
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Breaking the equilibrium limit: synthesis of diethyl carbonate from CO2 using regenerable bis-/tris-triethoxysilane substrates†
Wahyu S.
Putro
a,
Akira
Ikeda
a,
Toshihide
Yamamoto
b,
Satoshi
Hamura
b,
Jun-Chul
Choi
 *a and
Norihisa
Fukaya
*a
*a and
Norihisa
Fukaya
*a
aInterdisciplinary Research Center for Catalytic Chemistry, National Institute of Advanced Industrial Science and Technology (AIST), AIST Tsukuba Central 5, 1-1-1 Higashi, Tsukuba, 305-8565, Ibaraki, Japan. E-mail: junchul.choi@aist.go.jp; n.fukaya@aist.go.jp
bResearch and Development Planning, Tosoh Corporation, 3-8-2 Shiba, Minato-ku, Tokyo 105-8623, Japan
First published on 19th December 2024
Abstract
Breaking the equilibrium limit is necessary to promote the production of diethyl carbonate (DEC) from CO2 and alkoxysilanes. DEC yields are predicted to overcome the equilibrium limitation when substrates that generate oligomers as byproducts are used. In this study, we explored the catalytic synthesis of DEC using bis-/tris-triethoxysilane substrates over a Zr-based catalyst. Beyond-equilibrium DEC yields (>50% yield) are observed when typical substrates were used as the oligomer is obtained as a byproduct. For example, the isocyanate substrate solidified during DEC synthesis, yielding twice the amount of DEC generated from tetraethoxy orthosilicate. The isocyanate substrate was initially converted into an isocyanurate intermediate prior to polymerization to overcome the equilibrium limitation. The sustainability of this approach is highlighted by the feasibility of substrate regeneration from polymer byproducts. The demonstrated effectiveness of catalysis in promoting DEC from CO2 can drive scientific and industrial advancements while maintaining sustainability.
Sustainable spotlightThe advancement of CO2 utilization technologies is shattering the equilibrium limits and enabling us to transcend beyond previous boundaries, achieving higher yields of valuable products. The results demonstrate that diethyl carbonate (DEC) can be synthesized from carbon dioxide (CO2). The substrates generate oligomers as byproducts, breaking the equilibrium limitation. The DEC yield exceeded the limit (>50%) because the alkoxysilane by-product is solidified through oligomerization. The monomeric byproduct can be regenerated using ethanol in a circular reactor using a base catalyst. The conversion of CO2 coupled with byproduct regeneration is a pivotal technology for reducing CO2 emissions while promoting innovation in chemical manufacturing. This is consistent with global initiatives aimed at achieving SDG 11 (environmental sustainability) and SDG 13 (climate action). |
Introduction
CO2 emissions originating from anthropogenic activities, including coal combustion (43%), and oil (36%) and gas (20%) consumption, have a considerable impact on the elevation of global temperatures.1 Despite global CO2 emissions declining by 8.8% (approximately 1.5 Gt CO2) in the first half of 2020 due to the COVID-19 pandemic, the atmospheric concentration remained high (410 ppm).2 Efforts have been made to achieve a continual decline; CO2 utilization has recently garnered considerable attention as it can either contribute to CO2 mitigation or produce fine chemicals. However, the utilization of CO2 as a C1-building block is challenging due to its high thermodynamic stability. This is attributed to its low standard heat of formation (ΔHf = −394 kJ mol−1) and standard Gibbs energy of formation (ΔGf = −395 kJ mol−1).3 These properties make the activation and conversion of CO2 into valuable chemicals a significant challenge. Consequently, the use of CO2 as feedstock for organic reaction needs high energy input or reacts with a high free energy substrate to provide thermodynamically feasible process. As a C-1 chemical feedstock, CO2 can be converted to carbonates,4 carbamates,5 urea,6 isocyanate,7 and carboxylic acid,8 some of which have been commercialized. For example, 180 and 250 Mt of urea and carbonates, respectively, were produced from CO2 in 2016.9 The synthesis of dialkyl carbonates from CO2 has recently attracted significant attention in the chemical industry because of its environmentally friendly nature, and its strong potential for industry.Diethyl carbonate (DEC), a member of the dialkyl carbonate family, is an important green chemical because of its low toxicity and high biodegradability.10 It has wide applicability in the chemical, battery, and polymer industry; moreover, it has the potential to replace methyl tert-butyl ether (MTBE) as a fuel additive, which could increase the octane number of gasoline.11 Although several synthetic processes such as phosgenation (Bayer process),12 alcohol carbonylation (Enichem process),13,14 and indirect alcohol trans-esterification (Asahi Kasei process) have been industrialized,15,16 direct synthesis from the reaction of CO2 with alcohol seems more favorable for CO2 mitigation and reutilization of CO2 (Fig. 1).17 This direct method also generates water as the only byproduct. However, this reaction is limited by a chemical equilibrium; it is an unspontaneous reaction (ΔG298K = 35.85 kJ mol−1 > 0).18 The combination of a catalyst with an appropriate dehydrating agent is required to overcome the limitations associated with equilibrium. Various homogeneous and heterogeneous catalysts, such as Ti(i-OPr)4,19 Bu2SnO,20,21 Nb(OR)5,22 CeO2,23–26 and TixCe1−xO2 (ref. 27) have been extensively utilized for the direct synthesis of dialkyl carbonate from CO2 and alcohols in combination with dehydrating agents including molecular sieves (MS),28 acetal,21,29,30 orthoester,31 or 2-cyanopyridine.32,33 However, the development of an efficient process to regenerate the dehydrating agent remains challenging.
 | ||
| Fig. 1 Various routes for the synthesis of dialkyl carbonates. | ||
In our previous report, we identified a new method for direct DEC synthesis from the reaction of CO2 and ethoxysilane substrate with a Zr(OEt)4 catalyst.34,35 Instead of water, a disiloxane formed as a byproduct, which was regenerated into tetraethyl orthosilicate (TEOS), establishing a new waste-free synthesis method for DEC. The highest yield of DEC was approximately 50% due to the equilibrium (Fig. 2). The subsequent substrate scope investigation showed that the increasing number of ethoxy groups on the substrate gradually improved the DEC yield to the optimum 50% yield (Fig. S1†). Disiloxane was the only byproduct generated from these substrates. Based on these preliminary results, we hypothesized that the use of a substrate with a greater number of ethoxy moieties could promote polymerization. We further postulate that the equilibrium limit may be broken through the use of a substrate that generates not only a dimer but also an oligomer, resulting in a higher DEC yield. Herein, we report a catalytic DEC synthesis using regenerable substrates bearing multiple ethoxy groups, for example, bis-/tris-triethoxysilane or oligomer-generable substrates, to achieve a high DEC yield and present an ideal synthetic process to realize sustainability.
 | ||
| Fig. 2 Direct synthesis of DEC from TEOS and CO2. | ||
Experimental section
Materials & characterization
All compounds were purchased from Sigma-Aldrich, Alfa Aesar, TCI, Wako, and Gelest Inc. and used without further purification. 1H NMR spectra were recorded using a Bruker ADVANCE III HD (1H NMR at 400 MHz and 600 MHz, 13C NMR at 100.7 MHz, and 29Si NMR at 79.5 MHz). GC was performed using a Shimadzu GC-2014 instrument equipped with a flame ionization detector and a TC-1 column. Gas chromatography-mass spectrometry (GC-MS) was performed using a Shimadzu QP-2010 spectrometer with a TC-1 column. MALDI-TOF MS of the oligomers were performed using a Bruker Autoflex Speed. Samples for the MALDI-TOF MS were prepared by mixing the oligomer (20 mg mL−1, 10 μL), a matrix (DCTB, 50 mg mL−1, 50 μL), and a cationic agent (sodium trifluoroacetate or silver trifluoroacetate, 25 mg mL−1, 20 μL) in THF. GPC analysis was performed in THF (1.0 mL min−1) at 40 °C using a Shimadzu instrument equipped with a Shodex GPC KF-803 column (molecular weight range 1000–50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, exclusion limit, 7 × 104). Infrared (IR) spectra were recorded using a JASCO ATRS-100-CIR spectrophotometer. Elemental analyses were performed using a WDXRF Bruker AXS T8 Tiger spectrophotometer.
000, exclusion limit, 7 × 104). Infrared (IR) spectra were recorded using a JASCO ATRS-100-CIR spectrophotometer. Elemental analyses were performed using a WDXRF Bruker AXS T8 Tiger spectrophotometer.
DEC synthesis
DEC synthesis was carried out in an autoclave with an inner volume of 10 mL in a manner similar to a previous method using TEOS as the substrate.34 Approximately 0.3 g Zr(OEt)4 catalyst and 15.7 mmol of substrate 1a (3-(triethoxysilyl)propyl isocyanate) were put in the autoclave with a stirring bar. The reactor was subsequently pressurized and purged with CO2 to 5 MPa under ambient conditions. After CO2 insertion, the reactor was heated to 100–180 °C. The mixture was stirred continuously during the reaction. After the specified reaction time, the reactor was cooled to room temperature and 0.1 g of mesitylene was added to the liquid phase as an internal standard for quantitative analysis. Syntheses using other organosilicon substrates were conducted in a similar manner.The conversion, yield, material balance, and turnover number (TON) are calculated by following manner, when 1a, TEOS, or other alkoxysilanes were used as substrates.

| Conv. (%) = (1 − (mole of remaining substrate)/(initial mole of substrate)) × 100% |
| Yield (%) = ((mole of detected DEC × 2)/(initial mole of substrate)) × 100% |
| TON = (mole of DEC)/(mole of added catalyst) |
Synthesis of intermediate 1b from substrate 1a
Intermediate 1b was synthesized in an Erlenmeyer flask under a N2 atmosphere. The isocyanate 1a (10 mL) was stirred at 180 °C for 96 h without catalysts or other additives. Isocyanate 1b was subsequently used for DEC synthesis after the addition of Zr(OEt)4 catalysts. 1H NMR (400 MHz, C6D6): δ 3.68–3.78 (m, 6H + 18H), 1.90–1.78 (m, 6H), 1.16–1.07 (t, J = 14 Hz, 27H), 0.65–0.55 ppm (m, 6H). 13C NMR (100 MHz, C6D6): δ 148.6, 58.1, 45.0, 21.5, 18.1, 7.7 ppm. 29Si NMR (79.5 MHz, C6D6): δ −46.7 ppm. IR (ATR): 2973, 2928, 2886, 1698, 1455, 1390, 1313, 1167, 1067, 950, 761, 702, 539 cm−1.Results and discussion
The DEC formation reaction was initially explored at 140 °C under 5 MPa of initial CO2 pressure with 3-(triethoxysilyl)propyl isocyanate as substrate 1a. After screening for the optimum conditions (Table S1†), the highest yield of DEC (6 mmol; 75% yield) was obtained at 140 °C under 5 MPa of initial CO2 pressure. In Fig. 3A, the time profile of DEC produced from 1a is compared with the TEOS profile at their respective optimum conditions (1a at 140 °C and TEOS at 180 °C, Si/Zr = 14, 5 MPa of CO2 pressure). Compared with TEOS, substrate 1a was clearly superior for DEC production. At 6 h of reaction, 5 mmol of DEC was produced from 1a (Table S1†), which is almost a two-fold increase from the amount of DEC produced from TEOS at the same reaction time (2.9 mmol). The results show that the reaction with substrate 1a produced more than a 50% yield of DEC, even during the initial 6 h, indicating that this reaction is far beyond the chemical equilibrium. Increasing the reaction time to 96 h increased DEC formation to 7.4 mmol. We observed that polymerization proceeded during the synthesis of DEC, as indicated by the formation of a solid product after the reaction. Compared with TEOS, 1a produced a higher amount of DEC in the temperature and pressure screening tests (Fig. 3B). Substrate 1a followed a different trend, as decreasing the temperature from 180 °C to 140 °C resulted in a gradual increase of DEC, while further decreasing the temperature yielded less DEC product. Notably, a solid product was not obtained with the product evidently remaining in the liquid phase when the reaction proceeded at 180 °C. This indicates that polymerization hardly proceeded under harsh conditions, most likely due to a reverse reaction. A lower initial CO2 pressure (3 MPa) resulted in a lower DEC yield but produced a higher amount of DEC than TEOS. Moreover, these results suggest that the use of substrate 1a is more effective than previous reports for the direct synthesis of DEC from CO2 (Table S2†). | ||
| Fig. 3 (A) DEC produced from the reaction of 1a or TEOS with CO2. reaction conditions: 1a or TEOS, 15.7 mmol; CO2, 5 MPa at room temperature; Zr(OEt)4, 0.3 g (Si/Zr = 14); 140 °C. DEC yield and 1a conversion were determined by GC using mesitylene as an internal standard. (B) Profile of DEC produced from 1a and TEOS in (i) temperature screening (reaction conditions: 1a or TEOS, 15.7 mmol; CO2, 5 MPa at room temperature; Zr(OEt)4, 0.3 g (Si/Zr = 14)); and (ii) initial CO2 pressure screening (reaction conditions: 1a or TEOS, 15.7 mmol; Zr(OEt)4, 0.3 g; 180 °C). | ||
The above results led us to the hypothesis that the reaction may proceed through an intermediate product that drives the polymerization process. At the beginning of the reaction, substrate 1a was completely consumed with a 46% DEC yield (Table S3†). This yield gradually increased with the reaction time, indicating the formation of intermediate products during the reaction (Fig. S2†). Ganachaud et al. reported that metal alkoxides promote the trimerization of alkyl isocyanates to produce a cyclic isocyanurate moiety.36 In this reaction, cyclic isocyanurate was likely formed as an intermediate during the DEC synthesis. Assuming that the trimeric cyclic isocyanurate is formed, the nine ethoxy moieties present in isocyanurate raise the possibility of polymerization; thus, the polymer product is most likely the result of this polymerization. To verify the existence of a reaction intermediate in the DEC synthesis, we attempted to synthesize isocyanurate from the corresponding isocyanate 1a.
Synthesis of the isocyanurate was conducted by heating 1a at 180 °C under N2 atmosphere without a catalyst (Fig. 4). The reaction was complete after 96 h to produce tris[3-(trimethoxysilyl)propyl] isocyanurate (1b) exclusively. The chemical structure of the product was elucidated by spectroscopic analysis to confirm the formation of 1b. The proton nuclear magnetic resonance (1H NMR) spectrum showed a triplet at δ 3.23 ppm (N–CH2-3 of 1a) to δ 3.8 ppm, which overlapped with (O–CH2-4) (Fig. S3†). The results of carbon nuclear magnetic resonance spectroscopy (13C NMR) suggested that signal-6 at δ 122 ppm (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of isocyanate 1a) is shifted to δ 149 ppm (Fig. S4†). In the silicon nuclear magnetic resonance (29Si NMR) spectrum, the peak showed a slight up field shift from −46.5 to −46.7 ppm (Fig. S5†). The overlapped signals at δ 3.8 ppm were further confirmed by 2D (COSY) NMR. The signals CH2-3 and CH2-4 were correlated with CH2-2 and CH3-5, respectively (Fig. S6†). The 2D (HSQC) NMR spectrum reveals the correlation of the proton (CH2-3) at δ 3.8 ppm with the carbon (CH2-3) at δ 46 ppm and the proton (CH2-4) at δ 3.8 ppm with the carbon (CH2-4) at δ 57 ppm (Fig. 4A). The NMR data confirm the successful formation of tris[3-(trimethoxysilyl)propyl] isocyanurate 1b from substrate 1a.
O of isocyanate 1a) is shifted to δ 149 ppm (Fig. S4†). In the silicon nuclear magnetic resonance (29Si NMR) spectrum, the peak showed a slight up field shift from −46.5 to −46.7 ppm (Fig. S5†). The overlapped signals at δ 3.8 ppm were further confirmed by 2D (COSY) NMR. The signals CH2-3 and CH2-4 were correlated with CH2-2 and CH3-5, respectively (Fig. S6†). The 2D (HSQC) NMR spectrum reveals the correlation of the proton (CH2-3) at δ 3.8 ppm with the carbon (CH2-3) at δ 46 ppm and the proton (CH2-4) at δ 3.8 ppm with the carbon (CH2-4) at δ 57 ppm (Fig. 4A). The NMR data confirm the successful formation of tris[3-(trimethoxysilyl)propyl] isocyanurate 1b from substrate 1a.
 | ||
| Fig. 4 (A) 2D (HSQC) NMR result of isocyanurate 1b; (B) reaction pathway of DEC formation from CO2 and 1a through 1b intermediate product. Reaction conditions: 1a, 10 mL; N2, atmosphere; 180 °C; 96 h. Yield was determined by 1H NMR spectroscopy. | ||
Additionally, attenuated total reflection-FTIR (ATR-FTIR) analysis showed the disappearance of the –NCO band from isocyanate at 2267 cm−1 (Fig. S7†). In contrast, the new bands assigned as the CO bond of 1b and DEC were observed at 1698 cm−1 and 1746 cm−1, respectively.36 Furthermore, the matrix assisted laser desorption ionization coupled with time of flight mass spectrometry (MALDI-TOF MS) showed a single peak at m/z = 759, which closely agrees with the theoretical molecular weight of 1b (763; Fig. S8†). The Zr(OEt)4 catalyst accelerated the formation rate of 1b, and the reaction reached completion in 15 h. The spectroscopic 1H, 13C, and 27Si NMR patterns were identical to those of a previous reaction conducted without a catalyst (Fig. S9–S11†). These results indicate that 1b was formed during the synthesis of DEC.37,38
There is a significant ongoing effort to elucidate the formation of intermediate 1b during DEC synthesis and exploring its capability to produce DEC compared to substrate 1a is of considerable interest. DEC formation progress from 1b was observed under identical conditions (5 MPa of CO2, 180 °C, and Si/Zr ratio = 14) as in the DEC formation reaction from substrate 1a (Fig. S12†). The results showed that the rate of DEC formation from 1b was slightly higher than that from 1a, indicating that conversion of 1a to intermediate 1b is an initial step in the reaction. No induction period was detected during the early reaction stages, suggesting that 1b formed quickly. Additionally, 1b not only served as an important intermediate for triggering polymerization, but the high yield of DEC produced from 1b demonstrated that it is a promising reagent for the synthesis of DEC (Fig. 4B).
The solid product, referred to as polyisocyanurate 1c, formed during DEC synthesis was characterized after DEC extraction with tetrahydrofuran (THF) (Fig. S13†). It was identified by 29Si NMR, gel permeation chromatography (GPC), and MALDI-TOF MS. In 29Si NMR spectrum, two peaks appeared at −54.7 and −46.7 ppm that corresponded to Si–O bridge and terminal Si–O, respectively (Fig. S14†).39 Data from 29Si NMR analysis were utilized for determining average molecular weight 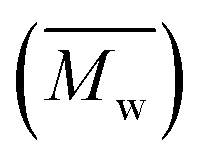 value of Si-based polymers.40 The average value of repeating unit (
value of Si-based polymers.40 The average value of repeating unit (![[n with combining macron]](https://www.rsc.org/images/entities/i_char_006e_0304.gif) ) of 1c after DEC synthesis can be estimated from the ratio of terminal-bridge Si–O bonds (Table S5†) and represents the degree of polymerization of 1c, which is then correlated with the DEC yield (Fig. S14†). For example, of 1c estimated from the reaction at 140 °C was 4, yielding 5.1 mmol of DEC, whereas the reaction at 180 °C gave equal to 3 and yielded 4.1 mmol DEC. However, this analysis is limited by the estimation of the average number of polymers dissolved in the deuterated solvent. Future studies could focus on improving solvent systems for 29Si NMR to enhance the accuracy of polymer dissolution and characterization. To overcome this limitation of 29Si NMR analysis, the distribution of Mw was determined by GPC and MALDI-TOF analysis.
) of 1c after DEC synthesis can be estimated from the ratio of terminal-bridge Si–O bonds (Table S5†) and represents the degree of polymerization of 1c, which is then correlated with the DEC yield (Fig. S14†). For example, of 1c estimated from the reaction at 140 °C was 4, yielding 5.1 mmol of DEC, whereas the reaction at 180 °C gave equal to 3 and yielded 4.1 mmol DEC. However, this analysis is limited by the estimation of the average number of polymers dissolved in the deuterated solvent. Future studies could focus on improving solvent systems for 29Si NMR to enhance the accuracy of polymer dissolution and characterization. To overcome this limitation of 29Si NMR analysis, the distribution of Mw was determined by GPC and MALDI-TOF analysis.
The Mw of 1c was in the range 800–7200, corresponding to the presence of monomers to oligomers with n > 5 (Fig. S15 and Table S6†). Monomer 1b was the dominant component (24%), indicating that 1b was not fully converted to 1c. However, the Mn values obtained from GPC did not precisely match the theoretical values that could support this claim. Thus, the Mw was further verified by MALDI-TOF MS measurements (Fig. 5). MALDI-TOF MS was performed using a 2-[(2E)-3-(4-tert-butylphenyl)-2-methylprop-2-enylidene] malononitrile (DCTB) matrix with Ag as the cation.41 The Mw of 1c obtained from MALDI-TOF MS agreed well with the calculated value, for which the structure varied from monomer to octamer (n = 1–8) with the mixture of linear and cyclic structures (Table S7†). These results strongly indicate that the reaction at 140 °C produced oligomer 1c in a broad range of Mw values, resulting in a high yield of the DEC product. Therefore, the use of a substrate that can form oligomers is a key factor in improving DEC yield.
 | ||
| Fig. 5 MALDI-TOF MS of oligomer 1c produced after DEC synthesis. | ||
Next, the substrate scope was explored. Other substrates that can produce oligomers, such as bis- and tris-triethoxysilane, were employed in the DEC reaction, and their performances were compared (Fig. 6). Generally, substrates bearing a high number of ethoxy groups yielded higher amounts of DEC than TEOS. For example, bis-(triethoxysilyl) substrates 2a–7a yielded 2.2–3.3 mmol DEC, an increase compared to the reaction with TEOS as the substrate. Tris-(triethoxysilyl) substrates (8a and 9a) gave 3.1 and 3.3 mmol of DEC, respectively. Similar to substrate 1a, the oligomer was observed as a byproduct of the bis- and tris-substrates. DEC synthesized from substrate 3a resulted in not only dimer by-products, but also monocyclic and bicyclic silsesquioxanes, along with a high DEC yield (Fig. S16†). Previously, the intramolecular condensation of bis-(trialkoxysilyl)alkanes to afford cyclic- and alkane-bridged trialkoxysilsesquioxanes (3b–3d) has been investigated extensively for the synthesis of sol–gel materials.42,43 Again, these results demonstrate that the utilization of substrates that can produce oligomers as byproducts can improve the yield of DEC. The use of intermediate 1b, which afforded the highest DEC yield (4.5 mmol), prompted us to explore the regeneration of 1b from byproduct 1c.
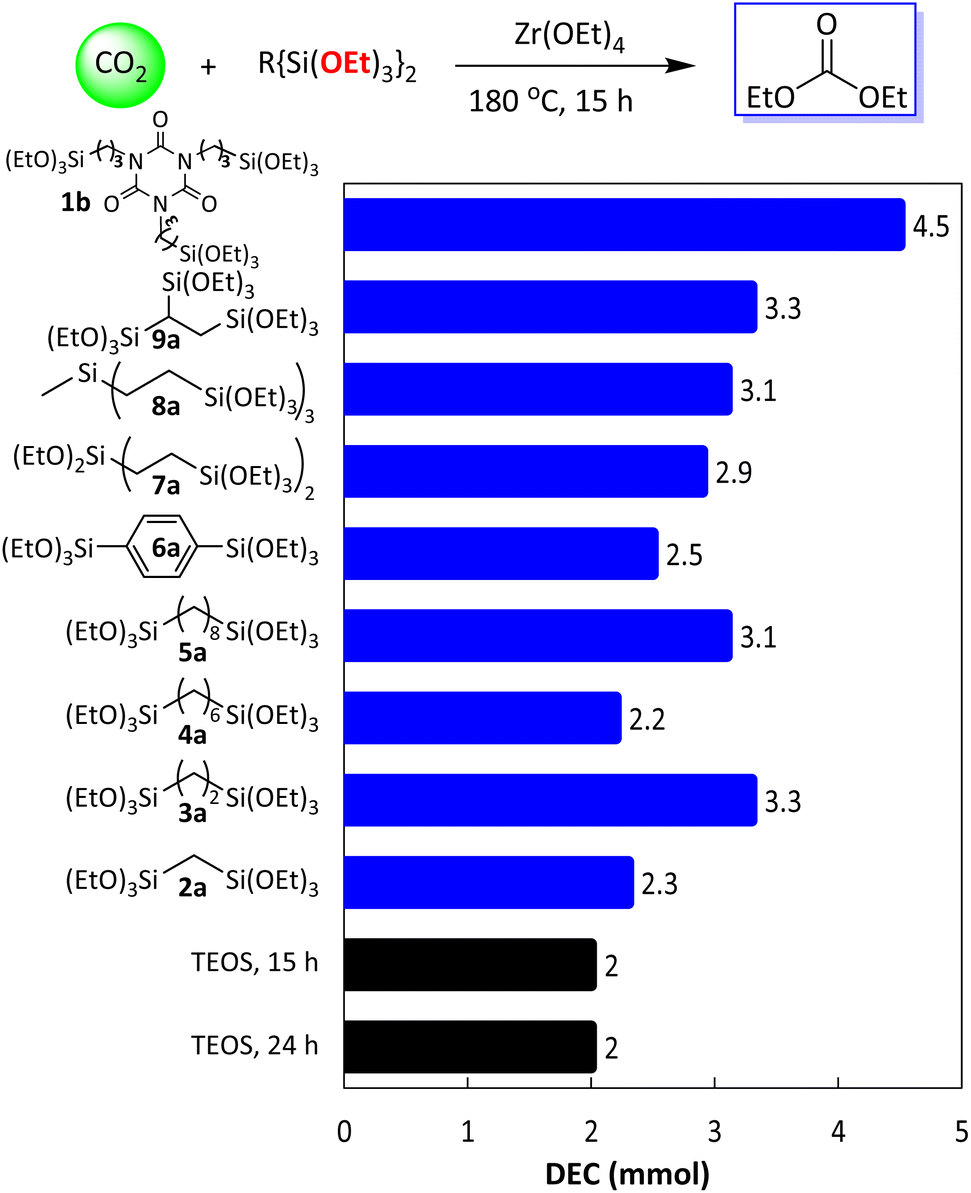 | ||
| Fig. 6 Substrate scope. Reaction conditions: Si amount = 7.8 mmol, Zr(OEt)4 = 0.3 g (Si/Zr = 7); CO2, 5 MPa at room temperature. Yield was determined by GC or 1H NMR analysis using mesitylene as an internal standard. | ||
Finally, to clarify the sustainability and potential industry applicability of this method, the regeneration of 1b from byproduct 1c (Fig. 7A) and potential scaling up were investigated. The scaling-up experiment was conducted under 5 MPa of CO2 at 180 °C using a 200 mL reactor (10 times larger than the normal scale). 96 mmol of DEC (61% yield) was achieved, which was slightly lower than the original scale (5 mmol or 64%) (Table S8 and Fig. S17†). The isolated yield of DEC was found to be identical to the GC yield (64%). The TON was increased to 41 by employing 0.2 g of catalyst over a 69 hours reaction. DEC was subsequently removed from the reaction system, followed by the addition of dry ethanol and potassium hydroxide (KOH) as catalyst to regenerate 1b from byproduct 1c (Scheme S1†). An autoclave equipped with MS 3A was installed in the circulating system (Fig. S18†). The successive DEC synthesis and regeneration process were monitored by 29Si NMR measurements to observe the compound 1c obtained after the DEC synthesis and the regeneration product 1b, as shown in Fig. 7B. The 29Si NMR spectrum for the product after regeneration exhibited the complete disappearance of the peak derived from bridged silicon at −54.7 ppm, indicating that the solid byproduct 1c was fully recyclable to liquid 1b after 16 h of reaction at 200 °C (Fig. 7B and S16†). Liquefaction of the solid of byproduct 1c indicating the depolymerization of byproduct 1c effectively proceeded over KOH catalyst. Collectively, these results demonstrate the potential industrial applicability of this method for the waste-free formation of DEC and its capability to achieve catalytic sustainability while producing high yields of DEC (Fig. 7C).
 | ||
| Fig. 7 (A) Reconversion of the oligomer byproduct 1c; (B) 29Si NMR results of 1b regeneration; and (C) schematic illustration of waste-free formation of DEC. | ||
Conclusion
The catalytic synthesis of DEC from CO2 and alkoxysilane was optimized to overcome equilibrium limitations. Bis-/tris-triethoxysilane substrates produced high amounts of oligomer byproducts that triggered high DEC formation. High DEC yields were obtained using isocyanate 1a through the intermediate isocyanurate 1b prior to the formation of solid polyisocyanurate 1c ranging from a monomer to an octamer. The other bis-/tris-triethoxysilane substrates produced higher amounts of DEC than TEOS. Additionally, the monomeric substrate 1b can be regenerated by simply reacting the oligomer byproduct 1c with ethanol. The development of an effective heterogeneous catalyst for this reaction could be ideal for future upcoming step for industrial applicability and sustainability of the process.44 Therefore, this method might provide the opportunity to obtain high DEC yields sustainably.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Wahyu S. Putro: conceptualization, investigation, writing, reviewing, and editing the original paper. Akira Ikeda: investigation, data validation, and formal analysis. Toshihide Yamamoto: writing, reviewing, and editing. Satoshi Hamura: writing, reviewing, and editing. Jun-Chul Choi: conceptualization, supervising, reviewing, and editing the original paper. Norihisa Fukaya: conceptualization, supervising, reviewing, and editing the original paper.Conflicts of interest
The authors declare no competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This study was based on the results obtained from project JPNP21021, subsidized by the New Energy and Industrial Technology Development Organization (NEDO). We thank Dr Tadahiro Nitta for his invaluable assistance with this experiment, and Dr Vladimir Ya. Lee for engaging in insightful discussions related to this research.References
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS.
- Z. Liu, P. Ciais, Z. Deng, R. Lei, S. J. Davis, S. Feng, B. Zheng, D. Cui, X. Dou, B. Zhu, R. Guo, P. Ke, T. Sun, C. Lu, P. He, Y. Wang, X. Yue, Y. Wang, Y. Lei, H. Zhou, Z. Cai, Y. Wu, R. Guo, T. Han, J. Xue, O. Boucher, E. Boucher, F. Chevallier, K. Tanaka, Y. Wei, H. Zhong, C. Kang, N. Zhang, B. Chen, F. Xi, M. Liu, F.-M. Bréon, Y. Lu, Q. Zhang, D. Guan, P. Gong, D. M. Kammen, K. He and H. J. Schellnhuber, Nat. Commun., 2020, 11, 5172 CrossRef CAS PubMed.
- A. Rehman, F. Saleem, F. Javed, A. Ikhlaq, S. W. Ahmad and A. Harvey, J. Environ. Chem. Eng., 2021, 9, 105113 CrossRef CAS.
- J.-C. Choi, L.-N. He, H. Yasuda and T. Sakakura, Green Chem., 2002, 4, 230–234 RSC.
- K. Takeuchi, M.-Y. Chen, H.-Y. Yuan, H. Koizumi, K. Matsumoto, N. Fukaya, Y.-K. Choe, S. Shigeyasu, S. Matsumoto, S. Hamura and J.-C. Choi, Chem.–Eur. J., 2021, 27, 18066–18073 CrossRef CAS PubMed.
- M. Xu, A. R. Jupp, M. S. E. Ong, K. I. Burton, S. S. Chitnis and D. W. Stephan, Angew. Chem., Int. Ed., 2019, 58, 5707–5711 CrossRef CAS.
- D. L. J. Broere, B. Q. Mercado and P. L. Holland, Angew. Chem., Int. Ed., 2018, 57, 6507–6511 CrossRef CAS.
- Y. Zhang, Y. Wang, Q. Qian, Y. Li, B. B. Asare Bediako, J. Zhang, J. Yang, Z. Li and B. Han, Green Chem., 2022, 24, 1973–1977 RSC.
- M. Aresta, A. Dibenedetto and E. Quaranta, J. Catal., 2016, 343, 2–45 CrossRef CAS.
- K. Shukla and V. C. Srivastava, RSC Adv., 2016, 6, 32624–32645 RSC.
- S. Huang, B. Yan, S. Wang and X. Ma, Chem. Soc. Rev., 2015, 44, 3079–3116 RSC.
- P. Kongpanna, V. Pavarajarn, R. Gani and S. Assabumrungrat, Chem. Eng. Res. Des., 2015, 93, 496–510 CrossRef CAS.
- K. Tomishige, T. Sakaihori, S. I. Sakai and K. Fujimoto, Appl. Catal., A, 1999, 181, 95–102 CrossRef CAS.
- M. A. Pacheco and C. L. Marshall, Energy Fuels, 1997, 11, 2–29 CrossRef CAS.
- S. Dabral and T. Schaub, Adv. Synth. Catal., 2019, 361, 223–246 CrossRef CAS.
- I. Omae, Coord. Chem. Rev., 2012, 256, 1384–1405 CrossRef CAS.
- K. Tomishige, Y. Gu, Y. Nakagawa and M. Tamura, Front. Energy Res., 2020, 8, 117 CrossRef.
- E. Leino, P. Mäki-Arvela, K. Eränen, M. Tenho, D. Y. Murzin, T. Salmi and J.-P. Mikkola, Chem. Eng. J., 2011, 176–177, 124–133 CrossRef CAS.
- K. Kohno, J.-C. Choi, Y. Ohshima, H. Yasuda and T. Sakakura, ChemSusChem, 2008, 1, 186–188 CrossRef CAS.
- J.-C. Choi, T. Sakakura and T. Sako, J. Am. Chem. Soc., 1999, 121, 3793–3794 CrossRef CAS.
- W. S. Putro, Y. Munakata, S. Shigeyasu, S. Hamur, S. Matsumoto, J.-C. Choi and N. Fukaya, Mendeleev Commun., 2022, 32, 54–56 CrossRef CAS.
- M. Aresta, A. Dibenedetto and C. Pastore, Inorg. Chem., 2003, 42, 3256–3261 CrossRef CAS.
- M. Honda, M. Tamura, Y. Nakagawa, K. Nakao, K. Suzuki and K. Tomishige, J. Catal., 2014, 318, 95–107 CrossRef CAS.
- Y. Gu, K. Matsuda, A. Nakayama, M. Tamura, Y. Nakagawa and K. Tomishige, ACS Sustain. Chem. Eng., 2019, 7, 6304–6315 CrossRef CAS.
- M. Honda, S. Kuno, N. Begum, K. I. Fujimoto, K. Suzuki, Y. Nakagawa and K. Tomishige, Appl. Catal., A, 2010, 384, 165–170 CrossRef CAS.
- M. Honda, S. Kuno, S. Sonehara, K. I. Fujimoto, K. Suzuki, Y. Nakagawa and K. Tomishige, ChemCatChem, 2011, 3, 365–370 CrossRef CAS.
- Y. Chen, H. Wang, Z. Qin, S. Tian, Z. Ye, L. Ye, H. Abroshan and G. Li, Green Chem., 2019, 21, 4642–4649 RSC.
- P. Kumar, P. With, V. C. Srivastava, R. Gläser and I. M. Mishra, J. Environ. Chem. Eng., 2015, 3, 2943–2947 CrossRef CAS.
- T. Chang, M. Yabushita, Y. Nakagawa, N. Fukaya, J.-C. Choi, T. Mishima, S. Matsumoto, S. Hamura and K. Tomishige, Catal. Sci. Technol., 2023, 13, 5084–5093 RSC.
- T. Chang, M. Tamura, Y. Nakagawa, N. Fukaya, J.-C. Choi, T. Mishima, S. Matsumoto, S. Hamura and K. Tomishige, Green Chem., 2020, 22, 7321–7327 RSC.
- W. S. Putro, Y. Munakata, S. Ijima, S. Shigeyasu, S. Hamura, S. Matsumoto, T. Mishima, K. Tomishige, J.-C. Choi and N. Fukaya, J. CO2 Util., 2022, 55, 101818 CrossRef CAS.
- M. Honda, M. Tamura, Y. Nakagawa, S. Sonehara, K. Suzuki, K. I. Fujimoto and K. Tomishige, ChemSusChem, 2013, 6, 1341–1344 CrossRef CAS.
- W. Sun, P. Li, M. Yabushita, Y. Nakagawa, Y. Wang, A. Nakayama and K. Tomishige, ChemSusChem, 2023, 16, e202300768 CrossRef CAS.
- W. S. Putro, A. Ikeda, S. Shigeyasu, S. Hamura, S. Matsumoto, V. Y. Lee, J.-C. Choi and N. Fukaya, ChemSusChem, 2021, 14, 842–846 CrossRef CAS PubMed.
- T. T. H. Nguyen, W. S. Putro, S. Hamura, M. Nakashige, J.-C. Choi, N. Fukaya, S. Taniguchi, T. Yamaki, N. Hara and S. Kataoka, J. Clean. Prod., 2023, 389, 136046 CrossRef CAS.
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS.
- M. A. Bahili, E. C. Stokes, R. C. Amesbury, D. M. C. Ould, B. Christo, R. J. Horne, B. M. Kariuki, J. A. Stewart, R. L. Taylor, P. A. Williams, M. D. Jones, K. D. M. Harris and B. D. Ward, Chem. Commun., 2019, 55, 7679–7682 RSC.
- H. A. Duong, M. J. Cross and J. Louie, Org. Lett., 2004, 6, 4679–4681 CrossRef CAS PubMed.
- H. El Rassy and A. C. Pierre, J. Non-Cryst. Solids, 2005, 351, 1603–1610 CrossRef CAS.
- D. A. Laude Jr and C. L. Wilkins, Macromolecules, 1986, 19, 2295–2300 CrossRef.
- K. Fuchise, K. Sato and M. Igarashi, Macromolecules, 2021, 54, 5204–5217 CrossRef CAS.
- D. A. Loy, J. P. Carpenter, T. M. Alam, R. Shaltout, P. K. Dorhout, J. Greaves, J. H. Small and K. J. Shea, J. Am. Chem. Soc., 1999, 121, 5413–5425 CrossRef CAS.
- R. Hayami, Y. Ideno, Y. Sato, H. Tsukagoshi, K. Yamamoto and T. Gunji, J. Polym. Res., 2020, 27, 316 CrossRef CAS.
- H. Nagae, H. Koizumi, K. Takeuchi, S. Hamura, T. Yamamoto, K. Matsumoto, Y. Kamimura, S. Kataoka, N. Fukaya and J.-C. Choi, ChemCatChem, 2024, e202400673 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00634h |
| This journal is © The Royal Society of Chemistry 2025 |