
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbohydrate-based alternatives to traditional synthetic plastic microbeads: a critical review
Amy
McMackin
 and
Sébastien
Cardinal
*
and
Sébastien
Cardinal
*
Département de Biologie, Chimie et Géographie, Université du Québec à Rimouski (UQAR), Rimouski, Québec, Canada. E-mail: sebastien_cardinal@uqar.ca
First published on 12th February 2025
Abstract
Microplastics in the environment threaten ecosystems around the world. Primary microplastics, including porous spherical particles known as microbeads, are actively produced by industry for use in cosmetics, exfoliants, household cleaning supplies, biomedical applications, and more. Not only do microbeads persist in the environment, leading to significant problems, but traditional plastic microbeads are commonly sourced from non-renewable resources and produced using toxic manufacturing processes. For these reasons, there is a push to develop environmentally friendly alternatives, notably from carbohydrate biopolymers. This paper reviews the carbohydrates used to prepare pure bioplastic microbeads. The results also compare the environmental impact, versatility, and capacity of these beads to perform the same functions as those of traditional plastic microbeads. Although we demonstrate that carbohydrate-based plastic microbeads pose a lesser environmental threat than conventional petroleum- or biobased synthetic options, this work concludes that the specific ecological impacts and potential applications vary widely. Among the biopolymers discussed within this review, we conclude that cellulose, chitin, or chitosan-based varieties hold considerable potential to provide an eco-friendly microbead for industry.
 Amy McMackin | Amy McMackin completed her bachelor's and master's degrees in chemistry at the Université du Québec à Rimouski, where she developed a strategy to prepare microbeads from brewer's spent grain under the supervision of Prof. Dr Sébastien Cardinal. She has since become a PhD candidate in food science in the Sustainable Food Processing group at ETH Zürich under the supervision of Prof. Dr-Ing. Alexander Mathys. Her research focuses on the relationships between natural deep eutectic solvents, polyphenolic compounds, and plant proteins in novel food processing strategies. |
 Sébastien Cardinal | Prof. Sébastien Cardinal was born in Chicoutimi (province of Quebec, Canada). He obtained his PhD in chemistry from Université Laval in 2016. He then joined Princeton University as a postdoctoral fellow to work under the supervision of Prof. David MacMillan to develop new metalaphotoredox-catalyzed reactions. After postdoctoral training, he joined Université du Québec à Rimouski (UQAR) in late 2018 as a faculty member. His group's research focuses on synthetic organic chemistry and biomass valorization. |
Sustainability spotlightTraditional synthetic plastic microbeads, linked to plastic pollution, particularly in marine environments, are banned in many countries due to pollution concerns. Industries are now relying on natural materials, particularly carbohydrate-based alternatives. However, these alternatives vary in chemical and mechanical properties, affecting their performance and environmental effects. This critical review synthesizes information on carbohydrate-based alternatives, consequently identifying ideal applications and potential drawbacks. Primarily aligned with the 12th Sustainable Development Goal of the United Nations, ensuring sustainable consumption and production patterns, this work also addresses the UN SDGs No. 14 and No. 15, focusing on conserving oceans and protecting terrestrial ecosystems. Given the link between plastic microbeads and environmental pollution, it supports UN SDG No. 6 by addressing concerns related to drinking water contamination. |
1 Introduction
The mid-twentieth century marks the beginning of life in the Anthropocene era and the proliferation of plastic in its many forms. For its manifold, robust, and inexpensive nature, petroleum-derived plastic has become abundant to the point of being the indicator of a new geological era.1 Synthetic polymers tend to be exceptionally durable, a characteristic that allows their use in many applications but also represents their persistence in the environment for hundreds, if not thousands, of years. Furthermore, most plastics never decompose but merely disintegrate into increasingly smaller pieces until undetectable.2 These tiny particles (microplastics and nanoplastics) are difficult, if not impossible, to entirely remove from the environment, can leach toxic additives, adsorb persistent organic pollutants, enter the food chain, and contaminate water, soil, and air.3 Moreover, these plastics' behaviors in marine environments remain misunderstood, especially in the case of nanoplastics.4,5 Despite these difficulties, plastic production could reach as much as 1900 million tonnes annually in 2050,6 representing an urgent need to replace petroleum-based plastics with more sustainable alternatives.7Microplastics are plastic particles with diameters ranging from 1 μm to 5 mm,8 regardless of their origin or shape. They originate from primary or secondary sources. Secondary microplastics form by plastic decomposition in the environment. Conversely, industries deliberately produce primary microplastics for cosmetics, exfoliants, household cleaning supplies, biomedical applications, and more.9 Primary microplastics account for a global market worth an estimated $3.5 billion in 2020.3 These are further classified as plastic pellets (plastic resin granules used as raw materials in the manufacturing of larger plastic products) or microbeads (spherical plastic particles, manufactured specifically for their size and shape). Microbeads are also characterized by their porous nature and their production from viscous polymer solutions.10 Like traditional petroleum-based macroplastics, they are commonly sourced from non-renewable resources, produced by toxic manufacturing processes, and pose significant environmental problems when it comes to their disposal.11 A significant fraction of marine pollution from microplastics derives from primary plastic microbeads, as they are too small to be fully recuperated by wastewater treatment facilities. In the USA, this is on the order of 8 trillion microbeads dumped into marine environments every day,6 roughly corresponding to 11.7 tonnes of plastics.12 All plastics may eventually break down into nanoplastics, particles less than 1 μm in size, which are even more difficult to recuperate from the environment.2
Replacing traditional plastic microbeads with biopolymers can make primary microplastics considerably more sustainable and viable.7 Biobased synthetic polymers, as the name would suggest, are manufactured from monomers isolated from renewable biomass raw materials, then synthetically polymerized. Elsewhere, naturally occurring biopolymers, notably carbohydrates, can be directly extracted from biomass, and then chemically altered to produce a wide variety of plastics with comparable mechanical properties to conventional petroleum-based or biobased synthetic plastics. Agriculture-based plants rich in carbohydrates, lignocellulosic plants unsuitable for human or animal consumption, algae, organic/food waste, or microbiota are commonly used as starting materials for biobased synthetic polymers or biopolymers.13,14
The definition of a material as a “biobased synthetic” or “biopolymer” is not mutually exclusive to biodegradability or green manufacturing techniques. Some biobased plastics or biopolymers are biodegradable while others are not, just as traditional synthetic plastics can be biodegradable in certain conditions.6,15 Similarly, some synthetic polymers' manufacturing processes are more energy-efficient or require fewer toxic solvents than those of their biobased counterparts.16 Different types of plastic are also better suited to different applications based on their unique characteristics. Although biobased plastics generally have a lesser environmental impact than conventional plastics, each specific type of polymer has its strengths and weaknesses.
This paper provides an overview of the conventional synthetic polymers used to produce primary microbeads alongside a comprehensive analysis of emerging carbohydrate-based biopolymer alternatives. Carbohydrate-based biopolymers are more commonly used than naturally occurring polyesters or protein-based options, and the scope of this critical review is consequently restricted to this category. Herein, we summarize and synthesize the advantages and disadvantages of these different polymers considering their environmental footprint, versatility, and performance regarding their respective applications.
First, we explain our chosen evaluation criteria and their order of importance. Then, we discuss common petrochemical-based materials and a popular bio-based synthetic (polylactic acid) used to create microbeads. Finally, we analyze different carbohydrate-based biopolymer alternatives, interspersed with explanations of their different design approaches and production methods. A thematic research strategy allowed us to search for, identify, and analyze articles pertinent to producing a comprehensive and detailed report. We restricted our literature review to English-language articles from peer-reviewed publications.
2 Evaluation guidelines
In this report, we evaluate different types of plastic microbeads for their environmental impact, versatility, and performance regarding their respective applications. The overall environmental impact is determined based on the Twelve Principles of Green Chemistry developed by Paul Anastas and John Warner, which are: waste prevention, atom economy, less hazardous synthesis, design of benign chemicals, use of benign solvents and auxiliaries, design for energy efficiency, use of renewable raw materials, reduction of derivatives, catalysis, design for degradation, real-time analysis for pollution prevention, and inherently benign chemistry for accident prevention.17In the context of this review, environmental impact is weighted towards two specific principles of Green Chemistry: use of renewable raw materials and design for degradation.17 In this review, we selected alternatives to conventional petrochemical plastic microbeads based on their origins from renewable raw materials. Within this, the nature of the renewable raw materials is another major factor to consider. Many agriculture-based plants rich in carbohydrates suitable for biobased plastic production or biopolymer isolation primarily serve as a food source for humans and animals. Likewise, lignocellulosic plants unsuitable for human or animal consumption are essential in the pulp and paper industry, as construction and building materials, and in the production of biofuels.18 Renewable raw materials sourced from microbiota or organic waste serve to avoid conflicts regarding securing food and housing supplies and prevent the destruction of rainforests and grasslands.16 Biobased synthetic plastics or biopolymers derived from organic waste also adhere to the first principle of Green Chemistry, waste prevention, by exploiting material that would otherwise end up in landfills.
Design for degradation (biodegradability) is an equally important criterion to evaluate the environmental friendliness of plastic microbeads, as plastic microbeads are commonly disposed of by being washed down the drain, accumulating in natural waters worldwide.9 Consequently, assessing the environmental impact of primary plastic microbeads requires particular attention to their biodegradability in aquatic settings. The persistence of non-biodegradable plastic microbeads prompts their consequences on marine life and ecosystems.19 In addition to their intrinsically hazardous nature, plastic particles in marine environments leach toxic additives and plasticizers and accumulate persistent organic pollutants, such as polychlorobiphenyls (PCBs). These plastics can contain 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 1000000 times more of these pollutants than the seawater of their surrounding environment.20 These particles then enter the marine food chain, affecting the biological activity, nutrient cycling, and the primary productivity of certain organisms.19 Claims that these microplastics and toxins pass through food webs are not substantiated yet. However, recent research identified microplastic contamination in 92% of tap water samples taken in the USA and 72% in Europe,6 inferring their direct ingestion by humans through drinking water. Rapid biodegradation of plastic microbeads in marine environments helps prevent their accumulation and associated consequences.
000 to 1000000 times more of these pollutants than the seawater of their surrounding environment.20 These particles then enter the marine food chain, affecting the biological activity, nutrient cycling, and the primary productivity of certain organisms.19 Claims that these microplastics and toxins pass through food webs are not substantiated yet. However, recent research identified microplastic contamination in 92% of tap water samples taken in the USA and 72% in Europe,6 inferring their direct ingestion by humans through drinking water. Rapid biodegradation of plastic microbeads in marine environments helps prevent their accumulation and associated consequences.
The other principles of Green Chemistry are mainly used to evaluate plastic microbeads' production, as some processes for creating plastic microbeads are more environmentally friendly than others.16 Plastic microbead manufacture often uses processes that depend on various toxic solvents to dissolve the polymers, such as emulsification, microfluidics, and precipitation.11 Biobased and biopolymer alternatives are no exception, as they often still require harsh chemicals and energetically inefficient processes in their production.3 Researchers are continuing to work on new green production processes and to find ways to make existing methods more environmentally friendly.
In addition to environmental impact, considering an alternative microbead's versatility and performance is crucial. Biobased or biopolymer-based plastics are more likely to be adopted if they are multipurpose or customizable. More importantly, biobased or biopolymer-based microbeads must match or exceed the efficiency of conventional plastic microbeads to be viable alternatives.
3 Synthetic polymers
Synthetic polymers are composed of repeating structural units (monomers) that are artificially prepared through chemical processes. They can be classified into two main categories – petrochemical- or biobased – depending on the type of monomer used in their production. The vast majority of petrochemical-based synthetic polymers are prepared using fossil fuel-derived monomers, such as those obtained through the hydrocarbon cracking of crude oil or natural gas.1 Although many petrochemical-based plastics exist, polyethylene (PE) and polypropylene (PP), and polyethylene terephthalate (PET) are the most common types of plastic microbeads found in the environment,21 and are the only types discussed in this report. Biobased synthetic polymers are prepared from monomers sourced from renewable biological resources, such as plants, algae, or microbial fermentation. Biobased synthetic polymers are often proposed as replacements for their petrochemical-derived counterparts, as their carbon footprint is potentially lower and their potential biodegradability more likely. Of the many biobased synthetic polymers that have been prepared in recent decades, polylactic acid (PLA) is the most widely used option in microbead production. As such, it is the only biobased synthetic polymer discussed in this report in reference to carbohydrate-based biopolymers.3.1 Petrochemical-based polymers
Conventional plastics, including those in microbead form, are easily functionalized, impermeable, inexpensive to produce, and have a high strength-to-weight ratio.15,22 Synthetic polymers are also very stable, which proves to be a double-edged sword. This stability indicates excellent mechanical properties, which explains the widespread use of plastic microbeads in various applications, including in cosmetics, coatings, plastics fabrication, industrial abrasives, wastewater remediation, and in chromatography applications.1 However, with greater stability comes greater resistance to environmental factors, chemicals, hydrolysis, and microorganisms, leading microbeads to accumulate in and pollute the environment.1,15Polyolefins, such as polyethylene (PE) and polypropylene (PP),23 and polyethylene terephthalate (PET), a polyester,24 are the most common types of plastic microbeads found in the environment.21 Unsurprising, as polyolefins are the most commonly produced plastics worldwide,25 and all three types are highly resistant to biodegradation.15 In the marine environment, microbeads of this nature can absorb and concentrate persistent organic pollutants (POPs) and leach toxic plasticizers, additives, and residual monomers.15 This has consequences on marine life and ecosystem regulation.19
Polyethylene is the result of the catalyzed (Cat.) addition polymerization reaction (with radical initiator, X*) of the monomer ethylene, a gaseous hydrocarbon with the chemical formula C2H4.23 The following equation describes the polymerization process:

Ethylene is generally produced from natural gas that contains relatively high proportions of ethane. Steam cracking is customary, which involves rapidly heating long-chain hydrocarbons and steam to 775–875 °C (typical outlet temperatures). This process results in short-chain, unsaturated hydrocarbons, which are then separated into their respective fractions by repeated compression and distillation. In petrochemistry, steam cracking is among the most energy- and emission-intensive processes.26
Other less common methods to produce ethylene include oxidative coupling of methane (using metal oxide catalysts at 700–900 °C), the Fischer–Tropsch synthesis, methanol to olefin conversion, and catalytic dehydrogenation.27 Like steam cracking, these processes rely on fossil fuels, release greenhouse gasses, and are energy-intensive. The subsequent polymerization of ethylene requires coordination catalysts, of which Ziegler catalysts or the hazardous Phillips catalyst are the most used. The temperatures and pressures used vary depending on the desired density of the polyethylene.23 Although this polymerization is highly exothermic, shaping the resulting polymer still requires large amounts of energy to produce the necessary heat and pressure.
Polypropylene is produced from catalyzed chain-growth polymerization (with radical initiator, X*) from the monomer propene (also known as propylene), as shown in the chemical equation below. It is slightly firmer and more heat-resistant than polyethylene.23 In microbead form, PP has an average hardness of 199 MPa.28

While many techniques exist, two main manufacturing processes produce PP.
• Bulk polymerization in liquid propene: this method uses liquid propene as a solvent with temperatures between 50 and 75 °C and pressures between 30 and 40 atm. After polymerization, the remaining unreacted monomer is flushed away.
• Gas phase propene polymerization: this method uses gaseous propylene, which is introduced to a solid catalyst, resulting in a fluidized-bed medium. Ziegler–Natta catalysts activate the reaction and control the tacticity of the resulting polymer.23
Propene (propylene) is mainly obtained from the steam cracking of naphtha, shale gas, or propane (fossil fuels) at 700–950 °C. In this process, propane dehydrogenates, resulting in unsaturated propene and hydrogen gas. Then, fractional distillation isolates and purifies propene.23 As previously mentioned, steam cracking requires high temperatures and pressures and is thus energy intensive.
Polyethylene terephthalate is a polyester resulting from a polycondensation reaction. Two processes commonly yield PET.
• The dimethyl terephthalate process: molten dimethyl terephthalate (150–160 °C) and ethylene glycol are reacted at 150–200 °C at ambient pressure in a nitrogen atmosphere in the presence of a catalyst. Here, many different catalysts are effective. Distillation removes methanol, which shifts the equilibrium of the reaction toward the products. Then, vacuum distillation removes excess ethylene glycol. The second catalyzed transesterification step occurs at 270–280 °C.24 Overall, this process can be described by the following:


• The terephthalic acid process: the esterification of ethylene glycol and terephthalic acid occurs by the chemical equation shown below at 220–260 °C and 2.7–5.4 atm. Water, the by-product of the reaction, is continuously removed by distillation, which drives the equilibrium forward.24

Both methods use ethylene glycol as a reactant. First, ethylene glycol is oxidized to provide the intermediate ethylene oxide. Ethylene oxide then reacts with water in the presence of an acid or base catalyst and heat to produce ethylene glycol:29
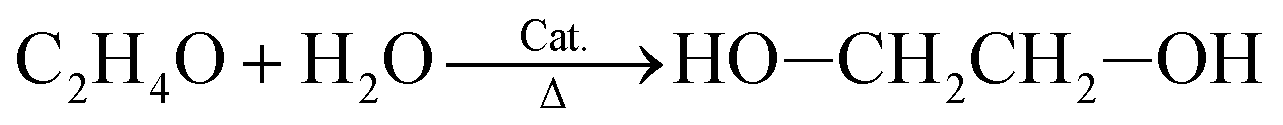
As previously mentioned, ethylene results from cracking long-chain hydrocarbons.27 Dimethyl terephthalate forms from the direct esterification of terephthalic acid in the presence of methanol.30 The Amoco process, which involves the catalytic oxidation of p-xylene, produces terephthalic acid. Acetic acid serves as the solvent, compressed air is the oxidant, and a cobalt–manganese–bromide complex is the catalyst.31 This reaction is highly corrosive. p-Xylene is another fossil fuel derivative, which as of recently, can be produced by toluene alkylation at 442.5 °C and 3.9 atm, with less of an environmental impact than traditional methods.32
PE, PP, and PET are products of the petrochemical industry, deriving from non-renewable resources. They result from energy-intensive, polluting methods, which also carry high levels of risk to environmental or human health in the case of a malfunction. As an upside, they are also all examples of thermoplastics (unless further chemically modified to become thermosetting), which melt when sufficiently heated and solidify when cooled.23,24 This property of reversible thermoplasticity allows for the simple shaping of uniform, spherical, smooth microbeads [Fig. 1] through mold injection, cutting, or dropping. Although this may be a beneficial aspect of these polymers, the high carbon footprint and likelihood of long-lasting environmental pollution associated with these plastics are notable downsides, especially when considering the availability of alternatives.
 | ||
| Fig. 1 Scanning electron microscopy image of a polyethylene microbead extracted from a cream-based personal hygiene product. (The original image has been cropped. Reproduced from ref. 33 under CC-BY.)33 | ||
3.2 Biobased synthetic polymers
Of the many recently-developed biobased synthetic polymers, polylactic acid (PLA) is the most commonly used in microbead production. PLA is a biobased, biodegradable thermoplastic aliphatic polyester. It is biocompatible, inexpensive, and has excellent mechanical properties controllable by external factors such as radiation and heat.11 PLA derives from lactic acid, which is produced by fermenting dextrose (D-glucose). Dextrose is a component of many raw materials, such as starch, lignocellulosic biomass, and agro-industrial wastes.34 Of these, agro-industrial waste is the most interesting, as it does not compete with other industries and would otherwise be unexploited. PLA production also has negative net CO2 emissions.35 PLA microbeads have potential applications in food packaging, tissue engineering, drug delivery, cosmetics and personal care products, cell delivery, plastic surgery, optics, and solid-phase extraction applications.11,36–41 Its versatility is largely owed to its three different stereoisomeric forms: poly(L-lactide acid) (PLLA), poly(D-lactide acid) (PDLA), and their atactic polymer, poly(DL-lactic acid) (PDLLA) [Fig. 2].42 The different crystallinities and melting points of these stereoisomers confer different mechanical properties to PLA-based microbeads.43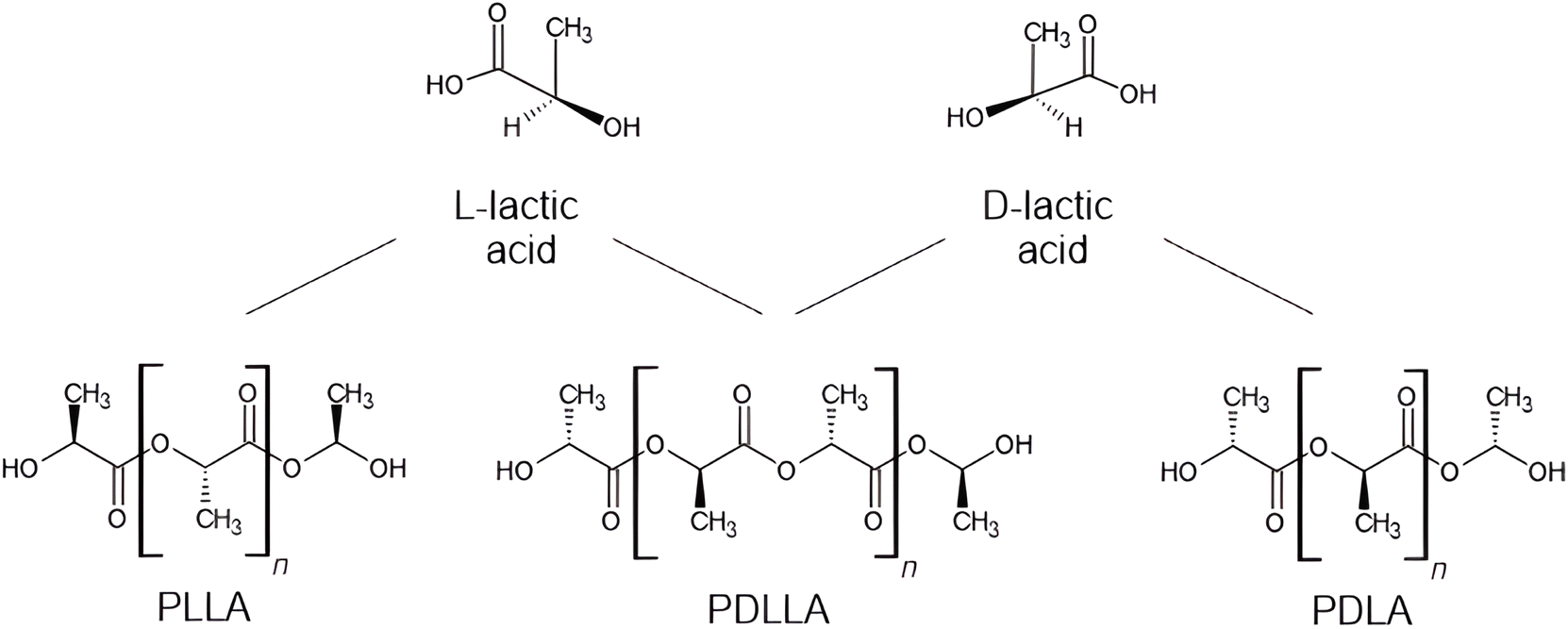 | ||
| Fig. 2 Molecular structures of PLA's monomeric units, L-lactic acid and D-lactic acid, and their arrangements as PLA's three different stereoisomeric forms: PLLA, PDLLA, and PDLA. | ||
In industry, lactic acid, the monomer of PLA, is produced by bacterial fermentation of carbohydrate-rich raw materials. Three methods are possible for the subsequent production of PLA: direct condensation polymerization, direct polycondensation in an azeotropic solution, and polymerization through lactide formation.35,42 While the first method is unfavorable for microbead production, the second has potential, using complex catalysts to produce high molecular weight polymers. The third is relatively eco-friendly, producing long polymer chains by removing water under mild conditions and without solvent. High temperatures remain necessary for purification and functionalization, with low-toxicity stannous octoate as an initiator35 or by applying a cost-effective Cs2CO3 catalyst.42 As PLA is a thermoplastic, the resulting pellets need only be melted down or solubilized to produce final products of varying forms. This characteristic also facilitates PLA recycling, similar to that of other melt-processable conventional plastics.41
Although PLA microbeads are theoretically biodegradable, the mechanism is not rapid enough to avoid a negative effect on marine ecosystems. Like conventional petroleum-derived plastics, PLA demonstrates no or very low degradation in the natural environment at ambient temperatures, instead breaking down into increasingly smaller particles.44 When integrated into sediments, PLA microbeads reduce microalgal biomass to the same extent as polyvinyl chloride (PVC) and high-density polyethylene (HDPE) microbeads. Due to its carbonyl and hydroxyl groups in proximity, PLA can also adsorb and sequester available nutrients, reducing concentrations of porewater ammonium necessary for the primary productivity of certain benthic organisms. Lugworms, for example, experience increased stress in the presence of PLA microbeads, yet lower rates than when exposed to PVC and HDPE.19 Several strategies can be used to improve PLA's degradation in the environment, such as blending with hydrophilic or hydrophobic polymers, mixing with other plasticizers, compounding with other non-plastic materials, or particle irradiation.11,44 However, especially when a microbead is designed for a single-use application, such as in wash-off personal hygiene products, it may be best to choose more readily biodegradable polymers.
Nam and Park (2020) report that PLA degrades under the influence of radiation. Radiation catalyzes a beta-elimination reaction, breaking the ester bonds between monomers of the polymer chain, resulting in polymers of lower molecular weight that are quicker to degrade in natural environments. Moreover, this rapid technique for controlling biodegradation rates does not require toxic oxidizing chemicals. In an aqueous environment, the polymer chains become hydrated, the ester bonds break, the resulting oligomers disperse, and the oligomers degrade. Biodegradation rates in seawater are slightly faster than in freshwater, as alkalinity reduces the hydrophobicity of the polymer. Anderson and Shive (1997) specify that additives and loaded therapeutic agents, depending on their pH, may significantly affect the degradation rate of PLA microbeads.36 For example, thioridazine, a tertiary amino compound with a pKa of 9.5, accelerates microbead degradation rates. Dimensions of the microbeads, their morphology, porosity, molecular weight, molecular weight distribution, water permeability, and aqueous solubility all affect the hydrolytic degradation mechanism.11
PLA microbeads can be prepared by an environmentally friendly melt electrospraying process, as described by Nam and Park (2020). Firstly, an electron beam at 500 kGy irradiates PLA chips to lower the number average molecular weight of the polymers from 110000 to 13000, improving their melt processability. The polymer chips are placed in a syringe and heated to produce a polymer liquid. When voltage is applied to the syringe, the polymer liquid sprays out of the tip of the needle into a coagulation bath of distilled water. This method facilitates the production of micro- and nanoparticles of homogenous size and shape, with smaller needle diameter and greater applied voltage resulting in smaller beads. The optimized parameters include a flow rate of polymer liquid into the coagulation bath of 5 mL per hour and a drop distance set at 10 cm. The resulting microbeads have a low specific surface area as they have smooth and non-porous exteriors and thus have low adsorption rates of POPs in the environment. They also demonstrate bulk degradation, losing mass and thermal and structural stability, and gaining crystallinity due to the degradation of the amorphous region first. Nam and Park (2020) elaborate that PLA microbeads are traditionally prepared by methods mostly requiring toxic, volatile, and harmful solvents, such as spray-drying, emulsification, ionic gelation, microfluidic techniques, and solution electrospraying.11 Electrospraying is also customary for the scalable production of cellulose and chitin microbeads.
A simple low-energy technique based on a single oil-in-water emulsion method using a simple fluidic device can also produce PLA microbeads.37,43 Although this method allows for highly customizable porous microbeads, conferring porosity to the material depends on the use of organic solvents and an alkane as the porogen (a substance used to make pores in molded structures, whose particles or individual molecules determine pore shape and size). When the solvent evaporates, the alkane undergoes spontaneous microphase separation, forming pores on the surface of the microbead. The concentration and the specific alkane used in the process control the pore size and distribution, as the van der Waals force of the alkane is the determinant for the morphology of the microbeads. Although this method is simple, fast, and uses little energy, using organic solvents and porogens increases its environmental footprint.
Elsewhere, a melt-homogenization process in silicon oil produces PLA microbeads for cosmetic and personal hygiene products. Homogenization creates an emulsion, and the beads are solidified at their desired dimensions by rapid cooling. While these beads meet the physical and chemical characteristics required of exfoliating microbeads in personal hygiene products, the study reiterates that these PLA microbeads do not degrade in aquatic environments.40
Hybrid PLA-poly caprolactone (PCL) microbeads require organic solvents (i.e., chloroform and dichloromethane) for their preparation and are resistant to degradation. Similarly to PLA alone, UV photoirradiation can improve degradation, although the environmental impact of the solvents used remains.45 Other PLA microbead preparation methods include casting, which involves evaporating an organic solvent such as chloroform38 or centrifuge-facilitated emulsification with a non-ionic surfactant and an organic solvent (i.e., Span® 80 and chloroform).46 In these cases, PLA is combined with alginate to encourage more rapid degradation but is otherwise offset by using a non-recyclable organic solvent. Other similar co-polymerizations of PLA, including that of poly(lactic-go-glycolic acid) (PLGA), achieve improved biodegradability while maintaining smooth surfaces and good compatibility with biomedical applications [Fig. 3]. However, the fabrication of these microbeads relies on organic solvents such as toxic dichloromethane.47
 | ||
| Fig. 3 Example images of PLGA microbeads for a biomedical application: (a) digital photograph and (b) SEM image. (The original image has been cropped. Reproduced from ref. 47 under CC-BY.)47 | ||
4 Carbohydrate-based biopolymers
Biopolymers used in microbead production fall into three categories: carbohydrates, proteins or polypeptides, and polyesters. Carbohydrate-based polymers are the most common choice for their ease of extraction and diverse chemical and mechanical properties. They include chitin and chitosan, cellulose, pectin, starch, alginate, agar and agarose, dextran, hyaluronic acid, and gellan gum, which we organize within this report according to their material of origin: animal tissues, plants, algae, or microorganisms. Other varieties of carbohydrates, such as pullulan, xanthan gum, galactomannans, or kappa-carrageenan have weak mechanical properties, currently limiting their use to hybrid microbeads combined with other polymers. We consequently exclude these varieties from our analysis.4.1 Animal-derived carbohydrate-based polymers
Depending on their natural source, chitin-based polymers can vary drastically in structure. Chitin occurs naturally in three allomorphs, with alpha and beta forms as the most abundant.59 Chitin is the result of the formation of linear polymers of N-acetyl-D-glucosamine monomers covalently bonded through beta-(1,4)-glycosidic linkages [Fig. 4] and their subsequent assembly into crystalline microfibrils of different length and diameter.59 Each allomorph is characterized by the orientation of these polymer chains within the structure of the fibril and behaves uniquely. Chitin-based bioplastics are mainly derived from the alpha allomorph of the polymer.59
 | ||
| Fig. 4 Molecular structure of chitin (>50% Ac as R groups) and chitosan (≤50% Ac as R groups). | ||
The industrial processing of chitin from crustacean shells or insect waste is reportedly time-consuming and energy-intensive. Chitin conventionally requires 1 to 24 hours in HCl to dissolve the raw material of choice to remove undesirable minerals and then 16 to 48 hours with NaOH for deproteinization. Recent work has sought to improve chemical methods for chitin isolation, mainly by using lower concentrations of HCl, substituting HCl for organic acids such as lactic or acetic acids, or using natural deep eutectic solvents.60–63 Another approach converts crustacean shells to chitin through the biological activity of Rhizopus arrhizus and Cunninghanella elegans, which would significantly improve the sustainability of chitin processing.50,60,64
King et al. (2017) confirm this, presenting another environmentally friendly method to produce opaque, porous chitin microbeads. First, 1-ethyl-3-methylimidazolium acetate dissolves crustacean shells. This ionic liquid, although toxic, allows for the extraction of high purity, high molecular weight chitin required to produce beads of homogeneous size distribution. The viscosity and temperature of the resulting solution control the polymer's chain entanglement, which in turn influences microbead size and shape. The source of chitin used and the rate of addition of the chitin solution to the coagulation medium also impact the process. The coagulation medium used is polypropylene glycol, which is non-irritating and has low volatility and toxicity. Beads can then be filtered from this medium and dried with supercritical CO2, resulting in a uniform batch of microbeads ideal for use in the cosmetics industry, in paint, or for the steady, prolonged release of active compounds. Air-drying is more environmentally friendly but collapses the beads and limits their applications. These beads reportedly biodegrade in marine environments within a few months.3
Like other biopolymer-based microbeads, low-energy and green solvent-compatible membrane emulsification techniques can yield chitin-based microbeads.65 This process allows to tailor bead size and porosity and is just as efficient at an industrial scale as in the lab. This method consists of forcing the polymeric solution through the pores of a microporous membrane into a continuous phase of the coagulating solution, as shown in Fig. 5. Drop extrusion techniques are equally effective and are compatible with benign solvents, such as an aqueous solution of NaOH and urea.66 A recent review on chitin microbeads by Liao et al. (2024) details the various solvents used to disperse chitin, compatible methods for microbead production, and popular gelation mechanisms. This work highlights the aforementioned advantages of emulsion and dropping methods and their notable drawbacks: for emulsion-based production random droplet coalescence is most problematic, while dropping is limited by low production rates and the need for additional extrusion equipment.57 Emulsification and dropping techniques are equally compatible with conventional organic solvents for solubilization and gelling, such as the N,N-dimethylacetamide and ethanol system used by Wei et al. (2023) to produce porous chitin microbeads for enzyme loading.51 Although organic solvent systems can help facilitate interesting microbead microstructures, they increase the overall environmental impact of the material and the risk of adverse health effects.
 | ||
| Fig. 5 Representation of membrane emulsification techniques for microbead production: (a) traditional membrane emulsification and (b) coarse membrane emulsification. | ||
Chitin must first undergo enzymatic or chemical deacetylation to produce chitosan to be effective in other applications.50 Chitin and chitosan's degree of deacetylation (% of R groups that are not Ac, Fig. 4) is directly associated with their chemical and mechanical properties.48,67 When the degree of deacetylation of chitin reaches about 50%, it is called chitosan [Fig. 4].59 Recent mechanochemistry approaches demonstrate how high molecular weight chitosan can be synthesized from chitin with minimal energy and solvent use,68 improving chitosan's overall sustainability.
Like chitin, chitosan is biocompatible, biodegradable, non-toxic, and compatible with other biopolymers (i.e., cellulose, starch, and pectin)69,70 to create functional composite microbeads. However, chitosan also has antimicrobial and anti-inflammatory properties.50 This bioactivity makes chitosan-based microbeads especially interesting for biomedical applications, especially drug delivery or cell therapies.69–71 Chitosan is also abundant, with studies estimating 2000 tonnes of the biopolymer obtained from seafood industry waste annually.52
A study by Moreno-Sader, Meramo-Hurtado, and Gonzalez-Delgado (2020) describes an environmentally friendly method for chitosan microbead production. Chitosan is dissolved in acetic acid and coagulated into microbeads using a solution of NaOH. The resulting microbeads are rinsed with distilled water and then dried to reduce moisture content. An aqueous solution of NaOH and sodium acetate is obtained as waste during rinsing. Their analysis concludes that the method is environmentally sustainable even in industrial scale-up conditions.72 The same year, Alinejad et al. described a similar gentle process for human cell encapsulation within 300–450 μm chitosan microbeads using a water-in-oil emulsion system. Here, an acidic chitosan solution gelled in a NaHCO3 and phosphate buffer or beta-glycerophosphate solution.71 A more recent study by Ju et al. (2021) describes a similar approach for chitosan microbead production using acetic acid as a solvent and NaOH as an anti-solvent, facilitated by a paraffin oil-based emulsion. These beads were small (280 μm), with a hardness of 128 MPa and rapidly biodegrade in seawater (93.2% after 1 month), meeting the criteria for exfoliating microbeads in cosmetics [Fig. 6].53 The sphericity, diameter, and polydispersity of these microbeads may be improved through microchannel emulsification, which facilitates inverse emulsions.55
 | ||
| Fig. 6 SEM image of chitosan microbeads destined for use in personal hygiene products. (The original image has been cropped. Reproduced from ref. 53 under CC-BY-NC.)53 | ||
Tedesco et al. (2018) report using droplet extrusion through a conical nozzle to produce soft chitosan microbeads, where flow rate, pressure, distance from the coagulation medium, coagulation medium surface tension, and the stirring rate of the coagulation medium determine the size, porosity, and shape of the resulting microbeads. This method also relies on an acetic acid–NaOH solvent/anti-solvent system.73 Elsewhere, an inverse emulsion with glutaraldehyde as a cross-linking agent yields chitosan microcapsules.74 Although these microbeads appear promising in the sustained release of encapsulated drugs, glutaraldehyde-based chemical cross-linking as a formation mechanism is undesirable from a Green Chemistry perspective, due to glutaraldehyde's toxicity.75 Alternatively, genipin is a novel alternative to glutaraldehyde in the emulsion cross-linking process, yielding resistant, biocompatible chitosan microbeads for biomedical or food applications.76–81
4.2 Plant-derived carbohydrate-based polymers
Cellulose is a homopolymer of glucose joined by beta-1,4 linkages [Fig. 7]. Linear cellulose chains are arranged as crystalline microfibrils, and microfibrils bundle together to form fibrils [Fig. 8]. In plants, these fibrils are often interwoven with lignin and hemicellulose, whereas bacterial cellulose is of high purity.86
 | ||
| Fig. 7 Molecular structure of cellulose. | ||
 | ||
| Fig. 8 Cellulose is naturally found in the cell walls of plant cells. These cell walls owe their rigidity to the crystallization of cellulose chains: linear cellulose chains are arranged as crystalline microfibrils, and microfibrils bundle together to form mechanically resistant fibrils. | ||
Cellulose has many practical derivatives in bioplastic production, such as micro- and nanocrystalline cellulose, produced by the acid treatment of cellulose. Cellulose nanocrystals (CNCs) can then be assembled into nanotubes (CNTs) or fibers (CNFs).87,88 Other publications describe cellulose acetate as equally important,18,89,90 and its production through reactions with acetic acid and acetic anhydride in the presence of sulfuric acid. Cellulose triacetate is an intermediate, which is then partially hydrolyzed to the desired degree of substitution. Ionic liquids or iodine catalysis in the presence of acetic anhydride can equally prepare cellulose acetate from cellulose. Elsewhere, carboxymethyl cellulose is a useful derivative.91
Besides its derivatization potential, cellulose is compatible with other natural polymers such as alginate, pectin, or hyaluronic acid.87,92,93 Otherwise, compounds such as carboxylated graphene oxide, nickel-iron layered double hydroxide (Ni–FeLDH), clays, or covalent organic nanosheets28,90,91,93 may be incorporated into cellulose microbeads for better control over their mechanical properties, improving their versatility. Cellulose's functionalization potential extends beyond this brief list. However, it is important to note these derivatives or additives' synthetic routes are often energy-intensive and rely on non-recyclable organic solvents.
Since the 1950s, various methods to produce cellulose microbeads have been developed, notably reporting different solvents and techniques for bead formation.10,28,39,85,92,94–98 Many of these are iterations of a dropping technique [Fig. 9]. These publications also describe the optimization parameters used to control some of the beads' properties, which shows that cellulose beads meet the criterion of versatility. The final product can be used in many applications, conducting the same functions as conventional plastic microbeads with comparable or superior efficiency. These include but are not limited to uses in cosmetics and scrubs, chromatography, foods, metal ion exchange, water treatment, protein immobilization, solid-phase synthesis release, dye adsorption, or drug, cell, or nutrient encapsulation.
 | ||
| Fig. 9 Illustration of various techniques used in the production of cellulose microbeads: (a) dropping, (b) jet cutting, (c) spinning drop atomization, and (d) spinning disc atomization. | ||
Cellulose-based plastic microbeads are relatively easy to make, with the manufacturing process generally involving the dissolution of cellulose pulp, and then the simultaneous shaping and solidification of the microbead.20 To start, cellulose is isolated from the raw material of choice and generally bleached.99 Then, as detailed below, various solvent systems can subsequently be used to solubilize the refined cellulose. Shaping and solidification are performed simultaneously and are achieved by exposing the polymer solution to a non-solvent, leading to polymer chain aggregation and a high level of inter-chain bonding, often according to cellulose fibers' typical hydrogen bonding network.20,83,100
Cellulose pulp is soluble in a variety of solvents, including several environmentally friendly options, such as reusable ionic liquids (i.e., 1-ethyl-3-methylimidazolium) or aqueous solutions of NaOH, NaOH–urea, NaOH–thiourea, and NaOH–ZnO, which also avoid the formation of undesirable derivatives.98,101–105 Cellulose dissolved in NaOH solutions can be solidified with HCl, HNO3, or distilled water,10 whereas ethanol can solidify ionic-liquid dissolved cellulose.98 Solutions of NaOH–ZnO have been proven to dissolve lignocellulosic material and beads can be regenerated in acidic solutions.106 When cellulose microbeads are produced using toxic organic solutions as the initial solvent, the dispersion medium, and for solidification, this amounts to the use of excess chemicals and the production of unnecessary waste. In this regard, aqueous solutions of NaOH would be preferable for microbead production.
Low-energy techniques, such as droplet extrusion (dropping) or dispersion in oil, can easily shape cellulose microbeads during the shaping/solidification step.10,89,91 Dropping can be coupled with jet cutting, spinning drop atomization, and spraying methods (i.e., electrospraying), slightly more energy-intensive techniques, to produce smaller microbeads.10,28,107 As with chitin, another possible method is membrane emulsification, a readily scalable process that continuously generates size-optimized microbeads.85,108 Elsewhere, ultrasonication is a low-energy method for cellulose microbead production.93
An important sustainability parameter to consider regarding using cellulose as a starting material for developing biopolymer microbeads is its origin. The primary source of cellulose is trees, which comes in competition with the building materials industry, followed by cotton, which is in high demand for textile fabrication and is associated with significant pesticide and fertilizer use, soil degradation, and high water consumption, possibly leading to desertification.109,110 Delignification is often necessary to obtain cellulose pulp from lignocellulosic biomasses, as plant cellulose is interwoven with hemicellulose and lignin (lignocellulosic fibers) [Fig. 8]. The two conventional and most widely used industrial processes are sulfite and alkaline pulping, which respectively involve treating the biomass with harsh acidic or alkaline conditions.96 Less popular options, known as solvent processes, rely on toxic organic solvents for delignification but are more effective, produce fewer by-products and emissions, and are less expensive.99 Both chemical and solvent processes have disadvantages regarding their environmental impact.
Cellulose can also be extracted from most agrifood biomass, especially agrifood residues. Although it is difficult to estimate the total of cellulosic agrifood waste generated annually, specific examples include up to 520 million tonnes of rice straw from rice cultivation, 279 million tonnes of sugarcane bagasse, or 39 million tonnes of spent grains.106,111 Sourcing cellulose from the non-edible parts of plants, including husks and stems or food waste, is a sustainable agrifood waste valorization strategy. In recent years, this approach has given rise to some publications.112–115 Our recent work even demonstrated that cellulose-based microbeads can be directly sourced from an agrifood residue (brewer's spent grain), without the need for cellulose delignification.106 The resulting microbeads were relatively large with a mean diameter of 1.25 mm and demonstrated an average hardness of 199 MPa, making them well-suited for applications in personal hygiene products.106 This can be compared to the hardness of pure cellulose microbeads, around 142 MPa, or that of composite cellulose-carbon nano-sheet microbeads, around 238 MPa, although these are able to be produced at smaller sizes (<1 mm) [Fig. 10].28,105

 | ||
| Fig. 11 Molecular structure of galacturonic acid. | ||
Pectin is sustainable and biodegradable but has relatively poor mechanical properties.117 Although pectin use in microbead production is environmentally friendly, its poor mechanical properties greatly limit its applications. Pectin microbeads are a popular choice for drug delivery applications, where their rapid degradation in physiological conditions is desirable.118,119 Otherwise, pectin microbeads may be coated with gelatin, shellac, chitosan, or a polymethacrylate-based copolymer (Eugradit®) to improve their mechanical characteristics according to a desired application in functional food preparation or biomedical therapies [Fig. 12].70,118–120
 | ||
| Fig. 12 SEM images of (a) pectin and (b) pectin–gelatin microbeads. (Reproduced from ref. 119 under CC-BY.)119 | ||
The degree of methoxylated esters in the polymer determines the physical and chemical characteristics of the resulting pectin bioplastic and the coagulating agent used in its production. While pectin with a high degree of esterification (greater than 50%) requires sugar and a hydrogen ion, pectin with a low degree of esterification (less than 50%) requires the presence of divalent ions (notably calcium).118,121 Control over methoxylated ester content in the polymer equally represents the ability to control pectin microbead degradation rates.
Pectin microbeads can encapsulate nanoparticles using microfluidics with biocompatible ingredients, as reported by Ogonczyk, Siek, and Garstecki (2011).121 Flow-focusing microfluidic chips, which can have several possible configurations [Fig. 13], break a continuous stream of a pectin and nanoparticle solution into droplets. The side inlets of the chip introduce the coagulating medium, which in this case is a mixture of rapeseed oil, calcium carbonate, and acetic acid. As the solutions travel along the microchip, protons diffuse into the droplets of the pectin solution, leading to their solidification by cross-linking. The chip extrudes the microbeads into distilled water, where they can finish solidifying. This technique produces highly uniform beads with non-toxic and renewable chemicals but is a relatively slow method for microbead production.121
 | ||
| Fig. 13 Representation of different designs for fluidic devices: (a) T-junction device, (b) flow-focusing device, and (c) co-flow device. | ||
To encapsulate bioactive compounds, pectin with a low degree of esterification is often combined with alginate or gellan gum. The most eco-friendly options to prepare such hybrid pectin microcapsules rely on microbead self-assembly in the presence of calcium ions over 8 hours of stirring.122 Elsewhere, automatic encapsulators provide better control of the process by dropping microbeads into the calcium-based solution.123 The pectin and alginate polymeric solution can be prepared using sodium hydroxide,123 sterile physiological water (9% sodium chloride), or sterile M17 broth with dextrose.124
The cross-linking mechanism exploited in microfluidics or automatic encapsulation can equally be extended to more straightforward techniques, such as water-in-oil emulsification and simple extrusion, so long as the chosen cross-linking agent coordinates with pectin's esterification degree.118 Other methods to prepare pectin microbeads include spray or aerosol drying, dehydration, or electrospraying. These methods avoid using cross-linking agents and can be adapted to pectin solutions with varying degrees of esterification.118,119 However, synthetic polymers such as polyethylene oxide or polypropylene oxide may need to be mixed with pectin to avoid microbead collapse or process discontinuity.119
 | ||
| Fig. 14 Molecular structure of amylose. | ||
Starch is easily isolated from its natural matrices. Starch-rich plants are dried, ground into flour, mixed with water, and filtered to remove undesirable fibers and debris. Most of the added water is decanted, and the remainder is evaporated in an oven. The resulting solid is ground and filtered to obtain a stable and uniform powder.127 However, the following disadvantages are associated with using starch in plastic microbead production: it competes with human and animal feedstocks, is not antimicrobial, and has poor mechanical properties.126 For these reasons, starch most commonly serves as a precursor for biobased synthetic polyester microbeads34 or is blended with other carbohydrates to create composite microbeads.96,127–131
Pure starch microbeads were prepared to encapsulate a model drug, methylene blue, using a water-in-oil emulsification approach with sodium trimetaphosphate as a crosslinking agent.132 This was eventually extended to other drugs or pharmaceutical natural products, such as curcumin in osteosarcoma treatment.133 Although curcumin is effectively cytotoxic towards cancer cells, it cannot sustain the physiological pH of the human body (average of 7.40) and is hydrophobic, which limits its use as a therapeutic agent.134 With curcumin-containing starch microbeads produced using a water-in-oil mini-emulsion technique, there is greater biocompatibility and increased cytotoxicity towards cancer cells through the sustained release mechanism. Starch is first dissolved into an aqueous NaOH solution and mixed with an aqueous NaCl solution. Then, this solution is mixed with sunflower oil and two surfactants: Span® 80 and Tween® 80. These solutions and surfactants are non-toxic and non-irritating. Ultrasonication, a low-energy technique, creates polymeric micelles [Fig. 15], which can then be separated from the solution, washed, and preserved. When soaked in a solution of curcumin and ethanol, curcumin adsorbs to the micellar microbeads and becomes encapsulated within the particles. This ultrasonic-assisted emulsion technique can equally produce composite starch and cellulose microbeads for dye and protein adsorption. However, this method requires the use of harmful organic solvents.96
 | ||
| Fig. 15 Schematic representation of the supramolecular structure of polymeric micelles. | ||
Luo et al. (2022) report a similar emulsion-based technique. This work describes the preparation of quinoa-starch microbeads by preparing a water-in-oil emulsion and then introducing epoxy chloropropane as a cross-linking agent to solidify the resulting micro-droplets. After characterization, the authors concluded that these microbeads showed promise as a drug carrier or in cosmetics, although their shape varied and adhesion between microparticles was unavoidable.135
Starch is commonly blended with sodium alginate to create mechanically resistant microbeads that can be useful in various applications. Jha and Bhattacharya (2009) report a simple and low-cost method to produce sweet potato starch-blended sodium alginate microbeads for controlled release drug administration by ionotropic gelation. This study uses external gelation (the ions are present in the external solution and gradually diffuse into the microbeads where they crosslink and gel the polysaccharide molecular chains) as opposed to internal gelation (the ions are already present within the beads as salts and are liberated by introducing the microbeads to an acidic medium) [Fig. 16].128,136 First, starch gelatinizes in water when heated. Then, starch is mixed with sodium alginate and the drug of choice. The resulting solution is dropped into a stirred coagulating solution of calcium chloride, barium chloride, or aluminum sulfate, of which calcium chloride is preferable for its relative non-toxicity.137In vitro, the resulting spherical and rigid microbeads biodegrade, slowly releasing the loaded bioactive compound encapsulated within. This approach has since been adopted in other works. Similarly, Rani et al. 2023 combines starch with sodium alginate to create fertilizer-loaded microbeads to enhance crop growth.131 Another protocol describes the same process but mixes starch with gelatin instead.127 Ionotropic gelation can also produce chitosan-starch microbeads for encapsulation, using a solution of sodium tripolyphosphate as a coagulation medium.138 This is equally non-toxic and environmentally friendly.
 | ||
| Fig. 16 Simplified representation of (a) external versus (b) internal ionotropic gelation using Ca2+ ions as an example. | ||
Okunlola, Odeku, and Patel (2012) propose a near-identical method for controlled-release drug administration through the production of loaded yam starch and sodium alginate microbeads. These microbeads are also produced by ionotropic gelation, which only differs from the previous method in that sodium bicarbonate is added to the gelatinized starch and sodium alginate blend, and the only coagulating solution tested is one of calcium chloride. Sodium bicarbonate is used to impart buoyancy to microbeads, and its alkalinity leads to the formation of a gel outer layer of increased resistance.129 Similarly, Olayemi, Apeji, and Isimi (2022) recently prepared tiger nut starch-alginate microbeads, solidified by ionotropic gelation, to encapsulate ibuprofen.139 Besides targeted drug delivery applications, Kozlowska, Prus, and Stachowiak (2019) report that starch-sodium alginate microbeads are also effective as cosmetic exfoliants and can be prepared using the BÜCHI B-395 Pro encapsulator with calcium chloride as a coagulating medium.130 This instrument automatizes the dropping technique, ensuring aseptic, easily reproducible conditions.140
Recent works also demonstrate how the fundamental backbone of starch microbeads is readily adaptable for more specific applications. For example, synthetic ligand-coated starch magnetic microbeads can successfully selectively extract silicon dioxide from commercially processed food, which has historically been used as a food additive but is now facing concerns about its nanoscale toxicity. In this case, silica-specific ligand protein-coated starch microbeads encapsulate dextran-coated iron oxide.141 Elsewhere, chitosan-coated gelatinized starch microbeads self-assemble during incubation with pullulanase, which debranches the polymer's amylopectin. In this work, the authors then coat the microbeads with silver nanoparticles through incubation in a HAuCl4 solution and use the microbeads in surface-enhanced Raman scattering applications.142 From these two examples, we get the sense of starch microbeads' improved versatility by incorporating substances within or coating their surfaces.
4.3 Algae-derived carbohydrate-based polymers
 | ||
| Fig. 17 Molecular structure of alginic acid with D-mannuronic acid (m) and L-guluronic acid (n). | ||
Alginate can be easily extracted from brown algae using a simple and eco-friendly method. Treating algae with sodium hydroxide extracts the alginate, then filtration removes debris. Then, adding sodium or calcium chloride precipitates alginate, resulting in sodium or calcium alginate that can be used in microbead production. Further treatment with dilute HCl produces alginic acid, an equally viable starting material.149 It is of high importance to use alginate of high purity for alginate microbeads for biomedical applications, as algae can contain toxins, heavy metals, and various other contaminants.100
Alginate derivatives can be combined with other carbohydrates to create composite microbeads, such as chitosan, starch, cellulose, carrageenan, pectin, or hyaluronic acid92,130,147,150,151 or with other polymers including gelatin, polyethylene imine, poly-L-ornithine, or phosphorous tetramethylmethyl sulfate as coatings.145,152–155 Otherwise, pure alginate microbeads present interesting mechanical properties. As a first step, sodium alginate dissolves in hot water alongside other additives, if desired. Then, the resulting solution is shaped according to a dropping, emulsification, or electrospraying technique and gelled using an ionic or chemical cross-linking mechanism [Fig. 18].150,153,155–157
 | ||
| Fig. 18 Schematization of the cross-linking mechanism: (a) ionic or (b) covalent. | ||
Alginate beads are promising for drug (i.e. icariin, mesalamine, guar gum succinate) or cell delivery in tissue and organ engineering or wound dressings.100,144,153,155,157–160 Alginate microbeads are the best material for cell encapsulation and drug delivery as they immobilize in isotonic solutions in conditions that mimic those of the human body.100 Additionally, dropping methods are ideal for creating spherical alginate microcapsules,151,153,157 meaning that automated sterile encapsulators (or electrosprayers) are compatible.144,146,147,158,160 Automatic encapsulators charge the surface of the beads that form as the polymer solution drops from a needle into a coagulation solution.161 These mechanisms can create matrix microcapsules, which contain encapsulated substances dispersed throughout the polymer matrix, or coaxial microcapsules, where the encapsulated substances are contained within a polymer shell [Fig. 19]. This method may lead to beads of poor polydispersity due to the coalescence of droplets during the gelation process and post-aggregation. Microfluidic emulsification, followed by a controlled shrinkage process and gelation in mineral oil containing Ca2+ ions, produces monodispersed microbeads of 5–30 μm instead.156 Whether dropping, emulsification, or electrospraying methods are used for shaping alginate microbeads, calcium chloride solutions are ideal for bead solidification through ionic gelation.145–147,150,151,154,157,159,160
 | ||
| Fig. 19 Representation of the mechanism of an automatic encapsulator for: (a) matrix or (b) coaxial microbeads. With matrix microbeads, the two (or more) components of the microbead are homogeneously mixed, and then extruded (green); with coaxial microbeads, the core material (blue) is encapsulated with a polymer shell (yellow) during extrusion. | ||
Other applications may use alginate microbeads. For example, Elzatahry et al. (2010) describe preparing microbeads for cationic dye absorption by automatic encapsulation and ionic gelation.161 As mentioned in Section 4.2.3 of this report, this method effectively produces composite starch-sodium alginate microbeads for cosmetics or encapsulation purposes.130,131 Alginate microbeads can also remove heavy metals.54 More recent work by Abdo et al. 2023 combines these properties with those of encapsulated silver nanoparticles and Chlorella minutissima, using alginate microbeads for municipal wastewater treatment (reducing bacterial count and removing undesirable nitrite, nitrate, ammonia, and phosphorous).162
Alginate microbeads for wastewater treatment may be prepared in an eco-friendly way by loading a sodium alginate solution into a pressure sprayer and solidifying with a CaCl2 solution.162 A similar ionotropic gelation method produces mechanically resistant sodium alginate-ulvan microbeads for personal hygiene products.163 Elsewhere, Sargin et al. (2016) report that bleached alginate can be combined with chitosan and acetic acid and dropped into a coagulating glutaraldehyde solution to form porous microbeads.54 Although these microbeads are highly effective in their application, bleaching alginate biomass requires harsh chemicals, and the cross-linking agent of choice, glutaraldehyde, is toxic. From an environmental perspective, ionic gelation mechanisms, such as the method elaborated by Abdo et al. 2023, are preferable to chemical cross-linking agents.162
 | ||
| Fig. 20 Molecular structure of the repeating disaccharide units for agarose (m) and agaropectin (n). | ||
Pure agar microbeads are commonly produced using a microchip-facilitated water-in-oil emulsion process. Microchips control bead dimensions and yield particles as small as 15 μm.168 In a method prepared by Kuroiwa et al. (2016), an agar–NaCl solution is the disperse phase, and isooctane containing 3 wt% Span® 85 is the continuous phase. The continuous phase is recovered for reuse, lessening the isooctane's environmental footprint. As agar is thermoresponsive, bead solidification is controllable with a thermoregulated microchannel device. Other studies have extended this process for enzyme immobilization in biocatalytic applications, notably for serine-proteases and maltase. These microbeads are recyclable and stable for relatively long periods.165,169 Mixing protease into a heated polymeric solution entraps the enzyme in the polymeric agar matrix. Then, the solution is poured onto Petri dishes, where it cools and solidifies. Microbeads are then cut from the solid plate using a metallic borer.165 Maltase entrapment uses the same method.169 It is preferable to use “top-down” approaches for enzyme entrapment by agar, as this limits the deactivation rate of the enzyme. On the other hand, bacterial encapsulation is achievable using a microfluidic chip, where heated polymeric solution forms uniform beads as it travels along a narrow channel and solidifies upon contact with a cooler continuous phase.170
Similarly, simple water-in-oil emulsion techniques can produce pure agar microbeads of varying sizes (10–165 μm). With this approach, an aqueous agar solution is the disperse phase, while a paraffin- or cyclohexane-Span® 85 solution is the continuous phase.171,172 Once again, temperature variation controls the microbeads' solidification. Further cross-linking with epichlorohydrin and 1,4-butanediol diglycidyl enhances the bead's mechanical and thermal stability but increases the method's human and environmental risks.171 These beads are an effective chromatography media or can be used to stabilize Pickering emulsions.171,172
Agar microbeads can also administer drugs when prepared with another carbohydrate for added robustness in physiological conditions (hybrid microbeads).166,167 For example, extrusion and dispersed phase solidification techniques can generate ibuprofen-loaded agar-cellulose microbeads. First, agar and cellulose are dissolved in distilled water and heated. Ibuprofen is added, and the resulting solution is extruded through a syringe into an ethyl acetate solidifying solution.166 Besides cellulose, agar is commonly mixed with agarose or alginate.166
Agarose is a version of purified agar with similar physical and chemical properties. Agarose microbeads are biodegradable in natural conditions but they do not degrade in mammalian bodies, fueling their potential in bioartificial implant therapies.100 Agarose microbeads can also be used to encapsulate and administer live cells. Konnova and Fakhrullin (2017) report preparing cell-doped microbeads with water-in-oil emulsion. They used an aqueous agarose solution containing live cells as the disperse phase and sunflower oil as the continuous phase. The two phases were combined, heated, and vortexed to form an emulsion. The resulting agarose beads were solidified by rapidly cooling the emulsion. The resulting microbeads are filtered from the solution and washed with ethanol.173 This method is rapid, easily scalable, biocompatible, and eco-friendly, all while providing encapsulated cells with a viable environment.
Likewise, Nguyen-Le et al. (2023) describe agarose microbeads as “mini-Petri dishes for bacterial co-cultivation”. A microfluidic system can prepare these microbeads with an aqueous agarose solution containing one or two types of bacteria as a dispersed phase and mineral oil as a continuous phase.174 Incubation proliferates the bacteria encapsulated within and allows researchers to study the competitive and cooperative interactions in bacterial communities under controllable conditions. Elsewhere, emulsification techniques produce agarose microbeads containing Fe3O4, using n-octane as a continuous phase. As with agar microbeads, agarose microbeads can be cross-linked with epichlorohydrin, improving their stability yet increasing the risk associated with their production.167
Higher concentrations of agarose lead to the formation of harder microbeads with smaller pores.100 Moreover, the types of agarose and their cross-linking degree influence the beads' pore structures. Higher molecular weight or increasingly cross-linked agarose leads to more porous beads with larger pores.175 Exploiting these relationships can produce customizable agarose microbeads useful for chromatography or protein purification and immobilization, among other applications. However, immobilized proteins, notably enzymes, affect the stability of agarose microbeads,176 and specific mixtures require further study before their use in biomedical applications.
4.4 Microorganism-derived carbohydrate-based polymers
 | ||
| Fig. 21 Molecular structure of dextran with branched (m) and linear (n) glucopyranose units. | ||
 | ||
| Fig. 22 SEM image of functionalized magnetic dextran microbeads. (The original image has been cropped. Reproduced from ref. 179 under CC-BY.)179 | ||
According to Ye et al. (2019), dextran-producing bacteria, such as Leuconostoc pseudomesenteroides, can be separated from fermented food products (such as fermented food wastes) through a series of serial dilutions and plating.180 Bacterial colonies deposited on the plate are incubated in the presence of sucrose. After dislodging the bacterial culture from the plate, ethanol precipitates and isolates dextran. Chromatography purifies the sample.
Dropping methods commonly produce dextran microbeads: the polymeric solution extrudes through a syringe into a stirred coagulating solution.181 Whereas potassium ions most efficiently solidify pure dextran microbeads,100 aluminum or sodium ions can solidify composite microbeads.181,182 In either case, these coagulating solutions are environmentally friendly and reusable. Before microbead extrusion, dextran can undergo methacrylation or other polymers can be added to the solution. Either approach increases the beads' stability. Elsewhere, water-in-water emulsions of dextran solutions allow the encapsulation of other water-soluble substances.183 Water-in-oil emulsification techniques are equally viable, using cyclohexane-Span® 80-Tween® 80 as a continuous phase. These beads can be readily functionalized within their matrix (i.e. designed to contain magnetic nanoparticles) or at their surface (i.e. polymer- or dendrimer-coated).179
Dextran microbeads are used in in vitro cell and drug delivery (with or without using liposomes as an encapsulated carrier system).100,178,183 Here, the encapsulated substances are released according to the degradation of the microbeads. The degradation rate of dextran microbeads increases with higher water content, methacrylate substitution, or by incorporating a hydrolytically sensitive spacer in the cross-links.183 Dextran–pullulan microbeads, cross-linked with sodium trimetaphosphate (STMP), can also provide a scaffold for in vivo bone repair. Increased cross-linking, induced using greater concentrations of STMP, decreases in vivo enzymatic degradability and increases bead swelling in physiological conditions.182 This indicates the beads' customizability to a specific patient's needs. Porous cellulose–dextran microbeads are also highly effective at cationic dye adsorption181 The ability of dextran to be functionalized through chemical modification or by mixing with other carbohydrates increases its versatility.100,181,183 However, research on dextran microbeads has declined in recent years in favor of more readily available carbohydrates.
000 repeating units of beta-1,4-D-glucuronic acid-beta-1,3-N-acetyl-D-glucosamine [Fig. 23]. Naturally found in many animal tissues, HA is biocompatible, non-cytotoxic, non-genotoxic, and non-immunogenic.184 It has excellent viscoelastic and lubricant properties and is biodegradable. HA used for microbead production is commonly sourced from genetically modified bacteria, such as Streptococcus equi, Streptococcus zooepidemicus, and Bacillus subtillis,100 or is extracted from animal waste (by-products of the meat industry).185 The latter process involves harsh extraction conditions with low yields and a high risk of viral and protein contamination but adds value to a waste product.
 | ||
| Fig. 23 Molecular structure of hyaluronic acid's repeating unit of beta-1,4-D-glucuronic acid and beta-1,3,-N-acetyl-D-glucosamine. | ||
Production by bacterial fermentation is the preferred route, producing desirable yields of relatively high-purity HA. The risk of bacterial endotoxin, protein, nucleic acid, or heavy metal contamination is small but ever-present, and HA still needs to be carefully monitored for purity. A third method involves the exploitation of isolated HA synthase to produce HA. However, this method needs further development before adoption by industry.185
To be used in pure microbead production, HA first must be chemically modified, notably through amidation with carbodiimides, methacrylation, or an addition reaction with bioactive ligands. Exposure to UV light can solidify methacrylated HA microbeads.100 In composite microbeads (i.e. mixed with alginate), HA does not require chemical modification.100,151
Li et al. (2020) reported dopamine-functionalized HA microbeads to be promising in novel cancer diagnosis techniques by capturing circulating tumor cells present in blood samples.186 The dopamine-functionalization method uses non-hazardous coupling reagents at slightly acidic conditions, and oxidation solidifies the resulting polymeric solution. Microfluidic chips can form microbeads, as described in Section 4.2.2 of this report. Similarly, maleimide-functionalized HA microbeads, proposed by Uthaman et al. (2016) as a therapeutic strategy against solid breast cancer tumors, use chemotactic and biological targeting mechanisms. Microfluidics can also prepare these microbeads, with solidification occurring via a Michael-type addition cross-linking between the functionalized HA and polyethylene glycol.187
HA microbeads are practical for immobilizing cells, particularly neural stem cells.100,188 According to Amirian et al. (2017), these microbeads can be prepared through a simple, low-energy spray method into a coagulating calcium chloride solution. A pressurized polymeric solution rapidly extrudes through a fine nozzle, and the microbead size is a function of the pressure applied.189 As the resulting microbeads are highly porous, soaking the beads in a cell-growth media encapsulates cells. Similarly, Hamilton et al. (2021) report the production of acrylated HA microcapsules containing stem cells using an automatic encapsulator and UV light for photo-crosslinking.188 Elsewhere, microfluidics and photo-crosslinking prepare and solidify bisphonate (alendronate)-conjugated methacrylated HA, which can control the delivery of bone morphogenetic proteins.190 Cell-encapsulating HA microbeads are non-inflammatory, pro-chondrogenic, and very smooth, making them viable injectable microcarriers for cartilage, bone, and kidney reconstruction [Fig. 24].100,190–192

 | ||
| Fig. 25 Molecular structure of gellan gum's tetrasaccharide unit. | ||
Moslemy, Guiot, and Neufeld (2002), describe that emulsion techniques commonly produce pure gellan gum microbeads. An aqueous gellan gum solution is heated, calcium chloride is added, and the mixture is cooled. Once cool, adding NaOH adjusts the pH to between 6.9 and 7.2. The solution is then emulsified with rapeseed oil in the presence of a surfactant. At this point, cells or drugs can be added to the solution for their encapsulation. Stirring and rapidly cooling the emulsion initiates the microbeads' solidification.193 Besides, extrusion into a solidifying solution (conventional dropping, vibrating nozzle-prilling, or electrostatic methods) or mechanical cutting can yield pure gellan gum microbeads.194,195 Gellan gum's solidification mechanism conventionally depends on heat and a multivalent cation (Zn2+, Al3+, Ca2+, Cd2+, Pb2+, etc.),193,195,196 similar to many other natural polymers. This similarity allows gellan gum's use in hybrid microbead preparation.197
Coutinho et al. (2012) report an automated technique to produce gellan gum-alginate microbeads. First, gellan gum powder dissolves in deionized water, and alginate dissolves in phosphate saline buffer. Then these two solutions are mixed. Microbeads can then be produced from these solutions using an automated system. A syringe pump disperses drops of the polymeric solution into a two-phase system, with hydrophobic mineral oil as the superior phase and hydrophilic aqueous calcium chloride as the inferior phase. This two-phase system is agitated, which decreases the size of the microbeads in the oil and brings about their perfect sphericity. These microbeads then pass through the oil-aqueous interface and solidify in the hydrophilic solution. The hydrophobic solution is removed and the microbeads are stored for future use.197 These microbeads are efficient at cell encapsulation through mixing with the original alginate solution and are thus important for biomedical therapies. Since this work, other research groups have built off this approach. For example, Park et al. (2021) describe similar gellan gum-alginate microcapsules containing hydrocarbon-degrading bacteria. An emulsification-internal gelation method produces these beads, using rapeseed oil as the continuous phase. A CaCl2 solution is added to the emulsion to induce the beads' gelation.198
Gellan gum is compatible with other naturally occurring polymers, such as whey protein isolate.199 In a method described by Kuhn et al. (2019) oil-in-water emulsions produce gellan microbeads, using flaxseed oil and a CaCl2 coagulating solution. The aqueous polymeric solution and flaxseed oil are vigorously shaken, and this emulsion is extruded through a nozzle into the coagulating solution.199 This method allows to encapsulate the bioactive compounds found in flaxseed oil, uses non-toxic products, few steps, and little energy. Gellan gum is also compatible with carboxymethyl tamarind gum to create microbeads for sustained drug release. Here, the polymer solution is emulsified and extruded into a solution of AlCl3.200 Gellan gum can also be mixed with synthetic polymers such as polyvinyl alcohol to the detriment of the material's biodegradability. An emulsion-dropping method is equally appropriate for these microbeads, which are then used for drug delivery.196 Similarly, emulsion-dropping techniques produce pure, size-controlled gellan gum microbeads using rapeseed oil.193
5 Conclusions
This work clearly demonstrates that carbohydrate-based plastic microbeads pose less of an environmental threat than conventional petroleum-based or popular biobased options, but that their environmental impact and application potential vary widely. We compile the different plastics presented in this report and their characteristics in Table 1.| Non-coveted source(s) | Waste valorization | Sustainable extraction & preparation | Potential fields of application | Key mechanical properties | Biodegradable in marine environments | |
|---|---|---|---|---|---|---|
| PE | No | No | No | Cosmetics, coatings, plastics, abrasives, filtration, chemistry | Hard, durable | No |
| PP | No | No | No | Cosmetics, coatings, plastics, abrasives, filtration | Hard, durable | No |
| PET | No | No | No | Cosmetics, coatings, plastics, abrasives, filtration, chemistry | Hard, durable | No |
| PLA | Possible | Possible | Yes, generally | Cosmetics, coatings, food, chemistry, biomedical | Hard, durable | No |
| Chitin | Yes | Yes | Yes, generally | Cosmetics, coatings, energy, biomedical, filtration | Hard, resistant | 1–2 months |
| Chitosan | Yes | Yes | Yes, generally | Cosmetics, coatings, food, biomedical, filtration | Hard, resistant | 1–2 months |
| Cellulose | Yes | Yes | Yes, generally | Cosmetics, food, chemistry, biomedical, filtration | Firm | 1–2 months |
| Pectin | Yes | Yes | Yes, generally | Biomedical | Soft | 1–2 months |
| Starch | No | No | No, generally | Biomedical & food | Soft, weak | Yes, unspecified |
| Alginate | Yes | No | No, generally | Biomedical & filtration | Firm | 1 month |
| Agar | Yes | No | Yes | Biomedical & food | Soft, weak | Yes, unspecified |
| Agarose | Yes | No | Yes | Biomedical | Soft, weak | Yes, unspecified |
| Dextran | Yes | Yes | Yes | Biomedical & filtration | Soft, weak | Yes, unspecified |
| HA | Yes | Possible | Yes, generally | Biomedical | Soft, weak | Yes, unspecified |
| Gellan gum | Yes | No | Yes | Biomedical & food | Soft, weak | 1 month |
Petrochemical-derived plastics (PE, PP, PET) clearly present desirable physical properties, lending to their immense popularity in recent decades in various application. However, their high carbon footprint, origins from non-renewable resources, and persistence in the environment provide ample justification for their discontinued use. Meanwhile, PLA as a biobased synthetic alternative in microbead production proves to be just as versatile, with good durability and strength among its many desirable mechanical properties. As a biobased synthetic plastic, PLA's monomers are successfully derived from renewable resources, including the possibility to use agrifood waste as a starting point. However, PLA biodegradability is so slow and condition-specific that it is increasingly considered practically non-biodegradable. Its misleading status as a durable biodegradable alternative to conventional plastics is especially concerning, as this may lead to their use in ecologically inappropriate applications (i.e., wastewater-destined personal hygiene products). Consequently, we wish to warn against the indiscriminate use of biobased synthetic plastics, including PLA, as a substitute for petrochemical plastics in microbead production.
Alternatively, cellulose, chitin, or chitosan-based microbead varieties are the most promising eco-friendly alternatives to synthetic plastics. They have similar or superior efficiency to traditional plastic microbeads in various applications, are highly adaptable, and should rapidly biodegrade in the environment (especially marine environments). Cellulose is abundant in renewable resources, including agrifood waste. However, its “greenness” depends on increasingly favoring novel environmentally-friendly methods over conventional extraction and purification techniques. Chitosan- or chitin microbeads are equally, if not more, promising. These polymers are sourced from the exoskeletons of arthropods (waste), and it is possible to produce chitosan- or chitin-microbeads with low-energy techniques and non-toxic solvents. Yet, further research is required to refine the environmental footprint of these polymers' extraction and purification, as well as their dissolution and shaping into functional microbeads.
Alginate, agar, dextran, and agarose-based microbeads pose a lesser environmental threat than traditional options, but each has weaknesses. Alginate microbead formation generally requires toxic solvents, and agar, agarose, and dextran have relatively weak mechanical properties that limit their applications. Pectin, gellan gum, and HA are similar, with eco-friendly extraction and microbead formation techniques but limited applications.
Starch is the least promising carbohydrate-based biopolymer. Starch-based microbeads are produced using relatively harsh methods, have poor mechanical properties, and limited applications. With an abundance of other carbohydrate options available, we recommend against using starch in most applications.
Generally, alternatives whose extraction and shaping methods are compatible with the principles of Green Chemistry correlate with poor mechanical properties and low versatility. To balance these problems, researchers must identify strategies to strengthen biopolymers' physical characteristics or develop more eco-friendly extraction and shaping techniques for those with greater strength and resistance.
Additionally, although biodegradable polymers such as carbohydrate-based biopolymers can contribute to closing carbon loops, the efficacy of this depends on specific polymer design, environmental conditions, and the ability of local ecosystems to assimilate the biopolymers' monomeric fractions. This information has generally been neglected in the study of novel biopolymer-based microbeads. To improve our commentary on the biodegradability of carbohydrate biopolymers, the degradation kinetics for each polymer type must be studied. It is crucial to evaluate the mechanisms of degradation of each type of plastic in a variety of environments, as under specific conditions (often in the absence of water and oxygen), certain plastics may lead to water-soluble or toxic metabolites, nanoparticle emissions, or provide a breeding ground for bacteria and spores.16 Understanding these mechanisms can lead to the development of intelligent, controlled biodegradation mechanisms that further limit the environmental impact of microbead use. Biopolymer design should be aligned with natural metabolic pathways.
It is equally important to ensure that the transition towards carbohydrate-based plastics evades undesirable socio-economic effects. Many of the alternatives described in this report compete with food sources, building materials, and the budding shift toward biofuels. Incorrect assessment of the choice of starting material when accounting for this parameter could lead to intensified farming, crop monocultures, the extensive use of fertilizers, deforestation, and grassland destruction and conversion. This, in turn, would result in increased greenhouse gas emissions and threats to biodiversity. A greater cost of food and housing could also ensue, negatively impacting already vulnerable people and pushing even more into poverty.
Finally, we believe that it would be worthwhile to critically review the biopolymers that lie outside the scope of this work (proteins, polypeptides, or naturally-occurring polyesters). This would effectively complete the comparison between biopolymer alternatives and traditional synthetic polymers for microbeads.
Despite these considerations, we strongly believe that carbohydrate-based plastic microbeads can represent a sustainable alternative to those resulting from petrochemical processes. They are generally more biodegradable and less toxic, produce less greenhouse gases, and can decrease manufacturers' dependence on non-renewable raw materials. Certain crops or wastes can gain added value through their use in the production of biopolymer-based plastics, which can be incentives for farmers and industries, especially in countries with surplus food production. Eliminating conventional microbeads to favor biopolymer options is a positive step in transitioning to a sustainable, circular economy.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
A. McMackin was responsible for identifying and reviewing pertinent articles cited in this work, developing the review methodology, writing the manuscript, and designing figures. S. Cardinal was responsible for project administration and reviewing and editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank MITACS (Accelerate program) for providing a graduate scholarship to A. McMackin. The authors also wish to acknowledge the use of Canva in the preparation of all schematic elements found in this manuscript, and ChemSketch Freeware for the preparation of molecular structure diagrams.Notes and references
- R. Geyer, J. Jambeck and K. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- C. Rhodes, Sci. Prog., 2019, 102, 218–248 CrossRef PubMed.
- C. King, J. Shamshine, O. Zavgorodnya, T. Cutfield, L. Block and R. Rogers, ACS Sustain. Chem. Eng., 2017, 5, 11660–11667 CrossRef CAS.
- D. Huang, J. Tao, M. Cheng, R. Deng, S. Chen, L. Yin and R. Li, J. Hazard. Mater., 2021, 407, 124399 CrossRef CAS PubMed.
- A. Pradel, C. Gatrouillet and J. Gigault, NanoImpact, 2023, 29, 100453 CrossRef CAS PubMed.
- C. Rhodes, Sci. Prog., 2018, 101, 207–260 CrossRef PubMed.
- R. Mori, RCS Sustainability, 2023, 1, 179–212 CAS.
- WHO, Microplastics in Drinking-Water, World health organization report, 2019 Search PubMed.
- H. Auta, C. Emenike and S. Fauziah, Environ. Int., 2017, 102, 165–176 CrossRef CAS PubMed.
- M. Gericke, J. Trygg and P. Fardim, Chem. Rev., 2013, 113, 4812–4836 CrossRef CAS PubMed.
- H. Nam and W. Park, ACS Biomater. Sci. Eng., 2020, 6, 2440–2449 CrossRef CAS PubMed.
- V. Ranjan, A. Joseph, S. Srivastava, H. Sharma, B. Biswas, S. Goel and S. Kumar, Waste Manag. Bulletin, 2024, 2, 229–240 CrossRef.
- N. Mostafa, A. Farag, H. Abo-dief and A. Tayeb, Arab. J. Chem., 2018, 11, 546–553 CrossRef CAS.
- M. Thielen, Bioplastics: Plants and Crops, Raw Mateirals, Products, 2014 Search PubMed.
- N. Raddadi and F. Fava, Sci. Total Environ., 2019, 679, 148–158 CrossRef CAS PubMed.
- R. Mülhaupt, Macromol. Chem. Phys., 2013, 214, 159–174 CrossRef.
- P. Anastas and J. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- H. Chen, Lignocellulose Biorefinery Engineering: Principles and Applications, Woodhead Publishing, Cambridge, 2015 Search PubMed.
- D. Green, B. Boots, J. Sigwart, S. Jiang and C. Rocha, Environ. Pollut., 2016, 208, 426–434 CrossRef CAS PubMed.
- D. Tristantini and A. Yunan, E3S Web Conf., 2018, 67, 04045 CAS.
- S. Agarwal, Macromol. Chem. Phys., 2020, 221, 2000017 CAS.
- R. Thompson, S. Swan, C. Moore and F. vom Saal, Philos. Trans. R. Soc. Lond. B Biol. Sci., 2007, 2(364), 1973–1976 Search PubMed.
- K. Whiteley, T. Heggs, H. Koch, R. Mawer and W. Immel, Polyolefins, 2005 Search PubMed.
- H. Köpnick, M. Schmidt, W. Brügging, J. Rüter and W. Kaminsky, Polyesters, 2012 Search PubMed.
- J. Prata, A. Silva, J. da Costa, C. Mouneyrac, T. Walker, A. Duarte and T. Rocha-Santos, Int. J. Environ. Res. Publ. Health, 2019, 16, 2411 CrossRef PubMed.
- L. Neal, V. Haribal and F. Li, iScience, 2019, 19, 894–904 CrossRef CAS PubMed.
- H. Zimmermann and R. Walzl, Ethylene, 2012 Search PubMed.
- S. Ryu, S. Kim, J. Jeong, D. Kim, M. Kim and H. Lee, Cellulose, 2024, 31, 2281–2293 CrossRef CAS.
- S. Rebsdat and D. Mayer, Ethylene Glycol, 2012 Search PubMed.
- NCBI, PubChem Compound Summary for CID 8441, Dimethyl terephthalate, 2020, https://pubchem.ncbi.nlm.nih.gov/compound/Dimethyl-terephthalate, (Visited on 26/06/2024).
- NCBI, PubChem Compound Summary for CID 7489, Terephthalic acid, 2020, https://pubchem.ncbi.nlm.nih.gov/compound/Terephthalic-acid, (Visited on 26/06/2024) Search PubMed.
- J. Liu, Y. Yang, S. Wei, W. Shen, N. Rakovitis and J. Li, Ind. Eng. Chem. Res., 2018, 57, 12829–12841 CrossRef CAS.
- S. Lee and T. Lee, Sci. Rep., 2021, 11, 18074 CrossRef CAS PubMed.
- S. Alhuthali, Polylactic Acid- An Emerging Bioplastic, 2023, https://www.ecomena.org/poly-lactic-acid/, (Visited on 26/06/2024) Search PubMed.
- M. Jamshidian, E. Tehrany, M. Imran, M. Jacquot and S. Desobry, Compr. Rev. Food Sci. Food Saf., 2010, 9, 552–571 CrossRef CAS PubMed.
- J. Anderson and M. Shive, Adv. Drug Delivery Rev., 1997, 28, 5–24 CrossRef CAS PubMed.
- S. Baek, S.-K. Moon, R. Kang, Y. Ah, H. Kim and S.-W. Choi, Macromol. Mater. Eng., 2018, 303, 223–229 CrossRef.
- D. Kostic, M. Vukasinovic-Sekulic, I. Armentano, L. Torre and B. Obradovic, Mater. Sci. Eng. C, 2019, 98, 1159–1168 CrossRef CAS PubMed.
- N. Rahim, F. Islahudin, N. Tahrim and M. Jasamai, Sains Malays., 2022, 51, 2495–2506 CrossRef CAS.
- H. Gan, T. Okada, S. Kimura, K. Kasuya and T. Iwata, Polym. Degrad. Stab., 2023, 208, 110239 CrossRef CAS.
- L. Antonelli, M. Frondaroli, M. De Cesaris, N. Felli, C. Dal Bosco, E. Lucci and A. Gentili, Mikrochim. Acta, 2024, 191, 251 CrossRef CAS PubMed.
- M. Ghadamyari, S. Chaemchuen, K. Zhou, M. Dusselier, B. Sels, B. Mousavi and F. Verpoort, Catal. Commun., 2018, 114, 33–36 CrossRef CAS.
- Suharman, W. Heah, H. Yamagishi and Y. Yamamoto, Nanoscale, 2023, 15, 19062–19068 RSC.
- W. Ali, H. Ali, S. Gillani, P. Zinck and S. Souissi, Environ. Chem. Lett., 2023, 21, 1761–1786 CrossRef CAS.
- T. Kim, S. Lee, S.-Y. Park and I. Chung, Polymers, 2021, 13, 3964 CrossRef CAS PubMed.
- E. Ibrahim, S. Ahmed, S. Abir, K. Taylor, V. Padilla-Gainza and K. Lozano, Int. J. Biol. Macromol., 2022, 220, 671–682 CrossRef CAS PubMed.
- J. Wang, J. Li and J. Ren, Materials, 2019, 12, 1959 CrossRef CAS PubMed.
- O. Amamou, J.-P. Denis, E. Heinen, T. Boubaker and S. Cardinal, Molecules, 2023, 28, 7294 CrossRef CAS PubMed.
- W. Tang, J. Fernandez, J. Sohn and C. Amemiya, Curr. Biol., 2015, 25, 897–900 CrossRef CAS PubMed.
- D. Elieh-Ali-Komi and M. Hamblin, Int. J. Adv. Res., 2016, 4, 411–427 CAS.
- G. Wei, C. Zhang, N. Zhou, B. Wu, H. Li, A. Zhang, P. Ouyang and K. Chen, Carbohydr. Polym., 2023, 321, 121322 CrossRef CAS PubMed.
- V. Santos, N. Marques, P. Maia, M. de Lima, L. de Oliveira Franco and G. de Campos-Takaki, Int. J. Mol. Sci., 2020, 21, 4290 CrossRef CAS PubMed.
- S. Ju, G. Shin, M. Lee, J. Koo, H. Jeon, Y. Ok, D. Hwang, S. Hwang, D. Oh and J. Park, Green Chem., 2021, 23, 6953–6965 RSC.
- I. Sargin, G. Arslan and M. Kaya, React. Funct. Polym., 2016, 98, 38–47 CrossRef CAS.
- T. Kuroiwa, H. Takada, A. Shogen, K. Saito, I. Kobayashi, K. Uemura and A. Kanazawa, Colloids Surf. A Physicochem. Eng. Asp., 2017, 514, 60–78 CrossRef.
- W.-L. Xu, H.-J. Chen, Y.-C. Wang, S. Liu, X.-Y. Wan, H.-L. Peng and K. Huang, Green Chem. Eng., 2022, 3, 267–279 CrossRef.
- J. Liao, Y. Zhou, X. Zhao, B. Hou, J. Zhang and H. Huang, Carbohydr. Polym., 2024, 121773 CrossRef CAS PubMed.
- A. Aldana, M. Strumia and M. Martinelli, Aust. J. Chem., 2015, 68, 1918–1925 CAS.
- M. Rinaudo, Prog. Polym. Sci., 2006, 603–632 CAS.
- L. Pighinelli, J. Broquá, B. Zanin, A. Flach, C. Mallmann, F. Taborda, L. Machado, S. Alves, M. Silva and R. Dias, Am. J. Biomed. Sci. Res., 2023, 3, 307–314 Search PubMed.
- M. Triunfo, E. Tafi, A. Guarnieri, R. Salvia, C. Scieuzo, T. Hahn, S. Zibek, A. Gagliardini, L. Panariello, M. Coltelli, A. De Bonis and P. Falabella, Sci. Rep., 2022, 12, 6613 CAS.
- T. Hahn, A. Roth, R. Ji, E. Schmitt and S. Zibek, J. Biomech., 2020, 310, 62–67 CAS.
- H. Al Hoqani, N. Al-Shaqsi, M. Hossain and M. Al Sibani, Carbohydr. Res., 2020, 492, 108001 Search PubMed.
- I. Younes and M. Rinaudo, Mar. Drugs, 2015, 13, 1133–1174 CAS.
- A. Wilson, E. Ekanem, D. Mattia, K. Edler and J. Scott, ACS Sustainable Chem. Eng., 2021, 9, 16617–16626 CAS.
- Y. Wang, Y. Li, S. Liu and B. Li, Carbohydr. Polym., 2015, 120, 53–59 CAS.
- O. Amamou, S. Kefil, J.-P. Denis, T. Boubaker and S. Cardinal, Molecules, 2024, 29, 7294 Search PubMed.
- T. Di Nardo, C. Hadad, A. Nguyen Van Nhien and A. Moores, Green Chem., 2019, 21, 3276–3285 CAS.
- Y. Li, X.-E. Luo, M.-J. Tan, F.-H. Yue, R.-U. Yao, X.-A. Zeng, M.-W. Woo, Q.-H. Wen and Z. Han, Int. J. Biol. Macromol., 2023, 30, 125716 Search PubMed.
- S. Sampathi, C. Haribhau, V. Kuchana, V. Junnuthula and S. Dyawanapelly, Carbohydr. Polym., 2023, 1, 121177 Search PubMed.
- Y. Alinejad, C. Bitar, K. Martinez Villegas, S. Perignon, C. Hoesli and S. Lerouge, ACS Biomater. Sci. Eng., 2020, 6, 288–297 CAS.
- K. Moreno-Sader, S. Meramo-Hurtado and A. Gonzalez-Delgado, Sustainable Chem. Pharm., 2020, 15, 100212 CrossRef.
- M. Tedesco, D. Di Lisa, P. Massobrio, N. Colistra, M. Pesce, T. Catelani, E. Dellacasa, R. Raiteri, S. Martinoia and L. Pastorino, Biomaterials, 2018, 156, 159–171 CrossRef CAS PubMed.
- N. Milašinovic, B. Calijac, B. Vidovic, M. Sakac, Z. Vujic and Z. Kneževic-Jugovic, J. Taiwan Inst. Chem. Eng., 2016, 60, 106–112 CrossRef.
- R. Beauchamp, M. St Clair, T. Fennell, D. Clarke, K. Morgan and F. Karl, Crit. Rev. Toxicol., 2008, 22, 143–174 CrossRef PubMed.
- K. Kamiński, K. Zazakowny, K. Szczubiałka and M. Nowakowska, Biomacromolecules, 2008, 9, 3127–3132 CrossRef PubMed.
- R. Harris, E. Lecumberri and A. Heras, Mar. Drugs, 2010, 8, 1750–1762 CAS.
- J. Karnchanajindanun, M. Srisa-ard and Y. Baimark, Carbohydr. Polym., 2011, 85, 674–680 CrossRef CAS.
- Y. Zhang, Y. Yang and T. Guo, Carbohydr. Polym., 2011, 83, 2016–2021 CAS.
- M. Luo, H. Peng, Z. Deng, Z. Yin, Q. Zhao and H. Xiong, Int. J. Food Eng., 2015, 11, 323–333 CrossRef CAS.
- E. Esparza-Flores, F. Dias Cardoso, L. Bertoldo Siquiera, P. Santagapita, P. Hertz and R. Rodrigues, Int. J. Biol. Macromol., 2023, 250, 126234 CAS.
- K. Gabov, T. Oja, T. Deguchi, A. Fallarero and P. Fardim, Cellulose, 2016, 24, 641–658 CrossRef.
- J. Trygg, P. Fardim, M. Gericke, E. Mäkilä and J. Salonen, Carbohydr. Polym., 2013, 93, 291–299 CAS.
- J. Boyd, Celluloid: The Eternal Substitute, 2011, https://www.sciencehistory.org/distillations/celluloid-the-eternal-substitute, (Visited on 26/06/2024) Search PubMed.
- J. OBrien, L. Torrente-Murciano, D. Mattia and J. Scott, ACS Sustainable Chem. Eng., 2017, 5, 5931–5939 Search PubMed.
- C. Brigham, Green Chem.: an Inclusive Approach, Elsevier, Amsterdam, 2017 Search PubMed.
- M. Goh and G. Tae, Carbohydr. Polym., 2022, 280, 119026 CAS.
- Y. Tanaka, N. Fukuda, N. Ranaivoarimanana, M. Hatakeyama and T. Kitaoka, Bioresources, 2023, 18, 1482–1492 CAS.
- C. Callaghan, D. Califano, M. Gomes, H. de Carvalho, K. Edler and D. Mattia, ACS Sustain. Chem. Eng., 2023, 11, 4749–4758 CAS.
- A. Omer, G. Elgarhy, G. El-Subruiti, E. El-Monaem and A. Eltaweil, Carbohydr. Polym., 2023, 311, 120771 CAS.
- A. Eltaweil, G. Elgarhy, G. El-Subruiti and A. Omer, Int. J. Biol. Macromol., 2020, 154, 307–318 CrossRef CAS PubMed.
- J. Kjesbu, D. Zaytseva-Zotova, S. Sämfors, P. Gatenholm, C. Troedsson, E. Thompson and B. Strand, Carbohydr. Polym., 2022, 286, 119284 CrossRef CAS PubMed.
- I. Bravo, L. Viejo, C. de Los Rios, E. Garcia-Frutos and M. Darder, Int. J. Biol. Macromol., 2023, 234, 123765 CrossRef CAS PubMed.
- J. Hua, R. Meng, T. Wang, H. Gao, Z. Luo, Y. Jin, L. Liu and J. Yao, Fibers Polym., 2019, 20, 794–803 CrossRef CAS.
- S. Nadiratuzzahra and D. Tristantini, AIP Conf. Proc., 2020, 2223, 050001 CrossRef CAS.
- S. Park, Y. Oh, J. Yun, E. Yoo, D. Jung, K. Oh and S. Lee, Cellulose, 2020, 27, 2757–2773 CrossRef CAS.
- G. Zavala, M.-P. Ramos, A. Figueroa-Valdés, P. Cistemas, U. Wyneken, M. Hernandez, P. Toa, B. Salmons, J. Dangerfield, W. Gunzberg and M. Khoury, Front. Pharmacol, 2020, 11, 679 CrossRef CAS PubMed.
- M. Gomes, C. Callaghan, A. Mendes, K. Edler, D. Mattia, Q. van Lier and H. de Carvalho, ACS Agric. Sci. Technol., 2022, 2, 340–348 CAS.
- R. Wool and X. Sun, Bio-Based Polymers and Composites, Academic Press, Cambridge, 2005 Search PubMed.
- B. Kupikowska-Stobba and D. Lewinska, Biomater. Sci., 2020, 8, 1536–1574 CAS.
- J. Zhou and L. Zhang, Polym. J., 2000, 32, 866–870 CrossRef CAS.
- L. Zhang, D. Ruan and S. Gao, J. Polym. Sci., 2002, 40, 1521–1529 CAS.
- P. Vandezande, L. Gevers, J. Vermant, J. Martens, P. Jacobs and I. Vankelecom, Chem. Mater., 2008, 20, 3457–3465 CAS.
- R. Sescousse and T. Budtova, Cellulose, 2009, 16, 417–426 CAS.
- S. Mohamed, K. Ganesan, B. Milow and L. Ratke, RSC Adv., 2015, 5, 90193–90201 RSC.
- A. McMackin, V. Banville and S. Cardinal, J. Polym. Sci., 2024, 62, 2270–2287 CrossRef CAS.
- C. Callaghan, J. L. Scott, K. Edler and D. Mattia, J. Colloid Interface Sci., 2022, 627, 1003–1010 CrossRef CAS PubMed.
- E. Ekanem, A. Sabirova, C. Callaghan, J. L. Scott, K. Edler, S. Nunes and D. Mattia, J. Membr. Sci., 2022, 2, 100024 Search PubMed.
- D. Lavanya, P. Kulkarni, M. Dixitt, P. Raavi and L. Krishna, Int. J. Drug Dev. Res., 2011, 2, 19–38 Search PubMed.
- M. Tausif, A. Jabbar, M. Naeem, A. Basit, F. Ahmad and T. Cassidy, Text. Prog., 2019, 50, 1–66 Search PubMed.
- M. Mujtaba, L. Fernandes Fraceto, M. Fazeli, S. Mukherjee, S. Savassa, G. Araujo de Medeiros, A. do Espírito Santo Pereira, S. Mancini, J. Lipponen and F. Vilaplana, J. Clean. Prod., 2023, 402, 136815 CrossRef CAS.
- P. Mishra, R. Wimmer and T. Gregor, Bioresources, 2017, 12, 107–116 CAS.
- M. Garnett, H. Kumar, B. Beckingham and S. Alexander, RSC Sustainability, 2024, 2, 170–178 RSC.
- R. Riseh, M. Vazvani, M. Hassanisaadi and V. Thakur, Ind. Crops, 2024, 208, 117904 CrossRef CAS.
- C. Rovera, D. Carullo, T. Bellesia, D. Büyükta, M. Ghaani, E. Caneva and S. Farris, Front. Sustain. Food Syst., 2023, 6, 1087867 CrossRef.
- E. Venkatanagaraju, N. Bharathi, R. Sindhuja, R. Cowdhury and Y. Sreelekha, in Extraction and Purification of Pectin from Agro-Industrial Wastes, IntechOpen, London (UK), 2019, book section 2, p. 85585 Search PubMed.
- C. Mellinas, M. Ramos, A. Jiménez and M. Garrigos, Materials, 2020, 13, 673 CrossRef CAS PubMed.
- K. Gutierrez-Alvarado, R. Chacon-Cerdas and R. Starbird-Perez, Chemistry, 2022, 4, 121–136 CrossRef CAS.
- A. Chai, K. Schmidt, G. Brewster, L. Xiong, B. Church, T. Wahl, S. Sadabadi and W. Zhang, Gels, 2023, 9, 707 CrossRef CAS PubMed.
- S. Molino, J. Henares and L. Gomez-Mascaraque, Food Struct., 2022, 32, 100256 CrossRef CAS.
- D. Ogonczyk, M. Siek and P. Garstecki, Biomicrofluidics, 2011, 5, 13405 CrossRef CAS PubMed.
- R. Sun, Z. Lv, Y. Wang, Y. Gu, Y. Sun, X. Zeng, Z. Gao, X. Zhao, Y. Yuan and T. Yue, Int. J. Biol. Macromol., 2024, 264, 130543 CAS.
- I. Osojnik Crnivec, K. Istenic, M. Skrt and N. Ulrih, Food Funct., 2020, 11, 1467–1477 RSC.
- M. Bekhit, L. Sanchez-Gonzalez, G. Messaoud and S. Desobry, J. Food Eng., 2016, 180, 1–9 CrossRef CAS.
- LibreTexts, Starch and Cellulose, 2019, https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Map%3A_Organic_Chemistry_(Smith)/Chapter_05%3A_Stereochemistry/5.01_Starch_and_Cellulose, (Visited on 26/06/2024) Search PubMed.
- A. Munoz-Bonilla, C. Echeverria, A. Sonseca, M. Arrieta and M. Fernandez-Garcia, Materials, 2019, 12, 641–691 CrossRef CAS PubMed.
- B. Vedha Hari, T. Praneetha, T. Prathyusha, K. Mounika and D. Ramya Devi, J. Adv. Pharm. Technol. Res., 2012, 3, 182–187 CrossRef CAS PubMed.
- A. Jha and A. Bhattacharya, Asian J. Pharm., 2009, 299–303 CrossRef.
- A. Okunlola, O. Odeku, R. Patel and J. Excip, Food Chem., 2012, 3, 19–25 Search PubMed.
- J. Kozlowska, W. Prus and N. Stachowiak, Int. J. Biol. Macromol., 2019, 129, 952–956 CrossRef CAS PubMed.
- N. Rani, G. Kaur, S. Kaur, S. Upadhyay and M. Tripathi, Int. J. Biol. Macromol., 2023, 15, 124381 CrossRef PubMed.
- Y.-Y. Fang, L.-J. Wang, D. Li, B.-Z. Li, B. Bhandari, X. Chen and Z.-H. Mao, J. Am. Sci., 2008, 74, 379–384 CAS.
- D. Dehghan-Baniani, P. Zahedifar, R. Bagheri and A. Solouk, Starch, 2019, 71, 1800305 Search PubMed.
- E. Hopkins, T. Sanvictores and S. Sharma, Physiology, Acid-Base Balance, 2022, https://www.ncbi.nlm.nih.gov/books/NBK507807/, (Visited on 09/01/2024) Search PubMed.
- Y. Luo, F. Ni, M. Guo, J. Liu, H. Chen, S. Zhang, Y. Li, G. Chen and G. Wang, Food Sci. Technol., 2022, 42, e126421 Search PubMed.
- L. Bebiano, R. Presa, F. Vieira, B. Lourenço and R. Pereira, Biomimetics, 2024, 9, 228 CAS.
- NCBI, PubChem, Explore Chemistry Compound Summary, 2024, https://pubchem.ncbi.nlm.nih.gov/, (Visited on 26/06/2024).
- J. Perez Bravo and N. François, J. Polym. Environ., 2020, 28, 2681–2690 CAS.
- O. Olayemi, Y. Apeji and C. Isimi, J. Pharm. Innovation, 2022, 17, 366–375 Search PubMed.
- BÜCHI, Encapsulator B-395 Pro, 2020 Search PubMed.
- J.-H. Lee, S.-M. You, K. Luo, J.-S. Ko, A.-H. Jo and Y.-R. Kim, Nanomaterials, 2021, 11, 532 CAS.
- K. Luo, K. Wang, M.-C. Lim, K.-B. Jeong and Y.-R. Kim, ACS Sustain. Chem. Eng., 2022, 10, 10268–10274 CAS.
- K. Lee and D. Mooney, Prog. Polym. Sci., 2012, 37, 106–126 CAS.
- S. Leslie, A. Nicolini, G. Sundaresan, J. Zweit, B. Boyan and Z. Schwartz, J. Mater. Chem. B, 2016, 4, 3411–3622 Search PubMed.
- F. Moghtader, S. Egri and E. Piskin, Artif. Cells, Nanomed. Biotechnol., 2017, 45, 357–363 CAS.
- S. Boi, N. Rouatbi, E. Dellacasa, D. Di Lisa, P. Bianchini, O. Monticelli and L. Pastorino, Int. J. Biol. Macromol., 2020, 156, 454–461 CAS.
- V. Dadwal, R. Joshi and M. Gupta, LWT, 2021, 152, 112290 CAS.
- N. Stachowiak, J. Kowalonek, J. Kozlowska and A. Burkowska-But, Polymers, 2023, 15, 2568 CAS.
- M. Rinaudo, Polym. Int., 2007, 57, 397–430 Search PubMed.
- S. Rotman, V. Post, A. Foster, R. Lavigne, J. Wagemans, A. Trampuz, M. Gonzalez Moreno, W.-J. Metsemakers, D. Grijpma, R. Richards, D. Eglin and T. Moriarty, J. Drug Deliv. Sci. Technol., 2023, 79, 103991 CAS.
- M. Ali, S. Kwak, J. Byeon and H. Choi, J. Funct. Biomater., 2023, 14, 403 CAS.
- O. Khanna, J. Larson, M. Moya, E. Opara and E. Brey, J. Vis. Exp., 2012, 66, e3388 Search PubMed.
- A. Eltaweil, M. Ahmed, G. El-Subruiti, R. Khalifa and A. Omer, Arab. J. Chem., 2023, 16, 104533 CrossRef CAS.
- S. Liang, W. Cai, C. Dang, X. Peng, Z. Luo and X. Wei, J. Environ. Chem. Eng., 2023, 11, 109317 CrossRef CAS.
- E. Julaeha, W. Puspita, N. Permadi, A. Harja, S. Nurjanah, T. Wahyudi and J. Al-Anshori, Carbohydr. Polym., 2024, 7, 100406 CAS.
- D. Yu, Z. Dong, H. Lim, Y.-F. Chen, Z. Ding, N. Sultana, J. Wu, B. Qin, J. Cheng and W. Li, RSC Adv., 2019, 9, 11101 RSC.
- S. Shaikh and A. Barik, J. Chem. Sci., 2023, 35, 39 Search PubMed.
- B. Chen, L. Benavente, M. Chittò, J. Wychowaniec, V. Post, M. D'Este, C. Constant, S. Zeiter, W. Feng, M. González Moreno, A. Trampuz, J. Wagemans, J. Onsea, R. Richards, R. Lavigne, T. Moriarty and W.-J. Metsemakers, J. Contr. Release, 2023, 364, 159–173 CAS.
- S. Somo, O. Khanna and E. Brey, Methods Mol. Biol., 2016, 1479, 217–224 Search PubMed.
- N. Uyen, Z. Hamid, N. Tram and N. Ahmad, Int. J. Biol. Macromol., 2016, 153, 1035–1046 CrossRef PubMed.
- A. A. Elzatahry, E. A. Soliman, M. S. Mohy Eldin and M. Elsayed Youssef, J. Am. Sci., 2010, 6, 846–851 Search PubMed.
- M. Abdo, M. Abdel-Hamid, I. El-Sherbiny, G. El-Sherbiny and E. Abdel-Al, Bioresour. Technol., 2023, 21, 101300 CrossRef CAS.
- N. Selvasudha, R. Goswami, M. Subi, S. Rajesh, K. Kishore and H. Vasanthi, Carbohydr. Polym., 2023, 6, 100342 CAS.
- D. McHugh, A Guide to the Seaweed Industry, Fao fisheries report, 2003 Search PubMed.
- H. Sattar, A. Aman and S. Qadar, Int. J. Biol. Macromol., 2018, 116, 917–922 CrossRef PubMed.
- K. Nayak, M. Moshra and G. Verma, Int. J. Adv. Pharm., 2017, 7, 11–15 Search PubMed.
- H. Chen, P. Wu, H. Xu and C. Wang, Front. Bioeng. Biotechnol., 2021, 9, 746609 CrossRef PubMed.
- T. Kuroiwa, T. Katsumata, Y. Sukeda, S. Warashina, I. Kobayashi, K. Uemura and A. Kanazawa, Jpn. J. Food Eng., 2016, 17, 11–19 CrossRef.
- M. Nawaz, A. Karim, A. Aman, R. Marchetti, S. Qader and A. Molinaro, Bioprocess Biosyst. Eng., 2015, 38, 631–638 CAS.
- A. Khater, O. Abdelrehim, M. Mohammadi, M. Azarmanesh, M. Janmaleki, R. Salahandish, A. Mohamad and A. Sanati-Nezhad, Lab Chip, 2020, 20, 2175–2187 CAS.
- C. Ge, Y. Hu, F. Zhang, Y. Lv and T. Tan, J. Sep. Sci., 2014, 37, 3253–3259 CAS.
- Z. Chen, Y. Xu, Z. Xiao, Y. Zhang, H. Weng, Q. Yang, Q. Xiao and A. Xiao, LWT, 2023, 181, 114751 CAS.
- S. Konnova and R. Fakhrullin, J. Bionanosci., 2017, 7, 75–77 Search PubMed.
- T. Nguyen-Le, X. Zhao, M. Bachmann, P. Ruelens, J. de Visser and L. Baraban, Micromachines, 2023, 14, 645 Search PubMed.
- L. Zhao, Y. Huang, K. Zhu, Z. Miao, J. Zhao, X. Che, D. Hao, R. Zhang and G. Ma, Eng. Life Sci., 2020, 20, 504–513 CAS.
- D. Gregurec, S. Velasco-Lozano, S. Moya, L. Vazquez and F. Lopez-Gallego, Soft Matter, 2016, 12, 8718–8725 CAS.
- N. Zoratto and P. Matricardi, in Semi-IPNs and IPN-Based Hydrogels, Woodhead, Cambridge, 2018, book section 4, pp. 91–124 Search PubMed.
- C. De Groot, M. Van Luyn, W. Van Dijk-Wolthuis, J. Cadée, J. Plantinga, W. Den Otter and W. Hennink, Biomaterials, 2001, 22, 1197–1203 CAS.
- J. Zhang, T. Zheng, S. Helalat, M. Yesibolati and Y. Sun, J. Colloid Interface Sci., 2024, 666, 603–614 CAS.
- G. Ye, G. Li, C. Wang, B. Ling, R. Yang and S. Huang, Carbohydr. Polym., 2019, 207, 218–223 CAS.
- T. Benhalima and H. Ferfera-Harrar, Int. J. Biol. Macromol., 2019, 132, 126–141 CAS.
- S. Lanouar, R. Aid-Launais, A. Oliveira, L. Bidault, B. Closs, M.-N. Labour and D. Letourneur, J. Mater. Sci. Mater. Med., 2018, 29, 77 CrossRef PubMed.
- R. Stenekes, A. Loebis, C. Fernandes, D. Crommelin and W. Hennink, Int. J. Pharm., 2001, 214, 17–20 CrossRef CAS PubMed.
- J.-T. Kim, D. Lee, T.-H. Kim, Y.-S. Song and N.-T. Cho, Met. Mater. Int., 2014, 20, 555–563 CrossRef CAS.
- C. Boeriu, J. Springer, F. Kooy, L. Van Den Broek and G. Eggink, Int. J. Carbohydr. Chem., 2013, 624967 Search PubMed.
- X. Li, T. Cui, W. Zhang, Z. Zhai, F. Wu, Y. Zhang, M. Yang, W. Zhong and W. Yue, Colloids Surf., B, 2020, 196, 111281 CrossRef CAS PubMed.
- S. Uthaman, S. Zheng, J. Han, Y. Choi, S. Cho, V. Nguyen, J.-O. Park, S.-H. Park, J.-J. Min, S. Park and I.-K. Park, Adv. Healthcare Mater., 2016, 5, 288–295 CrossRef CAS PubMed.
- M. Hamilton, S. Harrington, P. Dhar and L. Stehno-Bittel, ACS Biomater. Sci. Eng., 2021, 7, 3754–3763 CAS.
- J. Amirian, T. Van, S.-H. Bae, H.-I. Jung, H.-J. Choi, H.-D. Cho and B.-T. Lee, Int. J. Biol. Macromol., 2017, 105, 143–153 CrossRef CAS PubMed.
- R. Jenjob, H.-P. Nguyen, M.-K. Kim, Y. Jiang, J. Kim and S.-G. Yang, Biomacromolecules, 2021, 22, 4138–4145 CrossRef CAS PubMed.
- Y.-J. Seong, G. Lin, B. Kim, H.-E. Kim, S. Kim and S.-H. Jeong, ACS Omega, 2019, 4, 13834–13844 CrossRef CAS PubMed.
- J. Amirian, P. Makkar, G. Lee, K. Paul and B. Lee, Mater. Lett., 2020, 272, 127830 CrossRef CAS.
- P. Moslemy, S. Guiot and R. Neufeld, Enzyme Microb. Technol., 2002, 30, 10–18 CAS.
- A. Adrover, P. Paolicelli, S. Petralito, L. Di Muzio, J. Trilli, S. Cesa, I. Tho and M. Casadei, Pharmaceutics, 2019, 11, 187 CAS.
- P. Gadzinski, A. Froelich, B. Jadach, M. Wojtylko, A. Tatarek, A. Bialek, J. Krysztofiak, M. Gackowski, F. Otto and T. Osmalek, Pharmaceutics, 2023, 15, 108 CrossRef CAS PubMed.
- S. Jana and K. Sen, Mater. Today: Proc., 2019, 11, 614–619 CAS.
- D. Coutinho, A. Ahari, N. Kachouie, M. Gomes, N. Neves, R. Reis and A. Khademhosseini, Biofabrication, 2012, 4, 035003 Search PubMed.
- H. Park, H. Kim, G.-Y. Kim, M.-Y. Lee, Y. Kim and S. Kang, J. Hazard. Mater., 2021, 406, 124752 CAS.
- K. Kuhn, F. e Silva, F. Netto and R. da Cunha, J. Food Eng., 2019, 247, 104–114 CAS.
- S. Jana, R. Pramanik, A. Nayak and K. Sen, J. Drug Deliv. Sci. Technol., 2022, 69, 103034 CAS.
| This journal is © The Royal Society of Chemistry 2025 |