
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Medium-dependent regioselectivity of electrochemical bromination of methyl levulinate†
Dmitry A.
Pirgach
a,
Raghavendra
Meena
a,
Guanna
Li
 a,
Fedor M.
Miloserdov
b,
Daan S.
van Es
c,
Pieter C. A.
Bruijnincx
d and
Johannes H.
Bitter
*a
a,
Fedor M.
Miloserdov
b,
Daan S.
van Es
c,
Pieter C. A.
Bruijnincx
d and
Johannes H.
Bitter
*a
aBiobased Chemistry & Technology, Wageningen University and Research, Bornse Weilanden 9, 6708 WG Wageningen, The Netherlands. E-mail: harry.bitter@wur.nl
bLaboratory of Organic Chemistry, Wageningen University and Research, Stippeneng 4, 6708 WE Wageningen, The Netherlands
cFood & Biobased Research, Wageningen University and Research, Bornse Weilanden 9, 6708 WG Wageningen, The Netherlands
dOrganic Chemistry and Catalysis, Institute for Sustainable and Circular Chemistry, Faculty of Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands
First published on 7th April 2025
Abstract
An electrochemical method for the bromination of renewable methyl levulinate using ammonium bromide is reported. Regioselectivity depended on the solvent used: the formation of 5-bromolevulinate was favored in methanol and 3-bromolevulinate in a MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O mixture. To explain the observed change, different bromination mechanisms were proposed.
:H2O mixture. To explain the observed change, different bromination mechanisms were proposed.
Sustainability spotlightEstablishing the sustainable production of chemicals from renewable feedstocks is an important step towards achieving a circular economy. Bromo-derivatives of biomass-derived levulinic acid and its esters (3- and 5-bromolevulinates) serve as precursors for the production of biologically active compounds used in various market segments. Traditionally, these structures are synthesized by reaction with bromine – a hazardous and volatile chemical. In this work, we developed a method for electrochemical bromination of methyl levulinate using non-toxic ammonium bromide as a bromine source. This allowed to generate and convert bromine in situ, thereby reducing reliance on hazardous reagents. Implementing the developed green chemistry practices allows to minimize environmental harm in chemical manufacturing aligning with UN SDG 12 (Responsible Consumption and Production). |
Introduction
There is a clear and urgent need for the more sustainable production of energy carriers, food, and materials.1 In recent years, a significant number of environmentally friendly processes based on the utilization of renewable raw materials to replace fossil-based feedstocks has been described.2–4 As such, biomass has been identified as an interesting alternative to produce fuels and chemicals.5 Biobased chemicals are also already functionalized, so their use as building blocks may require fewer synthetic steps compared to starting from fossil-based feedstocks.6Levulinic acid (LA)7 has been identified as one of the most promising value-added platform chemicals that can be derived from biomass.8 Already currently, it had a global market size of $80 million in 2022, which is expected to grow at a compound annual growth rate (CAGR) of 8.2% from 2021 to 2030.9 LA and its esters can be produced using high-temperature acid hydrolysis or alcoholysis of carbohydrates such as glucose and sucrose but can also from cellulose present in wood and agricultural waste.10,11
Having both keto- and carboxylic functional groups, LA and its esters can be transformed into many chemicals relevant for different bulk and fine chemical markets.12,13 For example, alkyl levulinates find their application as green solvents and fuel additives,14 while LA is a starting material for γ-valerolactone (GVL) also used as green solvent and fuel additive,15,16 bioplastics,17,18 as well as succinic acid (used in the cosmetics and pharmaceutical industry).19,20 In addition, bromo-derivatives of levulinic acid (3- and 5-bromolevulinates) are of particular interest. These structures serve as versatile building blocks for the production of various biologically active compounds. For example, 5-bromolevulinic acid and its esters are the starting material for the synthesis of 5-aminolevulinic acid,10,21 which exhibits anti-cancer-,22–24 anti-bacterial activity,25 and is active as a herbicide.26 On the other hand, 3-bromolevulinic acid and its esters serve as precursors for the synthesis of heterocyclic compounds (thiazoles,27,28 pyridazines,29 pyridines,30,31etc.), which can be further used for the preparation of biologically active compounds (Scheme 1).
 | ||
| Scheme 1 Application of 3- and 5-bromolevulinates. | ||
3-Bromolevulinate 2 and 5-bromolevulinate 3 are traditionally synthesized via bromination of levulinic acid or its esters with molecular bromine (Scheme 2).32 Typically, a mixture of products 2 and 3 is formed in this reaction, after which the individual isomers are separated by distillation.33 The regioselectivity of bromination depends heavily on the reaction conditions: performing this reaction in acidic aqueous media or in ionic liquids favors the formation of the 3-brominated product 2 in a ratio of up to 1:6, while employing MeOH or CHCl3 as a solvent leads to 5-bromolevulinate 3 as the major product in a ratio of up to 3:1.29,32
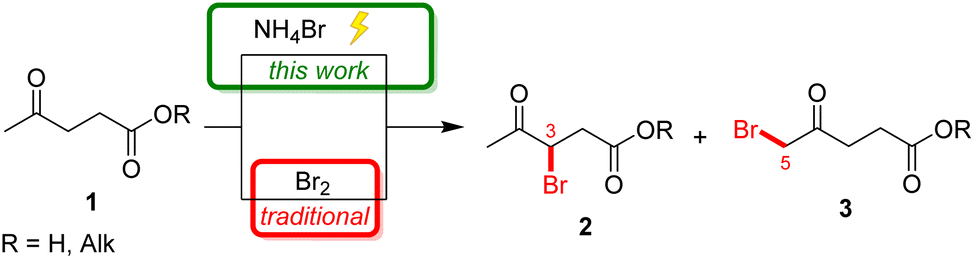 | ||
| Scheme 2 Bromination methods for levulinic acid and its esters. | ||
Although the use of molecular bromine in organic synthesis is common, due to its hazardous nature, it should be prevented or minimized.34 Indeed, from the perspective of green chemistry, developing more environmentally friendly and safer alternatives remains an important goal.35
Currently, due to its safety, controllability, and possibility to drive chemical reactions by renewable electricity, electrosynthesis is becoming a powerful alternative tool for organic synthesis.36–38 For example, it allows electrochemical halogenations by facile electrochemical oxidation of halide anions, as an alternative to methods using hazardous and volatile bromine or chlorine. Utilization of the non-toxic and inexpensive inorganic halides as halogen precursors, especially in combination with green electricity, is becoming a greener alternative not only to using halogens, but also to expensive and/or toxic halogenating agents used in conventional halogenation reactions, e.g., NBS (N-bromosuccinimide) or SOCl2.39,40 For LA or its esters, electrochemical bromination has, to our knowledge, not been reported yet, however.
Here, a convenient method for electrochemical bromination of methyl levulinate using non-toxic bromide salt as a bromine precursor to obtain 3- and 5-bromolevulinates as products will be presented. After preliminary optimization with respect to the type of Br-salt, temperature, substrate concentration, and current, the influence of solvent and addition of acid on substrate conversion and regioselectivity was studied. As bromo derivatives of levulinic acid are generally used as precursors in the form of esters,21,41,42 in this work, instead of LA, methyl levulinate (ML) was chosen as a model substrate, also allowing to avoid possible electrochemical side reactions with the unprotected carboxylic group of the free acid.43 The experiments were performed using Pt electrodes, as this electrode material was earlier reported to be used for electrochemical bromination of ketones.44,45 ICP-OES analysis of the reaction mixtures after electrolysis confirmed that no platinum leaching took place (additional information available from the ESI†), nevertheless, the use of non-CRM can be considered for future studies.
Results and discussion
First, preliminary studies were performed on the role of the nature of Br-salt, temperature, the concentration of ML, and current in methanol in an undivided cell. All the experimental details, as well as the characterization of the resulting reaction mixtures, are reported in the ESI (see Tables S1–S4 and Schemes S1–S4† for preliminary optimization). In all these experiments, the solutions turned slightly yellow from the beginning of the electrolysis which indicated the successful formation of bromine. However, since the intensity of the color (checked visually) did not increase, its concentration did not build up indicating that the bromine was further converted to products. Upon completion of electrolysis and after stirring at room temperature overnight, the reaction mixtures turned clear and colorless, indicating full consumption of electrochemically generated bromine. In these experiments, only 3-bromolevulinate methyl ester 2 and 5-bromolevulinate methyl ester 3 were found as products, while the formation of 2-bromolevulinate and 3,5-dibromolevulinate was never observed. Having carried out the preliminary optimization, studies were continued using the following conditions: NH4Br as bromine salt (15 mmol), 4 mmol (0.26 M) ML, 300 mA current (2F passed = 2573 s electrolysis time) and room temperature (Scheme 3). Under these conditions, products 2a and 3a were obtained with yields of 6% and 11%, respectively, at a conversion of 19%. | ||
| Scheme 3 Optimal conditions for the electrochemical bromination of methyl levulinate after preliminary optimization. | ||
Effect of acid
To increase reaction rate and selectivity, both strong and weak acids have been utilized in thermochemical bromination reactions.34 For example, acetic acid is often used as it stabilizes the intermediates and/or activates the bromine molecule.46 Among the others, such application of acetic acid as a solvent or an additive is also known for the thermochemical bromination of ketones and electrochemical bromination of arenes.47–50 On the other hand, strong acids are also commonly used for the thermochemical bromination of ketones as they are known to favor the bromination into the internal α-position,51 also for levulinic acid.29,52 This is explained by the fact that strong acids favor the formation of enol forms, more reactive towards bromination.53 To investigate whether acid addition would also benefit electrochemical bromination of methyl levulinate, the reaction was performed with acetic acid (CH3COOH), formic acid (HCOOH), and sulfuric acid (H2SO4) using MeOH as a solvent. In Table 1, the results on the influence of the type of acid and its concentration are compiled.| Entry | Solvent | Acid, (M) | Conversion 1a, % | Yield 2a, % | Yield 3a, % |
|---|---|---|---|---|---|
| a Reaction conditions: 0.54 g (4 mmol) 1a, 1.47 g (15 mmol) NH4Br, 12 ml MeOH, acid. 2F charge passed using Pt electrodes at rt (2573 s electrolysis time). The crude products were analyzed by 1H NMR using 1,4-dinitrobenzene as internal standard with typical error of 3%. | |||||
| 1 | MeOH | — | 19 | 6 | 11 |
| 2 | MeOH | AcOH (0.25 M) | 14 | 0 | 0 |
| 3 | MeOH | HCO2H (0.25 M) | 18 | 0 | 8 |
| 4 | MeOH | H2SO4 (0.25 M) | 85 | 16 | 52 |
| 5 | MeOH | H2SO4 (0.1 M) | 56 | 9 | 22 |
| 6 | MeOH | H2SO4 (0.5 M) | 92 | 24 | 66 |
| 7 | MeOH | H2SO4 (0.75 M) | 96 | 23 | 64 |
| 8 | MeOH | H2SO4 (1 M) | 96 | 15 | 39 |
In the absence of acid (Entry 1), 3- and 5-bromolevulinates 2a and 3a, with yields of 6% and 11%, respectively, were formed at 19% conversion (best performance obtained during preliminary optimization, given here for comparison). Acetic acid addition proved detrimental for ML bromination, as in 0.25 M acetic acid (Entry 2), the conversion was slightly lower (14%) compared to the experiment without acid, while products 2a and 3a were not observed. In the presence of 0.25 M formic acid (Entry 3), product 3a was formed with a yield of 8% at a conversion of 18%. Therefore, it can be concluded that the addition of these carboxylic acids was not beneficial for the electrochemical bromination of ML. In contrast, when using 0.25 M sulfuric acid (Entry 4), the conversion increased to 85%, yielding 16% for 2a and 52% for 3a. Moreover, in the presence of sulfuric acid, the electrochemically generated bromine was consumed immediately, as no coloration of the reaction mixture was observed. Although strong acidic media are known to favor bromination of the internal (in this case, C-3) alpha position,52 here, the product of C-5 bromination 3a was the major one formed (16% vs. 52% at 85% conversion).
After establishing that the addition of H2SO4 significantly improved bromination performance while maintaining good selectivity, the influence of the concentration of sulfuric acid was investigated. Decreasing its concentration to 0.1 M (Entry 5) resulted in a significant drop in both conversion and yield. Also, immediately after the start of electrolysis, the reaction mixture turned slightly yellow, meaning that bromine was no longer consumed immediately after generation, just like in a reaction without acid (Entry 1). When the acid concentration was increased to 0.5 M (Entry 6), ML conversion increased to 92% and yields of products 2a and 3a increased to 24 and 66%, respectively. Further increase in acid concentration to 0.75 M (Entry 7) showed a small decrease in products' yield at a small increase in the conversion. Selectivity dropped further upon further increase of acid concentration to 1 M (Entry 8), with yields of 2a and 3a to 15% and 39%, respectively, at 96% conversion. In the experiments where the total yield was significantly lower than conversion, the substrate and/or products were found to degrade under the electrochemical conditions (Entries 2, 3 and 8), individual products of these side reactions were not isolated. Thus, an 0.5 M sulfuric acid concentration proved most productive for 3a with faradaic efficiency of 90% at average cell voltage of 5 V (Entry 6).
Effect of solvent
Previously, it has been reported that performing the thermochemical bromination reaction in aqueous acidic media can favor the internal α-bromination with molecular bromine.29,54 Moreover, many studies on the electrochemical bromination are carried out in an aqueous–organic solvent mixtures,44,55 as the addition of water improves the conductivity and allows the dissolution of electrolytes. To further explore the dependence of regioselectivity on the reaction conditions, the effect of the addition of water was studied next.Interestingly, in MeOH:H2O (1:4 v/v) in the presence of 0.3 M sulfuric acid (Table 2, Entry 1), we now obtained 3-bromolevulinate 2a as the major product and 5-bromolevulinate 3a as the minor product with yields of 17% and 10%, respectively at 80% conversion. Even though the total product yield was only 27%, in this case, the selectivity has shifted towards the C-3 brominated product 2a in contrast to the reaction performed solely in MeOH (Table 1, Entry 4 vs.Table 2, Entry 1) albeit at a slightly different acid concentration. When the reaction was performed in MeCN:H2O (1:4 v/v) in the presence of 0.3 M sulfuric acid, a solvent mixture previously used for electrochemical bromination of ketones,44 selectivity towards the C-3-brominated product further increased, with product yields for 2a and 3a being 30% and 11%, respectively, at 43% conversion (Table 2, Entry 2 vs.Table 2, Entry 1).
| Entry | Solvent | Acid, (M) | Conversion 1a, % | Yield 2a, % | Yield 3a, % |
|---|---|---|---|---|---|
|
a Reaction conditions: 0.54 g (4 mmol) 1a, 9.6 g (98 mmol) NH4Br, organic solvent:water (4 + 16 ml), X ml H2SO4. 2F charge passed using Pt electrodes at rt (2573 s electrolysis time). The crude products were analyzed by 1H NMR using 1,4-dinitrobenzene as internal standard with typical error of 3%.
|
|||||
| 1 | MeOH–H2O | H2SO4 (0.3 M) | 82 | 17 | 10 |
| 2 | MeCN–H2O | H2SO4 (0.3 M) | 43 | 30 | 11 |
| 3 | MeCN–H2O | H2SO4 (1 M) | 81 | 59 | 20 |
| 4 | MeCN–H2O | H2SO4 (2 M) | 96 | 12 | 2 |
| 5 | MeCN–H2O | — | Trace | 0 | 0 |
| 6 | H2O | H2SO4 (0.3 M) | 90 | Trace | Trace |
Having noted the significant improvement of reaction performance in the acidic MeCN:H2O (1:4 v/v) media, the role of acid concentration using a MeCN:H2O mixture as a solvent was studied next. An increase in acid concentration to 1 M (Table 2, Entry 3) resulted in the increase of conversion to 81% while the high selectivity towards the product of C-3 bromination was maintained. In this experiment, the yields of products 2a and 3a increased further to 59% and 20%, respectively. When the acid concentration was further increased to 2 M (Table 2, Entry 4), the yield of products 2a and 3a significantly dropped while the conversion increased to 96%. Such an increase in conversion accompanied by the drop in the product yields can possibly be explained by the hydrolysis of the ester group of both substrate and products.
Control experiments in either MeCN:H2O without acid or in purely aqueous acidic medium showed that in the absence of sulfuric acid, the desired products were not formed (Entry 5), while in the absence of MeCN the desired products were formed in trace amounts at a ML conversion of 90% (Entry 6), probably due to a faster hydrolysis of the ester group in purely aqueous conditions.56 Thus, both sulfuric acid and MeCN were crucial to achieve successful C3-favored electrochemical bromination of methyl levulinate. Best performance was achieved using 1 M concentration of sulfuric acid (Entry 3), with faradaic efficiency of 79% at average cell voltage of 1.5 V. Under optimal conditions (for both MeCN:H2O and methanol), the reaction on the counter electrode (cathode) was hydrogen evolution, from either acid or solvent (water and MeOH). Formation of ammonia was also detected in trace amounts, but only in the experiments which were performed without acid. In presence of the acid, the electrochemically generated ammonia dissolved in the solvent, where under acidic conditions it again formed the ammonium cation. Upon completion of electrolysis, the ammonium salt was concentrated in the aqueous phase during extraction. Existing technologies (for example, membrane concentration) allow efficient and sustainable recovery of these salts from such solutions,57 with subsequent reuse allowing lower costs and also environmental impact.
Biobased solvents (EtOH, mTHF) were also tested in combination with water, however both of them demonstrated significantly lower performance compared to originally used MeCN:H2O system (detailed information available from p. 8 in the ESI†). Although MeCN is not considered as a green solvent, in this studies it was used as a co-solvent with volumetric concentration of 20%. At the same time, existing techniques allow to efficiently recover it (also from high-salt wastewater) thereby minimizing environmental impact.58,59
Rationalizing the regioselectivity
To explain the opposite trends in regioselectivity of electrochemical bromination using MeCN:H2O and MeOH as solvents, mechanisms are proposed in Scheme 4 (for acidic MeCN:H2O mixture) and in Scheme 5 (for acidic MeOH), based on our observations and literature data on (electrochemical) bromination of ketones under the same or similar reaction conditions. According to the proposed mechanism (Scheme 4), similar to electrochemical bromination of acetophenone,44 hypobromous acid (HOBr) is formed in situ from electrochemically generated bromine under aqueous conditions.60,61 Next, under acidic conditions, this intermediate undergoes protonation to form a more electrophilic H2OBr+ intermediate.62,63 At the same time, acidic conditions can promote the enolization of carbonyl compounds to form more reactive enol species.64 On the next step, H2OBr+ reacts with thermodynamic internal enol tautomer A of methyl levulinate,65 resulting in the formation of the 3-bromo derivative 2a as a major product due to the higher stability of internal enol A, as shown in Scheme 4 (thermodynamic stability of intermediates A and B was compared using DFT, see Table S5 in the ESI† for more information). This is in line with reported data for non-electrochemical bromination of levulinic acid with molecular bromine in purely aqueous acidic medium.29
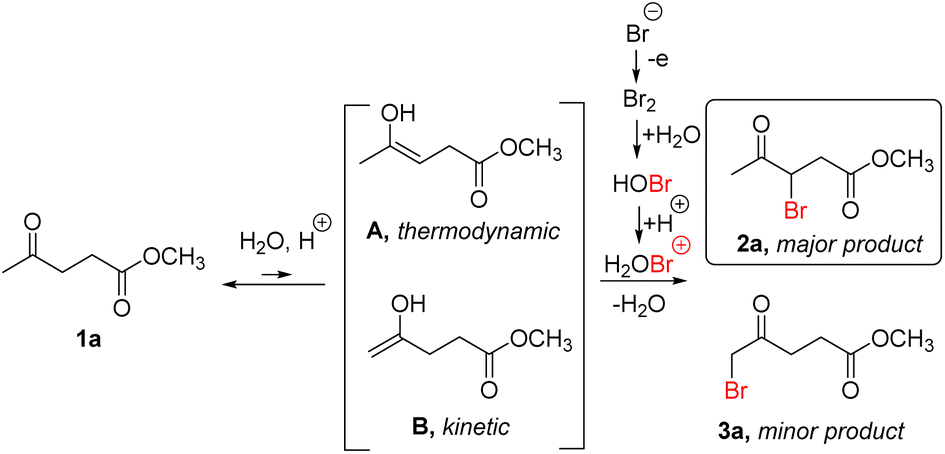 | ||
| Scheme 4 Proposed mechanism for the electrochemical bromination of ML in MeCN:H2O mixture. | ||
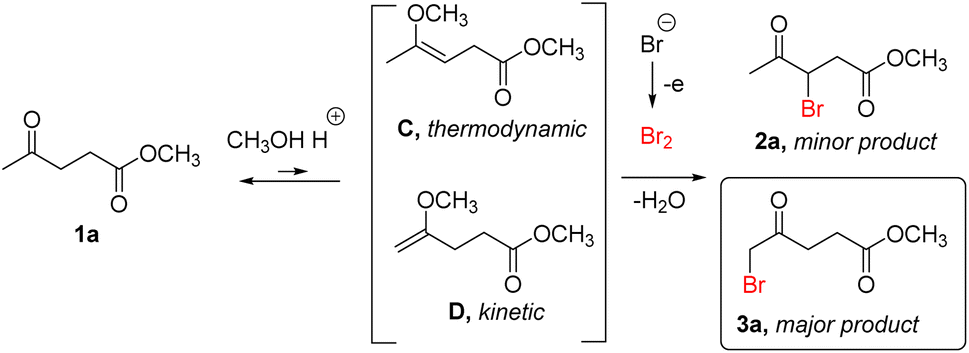 | ||
| Scheme 5 Proposed mechanism for the electrochemical bromination of ML in MeOH. | ||
On the other hand, that electrochemical LA bromination in methanol in the presence of sulfuric acid, yields the terminal 5-bromolevulinate 3a as a major product, is not common for ketones under acidic conditions.66 It has been observed, for thermochemical bromination of ketones in methanol without acid,21,67 however, the bromination of the α-CH3 group of ketones is usually performed solely in methanol without adding external acids.68–70 It is likely that in methanol, in the presence of strong acid, the ML 1a forms a mixture of enol ethers C and D (Scheme 5), as reported for various ketones.64,71,72 Under such conditions (in acidic methanol without water), a pathway of bromination via enol ether was dominating over the others in the studies of kinetics and mechanism for bromination of acetone.64 This indirectly suggests that ML can be brominated following the same pathway. Due to higher reactivity of enol ethers compared to corresponding enols,73 the kinetic factor can dominate and determine their reactivity because their reaction occurs faster than the equilibration of enol forms, defining the regioselectivity of the reaction. In the case of bromination in acidic methanol,74 unlike bromination in MeCN:H2O discussed above, the position where bromination of enol ether occurs is determined by the form that reacts most rapidly with the bromine, i.e., kinetically favored and sterically more accessible enol ether D, rather than the thermodynamically favored and sterically less accessible enol ether C (thermodynamic stability of enol ethers C and D was compared using DFT, see Table S6 in the ESI† for more information). This can explain the formation of the product of C-5 bromination 3a as a major one. In such a case, the observed immediate bromine consumption in the media of acidic methanol can be explained by the bromination proceeding via more reactive enol ethers, which indirectly confirms such a pathway. Faster reaction of enol ether compared to corresponding enol was also confirmed by kinetic studies on bromination of acetophenone.73 For bromination in methanol, in order to form the active enol ether, keto-group of the initial ketone first gets protonated, forming hemiketal and then enol ether.64,73 Most likely, strength of the tested organic acids was not enough to achieve this protonation resulting in considerably lower performance compared to strong sulfuric acid. An overview of 1H NMR spectra of crude products in acidic MeCN:H2O mixture and in acidic MeOH is available from the ESI (Fig. S1 and S2).†
Conclusions
Overall, we reported herein an electrochemical method for bromination of renewable methyl levulinate, where commercially available and non-toxic ammonium bromide was used as a bromine source. Having studied the influence of the media on reaction regioselectivity, we demonstrate that in the media of acidic MeCN:H2O mixture, the formation of C-3 bromolevulinate was favored. In contrast, performing the reaction in acidic methanol resulted in the formation of C-5 bromination product as a major. This can be possibly explained by different bromination pathways. According to the proposed mechanisms, both bromination reactions proceed through enolization. In an acidic MeCN:H2O mixture, more thermodynamically stable internal enol underwent the bromination resulting in the formation of a C-3 brominated product, while on the other hand, in the acidic methanol mixture, a more reactive terminal enol ether was converted into the C-5 brominated product. Unlike enol, enol ether reacted faster with bromine, forming the kinetic product of C-5 bromination. In the optimized conditions, upon passing 2F charge, the bromination products were formed in approximately 3:1 and 1:3 ratio with the total yield of 81 and 90%, respectively. As these compounds have application in different fields and quantities, more selective (electrochemical) synthesis is important for favoring formation of the desired isomer with higher yield.
Data availability
The data supporting this article is available from the ESI.†Author contributions
Dmitry A. Pirgach: investigation, conceptualization writing – original draft. Raghavendra Meena: software – DTF studies. Guanna Li: supervision. Fedor M. Miloserdov: writing – review & editing, supervision. Daan S. van Es: writing – review & editing, supervision. Pieter C. A. Bruijnincx: writing – review & editing, supervision. Johannes H. Bitter: writing – review & editing, project administrator, funding acquisition; supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from Wageningen University. Leane Gros is acknowledged for carrying out selected experiments.References
- L. Yang, X. C. Wang, M. Dai, B. Chen, Y. B. Qiao, H. J. Deng, D. F. Zhang, Y. Z. Zhang, C. M. V. B. de Almeida, A. S. F. Chiu, J. J. Klemes and Y. T. Wang, Energy, 2021, 228, 120533 Search PubMed.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 Search PubMed.
- L. Coniglio, J. A. P. Coutinho, J. Y. Clavier, F. Jolibert, J. Jose, I. Mokbel, D. Pillot, M. N. Pons, M. Sergent and V. Tschamber, Prog. Energy Combust. Sci., 2014, 43, 1–35 Search PubMed.
- H. R. Ghatak, Renewable Sustainable Energy Rev., 2011, 15, 4042–4052 Search PubMed.
- S. Dutta and N. S. Bhat, ChemCatChem, 2021, 13, 3202–3222 Search PubMed.
- K. Polman, Appl. Biochem. Biotechnol., 1994, 45–6, 709–722 Search PubMed.
- D. W. Rackemann and W. O. S. Doherty, Biofuels, Bioprod. Biorefin., 2011, 5, 198–214 Search PubMed.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass: Volume I – Results of Screening for Potential Candidates from Sugars and Synthesis Gas, United States, 2004 Search PubMed.
- Levulinic Acid Market Size, Share, Price|Global Industry Report, 2020, https://www.grandviewresearch.com/industry-analysis/levulinic-acid-market, accessed 24 December 2020 Search PubMed.
- J. J. Bozell, L. Moens, D. C. Elliott, Y. Wang, G. G. Neuenscwander, S. W. Fitzpatrick, R. J. Bilski and J. L. Jarnefeld, Resour., Conserv. Recycl., 2000, 28, 227–239 Search PubMed.
- D. Q. Ding, J. X. Xi, J. J. Wang, X. H. Liu, G. Z. Lu and Y. Q. Wang, Green Chem., 2015, 17, 4037–4044 Search PubMed.
- D. Di Menno Di Bucchianico, Y. Wang, J.-C. Buvat, Y. Pan, V. Casson Moreno and S. Leveneur, Green Chem., 2022, 24, 614–646 Search PubMed.
- G. Wu, C. Shen, S. S. Liu, Y. Huang, S. Zhang and H. Zhang, Green Chem., 2021, 23, 9254–9282 Search PubMed.
- L. Yan, Q. Yao and Y. Fu, Green Chem., 2017, 19, 5527–5547 Search PubMed.
- T. Raj, K. Chandrasekhar, R. Banu, J. J. Yoon, G. Kumar and S. H. Kim, Fuel, 2021, 303, 121333 Search PubMed.
- A. Démolis, N. Essayem and F. Rataboul, ACS Sustainable Chem. Eng., 2014, 2, 1338–1352 Search PubMed.
- G. C. Hayes and C. R. Becer, Polym. Chem., 2020, 11, 4068–4077 Search PubMed.
- L. Lenzi, M. Degli Esposti, S. Braccini, C. Siracusa, F. Quartinello, G. M. Guebitz, D. Puppi, D. Morselli and P. Fabbri, ACS Sustainable Chem. Eng., 2023, 11, 9455–9469 Search PubMed.
- D. Carnevali, M. G. Rigamonti, T. Tabanelli, G. S. Patience and F. Cavani, Appl. Catal., A, 2018, 563, 98–104 Search PubMed.
- L. Podolean, V. Kuncser, N. Gheorghe, D. Macovei, V. I. Parvulescu and S. M. Coman, Green Chem., 2013, 15, 3077–3082 Search PubMed.
- H. J. Ha, S. K. Lee, Y. J. Ha and J. W. Park, Synth. Commun., 1994, 24, 2557–2562 Search PubMed.
- N. Fotinos, M. A. Campo, F. Popowycz, R. Gurny and N. Lange, Photochem. Photobiol., 2006, 82, 994–1015 Search PubMed.
- M. H. Gold and M. P. Goldman, Dermatol. Surg., 2004, 30, 1077–1083 Search PubMed.
- Q. Peng, T. Warloe, K. Berg, J. Moan, M. Kongshaug, K. E. Giercksky and J. M. Nesland, Cancer-Am. Cancer Soc., 1997, 79, 2282–2308 Search PubMed.
- Y. Awa, N. Iwai, T. Ueda, K. Suzuki, S. Asano, J. Yamagishi, K. Nagai and M. Wachi, Biosci., Biotechnol., Biochem., 2005, 69, 1721–1725 Search PubMed.
- K. Sasaki, M. Watanabe, T. Tanaka and T. Tanaka, Appl. Microbiol. Biotechnol., 2002, 58, 23–29 Search PubMed.
- G. Guercio, D. Castoldi, N. Giubellina, A. Lamonica, A. Ribecai, P. Stabile, P. Westerduin, R. Dams, A. Nicoletti, S. Rossi, C. Bismara, S. Provera and L. Turco, Org. Process Res. Dev., 2010, 14, 1153–1161 Search PubMed.
- G. Westphal and H. Wasicki, Zeitschrift für Chemie, 1968, 8, 337 Search PubMed.
- N. Gouault, J. F. Cupif, M. Amoros and M. David, J. Chem. Soc., Perkin Trans. 1, 2002, 2, 2234–2236 Search PubMed.
- J. J. Kaminski, J. A. Bristol, C. Puchalski, R. G. Lovey, A. J. Elliott, H. Guzik, D. M. Solomon, D. J. Conn, M. S. Domalski and S. C. Wong, et al. , J. Med. Chem., 1985, 28, 876–892 Search PubMed.
- E. Abignente, F. Arena, E. Luraschi, C. Saturnino, E. Marmo, S. Russo and R. Magliulo, Farmaco, Ed. Sci., 1986, 41, 119–130 Search PubMed.
- A. G. Zavozin, N. E. Kravchenko, N. V. Ignat'ev and S. G. Zlotin, Tetrahedron Lett., 2010, 51, 545–547 Search PubMed.
- S. F. Macdonald, Can. J. Chem., 1974, 52, 3257–3258 Search PubMed.
- I. Saikia, A. J. Borah and P. Phukan, Chem. Rev., 2016, 116, 6837–7042 Search PubMed.
- S. A. Luan, T. Castanheiro and T. Poisson, Green Chem., 2024, 26, 3429–3434 Search PubMed.
- C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72–83 Search PubMed.
- A. Wiebe, T. Gieshoff, S. Mohle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew Chem. Int. Ed. Engl., 2018, 57, 5594–5619 Search PubMed.
- C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 Search PubMed.
- X. Shang, X. Liu and Y. J. Sun, Green Chem., 2021, 23, 2037–2043 Search PubMed.
- C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Green Chem., 2020, 22, 3358–3375 Search PubMed.
- R. Vallinayagam, H. Bertschy, Y. Berger, V. Wenger and R. Neier, Synthesis (Stuttg), 2007, 2007, 3731–3735 Search PubMed.
- C. Aggelidou, T. A. Theodossiou, A. R. Goncalves, M. Lampropoulou and K. Yannakopoulou, Beilstein J. Org. Chem., 2014, 10, 2414–2420 Search PubMed.
- T. R. dos Santos, P. Nilges, W. Sauter, F. Harnisch and U. Schröder, RSC Adv., 2015, 5, 26634–26643 Search PubMed.
- R. Jagatheesan, K. J. S. Raj, S. Lawrence and C. Christopher, RSC Adv., 2016, 6, 35602–35608 Search PubMed.
- R. S. Kumar, K. Kulangiappar and M. A. Kulandainathan, Synth. Commun., 2010, 40, 1736–1742 Search PubMed.
- J. Rajaram and J. Kuriacos, Aust. J. Chem., 1968, 21, 3069–3073 Search PubMed.
- N. P. Buu-Hoï and D. Lavit, J. Chem. Soc., 1955, 0, 18–20 Search PubMed.
- M. S. Shaikh, M. B. Palkar, H. M. Patel, R. A. Rane, W. S. Alwan, M. M. Shaikh, I. M. Shaikh, G. A. Hampannavar and R. Karpoormath, RSC Adv., 2014, 4, 62308–62320 Search PubMed.
- A. Olyaei, E. Feizy and A. Aghajanzadeh, J. Heterocycl. Chem., 2021, 58, 757–765 Search PubMed.
- G. Casalbore, M. Mastragostino and S. Valcher, J. Electroanal. Chem. Interfacial Electrochem., 1975, 61, 33–46 Search PubMed.
- P. G. Gassman, T. J. Van Bergen, D. P. Gilbert and B. W. Cue, J. Am. Chem. Soc., 1974, 96, 5495–5508 Search PubMed.
- N. A. Porter, D. M. Scott, I. J. Rosenstein, B. Giese, A. Veit and H. G. Zeitz, J. Am. Chem. Soc., 1991, 113, 1791–1799 Search PubMed.
- J. E. Dubois, M. Elalaoui and J. Toullec, J. Am. Chem. Soc., 1981, 103, 5393–5401 Search PubMed.
- M. Konishi, K. Saito, K. Numata, T. Tsuno and K. Asama, J. Antibiot., 1977, 30, 789–805 Search PubMed.
- R. Thasan and K. Kumarasamy, Korean J. Chem. Eng., 2014, 31, 365–373 Search PubMed.
- M. Balakrishnan, G. Venkoba Rao and N. Venkatasubramanian, Proceedings of the Indian Academy of Sciences - Section A, 1974, 80, 50–56 Search PubMed.
- Y. Ye, H. H. Ngo, W. Guo, Y. Liu, S. W. Chang, D. D. Nguyen, H. Liang and J. Wang, Bioresour. Technol., 2018, 268, 749–758 Search PubMed.
- Y. Wang, X. Mei, T. F. Ma, C. J. Xue, M. D. Wu, M. Ji and Y. G. Li, J. Cleaner Prod., 2018, 197, 742–749 Search PubMed.
- V. Sargsyan, Bulletin of High Technology, 2024, 39–50, DOI:10.56243/18294898-2024.2-39.
- C. M. Kelley and H. V. Tartar, J. Am. Chem. Soc., 1956, 78, 5752–5756 Search PubMed.
- B. N. Grgur, J. Electrochem. Soc., 2019, 166, E50–E61 Search PubMed.
- C.-Z. Dong, M. Julia and J. Tang, Eur. J. Org Chem., 1998, 1998, 1689–1696 Search PubMed.
- W. J. Wilson and F. G. Soper, J. Chem. Soc., 1949, 3376, 10.1039/jr9490003376.
- J. Toullec and J. E. Dubois, J. Am. Chem. Soc., 1976, 98, 5518–5524 Search PubMed.
- G. Rothenberg, R. M. H. Beadnall, J. E. McGrady and J. H. Clark, J. Chem. Soc., Perkin Trans. 2, 2002, 630–635, 10.1039/b108009a.
- J. Clayden, N. Greeves and S. Warren, Organic Chemistry, Oxford University Press, USA, 2012 Search PubMed.
- A. J. Manny, S. Kjelleberg, N. Kumar, R. deNys, R. W. Read and P. Steinberg, Tetrahedron, 1997, 53, 15813–15826 Search PubMed.
- W. Y. Wong, X. Z. Wang, Z. He, K. K. Chan, A. B. Djurisic, K. Y. Cheung, C. T. Yip, A. M. Ng, Y. Y. Xi, C. S. Mak and W. K. Chan, J. Am. Chem. Soc., 2007, 129, 14372–14380 Search PubMed.
- T. Mordhorst and U. Bickmeyer, Tetrahedron Lett., 2015, 56, 4363–4366 Search PubMed.
- R. K. Bressin, S. Osman, I. Pohorilets, U. Basu and K. Koide, J. Org. Chem., 2020, 85, 4637–4647 Search PubMed.
- A. Kankaanpera, P. Salomaa, P. Juhala, R. Aaltonen and M. Mattsen, J. Am. Chem. Soc., 1973, 95, 3618–3624 Search PubMed.
- E. W. Garbisch, J. Org. Chem., 1965, 30, 2109–2120 Search PubMed.
- J. Toullec and M. Elalaoui, J. Org. Chem., 1986, 51, 4054–4061 Search PubMed.
- F. Effenberger, Angew Chem. Int. Ed. Engl., 1969, 8, 295–312 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5su00037h |
| This journal is © The Royal Society of Chemistry 2025 |