
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Flexible fire-safe hybrid organic–inorganic cellulose aerogels from sol–gel casting†
Björn K.
Birdsong
a,
Antonio J.
Capezza
 a,
Rhoda Afriyie
Mensah
b,
Patric
Elf
a,
Mikael S.
Hedenqvist
a,
Fritjof
Nilsson
*a and
Richard T.
Olsson
*a
a,
Rhoda Afriyie
Mensah
b,
Patric
Elf
a,
Mikael S.
Hedenqvist
a,
Fritjof
Nilsson
*a and
Richard T.
Olsson
*a
aDepartment of Fibre and Polymer Technology, School of Chemical Science and Engineering, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden. E-mail: rols@kth.se; fritjofn@kth.se
bDepartment of Civil, Environmental and Natural Resources Engineering, Luleå University of Technology, 97187 Lulea, Sweden
First published on 10th January 2025
Abstract
The flexibility of hybrid silicon-oxide cellulose aerogels was achieved through the formation of thin, uniform silica coatings on cellulose fibres, or local regions of a classical spherical aerogel (Kistler aerogel) combined with areas of less coated cellulose fibres, making use of the flexible properties of the cellulose nanofibres. Furthermore, the inclusion of cellulose during the sol–gel formation allowed the use of traditional freeze-drying instead of CO2 critical point drying as a method for the removal of the liquid phase. The silicon oxide morphologies revealed the possibility of fine-tuning the coating's structure by the choice of the silicon-oxide precursors. Using methyltrimethoxysilane (MTMS) resulted in the formation of classical aerogel or spherical particles, while the use of tetraethyl orthosilicate (TEOS) yielded “pearl-necklace” fibres, and the mix of (3-aminopropyl)triethoxysilane (APTES) with MTMS yielded smooth uniform coatings. The prepared coating morphologies markedly influenced the aerogel's properties (mechanical stiffness/flexibility, flame resistance and hydrophilicity). The silica coatings endured high-temperature exposure and the thermal removal of the cellulose template without substantial morphological changes was confirmed, showing the possibility to use cellulose as an effective template for the synthesis of silicon-oxide nanofibres. The possibility to selectively control aerogel properties already at the synthesis stage, using abundant and renewable materials together with the possibility of using more energy-conservative freeze-drying (rather than critical point drying), is a promising method for more sustainable aerogel preparation towards high-end commercial applications such as electrical fuel cell insulation.
Sustainability spotlightInsulation materials are an essential part of creating sustainable cities and communities, as they allow for affordable and clean energy to be used (SDG 11 and SDG 7), as well as good health and well-being (SDG 3). Aerogels stand out among these materials for their exceptional thermal insulation and fire safety. However, traditional Kistler aerogels face limitations in large-scale applications due to their brittleness and the challenges associated with high-pressure critical point drying during production. These constraints have hindered their widespread commercial use. This work explored the use of cellulose nanofibres to enable the preparation of highly flexible, yet mechanically stable aerogels manufactured without the use of critical point drying due to the ability of cellulose to stabilise the gel nature of the aerogels during inexpensive freeze-drying operations. |
Introduction
Aerogels have, since their discovery, become sought-after materials for use in a wide range of applications from meteor dust collection to thermal insulation.1,2 What makes aerogels unique and of particular interest is related mainly to their exceptionally low density (ca. 0.003 g cm−3), thermal stability, and low thermal conductivity (ca. 0.005 W m−1 k−1), which originates from the microstructure and composition of the materials.3,4 However, despite these valuable properties, aerogels are also known for their brittleness, where even small deflections result in crack formation or even violent shattering.5,6Aerogels can be derived from a wide range of synthetic and biobased raw materials, all with their pros and cons. Silica aerogels are most common, due to their low density, excellent thermal insulation, good thermal stability and simple processing, but they are brittle, mechanically weak, and non-biocompatible and require expensive supercritical drying during their production.7 Cellulose aerogels are biobased, biodegradable, and biocompatible and have better mechanical properties than silica aerogels, but have inferior thermal stability, which limits their applications.8 Aerogels based on polymers, such as polystyrene and poly(2,6-dimethyl-1,4-phenylene)oxide, are cheaper and more flexible and have higher mechanical strength than silica aerogels but are less thermally stable and are difficult to recycle.9 Carbon based aerogels, often produced using pyrolysis of organic compounds or cellulose derivatives, are thermally stable and have very high electrical conductivity, but are expensive and show limited mechanical performance.10 Metal-oxide based aerogels (e.g. aluminium oxide or titanium oxide) have high thermal and chemical stability and show good catalytic performance but are mechanically brittle and more expensive.11 The aim of this study is to find a way to overcome the weaknesses of silica aerogels by making hybrid silica/cellulose aerogel materials with high performance.
The fragile properties of silicon oxide-based aerogels often originate from the delicate silica network structures formed during the synthesis of the material. A standard method of forming these networks is a process known as sol–gel, where precursors to silicon oxide are allowed to react in a liquid medium to form a gel network throughout the medium.6 The network is composed of nano- or micro-sized silicon oxide particles covalently bonded together. Even in the wet state, the formed gels are fragile due to the formed sensitive silicon oxide network, often being non-uniform in thickness, resulting in stress points around thinner regions.5 The formed silicon oxide particles are often large enough to prevent flexibility in the material, which is emphasized after the replacement of the liquid phase with air. The brittleness of aerogels is also a significant contributing factor to the challenges involved in any larger-scale manufacturing of aerogels. Due to their fragile nature, the method used for production often involves critical point drying (CPD), a technique to remove a liquid phase using high pressure without disturbing any solid material structures.12 Although the method is useful for smaller-scale production and eliminates the problems involved in aqueous removal involving water evaporation or sublimation, it is generally not considered scalable.13
To minimize the problems with the brittleness, doping aerogels with metal ions or using other materials such as cellulose, to either increase the mechanical strength or replace the silicon oxide network with other polymers, has been reported. However, these methods either do not allow for flexibility or high-temperature stable materials.14,15 In fact, the flexibility has been shown to rely on the unique formation of fine silica networks by using the stereochemistry of silanes such as methyltrimethoxysilane (MTMS) and (3-aminopropyl)triethoxysilane (APTES), which, due to the methyl or propan-1-amine functional group, restricts excessive and rapid particle growth, i.e., effectively limits the rate of condensation and the precursor conversion into its solid form. However, although these aerogels can be deformed without breaking, they are limited by their CH3-functional groups on the surface of the silicon oxide network, restricting hydrophilic modification of the surface chemistry, which may be regarded as a key functional feature with classical Kistler aerogels with accessible OH-functional surface groups. Lastly, no control over the direction of the formed network in the gel results in an isotropic material with no directional mechanical properties, which is often desired in materials prone to directional bending.16,17 A novel method that would enable flexible materials is here the inclusion of a mechanically strong and flexible scaffold material, which, even if cracks form, can hold the material together. The scaffold can function as a template, allowing for control over both the network formation and size, thickness, and fibre orientation, while allowing the use of a more comprehensive array of silicon oxide precursors.18–20
In this work, mechanically robust organic/inorganic hybrid cellulose aerogels were developed that were able to be bent and twisted, without permanent deformation occurring. Cellulose is the most abundant biopolymer on earth and is known to be chemically compatible with sol–gel reactions while also showing desired quality of insulation materials.20,21 Cellulose was previously used with the additional benefit of increasing the nucleation of metal ions as well as providing a template for silane deposition, enabling flexible hybrid organic/inorganic aerogels to be made.20–23 The possibility of fine-tuning coating morphology via the choice of the silicon oxide precursor allowed for the formation of spherical particles, “pearl-string” fibres, and more classical Kistler aerogel structures. Furthermore, the cellulose matrix incorporated during the sol–gel formation of the aerogel enabled the use of freeze drying, instead of critical point drying (CPD), demonstrating the possibility of producing aerogels at a larger scale. Lastly, the silicon oxide coatings were able to survive high-temperature thermal removal of the organic cellulose template without significant changes in the glass morphology, making cellulose an effective templating material for the synthesis of silica nanofibres.
Experimental
Materials
(3-Aminopropyl)triethoxysilane (APTES, ≥98%), methyltrimethoxysilane (MTMS, ≥95%), tetraethyl orthosilicate (TEOS, ≥98%), and ammonia hydroxide (aq. 28 wt%) were purchased from Sigma-Aldrich and used without further modification. Isopropanol (reagent grade, ≥99.8%) was purchased from VWR, and Milli-Q water (MQw) (18.2 MΩ, pH = 7.0) was used as the suspension medium. Bacterial cellulose (BC) was purchased from Monstra LLC (Thailand) as cubes with a volume of ca. 1 cm3 and was grown from strains of Acetobacter xylinum.Preparation of bacterial cellulose nanofibre
Bacterial cellulose (BC) was prepared by soaking the bacterial cellulose cubes as received in Milli-Q water (MQw) for 5 days before being treated with 10 vol% NaOH at 100 °C for 10 min, after which the cubes were rinsed for 24 h using deionized water to remove any remaining impurities. The purified BC cubes were shredded into a homogeneous slurry using a blender (Blendtec 625) before being diluted to a 0.3 ± 0.01 wt% suspension.Sol–gel reaction with incorporated bacterial cellulose
The BC suspension was prepared by the addition of 5 mL of isopropanol to 3 mL of BC and the suspension was thoroughly mixed before adding the silane amounts in Table 1. The suspension was poured into silicone moulds and 1 mL of ammonia was added to initiate and catalyse the silanisation reaction. The suspension was then left to age for 24 h. To replace the main alcohol solvent present in the formed gel with water, two solvent exchanges were used, first by immersing the formed gel in 40 mL of a 25/75 ethanol/MQw mixture and allowing it to reach an equilibrium in the gel for 24 h, after which the solution was replaced with 40 mL of pure MQw for 24 h as a final solvent exchange, resulting in a water-filled gel (Fig. 1). | ||
| Fig. 1 Methodology used for the development of flexible aerogels using gel casting to form the final material shape. | ||
| MTMS-BC | TEOS-BC | APTES/MTMS-BC | |
|---|---|---|---|
| APTES | — | — | 200 μL |
| MTMS | 1 mL | — | 800 μL |
| TEOS | — | 1 mL | — |
Solution removal and drying procedure
To replace the MQw medium in the BC gels with air, freeze-drying was used. The MQw was gently poured off from the BC-gels ensuring that a thin film (ca. 1 mm) of water remained on the surface of the gels. The gels were placed in a freezer at −30 °C for 2 h, ensuring complete freezing of the BC-gels, after which a HyperCOOL (HCC-3110, Republic of Korea) freeze dryer was used to remove the frozen water with a pressure of <0.1 mbar. To remove any potentially remaining surface-bound water from the BC-aerogels the materials were heated to 110 °C for 4 h using a Memmert UNB 100 oven. The samples were stored in polypropylene tubes until further use.Characterization
Thermogravimetric analysis (TGA)
The thermal stability of the BC-aerogels as well as the fraction of the materials that were composed of silicon oxide was determined by thermogravimetric analysis (TGA), using a Mettler Toledo thermal analyzer (TGA/DSC 3+). 5 to 10 mg of the BC-aerogel was placed in 70 μL alumina oxide (Al2O3) crucibles (ME-24123) and heated using a rate of 10 °C min−1 to 110 °C. The samples were kept at 110 °C for 5 min, followed by heating to 800 °C, using the same heating rate, where it was again kept for 5 min. The used purge gas was a 50 mL min−1 flow of O2.Contact angle measurements
The materials' wettability was investigated using droplets of Milli-Q water, using a CAM200 contact angle meter (KSV Instruments, Finland) with an automatic dispenser and a CCD camera. The sessile drop technique was used to measure the static water-material contact angle of the BC-aerogels, using 4 μL volume droplets from a 200 μL pipette tip. The measurements were conducted at 23 ± 1 °C and 50 ± 2% RH. Images of water droplets in contact with the BC-aerogels materials were recorded for 40 s at 1% FPS, using the software “Attension–Biolin Scientific”, while the Young–Laplace method was used to calculate the contact angle of the water-material interphase.Mechanical testing by 3-point bending
The stiffness of the aerogel materials was investigated using the 3-point bending technique, using an Instron 5566 tensile tester (Instron Corp., USA), equipped with a 10 kN load cell. The measurements were conducted on rectangular samples sized 70 mm, 4.5 mm, and 12 mm, using a support span of 40 mm, while the flexural strain rate was 0.02 mm mm−1 min−1.Cone calorimetry
The investigation of the aerogel's reaction-to-fire properties was done using a cone calorimeter (TCC 918, Germany) using 25 kW m−2 heat flux at the corresponding temperature of 618 °C. Due to sample preparation limitations, the measurements were conducted on 20 mm × 20 mm samples with a 25 mm distance between the material surface and the cone heater. To prevent excessive heat loss, the aerogels were surrounded and placed on an insulative material, leaving only the top surface exposed to the heat.Fourier-transform infrared spectroscopy (FTIR)
FTIR absorbance of the aerogel materials as well as freeze-dried virgin BC was done using a PerkinElmer Spectrum 100, equipped with an ATR accessory, an MIR TGS detector, and a Specac golden gate with a sapphire crystal. The measurements were performed with a resolution of 4 cm−1, using 16 consecutive scans ranging from 600 to 4000 cm−1.Thermal conductivity
A hot-plate method was used to calculate the thermal conductivity of the materials. The hot-plate, which was used to heat the ca. 4 mm thick sample, was heated to 80 °C before each experiment. A polished copper sheet was used to hinder infrared radiation from the plate and a thick HI 70 Styrofoam ring was applied to prevent convection from the part of the plate which was not covered by the sample. Each sample was first heated for 10 min to reach the equilibrium temperature, and then the temperature was measured every second minute for 10 minutes. For each sample, the average of five measurements was used, and for each material, the average of three samples was taken. The thermal conductivity k was computed as k = L × P/(A × dT), where L is the thickness of the sample, P is the effective heating effect applied on the sample, A is the sample area and dT is the difference in temperature between the plate and the sample surface. By calibrating the equipment with an EPS-sample with known thermal conductivity (k = 0.038 W m−1 K−1) and the same geometry, the parameters P and A were avoided.Molecular modelling
To calculate the molecular volumes of the functional groups for MTMS, TEOS, and APTES, atomistic structures of said molecules were generated with the simulation software Materials Studio®. The volume was defined as the region inside the molecule's Conolly surface, using the van der Waals radii of the individual atoms, and it was calculated using Monte-Carlo integration. To visualize the molecular systems and calculate the distances between their functional groups, a layer of crystalline monoclinic cellulose 1b was generated and MTMS, TEOS, and APTES molecules were attached to the accessible OH-groups of the cellulose crystal layer.24 Geometry optimization with the universal force field and the steepest descent and conjugated gradient optimization algorithms was applied.Results and discussion
Flexibility of the silane aerogel
Fig. 2a–c show MTMS-BC, TEOS-BC, and APTES/MTMS-BC at the maximum load recorded during 3-point bending. Although all the aerogels demonstrated bending ability, it was evident that the nature and amount of deflection without crack formation or breaking were different. The TEOS-BC aerogel was most sensitive to deformation in comparison to the others, even when handling, and formed an apparent crack after only 5 mm of deflection, as evidenced by the small load needed to break the aerogel (see Fig. 2f). In comparison both MTMS-BC and APTES/MTMS-BC were more flexible and capable of deflecting up to 10 mm and 7 mm respectively, without crack formation, and at this point slipping on the holding anvil occurred, see Fig. 2f. Furthermore, the materials APTES/MTMS-BC and MTMS-BC did not show any clear signs of crack formation or breaking; however, as seen in Fig. 2d, MTMS-BC maintained a slightly curved state while APTES/MTMS-BC returned to a flat state after the tests, further showing a difference in elasticity. | ||
| Fig. 2 MTMS-BC, TEOS-BC, and APTES/MTMS-BC aerogels at the maximum measured load during 3-point-bending (a–c), the mechanical recovery of the various aerogels (d), and the morphology of the aerogel materials (e). The 3-point bending plots correspond to the bending process of the aerogels, using two replicas of each sample (f). | ||
The differences in the mechanical deformation behaviour were suspected to arise from differences in silane coating morphology. The micrographs seen in Fig. 2e show that although the aerogels were visually similar to a white porous aerogel on the micro/millimetre scale, they showed very different fibre morphologies. The MTMS-BC aerogel comprised of fibrous areas and spherical particles, Fig. 2e top. The fibrous areas were suspected to be uncoated BC fibres, which would agree with the unique formation of typical spherical particles attributed to the precipitation of silane MTMS. In contrast, TEOS-BC showed a completely different morphology in which no visible BC fibres and no larger spherical particles were seen for TEOS-BC, Fig. 2e middle. Instead, TEOS had formed smaller spherical structures along the BC fibres creating a “pearl-necklace” structure. The thicker and more segmented TEOS coatings on the BC fibres were suspected to be the cause of the significantly more brittle nature of the hybrid fibres that were measured (Fig. 2f). The low load (2.1 ± 0.2 mN) needed to crack the TEOS-BC aerogel was suggested to be due to the “neck” regions between the thicker “pearls” along the fibres creating weak points, where the force was elevated due to the smaller fibre diameter.25 This neck morphology was only observed in the TEOS coatings, explaining why only the TEOS-BC aerogel cracked upon deformation. Lastly, the APTES/MTMS-BC aerogels showed a similar morphology to the MTMS-BC aerogels, where regions of BC fibres were visible, as well as clusters of spherical silane particles. It was suspected that APTES had formed a highly uniform and thin coating along the BC fibres, which were difficult to distinguish from the BC fibres. This thin uniform APTES coating would explain the significantly higher stiffness observed, needing roughly twice the load (5.5 ± 0.4 mN) to deform as that for the MTMS-BC aerogel (3.2 ± 0.2 mN), see Fig. 2f. Previous studies have demonstrated that APTES tends to form uniform coatings rather than spherical structures. To further investigate the specific silane coating formation, EDS was used.26,27
FTIR was used to analyse the chemical composition and performance of the materials (Fig. 3). The curve for MTMS/APTES-BC was very similar to that for MTMS-BC, which was expected due to the higher amount of MTMS used compared to APTES (80/20). The most striking observation from the FTIR-spectra was that all large peaks were associated with Si, whereas the peaks associated with cellulose (e.g. C–C, C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and OH-bonds) were in comparison much weaker. Cellulose based materials typically have such more pronounced peaks both in FTIR21 and other spectroscopic methods.28–30 This indicates that most of the cellulose surfaces were silanized. For TEOS-BC, the peaks around 1070 and 950 cm−1 were associated with the Si–O–Si bond and the Si–O silanol stretching (Si–O–H). For MTMS-BC, the peaks around 1270, 1100 + 1020 and 770 cm−1 were associated with (Si–CH3), (Si–O–Si) and (Si–C), respectively. Regardless of the silanes used, the formed bonds were primarily Si–O and Si–O–Si, typical for silica materials.31
O and OH-bonds) were in comparison much weaker. Cellulose based materials typically have such more pronounced peaks both in FTIR21 and other spectroscopic methods.28–30 This indicates that most of the cellulose surfaces were silanized. For TEOS-BC, the peaks around 1070 and 950 cm−1 were associated with the Si–O–Si bond and the Si–O silanol stretching (Si–O–H). For MTMS-BC, the peaks around 1270, 1100 + 1020 and 770 cm−1 were associated with (Si–CH3), (Si–O–Si) and (Si–C), respectively. Regardless of the silanes used, the formed bonds were primarily Si–O and Si–O–Si, typical for silica materials.31
 | ||
| Fig. 3 FTIR spectra of MTMS/APTES-BC, MTMS-BC and TEOS-BC. All large peaks are associated with Si-related bonds, whereas peaks associated with cellulose are nearly absent, e.g. OH-bond peaks (arrows). This indicates a nearly complete silanization of the cellulose surfaces. | ||
The much higher amount of silica-based material present in comparison to the BC meant that any BC signals were significantly lower and difficult to observe with FTIR.
Silane deposition and particle formation
Fig. 4a–c depict the aerogel materials using an optical microscope, and even at relatively low magnification there were observable differences between the MTMS-BC, TEOS-BC, and APTES/MTMS-BC aerogels. The microscope image of MTMS-BC aerogel (Fig. 4a) depicts a porous gel comprised of two regions, a highly thin fibre region and a thicker more solid-appearing region. Although the thin fibre regions did not exhibit any morphology suggesting gel formation, using higher magnification revealed that even at a 1 μm scale (see the inset) the BC fibres were surrounded by ca. 400 and 1500 nm spherical silane structures as seen in the inset image and particle size distribution. In contrast, more significant gel regions distinguished as uniform semi-translucent particles could be seen evenly distributed throughout the material. The formation of the two regions, fibre, and the gel was attributed to the impressive flexibility observed where the fibre regions were able to continue to exhibit flexible behaviour without disturbing the gel. In contrast, the TEOS-BC seen in Fig. 4b shows an overall different material structure, where instead of two distinct regions, there were only larger fibre structures observed. Using high magnification SEM (see the inset) it was apparent that the TEOS silane coating morphology was only composed of the “pearl-necklace” morphology previously described. The formed silane spherical “pearls” were estimated to be ca. 400 nm and were more uniform in size distribution in comparison to the MTMS-BC and APTES/MTMS-BC derived particles. The APTES/MTMS-BC aerogel seen in Fig. 4c showed similarities with the MTMS-BC aerogel in that there were seemingly uncoated regions of fine fibres as well as thicker regions with more visible material attached to the fibres. High magnification inset images revealed similar overall morphology to MTMS-BC where there were regions of seemingly uncoated fibres, as well as spherical silane particles ranging in diameter from ca. 300–1800 nm. | ||
| Fig. 4 The macrostructures of the MTMS-BC, TEOS-BC, and APTES/MTMS-BC aerogels (a–c), and elemental mapping of the silane coatings (d–f). | ||
The difference in sphere size was attributed to the various silanes used and their chemical structural differences. The more reactive TEOS, due to its additional OH group, was able to rapidly condense on the BC fibres forming a high number of nucleation sites that grew evenly in size on the said fibres.32 In contrast, both MTMS and APTES contain one less reactive OH group, possibly resulting in slightly lower reactivity with the BC fibres.32 Additionally, MTMS is often used for the formation of gels due to its tendency to self-condense resulting in spherical particles.33 In the case of APTES the presence of a somewhat bulky carbon chain results in less self-condensation and the formation of spherical particles, instead forming more uniform coatings on the surface of the BC fibres.34
Fig. 4d–f show images obtained from elemental mapping using EDS of MTMS-BC, TEOS-BC, and APTES/MTMS-BC aerogels. From the elemental mapping of the aerogel materials, it was clear that there was a significant amount of silicon oxide present throughout the materials, as seen by the Si and O distribution. However, when comparing the distribution of carbon, there was a slight difference observed in the MTMS-BC aerogels (Fig. 4d) in comparison to the other aerogel materials. The MTMS-BC material showed the clear presence of carbon within the aerogel material; more specifically the detection of carbon was seen between the silane spheres in regions with high amounts of seemingly uncoated BC fibres. This indicated that MTMS had a preference to self-condensate forming spherical structures and only some of the BC fibres were coated. In contrast, TEOS (Fig. 4e) showed that there was virtually no detectable carbon in the aerogel material. Interestingly, with the addition of APTES as in the case of APTES/MTMS-BC (Fig. 4f) there was a significant decrease in detected carbon on the observed fibre structures. The absence was attributed to the TEOS and APTES coatings hindering the BC carbon from being detected.35,36
The specific silane molecular structure was speculated to affect deposition and in turn the silane coating thickness and morphology. To gain a more fundamental understanding of the deposited silane and its effect on coating thickness, atomistic modelling was used (Fig. 5).
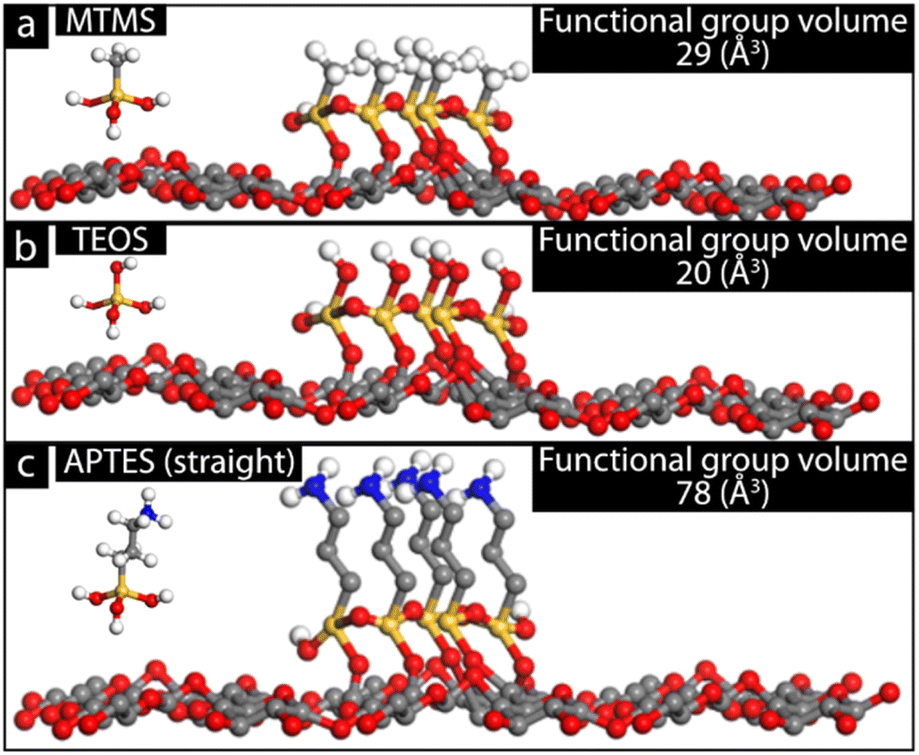 | ||
| Fig. 5 Modeling of MTMS (a), TEOS (b), and APTES in a straight conformation (c). Values were obtained using Materials Studio®. | ||
The characteristic functional groups of each silane result in differences in occupied space around parts of each silane molecule. The occupied space hinders the deposition of other silanes in the vicinity of the blocking functional group, leading to a reduction in silane deposition thickness.37 In the case of TEOS seen in Fig. 5b, the functional group OH, although occupying space, does not restrict any growth as it is able to further react with OH in other TEOS molecules. Furthermore, as each TEOS contains four OH groups, the deposition of TEOS on the BC fibre surface results in an increase in available OH functional groups available for further TEOS deposition. The increased amount of available OH groups results in a higher rate of deposition on a large number of areas on the BC fibres, which explains the smaller average silane sphere sizes observed in TEOS-BC. Replacing an OH group with CH3 as in the case for MTMS (Fig. 5a) may result in some restriction in thickness growth, as not only is the functional group (CH3) larger compared to OH (29 Å3), but also fewer OH groups are available to enable continued silane thickness growth.37 However, the deposition of one MTMS molecule on the BC fibre surface still results in two new available OH groups which can continue to grow in thickness. The competing MTMS depositions on the BC fibre and the already deposited MTMS were suspected to be the cause of the larger distribution in silane particle size. Lastly, Fig. 5c illustrates the APTES coating on the BC fibre and how the large carbon chain functional group (78 Å3) effectively prevents most growth out from the BC fibre template, resulting in smoother and more uniform silane deposition on the fibres, minimizing the risk of uneven growth and, in turn, leading to weak points due to “neck” formations.
It was of interest to investigate how the different silane coating structures and surface chemistry would affect wettability, thermal stability, and fire-resistance properties of the materials, all of which are of interest in materials for potential insulative purposes.38
Thermal stability and fire-safety
Fig. 6a depicts the aerogels after exposure to ca. 620 °C for 4 min in the cone calorimetry test. The appearance of the aerogels varied depending on the silane used. MTMS-BC formed a thin char layer on the exposed side, ca. 1 mm thick, while the remaining aerogel retained its white colour, but was somewhat more fragile compared to that before the heat exposure. In contrast, APTES-BC was more fragile after heat exposure than MTMS-BC, breaking, even with gentle handling, and a higher amount of char formed and also deeper into the material, even reaching the underside in some areas. TEOS-BC was pure white throughout the material, with no visible charring, and with seemingly similar mechanical behaviour to that before the heat exposure. The visual appearance and mechanical behaviour before and after the heat exposure were attributed to the high amount of silicon oxide present in the material. | ||
| Fig. 6 The various aerogels after cone calorimetry tests (4 min exposure to 618 °C) (a), thermal degradation of the aerogels between 200 and 800 °C (b), and the obtained water contact angle (c). The MTMS-BC, TEOS-BC, and APTES/MTMS-BC aerogel morphology after exposure to 800 °C is shown in (d), (e), and (f) respectively. | ||
To further investigate the thermal stability of the materials, TGA was performed. Fig. 6b shows the remaining mass of the various aerogels after exposure up to 800 °C. The plot was adjusted to account for moisture loss, which was found to be significant at the first 200 °C. All aerogels showed ca. 5 wt% mass loss between 300 and 350 °C, attributed to the thermal decomposition of the cellulose which is exposed to air.39 The high residual amount is due to that the silica glass is coating a very large fraction of the cellulose surface, which was also indicated by the FTIR-results. MTMS-BC experienced its most significant mass loss (ca. 6 wt%) at 520 °C, which was attributed to the loss of the CH3 functional groups from the MTMS silane. Lastly, a gradual mass loss was observed for APTES/MTMS-BC from ca. 530 to 650 °C which was attributed to the conversion from an imperfect Si–CH3 network into a more stable Si–O network.40
As both MTMS-BC and APTES/MTMS-BC contained CH3 functional groups known for their ability to alter the wettability of materials, the contact angle was used to investigate the wettability of the aerogel materials (Fig. 6c). Both TEOS-BC and APTES/MTMS-BC showed high affinity to water as they were immediately absorbed into the aerogel, suggesting that the presence of APTES was enough to prevent hydrophobicity from occurring for APTES/MTMS-BC.41 In contrast, MTMS-BC exhibited super-hydrophobicity with a contact angle exceeding 150°. The low wettability was attributed to a combination of the CH3 functional groups providing a low surface energy and the rough aerogel surface. From the literature it is well known that the hydrophobicity of a surface is strongly dependent on both the micro/nanostructure of the surface and the chemical composition of the material.42–44 The superhydrophobicity of our MTMS-BC hydrogel is clearly a combination of those two effects. Aerogels which are superhydrophobic or oleophobic can for instance be used for efficient oil/water separation42,43 [Fu 2020].45
To investigate the thermal stability of the silane coatings and to examine if the morphology of the silane coatings had changed after 800 °C, i.e. after thermal removal of the BC template, the remaining materials after the heat-exposure were imaged using SEM (Fig. 6d–f).
The higher magnification SEM revealed that there was virtually no change in the overall morphology of the nanomaterials. MTMS-BC showed the same spherical structures as before the template removal, where even thin BC fibres could be seen replicated in silicon oxide; however, there were slightly fewer fibres visible compared to before the heat treatment. In contrast, TEOS-BC showed no observable change in the structure, remaining composed solely of “pearl-necklace” fibre-like structures, which was expected due to the thick deposited TEOS coating forming a rigid silicon oxide network along the fibre. APTES/MTMS-BC proved most effective at replicating the BC fibre morphology in the silicon oxide material, as was observed by the intact fine fibre structures seen in Fig. 6f where the same observed fibre structures remained before and after the thermal removal of the BC template.
The high content of silicon oxide fibres in APTES/MTMS-BC suggested that using APTES enabled the replication of the fine BC fibre structures in silicon oxide. The presence of these fibres further indicates that thin and uniform silane coatings formed in the APTES/MTMS-BC aerogel and that these uniform coatings increased the stiffness compared to the uncoated BC fibres seen in MTMS-BC.
The peak heat release rate (PHRR) seen in Table 2 demonstrated that the thicker silane coatings obtained using TEOS as the aerogel precursor resulted in a lower PHRR compared to MTMS and the APTES/MTMS mixture. All the obtained aerogels exhibited a low PHRR and very low total heat release (THR), lower than 113.4 kW m−2 and 1.12 MJ m−2 respectively, making the materials promising candidates for fire-safe insulative materials.
The heat release was plotted as a function of time in Fig. 7a, highlighting the extremely low heat dissipation with TEOS. In Fig. 7b the smoke production rate was also shown as a function of time. For TEOS and MTMS, the signal was hardly detectable, whereas the APTES/MTMS based aerogel had a small peak (ca. 0.001 m2 s−1) at a time slightly after its maximum heat release rate.
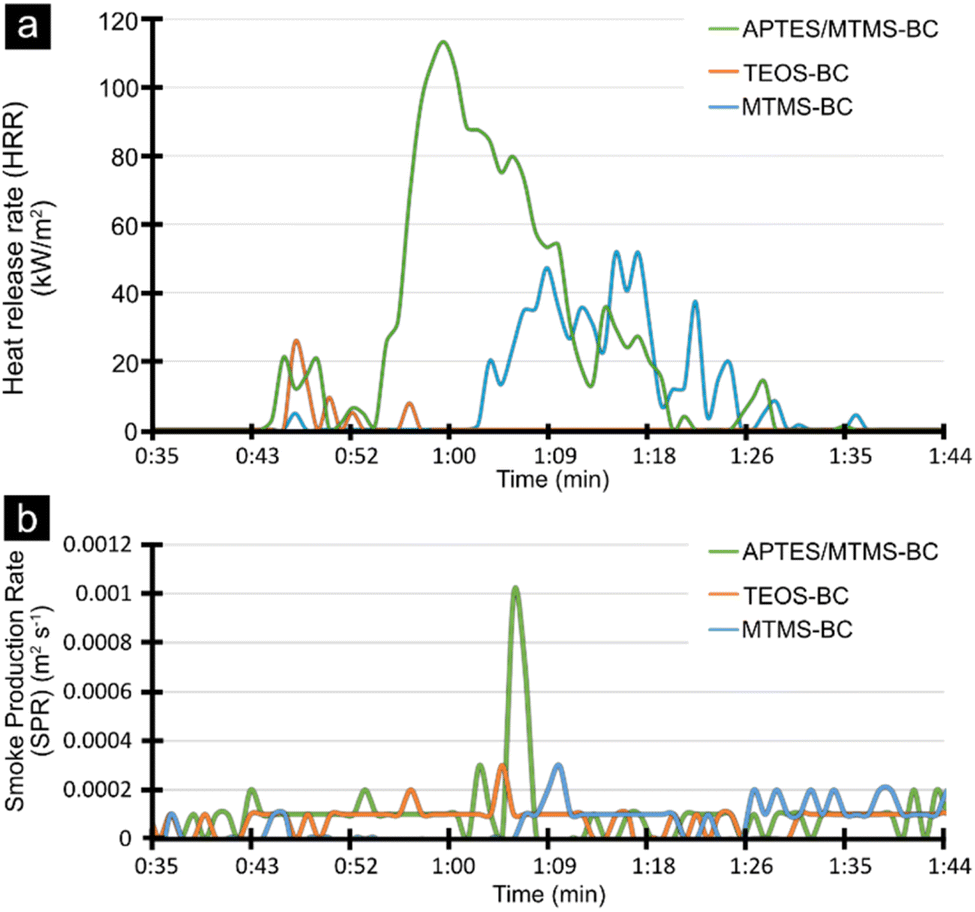 | ||
| Fig. 7 (a) Heat release rate as a function of time and (b) smoke production rate as a function of time. | ||
The main flame retarding mechanism for our hydrogels is that thermally stable silane coatings nearly completely cover the cellulose, thereby giving it excellent thermal properties. Many other promising flame retardant materials from the literature are based on slightly different mechanisms. For instance, adding flame retardant fillers to cellulose,46,47 applying graphene oxide coating on PDMS foam,46,47 or adding MXene/CNF to PDMS.48 Compared to most other foams, our materials have excellent flame retardant properties, including a low heat release rate and very high remaining mass fraction in TGA.
The densities and thermal conductivities (TCs) of the materials are summarized in Table 2. The densities for the aerogels (29–51 kg m−3) were approximately twice as high as that for EPS, whereas their thermal conductivities (0.014–0.020 W m−1 K−1) were roughly twice as low. These values are in the same range as those for other aerogels in the literature.21,49,50
Conclusions
The hybrid aerogels with MTMS or a combination of APTES and MTMS showed remarkable flexibility and elasticity and were able to bend to a high degree without permanent deformation or breaking. The choice of silane used was shown to have a significant effect on the obtained silane coating morphology and explained the variations in the mechanical properties of the aerogel materials. TEOS-BC showed the highest brittleness among the aerogels due to the formed “pearl-necklace” silane coating on the BC template. MTMS-BC showed the highest amount of flexibility due to the partially uncoated BC fibres that allowed for the flexibility of the BC fibres in the aerogel. Mixed APTES/MTMS-BC was able to flex to approximately the same degree as the MTMS-BC aerogel but required a higher amount of force due to the thin uniform APTES coatings formed on the BC fibres, increasing its stiffness. By using MTMS, it was possible to produce not only a flexible aerogel material but also an aerogel with apparent superhydrophobicity. Cone calorimetry and TGA revealed exceptionally low amounts of energy released when burned, as well as thermal stability up to ca. 600 °C. Since the thermal conductivities of the aerogels were also low (0.014–0.020 W m−1 K−1) and FTIR indicated that the silane-coatings protected the cellulose to a large extent, the new materials are also interesting for flame retardant applications.Data availability
Data will be made available on reasonable request.Author contributions
B. K. B. performed most of the laboratory work and characterization in this study. P. E. measured thermal conductivity and density. F. N. carried out theoretical simulations and calculations. B. K. B. wrote the manuscript. A. J. C., R. A. M., F. N., M. H., and R. T. O. reviewed the manuscript, with R. T. O. recognizing the concept of using bacterial cellulose as a template material as an aerogel scaffold.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the financial contributions from the Swedish Research Council (VR 2019-05650).Notes and references
- M. J. Burchell, G. Graham and A. Kearsley, Annu. Rev. Earth Planet. Sci., 2006, 34, 385–418 CrossRef CAS.
- R. Jin, Z. Zhou, J. Liu, B. Shi, N. Zhou, X. Wang, X. Jia, D. Guo and B. Xu, Gels, 2023, 9, 606 CrossRef CAS.
- H. Maleki, L. Durães, C. A. García-González, P. del Gaudio, A. Portugal and M. Mahmoudi, Adv. Colloid Interface Sci., 2016, 236, 1–27 CrossRef CAS.
- B. Hosticka, P. M. Norris, J. S. Brenizer and C. E. Daitch, J. Non-Cryst. Solids, 1998, 225, 293–297 CrossRef CAS.
- R. Ueoka, Y. Hara, A. Maeno, H. Kaji, K. Nakanishi and K. Kanamori, Nat. Commun., 2024, 15, 461 CrossRef CAS.
- T. Woignier, J. Primera, A. Alaoui, P. Etienne, F. Despestis and S. Calas-Etienne, Gels, 2015, 1, 256–275 CrossRef.
- F. Akhter, S. A. Soomro and V. J. Inglezakis, J. Porous Mater., 2021, 28, 1387–1400 CrossRef CAS.
- L.-Y. Long, Y.-X. Weng and Y.-Z. Wang, Polymers, 2018, 10(6), 623 CrossRef PubMed.
- B. Nagendra, P. Antico, C. Daniel, P. Rizzo and G. Guerra, Polymer, 2020, 210, 123073 CrossRef CAS.
- C. Wang, L. Bai, H. Xu, S. Qin, Y. Li and G. Zhang, Gels, 2024, 10, 286 CrossRef CAS PubMed.
- Y. Wu, X. Wang and J. Shen, J. Sol-Gel Sci. Technol., 2023, 106, 360–380 CrossRef CAS.
- İ. Şahin, Y. Özbakır, Z. İnönü, Z. Ulker and C. Erkey, Gels, 2017, 4, 3 CrossRef.
- R. Bhattacharya, S. Saha, O. Kostina, L. Muravnik and A. Mitra, Appl. Microsc., 2020, 50, 15 CrossRef CAS.
- H. Maleki, L. Durães, B. F. O. Costa, R. F. Santos and A. Portugal, Microporous Mesoporous Mater., 2016, 232, 227–237 CrossRef CAS.
- Q. Liao, X. Su, W. Zhu, W. Hua, Z. Qian, L. Liu and J. Yao, RSC Adv., 2016, 6, 63773–63781 RSC.
- X. Zhang, H. Wang, Z. Cai, N. Yan, M. Liu and Y. Yu, ACS Sustain. Chem. Eng., 2019, 7, 332–340 CrossRef CAS.
- A. Venkateswara Rao, S. D. Bhagat, H. Hirashima and G. M. Pajonk, J. Colloid Interface Sci., 2006, 300, 279–285 CrossRef PubMed.
- E. Utoiu, V. S. Manoiu, E. I. Oprita and O. Craciunescu, Gels, 2024, 10, 387 CrossRef CAS PubMed.
- E. Cuenca, V. Postolachi and L. Ferrara, Constr. Build. Mater., 2023, 374, 130785 CrossRef CAS.
- D. Liu, Q. Wu, R. L. Andersson, M. S. Hedenqvist, S. Farris and R. T. Olsson, J. Mater. Chem. A, 2015, 3, 15745–15754 RSC.
- B. K. Birdsong, Q. Wu, M. S. Hedenqvist, A. J. Capezza, R. L. Andersson, A. J. Svagan, O. Das, R. A. Mensah and R. T. Olsson, Mater. Adv., 2024, 5, 5041–5051 RSC.
- B. Hoogendoorn, M. Parra, A. Capezza, Y. Li, K. Forsberg, X. Xiao and R. Olsson, Mater. Adv., 2023, 4, 2683 RSC.
- A. L. S. Pereira, J. P. A. Feitosa, J. P. S. Morais and M. d. F. Rosa, Carbohydr. Polym., 2020, 250, 116927 CrossRef PubMed.
- S. Y. Oh, D. I. Yoo, Y. Shin, H. C. Kim, H. Y. Kim, Y. S. Chung, W. H. Park and J. H. Youk, Carbohydr. Res., 2005, 340, 2376–2391 CrossRef PubMed.
- A. S. Virk, W. Hall and J. Summerscales, Compos. Sci. Technol., 2010, 70, 995–999 CrossRef.
- B. K. Birdsong, B. W. Hoogendoorn, F. Nilsson, R. L. Andersson, A. J. Capezza, M. S. Hedenqvist, S. Farris, A. Guerrero and R. T. Olsson, Nanoscale, 2023, 15, 13037–13048 RSC.
- M. Sypabekova, A. Hagemann, D. Rho and S. Kim, Biosensors, 2022, 13, 36 CrossRef PubMed.
- M.-C. Brochier Salon, M. Abdelmouleh, S. Boufi, M. N. Belgacem and A. Gandini, J. Colloid Interface Sci., 2005, 289, 249–261 CrossRef.
- Z.-H. Wu, X.-L. Feng, Y.-X. Qu, L.-X. Gong, K. Cao, G.-D. Zhang, Y. Shi, J.-F. Gao, P. Song and L.-C. Tang, Compos. Commun., 2023, 37, 101402 CrossRef.
- J. Kim and J. Kim, J. Mater. Res. Technol., 2023, 26, 2782–2795 CrossRef.
- S. Azarshin, J. Moghadasi and Z. Arab Aboosadi, Energy Explor. Exploit., 2017, 35, 014459871771628 Search PubMed.
- A. A. Issa and A. S. Luyt, Polymers, 2019, 11, 537 CrossRef.
- N. H. Borzęcka, B. Nowak, J. M. Gac, T. Głaz and M. Bojarska, J. Non-Cryst. Solids, 2020, 547, 120310 CrossRef.
- M. Sypabekova, A. Hagemann, D. Rho and S. Kim, Biosensors, 2023, 13, 36 CrossRef CAS PubMed.
- D. E. Newbury and N. W. M. Ritchie, J. Anal. At. Spectrom., 2013, 28, 973–988 RSC.
- W. Giurlani and A. Lavacchi, Coatings, 2018, 8, 84 CrossRef.
- T. Keuter, N. H. Menzler, G. Mauer, F. Vondahlen, R. Vaßen and H. P. Buchkremer, J. Vac. Sci. Technol., A, 2014, 33, 01A104 CrossRef.
- A. M. Papadopoulos, Energy Build., 2005, 37, 77–86 CrossRef.
- W. Rukthong, P. Thanatawee, S. Sunphorka, P. Piumsomboon and B. Chalermsinsuwan, Eng. J., 2015, 19, 41–57 CrossRef.
- S. V S, P. Azeez, K. Warrier, B. Nair and U. S. Hareesh, ChemistrySelect, 2018, 3, 2989–2997 CrossRef.
- Y. Zhou, J. Zhang, Z. Wang, F. He, S. Peng and Y. Li, J. Membr. Sci., 2021, 618, 118703 CrossRef CAS.
- S. Chandrasekaran, A. Cruz-Izquierdo, R. Castaing, B. Kandola and J. L. Scott, Sci. Rep., 2023, 13, 3168 CrossRef CAS.
- L.-Q. Pan, Q.-N. Zheng, Q.-H. Feng, Y.-B. Shen, W.-Y. Hu, C.-F. Cao, G.-D. Zhang, J.-F. Gao, P. Song, Y.-Q. Shi and L.-C. Tang, Sep. Purif. Technol., 2025, 353, 128485 CrossRef CAS.
- A. I. Mendoza, P. Larroche, F. Nilsson, M. Hedenqvist, E. Strömberg, H. Hillborg and R. Moriana, Surf. Interfaces, 2023, 37, 102733 CrossRef CAS.
- B. Fu, Q. Yang and F. Yang, ACS Omega, 2020, 5, 8181–8187 CrossRef CAS.
- S. Chandrasekaran, A. Cruz-Izquierdo, R. Castaing, B. Kandola and J. L. Scott, Sci. Rep., 2023, 13, 3168 CrossRef CAS.
- F.-X. Shen, Y. Li, Z.-Y. Chen, C.-F. Cao, Y.-B. Shen, L.-T. Li, L.-Q. Pan, J.-Y. Li, G.-D. Zhang, J. Gao, Y. Shi, P. Song, J. Bae and L.-C. Tang, Prog. Org. Coat., 2024, 189, 108276 CrossRef CAS.
- H.-Y. Chen, Z.-Y. Chen, M. Mao, Y.-Y. Wu, F. Yang, L.-X. Gong, L. Zhao, C.-F. Cao, P. Song, J.-F. Gao, G.-D. Zhang, Y.-Q. Shi, K. Cao and L.-C. Tang, Adv. Funct. Mater., 2023, 33, 2304927 CrossRef CAS.
- B.-F. Guo, Y.-J. Wang, Z.-H. Qu, F. Yang, Y.-Q. Qin, Y. Li, G.-D. Zhang, J.-F. Gao, Y. Shi, P. Song and L.-C. Tang, Small, 2024, 20, 2309272 CrossRef CAS.
- E. Abraham, V. Cherpak, B. Senyuk, J. B. ten Hove, T. Lee, Q. Liu and I. I. Smalyukh, Nat. Energy, 2023, 8, 381–396 CrossRef CAS.
- M.-E. Li, H.-B. Zhao, J.-B. Cheng, T. Wang, T. Fu, A.-N. Zhang and Y.-Z. Wang, Engineering, 2022, 17, 151–160 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00568f |
| This journal is © The Royal Society of Chemistry 2025 |