogen from both polyethylene and a light alkane in a closed system, forming unsaturated intermediates along with Ir–H2. Subsequently, the olefin metathesis catalyst promotes the cleavage of alkenes, which leads to the fragmentation of polyethylene chains. The newly formed alkenes are then hydrogenated with Ir–H2, yielding saturated alkanes. This process of metathesis gradually reduces the molecular weight of PE as the internal double bonds in polyethylene-derived alkenes (PEAs) interact with the double bonds in light alkenes. With an excess of light alkanes, the intermediates undergo additional CAM cycles, continuously decomposing into shorter-sized hydrocarbons. Repeated repetitions of this process eventually convert polyethylene into short-chain hydrocarbons suitable for use as a transport fuel. For example, depolymerising commercial HDPE pellets produced 81% of waxes with a relatively high molecular weight (4390 g mol−1) after 2 hours, however, the same process resulted in 48% of waxes of low MW (1060 g mol−1) after 24 hours. Moreover, the choice of iridium catalyst also played a crucial role in regulating the yields of oils and waxes. The use of (MeO-i-PrPOCOP)Ir(C2H4)/g-Al2O3 dehydrogenation catalyst for degradation of various PEs constantly resulted in higher wax yields compared to the use of (t-Bu2PO-t-BuPOCOP)Ir(C2H4)/g-Al2O3. Moreover, the molecular weight of PE waxes, obtained by using (MeO-i-PrPOCOP)Ir(C2H4)/g-Al2O3, was almost double that obtained with (t-Bu2PO-t-BuPOCOP)Ir(C2H4)/g-Al2O3.674 A range of liquid n-alkane products were produced from PE degradation.674 In addition, all types of plastics predominantly produced alkanes within the diesel range as the primary products. Using the iridium catalyst (MeO-i-PrPOCOP)Ir(C2H4)/g-Al2O3, all polymers except LDPE, degraded into wax products with a narrow molecular weight distribution and comparatively lower molecular weights. However, changing the iridium catalyst from (MeO-i-PrPOCOP)Ir(C2H4)/g-Al2O3 to (t-Bu2PO-t-BuPOCOP)Ir(C2H4)/g-Al2O3, the resulting polyethylene wax products consistently exhibited broader molecular weight distributions (Fig. 29). The choice of iridium catalyst was critical to control the MW distribution of degradation products. Moreover, when this degradation process was applied to waste plastics with these catalysts, both the pincer Ir dehydrogenation and the Re metathesis catalysts demonstrated tolerance to the stabilisers typically found in commercial polyethylenes.Beckham's research group reported depolymerisation of PE feedstocks to a mixture of alkane products in the range of C3–C30.674 Polybutadiene's degradation has also been reported by some researchers in the recent literature.669,670,683 Depolymerisation of natural rubber using Grubbs 2nd generation catalyst with various chain transfer solvents resulted in telechelic oligomers.682 PB and SBS depolymerise to unsaturated diols and polyols via metathesis.683 Significant advancements led to enhanced efficiency and selectivity of various metathesis reactions, particularly with the development of next generation catalysts. These catalysts have improved the stability of catalysts but also afforded high selectivity for products. Plastic waste can be depolymerised using metathesis reactions, but there are certain thermodynamic and kinetic challenges to address. For example, latent catalysis has been used to recycle crosslinked PB rubbers, but it must be applied at relatively low temperatures to avoid catalyst deactivation and depolymerisation. Latent catalysis is an active area of research and the latest metathesis catalysts discovered, such as HeatMet will provide many prospects for in situ depolymerisation of cross-linked polymers. Tandem catalysis has been successfully applied for depolymerisation of polyolefins (PE, HDPE, LDPE) in different forms such as films, bottles, etc., however, the process is still at the lab scale. The tandem catalytic strategy enables high efficiency and selective conversion under relatively mild conditions and provides distinct advantages over traditional pyrolysis processes. However, further research is necessary in this area to fully understand the reaction mechanism, kinetics, optimum process parameters, and efficient catalysts to provide the best depolymerisation rates and tune product distribution. Noble metal catalysts and high loadings have been used in most of the studies; therefore, efforts must be focused on reducing catalyst loadings, catalyst regeneration and reusability, explore cheap metals to reduce the catalyst costs to translate this method for large scale applications.
Unfortunately, polyester plastics such as PET cannot be directly modified using metathesis due to the absence of reactive double bonds, the presence of polar ester groups, and the incompatibility of these groups with metathesis catalysts. Instead, alternative chemical recycling strategies have been developed, integrating multiple processes to overcome these challenges. For example, Foster et al. demonstrated an effective strategy for the decomposition of PET into various α,ω-dialkenyl terephthalates by organocatalysed transesterification using ω-unsaturated alcohols.684 These monomers were subsequently polymerised into long cycle polyesters through acyclic diene metathesis polymerisation (ADMET) under mild reaction conditions. The resulting semi-aromatic polyester materials possessed mechanical properties intermediate between polyethylene and polyethylene terephthalate, with their characteristics highly dependent on the monomer structure and the choice of a Ru-based catalyst. These polymers were derived from dialkenyl terephthalate monomers, obtained by organocatalytic deconstruction of PET.
Metathesis offers significant potential for industrial applications due to its use of cost effective paraffins as feedstock and its ability to generate stoichiometric hydrogen as a byproduct. However, the challenge involves the scaling up of the process to achieve pure chemicals at a low cost, while avoiding contamination by metals and ensuring high selectivity towards the desired products. Achieving these goals is crucial for making metathesis a viable and efficient option for large-scale industrial production.
10. Photocatalytic plastic recycling
Photocatalysis is a cost-efficient, highly promising and sustainable methodology to convert plastic waste under ambient conditions using renewable energy sources.685 Photocatalysis, utilising either limitless solar energy or low-energy artificial light sources like LEDs, is a key focus for green catalysis and sustainable development due to its mild operational conditions, cost-effectiveness, and environmental benefits. A photocatalytic reaction performed under mild conditions is expected to selectively and precisely activate specific chemical bonds, without affecting other functional groups, therefore achieving high selectivity of the desired products.686In traditional photocatalysis, most photodegradation reactions of polymers primarily produce CO2 and H2O.687 However, CO2 is a major greenhouse gas significantly contributing to global warming, making it an undesirable primary product, and the resulting H2O is often contaminated. Over time, researchers have been developing photocatalytic methods aimed at producing valuable products like H2 and carbon-based fuels from the photocatalytic upcycling of plastic waste. These innovative and efficient photocatalytic techniques add value to plastic waste upcycling and should remain a focus due to their importance and the urgent need to establish a circular economy.
10.1 Metal oxide catalysts
Various metal oxides including Ga2O3, Nb2O5 and TiO2 have been used for photocatalytic conversion of plastic.686,688 These catalysts have strong oxidation abilities and can convert polyolefins with robust C–C bonds including PE (HDPE, LDPE), PP and PVC. The activation of the photocatalyst depends on several important parameters, including specific energy and light intensity. Higher light energy results in a larger number of available photons, thereby accelerating the excitation of the photocatalyst and enhancing the generation of reactive particles. This photothermal synergy played a key role in lowering the energy barrier, facilitating the adsorption and activation of molecules.689 When exposed to light, these catalysts create highly oxidative photo-generated holes, which lead to the formation of hydroxyl radicals (˙OH). When used in an air or oxygen environment, photo-excited electrons in the conduction band typically react with O2, generating various reactive oxygen species including ˙OH, ˙O2 and ˙HO2. These reactive species initially oxidise polyolefins, forming carbonyl, hydroxyl, and carboxyl functionalities. Consequently, C–C and C–H bonds in polyolefins become polarised and considerably weakened, facilitating easier cleavage of these bonds and transforming these polymers into valuable chemicals and fuels.Gazi et al. synthesised a vanadium-based photocatalyst capable of converting polymers with hydroxyl terminals such as polyethylene glycol (PEG), polycaprolactone copolymers, and polyethylene into chemical building blocks and fuels, including formic acid and methyl formate.690 Under visible light, the catalyst facilitated the selective oxygenation of C–C bonds, enabling the efficient transformation of unactivated alcohols through a light driven cascade process. The hydroxyl polymer PEG 400 was completely converted in 2.5 days leading to formic acid as the major product, along with methyl formate and oligomeric products as minor products. The authors successfully achieved the selective cleavage of the aliphatic C–C bond under mild conditions with visible light for the environmental remediation of plastic pollutants. They converted the block polymer of PE and PEG (PE–PEG) and PE–monoalcohol to yield formic acid as the main product. Peng et al. fabricated nanofiber film Ni5P4/TiO2/C photocatalysts through electrospinning, hydrothermal treatment, and phosphating.691 The Ni5P4 addition onto TiO2/C NFs enhanced the light absorption efficiency of nanofibers and also improved their photocatalytic activity. This photocatalyst resulted in the degradation of PET microfibers into H2 which reached 76.66 μmol H2 per g sub in 12 h (3 times compared to TiO2/C NFs). Kang et al. developed a MoS2/g-C3N4 photocatalyst for upcycling of PET into high value chemicals, and the PET conversion mechanism was found to be concentration-dependent.692 The main product was acetate at an ethylene glycol (EG) concentration of 7.96 mM, whereas formate was obtained as the main product by increasing EG concentration to 300 mM. Interestingly, the hydrolysate from PET water bottles containing only 7.96 mM of EG produced 704.59 nmol of acetate, which is four times higher than the 174.50 nmol obtained by converting 300 mM of EG. However, increasing the EG concentration to 300 mM, which is 40 times higher, resulted in only approximately a threefold increase in formate production (179 nmol).
The Reisner group used a well-established Pt/TiO2 and cyanamide-functionalised carbon nitride powder (NCNCNx/Pt) as photocatalysts to convert ∼40% of PE to valuable chemicals.686 The major products from PE degradation were succinic acid (44%) and glutaric acid (22%), along with acetic acid, adipic acid, and propanoic acid with 21%, 12% and 4% yields, respectively, in the presence of HNO3 (6 wt%) as the oxidant at 180 °C under an inert atmosphere. The authors performed the photocatalytic experiments in a batch reactor using pure succinic acid obtained from oxidative PE treatment with a standard AM1.5G light source at pH 4 for 24 h. The major product was ethane for both Pt/TiO2 and NCNCNx/Pt, produced at rates of 56.3 and 7.2 μmol g−1 h−1, respectively.686 However, both Pt/TiO2 and NCNCNx/Pt gave propanoic acid as the intermediate product at 964.7 and 176.7 μmol g−1 h−1, and adipic acid as the minor product at 23.5 and 25.2 μmol g−1 h−1, respectively, as determined by HPLC. They also performed a photocatalysis reaction in a flow setup using a photocatalyst pane and obtained a constant production of ethane, ethylene, propane and propylene at 77.9 μmol m−2, 38.5 μmol m−2, 40.7 μmol m−2, and 19.1 μmol m−2, respectively, using NCNCNx/Pt over a period of 72 h.
A plasma treatment approach has been reported for pretreatment of polyolefins for partially breaking down C–C and C–H bonds, and generating oxygen functionalities in the backbone of polymers before light irradiation.693 The high energy plasma induced –OH, O–CO, and CO functionalities in the backbone of polyethylene were confirmed by FTIR and XPS spectra. Contact angle of plasma treated PE was gradually decreased by exposing it to water, indicating increased hydrophilicity. This enhanced hydrophilicity improves the dispersion of pretreated PE in aqueous solutions and enhances the photocatalytic activity. Pt nanoparticles (5–15 nm size) uniformly distributed on the P25–TiO2 catalyst was used as the co-catalyst to increase the activity for photocatalytic experiments.693 The H2 evolution initially increased and then decreased as the plasma treatment time on PE was extended. For pristine PE, the H2 production was 33.2 μmol g−1 h−1, which increased to 58.6 μmol g−1 h−1 for PE-10 and further to 108.95 μmol g−1 h−1 for PE-30. However, H2 production reduced significantly for longer pretreatment times due to excessive oxidation of PE, generating high amount of carbonyl and carboxyl functionalities. This approach was also applied for pretreatment of PP and PVC, producing H2 at 56.32 μmol g−1 h−1 and 69.64 μmol g−1 h−1, respectively.693 Hence, plasma treatment is considered an excellent strategy to enhance the depolymerisation rates of highly inert polyolefinic plastic waste. When the same photocatalyst (Pt/P25-TiO2) was subjected to hydrothermal pretreatment, the evolution rates of H2, C2H4, and C2H6 were significantly higher compared to plasma treatment. This increase is attributed to the fact that hydrothermal treatment converts most of the PE into C2–C6 carboxylic acids, which can be easily transformed into small molecules. In contrast, plasma treatment primarily introduces oxygen-containing functionalities, such as –OH, O–CO, and CO, to polyethylene without significantly breaking down the polymer backbones into smaller molecules.
Jiao et al. pioneered the highly selective approach for the photocatalytic conversion of polyolefins and PVC into CH3COOH without the need for a pre-treatment step.688 The study involved a sequential photoinduced C–C bond cleavage to produce CO2 followed by a coupling pathway to generate C2 fuels over the Nb2O5 photocatalyst. The selection of a photocatalyst is based on its suitable valence band (VB) and conduction band (CB) that should satisfy the redox potentials involved in the photocatalytic conversion reaction. Nb2O5 has a VB position at +2.5 V vs. NHE and its CB is positioned at −0.9 V vs. SHE at pH 7.694 Hence, Nb2O5 is capable of generating highly oxidative ˙OH radicals (+2.32 V) for plastic degradation, while simultaneously using photo-generated electrons to reduce CO2 (−0.6 V at pH 7). The photocatalytic reactions were conducted by dispersing plastic waste and catalyst in a 50:1 ratio in water, using a 300 W Xe lamp equipped with a AM 1.5G filter.688 An AgNO3 electron scavenger was used to detect the photooxidation products. By adding AgNO3 solution, only CO2 was identified which suggested the photooxidative C–C bond cleavage of polymers to produce CO2. The photocatalytic experiments established that Nb2O5 atomic layers degraded PE, PP and PVC in 40, 60 and 90 hours, respectively (Fig. 30a and b). The amount of carbon in the produced CO2 was nearly equivalent to the total carbon present in pure PE, PP or PVC. Moreover, the generation of CO2 gradually increased with time. Fig. 30c shows the CH3CO2H production from various plastics and its increase with time. The average CH3CO2H production from PE, PP, and PVC was analysed to be 47.4, 40.6, and 39.5 μg g−1 h−1, respectively (Fig. 30d). Electron spin resonance (ESR) spectroscopic studies revealed the formation of ˙OH and ˙O2 radicals during photoconversion of polyethylene as shown in Fig. 30e and f. Simultaneously, O2 underwent stepwise reduction to O2˙−, H2O2, and H2O. The produced CO2 upon photocatalytic C–C coupling resulted in the formation of CH3COOH as shown in Fig. 30e. In another study, Jiaqi et al. used Co–Ga2O3 nanosheets to convert plastics into syngas under ambient conditions.695 They crushed PET bottles, PE bags, and PP boxes into powders, and utilised Co–Ga2O3 or Ga2O3 catalysts to convert these powders using solar light under standard conditions. GC analysis revealed the presence of H2, CO and CO2, however a liquid product was not detected. The Co–Ga2O3 catalyst demonstrated excellent activity with H2 and CO generation rates at 647.8 mmol g−1 h−1 and 158.3 mmol g−1 h−1, respectively from PE conversion. These rates were almost 1.6 and 1.9 times more compared to those obtained using Ga2O3 alone. Additionally, the PE bags experienced a weight loss of 81% after 48 hours. H2 generation was found to originate from H2O rather than plastics.
 | ||
| Fig. 30 (a) Schematic representation of various plastics into C2 fuels; (b) CO2 production from pure PE, PP, and PVC using Nb2O5 atomic layers; (c) amount of CH3CO2H production; (d) CH3CO2H and CO generation rate from photoconversion of pure PE, PP, and PVC; (e) VB and CB potential of Nb2O5 and potential for CO2, H2O, H2O2, and O2; (f) possible two-step mechanism for C–C bond cleavage and C–C bond coupling to produce CH3CO2H. Adapted with permission from ref. 688. Copyright 2020, John Wiley & Sons. | ||
Tofa et al. utilised ZnO nanorods as photocatalysts for degradation of LDPE microplastic residues and observed a significant increase in the carbonyl index (30%) of residues, along with the formation of cracks and surface wrinkles.696 ZnO nanorods provided a greater surface area to produce ˙OH, and consequently enhanced the photocatalytic degradation activity. The presence of peroxides, unsaturated groups, carbonyl, and hydroperoxide was detected by time dependent FTIR studies that confirmed the degradation and oxidation of photocatalysed LDPE. The probable mechanism involved the initiation of the degradation process at the weak chromophoric groups by the ˙OH and O2 radicals leading to shorter PE alkyl radicals. This is followed by chain cleavage, oxidation, and cross-linking of LDPE. Next, removal of hydrogen atoms from polymer chains generates hydroperoxide groups. Finally, the catalyst generates ethanol and formaldehyde, which are eventually converted to CO2 and H2O.696 Jiao et al. in 2022 synthesised a charge asymmetrical dual active metallic site catalyst, Zr-doped CoFe2O4 quantum dots for direct photoreforming of PE into valuable C2 liquid fuels, particularly CH3COOH.697 This catalyst improved the adsorption and activation of the *CH2CH2 intermediate, and consequently increased the direct transformation of PE into C2 fuels under standard conditions. The doping of Zr atoms can shift their charge density to adjacent Fe or Co atoms, leading to the formation of asymmetrical Zr–Fe and Zr–Co sites for the *CH2CH2 adsorption. The Raman and X-ray photoelectron spectra of Zr-doped CoFe2O4 QDs revealed the incorporation of Zr atom, resulting in lattice distortion of CoFe2O4.697 The effect of H2O2 was also investigated, the CH3COOH production and weight loss of PE was significantly increased by increasing the H2O2 amount from 3% to 30%. When the reaction was performed under Ar, a similar amount of CH3COOH was produced with CoFe2O4 and Zr–CoFe2O4 catalysts, demonstrating the role of H2O2 as an oxidant.697
Qin et al. investigated the upcycling of PEG using Ag2O/Fe-MOF photocatalysts and produced formic acid coupled with hydrogen.698 The synthesised Ag2O/Fe-MOF heterojunction followed a type II charge transfer mechanism; the CB and VB of Ag2O were positioned above the CB and VB of Fe-MOF, which facilitated the transfer of electrons from the CB of Ag2O to the CB of Fe-MOF, and holes from the VB of Fe-MOF to the VB of Ag2O, thus reducing the charge recombination and increasing the photocatalytic activity. The Ag2O/Fe-MOF catalyst containing 0.2 wt% Ag2O showed the highest production of H2 (6.2 mmol g−1) from PEG microplastics. For PE and PET, the H2 production was 1.7 mmol g−1, 1.9 mmol g−1, respectively. Ag2O/Fe-MOF proved to be an exceptional photocatalyst compared to Ag2O, Fe-MOF, and TiO2.698 Pichler et al. used a similar approach for the photo-oxidative conversion of PE to fuels using Pt/TiO2 and NCN-functionalised GCN/Pt (NCNGCN/Pt) as photocatalysts.686 PE was initially transformed into dicarboxylic acids including glutaric acid (22%) and succinic acid (44%), with smaller amounts of adipic acid, acetic acid, and propanoic acid. Miao et al. used Ru/TiO2 catalyst for photothermal conversion of PE into hydrocarbons under concentrated sunlight or a Xe lamp.699 The Ru/TiO2 catalyst was heated in the presence of polyolefin plastics at 200–300 °C under the light irradiation which facilitated the melting of the polymer as well as cleavage of C–C and C–H bonds in the polymer backbone. The Ru/TiO2 catalyst absorbs light across UV, visible and near-IR ranges, and allows full-spectrum photothermal heating. Experimental investigation was performed using 80 mg of LDPE (Mw: 68.7 kDa) and 20 mg of catalyst in a reactor with photothermal heating at 250–300 °C and 1 bar H2/Ar atmosphere (30:70). The degradation of LDPE increased to 95% at 300 °C after 20 h, and the MW reduction was confirmed by gel permeation chromatography (GPC). Similarly, HDPE and PP were also degraded effectively by 87.8% and 93.9%, respectively. All the plastics gave gaseous products in 3–17% yield and liquid products in 91–97% yield. Moreover, CH4 selectivity was increased to 100% in the gaseous products by increasing the reaction time to 40 h at 300 °C.699
Li and Zhang used a tandem depolymerisation and photoreforming approach for conversion of PET to valuable chemicals, and achieved TPA monomers using a binuclear zinc catalyst through intramolecular hydrolysis under mild reaction conditions (COH− ≤ 0.1 M, ≤ 60 °C).700 They employed an ultrasmall carbon nitride nanosphere photocatalyst for reforming of PET to produce formic acid and H2 at the rate of 2000 μmol gca−1 h−1, which is approximately 5 times greater compared to the corresponding strong alkali pre-treatment system.
10.2 Metal sulphide based photocatalysts
The majority of metal sulphide-based photocatalysts used for plastic conversion are cadmium based catalysts due to their excellent light absorption, resulting from their suitable band gap and favourable conduction band (CB) positions, which allow for outstanding reduction abilities.701 The Reisner group introduced a novel approach utilising CdS/CdOx quantum dot (QD) photocatalysts to transform plastic waste to produce H2 and value-added organic building blocks using sunlight. They discovered that addition of CdS QDs to aq. NaOH formed a thin layer of cadmium oxide/hydroxide (CdOx), which helped prevent photo-corrosion of the catalyst. Ligand-free QDs have exposed surfaces hence they work with most substrates. Oleic acid-capped quantum dots (QDs) were effective only with PET, likely due to the hydrophobic effect enhancing the QD–substrate interaction. The authors utilised inexpensive CdS/CdOx quantum dots for photoreforming of polylactic acid, PET and polyurethane in aqueous alkaline solution under a nitrogen atmosphere.701 The process, conducted under ambient conditions, generated pure H2 and converted plastic waste into organic compounds including acetate, formate, and pyruvate. The CdS/CdOx quantum dots demonstrated H2 evolution rates of 0.85, 3.42 and 64.3 mmol g−1 h−1 for PUR, PET, and PLA, respectively. In comparison, 5% Pt/TiO2 exhibited much lower H2 generation rates, with only 0.011 and 0.074 mmol g−1 h−1 under the same conditions. However, only TiO2 showed no H2 production. Nagakawa et al. investigated the photocatalytic H2 generation from biomass waste and plastic waste including isoprene rubber (IR), PE, and PS, using a CdOx/CdS/SiC photocatalyst irradiating with a 300 W Xe lamp.702 The visible light successfully raised the reaction temperature. Moreover, there was reduction in charge recombination due to electron transfer between CdS and SiC, hence photocatalytic activity was increased. Biomass waste produced a higher amount of H2 due to the better hydrolysis rate, however for plastic waste, isoprene rubber exhibited the highest H2 generation. Furthermore, the rates of hydrolysis and H2 formation were observed to increase proportionally with both temperature and base concentration.Recently, Miao et al. reported a direct photoreforming process for PLA plastic using the Pd–CdS photocatalyst under visible light, and effectively produced H2 at the rate of 49.8 μmol gca−1 h−1 that continued for 100 h.703 The process showed excellent selectivity to pyruvic acid, achieving 95.9% in liquid products. In another study, Du et al. focused on photoreforming of PE, PET, and PLA to obtain valuable chemicals along with H2 production using MoS2/CdS photocatalyst (Fig. 31).704 PE has an inert and stable C–C linkage without any functional group and it is very difficult to induce some functionalisation or activation. Hence, the photoreforming of PE requires C–C cleavage into a short chain carboxylic acid. HNO3 was used as an oxidant to convert PE into a variety of carboxylic acids at 180 °C.704 After oxidation with HNO3, photocatalytic conversion of PET using the MoS2/CdS catalyst with 21.8 wt% MoS2 under solar irradiation resulted in evolution of significant amount of H2 gas (1.13 mmol g−1 h−1), even after 200 h (0.99 mmol g−1 h−1).704 Photoreforming of PLA in aqueous NaOH solution (10 M) using the MoS2/CdS catalyst showed the highest H2 evolution of 6.68 ± 0.10 mmol g−1 h−1 (Fig. 31b). Fig. 31c shows the effect of the amount of MoS2 loading on CdS; the highest H2 production was achieved with 21.8 wt% MoS2 loading, and it was achieved during the first 5 hours of the reaction (Fig. 31d). To further demonstrate the advantage of the MoS2/CdS structure, MoS2 sheets were selectively loaded on CdS nanorods with 21.1 wt% loading for use as the control model, represented as CdS@MoS2. However, this control sample showed a significantly lower photocatalytic H2 production rate. Fig. 31e shows the generation of the formate product at concentrations of 5.37 ± 0.67 mmol L−1 in 5 h photoreforming, along with reduction in lactate feedstock. The authors also investigated the photoreforming of PET with MoS2/CdS, which showed constant H2 production during the 25-hour period, achieving a rate of 3.90 mmol g−1 h−1. Moreover, pretreated PET produced higher amount of H2 compared to the PET bottle due to their small size (Fig. 31f). In addition to H2 evolution, formate, acetate, and glycolate were detected as photoreforming products, by 1H NMR analysis. Quantitative analysis using HPLC revealed that, after 5 hours of irradiation, formate and acetate accumulated to concentrations of 5.96 ± 0.02 mmol L−1 and 0.95 ± 0.01 mmol L−1, respectively (Fig. 31g). Similarly, Li et al. synthesised MoS2/Cd0.5Zn0.5S with 4.3 wt% MoS2 by combining the strategies of cocatalyst loading and band structure engineering.705 Raman and XPS spectra confirmed the strong electronic coupling between MoS2 and Cd0.5Zn0.5S. Control experiments showed that PET degradation and H2 production cannot occur without light, a photocatalyst, or NaOH. The 4.3 wt% MoS2 coupled Cd0.5Zn0.5S showed the highest H2 production rate (15.9 mmol g−1 h−1) compared to other CdxZn1−xS catalysts (x = 0.2, 0.4, 0.8, and 1). Excellent H2 production was achieved with 4.3 wt% MoS2/Cd0.5Zn0.5S using PET bottles. The pre-treated PET solution comprised EG, PTA and glycolate, which was confirmed by 1H-NMR spectroscopy analysis. After the reaction, the treated PET was finally oxidised to produce acetate, ethanol, formate, and methanol.
 | ||
| Fig. 31 (a) Elemental mapping of the MoS2/CdS photocatalyst, (b) photoreforming of pre-treated PLA in various KOH concentrations; (c) photoreforming of KOH (10 M) treated PLA using various MoS2 loadings; (d) H2 generation from pre-treated PLA; (e) formate and lactate concentration during 5 h duration, (f) H2 generation from pre-treated PET and PET bottles, (g) formate and lactate concentration during 5 h duration. Adapted with permission from ref. 704. Copyright 2022, American Chemical Society. | ||
Zhang et al. demonstrated the photoconversion of PET and PLA using a defect-rich chalcogenide photocatalyst, d-NiPS3/CdS.706 The catalyst was highly efficient in producing hydrogen in excellent yield and high stability for up to 100 h. Spectroscopic investigations revealed that the reaction was facilitated by the charge transfer mechanism, which involved the extraction of electrons from CdS by d-NiPS3, enabling the oxidation of PET and PLA to value added products. The photoreforming of PLA by d-NiPS3/CdS resulted in the formation of acetates and pyruvate-based compounds under alkaline conditions.706 The organic acid was produced in 78.1 μmol yield with H2 generation of ∼40 mmol gcat−1 h−1 in 9 h, proving the efficiency of d-NiPS3/CdS for photoreforming of plastic waste. PET photoreforming led to the formation of acetate, formate, and glycolate. These studies highlight the remarkable potential of metal sulfide-based photocatalysts for recycling pre-treated plastics into value-added chemicals and fuels. However, their toxicity, limited stability, and insufficient oxidation capacity pose serious limitations for large-scale applications.
10.3 Carbon nitride-based photocatalysts
Carbon nitride-based (CxNy) photocatalysts offer various advantages such as non-toxicity or low toxicity, earth abundance, cost-effectiveness, suitable band gap width for good light absorption, strong redox abilities, and excellent chemical stability.707,708 However, the moderate oxidation potential of CxNy makes the direct conversion of plastics into chemicals and fuels particularly challenging under ambient and anaerobic conditions. To overcome this challenge, Uekert et al. developed a highly efficient carbon nitride/nickel phosphide (CNx|Ni2P) catalyst for converting PET and PLA to hydrogen fuel and different organic compounds under alkaline conditions without producing CO2 or even CO32−.707 The oxidation process was proposed to be driven by direct transfer of photogenerated holes from the photocatalyst to the plastic substrate, resulting in the formation of useful organic chemicals such as acetate and formate. Ni2P, synthesised on cyanamide-functionalised carbon nitride, proved to be highly efficient and maintained photostability for 5 days. The photoreforming catalysts were further evaluated for microplastics and food-contaminated plastics, generating H2 as the major product. PET and PLA were initially pre-treated in aq. KOH solution at 40 °C to obtain the related monomers (EG and TPA for PET; lactate for PLA). Pre-treatment resulted in the release of 72% lactate from PLA, and 62% EG and 51% TPA from PET. Following this, the authors optimised the reaction conditions including Ni2P loading and KOH concentration to achieve the highest photocatalytic H2 generation. Pre-treated PET and PLA upon solar light irradiation on H2NCNx|Ni2P led to H2 generation of 82.5 ± 7.3 and 178 ± 12 mmol g−1. Furthermore, CNx|Ni2P can transform the pre-treated PLA into acetate and formate, and this photocatalyst also shows the possibility of photoreforming postconsumer polyester microfiber, PET bottles into H2 and a series of chemicals.In addition to H2 production, formic acid and acetic acid production in significant amounts has been reported by Han et al. using CPDs-CN for the photocatalytic conversion of PET.709 The study primarily focused on evaluating the yields of various photocatalytic products derived from ethylene glycol, one of the PET monomers. Initially, PET was transformed into TPA, EG, and small amounts of isophthalic acid. Upon extended irradiation with a 300 W Xe lamp, EG was converted into various products including acetic acid, formic acid, glycolic acid etc. CPDs-CN-7 performed better than other CPD-CN-x catalysts, producing acetic acid, glycolic acid, and formic acid in 554, 383, 139 μmol yields, respectively, along with ethanol (128 μmol), glycolaldehyde (110 μmol) and acetaldehyde (43 μmol) after 8 days of irradiation.709 Glycolic and acetic acids were obtained with the highest selectivity, whereas some variations were observed in the intermediate selectivity of other products.
Although photoreforming of untreated plastics using CxNy is very challenging at room temperature, Cao et al. successfully applied C3N4 for the photocatalytic conversion of PS into aromatic oxygenated products, such as acetophenone, benzaldehyde, and benzoic acid, at 80–150 °C via oxidative C–H activation followed by C–C bond scission using light irradiation.710 Authors have used various known photocatalysts such as TiO2, ZnO, ZnS and C3N4 to transform PS into oxygen containing aromatics at 80 °C using light under air, achieving conversions of 13%, 21%, 12%, and 46%, respectively. The selectivities for aromatic compounds were 15% (TiO2), 55% (ZnO), 64% (ZnS), and 60% (C3N4). Therefore, C3N4 based catalysts were synthesised by loading various metals, including Au (0.1%, 0.5%), Fe (0.5%), Pt (0.5%) and Cu (0.5%), onto C3N4. Although these metal loadings increased the conversion rates, they reduced selectivity. Subsequently, authors used g-C3N4 for the photocatalytic conversion of PS, achieving conversion rates of over 90% at 150 °C. The main products formed in the liquid fraction were acetophenone, benzaldehyde, and benzoic acid. The formation of different products occurred during a 3 to 24 hours period, with the first 3 hours serving as the induction period. Additionally, they demonstrated a stable production rate of different organic compounds (10 mg g−1 h−1) from oxidative photoreforming of PET pellets (500 mg), maintaining a selectivity of 76% in 18 cycles.
10.4 Composite photocatalysts
Composite photocatalysts designed for plastic recycling utilise two photon absorbers capable of capturing photons and generating photo-induced electrons and holes for plastic degradation. Qin et al. synthesised a zinc oxide composite, ZnO/UiO66-NH2, where ZnO nanoparticles (NPs) are encapsulated within the UiO66-NH2 framework, specifically targeting the photocatalytic conversion of PLA and PVC plastic waste (Fig. 32a).711 As shown in Fig. 32b, ZnO/UiO-66-NH2 exhibited a well-dispersed rhombic octahedral structure with a smooth surface. TEM analysis revealed a uniform particle size and even elemental distribution (Fig. 32c and d). In the valorisation of PLA, ZnO/UiO66-NH2 catalyst produced acetic acid in significantly higher yield (14.4%) compared to individual components ZnO (3.3%) and UiO66-NH2 (4.7%) (Fig. 32e). The acetic acid formation was low in the initial stage, hindered by the large particle size and low hydrophilicity of PLA, but gradually increased over time (Fig. 32f). However, as the reaction progressed, the large PLA particles were broken down into smaller, more hydrophilic particles, increasing the surface area and catalytic activity, which led to a steady rise in acetic acid production over time. The ZnO/UiO66-NH2 composite achieved an impressive 91.6% selectivity for acetic acid production during PLA conversion (Fig. 32g). In control experiments, acetic acid production was significantly lower with manually mixed ZnO/UiO66-NH2 (2.0%) and was negligible in the absence of a catalyst, light or PLA (Fig. 32h), highlighting the critical importance of the strong interaction between ZnO and UiO66-NH2. When photocatalytic PLA conversion was performed under a N2 atmosphere, no acetic acid was formed, emphasising the importance of O2 in the photocatalytic conversion process. Moreover, ZnO/UiO66-NH2 also demonstrated exceptional stability in the photocatalytic valorisation of PLA, successfully generating H2 with TON and TOF of 26.36 and 0.75 h−1, respectively (Fig. 32h). ZnO/UiO66-NH2 was also used for photocatalytic valorisation of PVC, producing acetic acid in 9.2% yield (Fig. 32i), with a TOF of 0.95 h−1 and TON of 33.13 for H2 production (Fig. 32j). The improved photocatalytic activity of ZnO/UiO66-NH2 compared to ZnO and UiO66-NH2 is attributed to its broad-spectrum absorption, effective charge separation and transfer, and the presence of highly accessible active sites.704 Moreover, the authors demonstrated the catalyst's performance in the photocatalytic valorisation of LDPE, PET, and commercial PLA bags and straws. In another study, the same research group embedded Ag2O nanoparticles in Fe-MOF.698 Initially, they synthesised an Fe-MOF, which was then subjected to post-synthetic modification to produce an Fe–Ag bimetallic MOF. Upon exposure to a light source, this resulted in the formation of Ag2O nanoparticle encapsulated Fe-MOF, represented as Ag2O/Fe-MOF. The framework of MOF efficiently restricted the growth of Ag2O nanoparticles, ensuring better dispersion and preventing agglomeration. The porous Ag2O/Fe-MOF exhibited a high surface area and more active sites, thereby expanding its light absorption range and improving photocatalytic performance. The Ag2O/Fe-MOF catalyst incorporating 0.2 wt% Ag2O achieved the highest photocatalytic degradation of PET microplastics, resulting in a weight loss of 27.5 mg within 3 h and generating 6.2 mmol g−1 of H2 in 2.5 h, whereas the catalyst with 0.05 wt% and 1 wt% Ag2O incorporation showed lower weight loss and H2 generation in photocatalytic PET microplastic transformation.698 This indicates that the optimal incorporation of Ag2O is critical to enhance the catalytic activity, whereas excessive amount may damage the Fe-MOF structure and reduce the active centres. In comparison, both Fe-MOF and bare Ag2O showed lower efficiency for PEG transformation due to unsuitable band gaps and limited light absorption. Moreover, Ag2O didn't generate H2 due to its lower conduction band (0.12 V vs. NHE) compared to H2 production (0 V vs. NHE). When Ag2O and Fe-MOF were physically mixed, the PEG conversion was minimal (6.1 mg) with low H2 generation (2.3 mmol g−1). The incorporation of small Ag2O nanoparticles into the Fe-MOF pores exposes more active sites, enhancing microplastic conversion. Acetic acid formation was also observed. Furthermore, Ag2O/Fe-MOF demonstrated enhanced photocatalytic H2 generation rates, achieving 1.7 mmol g−1 h−1 for PE and 1.9 mmol g−1 h−1 for PET microplastics. | ||
| Fig. 32 (a) Schematic diagram illustrating the synthesis of ZnO/UiO66-NH2; (b) SEM image; (c) TEM image; (d) HAADF-STEM image and elemental mapping; (e) acetic acid yield; (f) acetic acid generation at various reaction times; (g) selectivity of different reaction products; (h) TOF for generation of H2 in PLA conversion; (i and j) yield of H2 generated in PVC systems. Adapted with permission from ref. 711. Copyright 2023, John Wiley & Sons. | ||
Polyoxometalates (POMs) have been widely used in photo-/electrocatalysis due to their reversible multi-electron redox conversions. Xing et al. developed a heterostructure photocatalyst consisting of V-substituted phosphomolybdic acid clusters and g-C3N4 nanosheets (VPOM/CNNS) to convert plastic waste into formic acid by visible light.712 The VPOM/CNNS composite cluster preserved the robust redox potentials of both electrons and holes, enhancing its photocatalytic efficiency. Consequently, the VPOM/CNNS showed outstanding photocatalytic formic acid production from PE at the rate of 24.66 μmol h−1 g−1, which is nearly 262 times greater compared to pristine CNNS. Furthermore, the VPOM-CNNS composite demonstrated superior performance compared to mechanically mixed VPOM and CNNS. This composite was also effective in photocatalytic conversion of other plastic types, including polyacrylamide (PAM), PEG, PP, and PVC into formic acid. The formic acid production rate from photocatalytic upcycling of PAM and PEG was significantly higher than that from PP and PVC due to the polar groups in PEG and PAM, which facilitated their dissolution in polar solvents and the easier activation of C–O bonds. Furthermore, VPOM/CNNS was capable of selectively transforming real plastic waste, including polyethylene bags and polypropylene masks, into formic acid. Photoinduced holes (h+) and superoxide radicals (O2˙−) were identified as the primary reactive species for photocatalytic plastic conversion.712 In another study, Gong et al. synthesised a metal-free photocatalyst (CPD-CN) by combining carbonized polymer dots (CPDs) with graphitic carbon nitride (CN) for photoreforming of PET.713 Remarkably, carbon photodots (CPDs) played a crucial role in broad light absorbance, photoinduced electron transfer, and storage. The photocatalytic conversion of pretreated PET solution using CPD-CN resulted in the production of ethylene glycol derived chemical building blocks such as acetaldehyde, acetic acid, glycolic acid, etc. after 8 days of irradiation. The authors also explored the photocatalytic H2 generation in conjunction with hydrolysis of PET and PLA. Remarkably, even without the of use of a co-catalyst (Pt), CPDs-CN achieved a notable H2 generation of 298 ± 58 mmol g−1 h−1 with pre-treated PET. However, when Pt was loaded onto CPDs-CN, the H2 generation rates increased to 1034 ± 134 for pre-treated PET, and 1326 ± 181 mmol g−1 h−1 for pre-treated PLA. Mehta and co-workers synthesised a plastic waste derived fluorescent carbon (FC) based photocatalyst, FCs/CuO/TiO2, for H2 production.714 FC was synthesised from LDPE by treating it with sulphuric acid and nitric acid at 180 °C for 10 h. The photocatalytic reactions were performed using a 1.5 AM solar simulator, with both FCs and CuO being activated by visible light, leading to electronic excitations. Furthermore, FCs/CuO/TiO2 exhibited the highest H2 generation compared to other photocatalysts, such as FC, CuO, TiO2, and CuO@TiO2, demonstrating high stability for at least three reaction cycles. This investigation highlighted a compelling strategy to utilise plastic waste for synthesising a ternary photocatalyst with excellent performance in photocatalytic H2 production. Gogoi et al. synthesised polypyrrole using a template-free method, revealing the presence of residual α-Fe2O3 in the polymer matrix for photoreforming of PLA.715 The UV-vis spectrum of nPpy@Fe2O3 confirmed the π–π* transitions within the polypyrrole ring. Photoreforming involved alkaline conditions under a nitrogen atmosphere with lamp irradiation generating hydrogen at 65 mmol g−1 h−1 after 48 h. The hydrogen generation was found to be 77.5 and 78.6 mmol g−1 h−1 after 120 h and 168 h, respectively. CO2 is released as a by-product in the photoreforming of polycarbonate-based PLA, however the nPpy@Fe2O3 (1:5) photocatalyst didn't show CO2 formation, thereby increasing its advantage.716
Liu et al. in 2022 compared the glycolysis of PET to BHET by photothermal and conventional thermal catalysis.717 They modified the commercially available multiwalled carbon nanotubes (CNTs) with polydopamine (PDA) to enhance the catalyst dispersion in EG as compared to pure CNTs. PDA facilitated strong interaction between CNTs and EG, creating a bridging layer that improved dispersion compared to pure CNTs. These modified CNTs were used as light absorbers which showed an excellent solar thermal effect. Using 0.5 wt% CNTs, the temperature boosted to 180 °C within 30 min under a light intensity of 600 mW cm−2. Cholinium phosphate ([Ch]3[PO]4) ionic liquid was used as a photocatalyst with modified CNTs. [Ch]3[PO]4 increases the PET glycolysis by activating EG through intramolecular H-bonding. The nucleophilic oxygen in EG attacks the carbon of ester CO, which has been previously activated by [Ch]+ ions. The BHET formation reached 80% in 2.5 h via a photothermal method compared to 32% with the conventional thermal method, demonstrating the importance of a localised solar heating effect in photothermal catalysis.717 The same catalytic system converted PC to bisphenol A within 1 h under a light intensity of 400 mW cm−2. Furthermore, the scalability of the process was analysed using sunlight, and the authors created a solar flux by constructing a Fresnel lens as the optical concentrator. Upon light irradiation, the temperature reached 197 °C within 1 min and all the PET flakes were converted after 45 min producing 50 g of BHET. This demonstrates the potential for an industrialisation opportunity of this process. The same group later used an unsaturated Co single site catalyst (Co SSC) for photothermal glycolysis of polyester with improved catalytic activity and light absorption properties due to the high atomic utilisation, unique coordination state, and their local electronic structure.718 Co atoms were dispersed on the surface of PDA modified CNTs using a wet chemical impregnation method to synthesise Co SSC which was used for photothermal glycolysis of PET waste to form BHET. The reaction temperature reached 180 °C in 30 min with 0.75 wt% Co SSC loading in EG under a light intensity of 0.74 W cm−2. The solar power density was found to be directly proportional to PET conversion and BHET yield; increasing the solar power density from 0.52 to 0.74 W cm−2 increased PET conversion to 100% producing BHET in 82.6% yield. This catalyst system gave conversion and yield that were 5.4 and 6.6 times better than the same reaction performed in a thermal catalytic system, showing the higher performance of the photothermal system. Large-scale outdoor experiments under natural sunlight confirmed the industrial viability of Co SSC-catalysed photothermal PET glycolysis, achieving complete conversion within 40 minutes and yielding ∼8.5 g BHET per batch. Various photocatalysts investigated for recycling of different plastic feedstocks and their product distribution are summarised in Table 5.
| Feedstock | Catalyst | Reaction conditions | Products | Reference |
|---|---|---|---|---|
| PE (nitric acid treated) | Pt/TiO2 (1 wt%) | Simulated solar light, pH = 4, catalyst (4 mg), N2 atmosphere, 25 °C, 4 h | H2 (6.3 mmol gcat−1), C2H6 (0.25 mmol gcat−1), C3H8 (0.14 mmol gcat−1), CO2 (5.9 mmol gcat−1) | 686 |
| Commercial PE | Pt–P25–TiO2 | AM1.5 G, 100 mW cm−2, catalyst (30 mg), N2 atmosphere, 25 °C, 4 h | H2 (132.89 μmol gcat−1 h−1) in 4 h | 693 |
| H2 (521.72 μmol gcat−1 h−1) in 24 h | ||||
| Commercial PE-10 (plasma treated) | H2 (58.46 μmol gcat−1 h−1) | |||
| Commercial PE-20 (plasma treated) | H2 (73.36 μmol gcat−1 h−1) | |||
| Commercial PE-30 (plasma treated) | H2 (435.80 μmol gcat−1 h−1) | |||
| PP (plasma treated) | H2 (225.27 μmol gcat−1 h−1) | |||
| PV (plasma treated) | H2 (278.56 μmol gcat−1 h−1) | |||
| PE | Nb2O5 | 300 W xenon lamp, AM1.5G, 100 mW cm−2, catalyst (50 mg), 25 °C, 40, 60, and 90 hours | CH3CO2H (47.4 μg gcat−1 h−1) | 688 |
| Polymer: PE (150 mg), PP (150 mg), PVC (300 mg) | ||||
| PP | CH3CO2H (40.6 μg gcat−1 h−1) | |||
| PVC | CH3CO2H (39.5 μg gcat−1 h−1) | |||
| PLA pretreated in 10 M NaOH solution | 5% Pt/TiO2 | Simulated solar light, N2 atmosphere, catalyst (10 mg), 25 °C, 4 h | H2 (0.011 mmol gcat−1 h−1) | 701 |
| PET pretreated in 10 M NaOH solution | H2 (0.074 mmol gcat−1 h−1) | |||
| PET pretreated in 10 M NaOH solution | CdS/CdOx quantum dots | H2 (12.4 mmol gcat−1 h−1), acetate, ethanol, formate, glycolate, and lactate formed | ||
| PLA pretreated in 10 M NaOH solution | H2 (62.1 mmol gcat−1 h−1), pyruvate formed | |||
| PUR pretreated in 10 M NaOH solution | H2 (3.22 ± 0.13 mmol g−1 h−1) pyruvate, acetate, and formate formed | |||
| PET pretreated in 10 M NaOH solution | MoS2–Cd0.5Zn0.5S | 300 W xenon light, catalyst (10 mg), 4 h | H2 (15.90 mmol gcat−1 h−1), acetate, formate, CH3OH, and C2H5OH formed | 705 |
| PET pretreated in 10 M aqueous KOH | CNx/Ni2P | Solar light, catalyst (1.6 mg mL−1), N2 atmosphere, 25 °C, 20 h | H2 (111 μmol gsub−1 h−1), 50 h duration, acetate (190 nmol), formate (190 nmol), glyoxal (9.30 μmol), 5 days duration | 707 |
| PLA pretreated in 10 M aqueous KOH | H2 (211 μmol gsub−1 h−1), acetate (100 nmol), formate (95 nmol) | |||
| PET in 10 M aqueous KOH | CNx/Pt | H2 (104 μmol gsub−1 h−1), acetate (190 nmol), formate (190 nmol), glyoxal (9300 nmol) | ||
| PLA in 10 M aqueous KOH | H2 (314 μmol gsub−1 h−1), acetate (100 nmol), formate (95 nmol) | |||
| PET pretreated in 0.2 M aqueous KOH | CNx/Ni2P | Solar light, flow rate (2 mL min−1), 25 cm2 catalyst panel, 25 °C, N2 atmosphere, 20 h | H2 (52 mmol m−2 h−1) | 708 |
| PET treated in 5 M KOH at 70 °C | CN-CNT-NiMo | 500 W Xe lamp (95 mW cm−2), catalyst (10 mg), Ar atmosphere, 4 h | H2 (90 μmol gcat−1 h−1) | 713 |
| PS | g-C3N4 | 300 W xenon lamp, catalyst (50 mg), PS (20 mg) | C6H5COOH (39%), acetophenone (7%), C6H5CHO (2%) | 710 |
| 1 bar air, 150 °C, 24 h | ||||
| PET | Pt-cocatalyst-CPDs-CN-7 | 300 W xenon lamp, catalyst (20 mg), 40 °C, 22 h | HCOOH (139 μmol), CH3COOH (554 μmol), glycolaldehyde (110 μmol), glycolic acid (383 μmol), C2H5OH (128 μmol) | 709 |
| PET powder | TPA (304.7 ± 17.2 μmol) | |||
| PET film | TPA (201.1 ± 19.2 μmol) | |||
| PET pretreated in 5 M KOH | CPD-CN | 300 W xenon lamp, catalyst (20 mg), 40 °C, under vacuum, 22 h | H2 (1034 μmol g−1 h−1); C2H5OH (37%), glycolaldehyde (4%), glycolic acid (21%), HCOOH (12%), CH3CHO (12%), CH3COOH (14%) | 709 |
| PLA pretreated in 5 M KOH | H2 (1326 μmol g−1 h−1) | |||
| PET MPs | Ag2O/Fe-MOF | 300 W Xe lamp, catalyst (0.1 g), 25 °C, in air | H2 (1.9 mmol g−1 h−1) | 698 |
| PEG MPs | H2 (3.6 mmol g−1), 2.5 h, acetic acid (11.7 mg L−1), 5 h duration | |||
| PE MPs | H2 (1.7 mmol g−1 h−1) | |||
| PLA | ZnO/UiO66-NH2 | 300 W Xe lamp, catalyst (0.1 g), 1.0 g PLA, in air, 25 °C | CH3COOH (91.6% selectivity; 14.4% yield; TON = 17.92; TOF = 0.51 h−1). H2 evolution (TON = 26.36; TOF = 0.75 h−1) | 711 |
| PVC | CH3COOH (9.2% yield; TON = 0.90; TOF = 0.03 h−1). H2 generation (TON = 33.13; TOF = 0.95 h−1) | |||
| PE | VPOM/CNNS | 300 W Xe lamp, catalyst (10 mg), O2 atmosphere, 20–40 °C, 36 h | HCOOH (24.66 μmol g−1 h−1) | 712 |
| PEG | HCOOH (208.65 μmol g−1 h−1) | |||
| PP | HCOOH (208.65 μmol g−1 h−1) | |||
| PVC | HCOOH (29.85 μmol g−1 h−1) | |||
| PAA | (156.57 μmol g−1 h−1) | |||
| PE | 0.5 wt% Pt-CdOx/CdS/SiC | 300 W Xe lamp, catalyst (50 mg), 70 °C, Ar atmosphere, 3 h | H2 (25.0 μmol g−1 h−1) | 702 |
| PS | H2 (19.0 μmol g−1 h−1) | |||
| IR | H2 (36.7 μmol g−1 h−1) | |||
| PLA | d-NiPS3/CdS | 300 W Xe lamp, catalyst (1 mg), 9 h | Acetates (13.6 μmol) | |
| Pyruvates (64.5 μmol) | 706 | |||
| PET | Formate (23.6 μmol) | |||
| Acetate (13.8 μmol) | ||||
| Glycolate (25.0 μmol) | ||||
| PS | PSA-TiO2 | 370 nm LED, catalyst (10 mg), 1 bar O2, 4 h | Benzoic acid (43.5 mol%), acetophenone (1 mol%) | 719 |
| PS | DTSPA-TiO2 | Benzoic acid (40.5 mol%), acetophenone (0.8 mol%) | ||
| PEG-400 | Vanadium complex | White LED, catalyst (5 mol%), ambient temperature | Methyl formate (75 ± 4%) | 690 |
| PE–PEG | HCOOH (6 ± 1%) | |||
| PCL–PEG–PCL | HCOOH (70 ± 4%) |
11. Electrocatalytic plastic recycling
The electrocatalytic approach shows significant promise for recycling plastic waste and offers an eco-friendly solution for removing primary environmental contaminants due to its minimal operational requirements such as electricity derived from non-renewable resources. Despite its potential, this method has rarely been applied to plastic upcycling, with most applications focusing on the natural polymer lignin.720 Some reports have highlighted the transformation of synthetic plastic wastes into carbon-based materials for use in electrodes.721 Although this does not directly address plastic transformation, these materials are valuable for generating hydrogen fuel via electrocatalytic water splitting.722 This section will explore the latest studies on the electrocatalytic approach for plastic conversion.For electrochemical transformation of plastic waste, it is vital to develop a system that can effectively deliver charges to bulk solid plastics. To ensure the efficient diffusion of large molecular weight polymer molecules into the anode, highly porous materials are selected. Previously reported anodes for lignin upcycling can be adapted with necessary modifications to enhance conversion rates and efficiency. In classical electrocatalytic water splitting systems, hydrogen is generated at the cathode by the reduction of H+ ions, and oxygen is generated at the anode via the oxygen evolution reaction. The hydrogen evolution reaction (HER) is often limited by the thermodynamically less favourable oxygen evolution reaction (OER). To enhance the HER, researchers have been exploring replacement of the slow OER with simpler organic transformation reactions. One effective method is to replace the OER with plastic recycling processes, enabling the simultaneous production of hydrogen gas and valuable chemicals. This green approach not only provides an economical and sustainable solution to the growing plastic waste issue but also offers solution to meet the energy demands of industry. Additionally, this method eliminates the potential risk of generating a highly explosive H2/O2 mixture. Moreover, research efforts have begun to further increase energy efficiency, coupling a photoanode with the cathode to construct photoelectrochemical cells (PECs). This approach requires significantly less energy compared to conventional electrocatalytic systems, making the process more efficient and sustainable.
11.1 Electrocatalysts for polyester recycling
Chemical hydrolysis in alkaline solutions can readily extract terephthalic acid (TPA-K) and EG monomers from PET. However, the commercial use of this method is limited due to its complex nature and high energy consumption of the purification process. To enhance the purification process and obtain further advantages, electrochemical upcycling has been suggested as an additional approach. Zhou et al. successfully synthesised a series of cobalt and nickel phosphides on nickel foam (CoNixP/NF) as bifunctional catalysts for electrocatalytic transformation of PET, producing terephthalic acid (TPA), potassium diformate, and hydrogen fuel by water reduction.723 In this process, ethylene glycol is oxidised at the anode while H2 is evolved at the cathode. A three-electrode system was used with a saturated calomel reference electrode, and nickel foam as the working electrode as well the counter electrodes for electrodeposition. Among the catalysts, CoNi0.25P/NF exhibited the highest activity, upcycling PET to formate and hydrogen fuel with a current density of 500 mA cm−2, achieving over 80% faradaic efficiency and formate selectivity. In the electrochemical procedure, formic acid produced by the ethylene glycol oxidation reaction (EGOR) is neutralised in a potassium hydroxide electrolyte, leading to the precipitation of TPA with 94% yield. Further reduction of the electrolyte pH resulted in the formation of solid formate or potassium diformate. Behera et al. investigated the electrocatalytic upcycling of PET, focusing on the electrochemical oxidation of PET and its oligomer BHET using a cobalt-containing polymer with open metal sites as an electrocatalyst.724 During the electrocatalytic reforming process, the formic acid produced by the electrochemical oxidation of ethylene glycol (EGOR) is neutralised in a KOH electrolyte, causing terephthalic acid (TPA) to precipitate with 94% yield. Lowering the pH of the electrolyte further resulted in the formation of solid formate or potassium diformate (KDF). This study presented an effective and economical strategy for producing TPA, potassium diformate, and hydrogen fuel.Shi et al. pioneered an innovative electroreforming strategy to transform PET waste into terephthalate and carbonate using a palladium modified nickel foam catalyst.725 They synthesised various Pd/NF catalysts via a displacement reaction involving Ni foam in an H2PdCl4 aqueous solution, with Pd particles depositing onto the Ni foam. The Pd/NF-10 catalyst demonstrated the largest electrochemically active surface area, attributed to its porous network structure that provided more catalytic sites. The faradaic efficiency (FE) for H2 fuel generation was 98% at a cell voltage of 1.01 V. The efficiency of the hydrogen evolution reaction (HER) remained stable despite varying the concentration of EG. For EG oxidation, a peak current density of ∼880 mA cm−2 was recorded in 1 M KOH, which increased by increasing the KOH concentration up to 10 M, indicating the necessity of a highly alkaline environment for efficient EG oxidation. For PET electroreforming, a current density of approximately 400 mA cm−2 at 0.7 V vs. RHE was derived from cyclic voltammetry (CV) curves, with nearly complete PET conversion after 20 hours. The process predominantly produced terephthalate and carbonate, along with minor amounts of glyoxal. The catalyst demonstrated high stability, retaining 97% of its initial current density after four cycles. This electroreforming approach was also tested on real-world PET, achieving a current of 100 mA cm−2 at 1.01 V. Overall, the catalytic system demonstrated high activity for carbonate production with 95% selectivity, and 93% faradaic efficiency.725
Chen and colleagues recently reported the conversion of EG from PET waste into formate using a defective and anion–cation doped nickel sulfide.726 The oxidation of PET hydrolysate was performed in an H-type cell, simultaneously producing hydrogen. It was discovered that B and Co-doped Ni3S2 (B, Co–NiS) exhibited superior EGOR activity compared to samples doped with either Co or B alone. While B–NiS and Co–NiS required potentials of 1.364 V and 1.384 V, respectively, to achieve 100 mA cm−2, B,Co–NiS required only 1.341 V for EG oxidation. Moreover, the bifunctional B, Co–NiS catalyst facilitated the electrolysis of real PET waste hydrolysate, producing 15.24 mmol h−1 of formate. It also achieved a hydrogen production efficiency more than 70 times greater than that of conventional water electrolysis.
Wang et al. synthesised ultrathin CoNi0.2P nanosheets on Ni foam [CoNi0.2P-uNS/NF], an efficient and cost-effective electrocatalyst, using a template-post-phosphatisation methodology.727 These nanosheets demonstrated exceptional electroactivity for the HER, EG oxidation, and the oxidation of other biomass such as furfural and glycerol in alkaline environments. The CoNi0.2P-uNS/NF catalyst showed excellent HER activity and required only 43 mV to achieve 10 mA cm−2, outperforming other catalysts such as CoP-uNS/NF, CoNi0.1P-uNS/NF, and Ni2P-uNS/NF, which required 98 mV, 65 mV, and 124 mV potential, respectively. For practical applications, the authors assembled a CoNi0.2P-uNS/NF‖CoNi0.2P-uNS/NF electrolyser to oxidise PET hydrolysate into formate while co-producing hydrogen. Among all the tested catalysts, CoNi0.2P-uNS/NF showed the highest electroactivity due to the optimal inclusion of Ni, which positively influenced the Co active centre for EG oxidation. The reaction kinetics were assessed using Tafel plots, with CoNi0.2P-uNS/NF displaying the fastest EG oxidation kinetics and the lowest Tafel slope (39.9 mV dec−1) among the tested catalysts.
Ma et al. developed a bifunctional electrocatalyst, Ni3N/W5N4, directly grown on nickel foam (Fig. 33a), and used it for successful upcycling of prevalent PET microplastics (MPs), integrated with a counter cathodic HER.728 The Ni3N/W5N4 electrode demonstrated platinum-like hydrogen evolution reaction performance and remarkable stability, lasting 300 hours under current industrial conditions. The catalyst showed high selectivity for producing formic acid (HCOOH) with a faradaic efficiency of approx. 85% at a potential of 1.40–1.60 V vs. RHE (Fig. 33d). The current density at 1.60 V reached 120 mA cm−2, producing formate at 1.2 mmol cm−2 h−1. For PET oxidation, it attained a current density of 10 mA cm−2 at 1.33 V, which is significantly lower than the potential required for the OER, indicating that PET oxidation is an energy-efficient process (Fig. 33c). As shown in Fig. 33d, there was selective production of HCOOH during the PET upgradation, achieving a high Faradaic efficiency of ∼85% for Ni3N/W5N4 at 1.4–1.6 V to generate HCOOH. Moreover, the oxidation process at various potentials was found to be 3–5 times more efficient than the OER, enhancing the HER process at the cathode (Fig. 33e). Formic acid was identified as the primary product during PET upcycling, with its production increasing with the increase in voltage. Additionally, the catalyst demonstrated excellent recyclability, maintaining its activity without significant loss even after five cycles. In seawater containing PET hydrolysate, the current density increased sharply with potential, outperforming bare seawater due to the replacement of the energy-intensive chlorine evolution reaction (CER) with the plastic oxidation process. While the catalyst experienced severe corrosion in bare seawater after the anodic reaction, it exhibited minimal corrosion in the presence of PET hydrolysate. Overall, this electrocatalyst exhibited outstanding catalytic performance in harsh marine environments, showing great promise for practical electrocatalytic plastic upcycling under real-world conditions. Wang and co-workers developed an ordered macroporous superstructure using a Ni-modified CoP electrocatalyst (OMS-Ni1-CoP) for PET electrocatalytic upcycling.729 This catalyst was derived from Ni(II)/MSC-ZIF-67 which was synthesised from microporous ZIF-67 single-crystals. A three-electrode cell with 1 M KOH electrolyte was used to evaluate HER activity, performing comparative study using CoP, Ni1–CoP, and OMS-CoP catalysts. The electrocatalyst demonstrated a remarkable faradaic efficiency of up to 96% for formate synthesis in the ethylene glycol oxidation reaction. Using this electrocatalyst for simultaneous production of hydrogen and valuable chemicals, the researchers explored an integrated electrolysis system combining the HER and EGOR in PET plastic hydrolysate.
 | ||
| Fig. 33 (a) Schematic process for Ni3N/W5N4 electrocatalyst synthesis; (b) SEM image of Ni3N/W5N4. (c) LSV curves of the Ni3N/W5N4 Janus structure with and without plastic in deionized water; (d) rate of HCOOH formation and FA at different potentials. (e) The respective current densities at different potentials. Adapted with permission from ref. 728. Copyright 2022, Elsevier. | ||
Liu and co-workers harnessed the synergy between palladium (Pd) and nickel (Ni) to develop selective electrocatalysts for the electrochemical oxidation of ethylene glycol (EGOR), aiming to synthesise glycolate from PET-derived ethylene glycol.730 In this system, Ni(OH)2 efficiently oxidised –OH to *OH species on the Pd surface at a low potential. The resulting *OH species promoted the cleavage of EG bonds for a faster dehydrogenation process and facilitated the conversion of poisonous carbonyl intermediates, maintaining the catalyst's stability and effectiveness. The initial step involved the basic hydrolysis of PET pieces to form TPA and EG. For EG oxidation analysis, the Pd–Ni(OH)2/NF catalyst displayed a Tafel slope of 189 mV dec−1, significantly lower than the OER at 259 mV dec−1, confirming the kinetic favourability of EG oxidation over the OER. As electrolysis proceeds, glycolic acid production increases linearly with EG consumption. The Pd–Ni(OH)2/NF catalyst exhibited excellent performance, achieving faradaic efficiency (FE) greater than 85% at various voltages, glycolic acid selectivity of 91.6%, and EG conversion rate of 93.2%. This performance was significantly superior to that of Pd/NF (55%) and Pd/C (16%), highlighting the higher stability of the catalyst in the EGOR. The faradaic efficiency for glycolate exceeded 90% across a wide potential range (0.7–1.2 V vs. RHE). For hydrogen evolution, the catalyst showed outstanding activity, producing hydrogen at 0.93 V, significantly lower than the conventional 1.88 V required for water splitting in a KOH and EG electrolyte solution. Wang et al. used a unique approach for simultaneous upcycling of PET and CO2 reduction, producing formic acid (HCOOH) at both the cathode and anode using a NiCo2O4 electrocatalyst.731 In the context of CO2 reduction reactions, they reduced energy consumption and enhanced the formation of valuable products at the anode by utilising small molecular organic reactions to substitute the sluggish OER. The electrocatalyst demonstrated 90% faradaic efficiency for formic acid production, showcasing its outstanding selectivity for the oxidation of PET hydrolysate. By combining PET hydrolysate oxidation with the CO2 reduction reaction, the electrolyser operated at 1.55 V to facilitate the combined half-reactions. Moreover, when operating at 1.90 V, Faraday efficiency of up to 155% could be achieved for formic acid production, indicating a highly efficient process for converting both PET and CO2 into valuable chemicals. A similar strategy was also investigated by Kilaparthi et al. who also coupled the CO2 reduction process with PET recycling.732 The authors used reduced graphene oxide (rGO) loaded with bismuth oxide carbonate (BOC) and CuCoO as the anode and cathode, respectively. Formate was concurrently produced at both electrodes. The CuCoO@rGO catalyst demonstrated higher electroactivity, achieving a significant FE of 85.7% at 1.5 V. In contrast, BOC@rGO catalyst demonstrated an impressive FE of 97.4% at −0.8 V, effectively driving formate production via the CO2 reduction reaction. Using this system in an electrolyser, the system was able to produce formic acid at a low voltage of 1.9 V and current density of 10 mA cm−2, achieving a remarkable FE of 151.8% for formate production.
Mao et al. introduced a Mn0.1Ni0.9Co2O4−δ rod-shaped fibre electrocatalyst for transformation of PET microplastics (MPs) into formate, terephthalic acid (TPA), and potassium sulfate (K2SO4).733 The catalyst preparation involved electrospinning, vacuum drying, and calcination process. The introduction of manganese (Mn) into the NiCo2O4 structure was believed to enhance its electrocatalytic performance by altering its electronic structure. Initially, 2 M KOH solution was used to hydrolyse PET into TPA and EG, which served as the electrolyte for formate production at the anode. The Mn0.1Ni0.9Co2O4−δ electrocatalyst achieved an FE of 95% at 1.42 V vs. RHE for oxidation of PET hydrolysate. The oxidation of EG to formate and carbonate involved a two-electron transfer process and proceeded by two pathways: (i) EG conversion to glycolaldehyde and its oxidation to glycolic acid, which then underwent C–C bond cleavage to produce formate and carbonate; (ii) glycolaldehyde oxidation to glyoxal, followed by C–C bond cleavage to generate formate.
Ren et al. recently developed a process coupling the nitrate reduction reaction (NO3RR) with the oxidation of PET hydrolysate.734 In this process, PET hydrolysate was converted into formate while concurrently producing ammonia from nitrate wastewater. They used a bifunctional catalyst, (CoRuMOF/NF), developed by growing Ru-incorporated Co-based MOF (CoRuMOF) on nickel foam. Excitingly, upon applying the external potential CoRuMOF/NF underwent in situ transformation, forming Ru–Co(OH)2/NF and Ru–CoOOH/NF at the cathode and anode, respectively. These reconstructed catalysts proved highly active for the NO3RR and oxidation of PET hydrolysate.734 In evaluating the NO3RR activity, linear sweep voltammetry curves showed a high current density in 1 M KOH solution with 200 ppm of KNO3–N compared to pure 1 M KOH solution, confirming the catalyst's efficiency in driving nitrate reduction.734 For PET hydrolysate oxidation, Ru–CoOOH/NF showed significant advantages, lowering the required potential by 220 mV at 100 mA cm−2, revealing the easier oxidation of PET hydrolysate than the OER. Additionally, the system achieved a higher current density of 50 mA cm−2 at 1.5 V, enabling efficient co-production of NH3 and formate.
Noble metal-based catalysts are known for their good selectivity in preserving C–C bonds during the electrooxidation of alcohols.735 In a study by Li et al., gold particles were electrodeposited on Ni(OH)2 for 600 seconds at −1 V vs. Ag/AgCl, using Ag/AgCl, Pt foil, and nickel foam-supported Ni(OH)2 nanosheets as the reference electrode, counter electrode, and working electrode, respectively.735 Using Au/Ni(OH)2, glycolic acid was produced from ethylene glycol at the rate of 2.24 mmol−2 h−1 with 91% selectivity at 1.15 V. The transformation of ethylene glycol into glycolic acid (GA) was found to involve an initial electrooxidation of EG to glycolaldehyde and its subsequent conversion to an enol form, followed by nucleophilic dehydrogenation to produce GA. It was found that EG molecules with adjacent hydroxyl (OH) groups were more prone to form alkoxides, thus enhancing the catalytic activity. These EG alkoxides were concentrated on the surface of an Au/Ni(OH)2 catalyst through σ interactions between the alkoxides and Au, while H-bonding between the adjacent OH groups and Ni(OH)2 further increased local concentrations and current densities. The optimised electrolyser yielded 36.8 mmol h−1 of glycolic acid with 94% selectivity and faradaic efficiency of 96%, along with hydrogen production. To improve the process's applicability to consumer plastic waste, a membrane-free flow electrolyser using PET bottles produced 81.6% glycolic acid and 9.4 Litres of H2.736
Liu et al. recently applied an ultrafast electro-corrosion approach, utilising chloride ions (Cl−), to activate Ni foam into a highly effective electrocatalyst for converting polybutylene terephthalate (PBT).737 Chloride ions are known for their corrosion acceleration properties due to their strong penetration and depassivation potential, which increase the corrosion process.738 The corrosion process can create active layers on the surface of metal substrates, which act as catalytic species. The electro-corrosion reaction resulted in the increase of catalytic activity of Ni foam by 150 times. First, PBT under basic conditions resulted in the formation of TPA and 1,4-butanediol monomers. Fig. 34a shows the electro-corrosion method of treated Ni foam (T-NF), and Fig. 34c illustrates the schematic process of PBT electro-reforming into TPA and succinate. The treated Ni foam maintained its 3D skeletal structure even after undergoing electro-corrosion treatment with NaCl (Fig. 34b). A long-term electrolysis test was performed to access the durability of T-NF (Fig. 34d). The electrolysis current showed only a slight decrease, possibly due to reagent depletion. Furthermore, the LSV curve exhibited minimal variation after electrolysis, indicating stable performance. The T-NF demonstrated excellent catalytic activity in the oxidation of 1,4-butanediol (BDO) and achieved a current density of 50 mA cm−2 at 1.36 V potential, as TPA did not involve in the oxidation process (Fig. 34e).737 Moreover, BDO was converted into succinate with an FE of 93% at 1.45 V. This reaction involved different intermediates including 4-hydroxybutanal, succinaldehyde, and 4-hydroxybutanoic acid. Fig. 34f shows the 1H NMR spectra of PBT before and after upcycling, with the succinate peak observable in the spectrum after upcycling. Recently, Du et al. used a similar corrosion strategy to prepare Pt/γ-NiOOH/NF electrocatalyst comprising Pt nanoparticles hybridised with γ-NiOOH nanosheets supported on Ni foam, specifically for the transformation of real-world PET hydrolysate.739 This catalyst achieved over 90% selectivity and faradaic efficiency for glycolate across a wide range of reactant (ethylene glycol) concentrations under a voltage of 0.55 V, while simultaneously producing hydrogen as fuel.
 | ||
| Fig. 34 (a) Diagram showing the preparation of T-NF via the electro-corrosion method; (b) SEM image of T-NF; (c) schematic illustration of electro-reforming of PBT to TPA and succinate; (d) LSV curves for long term stability of T-NF; (e) LSV curves for the T-NF in PBT hydrolysate and 1.0 M KOH; (f) 1H NMR spectra of products of PBT conversion. Adapted with permission from ref. 737. Copyright 2023, Elsevier. | ||
11.2 Electrocatalysts for other plastics
Although electrocatalysis has mainly been studied for polyethylene terephthalate plastics, there are a number of studies currently focused on the electrocatalytic decomposition of other plastics. These studies are aimed at solving problems related to the variety of chemical structure and stability of various polymers. Electrocatalysts offer a promising approach to recycle these materials into valuable chemical raw materials or less harmful by-products, using electrochemical processes to achieve selective bond cleavage under mild conditions.Bifunctional electrocatalysts, which function efficiently as both cathode and anode, are key materials in electrocatalysis. Li and coworkers have reported a novel bifunctional CoSe2/NF electrocatalyst, developed through a hydrothermal process followed by post-selenisation treatment.740 They synthesised CoSe2/NF catalysts with different amounts of selenium (0.02, 0.1, and 0.5 g) and used them in the reforming of PLA to acetic acid, coupled with H2 gas evolution. The CoSe2 electrocatalyst revealed a large surface area, high conductivity, and ample active sites. Linear sweep voltammetry (LSV) curves indicated that CoSe2/NF showed excellent activity for the HER in 1 M KOH. Particularly, the 0.1-CoSe2/NF catalyst demonstrated a potential of 76 mV, indicating good HER activity.740 For the PLA oxidation reaction, the 0.1-CoSe2/NF catalyst needed only 288 mV of overpotential, which was lower than the overpotential required for water-splitting (361 mV). The oxidation of PLA at a potential of 1.5 V over 30 hours resulted in the production of 80.73 mM acetic acid, translating to an 87% conversion rate. Moreover, PLA oxidation required a relatively lower potential (1.37 V) compared to water splitting (1.63 V), underscoring the superior performance of the 0.1-CoSe2/NF catalyst over its pristine counterpart. This research highlights CoSe2/NF as an exceptional bifunctional electrocatalyst for transforming PLA into value-added chemicals and hydrogen production. Li et al. developed a highly efficient bifunctional electrocatalyst, Co–Ni2P/NF, using a hydrothermal and phosphidation process, for the oxidation of PET hydrolysate and hydrogen evolution reaction.741 Initially, they produced cobalt-modified nickel hydroxide nanosheets on nickel foam [CoNi(OH)2/NF] followed by phosphidation of these arrays using NaH2PO2·H2O to form the final Co–Ni2P/NF catalyst. SEM images revealed the uniform distribution of nanosheets across the 3D porous nickel foam. The catalyst demonstrated remarkable performance, requiring only 148 mV of overpotential to achieve 50 mA cm−2 for the HER. When coupled with PET oxidation, the electrolyser, utilising Co–Ni2P/NF as the catalyst, required just 1.43 V to achieve 10 mA cm−2, significantly lower than the 1.55 V needed for pure water splitting. The Co0.6Ni2P/NF variant showed the highest HER activity, achieving 10 mA cm−2 at 69 mV. For EG oxidation, the catalyst required just 90 mV to reach 50 mA cm−2. CV curves indicated that the Co0.6Ni2P/NF attained an electro-oxidation current density of 183 mA cm−2 at 1.5 V vs. RHE for PET hydrolysate, compared to 46 mA cm−2 for the OER.741 This demonstrates the favourable thermodynamics for EG oxidation compared to the OER. The study identified Co0.6Ni2P/NF as an outstanding bifunctional catalyst, achieving 85% faradaic efficiency (FE) and 80% TPA recovery in a 1 M NaOH electrolyte.741
Liu et al. reported a novel Ni foam-based electrocatalyst that employed a straightforward strategy for upcycling polyester plastic wastes (PET, PBT, PLA, and polytrimethylene terephthalate (PTT)) into value-added chemicals.742 This study involved a wide range of polyester-based plastics as model substrates for electrocatalytic plastic upcycling, thereby expanding the substrate scope in electrocatalysis. The authors synthesised earth-abundant CuCo2O4 spinel oxide nanowire arrays grown on Ni foam using the hydrothermal method. Morphological analysis revealed uniform distribution of CuCo2O4 nanowires on the Ni foam surface. PET was efficiently converted into terephthalate and formate, demonstrating excellent efficiency and selectivity of over 86%.742 The CuCo2O4 catalyst showed remarkable activity for upcycling of different polyester plastics including PBT, PET, PLA, and PTT, with working potentials of 1.23 V, 1.29 V, 1.34 V, and 1.16 V at 10 mA cm−2, respectively. This strategy led to the conversion of PBT into terephthalic acid (TPA) and 4-hydroxybutyraldehyde, PLA into pyruvic acid, and PTT into TPA and 3-hydroxypropionaldehyde (HPA). These chemicals have various applications, for example, pyruvic acid is commonly used in cosmetic applications and for synthesis of alanine, HPA is a food preservative and precursor for chemicals, and 4-hydroxybutyraldehyde is used for the synthesis of deoxyketoses, acetonides, etc. Yan et al. further advanced the research by employing a gold electrocatalyst supported on Ni(OH)2 [Au/Ni(OH)2] to convert ethylene glycol, derived from the basic hydrolysis of PET, into glycolic acid while simultaneously producing hydrogen.736
Botte et al. introduced an innovative electrochemical method to modify LDPE surfaces in a water-based solution under ambient conditions.743 In this process, LDPE particles were placed between two metal electrodes (such as Cu, Ni, or stainless steel), which were immersed in electrolytes containing the respective metal ions. The LDPE surface underwent oxidation by changing the voltage from 1 V to −1 V, particularly with copper electrode/Cu2+ and nickel electrode/Ni2+. The oxidation of the polymer surface was confirmed by the presence of C–O and CO bonds. The most prominent changes were observed when copper electrodes were used, as evidenced by the formation of new absorption peaks at 1150 and 1230 cm−1 in FTIR spectra, corresponding to the formation of C–O bonds in ester and ether groups. Electrolysis using nickel broadened the peak at 1080 cm−1, which corresponds to alcohol and peroxide functionalities. Significantly, the peak at 1735 cm−1 only appeared for the LDPE sample treated with copper, revealing the presence of the CO bond. GC-MS analyses further revealed the presence of fatty acids and hydrocarbons in LDPE samples treated with copper and nickel, indicating the chain scission.743 Moreover, the presence of Cu and Ni nanoparticles in the treated LDPE indicated that metal–polymer interaction contributed to the electrooxidation process. Brantley et al. developed an electrochemical method for degradation and modification of polymer backbones.744 They used a dichloromethane solution containing 0.2 M N(Bu)4BF4 as the supporting electrolyte to incorporate polymers into the electrocatalytic reaction. They opted for a graphite/zinc (anode/cathode) combination instead of the RVC/Pt setup due to its cost-effectiveness, ease of cleaning, and effective control over degradation reactions. A constant potential was applied to detect the oxidation of polynorbornene at different intervals. Gel Permeation Chromatography analysis revealed a significant reduction confirming the degradation and shift to lower molecular weights in samples. The number average molecular weight (Mn) was found to be −207 kDa after 16 hours, and the polynorbornene was further reduced to 805 kDa by prolonging the reaction duration to 36 hours.744 The authors suggested that the formation of radical cations produced during the anodic process could be suppressed by introducing a hydrogen atom transfer (HAT) reagent, thereby reducing chain scission. Consequently, the addition of excess 9,10-dihydroanthracene considerably reduced the degradation rate of the polymer. They degraded polynorbornene by this electrochemical approach and successfully appended an azide group. This azide group was then further reacted with ethynyl pyrene through click chemistry, resulting in the addition of a strong UV-absorbing group. These findings concluded that coupling of polymer degradation with functionalisation (e.g., azidation) could afford new chemical structures with potential to serve as macromonomers.744
Despite the numerous advantages of using electrocatalysts for plastic waste recycling, there are significant challenges hindering their practical application. One of the main challenges in global plastic treatment methods is the economic feasibility of these processes. Applying these methods on an industrial scale faces challenges due to the high costs associated with substantial energy requirements and the necessity for effective sorting of plastic waste.
From a plastic recycling perspective, it is crucial to develop electrocatalysts that can facilitate the production of valuable chemicals through partial oxidation reactions. Therefore, advancing electrocatalysts that can selectively oxidise EG without generating CO2 is essential for efficient and environmentally friendly plastic upcycling. Table 6 provides a summary of various studies on electrocatalytic conversion of plastic wastes.
| Catalyst | Feedstock | Electrolyte | Product | Reference |
|---|---|---|---|---|
| CoNi0.25P/NF | PET | 1 M KOH | Formate (FE: >80%; selectivity: >80%) | 723 |
| Co-based 1D coordination polymer | PET | 1 M KOH | Potassium diformate (77%; selectivity: ∼80%) | 724 |
| BHET (100%) | ||||
| Pd/Ni | PET | 10 M KOH | Carbonate (FE: 93%; selectivity: 95%) | 725 |
| H2 (FE: 98%) | ||||
| CoSe2/NF | PLA | 1 M KOH | Acetic acid (FE: 97%) | 740 |
| Co0.6Ni2P/NF | PET | 1 M NaOH | Formate (FE: 85%) | 741 |
| TPA recovery: 80% | ||||
| CoNi0.2P-uNS/NF | PET | 1 M KOH | Formate (FE: >90%) | 727 |
| Ni3N/W5N4 | PET | 1 M KOH | Formate (FE: 85%) | 728 |
| Atomic Ni-modified OMS-Ni1-CoP | PET | 1 M KOH | Formate (FE: 96%) | 729 |
| Pd–Ni(OH)2 | PET | 1 M KOH | Glycolic acid (FE: >85%) | 730 |
| H2 (FE: 98%) | ||||
| NiCo2O4 | PET | 1 M NaOH | Formic acid (FE: 90%) | 731 |
| CuCoO@rGO | PET | 1 M KOH | Formate (FE: 87.5%) | 732 |
| Mn0.1Ni0.9Co2O4−δ RSF spinel | PET | 2 M KOH | Formic acid (FE: >95%) | 733 |
| H2 (0.226 kg per ton PET) | ||||
| Cl− activated-NF | PBT | 1 M KOH | Succinate (FE: >93%) | 737 |
| (Pt/γ-NiOOH/NF) | PET | 1 M KOH | Glycolate (FE: >90%) | 739 |
| CuCo2O4/Ni | PET | 5 M KOH | Formate (FE: >93%) | 742 |
| H2 (1.89 L: 5 g PET) | ||||
| Au/Ni(OH)2 | PET | 5 M KOH | Glycolic acid (81.6% yield) | 736 |
| H2 (9.4 L: 70 g PET) | ||||
| Ru–CoOOH/NF | PET | 1 M KOH | Formate (FE: 95.53%) | 734 |
| Cu or Ni foil | LDPE | CuSO4 or NiSO4 solution | Oxidized LDPE or depolymerisation | 743 |
| B, Co–NiS | PET | PET in 1 M KOH | Formate (FE: >93%; selectivity: >92%) | 726 |
| NiCo2O4 | PEF | 1 M KOH | Formic acid (FE: 98%; selectivity: 85.8%) | 745 |
12. Biocatalytic plastic recycling
The use of biocatalysts in plastic recycling has attracted considerable attraction in recent years due to its potential for highly selective reactions under mild conditions, without generating toxic waste. Biocatalysis relies on enzymes derived from renewable sources that are biodegradable, making the process more environmentally friendly and cost-effective than traditional chemical methods.746 Some reviews have highlighted the biological recycling and valorisation of plastic waste using microorganisms and enzymes.747–749 The physicochemical properties of polymers such as crystallinity, hydrophilicity, molecular weight, and availability of functional groups, significantly influence the biodegradation process.750 For instance, plastics with an amorphous structure are more susceptible to enzymes, whereas highly crystalline polymers are resistant to biodegradation. The flexibility of amorphous polymer chains enhances enzyme accessibility, particularly at temperatures above 65 °C (Tg of PET), where the material's slight water absorbency (0.1–1.0%) facilitates the process. However, a study found that aging PET at 70 °C leads to reorganisation of its amorphous regions into more ordered microstructures, making them less vulnerable to hydrolysis.751 The lower crystallinity in PET also improves depolymerisation activity, with crystallinity levels varying across different products: 20–30% in bottles, up to 40% in textiles, and about 8% in packaging.752 Additionally, reducing PET to micron-sized particles significantly enhances the reaction rate by maximising the surface area available to enzymes.750 Consequently, pretreatment units designed to reduce crystallinity and particle size may be required in biocatalytic recycling facilities to optimise the efficiency of the recycling process.Polymers featuring hydrolysable functional groups, such as ester linkages in PET and PUR, are more appropriate for enzymatic depolymerisation, whereas, polymers with C–C bond backbones, including PE, PP, and PS, are more resistant to enzymatic breakdown.753 Enzymes generally do not tolerate harsh reaction conditions and perform best under aqueous environments, where an interfacial mechanism is required for deconstruction of water-insoluble polymers. Current efforts are focused on improving enzyme stability and increasing the polymer's accessible surface area to improve the efficiency of enzymatic depolymerisation. The success of biocatalytic recycling depends on processing conditions and intrinsic properties of the polymer substrate, such as molecular weight, glass transition temperature, and melting temperature.
Two main methods of using enzymes are described in the literature, the first involves the surface modification for increasing the hydrophilicity of the polymer without significantly changing its morphology, while the second involves deeper degradation, resulting in substantial changes in the morphology of the polymer.754 A change in the surface properties of PET-fabrics was achieved using single and double T. fusca cutinases. One of the enzymes created an accessible space around the active centre and the other increased the hydrophobic properties in the binding area, improving substrate attachment. This use of enzyme combination increased the rate of decomposition, resulting in higher yields of TPA in the reaction mixture.755
Nearly five decades ago, Aureobasidium pullulans was the first microorganism identified with the ability to degrade synthetic polymers, specifically polycaprolactone.756 However, significant research in this area remained limited until 2000.756 In the past two decades, driven by fossil fuel constraints and the plastic pollution crisis, there is significant increase in interest for enzymes and microorganisms that can degrade synthetic polymers. Enzyme-mediated depolymerisation is now considered a promising and sustainable approach for plastic recycling,757 though its current application is primarily limited to certain polyesters such as PET and PLA.748 Identifying microorganisms capable of degrading plastics is essential for discovering depolymerases and other enzymes critical to plastic decomposition.758–761 However, finding effective enzymes for the breakdown of more resistant plastics, such as PA, PE, PP, and PVC, remains a significant challenge that needs to be addressed. This section covers methods for polymer depolymerisation and controlled degradation using various classes of enzymes, including serine hydrolases (cutinases, lipases, and carboxyl ester hydrolases) and oxidases (laccases and peroxidases), tailored to the specific polymers and catalytic reactions involved.
12.1 Polyethylene terephthalate
PET was considered non-degradable until 1977 when lipase enzymes were discovered for breaking down aliphatic esters.762 Similarly, aromatic esters were initially believed to resist enzymatic degradation, however, this perception changed with the identification of various lipases capable of modifying the surface of PET or degrading the polymer bulk.763 A significant breakthrough in enzymatic PET depolymerisation came with the discovery of cutinase TfH from the bacterium actinomycete Thermobifida fusca.764 This enzyme hydrolysed PET bottle films at 55 °C, achieving 40–50% weight loss of the polyester within three weeks. Since then, T. fusca has become a benchmark organism for discovering active enzymes.765 Consequently, several enzymatic catalysts capable of depolymerising PET have been identified.750,751,766–768 Among the new enzyme catalysts, TfCut2 catalyst demonstrated 50% weight loss in post-consumer PET packaging degradation at 70 °C in 96 h.751 Addition of Ca2+ and Mg2+ increased the enzyme's thermal stability by 10 °C, allowing a range of enzymes useable at or above the Tg of PET.769 Another cutinase enzyme “HiC”, performed better than T. fusca-based enzymes for low-crystallinity PET and achieved almost complete degradation under similar conditions.770 However, PET bottles having higher crystallinity didn't degrade, highlighting the method's sensitivity to PET's structural properties.Wei et al. demonstrated that Thermobifida fusca cutinase could degrade low crystallinity PET films (up to 7%) and achieve weight losses up to 97%.751 Moreover, the enzyme was able to degrade two samples from post-consumer PET with low crystallinity (5% and 6%), achieving 50.5% and 56.6% weight loss, respectively, at 70 °C within 120 hours. LCC and PES-H1 (PHL7), as well as HiC from the thermophilic fungus Thermomyces insolens can effectively decompose PET at 70 °C. Leaf branch compost cutinase (LCC) is a thermally stable protein composed of 259 amino acids with an α/β-hydrolase fold, capable of decomposing 40% of low-crystalline PET within 24 hours at 70 °C, whereas IsPETase decomposes only 1% at 30 °C.771 Wei et al. utilised a thermostable hydrolase, expressed in Bacillus subtilis, to degrade post-consumer PET food packaging containers.772 Their results showed that PET lost more than 50% of its weight after 96 hours of incubation at 70 °C using TfCut2 from Thermobifida fusca. Xue et al. also employed LCC for the enzymatic degradation of PET waste, achieving approximately 84% conversion into solid hydrated calcium terephthalate (CaTP·3H2O), which was then used for producing battery electrodes.773 Tarazona et al. investigated the enzymatic degradation of amorphous PET nanofilms using a thermally stabilised IsPETase triple mutant (Tm = 56.6 °C).774 The nanofilms, with a porous structure and reduced glass transition temperatures (Tg = 40–44 °C), exhibited Tg values over 20 °C lower than bulk amorphous PET. Using a dual-enzyme system, composed of thermostabilised variants of IsPETase and MHETase, they achieved up to 70% depolymerisation within 1 hour at 50 °C. The study highlights how increased surface area, reduced Tg, and enhanced amorphisation collectively accelerate PET hydrolysis and lower the onset of degradation-induced crystallisation.
Tourneir et al. reported the degradation of post-consumer PET using LCC enzyme and achieved 85% conversion at 72 °C in 15 hours.765 This LCC enzyme showed superior performance compared to benchmark enzymes for depolymerising amorphous PET at 65 °C, however, it was deactivated in three days when used with bottle-grade PET. To improve the enzyme's performance and thermal stability, a disulfide bridge was incorporated in wild-type LCC that improved the thermal stability by 9–14 °C. In addition, two mutations were introduced to enhance the active site's specific activity. The combined modifications resulted in the production of two variant strains, ICCG and WCCG. The LCCICCG variant successfully depolymerised 90% of PET solution (200 g L−1) within 10 hours, an optimal timeframe to prevent PET recrystallisation. However, when the depolymerisation was conducted at 75 °C, the depolymerisation yield was capped at 55%, as recrystallisation rate outpaced depolymerisation. Within 6 hours, the crystallinity exceeded 40%, halting the reaction before reaching high conversion levels. Kim and coworkers reported the first effort towards microbial refinery transforming PET waste into aromatic compounds.775
Polymer molecules typically exhibit a non-uniform packing, comprising both ordered crystalline regions and disordered amorphous domains. In amorphous domains, the polymer chains are loosely packed compared to denser crystalline domains, making low-crystallinity PET (lcPET), with its higher proportion of amorphous regions, more vulnerable to enzymatic degradation. Enzymatic hydrolysis of PET is more likely to occur near its glass transition temperature (65–75 °C).750 Kawai et al. categorised PET hydrolases into two groups: thermophilic cutinases and mesophilic cutinases, with IsPETase from a mesophilic bacterium as a key example.776 PET hydrolysis is most efficient near its Tg, rendering mesophilic cutinases less effective for PET recycling due to their significantly lower degradation, typically 1–3 times lower thermophilic cutinases. Therefore, a comparative evaluation of thermophilic and mesophilic cutinases is essential to determine their activity. The biological pretreatment of PET-fibres in combination with hydrolysis has also demonstrated good results. PET oligomers produced during hydrolysis were further hydrolysed to disodium terephthalate and EG using wild type Humicola insolens cutinase. The results showed a TPA yield of 97%, which was significantly higher than neutral hydrolysis without enzymes.777Clostridium thermocellum bacteria were engineered to achieve high-level secretory expressions of LC-cutinase. This modified bacterium effectively expressed LCC and degraded commercial PET films at 60 °C, achieving more than 60% conversion into soluble monomers, after 14 days of incubation.778 Moreover, C. thermocellum exhibited the additional ability to hydrolyse cellulose at 60 °C, making it highly effective to process textiles consisting of both PET and cellulose.746
Kawai et al. discovered that surface modification of PET with enzymes led to hydrolysis of ester bonds on the polymer's surface, producing hydroxyl and carboxy groups, while leaving the inner bulk intact.750,779 In a review on PET biodegradation, Kawai and colleagues categorised hydrolases with moderate activity that act on the surface of PET as surface-modifying enzymes.750 Conversely, enzymes capable of extensively hydrolysing PET's inner bulk were named PET hydrolases.750 Yoshida et al. isolated an Ideonella sakaiensis 201-F6 bacterium that could degrade PET films of low-crystallinity (1.9%) at room temperature.780 They identified IsPETase, a PET-hydrolysing enzyme, from this bacterium. IsPETase demonstrated higher PET degradation activity at 30 °C compared to other PET hydrolases.768 This bacterium produces enzymes that break down both PET and the intermediate mono(2-hydroxyethyl) terephthalic acid (MHET) when grown on PET. However, IsPETase demonstrated a limited degradation efficiency, achieving only 1% weight loss of low-crystallinity PET (lcPET) at 30 °C over 24 hours, significantly lower than the degradation achieved by other PET hydrolases at 50–70 °C.768 MHET was identified as the predominant product, with a minor amount of TPA. However, these PET hydrolases could degrade lcPET but not high-crystallinity PET (hcPET).781 Though their properties are similar to lipases and cutinases, PETase has successfully resulted in creation of a new family of PET-hydrolysing enzymes that are currently gaining significant interest.
Different research groups have investigated the structure of PETase, providing valuable insights for enhancing its performance.782 Han et al. found the S185 amino acid residue as a key feature of PETase.782 In another study, Kim and coworkers reported the structure of IsPETase (Fig. 35a), and proposed that PETase's substrate-binding site consists of two subsites: subsite I, which binds a single MHET moiety, and subsite II, which accommodates three MHET moieties (Fig. 35b).783 They attributed PETase's superior performance to structural variations in subsite II and the formation of the disulfide bond (Fig. 35c and d). Additionally, they suggested that modifying a residue located 23 Å from the catalyst site could enhance the enzyme's ability for PET substrate's accommodation in the active site. This led to the synthesis of a modified PETase with enhanced efficiency. They also predicted similar PET-degrading activities for other type IIb enzymes. The structural analysis of PETase showed that it retained the α/β-hydrolase bend but with more exposed active sites.784 Introducing mutations in two active site residues significantly enhanced the degradation efficiency of bottle-grade PET, resulting in a marked increase in MHET production.
 | ||
| Fig. 35 (a) Crystal structure of IsPETase; (b) active site of IsPETase; (c) structural comparison of IsPETase and TfCut2; (d) PETase activity of IsPETase proteins using the PET film as a substrate. Adapted from ref. 783. Copyright 2018, Springer Nature. | ||
Ideonella sakaiensis produces two key enzymes, PETase and MHETase, which are capable of breaking down PET into its monomers. At first, PETase hydrolyses the PET polymer into MHET and then MHETase hydrolyses it into TPA and EG.755 The bacterium later uses these products as a food source. It was demonstrated that a consortium of five bacteria (two Bacillus and three Pseudomonas species) can cooperatively decompose PET.785 Orange peel ash catalysed the glycolysis of PET using EG as the solvent, and resulted in BHET formation with 90.21% yield, achieving complete PET depolymerisation.786 The resulting glycolysis products were further biodegraded by 62.63% within two weeks by the bacterial consortium. Bell et al. engineered a thermostable version of PETase, known as HotPETase (Tm 82.5 °C), which can more rapidly depolymerise semicrystalline PET compared to other PETases.787 This enzyme efficiently degrades commercial grade PET bottles selectively into PET/PE laminated packaging materials. Additionally, the esterase enzyme family, such as p-nitrobenzylesterase from Bacillus subtilis, has demonstrated effective hydrolysis of PET into TPA and MHET.755 Lipases are highly versatile enzymes available from animal, plant, and microbial sources. They can hydrolyse long-chain triglycerides into fatty acids, and are also active in transesterification, esterification, aminolysis, and alcoholysis processes.788 Lipases have been reported to degrade PET fibres, enhancing their wettability, dyeability, and absorbency.755 Carniel et al.789 and de Castro et al.790 combinedly used lipase from Candida antarctica and HiC, which significantly improved the hydrolysis of PET into TPA. Zhou et al. developed an efficient PET hydrolysis strategy using a dual-function hydrolase, IsPETasePA, which exhibits balanced activity toward both PET and its intermediate, MHET.791 They observed that the accumulation of TPA during the reaction lowered the pH, impairing the enzyme's ability to convert MHET to TPA. Mechanistic analysis revealed that protonation of His208 in the catalytic triad under acidic conditions disrupted the enzyme–substrate interaction. By implementing a pH control strategy, the single-enzyme system delivered enhanced performance, enabling the recovery of high-purity TPA (>99%).
Sales et al. investigated the use of watermelon peels as a supplement to enhance the production of lipase and esterase enzymes by Yarrowia lipolytica during solid-state fermentation, with the aim of applying these enzymes in PET depolymerisation reactions.792Yarrowia lipolytica can produce numerous metabolites, including lipases and esterases, which serve as crucial industrial biocatalysts. The study compared enzyme production using soybean bran alone versus soybean bran supplemented with varying amounts of watermelon peels. The enzyme extracts were then tested for PET hydrolysis. The results showed that supplementing the culture media with the lowest content of watermelon peels increased lipase activity by nearly 31%. Esterase production increased by 1.5 times by adding 20 wt% of watermelon peels. The study revealed that peak lipase activity occurred at 14 hours, while esterase activity remained high at both 14 and 20 hours of fermentation. This research highlights the potential for generating enzymes using inexpensive fermentation media, making them effective biocatalysts for PET hydrolysis reactions.792
12.2 Polystyrene
Mealworms have been reported to rapidly digest polystyrene foam, achieving up to 50% weight loss within 24 hours. This degradation was confirmed by noticeable changes in the chemical composition and reduced molecular weight after the polymer foam passed through their intestinal tract.793 Yang et al. replicated these results using mealworms from 22 locations globally, all of which exhibited efficient PS degradation using the same experimental protocol.794 Similarly, Peng et al. studied other insect species such as dark mealworms for PS degradation, and found that PS degraded at a faster rate within the gut of T. obscurus than dark mealworms (T. molitor).795 Yang's group reported that superworms were also capable of degrading PS.796 Superworms consumed Styrofoam at the rate of 0.58 mg per day per superworm, 4 times higher compared to mealworms. The swallowed long-chain PS molecules were broken down into low molecular-weight products after passing through their guts. Greater wax moths (Galleria mellonella) and superworm (Zophobas atratus) have demonstrated the highest PS consumption capacity, survival rate, and capability to convert PS into lower molecular weight constituents. Yellow mealworms were particularly effective in depolymerising PS by breaking down the benzene ring. After PS ingestion, the microbial diversity in superworms and yellow mealworms was decreased, but greater wax moths exhibited increased diversity. The Mw and Mn of the PS in frass was found to decrease by 26% and 34%, respectively, compared to the initial PS foam (Styrofoam). Mechanical action such as mastication likely contributed to the reduction in the molecular weight of PS, although enzyme involvement remains uncertain. Similar findings were reported by Song et al. in the feces of the snail Achatina fulica, showing a 31% PS mass loss with reduced molecular weight of PS.797 However, despite the promising degradation results, PS feeding in superworms also caused decline in gut microbial diversity and the emergence of opportunistic pathogens.798 Recently, Liberto et al. studied the biodegradation of PS using Tenebrio molitor and Zophobas morio larvae, applying a pretreatment with H2O2 to facilitate the degradation.799 Over 30 days, the Mw and Mn of PS in the frass of both enzymes reduced, demonstrating effective PS biodegradation.Recent genome mining efforts have identified potential PS-degrading microorganisms, particularly highlighting enzymes like cytochrome P450s, and monooxygenases due to their capability to break C–C bonds.792 However, these genomic studies could not confirm whether these enzymes have the ability to degrade insoluble substrates like PS outside the cell. Kim et al. investigated the degradation of PS beads (Mw: 371, 1500 μm diameter) using a bacterial Pseudomonas aeruginosa strain DSM 50071, isolated from superworm's gut.800 The strain reduced the PS weight by 2.6% reduction over 15 days, with increased carbonyl and hydroxyl groups, indicating successful degradation. The study revealed that serine hydrolase and S-formylglutathione hydrolase enzymes were upregulated during PS degradation, while other enzymes were downregulated, possibly to conserve energy. Inhibition of SH completely blocked PS biodegradation, indicating its critical role in the degradation process.
12.3 Polyethylene
Although various enzymes capable of degrading polyolefins (PO) have been investigated, the evidence supporting their effective degradation is often inconclusive. For example, some studies include additional carbon sources besides the plastic or lack evidence of monomer formation during degradation. In many cases, abiotic pretreatments such as UV irradiation oxidation are employed to introduce reactive groups like carbonyl, hydroxyl, or double bonds into the polymer structure before attempting enzymatic or microbial degradation. It is critical to recognise that the presence of enzymes or microbes colonizing PO-based plastics does not necessarily confirm polymer degradation, as biodegradable additives in these plastics might be preferentially consumed.748,757 Thus, the enzymatic breakdown of petroleum-based polymers remains a significant challenge. While there are efforts to produce ethylene and propylene from bio derived sources with the aim to lessen the environmental impact, these approaches do not improve their biodegradability.Several studies have been focused on microbial degradation of polyethylene, with most experiments involving PE films or powders. A study demonstrated that LDPE films experienced significant biodegradation by two Aspergillus strains within 10 days, with SEM revealing surface damage in the form of holes and cracks. CO2 assays suggested around 80% biodegradation, however, this conversion efficiency might be inflated since only surface modifications were visible under SEM. Similarly, another study found a 46% weight reduction in PE films after six months of exposure to Streptomyces strain.801 Furthermore, a consortium of Bacillus cereus, Bacillus pumilus, and Arthrobacter sp. achieved a 22% weight reduction in both LDPE and HDPE after a two-week incubation period.802
Long-chain polyethylene (PE), with a molecular weight greater than 2000 Da, has shown to be degradable by naturally occurring microbial organisms. Several strains capable of breaking down untreated PE have been isolated from diverse environments, such as marine water, sewage sludge, and landfills. For instance, Azeko et al. observed a 36% weight loss in PE degraded by Serratia marcescens strain over a period of 70 days.803 Similarly, Phormidium lucidum and Oscillatoria subbrevis species were found to degrade 30% of polyethylene within 42 days.804 Studies have also reported that waxworms, which naturally digest beeswax, can consume PE films.805,806 In particular, the biodegradation of PE has been observed in the presence of the waxworm Galleria mellonella807 and the waxworm Achroia grisella.805 Moreover, Enterobacter asburiae YT1 and Bacillus sp. bacterial strains, from Plodia interpunctella, were shown to degrade PE over 60 days incubation.808 These results indicate that bacteria associated with waxworms hold promise for identifying more PE-degrading microorganisms. Streptomyces sp. exhibited 47% weight reduction of LDPE pellets after six months incubation.809 The fungus Zalerion maritimum achieved a remarkable 57% weight reduction of PE pellets within 14 days.810 Muhonja et al. studied various bacterial strains belonging to Bacillus, Cellulosimicrobium, Brevibacillus, , Pseudomonas, and Aspergillus for degradation of LDPE film, and achieved 36% weight loss after three months with Aspergillus oryzae.811 DSouza et al. used a fungal consortium consisting of Aspergillus niger, A. flavus, and A. oryzae for degradation of LDPE bags, resulting in 26% weight loss after 55 days, which was higher than the loss observed with individual species.812 When only LDPE was used as the carbon source, the weight loss was 15%, indicating that the availability of additional carbon sources can enhance degradation. Dang et al. studied the degradation of three types of plastic bags, HL (with nano-additives), VHL (contains LLDPE > 70% and HDPE < 30%), and VN1, using Bacillus sp. BCBT21 at 55 °C for 30 days.813 The weight losses were 61%, 11%, and 4%, respectively. The molecular weight (Mv) of pretreated plastic waste was also considerably reduced by 43%, from 205 kg mol−1 to 116.8 kg mol−1. Additionally, notable changes were observed in the morphology and properties.
The gut microbiome of some wax worms uses plastic substrates as the only carbon source. Thus, microorganisms of Lumbricus terrestris significantly decreased the particle size of LDPE microplastics. Ceres and Demetra enzymes present in the gut microbiota and saliva of Galleria mellonella wax worms have been found to oxidise and depolymerise polyethylene, at neutral pH and room temperature.814,815 A consortium of Pseudomonas aeruginosa and Enterobacter cloacae, formulated from cow dung, was able to depolymerise polyethylene and polypropylene by 64.25% and 63%, respectively, in 160 days.816 HDPE film upon colonization with Achromobacter xylosoxidans resulted in 9% weight reduction.817 Notably, Penicillium oxalicum and Penicillium chrysogenum fungal strains demonstrated more effective degradation of HDPE than LDPE, although specific details on the thickness or molecular weight of the films were not provided.818Penicillium oxalicum caused a weight loss of 55% in HDPE and 37% in LDPE after a 90 days incubation period. Similarly, Penicillium chrysogenum resulted in weight losses of 59% for HDPE and 34% for LDPE under the same conditions. Microbial enzymes such as manganese peroxidase and soybean peroxidase have been reported to reduce the molecular weight and tensile strength of polyethylene films.761,819 Jeon et al. identified alkB, alkB1, and alkB2 genes which play a critical role in the degradation of low molecular-weight PE.819 Furthermore, a study by Xu et al. suggested that oxidases or oxygenases could enzymatically cleave C–C bonds in polyolefins.820
Sullivan et al. developed an innovative hybrid process that converts mixed plastic waste into various platforms and specialty chemicals by integrating chemical recycling and engineered bacteria.635,821 This method, designed for mixed plastics (HDPE, PS, and PET), is particularly promising for industrial applications as it eliminates the need for a costly sorting step. The process begins with the chemical oxidation of polymer blends using a Co–Mn–Br catalytic system under ambient air, breaking down the plastics into oxygenated compounds, such as benzoic acid from PS and dicarboxylic acids from HDPE. These oxidised products are then processed by genetically engineered Pseudomonas putida bacteria. The authors used advanced genetic engineering for processing carboxylic acid mixtures. One strain was engineered to produce poly(hydroxyalkanoates), while the other converts carboxylic acids into β-ketoadipate, a key precursor for high performance nylons. This tandem approach of chemical oxidation followed by biocatalytic conversion shows potential for recycling multilayered packaging and textile materials. Although the process offers a novel solution for complex plastic waste, its economic feasibility for large-scale industrial application still requires detailed investigation.
12.4 Polypropylene
Although polypropylene is more susceptible to oxidation than polyethylene, its higher crystallinity and melting temperature (Tm) have resulted in fewer studies on its biodegradation. When Rhodococcus sp. and Bacillus sp. were used to degrade polypropylene microplastics, they caused slight weight losses of 6% and 4%, respectively, after 40 days of incubation.39 Jain et al. demonstrated the degradation of polypropylene (PP) and poly-L-lactide blends (PLLA) using Bacillus species isolated from compost samples, providing promising evidence for the environmental breakdown of these materials.822 The isolates P8 and P6 achieved biodegradation rates of 12% and 10%, respectively. Yang et al. investigated the use of Tenebrio molitor and Zophobas atratus for degradation of PP foam over a 35-day incubation period.823 Pires et al. used an enzymatic additive to explore the degradation of PP in soil over six months, comparing its effect with organic additives. The enzymatic additive significantly enhanced the degradation process, resulting in a substantial increase in the carbonyl index (3693%), changes in crystallinity (18.7%), and structural characteristics.824 The study indicated the great influence of enzymatic additives on the degradation. Auta et al. reported that Bacillus sp. strain 27 and Rhodococcus sp. strain 36 achieved a 4.0–6.4% weight loss in PP microplastics after 40 days, when grown in aqueous synthetic medium with PP microplastics.825 Another study identified a mesophilic strain, Stenotrophomonas panacihumi PA3-2, which was capable of degrading both low molecular weight PP (Mw = 2800–3600 Da) and high molecular weight PP (Mw = 44000 Da).819 This strain achieved degradation rates of 12.7–20.3% based on CO2 release after 90 days of incubation. Skariyachan et al. reported a consortium of Pseudomonas aeruginosa and Enterobacter cloacae bacterial strains, degrading PP with 63% weight reduction after 160 days.816 FTIR and SEM analyses revealed the formation of new functional groups, and cracks on the polymer surface, respectively. These studies highlighted the potential of various microorganisms in the biodegradation of PP.
12.5 Polyvinyl chloride
PVC plastic is formulated with higher plasticiser content compared to other synthetic plastics which makes it difficult for degradation. However, many microorganisms such as bacteria and fungi can utilise plasticisers as a nutrient source, making plasticised PVC prone to bacterial or fungal degradation. Various fungal species isolated from different environmental samples, such as plasticised PVC films buried in the grassland soil826 and plastic waste disposal sites,827 have exhibited the ability to degrade the plasticised PVC. Ali and coworkers identified various fungal strains on PVC films buried in soil for 10 months.826 They identified P. chrysosporium PV1, L. tigrinus PV2, A. niger PV3, and A. sydowii PV4 strains through 18rRNA gene sequencing, morphology, and phylogenetic analysis. The authors also evaluated the biodegradability of these strains for thin PVC films in shake flask experiments, where significant colour change, and surface deterioration of PVC films were observed visually and further confirmed by Scanning Electron Microscopy. Zhang et al. recently reported the ability of a bacterial strain Klebsiella sp. EMBL-1, from the insect Spodoptera frugiperda, to efficiently decompose PVC.828Various bacterial strains, isolated from marine atmospheres and waste disposal sites, have shown potential as effective agents for degrading plasticised PVC.827,829,830 However, it has been observed that these PVC-degrading microorganisms primarily degrade components of the plasticiser, such as bis(2-ethylhexyl) phthalate (DEHP) or surface of PVC, rather than breaking down the PVC polymer backbone.829–832 For example, Micrococcus luteus was able to achieve only 9% mineralisation of PVC after 70 days. Additionally, PVC concentrations above 5 g L−1 inhibit microbial growth. An important report revealed that employing a microbial consortium reduced PVC's molecular weight from 70 kg mol−1 to 16 kg mol−1 over a period of 9 months.833 Alternatively, a study by Peng et al. found the larvae of Tenebrio molitor exhibited efficient depolymerisation of PVC and frass contained only 35% residual PVC.834 To date, no microorganism has been discovered that can effectively degrade both PVC and its plasticisers. Therefore, the specific enzymes responsible for PVC degradation remain unidentified, representing a key hurdle in advancing microbial strategies for complete PVC recycling.
12.6 Polyurethane
After the first report of polyurethane degradation in 1968 using fungi, various enzymes that decompose or reduce the weight of polyurethane have been identified and characterised, such as promiscuous esterase, urease, protease, amidase, cutinases, proteases, amidases, and oxidases.761,835 Esterases are known to hydrolyse ester bonds and can destroy liquid dispersion as well as bulk polyester polyurethanes, but their ability to degrade urethane bonds is not evident.836 Schmidt et al. isolated four polyester hydrolases: LC cutinase, Tcur0390, Tcur1278, and TfCut2, capable of degrading emulsified PUR.836 Among these enzymes, LC cutinase showed the highest activity, leading to weight reductions of 4.9% and 4.1% in two commercial PUR polyesters (Elastollan B85A-10 and C85A-10) after 200 hours at 70 °C. Proteases and amylases are able to hydrolyse both urethane and ester bonds, but the degradation rate on PUR films or granules remains very low. Using a complex of esterase and amidase enzymes, 33% of polyurethane was decomposed within 51 days.837 It is known that Tenebrio molitor larvae are able to consume polyurethane foams with a 65% weight loss of the initial polymer after 35 days. Alternaria sp. strain PURDK2 and Cladosporium fungal strains degraded polyurethane polymers by 27.5% and 65%, respectively.835Pellis' group in 2022 reported the use of Humicola insolens cutinase (HiC), specifically Novozym 51032, to investigate the hydrolytic degradation of polyurethane–polyester (PU–PE) copolymer.838 The polyester–PU film was incubated with HiC at 50 °C for 7 days, resulting in substantial weight reduction after 3 days. The number-average molecular weight (Mn) was decreased by 84%, from 22 to 3.4 kg mol−1, whereas the weight-average molecular weight (Mw) decreased by 42%, from 108 to 63 kg mol−1. FTIR and NMR analyses confirmed the hydrolysis of ester bonds, and SEM images identified cracks on the PU films due to enzymatic erosion.838 GCMS analysis identified 3,3′-methylendianiline (MDA) in the degradation products, providing evidence that the enzyme was capable of cleaving urethane bonds in addition to ester bonds. Magnin et al. synthesised four PUs with different molar masses and investigated their degradation using laccase, with HBT as a mediator.839 The TDI-based PU foam resulted in the highest weight loss of 25% after 24 h at 37 °C, compared to only 3% in the control. These results marked a significant milestone as the first complete degradation of an entire PU component, producing 6-hydroxycaproic acid and a small chain acid-terminated diurethane. Furthermore, the authors studied the feasibility of recycling the degraded monomers. By combining 50% recycled monomers and building blocks with 50% 6-hydroxycaproic acid, they synthesised a high molecular weight polymer (74 kg mol−1) without using toxic polyisocyanates. However, using only recycled building blocks proved ineffective due to the presence of trimethylolpropane, a byproduct from the hydrolysis of PCL triol segments, leading to undesired crosslinking.
In addition to the hydrolysis of PET and polystyrene, research on the hydrolysis of polycaprolactone,840 polyethylene furanoate,841 polybutylene succinate,842 and polylactic acid843 has also been explored. For example, a PLA film (PLA 2002D) experienced a 71% weight loss by incubating with Pseudonocardia sp. RM423 at 30 °C for 4 weeks. Another Amycolatopsis strain, isolated from soil, significantly degraded the PLA polymer resulting in a 36% weight loss in 30 days under mesophilic conditions at 30 °C.844 Despite promising development in the field of plastic biodegradation, most enzymes do not have properties that enable them for direct use in industrial processes due to low yields of monomers and the longer degradation times. The diversity of enzymes in nature is vast, and the biocatalysts studied for plastic processing represent only a small part of this diversity. It follows that success in industrial applications will depend on identifying stable and highly active enzyme catalysts. Various microorganisms tested for degradation of polymers are listed in Table 7.
| Enzyme | Feedstock | Reaction conditions | Product | Conversion | Reference |
|---|---|---|---|---|---|
| a MHET: mono-(2-hydroxyethyl) terephthalate; SCB: sugarcane bagasse. | |||||
| TfCut2 from Thermobifida fusca | PET | 70 °C, 96 h, K2HPO4/Cl | Terephthalic acid | 92.3% | 751 |
| Hydrolase from Thermobifida fusca | PET | 55 °C, 21 days, phosphate buffer | — | 50% | 764 |
| Leaf-branch carbon cutinase | PET | 65 °C, 10 h, phosphate buffer pH, 7 | Terephthalic acid, ethylene glycol | 90% | 765 |
| Ca2+/Mg2+ sites on hydrolase from T. fusca | PET | 65 °C, 48 h, tris buffer | — | 12.90% weight loss | 769 |
| LC-cutinase | Waste PET | 80 °C, 12 h, pH 8 | Calcium terephthalate (CaTP·3H2O) | 88% | 773 |
| p-Hydroxybenzoate hydroxylase (PobA mutated) | PET hydrosylate | 30 °C, 72 h | Gallic acid | 74.3% | 775 |
| Cutinase from Humicola insolens | PET | 50 °C, 24 h, Tris–HCl buffer, pH 8 | Terephthalic acid | 97% | 777 |
| PETase from Ideonellasakaiensis | PET | 30 °C, pH 7, 45 days | Terephthalic acid | 60% | 780 |
| PETase from Ideonellasakaiensis | PET | 30 °C, 36 h | Terephthalic acid, mono(hydroxyethyl)terephthalate, ethylene glycol | — | 783 |
| Consortia of five bacteria (3 species of Pseudomonas and 2 species of Bacillus) | PET | 30 °C, 14 days | BHET, terephthalic acid | BHET (92.21%). Final TPA (62.63%) | 786 |
| Lipase from Candida antarctica | PET film | 60 °C, 21 days, phosphate buffer, pH 7 | Terephthalic acid | 40% weight loss (max 956 μM TPA) | 789 |
| p-Nitrobenzylesterase from Bacillus subtilis | PET | 40–45 °C, 72 h, pH 7 | Terephthalic acid, benzoic acid, MHET | — | 845 |
| Yellow mealworms | PS foam | 25–26 °C, 34 days | — | Up to 46% weight loss | 794 |
| Tenebrio obscurus | PS | 25 °C, 70% humidity, 31 days | — | Up to 26% weight loss | 795 |
| Achatina fulica snails | PS foam | 28 days | — | 30.7% weight loss | 797 |
| Bacillus species (P1 to P16) | PP and polylactide blends | 37 °C, 360 h | — | 10–12% weight loss | 822 |
| Tenebrio molitor and Zophobasatratus | PP foam | 25 °C, 840 h | — | 32% weight loss | 823 |
| Phanerochaete chrysosporium | Blend of LDPE/SCB | 35 °C, 768 h, phosphate buffer, pH 7 | — | 16% weight loss | 846 |
| Brevibacillus sp. and Aneurinibacillus sp | HDPE | 50 °C, 3360 h | Methyl and aldehyde groups | 46.6% | 847 |
| LDPE | 58.21% | ||||
| PP pellets and film | 56.3% | ||||
| Bacillus sp. strain 27 | PP microplastic | 29 °C, 672 h, nutrient agar (NA) plates | — | 4% weight loss | 825 |
| Rhodococcus sp. strain 36 | 6.4% weight loss | ||||
| Fungal lignin peroxidase | Plasticised PVC | 25 °C, pH 5, 28 days | — | 31% weight loss | 827 |
| Pseudomonas citronellolis and Bacillus flexus | Plasticised PVC | 45 °C under aerobic conditions | — | 19% weight loss | 829 |
| Tenebrio molitor | PVC microplastic | 25 °C, 6 days | — | M w reduced by 33.4 ± 2.6% | 834 |
| Esterase (E3576) | Waterborne polyester polyurethane dispersion | 37 °C, phosphate buffer, 51 days | — | 33% weight loss | 834 |
| Laccase from Trametes versicolor | Polyether-based PU foam | 37 °C, sodium acetate buffer, pH 4.5, 18 days | — | 25% weight loss | 839 |
| Pseudonocardia sp. RM423 | PLA film | 30 °C, 28 days | — | 71% weight loss | 848 |
| Amycolatopsis | PLA | 30 °C, 30 days | — | 36% weight loss | 844 |
| Ideonellasakaiensis PETase and MHETase | PET nanofilm | 50 °C, 1 h | 70% weight loss | 774 | |
13. Conclusion, challenges and future perspectives
Establishing a plastic circular economy is crucial for achieving environmental sustainability and fostering sustainable energy markets. Despite extensive global efforts, the plastics economy remains mostly linear, with only 9% of global plastic waste being recycled. Various catalytic approaches, including pyrolysis, hydrocracking, chemolysis, metathesis, hydrogenolysis, photo-, electro and biocatalysis, offer unique advantages in converting plastic waste into value-added chemicals and fuels, thus promoting a more sustainable energy paradigm. However, all recycling processes contribute only 10–20% of plastic waste management. Consequently, the current situation of the plastic economy is quite far from circularity. The development and optimisation of catalytic processes highlight the potential for transforming plastic waste management from a linear to a circular economy, where waste becomes a resource for sustainable energy production. Although feedstock recycling processes have significant potential to transform plastic waste into non-plastic products, their large-scale deployment is hindered due to high economic costs. Industrial-scale implementation of various processes incurs substantial costs, primarily due to high energy requirements and the need for efficient sorting of plastic waste. Moreover, the presence of contaminants and other materials in the waste stream further complicates the recycling process as these must be removed before the plastics can undergo catalytic processing. There are several challenges that must be addressed to enable these processes for large scale deployment, as summarised below.13.1 Pyrolysis
Catalytic pyrolysis technology has substantial potential for practical applications and offers a sustainable and clean solution to address the plastic waste problem. It offers a flexible pathway to convert plastic waste to products with required selectivity and good quality by careful tuning of the process parameters. Based on the catalyst type, petroleum components can be achieved in good quality and selectivity, which can serve as fuel or feedstock for making chemicals and plastics. Typically, high olefinic fractions are present in the pyrolytic oil, therefore, it is mostly subjected to hydrogenation to enhance the quality of resultant petroleum fractions. The reactor design and process configuration are important factors that influence the pyrolysis efficiency. Among various type of reactors, fluidised bed reactors have proven to be the most suitable reactors for catalytic pyrolysis due to their ability to facilitate better interaction between catalysts and feed, ensure uniform temperature distribution, and minimise coke formation on the catalyst. It is crucial to select the reaction temperature carefully as it significantly affects the quality and quantity of liquid fuel. The selection of plastic feedstock is also critical as it has strong influence on the liquid oil output. Plastics such as LDPE, HDPE, PS, and PP are the most suitable plastics for pyrolysis because PET and PVC have been found to produce low liquid yields. PVC is usually unsuitable for the pyrolysis process because it generates harmful byproducts, such as HCl and chlorinated organic compounds, which are detrimental both for the environment and the oil quality.Catalyst selection plays an important role in catalytic pyrolysis as it can significantly improve the process efficiency by increasing the quality of liquid oils while reducing the activation energy, reaction time, and temperature. It is essential to evaluate catalyst stability, deactivation, long-term reusability, and the type of catalyst supports for optimal catalyst selection. Commonly used catalysts for plastic pyrolysis include activated carbon, FCC catalysts, amorphous silica, and zeolites. Among these, zeolite catalysts are known for producing high-quality fuel with good conversion rates. Particularly, zeolite Y is widely utilised and has been reported to generate oil yields of <70 wt%, with a higher proportion of gasoline-range hydrocarbons and significant aromatic content. The relay catalysis method, which leverages the highly acidic nature of ZSM-5, has also proven effective in producing naphtha-range oil. However, highly active zeolite catalysts typically result in high gas yields. To address this issue, combinations of FCC, zeolite, and silica catalysts have been explored, successfully producing high-quality oil with good yields. Activated carbon catalysts have also reported to deliver better results. Recently, composite catalysts with micro- and mesoporous structures have gained interest due to their superior cracking abilities and capacity to achieve higher oil yields.
Plastic pyrolysis oil has been identified as more suitable for use in compression ignition engines than spark ignition engines, demonstrating improved engine performance. With the increasing research interests in producing sustainable jet fuel, the development of more advanced catalyst systems has the potential to enhance catalytic pyrolysis processes for efficient production of aviation fuel. The following investigation should be performed for commercial applications:
• Development of highly stable, recyclable, and cost-effective catalysts.
• Developing reactor design and process to recycle medical waste.
• Exploiting the synergistic effects of combining different plastics.
• Conducting comprehensive life cycle studies of plastics, pyrolysis processes, and the oil products to evaluate the economic and environmental feasibility.
• Assess the potential applications of gas and char by-products from pyrolysis for improving the economic sustainability of the process.
Catalytic pyrolysis of plastic waste represents a promising strategy for addressing Scope 3 emissions. This process transforms non-recyclable plastics into valuable fuel products, thus lessening reliance on virgin fossil fuels and minimising emissions associated with waste disposal. Scope 3 emissions encompass indirect greenhouse gas (GHG) emissions arising from activities such as waste disposal, transportation, and supply chain operations that fall outside an organisation's direct oversight. Conventional plastic waste management methods, including landfilling and incineration, significantly contribute to these emissions by generating methane in landfills and emitting carbon dioxide during incineration. Moreover, catalytic pyrolysis aids in the reduction of Scope 3 emissions by creating a closed loop in plastic production. The pyrolysis process generates various hydrocarbons that can be converted into synthetic fuels or utilised as feedstock in petrochemical processes, thereby lessening dependence on finite resources and the production of virgin fossil fuels. This closed-loop approach prevents plastics from ending up in landfills and supplies alternative fuels and raw materials for industries, reducing their reliance on conventional oil and gas sources. Industries can foster a circular economy by incorporating catalytic pyrolysis into the value chain, thus reducing upstream and downstream Scope 3 emissions. Finally, catalytic pyrolysis is vital in achieving sustainability goals by enhancing resource efficiency and enabling organisations to comply with regulatory and consumer expectations for lower carbon footprints. Since Scope 3 emissions present some of the most significant challenges, adopting innovative waste-to-fuel technologies like catalytic pyrolysis can provide an effective solution. This approach signifies a commitment to sustainable practices and environmental stewardship and substantially diminishes the overall ecological impact.
13.2 Hydrocracking
Hydrocracking is a highly effective method to produce high quality liquid fuel from waste plastics. This process can be carried out using either a direct liquefaction or a two-stage liquefaction process. Optimising reaction parameters is essential for achieving the desired product distribution during hydrocracking. The type of plastic feed is a primary consideration, with HDPE, LDPE, PP, and PS being the most suitable, though PET can also be used. For PVC, a two-stage liquefaction process with dechlorination at the first stage is recommended. Another important parameter is the reaction temperature which must be optimised as it strongly affects the process outcome. The optimum temperature range varies depending on the catalyst used. Bifunctional catalysts, such as metals incorporated on acidic supports, typically require 350–375 °C for complete plastic conversion, whereas zeolites without metals usually require higher temperatures, around 400–425 °C. Temperature variation also significantly affects the product distribution.Reaction time is another key factor in the hydrocracking process. Increasing reaction time generally enhances conversion and liquid yield, but beyond a certain optimised duration, further increase has minimal or no additional effect on conversion. However, it can shift the product distribution towards lighter fractions. Additionally, selecting and optimising catalyst support, metal type, and metal loading are also essential and must be carefully considered. Promising catalyst supports include zeolites such as HBeta, HY, and HZSM-5, with metals like Pt, Pd, and Ni delivering excellent results on highly acidic supports. Bifunctional catalysts enhance both conversion efficiency rates and product selectivity, achieving optimal performance at comparatively lower temperatures.
To reduce the reliance on costly noble metal hydrocracking catalysts, the research should be focused on developing alternative catalysts that use minimal metals or entirely non-noble metals. Developing single atom metal alloy catalysts offers significant potential for practical applications. While non-noble metal catalysts, such as nickel incorporated on silica and zeolites, have been developed for hydrocracking of polyolefins, they continue to exhibit lower performance compared to noble metal catalysts. Improving the efficiency of non-noble metal-based hydroconversion catalysts requires careful selection of metals, promoters, and supports, along with precise control over the catalyst structure. Additionally, insights from previous studies on small hydrocarbon conversion can help design more effective non-precious metal catalysts.
The breakdown of stable C–C bonds through a thermodynamic process currently demands significant external energy, leading to severe reaction conditions that are unsustainable for large scale plastic waste degradation. A key challenge is to reduce the activation energy barrier for cleaving inert C–C bonds. Tandem catalysis presents a promising approach by facilitating polymer degradation under moderate conditions. For example, a tandem cracking–alkylation mechanism process can create a highly ionic reactive environment, increasing C–C bond reactivity and lowering the energy required for ionic transition states. The future efforts could be focused on developing additional tandem catalytic strategies. Hydrocracking yields high value hydrocarbon products, however, it requires hydrogen stream which is more expensive than other fluidising gases, such as nitrogen used in the pyrolysis. In addition, the high-pressure conditions required in hydrocracking increase equipment costs and operational complexity. As a result, hydrocracking is a less popular method for converting plastic waste into hydrocarbons compared to pyrolysis.
The hydrocracking process has the potential to significantly lower Scope 3 emissions by substituting virgin fossil fuels with fuels derived from plastic waste. This shift diminishes the need to extract and refine primary fossil resources. Hydrocracking effectively converts difficult-to-recycle plastics into valuable fuel, which also helps to mitigate emissions associated with plastic waste disposal and incineration. Turning plastics into useable fuel reduces dependence on conventional disposal methods that significantly contribute to Scope 3 emissions. Additionally, employing synthetic fuels generated from hydrocracking in industrial applications can reduce the dependence on fossil-fuel-based energy for energy-intensive processes, thereby decreasing Scope 3 emissions linked to fuel combustion. Incorporating hydrocracking technology into supply chains offers a viable strategy for reducing carbon footprints in downstream activities, especially in sectors reliant on fossil fuel energy. This technology enables industries to simultaneously tackle waste management and emission reduction objectives, presenting a practical approach to enhancing the overall sustainability performance.
13.3 Depolymerisation
The diverse chemical structures of polymers make the depolymerisation of plastic waste a highly complex and technically challenging process, especially when dealing with mixed waste streams. Different plastics require distinct reaction conditions and catalysts for efficient conversion, further complicating the process. The presence of additives and contaminants exacerbates these difficulties, making the selective depolymerisation of waste plastics into valuable chemicals even more challenging. Additionally, the high energy input and stringent reaction conditions often required for depolymerisation make it less economically viable compared to other recycling methods. The development of catalysts that can operate efficiently under milder conditions and with high selectivity remains a significant hurdle. Moreover, the potential deactivation and loss of catalyst activity over time pose further operational challenges, necessitating frequent catalyst regeneration or replacement, which adds to the overall cost and complexity of the process. Furthermore, due to the varying chemical properties of different plastics, a single solution or process for each plastic type is impractical. Therefore, technologies must be specifically tailored to each type of polymer to ensure sufficient and practical depolymerisation.Catalytic depolymerisation offers a way to convert polymers into monomers, valuable hydrocarbons like BTX, BHET, and other small molecules. This process can enable the recovery and purification of monomers, which can then be re-polymerised. However, depolymerisation is slow under moderate conditions, necessitating the development of efficient catalysts. Ideal depolymerisation catalysts should be cost-effective, competitive with virgin material prices, and stable in the presence of air, moisture, and contaminants. Additionally, these catalysts must exhibit high selectivity in heterogeneous mixtures, efficiently converting polymers into monomers. Despite these challenges, the future of plastic recycling by depolymerisation is promising, driven by ongoing advancements in catalyst technology and process optimisation. Research is increasingly focused on developing robust and versatile catalysts that can handle a wide range of plastic types and impurities while operating under milder conditions. Innovations such as single-atom catalysts, nanocatalysts, and hybrid catalysts are being explored to enhance efficiency and selectivity. Furthermore, integrating depolymerisation processes with renewable energy sources could significantly lower the energy footprint, making the process more sustainable.
The catalytic depolymerisation process effectively decreases Scope 3 emissions linked to plastic production and waste disposal by converting end-of-life plastics into raw materials, thus diminishing the need for new fossil feedstocks. Chemically recycling plastics into high-quality chemical feedstocks prevents these materials from being discarded in landfills or incinerated, significantly reducing the contribution to Scope 3 emissions. Moreover, it promotes a circular economy, facilitating the continuous recycling of plastic materials and, in turn, lowering emissions related to the extraction and manufacture of new resources. Catalytic depolymerisation's efficacy further helps mitigate Scope 3 emissions by supplying industries with lower-carbon raw materials. This method reduces upstream emissions associated with the production of plastics from virgin resources, as recycled monomers require less energy and fewer inputs than resources acquired through extraction. By adopting catalytic depolymerisation, the management of end-of-life plastic materials can be conducted in an environmentally responsible manner, aligning with sustainability goals and decreasing greenhouse gas emissions throughout the lifecycle of plastic products. Depolymerisation has the potential to convert plastic waste into high-value and circular fuels, chemicals, and plastics for enhancing plastic circularity.
13.4 Metathesis
Significant advancements have been made in improving the efficiency and recyclability of various metathesis reactions, which can be used for the depolymerisation of waste plastic. However, certain thermodynamic and kinetic limitations exist, such as the recycling and removal of cross-linked PB rubbers, which have been successfully addressed using latent catalysis. However, to prevent premature catalyst deactivation and ensure efficient depolymerisation, it is essential that catalysis occurs at moderate or low temperatures. Latent catalysis is an evolving field, and with the discovery of new metathesis catalysts and their increasing commercial availability, there is a great potential for further advancements for the in situ depolymerisation of cross-linked polymers. Additionally, the variability in plastic feedstock, which includes differences in polymer types, additives, and contaminants, complicates the recycling process. Ensuring consistent catalyst performance and selectivity across diverse plastic waste streams remains a complex task. Furthermore, understanding the detailed mechanisms and kinetics of metathesis reactions is still in its infancy, necessitating extensive research to optimise reaction conditions, maximise yield and control product distributions.Plastic recycling by metathesis faces several significant challenges. One of the primary obstacles is the high cost and limited availability of efficient metathesis catalysts, particularly those based on noble metals. These catalysts often require high loadings and are prone to deactivation, making the process economically unfeasible for industrial scale applications. Additionally, the variability in plastic feedstock, which includes differences in polymer types, additives, and contaminants, complicates the recycling process. Ensuring consistent catalyst performance and selectivity across diverse plastic waste streams remains a complex task. Metathesis holds promise for recycling and degrading waste plastics, but scaling the process for industrial use remains a challenge. This involves producing pure chemicals at low costs without metal contamination and achieving high selectivity for desired products.
By employing metathesis to decompose plastic waste, this method significantly reduces the reliance on newly sourced fossil fuels, presenting an environmentally friendly solution for creating energy precursors and high-value hydrocarbons. These outputs can be used as feedstocks in various fuels and industrial processes, aiding the shift towards more sustainable energy sources while simultaneously addressing the challenge of plastic waste management. The role of metathesis in sustainable energy markets is underscored by its capacity to generate high-quality feedstocks that diminish environmental impacts while preserving energy efficiency. Given that metathesis reactions are predominantly catalytic, they can be tailored to lower energy demands, further curbing the carbon footprint associated with recycled plastic products. This approach aligns harmoniously with sustainable energy objectives by introducing lower-carbon feedstocks into fuel production processes while also steering plastic waste away from detrimental disposal practices. Consequently, metathesis plays a vital role in fostering a more sustainable and resource-efficient energy market by converting plastic waste into products that bolster cleaner energy generation and industrial applications, thus significantly reducing Scope 3 emissions.
13.5 Photo and electrocatalysis
Chemical recycling of plastic waste using alternative and renewable energy sources (photocatalysis and electrocatalysis) faces significant challenges, primarily due to economic factors. The high costs associated with the energy demands and sorting of plastic waste make it difficult to deploy these methods on an industrial scale. Additionally, contaminants in the waste stream must be separated before catalytic processing, further complicating the process. Key challenges include designing and synthesising inexpensive, less toxic photocatalysts with high performance and recyclability, as well as dealing with the high costs of pretreatment and separation of photocatalytically derived products. Furthermore, electrocatalytic recycling is less developed than photocatalysis, with limited technology for efficient plastic waste recycling under ambient conditions. The large molecular weights of plastic molecules also pose difficulties in designing highly porous anodes for efficient diffusion. Exploring a wider range of substrates and developing novel catalytic systems can improve the scope and efficiency of both photocatalytic and electrocatalytic approaches. Emphasising the use of real-world plastic waste and scaling up reaction systems will be crucial for industrial implementation. Utilising alternative energy sources for plastic upcycling offers a promising approach to managing primary pollutants and enhancing sustainability.Photo- and electrocatalytic recycling of plastic waste offer innovative, energy-efficient solutions to mitigate Scope 3 emissions by converting waste plastics into valuable resources. The scalability of these technologies holds promise to reduce the carbon footprint associated with the entire lifecycle of plastic products, from raw material extraction to end-of-life disposal. Additionally, as photo- and electrocatalytic systems can be powered by renewable energy, they support a circular economy model with minimal environmental impact while addressing the indirect emissions (Scope 3).
13.6 Biocatalysis
Enzyme biocatalysis presents a promising alternative for plastic waste biodegradation, potentially reducing energy consumption, greenhouse gas emissions, and pollutants compared to traditional methods like hydrocracking, pyrolysis or chemical depolymerisation. This method can potentially be integrated into existing recycling processes or replace current chemical recycling techniques. While these processes tend to be slower, the use of biocatalysts can accelerate reaction rates and reduce processing times. Rapid advancements in synthetic biology and protein engineering offer a range of tools for discovering, characterising, and optimising plastic-degrading enzymes, creating new opportunities to develop efficient and cost-effective biocatalysts for plastic depolymerisation. However, several challenges persist, such as the integration of separation stages for biodegradation products and the high initial infrastructure costs. Future research should focus to target the development of these recycling methods for large scale deployment and gain a deeper understanding of new bioprocess depolymerisation mechanisms. Such efforts will enable economic impact assessments to support wider adoption of biodegradation technologies and fostering a more sustainable approach to managing global plastic waste.Innovations in catalyst design, such as high-throughput screening methods and advanced platform techniques, are necessary to identify and develop efficient plastic-degrading enzymes. Advanced techniques such as cell-as-compartment, micro-droplet, and pico-droplet-based screening have the potential to significantly improve the efficiency and precision of enzyme discovery. However, a key challenge lies in finding suitable substrates that work effectively with these advanced screening platforms. The recent advancements in chromogenic and fluorogenic substrates for high-throughput analysis of plastic-degrading enzymes offer valuable insights that could drive future substrate innovations and significantly enhance the screening process for more efficient enzyme discovery.
13.7 Artificial intelligence
The application of artificial intelligence (AI) and machine learning to optimise reaction parameters and predict catalyst performance shows significant potential for improving the scalability and efficiency of plastic recycling processes. The integration of machine learning can help in understanding reaction dynamics and optimising reaction parameters. AI is instrumental in sorting plastic waste to maximise the recycling potential of plastics. Machine learning methods are usually used in various types of pyrolysis, such as catalytic pyrolysis, co-pyrolysis of plastic waste with biomass, and microwave pyrolysis. Among these, co-pyrolysis has emerged as the most popular type of pyrolysis modelled by machine learning methods, followed by microwave supported pyrolysis. Integrating machine learning methods with other optimisation algorithms offers valuable insights that can guide experimental research and process development. Future research should focus on key areas such as catalyst synthesis, selecting suitable additives, reaction kinetics, understanding catalyst deactivation mechanisms, and designing renewable energy-powered systems.Future perspectives include the development of closed-loop recycling systems, where depolymerised monomers are reused to produce new plastics, thus significantly minimising waste and resource consumption. Achieving this requires collaborative efforts among academia, industries, and government bodies to drive innovation and establish regulatory frameworks that support the commercialisation of advanced depolymerisation technologies. As research continues to overcome the current limitations, depolymerisation has the potential to become a cornerstone of sustainable plastic recycling, contributing significantly to the reduction of plastic waste and the establishment of a circular economy for meeting the growing energy demand.
13.8 Polymer types
While thermal pyrolysis remains a widely used recovery method, its broad product distribution and high impurity levels hinder its scalability. Catalytic pyrolysis and solvolysis offer improved product selectivity, though the development of low-cost, robust catalysts remains a key research priority. Subsequent catalytic and steam cracking processes are crucial for producing olefins, the fundamental building blocks of plastics, from pyrolysis oils, thus enabling a closed-loop recycling system.
To truly realise circularity, chemical recycling must shift from merely producing fuels, where carbon is lost, to generating high-value, reusable feedstocks such as lubricants or naphtha, which can re-enter the polymer production cycle. This requires refined control over product quality, including impurity management and molecular composition, as well as integration with existing refinery infrastructure. Moving forward, a coordinated effort is essential to develop robust, selective catalysts and reactor systems that operate effectively with real-world waste streams.
Innovative advancements in catalyst design are needed to enable efficient PS depolymerisation at lower temperatures with improved product selectivity. This includes developing highly dispersed, surface-engineered catalysts that offer better polymer–catalyst contact and allow operation under milder conditions, thereby reducing energy input. Moreover, integrating optimised process conditions, such as minimised reaction temperatures with extended residence times, may offer energy savings without compromising the yield. Industrial viability also depends on regenerable catalysts with stable long-term performance, as well as scalable, cost-effective synthesis methods. Future research must bridge the gap between material innovation and process integration to realise scalable, low-energy PS recycling technologies aligned with circular economy goals.
Catalysis is central to advancing PET depolymerisation, with heterogeneous systems offering advantages such as enhanced stability, reusability, and lower toxicity compared to their homogeneous counterparts. Innovations in catalytic glycolysis and methanolysis have enabled the production of high-purity monomers like BHET and DMT, although careful tuning of catalyst properties is required to minimise side reactions and prevent deactivation by impurities. Novel catalytic materials, including nanocatalysts, ionic liquids, and deep eutectic solvents, have shown promise in improving efficiency and environmental performance. Reductive depolymerisation offers access to high-value chemicals but remains underdeveloped due to its harsh conditions and limited catalyst availability. Catalytic pyrolysis provides an alternative route to aromatic hydrocarbons, with product selectivity influenced by catalyst acidity/basicity and process parameters. However, challenges such as catalyst deactivation, coke formation, and dependence on noble metals continue to limit process scalability and sustainability. Consequently, future research should focus on developing atom-efficient catalysts based on earth-abundant elements, such as single-atom catalysts, through cost-effective synthesis strategies, to advance scalable, energy-efficient, and economically viable PET chemical recycling technologies.
Further research should focus on developing efficient dechlorination methods, such as catalysts or solvents capable of working under mild conditions. Innovations in closed-loop recycling that convert PVC into reusable monomers, along with safer additives and design-for-recycling approaches, are also critical.
Despite recent progress, industrial-scale implementation of BPA-PC recycling faces key challenges. Methanolysis, while effective in yielding BPA and DMC, requires energy-intensive separation due to DMC's low boiling point. Ethanolysis offers some improvements, though azeotrope formation still complicates purification. Aminolysis produces diverse products that demand complex extraction or chromatographic steps. To overcome these limitations, future research should explore advanced techniques such as membrane separation and continuous flow systems to enhance separation efficiency and minimise product cross-contamination. Future research must focus on low-energy, cost-effective depolymerisation using recyclable heterogeneous catalysts, addressing both catalytic efficiency and downstream purification to enable scalable and sustainable BPA-PC recycling.
Despite progress, PU recycling faces significant challenges due to the material's structural complexity, ranging from variability in chemical composition and crosslinking density to diverse product forms such as foams, elastomers, and bulk materials. This diversity necessitates tailored recycling approaches rather than a universal solution. Less mature methods, including pyrolysis, gasification, and biodegradation, remain confined to the laboratory stage, hindered by emission control, long degradation times, and limited microbial efficacy. Moving forward, targeted strategies such as selective waste collection, advanced process optimisation, and metagenomic tools to enhance biodegradation hold promise. To enable widespread adoption, future efforts must focus on improving scalability, reducing environmental impact, and establishing a structured market for PU recyclates, thereby supporting circular economy integration.
Despite the diversity of chemical recycling methods, most require harsh reaction conditions, including high temperatures, elevated pressures, or strong reagents, which limits their industrial scalability. Biological degradation, although environmentally benign, is constrained by the limited availability of microorganisms or enzymes capable of depolymerising high-molecular-weight polyamides. Enzymatic processes are limited to surface degradation or low-molecular-weight substrates. To address these challenges, future research must focus on developing mild, energy-efficient recycling methods; engineering enzymes capable of depolymerising bulk polyamides; and designing bio-based or copolymerised polyamides with intrinsic biodegradability. These innovations will be critical in shifting the lifecycle of polyamides toward a circular and sustainable model.
Data availability
No primary research results, software or code has been included and no new data were generated or analysed as part of this review.Author contributions
M. Anwar and S. Dastgir created the concept, acquired funding, managed the project, carried out statistic data curation, wrote the original draft, reviewed and edited the final manuscript. M. E. Konnova acquired data, wrote sections of the manuscript, reviewed and edited the final draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Qatar National Research Fund (QNRF) for financial support (NPRP14S-0406-210154). Fig. 4–7 and the graphical abstract image were generated using iStock AI Image Generator.References
- M. K. Eriksen, K. Pivnenko, G. Faraca, A. Boldrin and T. F. Astrup, Environ. Sci. Technol., 2020, 54, 16166–16175 CrossRef CAS PubMed.
- Z. Tang, W. Li, V. W. Y. Tam and C. Xue, Resour. Conserv. Recycl. X, 2020, 6, 100036 Search PubMed.
- M. C. Heller, M. H. Mazor and G. A. Keoleian, Environ. Res. Lett., 2020, 15, 094034 CrossRef CAS.
- G. Bishop, D. Styles and P. N. L. Lens, Environ. Int., 2020, 142, 105893 CrossRef CAS PubMed.
- Statista, Annual production of plastics worldwide from 1950 to 2022, https://www.statista.com/statistics/282732/global-production-of-plastics-since-1950/, accessed 19 September, 2023.
- Plastics – the Facts 2021, An Analysis of European Plastics Production, Demand and Waste Data, 2021 Search PubMed.
- B. C. Gibb, Nat. Chem., 2019, 11, 394–395 CrossRef CAS PubMed.
- M. Jaganmohan, Plastic production forecast worldwide 2025–2050, Statista Research Department, 2023, https://www.statista.com/statistics/664906/plastics-production-volume-forecast-worldwide/ Search PubMed.
- J. Zheng and S. Suh, Nat. Clim. Change, 2019, 9, 374–378 CrossRef.
- PlasticsEurope, Plastics – the Facts 2022, Plastics Europe, 2022 Search PubMed.
- C. Areeprasert, J. Asingsamanunt, S. Srisawat, J. Kaharn, B. Inseemeesak, P. Phasee, C. Khaobang, W. Siwakosit and C. Chiemchaisri, Energy Procedia, 2017, 107, 222–226 CrossRef CAS.
- G. Leone, J. M. Borras and E. D. V. Martin, The New Plastic Economy: Rethinking the Future of Plastics & Catalysing Action, Ellen MacArthur Foundation, 2017 Search PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv. Mater., 2017, 3, e1700782 Search PubMed.
- A. J. Martín, C. Mondelli, S. D. Jaydev and J. Pérez-Ramírez, Chem, 2021, 7, 1487–1533 Search PubMed.
- D. E. MacArthur, D. Waughray and M. R. Stuchtey, The New Plastic Economy: Rethinking the Future of Plastics, 2016 Search PubMed.
- N. J. Themelis and C. Mussche, 2014 Energy and Economic Value of Municipal Solid Waste (MSW), Including Non-Recycled Plastics (NRP), Currently Landfilled in the Fifty States, Earth Engineering Center, Columbia University, 2014 Search PubMed.
- J. Raynaud, Valuing Plastic: the Business Case for Measuring Managing and Disclosing Plastic Use in the Consumer Goods Industry, 2014 Search PubMed.
- C. M. Rochman, M. A. Browne, B. S. Halpern, B. T. Hentschel, E. Hoh, H. K. Karapanagioti, L. M. Rios-Mendoza, H. Takada, S. Teh and R. C. Thompson, Nature, 2013, 494, 169–171 CrossRef CAS PubMed.
- OECD, Global Plastic Outllook: Economic Drivers, Environmental Impacts and Policy Options, 2022 Search PubMed.
- SYSTEMIQ, ReShaping Plastics: Pathways to a Circular, Climate Neutral Plastics System in Europe, 2022 Search PubMed.
- Material Economics. The Circular Economy – a Powerful Force for Climate Mitigation, 2018 Search PubMed.
- U. R. Gracida-Alvarez, P. T. Benavides, U. Lee and M. Wang, J. Cleaner Prod., 2023, 425, 138867 CrossRef CAS.
- K. R. Vanapalli, H. B. Sharma, V. P. Ranjan, B. Samal, J. Bhattacharya, B. K. Dubey and S. Goel, Sci. Total Environ., 2021, 750, 141514 CrossRef CAS PubMed.
- J. Sills and T. M. Adyel, Science, 2020, 369, 1314–1315 CrossRef PubMed.
- M. Ilyas, W. Ahmad, H. Khan, S. Yousaf, K. Khan and S. Nazir, Rev. Environ. Health, 2018, 33, 383–406 CrossRef PubMed.
- J. M. Garcia and M. L. Robertson, Science, 2017, 358, 870–872 CrossRef CAS PubMed.
- J. N. Hahladakis, C. A. Velis, R. Weber, E. Iacovidou and P. Purnell, J. Hazard. Mater., 2018, 344, 179–199 CrossRef CAS PubMed.
- European Commission, European Strategy for Plastics in a Circular Economy, Communication from the Commission to the European Parliament, The Council, the European Economic and Social Committee and the Committee of the Regions, 2018, Secretariat-GeneralLink to the https://eur-lex.europa.eu/legal-content/EN/ALL/?uri=CELEX:52018DC0028A Search PubMed.
- F. Zhang, Y. Zhao, D. Wang, M. Yan, J. Zhang, P. Zhang, T. Ding, L. Chen and C. Chen, J. Cleaner Prod., 2021, 282, 124523 CrossRef CAS.
- T. Thiounn and R. C. Smith, J. Polym. Sci., 2020, 58, 1347–1364 CrossRef CAS.
- H. Ritchie, V. Samborska and M. Roser, Plastic Pollution-Our World in Data, https://ourworldindata.org/plastic-pollution.
- A. G. J. Driedger, H. H. Dürr, K. Mitchell and P. Van Cappellen, J. Great Lakes Res., 2015, 41, 9–19 CrossRef CAS.
- R. C. P. Monteiro, J. A. Ivar do Sul and M. F. Costa, Environ. Pollut., 2018, 238, 103–110 CrossRef CAS PubMed.
- C. V. Eyzaguirre, D. V. Eugui, H. Pacini, D. P. Rial, K. Spur and K. Attafuah-Wadee, Presented in Part at the United Nations Conference on Trade and Development, Geneva, Switzerland, 2023 Search PubMed.
- M. Allsopp, A. Walters, D. Santillo and P. Johnston, Plastic Debris in the World's Oceans, 2006 Search PubMed.
- Plastic Debris in the Ocean: The Characterization of Marine Plastics and their Environmental Impacts, Situation Analysis Report, ed. F. Thevenon, C. Carroll and J. Sousa, IUCN, Gland, Switzerland, 2014, p. 52 Search PubMed.
- L. A. Hamilton, S. Feit, C. Muffett, M. Kelso, S. M. Rubright, C. Bernhardt, E. Schaeffer, D. Moon, J. Morris and R. Labbe-Bellas, Plastic & Climate: The Hidden Costs of a Plastic Planet, 2019 Search PubMed.
- E. J. Carpenter and K. L. Smith, Science, 1972, 175, 1240–1241 CrossRef CAS PubMed.
- H. S. Auta, C. U. Emenike and S. H. Fauziah, Environ. Int., 2017, 102, 165–176 CrossRef CAS PubMed.
- A. L. Andrady, Mar. Pollut. Bull., 2011, 62, 1596–1605 CrossRef CAS PubMed.
- K. T. T. Amesho, C. Chinglenthoiba, M. S. A. B. Samsudin, M. N. Lani, A. Pandey, M. N. M. Desa and V. Suresh, J. Environ. Manage., 2023, 344, 118713 CrossRef CAS PubMed.
- D. Yang, H. Shi, L. Li, J. Li, K. Jabeen and P. Kolandhasamy, Environ. Sci. Technol., 2015, 49, 13622–13627 CrossRef CAS PubMed.
- S. Chatterjee and S. Sharma, Field Actions Sci. Rep., 2019, 19, 54–61 Search PubMed.
- U. Subaramaniyam, R. Allimuthu, S. Vappu, D. Ramalingam, R. B, B. Paital, N. Panda, P. K. Rath, N. Ramalingam and D. Sahoo, Front. Physiol., 2023, 14, 1217666 CrossRef PubMed.
- Y. Gao and Y. Liu, J. Environ. Chem. Eng., 2022, 10, 108197 CrossRef CAS.
- S. C. Finney and L. E. Edwards, GSA Today, 2016, 29, 3–4 Search PubMed.
- R. C. Thompson, C. J. Moore, F. S. vom Saal and S. H. Swan, Philos. Trans. R. Soc., B, 2009, 364, 2153–2166 CrossRef CAS PubMed.
- J. Oehlmann, U. Schulte-Oehlmann, W. Kloas, O. Jagnytsch, I. Lutz, K. O. Kusk, L. Wollenberger, E. M. Santos, G. C. Paull, K. J. W. Van Look and C. R. Tyler, Philos. Trans. R. Soc., B, 2009, 364, 2047–2062 CrossRef CAS PubMed.
- M. M. A. Allouzi, D. Y. Y. Tang, K. W. Chew, J. Rinklebe, N. Bolan, S. M. A. Allouzi and P. L. Show, Sci. Total Environ., 2021, 788, 147815 CrossRef CAS PubMed.
- T. Zhao, Y. M. Lozano and M. C. Rillig, Front. Environ. Sci., 2021, 9, 675803 CrossRef.
- United Nations, Department of Economic and Social Affairs, Population Division, Word Population Prospects 2019: Highlights (ST/ESA.SER.A/423), 2019 Search PubMed.
- R. C. Valencia, The Future of the Chemical Industry by 2050, Wiley-VCH Verlag GmbH & Co, 2013 Search PubMed.
- S. J. Bennett, in Handbook of Climate Change Mitigation, ed. W.-Y. Chen, J. Seiner, T. Suzuki and M. Lackner, Springer US, New York, NY, 2012, pp. 319–357, DOI:10.1007/978-1-4419-7991-9_10.
- M. Gordon, Sustainability shift: Oil's future in the energy transition, S&P Global Platts, 2020 Search PubMed.
- R. Miandad, M. Rehan, A.-S. Nizami, M. A. El-Fetouh Barakat and I. M. Ismail, in Recycling of Solid Waste for Biofuels and Bio-Chemicals, ed. O. P. Karthikeyan, K. Heimann and S. S. Muthu, Springer Singapore, Singapore, 2016, pp. 333–355, DOI:10.1007/978-981-10-0150-5_12.
- S. Evangelisti, P. Lettieri, D. Borello and R. Clift, Waste Manage., 2014, 34, 226–237 CrossRef CAS PubMed.
- A. F. Pales, P. Levi, S. Bennett, J. Elliott, T.-Y. Kim, K. Petrosyan, J. Ritchie, A. V. Bohemen, T. Vass, M. A. Walton and K. West, The Future of Petrochemicals: Towards more sustainable plastics and fertilisers, 2018 Search PubMed.
- E. M. Foundation, New Plastics Economy Global Commitment, 2020 Search PubMed.
- J. A. Du, Pisani Professor of History, Environ. Sci., 2006, 3, 83–96 Search PubMed.
- V. Sima, I. G. Gheorghe, J. Subić and D. Nancu, Sustainability, 2020, 120, 1–28 Search PubMed.
- G. Michelini, R. N. Moraes, R. N. Cunha, J. M. H. Costa and A. R. Ometto, Proced. CIRP, 2017, 64, 2–6 CrossRef.
- J. Aguado, D. P. Serrano, M. J. Braithwaite, S. M. Hassur and T. Papenfuhs, Feedstock Recycling of Plastic Wastes, The Royal Society of Chemistry, Cambridge, UK, 1999 Search PubMed.
- T. Hundertmark, M. Mayer, C. McNally, T. J. Simons and C. Witte, How plastics waste recycling could transform the chemical industry, https://www.mckinsey.com/industries/chemicals/our-insights/, accessed 14 December, 2023.
- D. A. Tsiamis and M. J. Castaldi, Determining Accurate Heating Values of Nonrecycled Plastics (NRP), American Chemistry Council, 2016 Search PubMed.
- H. Sardon and A. P. Dove, Science, 2018, 360, 380–381 CrossRef CAS PubMed.
- S. D. Anuar Sharuddin, F. Abnisa, W. M. A. Wan Daud and M. K. Aroua, Energy Convers. Manage., 2017, 148, 925–934 CrossRef CAS.
- K. Kaiser, M. Schmid and M. Schlummer, Recycling, 2018, 3, 1 CrossRef.
- Z. O. G. Schyns and M. P. Shaver, Macromol. Rapid Commun., 2021, 42, 2000415 CrossRef CAS PubMed.
- S. Kumar, A. K. Panda and R. K. Singh, Resour. Conserv. Recycl., 2011, 55, 893–910 CrossRef.
- M. D. Jagtap, S. Khatavkar and T. Quazi, Int. J. Eng. Technol. Mech. Electr. Eng., 2015, 2, 120–122 Search PubMed.
- D. E. MacArthur, Science, 2017, 358, 843 CrossRef CAS PubMed.
- C. Mohanraj, T. Senthilkumar and M. Chandrasekar, Int. J. Energy Res., 2017, 41, 1534–1552 CrossRef CAS.
- S. L. Wong, N. Ngadi, T. A. T. Abdullah and I. M. Inuwa, Renewable Sustainable Energy Rev., 2015, 50, 1167–1180 CrossRef CAS.
- C. I. Idumah, J. Therm. Anal. Calorim., 2022, 147, 3495–3508 CrossRef CAS.
- K. M. Rajendran, V. Chintala, S. Amit, S. Pal, J. K. Pandey and P. Ghodke, Mater. Today Commun., 2020, 24, 100982 CrossRef.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- S. H. Park and S. H. Kim, Fash. Text., 2014, 1, 1 CrossRef.
- A. Lee and M. S. Liew, J. Mater. Cycles Waste Manage., 2021, 23, 32–43 CrossRef.
- M. Okan, H. M. Aydin and M. Barsbay, J. Chem. Technol. Biotechnol., 2019, 94, 8–21 CrossRef CAS.
- Q. Li, A. Faramarzi, S. Zhang, Y. Wang, X. Hu and M. Gholizadeh, Energy Convers. Manage., 2020, 226, 113525 CrossRef CAS.
- H. K. Webb, J. Arnott, R. J. Crawford and E. P. Ivanova, Polymers, 2013, 5, 1–18 CrossRef.
- S. M. Al-Salem, A. Antelava, A. Constantinou, G. Manos and A. Dutta, J. Environ. Manage., 2017, 197, 177–198 CrossRef CAS PubMed.
- K.-H. Lee, S.-G. Jeon, K.-H. Kim, N.-S. Noh, D.-H. Shin, J. Park, Y. Seo, J.-J. Yee and G.-T. Kim, Korean J. Chem. Eng., 2003, 20, 693–697 CrossRef CAS.
- G. Lopez, M. Artetxe, M. Amutio, J. Bilbao and M. Olazar, Renewable Sustainable Energy Rev., 2017, 73, 346–368 CrossRef CAS.
- A. Chaala, H. Darmstadt and C. Roy, J. Anal. Appl. Pyrolysis, 1997, 39, 79–96 CrossRef CAS.
- M. Arabiourrutia, G. Elordi, G. Lopez, E. Borsella, J. Bilbao and M. Olazar, J. Anal. Appl. Pyrolysis, 2012, 94, 230–237 CrossRef CAS.
- M. Predel and W. Kaminsky, Polym. Degrad. Stab., 2000, 70, 373–385 CrossRef CAS.
- B.-S. Kang, S. G. Kim and J.-S. Kim, J. Anal. Appl. Pyrolysis, 2008, 81, 7–13 CrossRef CAS.
- W. Kaminsky, M. Predel and A. Sadiki, Polym. Degrad. Stab., 2004, 85, 1045–1050 CrossRef CAS.
- D. P. Serrano, J. Aguado, J. M. Escola, E. Garagorri, x Rodrı, J. M. guez, L. Morselli, G. Palazzi and R. Orsi, Appl. Catal., B, 2004, 49, 257–265 CrossRef CAS.
- P. Kasar, D. K. Sharma and M. Ahmaruzzaman, J. Cleaner Prod., 2020, 265, 121639 CrossRef CAS.
- K. Ragaert, L. Delva and K. Van Geem, Waste Manage., 2017, 69, 24–58 CrossRef CAS PubMed.
- M. Roosen, N. Mys, M. Kusenberg, P. Billen, A. Dumoulin, J. Dewulf, K. M. Van Geem, K. Ragaert and S. De Meester, Environ. Sci. Technol., 2020, 54, 13282–13293 CrossRef CAS PubMed.
- K. Kleinhans, M. Hallemans, S. Huysveld, G. Thomassen, K. Ragaert, K. M. Van Geem, M. Roosen, N. Mys, J. Dewulf and S. De Meester, Waste Manage., 2021, 120, 290–302 CrossRef PubMed.
- S. Serranti and G. Bonifazi, in Use of Recycled Plastics in Eco-Efficient Concrete, ed. F. Pacheco-Torgal, J. Khatib, F. Colangelo and R. Tuladhar, Woodhead Publishing, 2019, pp. 9–37, DOI:10.1016/B978-0-08-102676-2.00002-5.
- H. Wang, Y. Zhang and C. Wang, Sep. Purif. Technol., 2019, 226, 75–94 CrossRef CAS.
- E. Dijkgraaf and R. Gradus, Environ. Resour. Econ., 2020, 77, 127–142 CrossRef.
- G. Wu, J. Li and Z. Xu, Waste Manage., 2013, 33, 585–597 CrossRef PubMed.
- T. Sykes, Sorting the Plastic Recycling Problem, https://packagingeurope.com/sorting-the-plastic-recycling-problem/6666.article, accessed 11 March, 2024.
- M. Naitove, Digital Watermarking Consortium Advances Technology for Sorting & Recycling Plastics, https://www.ptonline.com/news/digital-watermarking-consortium-advances-technology-for-sorting-recycling-plastics.
- C. Pahl, How Machine Learning and Robotics are Solving the Plastic Sorting Crisis, https://www.plugandplaytechcenter.com/insights.
- M. Strangl, E. Ortner, T. Fell, T. Ginzinger and A. Buettner, J. Cleaner Prod., 2020, 260, 121104 CrossRef CAS.
- D. Xia and F.-S. Zhang, J. Cleaner Prod., 2018, 171, 1472–1480 CrossRef CAS.
- V. K. Soni, G. Singh, B. K. Vijayan, A. Chopra, G. S. Kapur and S. S. V. Ramakumar, Energy Fuels, 2021, 35, 12763–12808 CrossRef CAS.
- R. Miandad, M. A. Barakat, A. S. Aburiazaiza, M. Rehan and A. S. Nizami, Process Saf. Environ. Prot., 2016, 102, 822–838 CrossRef CAS.
- F. Obeid, J. Zeaiter, A. a. H. Al-Muhtaseb and K. Bouhadir, Energy Convers. Manage., 2014, 85, 1–6 CrossRef CAS.
- I. Velghe, R. Carleer, J. Yperman and S. Schreurs, J. Anal. Appl. Pyrolysis, 2011, 92, 366–375 CrossRef CAS.
- G. Manos, I. Y. Yusof, N. H. Gangas and N. Papayannakos, Energy Fuels, 2002, 16, 485–489 CrossRef CAS.
- G. Fadillah, I. Fatimah, I. Sahroni, M. M. Musawwa, T. M. Mahlia and O. Muraza, Catalysts, 2021, 11, 837 CrossRef CAS.
- R. Mishra, A. Kumar, E. Singh and S. Kumar, ACS Sustainable Chem. Eng., 2023, 11, 2033–2049 CrossRef CAS.
- B. Singh and N. Sharma, Polym. Degrad. Stab., 2008, 93, 561–584 CrossRef CAS.
- C. Wu and P. T. Williams, Int. J. Hydrogen Energy, 2010, 35, 949–957 CrossRef CAS.
- K.-B. Park, M.-J. Choi, D.-Y. Chae, J. Jung and J.-S. Kim, Energy, 2022, 244, 122583 CrossRef CAS.
- J. M. Saad and P. T. Williams, Energy Fuels, 2016, 30, 3198–3204 CrossRef CAS.
- S. V. Papuga, P. M. Gvero and L. M. Vukić, Therm. Sci., 2016, 20, 731–774 CrossRef.
- M. S. Renzini, L. C. Lerici, U. Sedran and L. B. Pierella, J. Anal. Appl. Pyrolysis, 2011, 92, 450–455 CrossRef CAS.
- M. S. Renzini, U. Sedran and L. B. Pierella, J. Anal. Appl. Pyrolysis, 2009, 86, 215–220 CrossRef CAS.
- S. M. Al-Salem and A. Dutta, Ind. Eng. Chem. Res., 2021, 60, 8301–8309 CrossRef CAS.
- R. Bagri and P. T. Williams, J. Anal. Appl. Pyrolysis, 2002, 63, 29–41 CrossRef CAS.
- S.-H. Jung, M.-H. Cho, B.-S. Kang and J.-S. Kim, Fuel Process. Technol., 2010, 91, 277–284 CrossRef CAS.
- S. D. Anuar Sharuddin, F. Abnisa, W. M. A. Wan Daud and M. K. Aroua, Energy Convers. Manage., 2016, 115, 308–326 CrossRef CAS.
- M. S. Qureshi, A. Oasmaa, H. Pihkola, I. Deviatkin, A. Tenhunen, J. Mannila, H. Minkkinen, M. Pohjakallio and J. Laine-Ylijoki, J. Anal. Appl. Pyrolysis, 2020, 152, 104804 CrossRef CAS.
- P. N. Sharratt, Y. H. Lin, A. A. Garforth and J. Dwyer, Ind. Eng, Ind. Eng. Chem. Res., 1997, 36, 5118–5124 CrossRef CAS.
- G. Luo, T. Suto, S. Yasu and K. Kato, Polym. Degrad. Stab., 2000, 70, 97–102 CrossRef CAS.
- Y. H. Lin and H. Y. Yen, Polym. Degrad. Stab., 2005, 89, 101–108 CrossRef CAS.
- M. L. Mastellone, F. Perugini, M. Ponte and U. Arena, Polym. Degrad. Stab., 2002, 76, 479–487 CrossRef CAS.
- F. J. Mastral, E. Esperanza, P. García and M. Juste, J. Anal. Appl. Pyrolysis, 2002, 63, 1–15 CrossRef CAS.
- J. F. Mastral, C. Berrueco, M. Gea and J. Ceamanos, Polym. Degrad. Stab., 2006, 91, 3330–3338 CrossRef CAS.
- A. Miriam, E. Gorka, O. Martin and B. Javier, in Pyrolysis, ed. S. Mohamed, IntechOpen, Rijeka, 2017, ch. 12, DOI:10.5772/67706.
- H. S. Fogler, Elements of Chemical Reaction Engineering, Pearson Education Incorporated, 4th edn, 2010 Search PubMed.
- D. Czajczyńska, L. Anguilano, H. Ghazal, R. Krzyżyńska, A. J. Reynolds, N. Spencer and H. Jouhara, Therm. Sci. Eng. Prog., 2017, 3, 171–197 CrossRef.
- G. Elordi, M. Olazar, G. Lopez, M. Amutio, M. Artetxe, R. Aguado and J. Bilbao, J. Anal. Appl. Pyrolysis, 2009, 85, 345–351 CrossRef CAS.
- G. Elordi, M. Olazar, P. Castaño, M. Artetxe and J. Bilbao, Ind. Eng. Chem. Res., 2012, 51, 14008–14017 CrossRef CAS.
- G. Elordi, M. Olazar, G. Lopez, M. Artetxe and J. Bilbao, Ind. Eng. Chem. Res., 2011, 50, 6650–6659 CrossRef CAS.
- M. Artetxe, G. Lopez, M. Amutio, G. Elordi, J. Bilbao and M. Olazar, Ind. Eng. Chem. Res., 2013, 52, 10637–10645 CrossRef CAS.
- Y. Zhang, G. Ji, C. Chen, Y. Wang, W. Wang and A. Li, Fuel Process. Technol., 2020, 206, 106455 CrossRef CAS.
- W. Kaminsky and J.-S. Kim, J. Anal. Appl. Pyrolysis, 1999, 51, 127–134 CrossRef CAS.
- E. Butler, G. Devlin and K. McDonnell, Waste Biomass Valorization, 2011, 2, 227–255 CrossRef CAS.
- J. Scheirs and W. Kaminsky, Feedstock Recycling and Pyrolysis of Waste Plastics: Converting Waste Plastics into Diesel and Other Fuels, John Wiley & Sons, Ltd, 2006 Search PubMed.
- S. S. Lam and H. A. Chase, Energies, 2012, 5, 4209–4232 CrossRef CAS.
- F. Motasemi and M. T. Afzal, Renewable Sustainable Energy Rev., 2013, 28, 317–330 CrossRef CAS.
- S. Anis, L. Shahadati, W. Sumbodo and Wahyudi, AIP Conf. Proc., 2017, 1818, 020003 CrossRef.
- C. Ludlow-Palafox and H. A. Chase, Ind. Eng. Chem. Res., 2001, 40, 4749–4756 CrossRef CAS.
- P. H. M. Putra, S. Rozali, M. F. A. Patah and A. Idris, J. Environ. Manage., 2022, 303, 114240 CrossRef CAS PubMed.
- A. Domínguez, Y. Fernández, B. Fidalgo, J. J. Pis and J. A. Menéndez, Chemosphere, 2008, 70, 397–403 CrossRef PubMed.
- Z. Song, Y. Yang, X. Zhao, J. Sun, W. Wang, Y. Mao and C. Ma, J. Anal. Appl. Pyrolysis, 2017, 123, 152–159 CrossRef CAS.
- X. Jing, Y. Zhao, H. Wen and Z. Xu, Energy Fuels, 2017, 31, 2052–2062 CrossRef CAS.
- S. Hassanzadeh, N. Aminlashgari and M. Hakkarainen, ACS Sustainable Chem. Eng., 2015, 3, 177–185 CrossRef CAS.
- A. Undri, L. Rosi, M. Frediani and P. Frediani, J. Anal. Appl. Pyrolysis, 2014, 110, 55–65 CrossRef CAS.
- E. Khaghanikavkani, M. M. Farid, J. Holdem and A. Williamson, J. Chem. Eng. Process Technol., 2013, 4, 150 Search PubMed.
- Y. Fernández and J. A. Menéndez, J. Anal. Appl. Pyrolysis, 2011, 91, 316–322 CrossRef.
- J. Fan, P. S. Shuttleworth, M. Gronnow, S. W. Breeden, J. H. Clark, D. J. Macquarrie and V. L. Budarin, ACS Sustainable Chem. Eng., 2018, 6, 2916–2920 CrossRef CAS.
- D. V. Suriapparao, R. Vinu, A. Shukla and S. Haldar, Bioresour. Technol., 2020, 302, 122775 CrossRef CAS PubMed.
- A. Undri, L. Rosi, M. Frediani and P. Frediani, in Microwave Heating, ed. U. Chandra, 2011, p. 384, DOI:10.5772/825.
- K. Ding, S. Liu, Y. Huang, S. Liu, N. Zhou, P. Peng, Y. Wang, P. Chen and R. Ruan, Energy Convers. Manage., 2019, 196, 1316–1325 CrossRef CAS.
- S. M. R. Mirkarimi, S. Bensaid and D. Chiaramonti, Appl. Energy, 2022, 327, 120040 CrossRef CAS.
- P. D. Muley, C. Henkel, K. K. Abdollahi and D. Boldor, Energy Fuels, 2015, 29, 7375–7385 CrossRef CAS.
- Y. Xue, Y. Zhou, J. Liu, Y. Xiao and T. Wang, Waste Manage., 2021, 120, 513–521 CrossRef CAS PubMed.
- W. M. Lewandowski, K. Januszewicz and W. Kosakowski, J. Anal. Appl. Pyrolysis, 2019, 140, 25–53 CrossRef CAS.
- H. S. Fogler, Essentials of Chemical Reaction Engineering, Pearson Education Inc., 2011 Search PubMed.
- M. S. Abbas-Abadi, M. N. Haghighi and H. Yeganeh, Fuel Process. Technol., 2013, 109, 90–95 CrossRef CAS.
- K.-B. Park, Y.-S. Jeong, B. Guzelciftci and J.-S. Kim, Appl. Energy, 2020, 259, 114240 CrossRef CAS.
- J. M. Saad and P. T. Williams, Waste Manage., 2016, 58, 214–220 CrossRef CAS PubMed.
- D. Chen, L. Yin, H. Wang and P. He, Waste Manage., 2014, 34, 2466–2486 CrossRef CAS PubMed.
- P. M. Witt and D. A. Hickman, AIChE J., 2022, 68, e17803 CrossRef CAS.
- W. Kaminsky, Fuel Commun., 2021, 8, 100023 CrossRef.
- H. Jouhara, D. Ahmad, I. van den Boogaert, E. Katsou, S. Simons and N. Spencer, Therm. Sci. Eng. Prog., 2018, 5, 117–143 CrossRef.
- V. K. Guda, P. H. Steele, V. K. Penmetsa and Q. Li, in Recent Advances in Thermo-Chemical Conversion of Biomass, ed. A. Pandey, T. Bhaskar, M. Stöcker and R. K. Sukumaran, Elsevier, Boston, 2015, pp. 177–211, DOI:10.1016/B978-0-444-63289-0.00007-7.
- M. I. Jahirul, M. G. Rasul, A. A. Chowdhury and N. Ashwath, Energies, 2012, 5, 4952–5001 CrossRef CAS.
- D. S. Achilias, C. Roupakias, P. Megalokonomos, A. A. Lappas and E. V. Antonakou, J. Hazard. Mater., 2007, 149, 536–542 CrossRef CAS PubMed.
- A. K. Panda, R. K. Singh and D. K. Mishra, Renewable Sustainable Energy Rev., 2010, 14, 233–248 CrossRef CAS.
- H. Ke, T. Li-hua, Z. Zi-bin and Z. Cheng-fang, Polym. Degrad. Stab., 2005, 89, 312–316 CrossRef.
- M. Chandran, S. Tamilkolundu and C. Murugesan, in Plastic Waste and Recycling, ed. T. M. Letcher, Academic Press, 2020, pp. 385–399, DOI:10.1016/B978-0-12-817880-5.00014-1.
- S. Kumar, E. Singh, R. Mishra, A. Kumar and S. Caucci, ChemSusChem, 2021, 14, 3985–4006 CrossRef CAS PubMed.
- N. Miskolczi, F. Ateş and N. Borsodi, Bioresour. Technol., 2013, 144, 370–379 CrossRef CAS PubMed.
- A. Lopez-Urionabarrenechea, I. de Marco, B. M. Caballero, M. F. Laresgoiti and A. Adrados, J. Anal. Appl. Pyrolysis, 2012, 96, 54–62 CrossRef CAS.
- M. Brebu, T. Bhaskar, K. Murai, A. Muto, Y. Sakata and M. A. Uddin, Polym. Degrad. Stab., 2005, 87, 225–230 CrossRef CAS.
- A. Marcilla, M. I. Beltrán and R. Navarro, Appl. Catal. B Environ., 2009, 86, 78–86 CrossRef CAS.
- M. A. Uddin, K. Koizumi, K. Murata and Y. Sakata, Polym. Degrad. Stab., 1997, 56, 37–44 CrossRef CAS.
- Y. Sonawane, M. Shindikar and M. Khaladkar, Int. J. Sci. Environ. Technol., 2016, 5, 1421–1425 Search PubMed.
- A. K. Panda and R. K. P. Singh, J. Fuel Chem. Technol., 2011, 39, 198–202 CrossRef CAS.
- S. Gopinath, P. K. Devan and K. Pitchandi, RSC Adv., 2020, 10, 37266–37279 RSC.
- M. L. Pennel, A. K. Maurya, A. M. Ebrahim, C. J. Tassone and M. Cargnello, ACS Sustainable Chem. Eng., 2023, 11, 12623–12630 CrossRef CAS.
- A. López, I. de Marco, B. M. Caballero, M. F. Laresgoiti, A. Adrados and A. Aranzabal, Appl. Catal., B, 2011, 104, 211–219 CrossRef.
- A. S. Nizami, O. K. M. Ouda, M. Rehan, A. M. O. El-Maghraby, J. Gardy, A. Hassanpour, S. Kumar and I. M. I. Ismail, Energy, 2016, 108, 162–171 CrossRef CAS.
- IZA-SC, Database of Zeolite Structures, https://izasc.fos.su.se/IZA-SC/ftc_table.php.
- A. A. Garforth, Y. H. Lin, P. N. Sharratt and J. Dwyer, Appl. Catal., A, 1998, 169, 331–342 CrossRef CAS.
- E. K. L. Morais, S. Jiménez-Sánchez, H. Hernando, C. Ochoa-Hernández, P. Pizarro, A. S. Araujo and D. P. Serrano, Ind. Eng. Chem. Res., 2019, 58, 6243–6254 CrossRef CAS.
- Y.-H. Seo, K.-H. Lee and D.-H. Shin, J. Anal. Appl. Pyrolysis, 2003, 70, 383–398 CrossRef CAS.
- M. del Remedio Hernández, Á. N. García and A. Marcilla, J. Anal. Appl. Pyrolysis, 2007, 78, 272–281 CrossRef.
- A. A. Ajibola, J. A. Omoleye and V. E. Efeovbokhan, Appl. Petrochem. Res., 2018, 8, 211–217 CrossRef CAS.
- J. V. Milato, R. J. França and M. R. C. Marques Calderari, J. Environ. Chem. Eng., 2020, 8, 103805 CrossRef CAS.
- A. A. P. Susastriawan, Purnomo and A. Sandria, Therm. Sci. Eng. Prog., 2020, 17, 100497 CrossRef.
- E. Erawati, H. Hamid and A. A. Ilma, Molekul, 2018, 13, 106–113 CrossRef CAS.
- Hendrawati, A. R. Liandi, M. a. Solehah, M. H. Setyono, I. Aziz and Y. D. I. Siregar, Case Stud. Chem. Environ. Eng., 2023, 7, 100290 CrossRef CAS.
- K. Sivagami, K. V. Kumar, P. Tamizhdurai, D. Govindarajan, M. Kumar and I. Nambi, RSC Adv., 2022, 12, 7612–7620 RSC.
- M. Rehan, R. Miandad, M. A. Barakat, I. M. I. Ismail, T. Almeelbi, J. Gardy, A. Hassanpour, M. Z. Khan, A. Demirbas and A. S. Nizami, Int. Biodeterior. Biodegrad., 2017, 119, 162–175 CrossRef CAS.
- N. Zhou, L. Dai, Y. Lyu, Y. Wang, H. Li, K. Cobb, P. Chen, H. Lei and R. Ruan, Chem. Eng. J., 2022, 440, 135836 CrossRef CAS.
- D. P. Serrano, J. Aguado, J. M. Escola and J. M. Rodríguez, in Studies in Surface Science and Catalysis, ed. R. Aiello, G. Giordano and F. Testa, Elsevier, 2002, vol. 142, pp. 77–84 Search PubMed.
- D. P. Serrano, J. Aguado, J. M. Escola and E. Garagorri, Appl. Catal., B, 2003, 44, 95–105 CrossRef CAS.
- J. Aguado, J. L. Sotelo, D. P. Serrano, J. A. Calles and J. M. Escola, Energy Fuels, 1997, 11, 1225–1231 CrossRef CAS.
- S. Colantonio, L. Cafiero, D. De Angelis, N. M. Ippolito, R. Tuffi and S. V. Ciprioti, Front. Chem. Sci. Eng., 2020, 14, 288–303 CrossRef CAS.
- T. Liu, Y. Li, Y. Zhou, S. Deng and H. Zhang, Catalysts, 2023, 13, 382 CrossRef CAS.
- S. L. Wong, S. Armenise, B. B. Nyakuma, A. Bogush, S. Towers, C. H. Lee, K. Y. Wong, T. H. Lee, E. Rebrov and M. Muñoz, J. Anal. Appl. Pyrolysis, 2023, 169, 105793 CrossRef CAS.
- E. T. C. Vogt and B. M. Weckhuysen, Chem. Soc. Rev., 2015, 44, 7342–7370 RSC.
- C. R. Marcilly, Top. Catal., 2000, 13, 357–366 CrossRef CAS.
- J. A. Onwudili, C. Muhammad and P. T. Williams, J. Energy Inst., 2019, 92, 1337–1347 CrossRef CAS.
- E. T. Aisien, I. C. Otuya and F. A. Aisien, Environ. Technol. Innovation, 2021, 22, 101455 CrossRef CAS.
- F. Aisien and E. Aisien, Detritus, 2022, 19, 75–83 CrossRef.
- S. Orozco, L. Santamaria, M. Artetxe, J. Alvarez, J. Bilbao, M. Olazar and G. Lopez, Chem. Eng. J., 2023, 472, 144947 CrossRef CAS.
- H. Zhang, J. Nie, R. Xiao, B. Jin, C. Dong and G. Xiao, Energy Fuels, 2014, 28, 1940–1947 CrossRef CAS.
- S. Streiff, M. Piccinini and A. Corma, WO2017103018A1, 2017.
- D. K. Ratnasari, M. A. Nahil and P. T. Williams, J. Anal. Appl. Pyrolysis, 2017, 124, 631–637 CrossRef CAS.
- M. Genç, A. Gül, S. Avcilar, B. Öztürk, M. F. Önen and E. Akkuş, Hydrocarbon Process., 2019, 1–14 Search PubMed.
- K. Akubo, M. A. Nahil and P. T. Williams, J. Energy Inst., 2019, 92, 195–202 CrossRef CAS.
- S. Streiff, M. Piccinini, A. Corma, M. Cerro-Alarcon and J. Mengual, WO2017103010A1, 2017.
- Y. Wang, L. Cheng, J. Gu, Y. Zhang, J. Wu, H. Yuan and Y. Chen, ACS Omega, 2022, 7, 2752–2765 CrossRef CAS PubMed.
- T. Nandakumar, U. Dwivedi, K. K. Pant, S. Kumar and E. Balaraman, Catal. Today, 2023, 408, 111–126 CrossRef CAS.
- M. R. Yousefi, S. Rahimi and M. Rostamizadeh, J. Anal. Appl. Pyrolysis, 2021, 156, 105108 CrossRef CAS.
- S. A. H. Seyed Mousavi, S. M. Sadrameli and A. H. Saeedi Dehaghani, Process Saf. Environ. Prot., 2022, 164, 449–467 CrossRef CAS.
- S. A. H. S. Mousavi and A. H. S. Dehaghani, Energy Convers. Manage., 2024, 299, 117825 CrossRef CAS.
- Y. Zang, J. Wang, J. Gu, J. Qu, F. Gao and M. Li, J. Solid State Chem., 2020, 291, 121643 CrossRef CAS.
- M. J. B. de Souza, T. H. A. Silva, T. R. S. Ribeiro, A. O. S. da Silva and A. M. G. Pedrosa, J. Therm. Anal. Calorim., 2020, 140, 167–175 CrossRef CAS.
- Z. Li, Z. Zhong, B. Zhang, W. Wang, G. V. S. Seufitelli and F. L. P. Resende, Waste Manage., 2020, 102, 561–568 CrossRef CAS PubMed.
- Y. Zhang, D. Duan, H. Lei, E. Villota and R. Ruan, Appl. Energy, 2019, 251, 113337 CrossRef CAS.
- E. Huo, H. Lei, C. Liu, Y. Zhang, L. Xin, Y. Zhao, M. Qian, Q. Zhang, X. Lin, C. Wang, W. Mateo, E. M. Villota and R. Ruan, Sci. Total Environ., 2020, 727, 138411 CrossRef CAS PubMed.
- K. Wan, H. Chen, F. Zheng, Y. Pan, Y. Zhang and D. Long, Ind. Eng. Chem. Res., 2020, 59, 17451–17461 CrossRef CAS.
- X. Lin, H. Lei, E. Huo, M. Qian, W. Mateo, Q. Zhang, Y. Zhao, C. Wang and E. Villota, Energy Convers. Manage., 2020, 211, 112757 CrossRef CAS.
- Z. Ali, P. Rathnakumar, M. Ashfaq Hussain, E. Roma, M. Nagaral and M. Umar, Mater. Today: Proc., 2022, 52, 716–723 CAS.
- D. Duan, Z. Feng, X. Dong, X. Chen, Y. Zhang, K. Wan, Y. Wang, Q. Wang, G. Xiao, H. Liu and R. Ruan, Energy, 2021, 232, 121090 CrossRef CAS.
- S. Fan, Y. Zhang, T. Liu, W. Fu and B. Li, J. Anal. Appl. Pyrolysis, 2022, 162, 105425 CrossRef CAS.
- C. Li, C. Zhang, M. Gholizadeh and X. Hu, J. Hazard. Mater., 2020, 399, 123075 CrossRef CAS PubMed.
- C. Wang, H. Lei, M. Qian, E. Huo, Y. Zhao, Q. Zhang, W. Mateo, X. Lin, X. Kong, R. Zou and R. Ruan, Sustainable Energy Fuels, 2020, 4, 4614–4624 RSC.
- M. V. Singh, ChemistrySelect, 2023, 8, e202204400 CrossRef CAS.
- K. Zoroufchi Benis, A. Sokhansanj, J. Norberto, K. N. McPhedran and J. Soltan, Chem. Eng. J., 2022, 446, 137024 CrossRef CAS.
- D. Yao, H. Li, Y. Dai and C.-H. Wang, Chem. Eng. J., 2021, 408, 127268 CrossRef CAS.
- W. Luo, Q. Hu, Z.-y. Fan, J. Wan, Q. He, S.-x. Huang, N. Zhou, M. Song, J.-c. Zhang and Z. Zhou, Energy, 2020, 213, 119080 CrossRef CAS.
- T. S. Singh, T. N. Verma and H. N. Singh, Fuel, 2020, 277, 118176 CrossRef CAS.
- D. P. Serrano, J. Aguado and J. M. Escola, ACS Catal., 2012, 2, 1924–1941 CrossRef CAS.
- A. De Stefanis, P. Cafarelli, F. Gallese, E. Borsella, A. Nana and G. Perez, J. Anal. Appl. Pyrolysis, 2013, 104, 479–484 CrossRef CAS.
- K. Li, J. Lei, G. Yuan, P. Weerachanchai, J.-Y. Wang, J. Zhao and Y. Yang, Chem. Eng. J., 2017, 317, 800–809 CrossRef CAS.
- K. Li, Y. Wang, W. Zhou, T. Cui, J. Yang, Z. Sun, Y. Min and J.-M. Lee, Chemosphere, 2022, 299, 134440 CrossRef CAS PubMed.
- Y. Wang and Z. Sun, IOP Conf. Ser. Earth Environ. Sci., 2021, 770, 012055 CrossRef.
- J. G. Faillace, C. F. de Melo, S. P. L. de Souza and M. R. de Costa Marques, J. Anal. Appl. Pyrolysis, 2017, 126, 70–76 CrossRef CAS.
- K. Li, C. Cai, W. Zhou, Y. Wang, T. G. Y. Amy, Z. Sun and Y. Min, J. Hazard. Mater., 2024, 465, 133231 CrossRef CAS PubMed.
- G. Xu, X. Zhu, X. Niu, S. Liu, S. Xie, X. Li and L. Xu, Microporous Mesoporous Mater., 2009, 118, 44–51 CrossRef CAS.
- Z. Obalı, N. A. Sezgi and T. Doğu, Chem. Eng. J., 2011, 176–177, 202–210 CrossRef.
- A. K. Panda and R. Singh, J. Fuel Chem. Technol., 2011, 39, 198–202 CrossRef CAS.
- A. Marcilla, A. Gómez-Siurana and D. Berenguer, Appl. Catal., A, 2006, 301, 222–231 CrossRef CAS.
- Z. Obalı, N. A. Sezgi and T. Doğu, Chem. Eng. J., 2012, 207–208, 421–425 CrossRef.
- J. Xu, X. Tian, W. Huang, L. Ke, L. Fan, Q. Zhang, X. Cui, Q. Wu, Y. Zeng, K. Cobb, Y. Liu, R. Ruan and Y. Wang, Sci. Total Environ., 2023, 899, 165597 CrossRef CAS PubMed.
- L. Dai, N. Zhou, Y. Lv, K. Cobb, Y. Cheng, Y. Wang, Y. Liu, P. Chen, R. Zou, H. Lei and R. Ruan, Energy Convers. Manage., 2021, 245, 114578 CrossRef CAS.
- R. G. Kukushkin, P. M. Eletskii, O. O. Zaikina, G. A. Sosnin, O. A. Bulavchenko and V. A. Yakovlev, Catal. Ind., 2018, 10, 344–352 CrossRef.
- Y. Zhao, W. Wang, X. Jing, X. Gong, H. Wen and Y. Deng, J. Anal. Appl. Pyrolysis, 2020, 146, 104755 CrossRef CAS.
- D. P. Serrano, J. Aguado, J. M. Escola, E. Garagorri, x Rodrı, J. M. guez, L. Morselli, G. Palazzi and R. Orsi, Appl. Catal., B, 2004, 49, 257–265 CrossRef CAS.
- C. Ma, J. Yu, Q. Yan, Z. Song, K. Wang, B. Wang and L. Sun, Polym. Degrad. Stab., 2017, 146, 1–12 CrossRef CAS.
- J. Gong, J. Liu, Z. Jiang, J. Feng, X. Chen, L. Wang, E. Mijowska, X. Wen and T. Tang, Appl. Catal., B, 2014, 147, 592–601 CrossRef CAS.
- K. Li, S. W. Lee, G. Yuan, J. Lei, S. Lin, P. Weerachanchai, Y. Yang and J.-Y. Wang, Energies, 2016, 9, 431 CrossRef.
- S. R. Juliastuti, M. I. Hisbullah and M. Abdillah, IOP Conf. Ser.: Mater. Sci. Eng., 2018, 334, 012015 Search PubMed.
- P. Rex, I. P. Masilamani and L. R. Miranda, J. Energy Inst., 2020, 93, 1819–1832 CrossRef CAS.
- Y. H. Lin, M. H. Yang, T. F. Yeh and M. D. Ger, Polym. Degrad. Stab., 2004, 86, 121–128 CrossRef CAS.
- A. Salmiaton and A. A. Garforth, Waste Manage., 2011, 31, 1139–1145 CrossRef CAS PubMed.
- J. Mertinkat, A. Kirsten, M. Predel and W. Kaminsky, J. Anal. Appl. Pyrolysis, 1999, 49, 87–95 CrossRef CAS.
- S. Chaianansutcharit, R. Katsutath, A. Chaisuwan, T. Bhaskar, A. Nigo, A. Muto and Y. Sakata, J. Anal. Appl. Pyrolysis, 2007, 80, 360–368 CrossRef CAS.
- L. Fan, Y. Zhang, S. Liu, N. Zhou, P. Chen, Y. Liu, Y. Wang, P. Peng, Y. Cheng, M. Addy, H. Lei and R. Ruan, Energy Convers. Manage., 2017, 149, 432–441 CrossRef CAS.
- J. Nisar, G. Ali, A. Shah, M. R. Shah, M. Iqbal, M. N. Ashiq and H. N. Bhatti, Energy Fuels, 2019, 33, 12666–12678 CrossRef CAS.
- N. Miskolczi, N. Gao and C. Quan, J. Energy Inst., 2023, 108, 101233 CrossRef CAS.
- L. Quesada, M. Calero de Hoces, M. A. Martín-Lara, G. Luzón and G. Blázquez, Sustainability, 2020, 12, 5482 CrossRef CAS.
- L. S. Diaz-Silvarrey, A. McMahon and A. N. Phan, J. Anal. Appl. Pyrolysis, 2018, 134, 621–631 CrossRef CAS.
- A. I. Eldahshory, K. Emara, M. S. Abd-Elhady and M. A. Ismail, Sci. Rep., 2023, 13, 11766 CrossRef CAS PubMed.
- N. Cai, X. Li, S. Xia, L. Sun, J. Hu, P. Bartocci, F. Fantozzi, P. T. Williams, H. Yang and H. Chen, Energy Convers. Manage., 2021, 229, 113794 CrossRef CAS.
- Q.-L. Li, R. Shan, J. Zhang, M. Lei, H.-R. Yuan and Y. Chen, J. Anal. Appl. Pyrolysis, 2023, 169, 105829 CrossRef CAS.
- P. Gaurh and H. Pramanik, Waste Manage., 2018, 77, 114–130 CrossRef CAS PubMed.
- R. K. Singh, B. Ruj, A. K. Sadhukhan and P. Gupta, J. Energy Inst., 2020, 93, 1020–1035 CrossRef CAS.
- J. Lai, Y. Meng, Y. Yan, E. Lester, T. Wu and C. H. Pang, Korean J. Chem. Eng., 2021, 38, 2235–2246 CrossRef CAS PubMed.
- A. Khan, N. Iqbal, T. Noor, N. Zaman and S. R. Khan, Sustainable Energy Fuels, 2023, 7, 4935–4954 RSC.
- M. Valášková, J. Madejová, A. Inayat, L. Matějová, M. Ritz, A. Martaus and P. Leštinský, Appl. Clay Sci., 2020, 192, 105639 CrossRef.
- S. L. Wong, N. Ngadi, T. A. T. Abdullah and I. M. Inuwa, Renewable Sustainable Energy Rev., 2015, 50, 1167–1180 CrossRef CAS.
- F. Bai, C.-C. Zhu, Y. Liu, P.-Q. Yuan, Z.-M. Cheng and W.-K. Yuan, Fuel Process. Technol., 2013, 106, 267–274 CrossRef CAS.
- X. Su, Y. Zhao, R. Zhang and J. Bi, Fuel Process. Technol., 2004, 85, 1249–1258 CrossRef CAS.
- Z. Fang, R. L. Smith, H. Inomata and K. Arai, J. Supercrit. Fluids, 2000, 16, 207–216 CrossRef CAS.
- P. Zhao, Z. Yuan, J. Zhang, X. Song, C. Wang, Q. Guo and A. J. Ragauskas, Sustainable Energy Fuels, 2021, 5, 575–583 RSC.
- A. Queiroz, G. B. Pedroso, S. N. Kuriyama and A. A. Fidalgo-Neto, Curr. Opin. Green Sustainable Chem., 2020, 25, 100364 CrossRef.
- B. Bai, Y. Liu, H. Zhang, F. Zhou, X. Han, Q. Wang and H. Jin, Fuel, 2020, 262, 116630 CrossRef CAS.
- L. Dai, N. Zhou, H. Li, Y. Wang, Y. Liu, K. Cobb, Y. Cheng, H. Lei, P. Chen and R. Ruan, Sci. Total Environ., 2021, 771, 144995 CrossRef CAS PubMed.
- L. Dai, N. Zhou, Y. Lv, Y. Cheng, Y. Wang, Y. Liu, K. Cobb, P. Chen, H. Lei and R. Ruan, Sci. Total Environ., 2021, 782, 146897 CrossRef CAS.
- O. Y. Yansaneh and S. H. Zein, Processes, 2022, 10, 332 CrossRef CAS.
- G. Lopez, M. Olazar, R. Aguado, G. Elordi, M. Amutio, M. Artetxe and J. Bilbao, Ind. Eng. Chem. Res., 2010, 49, 8990–8997 CrossRef CAS.
- V. Krishnamoorthy, Y. D. Yeboah and S. V. Pisupati, Energies, 2019, 12, 107 CrossRef CAS.
- D. G. Kulas, A. Zolghadr and D. Shonnard, ACS Sustainable Chem. Eng., 2021, 9, 14443–14450 CrossRef CAS.
- D. G. Kulas, A. Zolghadr and D. R. Shonnard, J. Anal. Appl. Pyrolysis, 2022, 166, 105601 CrossRef CAS.
- S. Streiff, D. Balthasart, M. Piccinini, M. Garrait, A. Corma, M. Cerro-Alarcon and J. Mengual, US20190119191A1, 2019.
- Z. Alhulaybi and I. Dubdub, Polymers, 2023, 15, 3010 CrossRef CAS PubMed.
- A. F. Anene, S. B. Fredriksen, K. A. Sætre and L.-A. Tokheim, Sustainability, 2018, 10, 3979 CrossRef CAS.
- L. Contat-Rodrigo, A. Ribes-Greus and C. T. Imrie, J. Appl. Polym. Sci., 2002, 86, 764–772 CrossRef CAS.
- E. Esmizadeh, C. Tzoganakis and T. H. Mekonnen, Polymers, 2020, 12, 1627 CrossRef CAS PubMed.
- J. A. Onwudili, N. Insura and P. T. Williams, J. Anal. Appl. Pyrolysis, 2009, 86, 293–303 CrossRef CAS.
- K. I. Dement'ev, S. P. Bedenko, Y. D. Minina, A. A. Mukusheva, O. A. Alekseeva and T. A. Palankoev, Polymers, 2023, 15, 290 CrossRef PubMed.
- Y. Peng, L. Dai, A. Dai, Q. Wu, R. Zou, Y. Liu, R. Ruan and Y. Wang, J. Anal. Appl. Pyrolysis, 2022, 168, 105719 CrossRef CAS.
- G. Özsin, M. Alpaslan Takan, A. Takan and A. E. Pütün, Int. J. Energy Res., 2022, 46, 16959–16978 CrossRef.
- I. Mohan, A. Sahoo, A. K. Panda, S. Mandal and S. Kumar, J. Environ. Chem. Eng., 2023, 11, 111039 CrossRef CAS.
- C. Kassargy, S. Awad, G. Burnens, K. Kahine and M. Tazerout, Fuel, 2018, 224, 764–773 CrossRef CAS.
- M. F. Paucar-Sánchez, M. Calero, G. Blázquez, R. R. Solís, M. J. Muñoz-Batista and M. Á. Martín-Lara, Process Saf. Environ. Prot., 2022, 168, 1201–1211 CrossRef.
- K. Sivagami, G. Divyapriya, R. Selvaraj, P. Madhiyazhagan, N. Sriram and I. Nambi, Process Saf. Environ. Prot., 2021, 149, 497–506 CrossRef CAS.
- S. Budsaereechai, A. J. Hunt and Y. Ngernyen, RSC Adv., 2019, 9, 5844–5857 RSC.
- A. Ochoa, J. Bilbao, A. G. Gayubo and P. Castaño, Renewable Sustainable Energy Rev., 2020, 119, 109600 CrossRef CAS.
- A. Tukker, H. de Groot, L. Simons and S. Wiegersma, Chemical Recycling of Plastic Waste: PVC and Other Resins, Report STB-99-55 Final, TNO Institute of Strategy, Technology and Policy, Delft, Netherland, 1999 Search PubMed.
- M. Kusenberg, A. Eschenbacher, L. Delva, S. De Meester, E. Delikonstantis, G. D. Stefanidis, K. Ragaert and K. M. Van Geem, Fuel Process. Technol., 2022, 238, 107474 CrossRef CAS.
- Quantafuel, Pyrolysis Technology, https://www.quantafuel.com/our-solution/technology, accessed 6 August, 2024.
- A. Maisels, A. Hiller and F.-G. Simon, ChemBioEng Rev., 2022, 9, 541–555 CrossRef CAS.
- M. Solis and S. Silveira, Waste Manage., 2020, 105, 128–138 CrossRef CAS PubMed.
- Plaxx, Recycling Technologies: Solving the problem of plastic waste, https://recyclingtechnologies.co.uk/technology/, accessed 11 February, 2024.
- M. Bricker, V. Thakkar and J. Petri, in Handbook of Petroleum Processing, ed. S. A. Treese, P. R. Pujadó and D. S. J. Jones, Springer International Publishing, Cham, 2015, pp. 317–359, DOI:10.1007/978-3-319-14529-7_3.
- D. Munir, M. F. Irfan and M. R. Usman, Renewable Sustainable Energy Rev., 2018, 90, 490–515 CrossRef CAS.
- D. Yao, H. Yang, H. Chen and P. T. Williams, Appl. Catal., B, 2018, 227, 477–487 CrossRef CAS.
- N. Gama, B. Godinho, G. Marques, R. Silva, A. Barros-Timmons and A. Ferreira, Chem. Eng. J., 2020, 395, 125102 CrossRef CAS.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS PubMed.
- D. P. Serrano, J. M. Escola, L. Briones and M. Arroyo, Fuel, 2017, 206, 190–198 CrossRef CAS.
- A. A. Garforth, S. Ali, J. Hernández-Martínez and A. Akah, Curr. Opin. Solid State Mater. Sci., 2004, 8, 419–425 CrossRef CAS.
- A. Brems, J. Baeyens and R. Dewil, Therm. Sci., 2012, 16, 669–685 CrossRef.
- S. Karagöz, T. Karayildirim, S. Uçar, M. Yuksel and J. Yanik, Fuel, 2003, 82, 415–423 CrossRef.
- V. P. S. Caldeira, A. G. D. Santos, D. S. Oliveira, R. B. Lima, L. D. Souza and S. B. C. Pergher, J. Therm. Anal. Calorim., 2017, 130, 1939–1951 CrossRef CAS.
- A. Nanduri and P. L. Mills, Fuel, 2020, 278, 118117 CrossRef.
- L. Hauli, K. Wijaya and A. Syoufian, Orient. J. Chem., 2019, 35, 128–133 CAS.
- P. S. F. Mendes, J. M. Silva, M. F. Ribeiro, C. Bouchy and A. Daudin, J. Ind. Eng. Chem., 2019, 71, 167–176 CrossRef CAS.
- U. J. Etim, B. Xu, Z. Zhang, Z. Zhong, P. Bai, K. Qiao and Z. Yan, Fuel, 2016, 178, 243–252 CrossRef CAS.
- J. Lee, M. Young Kim, J. Hong Jeon, D. H. Lee, K. N. Rao, D. G. Oh, E. Jeong Jang, E. Kim, S. C. Na, H. S. Han and J. H. Kwak, Appl. Catal., B, 2020, 260, 118098 CrossRef CAS.
- N. Priharto, F. Ronsse, W. Prins, I. Hita, P. J. Deuss and H. J. Heeres, Biomass Bioenergy, 2019, 126, 84–93 CrossRef CAS.
- Z. Pan, X. Xue, C. Zhang, D. Wang, Y. Xie and R. Zhang, J. Anal. Appl. Pyrolysis, 2018, 136, 208–214 CrossRef CAS.
- J. Shabtai, X. Xiao and W. Zmierczak, Energy Fuels, 1997, 11, 76–87 CrossRef CAS.
- K. Faust, P. Denifl and M. Hapke, ChemCatChem, 2023, 15, e202300310 CrossRef CAS.
- D. Munir, Abdullah, F. Piepenbreier and M. R. Usman, Powder Technol., 2017, 316, 542–550 CrossRef CAS.
- K. Zhang, E.-H. Yuan, L.-L. Xu, Q.-S. Xue, C. Luo, B. Albela and L. Bonneviot, Eur. J. Inorg. Chem., 2012, 2012, 4183–4189 CrossRef CAS.
- H. Kim, S. J. Choi, J. M. Kim, J.-K. Jeon, S. H. Park, S.-C. Jung, S. C. Kim and Y.-K. Park, Mater. Res. Bull., 2016, 82, 61–66 CrossRef CAS.
- Y. Liu, Reactions, 2020, 1, 195–209 CrossRef.
- D. Munir and M. R. Usman, J. Anal. Appl. Pyrolysis, 2018, 135, 44–53 CrossRef CAS.
- M. F. Hanafi and N. Sapawe, Mater. Today: Proc., 2020, 31, 266–268 CAS.
- J. Liu, N. Liu, K. Ren, L. Shi and X. Meng, Ind. Eng. Chem. Res., 2017, 56, 7693–7699 CrossRef CAS.
- A. K. Aboul-Gheit, F. K. Gad, G. M. Abdel-Aleem, D. S. El-Desouki, S. M. Abdel-Hamid, S. A. Ghoneim and A. H. Ibrahim, Egypt. J. Pet., 2014, 23, 303–314 CrossRef.
- G. D. Yadav and J. J. Nair, Microporous Mesoporous Mater., 1999, 33, 1–48 CrossRef CAS.
- M. Utami, K. Wijaya and W. Trisunaryanti, Mater. Chem. Phys., 2018, 213, 548–555 CrossRef CAS.
- A. K. Amin, K. Wijaya and W. Trisunaryanti, Orient. J. Chem., 2018, 34, 3070–3078 CAS.
- E. Dahdah, J. Estephane, C. Gennequin, A. Aboukaïs, E. Abi-Aad and S. Aouad, Int. J. Hydrogen Energy, 2020, 45, 4457–4467 CrossRef CAS.
- A. Bin Jumah, V. Anbumuthu, A. A. Tedstone and A. A. Garforth, Ind. Eng. Chem. Res., 2019, 58, 20601–20609 CrossRef CAS.
- A. b. Jumah, A. A. Tedstone and A. A. Garforth, Microporous Mesoporous Mater., 2021, 315, 110912 CrossRef CAS.
- C. Kassargy, S. Awad, G. Burnens, G. Upreti, K. Kahine and M. Tazerout, Appl. Catal., B, 2019, 244, 704–708 CrossRef CAS.
- M. Sun, L. Zhu, W. Liu, X. Zhao, Y. Zhang, H. Luo, G. Miao, S. Li, S. Yin and L. Kong, Sustainable Energy Fuels, 2022, 6, 271–275 RSC.
- W. Ding, J. Liang and L. L. Anderson, Energy Fuels, 1997, 11, 1219–1224 CrossRef CAS.
- W.-T. Lee, A. van Muyden, F. D. Bobbink, M. D. Mensi, J. R. Carullo and P. J. Dyson, Nat. Commun., 2022, 13, 4850 CrossRef CAS PubMed.
- D. Munir, H. Amer, R. Aslam, M. Bououdina and M. R. Usman, Mater. Renew. Sustain. Energy, 2020, 9, 9 CrossRef.
- S. Liu, P. A. Kots, B. C. Vance, A. Danielson and D. G. Vlachos, Sci. Adv., 2021, 7, eabf8283 CrossRef CAS PubMed.
- A. L. Figueiredo, A. S. Araujo, M. Linares, Á. Peral, R. A. García, D. P. Serrano and V. J. Fernandes, J. Anal. Appl. Pyrolysis, 2016, 117, 132–140 CrossRef CAS.
- M. P. González-Marcos, E. G. Fuentes-Ordóñez, J. A. Salbidegoitia and J. R. González-Velasco, Top. Catal., 2021, 64, 224–242 CrossRef.
- B. J. B. Silva, L. V. Sousa, L. R. A. Sarmento, S. L. Alencar, P. H. L. Quintela and A. O. S. Silva, Appl. Catal., B, 2020, 267, 118699 CrossRef.
- J. Tekla, L. Lakiss, V. Valchev, K. A. Tarach, M. Jabłońska, V. Girman, A. Szymocha, A. Kowalczyk, K. Góra-Marek and J.-P. Gilson, Microporous Mesoporous Mater., 2020, 299, 110088 CrossRef CAS.
- A. Galadima and O. Muraza, J. Ind. Eng. Chem., 2018, 61, 265–280 CrossRef CAS.
- A. b. Jumah, A. A. Tedstone and A. A. Garforth, Microporous Mesoporous Mater., 2021, 315, 110912 CrossRef CAS.
- A. A. Tedstone, A. Bin Jumah, E. Asuquo and A. A. Garforth, R. Soc. Open Sci., 2022, 9, 211353 CrossRef CAS PubMed.
- R. Ochoa, H. Van Woert, W. H. Lee, R. Subramanian, E. Kugler and P. C. Eklund, Fuel Process. Technol., 1996, 49, 119–136 CrossRef CAS.
- K. R. Venkatesh, J. Hu, W. Wang, G. D. Holder, J. W. Tierney and I. Wender, Energy Fuels, 1996, 10, 1163–1170 CrossRef CAS.
- I. Nakamura and K. Fujimoto, Catal. Today, 1996, 27, 175–179 CrossRef CAS.
- N. Shah, J. Rockwell and G. P. Huffman, Energy Fuels, 1999, 13, 832–838 CrossRef CAS.
- Z. Feng, J. Zhao, J. Rockwell, D. Bailey and G. Huffman, Fuel Process. Technol., 1996, 49, 17–30 CrossRef CAS.
- C. S. Costa, M. Muñoz, M. R. Ribeiro and J. M. Silva, Catal. Today, 2021, 379, 192–204 CrossRef CAS.
- A. Gala, M. Guerrero, B. Guirao, M. E. Domine and J. M. Serra, Energy Fuels, 2020, 34, 5969–5982 CrossRef CAS.
- K. A. Tarach, K. Pyra, S. Siles, I. Melián-Cabrera and K. Góra-Marek, ChemSusChem, 2019, 12, 633–638 CrossRef CAS PubMed.
- A. Akaha, J. Hernandez-Martinezb, C. Rallanb and A. A. Garforth, Chem. Eng. Trans., 2015, 43, 2395–2400 Search PubMed.
- M. Kusenberg, A. Zayoud, M. Roosen, H. D. Thi, M. S. Abbas-Abadi, A. Eschenbacher, U. Kresovic, S. De Meester and K. M. Van Geem, Fuel Process. Technol., 2022, 227, 107090 CrossRef CAS.
- D. Damodharan, A. P. Sathiyagnanam, D. Rana, S. Saravanan, B. Rajesh Kumar and B. Sethuramasamyraja, Energy Convers. Manage., 2018, 166, 81–97 CrossRef CAS.
- K. Pyra, K. A. Tarach, A. Śrębowata, I. Melián-Cabrera and K. Góra-Marek, Appl. Catal., B, 2020, 277, 119070 CrossRef CAS.
- L. Y. Jia, A. Farouha, L. Pinard, S. Hedan, J. D. Comparot, A. Dufour, K. Ben Tayeb, H. Vezin and C. Batiot-Dupeyrat, Appl. Catal., B, 2017, 219, 82–91 CrossRef CAS.
- M. Usman Azam, A. Fernandes, I. Graça and W. Afzal, Fuel, 2023, 349, 128704 CrossRef CAS.
- L. Hauli, K. Wijaya and A. Syoufian, Indones. J. Chem., 2020, 20, 422–429 CrossRef.
- X. Zhang, H. Lei, L. Zhu, M. Qian, G. Yadavalli, J. Wu and S. Chen, Fuel, 2017, 188, 28–38 CrossRef CAS.
- Z. Qiu, S. Lin, Z. Chen, A. Chen, Y. Zhou, X. Cao, Y. Wang and B.-L. Lin, Sci. Adv., 2023, 9, eadg5332 CrossRef CAS PubMed.
- M. Utami, W. Trisunaryanti, K. Shida, M. Tsushida, H. Kawakita, K. Ohto, K. Wijaya and M. Tominaga, RSC Adv., 2019, 9, 41392–41401 RSC.
- M. S. Al-Iessa, B. Y. Al-Zaidi, R. S. Almukhtar, Z. M. Shakor and I. Hamawand, Energies, 2023, 16, 4871 CrossRef CAS.
- G. Lopez, M. Artetxe, M. Amutio, J. Alvarez, J. Bilbao and M. Olazar, Renewable Sustainable Energy Rev., 2018, 82, 576–596 CrossRef CAS.
- M. Artetxe, G. Lopez, G. Elordi, M. Amutio, J. Bilbao and M. Olazar, Ind. Eng. Chem. Res., 2012, 51, 13915–13923 CrossRef CAS.
- D. S. N. Parker, R. I. Kaiser, T. P. Troy and M. Ahmed, Angew. Chem., Int. Ed., 2014, 53, 7740–7744 CrossRef CAS PubMed.
- G. Ruoppolo, P. Ammendola, R. Chirone and F. Miccio, Waste Manage., 2012, 32, 724–732 CrossRef CAS PubMed.
- L. Devi, K. J. Ptasinski and F. J. J. G. Janssen, Biomass Bioenergy, 2003, 24, 125–140 CrossRef CAS.
- G. Guan, M. Kaewpanha, X. Hao and A. Abudula, Renewable Sustainable Energy Rev., 2016, 58, 450–461 CrossRef CAS.
- H. Shi, W. Si and X. Li, Energies, 2016, 9, 67 CrossRef.
- X. Wang, A. Panahi, H. Qi, M. Zhai, P. Dong and Y. A. Levendis, J. Energy Eng., 2020, 146, 04020049 CrossRef.
- M. Cortazar, L. Santamaria, G. Lopez, J. Alvarez, L. Zhang, R. Wang, X. Bi and M. Olazar, Energy Convers. Manage., 2023, 276, 116496 CrossRef CAS.
- H. H. Shah, M. Amin, A. Iqbal, I. Nadeem, M. Kalin, A. M. Soomar and A. M. Galal, Front. Chem., 2022, 10, 960894 CrossRef CAS PubMed.
- M.-H. Cho, T.-Y. Mun and J.-S. Kim, Energy, 2013, 53, 299–305 CrossRef CAS.
- M.-H. Cho, Y.-K. Choi and J.-S. Kim, Energy, 2015, 87, 586–593 CrossRef CAS.
- B. Ciuffi, D. Chiaramonti, A. M. Rizzo, M. Frediani and L. Rosi, Appl. Sci., 2020, 10, 6307 CrossRef CAS.
- S. A. Salaudeen, P. Arku and A. Dutta, in Plastics to Energy: Fuel, Chemicals, and Sustainability Implications, ed. S. M. Al-Saleem, 2019, p. 269 Search PubMed.
- S. A. Salaudeen, P. Arku and A. Dutta, in Plastics to Energy, ed. S. M. Al-Salem, William Andrew Publishing, 2019, pp. 269–293, DOI:10.1016/B978-0-12-813140-4.00010-8.
- U. Lee, J. N. Chung and H. A. Ingley, Energy Fuels, 2014, 28, 4573–4587 CrossRef CAS.
- J. Wang, G. Cheng, Y. You, B. Xiao, S. Liu, P. He, D. Guo, X. Guo and G. Zhang, Int. J. Hydrogen Energy, 2012, 37, 6503–6510 CrossRef CAS.
- A. Ongen, Clean Technol. Environ. Policy, 2016, 18, 915–924 CrossRef CAS.
- I. M. Maafa, Polymers, 2021, 13, 225 CrossRef CAS PubMed.
- P. Straka and O. Bičáková, Int. J. Hydrogen Energy, 2014, 39, 10987–10995 CrossRef CAS.
- I. I. Ahmed, N. Nipattummakul and A. K. Gupta, Appl. Energy, 2011, 88, 165–174 CrossRef CAS.
- X. Guo, W. Zhang, L. Wang and J. Hao, J. Anal. Appl. Pyrolysis, 2016, 120, 144–153 CrossRef CAS.
- Z. A. B. Z. Alauddin, P. Lahijani, M. Mohammadi and A. R. Mohamed, Renewable Sustainable Energy Rev., 2010, 14, 2852–2862 CrossRef CAS.
- E. Madadian, C. Crowe and M. Lefsrud, J. Cleaner Prod., 2017, 164, 137–145 CrossRef CAS.
- A. Ponzio, S. Kalisz and W. Blasiak, Fuel Process. Technol., 2006, 87, 223–233 CrossRef CAS.
- P. McKendry, Bioresour. Technol., 2002, 83, 55–63 CrossRef CAS PubMed.
- Y. Liu, J. Qian and J. Wang, Fuel Process. Technol., 2000, 63, 45–55 CrossRef CAS.
- U. Arena and F. Di Gregorio, Energy, 2014, 68, 735–743 CrossRef CAS.
- V. Wilk and H. Hofbauer, Fuel, 2013, 107, 787–799 CrossRef CAS.
- P. Brachi, R. Chirone, F. Miccio, M. Miccio, A. Picarelli and G. Ruoppolo, Fuel, 2014, 128, 88–98 CrossRef CAS.
- J. Makibar, A. R. Fernandez-Akarregi, I. Alava, F. Cueva, G. Lopez and M. Olazar, Chem. Eng. Process., 2011, 50, 790–798 CrossRef CAS.
- J. Alvarez, M. Amutio, G. Lopez, I. Barbarias, J. Bilbao and M. Olazar, Chem. Eng. J., 2015, 273, 173–183 CrossRef CAS.
- A. R. Fernandez-Akarregi, J. Makibar, G. Lopez, M. Amutio and M. Olazar, Fuel Process. Technol., 2013, 112, 48–56 CrossRef CAS.
- J. Makibar, A. R. Fernandez-Akarregi, M. Amutio, G. Lopez and M. Olazar, Fuel Process. Technol., 2015, 137, 283–289 CrossRef CAS.
- P. G. Rutberg, V. A. Kuznetsov, E. O. Serba, S. D. Popov, A. V. Surov, G. V. Nakonechny and A. V. Nikonov, Appl. Energy, 2013, 108, 505–514 CrossRef CAS.
- T. R. Praveenkumar, M. Sekar, R. R. Pasupuleti, B. Gavurová, G. Arun Kumar and M. Vignesh Kumar, Fuel, 2024, 357, 129379 CrossRef CAS.
- Gasification Technologies, https://www.eep.ebara.com/en/business_technology/technology_3.html, accessed 24 May, 2024.
- C. Ducharme, MS thesis, Columbia University, 2010.
- R. P. Lee, L. G. Seidl, Q.-l. Huang and B. Meyer, J. Fuel Chem. Technol., 2021, 49, 1057–1076 CrossRef CAS.
- D. A. Tsiamis and M. J. Castaldi, The Effects of Non-recyled Plastic on Gasification,:a Quantitaive Asssessment, Earth Engineering Center, City University of New York, 2018 Search PubMed.
- S. M. Santos, A. C. Assis, L. Gomes, C. Nobre and P. Brito, Waste, 2023, 1, 140–165 CrossRef.
- D. Maggio, Steel made from plastics. Projects in the steelmaking industry, https://www.expometals.net/en/news/steel-made-from-plastics-projects-in-the-steelmaking-industry-id28875, accessed 19 March, 2024.
- R. A. M. Meneses, G. Cabrera-Papamija, F. Machuca-Martínez, L. A. Rodríguez, J. E. Diosa and E. Mosquera-Vargas, Heliyon, 2022, 8, e09028 CrossRef PubMed.
- E. V. Antonakou and D. S. Achilias, Waste Biomass Valorization, 2013, 4, 9–21 CrossRef CAS.
- S. Makkam and W. Harnnarongchai, Energy Procedia, 2014, 56, 547–553 CrossRef CAS.
- C. Jehanno, M. M. Pérez-Madrigal, J. Demarteau, H. Sardon and A. P. Dove, Polym. Chem., 2019, 10, 172–186 RSC.
- A. B. Raheem, Z. Z. Noor, A. Hassan, M. K. Abd Hamid, S. A. Samsudin and A. H. Sabeen, J. Cleaner Prod., 2019, 225, 1052–1064 CrossRef CAS.
- L. Monsigny, J.-C. Berthet and T. Cantat, ACS Sustainable Chem. Eng., 2018, 6, 10481–10488 CrossRef CAS.
- S. K. Das, S. K. Eshkalak, A. Chinnappan, R. Ghosh, W. A. D. M. Jayathilaka, C. Baskar and S. Ramakrishna, Mater. Circ. Econ., 2021, 3, 9 CrossRef.
- Z. Laldinpuii, S. Lalhmangaihzuala, Z. Pachuau and K. Vanlaldinpuia, Waste Manage., 2021, 126, 1–10 CrossRef CAS PubMed.
- N. E. Kamber, Y. Tsujii, K. Keets, R. M. Waymouth, R. C. Pratt, G. W. Nyce and J. L. Hedrick, J. Chem. Educ., 2010, 87, 519–521 CrossRef CAS.
- N. D. Pingale and S. R. Shukla, Eur. Polym. J., 2008, 44, 4151–4156 CrossRef CAS.
- S. Baliga and W. T. Wong, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 2071–2082 CrossRef CAS.
- A. Sangalang, L. Bartolome and D. H. Kim, Polym. Degrad. Stab., 2015, 115, 45–53 CrossRef CAS.
- R. Esquer and J. J. García, J. Organomet. Chem., 2019, 902, 120972 CrossRef CAS.
- H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018, 20, 1929–1961 RSC.
- K. Fukushima, O. Coulembier, J. M. Lecuyer, H. A. Almegren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. McNeil, P. Dubois, R. M. Waymouth, H. W. Horn, J. E. Rice and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1273–1281 CrossRef CAS.
- K. Fukushima, D. J. Coady, G. O. Jones, H. A. Almegren, A. M. Alabdulrahman, F. D. Alsewailem, H. W. Horn, J. E. Rice and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1606–1611 CrossRef CAS.
- Z. Wang, Y. Jin, Y. Wang, Z. Tang, S. Wang, G. Xiao and H. Su, ACS Sustainable Chem. Eng., 2022, 10, 7965–7973 CrossRef CAS.
- I. Olazabal, E. J. Luna Barrios, S. De Meester, C. Jehanno and H. Sardon, ACS Appl. Polym. Mater., 2024, 6, 4226–4232 CrossRef CAS PubMed.
- J. Xin, Q. Zhang, J. Huang, R. Huang, Q. Z. Jaffery, D. Yan, Q. Zhou, J. Xu and X. Lu, J. Environ. Manage., 2021, 296, 113267 CrossRef CAS PubMed.
- A. M. Al-Sabagh, F. Z. Yehia, G. Eshaq, A. M. Rabie and A. E. ElMetwally, Egypt. J. Pet., 2016, 25, 53–64 CrossRef.
- S. Wang, C. Wang, H. Wang, X. Chen and S. Wang, Polym. Degrad. Stab., 2015, 114, 105–114 CrossRef CAS.
- Y. Geng, T. Dong, P. Fang, Q. Zhou, X. Lu and S. Zhang, Polym. Degrad. Stab., 2015, 117, 30–36 CrossRef CAS.
- P. Fang, S. Xia and X. Lu, J. Environ. Chem. Eng., 2022, 10, 107823 CrossRef CAS.
- Z. Wang, Y. Wang, S. Xu, Y. Jin, Z. Tang, G. Xiao and H. Su, Polym. Degrad. Stab., 2021, 190, 109638 CrossRef CAS.
- D. Simón, J. F. Rodríguez, M. Carmona, A. Serrano and A. M. Borreguero, Chem. Eng. J., 2018, 350, 300–311 CrossRef.
- X. Wang, H. Chen, C. Chen and H. Li, Fibers Polym., 2011, 12, 857–863 CrossRef CAS.
- D. Simón, M. T. García, A. de Lucas, A. M. Borreguero and J. F. Rodríguez, Polym. Degrad. Stab., 2013, 98, 144–149 CrossRef.
- A. Aguado, L. Martínez, A. Moral, J. Fermoso and R. Irusta, Chem. Eng. Trans., 2011, 24, 1069–1074 Search PubMed.
- D. Simón, A. M. Borreguero, A. de Lucas and J. F. Rodríguez, Waste Manage., 2018, 76, 147–171 CrossRef PubMed.
- E. Quaranta, C. C. Minischetti and G. Tartaro, ACS Omega, 2018, 3, 7261–7268 CrossRef CAS PubMed.
- A. J. Spicer, A. Brandolese and A. P. Dove, ACS Macro Lett., 2024, 13, 189–194 CrossRef CAS PubMed.
- I. Olazabal, E. Luna, S. De Meester, C. Jehanno and H. Sardon, Polym. Chem., 2023, 14, 2299–2307 RSC.
- H. Wang, Z. Li, Y. Liu, X. Zhang and S. Zhang, Green Chem., 2009, 11, 1568–1575 RSC.
- Q. Wang, X. Lu, X. Zhou, M. Zhu, H. He and X. Zhang, J. Appl. Polym. Sci., 2013, 129, 3574–3581 CrossRef CAS.
- A. M. Al-Sabagh, F. Z. Yehia, A.-M. M. F. Eissa, M. E. Moustafa, G. Eshaq, A.-R. M. Rabie and A. E. ElMetwally, Ind. Eng. Chem. Res., 2014, 53, 18443–18451 CrossRef CAS.
- C. S. Nunes, M. J. Vieira da Silva, D. Cristina da Silva, A. d. R. Freitas, F. A. Rosa, A. F. Rubira and E. C. Muniz, RSC Adv., 2014, 4, 20308–20316 RSC.
- C. Jehanno, I. Flores, A. P. Dove, A. J. Müller, F. Ruipérez and H. Sardon, Green Chem., 2018, 20, 1205–1212 RSC.
- Q. F. Yue, L. F. Xiao, M. L. Zhang and X. F. Bai, Polymers, 2013, 5, 1258–1271 CrossRef.
- Q. Wang, X. Yao, Y. Geng, Q. Zhou, X. Lu and S. Zhang, Green Chem., 2015, 17, 2473–2479 RSC.
- B. Liu, W. Fu, X. Lu, Q. Zhou and S. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 3292–3300 CrossRef CAS.
- L. Wang, G. A. Nelson, J. Toland and J. D. Holbrey, ACS Sustainable Chem. Eng., 2020, 8, 13362–13368 CrossRef CAS.
- M. A. Alnaqbi, M. A. Mohsin, R. M. Busheer and Y. Haik, J. Appl. Polym. Sci., 2015, 132, 41666 CrossRef.
- M. Liu, J. Guo, Y. Gu, J. Gao and F. Liu, Polym. Degrad. Stab., 2018, 157, 9–14 CrossRef CAS.
- F. Liu, J. Guo, P. Zhao, Y. Gu, J. Gao and M. Liu, Polym. Degrad. Stab., 2019, 167, 124–129 CrossRef CAS.
- M. Liu, J. Guo, Y. Gu, J. Gao and F. Liu, ACS Sustainable Chem. Eng., 2018, 6, 15127–15134 CrossRef CAS.
- A. M. Al-Sabagh, F. Z. Yehia, G. Eshaq and A. E. ElMetwally, Ind. Eng. Chem. Res., 2015, 54, 12474–12481 CrossRef CAS.
- R. Wang, T. Wang, G. Yu and X. Chen, Polym. Degrad. Stab., 2021, 183, 109463 CrossRef CAS.
- T. Wang, Y. Zheng, G. Yu and X. Chen, Eur. Polym. J., 2021, 155, 110590 CrossRef CAS.
- Y. Wang, T. Wang, L. Zhou, P. Zhang, Z. Wang and X. Chen, Eur. Polym. J., 2023, 201, 112578 CrossRef CAS.
- H. Qiu, X. Du, J. Zhao, Y. Wang, J. Ju, Z. Chen, Z. Hu, D. Yan, X. Zhou and G. Cui, Nat. Commun., 2019, 10, 5374 CrossRef PubMed.
- N. George and T. Kurian, Ind. Eng. Chem. Res., 2014, 53, 14185–14198 CrossRef CAS.
- S. Sirohi, S. Dobhal, M. Doshi, R. Nain, K. Dutt and B. Pani, Indian Chem. Eng., 2019, 61, 206–217 CrossRef CAS.
- M. Imran, D. H. Kim, W. A. Al-Masry, A. Mahmood, A. Hassan, S. Haider and S. M. Ramay, Polym. Degrad. Stab., 2013, 98, 904–915 CrossRef CAS.
- C. A. Fuentes, M. V. Gallegos, J. R. García, J. Sambeth and M. A. Peluso, Waste Biomass Valorization, 2020, 11, 4991–5001 CrossRef CAS.
- L. Bartolome, M. Imran, K. G. Lee, A. Sangalang, J. K. Ahn and D. H. Kim, Green Chem., 2014, 16, 279–286 RSC.
- J.-M. Jeong, S. B. Jin, S. G. Son, H. Suh, J.-M. Moon and B. G. Choi, React. Chem. Eng., 2021, 6, 297–303 RSC.
- A. M. Al-Sabagh, F. Z. Yehia, D. R. K. Harding, G. Eshaq and A. E. ElMetwally, Green Chem., 2016, 18, 3997–4003 RSC.
- G. R. Lima, W. F. Monteiro, R. Ligabue and R. M. C. Santana, Mater. Res., 2017, 20, 588–595 CrossRef.
- G. Park, L. Bartolome, K. G. Lee, S. J. Lee, D. H. Kim and T. J. Park, Nanoscale, 2012, 4, 3879–3885 RSC.
- S. B. Jin, J.-M. Jeong, S. G. Son, S. H. Park, K. G. Lee and B. G. Choi, Mater. Today Commun., 2021, 26, 101857 CrossRef CAS.
- J.-M. Jeong, S. B. Jin, H. J. Park, S. H. Park, H. Jeon, H. Suh, Y.-J. Park, D. Seo, S. Y. Hwang, D. H. Kim and B. G. Choi, Adv. Mater. Interfaces, 2020, 7, 2000599 CrossRef CAS.
- M. R. Nabid, Y. Bide and M. Jafari, Polym. Degrad. Stab., 2019, 169, 108962 CrossRef CAS.
- Z. Fehér, J. Kiss, P. Kisszékelyi, J. Molnár, P. Huszthy, L. Kárpáti and J. Kupai, Green Chem., 2022, 24, 8447–8459 RSC.
- H. Yao, L. Liu, D. Yan, Q. Zhou, J. Xin, X. Lu and S. Zhang, Chem. Eng. Sci., 2022, 248, 117109 CrossRef CAS.
- I. E. Wachs and K. Routray, ACS Catal., 2012, 2, 1235–1246 CrossRef CAS.
- M. R. Othman, Chem. Eng. Sci., 2009, 64, 925–929 CrossRef CAS.
- A. P. Arcanjo, D. O. Liborio, S. Arias, F. R. Carvalho, J. P. Silva, B. D. Ribeiro, M. L. Dias, A. M. Castro, R. Fréty, C. M. B. M. Barbosa and J. G. A. Pacheco, Polymers, 2023, 15, 3274 CrossRef CAS PubMed.
- F. Chen, G. Wang, W. Li and F. Yang, Ind. Eng. Chem. Res., 2013, 52, 565–571 CrossRef CAS.
- Z. Guo, E. Adolfsson and P. L. Tam, Waste Manage., 2021, 126, 559–566 CrossRef CAS PubMed.
- W. Y. Hernández, J. Lauwaert, P. Van Der Voort and A. Verberckmoes, Green Chem., 2017, 19, 5269–5302 RSC.
- F. Chen, F. Yang, G. Wang and W. Li, J. Appl. Polym. Sci., 2014, 131, 41053 CrossRef.
- G. Eshaq and A. E. ElMetwally, J. Mol. Liq., 2016, 214, 1–6 CrossRef CAS.
- M. Zhu, S. Li, Z. Li, X. Lu and S. Zhang, Chem. Eng. J., 2012, 185–186, 168–177 CrossRef CAS.
- E. Borsella, I. De Bari, P. Colucci, S. Mastrolitti, F. Liuzzi, A. De Stefanis, V. Valentini, F. Gallese and G. Perez, Energy Technol., 2020, 8, 2000633 CrossRef CAS.
- Y. Wang, L. Wei, Q. Hou, Z. Mo, X. Liu and W. Li, Fermentation, 2023, 9, 386 CrossRef CAS.
- R. Roy, M. S. Rahman, T. A. Amit and B. Jadhav, Biomass, 2022, 2, 130–154 CrossRef CAS.
- G. Jeya, H. Ilbeygi, D. Radhakrishnan and V. Sivamurugan, Adv. Porous Mater., 2017, 5, 128–136 CrossRef.
- S. Bhandari and P. Gupta, in Recycling of Polyurethane Foams, ed. S. Thomas, A. V. Rane, K. Kanny, V. K. Abitha and M. G.Thomas, William Andrew Publishing, 2018, pp. 77–87, DOI:10.1016/B978-0-323-51133-9.00007-3.
- S. Ghorbantabar, M. Ghiass, N. Yaghobi and H. Bouhendi, J. Mater. Cycles Waste Manage., 2021, 23, 526–536 CrossRef CAS.
- K. Fukushima, J. M. Lecuyer, D. S. Wei, H. W. Horn, G. O. Jones, H. A. Al-Megren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. McNeil, J. E. Rice and J. L. Hedrick, Polym. Chem., 2013, 4, 1610–1616 RSC.
- Z. Leng, R. K. Padhan and A. Sreeram, J. Cleaner Prod., 2018, 180, 682–688 CrossRef CAS.
- F. Gardea, J. M. Garcia, D. J. Boday, K. M. Bajjuri, M. Naraghi and J. L. Hedrick, Macromol. Chem. Phys., 2014, 215, 2260–2267 CrossRef CAS.
- M. E. Tawfik and S. B. Eskander, Polym. Degrad. Stab., 2010, 95, 187–194 CrossRef CAS.
- S. A. Holmes, J. Appl. Polym. Sci., 1996, 61, 255–260 CrossRef CAS.
- Z. Syeda, H.-y. Chen and M.-k. Leung, Polym. Degrad. Stab., 2023, 214, 110387 CrossRef CAS.
- E. Selvam, Y. Luo, M. Ierapetritou, R. F. Lobo and D. G. Vlachos, Catal. Today, 2023, 418, 114124 CrossRef CAS.
- N. D. Pingale and S. R. Shukla, Eur. Polym. J., 2009, 45, 2695–2700 CrossRef CAS.
- N. George and T. Kurian, Prog. Rubber, Plast. Recycl. Technol., 2016, 32, 153–168 CrossRef.
- E. Bäckström, K. Odelius and M. Hakkarainen, Eur. Polym. J., 2021, 151, 110441 CrossRef.
- K. P. Blackmon, D. W. Fox and S. J. Shafer, US4973746A, 1990.
- P. Gupta and S. Bhandari, in Recycling of Polyethylene Terephthalate Bottles, ed. S. Thomas, A. V. Rane, K. Kanny, V. K. Abitha and M. G.Thomas, William Andrew Publishing, NY, USA, 2019 Search PubMed.
- J. Demarteau, I. Olazabal, C. Jehanno and H. Sardon, Polym. Chem., 2020, 11, 4875–4882 RSC.
- G. Pastore, R. Giacomantonio, G. Lupidi, F. Stella, R. Risoluti, E. Papa, R. Ballini, F. Sarasini, J. Tirillò, E. Marcantoni and S. Gabrielli, Front. Chem., 2023, 11, 1234763 CrossRef CAS PubMed.
- S. Singh, Y. Lei and A. Schober, RSC Adv., 2015, 5, 3454–3460 RSC.
- E. Quaranta, A. Dibenedetto, F. Nocito and P. Fini, J. Hazard. Mater., 2021, 403, 123957 CrossRef CAS PubMed.
- Y.-C. Huang, Y.-H. Huang, L.-Y. Chen, C.-A. Dai, S. A. Dai, Y.-H. Chen, C.-H. Wu and R.-J. Jeng, Polymer, 2021, 212, 123296 CrossRef CAS.
- Z. Wang, R. Yang, G. Xu, T. Liu and Q. Wang, ACS Sustainable Chem. Eng., 2022, 10, 4529–4537 CrossRef CAS.
- T. Spychaj, in Handbook of Thermoplastic Polyesters, ed. S. Fakirov, 2002, ch. 27, pp. 1252–1290, DOI:10.1002/3527601961.
- G. P. Karayannidis, A. P. Chatziavgoustis and D. S. Achilias, Adv. Polym. Technol., 2002, 21, 250–259 CrossRef CAS.
- P. Benyathiar, P. Kumar, G. Carpenter, J. Brace and D. K. Mishra, Polymers, 2022, 14, 2366 CrossRef CAS PubMed.
- W. Yang, J. Wang, L. Jiao, Y. Song, C. Li and C. Hu, Green Chem., 2022, 24, 1362–1372 RSC.
- H. Abedsoltan and M. R. Coleman, J. Appl. Polym. Sci., 2022, 139, e52451 CrossRef CAS.
- S. Mishra, A. S. Goje and V. S. Zope, Polym. React. Eng., 2003, 11, 79–99 CrossRef CAS.
- S. Ügdüler, K. M. Van Geem, R. Denolf, M. Roosen, N. Mys, K. Ragaert and S. De Meester, Green Chem., 2020, 22, 5376–5394 RSC.
- M. Han, in Recycling of Polyethylene Terephthalate Bottles, ed. S. Thomas, A. Rane, K. Kanny, V. K. Abitha and M. G.Thomas, William Andrew Publishing, 2019, pp. 85–108, DOI:10.1016/B978-0-12-811361-5.00005-5.
- M. Pohjakallio, T. Vuorinen and A. Oasmaa, in Plastic Waste and Recycling, ed. T. M. Letcher, Academic Press, 2020, pp. 359–384, DOI:10.1016/B978-0-12-817880-5.00013-X.
- M. N. Siddiqui, D. S. Achilias, H. H. Redhwi, D. N. Bikiaris, K. A. G. Katsogiannis and G. P. Karayannidis, Macromol. Mater. Eng., 2010, 295, 575–584 CrossRef CAS.
- L. Zhang, Eur. Polym. J., 2014, 60, 1–5 CrossRef CAS.
- H. I. Khalaf and O. A. Hasan, Chem. Eng. J., 2012, 192, 45–48 CrossRef CAS.
- S. Motokucho, Y. Nakayama, H. Morikawa and H. Nakatani, J. Appl. Polym. Sci., 2018, 135, 45897 CrossRef.
- Y. Hirota, K. Hayashi, T. Kawanishi and N. Takiguchi, J. Chem. Eng. Jpn., 2020, 53, 267–272 CrossRef CAS.
- N. Kozlow, G. Korotusko, A. Kashinskii and N. Gavrilenco, Vestsi Akad. Navuk BSSR, Ser. Khim. Navuk, 1984, 5, 91–93 Search PubMed.
- T. Yoshioka, M. Ota and A. Okuwaki, Ind. Eng. Chem. Res., 2003, 42, 675–679 CrossRef CAS.
- S. Zhang, Y. Xue, Y. Wu, Y.-X. Zhang, T. Tan and Z. Niu, Chem. Sci., 2023, 14, 6558–6563 RSC.
- S. Zhang, Q. Hu, Y.-X. Zhang, H. Guo, Y. Wu, M. Sun, X. Zhu, J. Zhang, S. Gong, P. Liu and Z. Niu, Nat. Sustainability, 2023, 6, 965–973 CrossRef.
- Z. Zhao, J. Bai, H. Tao, S. Wang, K. Wang, W. Lin, L. Jiang, H. Li and C. Wang, Green Chem. Eng., 2024 DOI:10.1016/j.gce.2024.12.001.
- V. A. Kosmidis, D. S. Achilias and G. P. Karayannidis, Macromol. Mater. Eng., 2001, 286, 640–647 CrossRef CAS.
- N. R. Paliwal and A. K. Mungray, Polym. Degrad. Stab., 2013, 98, 2094–2101 CrossRef CAS.
- H. J. Glatzer and L. K. Doraiswamy, Chem. Eng. J., 2000, 55, 5149–5160 CAS.
- A. Barredo, A. Asueta, I. Amundarain, J. Leivar, R. Miguel-Fernández, S. Arnaiz, E. Epelde, R. López-Fonseca and J. I. Gutiérrez-Ortiz, J. Environ. Chem. Eng., 2023, 11, 109823 CrossRef CAS.
- C.-Y. Kao, B.-Z. Wan and W.-H. Cheng, Ind. Eng. Chem. Res., 1998, 37, 1228–1234 CrossRef CAS.
- V. de Paula, S. V. Pandeirada, P. J. A. Ribeiro-Claro, A. J. D. Silvestre and A. F. Sousa, ACS Sustainable Chem. Eng., 2025, 13, 3577–3587 CrossRef CAS.
- S. Teke, S. Saud, R. M. Bhattarai, A. Ali, L. Nguyen, A. Denra, D. B. Nguyen and Y. S. Mok, Chemosphere, 2024, 365, 143391 CrossRef CAS PubMed.
- W. Wu, H. Zhai, K. Wu, X. Wang, W. Rao, J. Ding and L. Yu, Chem. Eng. J., 2024, 480, 148131 CrossRef CAS.
- M. Azeem, O. A. Attallah, C. E. Tas and M. B. Fournet, J. Polym. Environ., 2024, 32, 303–315 CrossRef CAS.
- J. Benninga, B. Gebben, R. Folkersma, V. S. D. Voet and K. Loos, J. Am. Chem. Soc., 2025, 147, 7191–7195 CrossRef CAS PubMed.
- P. Pereira, P. E. Savage and C. W. Pester, ACS Sustainable Chem. Eng., 2023, 11, 7203–7209 CrossRef CAS.
- C. N. Onwucha, C. O. Ehi-Eromosele, S. O. Ajayi, M. Schaefer, S. Indris and H. Ehrenberg, Ind. Eng. Chem. Res., 2023, 62, 6378–6385 CrossRef CAS.
- S. Motokucho, A. Yamaguchi, Y. Nakayama, H. Morikawa and H. Nakatani, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2004–2010 CrossRef CAS.
- P. Van Wouwe, M. Dusselier, E. Vanleeuw and B. Sels, ChemSusChem, 2016, 9, 907–921 CrossRef CAS PubMed.
- G. Kale, R. Auras, S. P. Singh and R. Narayan, Polym. Test., 2007, 26, 1049–1061 CrossRef CAS.
- V. Piemonte and F. Gironi, J. Polym. Environ., 2013, 21, 313–318 CrossRef CAS.
- K. Hirao, Y. Shimamoto, Y. Nakatsuchi and H. Ohara, Polym. Degrad. Stab., 2010, 95, 86–88 CrossRef CAS.
- X. Song, H. Wang, X. Yang, F. Liu, S. Yu and S. Liu, Polym. Degrad. Stab., 2014, 110, 65–70 CrossRef CAS.
- F. Codari, S. Lazzari, M. Soos, G. Storti, M. Morbidelli and D. Moscatelli, Polym. Degrad. Stab., 2012, 97, 2460–2466 CrossRef CAS.
- S. J. de Jong, E. R. Arias, D. T. S. Rijkers, C. F. van Nostrum, J. J. Kettenes-van den Bosch and W. E. Hennink, Polymer, 2001, 42, 2795–2802 CrossRef CAS.
- M. Niaounakis, Eur. Polym. J., 2019, 114, 464–475 CrossRef CAS.
- NatureWorks, Chemical Recycling, https://www.natureworksllc.com/sustainability/end-of-life-opportunities/chemical-recycling.
- M. Watanabe, Y. Matsuo, T. Matsushita, H. Inomata, T. Miyake and K. Hironaka, Polym. Degrad. Stab., 2009, 94, 2157–2162 CrossRef CAS.
- E. Quaranta, Appl. Catal., B, 2017, 206, 233–241 CrossRef CAS.
- E. Quaranta, E. Mesto, M. Lacalamita, C. Malitesta, E. Mazzotta, E. Scelsi and E. Schingaro, Waste Manage., 2021, 120, 642–649 CrossRef CAS PubMed.
- M. Sun, Z. Xu, N. E. Munyaneza, Y. Zhang, C. Posada and G. Liu, Polym. Chem., 2023, 14, 1915–1922 RSC.
- J. J. Rubio Arias, E. Barnard and W. Thielemans, ChemSusChem, 2022, 15, e202200625 CrossRef CAS PubMed.
- C. Alberti, N. Damps, R. R. R. Meißner and S. Enthaler, ChemistrySelect, 2019, 4, 6845–6848 CrossRef CAS.
- C. Alberti and S. Enthaler, Asian J. Org. Chem., 2020, 9, 359–363 CrossRef CAS.
- C. Alberti, H. R. Kricheldorf and S. Enthaler, ChemistrySelect, 2020, 5, 12313–12316 CrossRef CAS.
- K. Ikenaga, K. Higuchi, S. Kohri and K. Kusakabe, IOP Conf. Ser.: Mater. Sci. Eng., 2018, 012037 Search PubMed.
- J. Scheirs and T. E. Long, Modern Polyesters: Chemistry and Technology of Polyesters and Copolyesters, John Wiley and Sons, 2005 Search PubMed.
- H. Kurokawa, M.-a. Ohshima, K. Sugiyama and H. Miura, Polym. Degrad. Stab., 2003, 79, 529–533 CrossRef CAS.
- Z. T. Laldinpuii, V. Khiangte, S. Lalhmangaihzuala, C. Lalmuanpuia, Z. Pachuau, C. Lalhriatpuia and K. Vanlaldinpuia, J. Polym. Environ., 2022, 30, 1600–1614 CrossRef CAS.
- P. McKeown, M. Kamran, M. G. Davidson, M. D. Jones, L. A. Román-Ramírez and J. Wood, Green Chem., 2020, 22, 3721–3726 RSC.
- C. Li, G. Yan, Z. Dong, G. Zhang and F. Zhang, Nat. Commun., 2025, 16, 2482 CrossRef CAS PubMed.
- V. Sinha, M. R. Patel and J. V. Patel, J. Polym. Environ., 2010, 18, 8–25 CrossRef CAS.
- L. Bartolome, M. Imran, B. G. Cho, W. A. Al-Masry and D. H. Kim, in Material Recycling – Trends and Perpective, ed. D. S. Achilias, 2012, pp. 65–84, DOI:10.5772/33800.
- X. Song, X. Zhang, H. Wang, F. Liu, S. Yu and S. Liu, Polym. Degrad. Stab., 2013, 98, 2760–2764 CrossRef CAS.
- F. M. Lamberti, L. A. Román-Ramírez, A. P. Dove and J. Wood, Polymers, 2022, 14, 1763 CrossRef CAS PubMed.
- E. Cheung, C. Alberti and S. Enthaler, ChemistryOpen, 2020, 9, 1224–1228 CrossRef CAS PubMed.
- D. D. Pham and J. Cho, Green Chem., 2021, 23, 511–525 RSC.
- D. Paszun and T. Spychaj, Ind. Eng. Chem. Res., 1997, 36, 1373–1383 CrossRef CAS.
- M. Muszyński, J. Nowicki, M. Zygadło and G. Dudek, Molecules, 2023, 28, 6385 CrossRef PubMed.
- R. L. Anderson and D. L. Sikkenga, WO2007076384, 2007.
- F. Liu, J. Guo, P. Zhao, M. Jia, M. Liu and J. Gao, Polym. Degrad. Stab., 2019, 169, 108996 CrossRef.
- F. Liu, L. Li, S. Yu, Z. Lv and X. Ge, J. Hazard. Mater., 2011, 189, 249–254 CrossRef CAS PubMed.
- L. Li, F. Liu, Z. Li, X. Song, S. Yu and S. Liu, Fibers Polym., 2013, 14, 365–368 CrossRef CAS.
- M. Liu, J. Guo, Y. Gu, J. Gao, F. Liu and S. Yu, ACS Sustainable Chem. Eng., 2018, 6, 13114–13121 CrossRef CAS.
- S. Tanaka, M. Koga, T. Kuragano, A. Ogawa, H. Ogiwara, K. Sato and Y. Nakajima, ACS Mater. Au, 2024, 4, 335–345 CrossRef CAS PubMed.
- F. D'Anna, M. Sbacchi, G. Infurna, N. T. Dintcheva and S. Marullo, Green Chem., 2021, 23, 9957–9967 RSC.
- A. K. Manal, G. Saini and R. Srivastava, Green Chem., 2024, 26, 3814–3831 RSC.
- W. Huang, H. Wang, W. Hu, D. Yang, S. Yu, F. Liu and X. Song, RSC Adv., 2021, 11, 1595–1604 RSC.
- E. Quaranta, D. Sgherza and G. Tartaro, Green Chem., 2017, 19, 5422–5434 RSC.
- T. Do, E. R. Baral and J. G. Kim, Polymer, 2018, 143, 106–114 CrossRef CAS.
- D. Parida, A. Aerts, L. Vargas Perez, C. Marquez, S. Vloemans, K. Vanbroekhoven, E. Feghali and K. Elst, Chem. Eng. J., 2024, 497, 154390 CrossRef CAS.
- P. A. Krisbiantoro, M. Sato, T.-M. Lin, Y.-C. Chang, T.-Y. Peng, Y.-C. Wu, W. Liao, Y. Kamiya, R. Otomo and K. C. W. Wu, Langmuir, 2024, 40, 5338–5347 CrossRef CAS PubMed.
- Y. Xu, Y. Ji, Y. Liu, W. Deng, F. Huang and F. Zhang, ChemCatChem, 2024, 16, e202301763 CrossRef CAS.
- M. Hofmann, J. Sundermeier, C. Alberti and S. Enthaler, ChemistrySelect, 2020, 5, 10010–10014 CrossRef CAS.
- Y. Peng, J. Yang, C. Deng, J. Deng, L. Shen and Y. Fu, Nat. Commun., 2023, 14, 3249 CrossRef CAS PubMed.
- T. Li, S. Menegatti and N. Crook, AIChE J., 2023, 69, e18228 CrossRef CAS.
- R.-J. Mueller, Process Biochem., 2006, 41, 2124–2128 CrossRef CAS.
- S. Lu, Y. Jing, B. Feng, Y. Guo, X. Liu and Y. Wang, ChemSusChem, 2021, 14, 4242–4250 CrossRef CAS PubMed.
- S. Westhues, J. Idel and J. Klankermayer, Sci. Adv., 2018, 4, eaat9669 CrossRef CAS PubMed.
- E. M. Krall, T. W. Klein, R. J. Andersen, A. J. Nett, R. W. Glasgow, D. S. Reader, B. C. Dauphinais, S. P. Mc Ilrath, A. A. Fischer, M. J. Carney, D. J. Hudson and N. J. Robertson, Chem. Commun., 2014, 50, 4884–4887 RSC.
- E. Balaraman, E. Khaskin, G. Leitus and D. Milstein, Nat. Chem., 2013, 5, 122–125 CrossRef CAS PubMed.
- J. A. Fuentes, S. M. Smith, M. T. Scharbert, I. Carpenter, D. B. Cordes, A. M. Z. Slawin and M. L. Clarke, Chem.–Eur. J., 2015, 21, 10851–10860 CrossRef CAS PubMed.
- E. Feghali and T. Cantat, ChemSusChem, 2015, 8, 980–984 CrossRef CAS PubMed.
- M. Meuresch, S. Westhues, W. Leitner and J. Klankermayer, Angew. Chem., Int. Ed., 2016, 55, 1392–1395 CrossRef CAS PubMed.
- A. Kumar, N. von Wolff, M. Rauch, Y.-Q. Zou, G. Shmul, Y. Ben-David, G. Leitus, L. Avram and D. Milstein, J. Am. Chem. Soc., 2020, 142, 14267–14275 CrossRef CAS PubMed.
- Y. Kratish, J. Li, S. Liu, Y. Gao and T. J. Marks, Angew. Chem., Int. Ed., 2020, 59, 19857–19861 CrossRef CAS PubMed.
- S. Hongkailers, Y. Jing, Y. Wang, N. Hinchiranan and N. Yan, ChemSusChem, 2021, 14, 4330–4339 CrossRef CAS PubMed.
- P. Wu, G. Lu and C. Cai, Green Chem., 2021, 23, 8666–8672 RSC.
- Y. Jing, Y. Wang, S. Furukawa, J. Xia, C. Sun, M. J. Hülsey, H. Wang, Y. Guo, X. Liu and N. Yan, Angew. Chem., Int. Ed., 2021, 60, 5527–5535 CrossRef CAS PubMed.
- J. Li, Z. An, Y. Kong, L. Zhang, J. Yang, X. Wang, J. Wang, D. Duan, Q. Zhang, R. Long, D. G. Vlachos and Z. Li, Appl. Catal., B, 2024, 357, 124307 CrossRef CAS.
- J. E. Rorrer, G. T. Beckham and Y. Román-Leshkov, JACS Au, 2021, 1, 8–12 CrossRef CAS PubMed.
- J. E. Rorrer, C. Troyano-Valls, G. T. Beckham and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2021, 9, 11661–11666 CrossRef CAS.
- Y. Nakaji, M. Tamura, S. Miyaoka, S. Kumagai, M. Tanji, Y. Nakagawa, T. Yoshioka and K. Tomishige, Appl. Catal., B, 2021, 285, 119805 CrossRef CAS.
- K. Saito, T. Masuyama, K. Oyaizu and H. Nishide, Chem.–Eur. J., 2003, 9, 4240–4246 CrossRef CAS PubMed.
- Y. Shimoyama and Y. Nakajima, ChemSusChem, 2023, 16, e202300684 CrossRef CAS PubMed.
- D. H. Kim, A. Yu and M. Goh, J. Ind. Eng. Chem., 2021, 96, 76–81 CrossRef CAS.
- G. Lyu, C. G. Yoo and X. Pan, Biomass Bioenergy, 2018, 108, 7–14 CrossRef CAS.
- C. Crestini, M. Crucianelli, M. Orlandi and R. Saladino, Catal. Today, 2010, 156, 8–22 CrossRef CAS.
- H. Lange, S. Decina and C. Crestini, Eur. Polym. J., 2013, 49, 1151–1173 CrossRef CAS.
- L. G. Akim, J. L. Colodette and D. S. Argyropoulos, Can. J. Chem., 2001, 79, 201–210 CAS.
- F. Sadaka, I. Campistron, A. Laguerre and J.-F. Pilard, Polym. Degrad. Stab., 2012, 97, 816–828 CrossRef CAS.
- X. Luo, J. Zhan, Q. Mei and S. Zhang, Green Chem., 2023, 25, 6717–6727 RSC.
- E. Bäckström, K. Odelius and M. Hakkarainen, Ind. Eng. Chem. Res., 2017, 56, 14814–14821 CrossRef.
- E. Bäckström, K. Odelius and M. Hakkarainen, ACS Sustainable Chem. Eng., 2019, 7, 11004–11013 CrossRef.
- V. S. Nguyen, Y. Chang, E. V. Phillips, J. A. DeWitt and C. Sievers, ACS Sustainable Chem. Eng., 2023, 11, 7617–7623 CrossRef CAS.
- S. N. Firmansyah, H. Sun, C.-J. Yoo and J. K. Ko, Chem. Eng. J., 2025, 504, 158823 CrossRef CAS.
- K. Wang, R. Jia, P. Cheng, L. Shi, X. Wang and L. Huang, Angew. Chem., Int. Ed., 2023, 62, e202301340 CrossRef CAS PubMed.
- Q. Zhang, J. He, X. Wei, C. Shen, P. Ye, W. An, X. Liu, H. Li, S. Xu, Z. Su and Y.-Z. Wang, Angew. Chem., Int. Ed., 2024, 63, e202407510 CrossRef CAS PubMed.
- Y. Chang, Y. Xiao, M. Sun, W. Gao, L. Zhu, Q. Wang, W.-J. Wang, B.-G. Li and P. Liu, Macromolecules, 2024, 57, 9943–9949 CrossRef CAS.
- Z. Xu, N. E. Munyaneza, Q. Zhang, M. Sun, C. Posada, P. Venturo, N. A. Rorrer, J. Miscall, B. G. Sumpter and G. Liu, Science, 2023, 381, 666–671 CrossRef CAS.
- A. Ong, J. Y. Q. Teo, Z. Feng, T. T. Y. Tan and J. Y. C. Lim, ACS Sustainable Chem. Eng., 2023, 11, 12514–12522 CrossRef CAS.
- C. Sun, Y. Guo, X. Liu and Y. Wang, Catal. Sci. Technol., 2024, 14, 6584–6591 RSC.
- Q. Chen, H. Yan, K. Zhao, S. Wang, D. Zhang, Y. Li, R. Fan, J. Li, X. Chen, X. Zhou, Y. Liu, X. Feng, D. Chen and C. Yang, Nat. Commun., 2024, 15, 10732 CrossRef CAS PubMed.
- Y. Yu, Y. Qi, J. Tang, B. Yan, L. Lou, W. Wu and Q. Mei, Chem. Eng. J., 2025, 508, 160970 CrossRef CAS.
- K. P. Sullivan, A. Z. Werner, K. J. Ramirez, L. D. Ellis, J. R. Bussard, B. A. Black, D. G. Brandner, F. Bratti, B. L. Buss, X. Dong, S. J. Haugen, M. A. Ingraham, M. O. Konev, W. E. Michener, J. Miscall, I. Pardo, S. P. Woodworth, A. M. Guss, Y. Román-Leshkov, S. S. Stahl and G. T. Beckham, Science, 2022, 378, 207–211 CrossRef CAS PubMed.
- C. Rabot, Y. Chen, S. Bijlani, Y.-M. Chiang, C. E. Oakley, B. R. Oakley, T. J. Williams and C. C. C. Wang, Angew. Chem., Int. Ed., 2023, 62, e202214609 CrossRef CAS PubMed.
- A. C. Fernandes, ChemSusChem, 2021, 14, 4228–4233 CrossRef CAS PubMed.
- C. Berti, E. Binassi, A. Celli, M. Colonna, M. Fiorini, P. Marchese, E. Marianucci, M. Gazzano, F. Di Credico and D. J. Brunelle, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 619–630 CrossRef CAS.
- A. B. Lende, S. Bhattacharjee and C.-S. Tan, Catal. Today, 2022, 388, 117–124 CrossRef.
- A. B. Lende, S. Bhattacharjee and C.-S. Tan, ACS Sustainable Chem. Eng., 2021, 9, 7224–7234 CrossRef CAS.
- B. F. S. Nunes, M. C. Oliveira and A. C. Fernandes, Green Chem., 2020, 22, 2419–2425 RSC.
- M. Kobylarski, J.-C. Berthet and T. Cantat, Chem. Commun., 2022, 58, 2830–2833 RSC.
- M. Kobylarski, L. J. Donnelly, J.-C. Berthet and T. Cantat, Green Chem., 2022, 24, 6810–6815 RSC.
- A. Maisels, A. Hiller and F.-G. Simon, ChemBioEng Rev., 2022, 9, 541–555 CrossRef CAS.
- Axens, Rewind PET Process, https://www.axens.net/resources-events/news/axens-ifpen-and-jeplan-announce-start-operation-rewind-pet-semi-industrial-japan, accessed 9 September, 2024.
- Ioniqa, Ioniqa's Circular Solution for PET, https://ioniqa.com/applications/.
- GR3N, Turning unrecyclable plastic waste PET into 100% virgin material for a full circular economy, https://gr3n-recycling.com/project-1.html.
- LoopIndustries, Leading the Sustainable Plastic Revolution, https://www.loopindustries.com/en/tech.
- H. Essaddam, WO2017007965A1, 2017.
- GABRO, ChemPET, https://garbo.it/en/chempet/, accessed 18 Januray, 2024.
- Y. Wang, Y. Zhang, H. Song, Y. Wang, T. Deng and X. Hou, J. Cleaner Prod., 2019, 208, 1469–1475 CrossRef CAS.
- R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, S. Arnaiz and J. I. Gutiérrez-Ortiz, Polym. Degrad. Stab., 2010, 95, 1022–1028 CrossRef.
- L. Zhang, J. Gao, J. Zou and F. Yi, J. Appl. Polym. Sci., 2013, 130, 2790–2795 CrossRef CAS.
- P. Fang, B. Liu, J. Xu, Q. Zhou, S. Zhang, J. Ma and X. lu, Polym. Degrad. Stab., 2018, 156, 22–31 CrossRef CAS.
- A. M. Al-Sabagh, F. Z. Yehia, A. M. F. Eissa, M. E. Moustafa, G. Eshaq, A. M. Rabie and A. E. ElMetwally, Polym. Degrad. Stab., 2014, 110, 364–377 CrossRef CAS.
- X.-K. Li, H. Lu, W.-Z. Guo, G.-P. Cao, H.-L. Liu and Y.-H. Shi, AIChE J., 2015, 61, 200–214 CrossRef CAS.
- R. López-Fonseca, J. R. González-Velasco and J. I. Gutiérrez-Ortiz, Chem. Eng. J., 2009, 146, 287–294 CrossRef.
- H. S. Joo and J. A. Guin, Energy Fuels, 1997, 11, 586–592 CrossRef CAS.
- J.-M. Basset, C. Coperet, D. Soulivong, M. Taoufik and J. T. Cazat, Acc. Chem. Res., 2010, 43, 323–334 CrossRef CAS PubMed.
- M. K. Samantaray, R. Dey, E. Abou-Hamad, A. Hamieh and J.-M. Basset, Chem.–Eur. J., 2015, 21, 6100–6106 CrossRef CAS PubMed.
- M. K. Samantaray, S. Kavitake, N. Morlanés, E. Abou-Hamad, A. Hamieh, R. Dey and J.-M. Basset, J. Am. Chem. Soc., 2017, 139, 3522–3527 CrossRef CAS PubMed.
- M. K. Samantaray, E. Callens, E. Abou-Hamad, A. J. Rossini, C. M. Widdifield, R. Dey, L. Emsley and J.-M. Basset, J. Am. Chem. Soc., 2014, 136, 1054–1061 CrossRef CAS PubMed.
- W. Wackerow, M. K. Samantaray and J.-M. Basset, ChemCatChem, 2020, 12, 5627–5631 CrossRef CAS.
- N. Morlanés, S. G. Kavitake, D. C. Rosenfeld and J.-M. Basset, ACS Catal., 2019, 9, 1274–1282 CrossRef.
- M. C. Haibach, S. Kundu, M. Brookhart and A. S. Goldman, Acc. Chem. Res., 2012, 45, 947–958 CrossRef CAS PubMed.
- H. Özer, D. Arslan and B. Ö. Öztürk, New J. Chem., 2021, 45, 5992–6000 RSC.
- D. Soulivong, C. Copéret, J. Thivolle-Cazat, J.-M. Basset, B. M. Maunders, R. B. A. Pardy and G. J. Sunley, Angew. Chem., Int. Ed., 2004, 43, 5366–5369 CrossRef CAS PubMed.
- C. Megías-Sayago, I. Centeno-Vega, L. F. Bobadilla, S. Ivanova, N. Rendón and A. Suarez, Appl. Catal., B, 2023, 338, 123002 CrossRef.
- A. Dewaele, T. Renders, B. Yu, F. Verpoort and B. F. Sels, Catal. Sci. Technol., 2016, 6, 7708–7717 RSC.
- J. A. Herman, M. E. Seazzu, L. G. Hughes, D. R. Wheeler, C. M. Washburn and B. H. Jones, ACS Appl. Polym. Mater., 2019, 1, 2177–2188 CrossRef CAS.
- L. D. Ellis, S. V. Orski, G. A. Kenlaw, A. G. Norman, K. L. Beers, Y. Román-Leshkov and G. T. Beckham, ACS Sustainable Chem. Eng., 2021, 9, 623–628 CrossRef CAS PubMed.
- W. Kaminsk, Polyolefins: 50 years after Ziegler and Natta I: Polyethylene and Polypropylene, Springer, Heidelberg, 2013 Search PubMed.
- D. Nwabunma and T. Kyu, Polyolefin Blends, John Wiley & Sons, Inc., 2007 Search PubMed.
- X. Jia, C. Qin, T. Friedberger, Z. Guan and Z. Huang, Sci. Adv., 2016, 2, e1501591 CrossRef PubMed.
- V. Vidal, A. Théolier, J. Thivolle-Cazat and J.-M. Basset, Science, 1997, 276, 99–102 CrossRef CAS PubMed.
- R. L. Burnett and T. R. Hughes, J. Catal., 1973, 31, 55–64 CrossRef CAS.
- Z. Huang, E. Rolfe, E. C. Carson, M. Brookhart, A. S. Goldman, S. H. El-Khalafy and A. H. R. MacArthur, Adv. Synth. Catal., 2010, 352, 125–135 CrossRef CAS.
- F. Zhang, M. Zeng, R. D. Yappert, J. Sun, Y.-H. Lee, A. M. LaPointe, B. Peters, M. M. Abu-Omar and S. L. Scott, Science, 2020, 370, 437–441 CrossRef CAS PubMed.
- X. Michel, S. Fouquay, G. Michaud, F. Simon, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Eur. Polym. J., 2017, 96, 403–413 CrossRef CAS.
- A. Mouawia, A. Nourry, A.-C. Gaumont, J.-F. Pilard and I. Dez, ACS Sustainable Chem. Eng., 2017, 5, 696–700 CrossRef CAS.
- C. Ai, G. Gong, X. Zhao and P. Liu, Polym. Test., 2017, 60, 250–252 CrossRef CAS.
- G. Hu, S. Lin, B. Zhao and Q. Pan, J. Appl. Polym. Sci., 2021, 138, 49899 CrossRef CAS.
- M. Burelo, S. Gutiérrez, C. D. Treviño-Quintanilla, J. A. Cruz-Morales, A. Martínez and S. López-Morales, Polymers, 2022, 14, 4973 CrossRef CAS PubMed.
- J. C. Foster, J. Zheng, M. Arifuzzaman, M. A. Rahman, J. T. Damron, C. Guan, I. Popovs, N. Galan, Z. Demchuk and T. Saito, Cell Rep. Phys. Sci., 2023, 4, 101734 CrossRef CAS.
- W. K. Fan and M. Tahir, Energy Convers. Manage., 2022, 253, 115180 CrossRef CAS.
- C. M. Pichler, S. Bhattacharjee, M. Rahaman, T. Uekert and E. Reisner, ACS Catal., 2021, 11, 9159–9167 CrossRef CAS PubMed.
- Z. Wang, V. Srivastava, S. Wang, H. Sun, S. K. Thangaraj, J. Jänis and M. Sillanpää, J. Colloid Interface Sci., 2020, 562, 461–469 CrossRef CAS PubMed.
- X. Jiao, K. Zheng, Q. Chen, X. Li, Y. Li, W. Shao, J. Xu, J. Zhu, Y. Pan, Y. Sun and Y. Xie, Angew. Chem., Int. Ed., 2020, 59, 15497–15501 CrossRef CAS PubMed.
- H. Zhou, H. Wang, C. Yue, L. He, H. Li, H. Zhang, S. Yang and T. Ma, Appl. Catal., B, 2024, 344, 123605 CrossRef CAS.
- S. Gazi, M. Đokić, K. F. Chin, P. R. Ng and H. S. Soo, Adv. Sci., 2019, 6, 1902020 CrossRef CAS PubMed.
- G. Peng, X. Qi, W. Qu, X. Shao, L. Song, P. Du and J. Xiong, Catal. Sci. Technol., 2023, 13, 5868–5879 RSC.
- H. Kang, A. Washington, M. D. Capobianco, X. Yan, V. V. Cruz, M. Weed, J. Johnson, G. Johns III, G. W. Brudvig, X. Pan and J. Gu, ACS Mater. Lett., 2023, 5, 3032–3041 CrossRef CAS PubMed.
- Y. Jiang, H. Zhang, L. Hong, J. Shao, B. Zhang, J. Yu and S. Chu, ChemSusChem, 2023, 16, e202300106 CrossRef CAS PubMed.
- A. E. Nogueira, O. F. Lopes, A. B. S. Neto and C. Ribeiro, Chem. Eng. J., 2017, 312, 220–227 CrossRef CAS.
- J. Xu, X. Jiao, K. Zheng, W. Shao, S. Zhu, X. Li, J. Zhu, Y. Pan, Y. Sun and Y. Xie, Natl. Sci. Rev., 2022, 9, nwac011 CrossRef CAS PubMed.
- T. S. Tofa, K. L. Kunjali, S. Paul and J. Dutta, Environ. Chem. Lett., 2019, 17, 1341–1346 CrossRef CAS.
- X. Jiao, Z. Hu, K. Zheng, J. Zhu, Y. Wu, X. Zhang, J. Hu, W. Yan, J. Zhu, Y. Sun and Y. Xie, Nano Lett., 2022, 22, 10066–10072 CrossRef CAS PubMed.
- J. Qin, Y. Dou, F. Wu, Y. Yao, H. R. Andersen, C. Hélix-Nielsen, S. Y. Lim and W. Zhang, Appl. Catal., B, 2022, 319, 121940 CrossRef CAS.
- Y. Miao, Y. Zhao, G. I. N. Waterhouse, R. Shi, L.-Z. Wu and T. Zhang, Nat. Commun., 2023, 14, 4242 CrossRef CAS PubMed.
- M. Li and S. Zhang, ACS Catal., 2024, 14, 2949–2958 CrossRef CAS.
- T. Uekert, M. F. Kuehnel, D. W. Wakerley and E. Reisner, Energy Environ. Sci., 2018, 11, 2853–2857 RSC.
- H. Nagakawa and M. Nagata, ACS Appl. Mater. Interfaces, 2021, 13, 47511–47519 CrossRef CAS PubMed.
- Y. Miao, Y. Zhao, J. Gao, J. Wang and T. Zhang, J. Am. Chem. Soc., 2024, 146, 4842–4850 CrossRef CAS PubMed.
- M. Du, Y. Zhang, S. Kang, X. Guo, Y. Ma, M. Xing, Y. Zhu, Y. Chai and B. Qiu, ACS Catal., 2022, 12, 12823–12832 CrossRef CAS.
- Y. Li, S. Wan, C. Lin, Y. Gao, Y. Lu, L. Wang and K. Zhang, Sol. RRL, 2021, 5, 2000427 CrossRef CAS.
- S. Zhang, H. Li, L. Wang, J. Liu, G. Liang, K. Davey, J. Ran and S.-Z. Qiao, J. Am. Chem. Soc., 2023, 145, 6410–6419 CrossRef CAS PubMed.
- T. Uekert, H. Kasap and E. Reisner, J. Am. Chem. Soc., 2019, 141, 15201–15210 CrossRef CAS PubMed.
- T. Uekert, M. A. Bajada, T. Schubert, C. M. Pichler and E. Reisner, ChemSusChem, 2021, 14, 4190–4197 CrossRef CAS PubMed.
- M. Han, S. Zhu, C. Xia and B. Yang, Appl. Catal., B, 2022, 316, 121662 CrossRef CAS.
- R. Cao, M.-Q. Zhang, C. Hu, D. Xiao, M. Wang and D. Ma, Nat. Commun., 2022, 13, 4809 CrossRef CAS PubMed.
- J. Qin, Y. Dou, J. Zhou, V. M. Candelario, H. R. Andersen, C. Hélix-Nielsen and W. Zhang, Adv. Funct. Mater., 2023, 33, 2214839 CrossRef CAS.
- C. Xing, G. Yu, J. Zhou, Q. Liu, T. Chen, H. Liu and X. Li, Appl. Catal., B, 2022, 315, 121496 CrossRef CAS.
- X. Gong, F. Tong, F. Ma, Y. Zhang, P. Zhou, Z. Wang, Y. Liu, P. Wang, H. Cheng, Y. Dai, Z. Zheng and B. Huang, Appl. Catal., B, 2022, 307, 121143 CrossRef CAS.
- A. Mehta, R. A. Rather, B. Belec, S. Gardonio, M. Fang and M. Valant, Energies, 2022, 15, 1734 CrossRef CAS.
- R. Gogoi, A. Singh, V. Moutam, L. Sharma, K. Sharma, A. Halder and P. F. Siril, J. Environ. Chem. Eng., 2022, 10, 106649 CrossRef CAS.
- X.-G. Li, A. Li, M.-R. Huang, Y. Liao and Y.-G. Lu, J. Phys. Chem. C, 2010, 114, 19244–19255 CrossRef CAS.
- Y. Liu, Q. Zhong, P. Xu, H. Huang, F. Yang, M. Cao, L. He, Q. Zhang and J. Chen, Matter, 2022, 5, 1305–1317 CrossRef CAS.
- Y. Liu, X. Wang, Q. Li, T. Yan, X. Lou, C. Zhang, M. Cao, L. Zhang, T.-K. Sham, Q. Zhang, L. He and J. Chen, Adv. Funct. Mater., 2023, 33, 2210283 CrossRef CAS.
- Z. Peng, R. Chen and H. Li, ACS Sustainable Chem. Eng., 2023, 11, 10688–10697 CrossRef CAS.
- D. Ouyang, D. Gao, Y. Qiang and X. Zhao, Appl. Catal., B, 2023, 328, 122491 CrossRef CAS.
- N. Cai, S. Xia, X. Zhang, Z. Meng, P. Bartocci, F. Fantozzi, Y. Chen, H. Chen, P. T. Williams and H. Yang, ChemSusChem, 2020, 13, 938–944 CrossRef CAS PubMed.
- R. A. Mir and O. P. Pandey, J. Cleaner Prod., 2019, 218, 644–655 CrossRef CAS.
- H. Zhou, Y. Ren, Z. Li, M. Xu, Y. Wang, R. Ge, X. Kong, L. Zheng and H. Duan, Nat. Commun., 2021, 12, 4679 CrossRef CAS PubMed.
- S. Behera, S. Dinda, R. Saha and B. Mondal, ACS Catal., 2023, 13, 469–474 CrossRef CAS.
- R. Shi, K.-S. Liu, F. Liu, X. Yang, C.-C. Hou and Y. Chen, Chem. Commun., 2021, 57, 12595–12598 RSC.
- Z. Chen, W. Wei, Y. Shen and B.-J. Ni, Green Chem., 2023, 25, 5979–5988 RSC.
- X.-H. Wang, Z.-N. Zhang, Z. Wang, Y. Ding, Q.-G. Zhai, Y.-C. Jiang, S.-N. Li and Y. Chen, Chem. Eng. J., 2023, 465, 142938 CrossRef CAS.
- F. Ma, S. Wang, X. Gong, X. Liu, Z. Wang, P. Wang, Y. Liu, H. Cheng, Y. Dai, Z. Zheng and B. Huang, Appl. Catal. B Environ., 2022, 307, 121198 CrossRef CAS.
- N. Wang, X. Li, M.-K. Hu, W. Wei, S.-H. Zhou, X.-T. Wu and Q.-L. Zhu, Appl. Catal., B, 2022, 316, 121667 CrossRef CAS.
- F. Liu, X. Gao, R. Shi, Z. Guo, E. C. M. Tse and Y. Chen, Angew. Chem., Int. Ed., 2023, 62, e202300094 CrossRef CAS PubMed.
- J. Wang, X. Li, M. Wang, T. Zhang, X. Chai, J. Lu, T. Wang, Y. Zhao and D. Ma, ACS Catal., 2022, 12, 6722–6728 CrossRef CAS.
- S. K. Kilaparthi, A. Addad, A. Barras, S. Szunerits and R. Boukherroub, J. Mater. Chem. A, 2023, 11, 26075–26085 RSC.
- Y. Mao, S. Fan, X. Li, J. Shi, M. Wang, Z. Niu and G. Chen, J. Hazard. Mater., 2023, 457, 131743 CrossRef CAS PubMed.
- T. Ren, Z. Yu, H. Yu, K. Deng, Z. Wang, X. Li, H. Wang, L. Wang and Y. Xu, ACS Nano, 2023, 17, 12422–12432 CrossRef CAS PubMed.
- T. Li and D. A. Harrington, ChemSusChem, 2021, 14, 1472–1495 CrossRef CAS PubMed.
- Y. Yan, H. Zhou, S.-M. Xu, J. Yang, P. Hao, X. Cai, Y. Ren, M. Xu, X. Kong, M. Shao, Z. Li and H. Duan, J. Am. Chem. Soc., 2023, 145, 6144–6155 CrossRef CAS PubMed.
- X. Liu, J. Wang, Z. Fang, S. Gong, D. Xiong, W. Chen, D. Wu and Z. Chen, Appl. Catal., B, 2023, 334, 122870 CrossRef CAS.
- S. Hao, L. Chen, C. Yu, B. Yang, Z. Li, Y. Hou, L. Lei and X. Zhang, ACS Energy Lett., 2019, 4, 952–959 CrossRef CAS.
- M. Du, Y. Zhang, S. Kang, C. Xu, Y. Ma, L. Cai, Y. Zhu, Y. Chai and B. Qiu, Small, 2023, 19, 2303693 CrossRef CAS PubMed.
- Y. Li, Y. Zhao, H. Zhao, Z. Wang, H. Li and P. Gao, J. Mater. Chem. A, 2022, 10, 20446–20452 RSC.
- Y. Li, L. Q. Lee, Z. G. Yu, H. Zhao, Y.-W. Zhang, P. Gao and H. Li, Sustainable Energy Fuels, 2022, 6, 4916–4924 RSC.
- F. Liu, X. Gao, R. Shi, E. C. M. Tse and Y. Chen, Green Chem., 2022, 24, 6571–6577 RSC.
- R. Rahimzadeh, J. Ortega-Ramos, Z. Haque and G. G. Botte, ChemElectroChem, 2023, 10, e202300021 CrossRef CAS.
- A. D. Fried, B. J. Wilson, N. J. Galan and J. N. Brantley, J. Am. Ceram. Soc., 2022, 144, 8885–8891 CrossRef CAS PubMed.
- L. Ren, S. Yang, J. Wang, T. Zhang, X. Li, T. Wang and Y. Zhao, React. Chem. Eng., 2023, 8, 1937–1942 RSC.
- R. A. Sheldon and D. Brady, ChemSusChem, 2022, 15, e202102628 CrossRef CAS PubMed.
- V. Tournier, S. Duquesne, F. Guillamot, H. Cramail, D. Taton, A. Marty and I. André, Chem. Rev., 2023, 123, 5612–5701 CrossRef CAS PubMed.
- J. Chow, P. Perez-Garcia, R. Dierkes and W. R. Streit, Microbiol. Biotechnol., 2023, 16, 195–217 CrossRef CAS PubMed.
- Z.-H. Qin, J.-H. Mou, C. Y. H. Chao, S. S. Chopra, W. Daoud, S.-y. Leu, Z. Ning, C. Y. Tso, C. K. Chan, S. Tang, Z. J. Hathi, M. A. Haque, X. Wang and C. S. K. Lin, ChemSusChem, 2021, 14, 4103–4114 CrossRef CAS PubMed.
- F. Kawai, T. Kawabata and M. Oda, Appl. Microbiol. Biotechnol., 2019, 103, 4253–4268 CrossRef CAS PubMed.
- R. Wei, D. Breite, C. Song, D. Gräsing, T. Ploss, P. Hille, R. Schwerdtfeger, J. Matysik, A. Schulze and W. Zimmermann, Adv. Sci., 2019, 6, 1900491 CrossRef PubMed.
- Y. Nakaji, Y. Nakagawa, M. Tamura and K. Tomishige, Fuel Process. Technol., 2018, 176, 249–257 CrossRef CAS.
- R. Wei and W. Zimmermann, Microbiol. Biotechnol., 2017, 10, 1308–1322 CrossRef CAS PubMed.
- A. Carniel, V. d. A. Waldow and A. M. d. Castro, Biotechnol. Adv., 2021, 52, 107811 CrossRef CAS PubMed.
- N. A. Samak, Y. Jia, M. M. Sharshar, T. Mu, M. Yang, S. Peh and J. Xing, Environ. Int., 2020, 145, 106144 CrossRef CAS PubMed.
- R. D. Fields, F. Rodriguez and R. K. Finn, J. Appl. Polym. Sci., 1974, 18, 3571–3579 CrossRef CAS.
- R. Wei, T. Tiso, J. Bertling, K. O'Connor, L. M. Blank and U. T. Bornscheuer, Nat. Catal., 2020, 3, 867–871 CrossRef CAS.
- K. M. Akato, N. A. Nguyen, P. V. Bonnesen, D. P. Harper and A. K. Naskar, ACS Omega, 2018, 3, 10709–10715 CrossRef CAS PubMed.
- D. Danso, J. Chow and R. Streit Wolfgang, Appl. Environ. Microbiol., 2019, 85, e01095–e01019 CrossRef CAS PubMed.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- J. Ru, Y. Huo and Y. Yang, Front. Microbiol., 2020, 11, 442 CrossRef PubMed.
- Y. Tokiwa and T. Suzuki, Nature, 1977, 270, 76–78 CrossRef CAS PubMed.
- M. A. M. E. Vertommen, V. A. Nierstrasz, M. v. d. Veer and M. M. C. G. Warmoeskerken, J. Biotechnol., 2005, 120, 376–386 CrossRef CAS PubMed.
- R.-J. Müller, H. Schrader, J. Profe, K. Dresler and W.-D. Deckwer, Macromol. Rapid Commun., 2005, 26, 1400–1405 CrossRef.
- V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M. L. Desrousseaux, H. Texier, S. Gavalda, M. Cot, E. Guémard, M. Dalibey, J. Nomme, G. Cioci, S. Barbe, M. Chateau, I. André, S. Duquesne and A. Marty, Nature, 2020, 580, 216–219 CrossRef CAS PubMed.
- A. Farzi, A. Dehnad and A. F. Fotouhi, Biocatal. Agric. Biotechnol., 2019, 17, 25–31 CrossRef.
- K. Hiraga, I. Taniguchi, S. Yoshida, Y. Kimura and K. Oda, EMBO Rep., 2019, 20, e49365 CrossRef CAS PubMed.
- I. Taniguchi, S. Yoshida, K. Hiraga, K. Miyamoto, Y. Kimura and K. Oda, ACS Catal., 2019, 9, 4089–4105 CrossRef CAS.
- J. Then, R. Wei, T. Oeser, M. Barth, M. R. Belisário-Ferrari, J. Schmidt and W. Zimmermann, Biotechnol. J., 2015, 10, 592–598 CrossRef CAS PubMed.
- Å. M. Ronkvist, W. Xie, W. Lu and R. A. Gross, Macromolecules, 2009, 42, 5128–5138 CrossRef.
- J. Mican, D. s. M. M. Jaradat, W. Liu, G. Weber, S. Mazurenko, U. T. Bornscheuer, J. Damborsky, R. Wei and D. Bednar, Appl. Catal., B, 2024, 342, 123404 CrossRef CAS.
- S. Brott, L. Pfaff, J. Schuricht, J. N. Schwarz, D. Böttcher, C. P. S. Badenhorst, R. Wei and U. T. Bornscheuer, Eng. Life Sci., 2022, 22, 192–203 CrossRef CAS PubMed.
- R. Xue, C. Qiu, X. Zhou, Y. Cheng, Z. Zhang, Y. Zhang, U. Schröder, U. T. Bornscheuer, W. Dong, R. Wei and M. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202313633 CrossRef CAS PubMed.
- N. A. Tarazona, R. Wei, S. Brott, L. Pfaff, U. T. Bornscheuer, A. Lendlein and R. Machatschek, Chem Catal., 2022, 2, 3573–3589 CrossRef CAS PubMed.
- H. T. Kim, J. K. Kim, H. G. Cha, M. J. Kang, H. S. Lee, T. U. Khang, E. J. Yun, D.-H. Lee, B. K. Song, S. J. Park, J. C. Joo and K. H. Kim, ACS Sustainable Chem. Eng., 2019, 7, 19396–19406 CrossRef CAS.
- F. Kawai, Catalysts, 2021, 11, 206 CrossRef CAS.
- F. Quartinello, S. Vajnhandl, J. Volmajer Valh, T. J. Farmer, B. Vončina, A. Lobnik, E. Herrero Acero, A. Pellis and G. M. Guebitz, Microbiol. Biotechnol., 2017, 10, 1376–1383 CrossRef CAS PubMed.
- F. Yan, R. Wei, Q. Cui, U. T. Bornscheuer and Y.-J. Liu, Microbiol. Biotechnol., 2021, 14, 374–385 CrossRef CAS PubMed.
- F. Kawai, M. Oda, T. Tamashiro, T. Waku, N. Tanaka, M. Yamamoto, H. Mizushima, T. Miyakawa and M. Tanokura, Appl. Microbiol. Biotechnol., 2014, 98, 10053–10064 CrossRef CAS PubMed.
- S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara, K. Miyamoto, Y. Kimura and K. Oda, Science, 2016, 351, 1196–1199 CrossRef CAS PubMed.
- R. Wei, C. Song, D. Gräsing, T. Schneider, P. Bielytskyi, D. Böttcher, J. Matysik, U. T. Bornscheuer and W. Zimmermann, Nat. Commun., 2019, 10, 5581 CrossRef CAS PubMed.
- X. Han, W. Liu, J.-W. Huang, J. Ma, Y. Zheng, T.-P. Ko, L. Xu, Y.-S. Cheng, C.-C. Chen and R.-T. Guo, Nat. Commun., 2017, 8, 2106 CrossRef PubMed.
- S. Joo, I. J. Cho, H. Seo, H. F. Son, H.-Y. Sagong, T. J. Shin, S. Y. Choi, S. Y. Lee and K.-J. Kim, Nat. Commun., 2018, 9, 382 CrossRef PubMed.
- H. P. Austin, M. D. Allen, B. S. Donohoe, N. A. Rorrer, F. L. Kearns, R. L. Silveira, B. C. Pollard, G. Dominick, R. Duman, K. El Omari, V. Mykhaylyk, A. Wagner, W. E. Michener, A. Amore, M. S. Skaf, M. F. Crowley, A. W. Thorne, C. W. Johnson, H. L. Woodcock, J. E. McGeehan and G. T. Beckham, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E4350–e4357 CrossRef CAS PubMed.
- C. Roberts, S. Edwards, M. Vague, R. León-Zayas, H. Scheffer, G. Chan, A. Swartz Natasja and L. Mellies Jay, mSphere, 2020, 5, e01151–e01120 CrossRef CAS PubMed.
- D. Shingwekar, H. Laster, H. Kemp and J. L. Mellies, Bioengineering, 2023, 10, 1253 CrossRef CAS PubMed.
- E. L. Bell, R. Smithson, S. Kilbride, J. Foster, F. J. Hardy, S. Ramachandran, A. A. Tedstone, S. J. Haigh, A. A. Garforth, P. J. R. Day, C. Levy, M. P. Shaver and A. P. Green, Nat. Catal., 2022, 5, 673–681 CrossRef CAS.
- P. Chandra, Enespa, R. Singh and P. K. Arora, Microb. Cell Fact., 2020, 19, 169 CrossRef CAS PubMed.
- A. Carniel, É. Valoni, J. Nicomedes, A. d. C. Gomes and A. M. d. Castro, Process Biochem., 2017, 59, 84–90 CrossRef CAS.
- A. M. de Castro, A. Carniel, J. Nicomedes Junior, A. da Conceição Gomes and É. Valoni, J. Ind. Microbiol. Biotechnol., 2017, 44, 835–844 CrossRef PubMed.
- Y. Zhou, J. Zhang, S. You, W. Lin, B. Zhang, M. Wang, R. Su and W. Qi, Bioresour. Technol., 2024, 413, 131461 CrossRef CAS PubMed.
- J. C. S. Sales, A. M. de Castro, B. D. Ribeiro and M. A. Z. Coelho, Biocatal. Biotransform., 2020, 38, 457–468 CrossRef CAS.
- Y. Yang, J. Yang, W.-M. Wu, J. Zhao, Y. Song, L. Gao, R. Yang and L. Jiang, Environ. Sci. Technol., 2015, 49, 12080–12086 CrossRef CAS PubMed.
- S.-S. Yang, W.-M. Wu, A. M. Brandon, H.-Q. Fan, J. P. Receveur, Y. Li, Z.-Y. Wang, R. Fan, R. L. McClellan, S.-H. Gao, D. Ning, D. H. Phillips, B.-Y. Peng, H. Wang, S.-Y. Cai, P. Li, W.-W. Cai, L.-Y. Ding, J. Yang, M. Zheng, J. Ren, Y.-L. Zhang, J. Gao, D. Xing, N.-Q. Ren, R. M. Waymouth, J. Zhou, H.-C. Tao, C. J. Picard, M. E. Benbow and C. S. Criddle, Chemosphere, 2018, 212, 262–271 CrossRef CAS PubMed.
- B.-Y. Peng, Y. Su, Z. Chen, J. Chen, X. Zhou, M. E. Benbow, C. S. Criddle, W.-M. Wu and Y. Zhang, Environ. Sci. Technol., 2019, 53, 5256–5265 CrossRef CAS PubMed.
- Y. Yang, J. Wang and M. Xia, Sci. Total Environ., 2020, 708, 135233 CrossRef CAS PubMed.
- Y. Song, R. Qiu, J. Hu, X. Li, X. Zhang, Y. Chen, W.-M. Wu and D. He, Sci. Total Environ., 2020, 746, 141289 CrossRef CAS PubMed.
- J. Sun, A. Prabhu, S. T. N. Aroney and C. Rinke, Microb. Genomes, 2022, 8, 842–861 CAS.
- E. A. Di Liberto, G. Battaglia, R. Pellerito, G. Curcuruto and N. T. Dintcheva, Polymers, 2024, 16, 1404 CrossRef CAS PubMed.
- H. R. Kim, H. M. Lee, H. C. Yu, E. Jeon, S. Lee, J. Li and D.-H. Kim, Environ. Sci. Technol., 2020, 54, 6987–6996 CrossRef CAS PubMed.
- R. Usha, T. Sangeetha and P. Muthusamy, Libyan Agric. Res. Cent. J. Int., 2011, 2, 200–204 Search PubMed.
- A. Satlewal, S. Ravindra, Z. Mgh and S. Yogesh, J. Microbiol. Biotechnol., 2008, 18, 477–482 CAS.
- S. T. Azeko, G. A. Etuk-Udo, O. S. Odusanya, K. Malatesta, N. Anuku and W. O. Soboyejo, Waste Biomass Valorization, 2015, 6, 1047–1057 CrossRef CAS.
- P. Sarmah and J. Rout, Environ. Sci. Pollut. Res., 2018, 25, 33508–33520 CrossRef CAS PubMed.
- H. Kundungal, M. Gangarapu, S. Sarangapani, A. Patchaiyappan and S. P. Devipriya, Environ. Sci. Pollut. Res. Int., 2019, 26, 18509–18519 CrossRef CAS PubMed.
- A. Chalup, M. M. Ayup, A. C. Monmany Garzia, A. Malizia, E. Martin, R. De Cristóbal and A. Galindo-Cardona, J. Apic. Res., 2018, 57, 569–571 CrossRef.
- P. Bombelli, C. J. Howe and F. Bertocchini, Curr. Biol., 2017, 27, R292–R293 CrossRef CAS PubMed.
- Y. Yang, J. Chen, W. M. Wu, J. Zhao and J. Yang, J. Biotechnol., 2015, 200, 77–78 CrossRef CAS PubMed.
- S. Deepika and R. J. Madhuri, J. Exp. Biol. Agric. Sci., 2015, 3, 15–21 CAS.
- A. Paço, K. Duarte, J. P. da Costa, P. S. M. Santos, R. Pereira, M. E. Pereira, A. C. Freitas, A. C. Duarte and T. A. P. Rocha-Santos, Sci. Total Environ., 2017, 586, 10–15 CrossRef PubMed.
- C. N. Muhonja, H. Makonde, G. Magoma and M. Imbuga, PLoS One, 2018, 13, e0198446 CrossRef PubMed.
- G. C. Dsouza, R. S. Sheriff, V. Ullanat, A. Shrikrishna, A. V. Joshi, L. Hiremath and K. Entoori, Heliyon, 2021, 7, e07008 CrossRef CAS PubMed.
- T. C. H. Dang, D. T. Nguyen, H. Thai, T. C. Nguyen, T. T. Hien Tran, V. H. Le, V. H. Nguyen, X. B. Tran, T. P. Thao Pham, T. G. Nguyen and Q. T. Nguyen, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2018, 9, 015014 Search PubMed.
- S. Veluru and R. Seeram, Circ. Econ., 2024, 3, 100077 Search PubMed.
- A. Sanluis-Verdes, P. Colomer-Vidal, F. Rodriguez-Ventura, M. Bello-Villarino, M. Spinola-Amilibia, E. Ruiz-Lopez, R. Illanes-Vicioso, P. Castroviejo, R. Aiese Cigliano, M. Montoya, P. Falabella, C. Pesquera, L. Gonzalez-Legarreta, E. Arias-Palomo, M. Solà, T. Torroba, C. F. Arias and F. Bertocchini, Nat. Commun., 2022, 13, 5568 CrossRef CAS PubMed.
- S. Skariyachan, N. Taskeen, A. P. Kishore, B. V. Krishna and G. Naidu, J. Environ. Manage., 2021, 284, 112030 CrossRef CAS PubMed.
- A. Kowalczyk, M. Chyc, P. Ryszka and D. Latowski, Environ. Sci. Pollut. Res. Int., 2016, 23, 11349–11356 CrossRef CAS PubMed.
- N. Ojha, N. Pradhan, S. Singh, A. Barla, A. Shrivastava, P. Khatua, V. Rai and S. Bose, Sci. Rep., 2017, 7, 39515 CrossRef CAS PubMed.
- H. J. Jeon and M. N. Kim, Int. Biodeterior. Biodegrad., 2016, 114, 202–208 CrossRef CAS.
- J. Xu, Z. Cui, K. Nie, H. Cao, M. Jiang, H. Xu, T. Tan and L. Liu, Front. Microbiol., 2019, 10, 489 CrossRef PubMed.
- N. Yan, Science, 2022, 378, 132–133 CrossRef CAS PubMed.
- K. Jain, H. Bhunia and M. Sudhakara Reddy, Biorem. J., 2018, 22, 73–90 CrossRef CAS.
- S.-S. Yang, M.-Q. Ding, L. He, C.-H. Zhang, Q.-X. Li, D.-F. Xing, G.-L. Cao, L. Zhao, J. Ding, N.-Q. Ren and W.-M. Wu, Sci. Total Environ., 2021, 756, 144087 CrossRef CAS PubMed.
- J. P. Pires, G. M. Miranda, G. L. de Souza, F. Fraga, A. da Silva Ramos, G. E. de Araújo, R. A. Ligabue, C. M. N. Azevedo, R. V. Lourega and J. E. A. de Lima, Iran. Polym. J., 2019, 28, 1045–1055 CrossRef CAS.
- H. S. Auta, C. U. Emenike, B. Jayanthi and S. H. Fauziah, Mar. Pollut. Bull., 2018, 127, 15–21 CrossRef CAS PubMed.
- M. I. Ali, S. Ahmed, G. Robson, I. Javed, N. Ali, N. Atiq and A. Hameed, J. Basic Microbiol., 2014, 54, 18–27 CrossRef CAS PubMed.
- N. Khatoon, A. Jamal and M. I. Ali, Environ. Technol., 2019, 40, 1366–1375 CrossRef CAS PubMed.
- Z. Zhang, H. Peng, D. Yang, G. Zhang, J. Zhang and F. Ju, Nat. Commun., 2022, 13, 5360 CrossRef CAS PubMed.
- L. Giacomucci, N. Raddadi, M. Soccio, N. Lotti and F. Fava, N. Biotechnol., 2019, 52, 35–41 CrossRef CAS PubMed.
- A. Kumari, D. R. Chaudhary and B. Jha, Environ. Sci. Pollut. Res. Int., 2019, 26, 1507–1516 CrossRef CAS PubMed.
- L. Giacomucci, N. Raddadi, M. Soccio, N. Lotti and F. Fava, Mar. Environ. Res., 2020, 158, 104949 CrossRef CAS PubMed.
- T. Sumathi, B. Viswanath, A. Sri Lakshmi and D. V. R. SaiGopal, Biochem. Res. Int., 2016, 2016, 9519527 Search PubMed.
- M. S. Anwar, A. Kapri, V. Chaudhry, A. Mishra, M. W. Ansari, Y. Souche, C. S. Nautiyal, M. G. H. Zaidi and R. Goel, Protoplasma, 2016, 253, 1023–1032 CrossRef CAS PubMed.
- B.-Y. Peng, Z. Chen, J. Chen, H. Yu, X. Zhou, C. S. Criddle, W.-M. Wu and Y. Zhang, Environ. Int., 2020, 145, 106106 CrossRef CAS PubMed.
- M. Orlando, G. Molla, P. Castellani, V. Pirillo, V. Torretta and N. Ferronato, Int. J. Mol. Sci., 2023, 24, 3877 CrossRef CAS PubMed.
- J. Schmidt, R. Wei, T. Oeser, L. A. Dedavid e Silva, D. Breite, A. Schulze and W. Zimmermann, Polymers, 2017, 9, 65 CrossRef PubMed.
- A. Magnin, E. Pollet, R. Perrin, C. Ullmann, C. Persillon, V. Phalip and L. Avérous, Waste Manage., 2019, 85, 141–150 CrossRef CAS PubMed.
- F. Di Bisceglie, F. Quartinello, R. Vielnascher, G. M. Guebitz and A. Pellis, Polymers, 2022, 14, 411 CrossRef CAS PubMed.
- A. Magnin, L. Entzmann, E. Pollet and L. Avérous, Waste Manage., 2021, 132, 23–30 CrossRef CAS PubMed.
- A. O. Adıgüzel and M. Tunçer, Prep. Biochem. Biotechnol., 2017, 47, 925–933 CrossRef PubMed.
- S. Weinberger, J. Canadell, F. Quartinello, B. Yeniad, A. Arias, A. Pellis and G. M. Guebitz, Catalysts, 2017, 7, 318 CrossRef.
- X. Hu, Z. Gao, Z. Wang, T. Su, L. Yang and P. Li, Polym. Degrad. Stab., 2016, 134, 211–219 CrossRef CAS.
- T. Miyakawa, H. Mizushima, J. Ohtsuka, M. Oda, F. Kawai and M. Tanokura, Appl. Microbiol. Biotechnol., 2015, 99, 4297–4307 CrossRef CAS PubMed.
- F. Decorosi, M. L. Exana, F. Pini, A. Adessi, A. Messini, L. Giovannetti and C. Viti, Microorganisms, 2019, 7, 590 CrossRef CAS PubMed.
- D. Ribitsch, S. Heumann, E. Trotscha, E. Herrero Acero, K. Greimel, R. Leber, R. Birner-Gruenberger, S. Deller, I. Eiteljoerg, P. Remler, T. Weber, P. Siegert, K.-H. Maurer, I. Donelli, G. Freddi, H. Schwab and G. M. Guebitz, Biotechnol. Prog., 2011, 27, 951–960 CrossRef CAS PubMed.
- A. Manzur, F. Cuamatzi and E. Favela, J. Appl. Polym. Sci., 1997, 66, 105–111 CrossRef CAS.
- S. Skariyachan, A. A. Patil, A. Shankar, M. Manjunath, N. Bachappanavar and S. Kiran, Polym. Degrad. Stab., 2018, 149, 52–68 CrossRef CAS.
- T. Apinya, N. Sombatsompop and B. Prapagdee, Int. Biodeterior. Biodegrad., 2015, 99, 23–30 CrossRef CAS.
Footnote |
| † Current address: Green Global Group of Companies, 3183 Wilshire Boulevard, Los Angeles, CA 90010, USA. Email: E-mail: sarim.dastgir@lmh.oxon.org. |
| This journal is © The Royal Society of Chemistry 2025 |