
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Low-data machine learning models for predicting thermodynamic properties of solid–solid phase transformations in plastic crystals†
Tzu-Hsuan
Chao
a,
Alexander
Foncerrada
b,
Patrick J.
Shamberger
 *b and
Daniel P.
Tabor
*a
*b and
Daniel P.
Tabor
*a
aDepartment of Chemistry, Texas A&M University, College Station, TX 77843, USA. E-mail: daniel_tabor@tamu.edu
bDepartment of Material Science and Engineering, Texas A&M University, College Station, TX 77843, USA. E-mail: patrick.shamberger@tamu.edu
First published on 25th June 2025
Abstract
Plastic crystals, many of which are globular small molecules that exhibit transitions between rotationally ordered and rotationally disordered states, represent an important subclass of colossal barocaloric effect materials. The known set of plastic crystals is notably sparse, which presents a challenge to developing predictive thermodynamic models to describe new molecular structures. To predict the transformation entropy of plastic crystals, we developed a comprehensive database of tetrahedral plastic crystal molecules (neopentane analogs) and used several types of features, including chemical functional groups, molecular symmetry, DFT-calculated vibrational entropy, and energy decomposition analysis to train a machine learning model. To select the most relevant features, we used a correlation matrix to screen out highly correlated features and ran sure independence screening and sparsifying operator (SISSO) regression on the remaining features. The SISSO regression samples over combinatorial spaces, including operations and features, to find the relationship between material properties. Using a dataset of 49 plastic crystals and 37 non-plastic crystals based on a common tetrahedral geometry, we have demonstrated the effectiveness of this strategy. Furthermore, we applied this strategy to develop a regression model to predict transition entropy and enthalpy. The top 100 models from the operation space showed that the overall distribution of performance became narrower, sacrificing the top-performing model but avoiding the worst models. Using this approach, we identified the top-performing descriptors to further clarify the underlying mechanisms of the plastic crystal transformation.
1 Introduction
Modern refrigeration technologies rely largely on fluorinated vapor-phase refrigerants, which, when they escape into the atmosphere, can contribute significantly to climate change,1 as well as other potential health and environmental effects associated with per- and poly-fluoroalkyl substances (PFAS). In contrast, solid state caloric effect materials offer an alternative strategy to eliminate the use of vapor phase refrigerants.2 In ferroic caloric effect materials, the application of external fields such as magnetic fields,3 electric fields,4 and pressure,5,6 can induce a phase transition, which results in a change in the internal state variable (entropy or temperature) of the material, forming the basis for a refrigeration cycle. Among these, plastic crystal phases7–10 exhibit colossal barocaloric effects,5,6,11–13 resulting in a comparatively larger entropy change during phase transition per unit mass for an attainable field change. The required “fields” for phase transformations are generally quite high for their domain: 2 T (for magnetocaloric materials), 1 kV m−1 (for electrocaloric), or 700 MPa (for elastocaloric materials). In contrast, barocaloric materials usually only require about 200 MPa of pressure.5The mechanism of plastic crystal transformations14,15 in the model system neopentyl glycol (NPG) has been investigated using ab initio methods,16 demonstrating that intermolecular hydrogen bonds play a key role in regulating phase stability in NPG. Upon treatment with external pressure, these hydrogen bonds are further strengthened, influencing the thermodynamics of order-to-disorder transition. When the temperature increases, the rotational entropy contribution becomes larger, and the disordered cubic phase becomes more favorable in terms of ΔG. Other plastic crystals that lack hydrogen bonds (e.g., CCl4) show a similar mechanism, but the interactions among molecules are not as strong as in neopentyl glycol (NPG). These molecules have been shown to exhibit many orientations.17 While some orientations are more energetically favored, many of the others are still accessible in the disordered phase.
The broader goal of developing barocaloric phases with specifically targeted properties (transformation temperature, enthalpy of transition, etc.) requires a strategy to map the effect of molecular chemistry onto the corresponding phase transition behavior. Due to the large combinatorial space consisting of a parent globular molecule (e.g., NPG) with chemically allowable substituted functional groups, sequential synthesis and characterization of individual molecules is practically infeasible. Thus, developing an accurate predictive strategy offers significant advantages. To achieve this aim, one could turn toward machine learning techniques to both predict the properties of future plastic crystals and understand the thermodynamics in existing systems. Since the 1960s, 27 plastic crystal molecular types have been reported.10,18 Here, we seek to assess the predictive ability of trained models based on the sparse known set of tetrahedral plastic crystalline phases. A chief objective of this work is to determine whether this current data set is sufficient to predict whether a related unknown compound will exhibit a plastic crystalline state, as well as the magnitude of entropy and enthalpy changes and the equilibrium transformation temperature.
To address this question, we systematically assessed previously reported experimental data on tetrahedral carbon-centered small molecules, a class that includes a number of compounds (including methane analogs and neopentane analogs) known to exhibit plastic crystalline states, as well as compounds with similar geometries in which this state is not observed. To develop a machine learning model capable of predicting phase transition properties, we aim to use descriptors with high correlation to relevant properties. The descriptor design focuses on two key aspects: molecular shapes and interactions. Overall, we developed five descriptor calculation methods: group contribution analysis, molecular symmetry, hydrogen bond strength (both donors and acceptors), DFT-derived molecular properties, and intermolecular energy decomposition analysis terms.
Molecular symmetry has been shown to correlate strongly with the classification of plastic versus non-plastic crystals through interaction with environmental orientation.19 This symmetry also correlates with rotational entropy as described by the equation ΔSrotm = R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnσ.20 Given the transformation's reliance on rotational degrees of freedom, sphericity—derived from the moment of inertia tensor21—is included as a molecular symmetry descriptor. To quantify molecular interactions, a previously calculated empirical table correlating hydrogen bond strength with functional groups22 was utilized. Furthermore, descriptors such as single-molecule vibrational entropy, intermolecular interaction energy, and single-molecule volume, all calculated using DFT, were incorporated as molecular properties.
lnσ.20 Given the transformation's reliance on rotational degrees of freedom, sphericity—derived from the moment of inertia tensor21—is included as a molecular symmetry descriptor. To quantify molecular interactions, a previously calculated empirical table correlating hydrogen bond strength with functional groups22 was utilized. Furthermore, descriptors such as single-molecule vibrational entropy, intermolecular interaction energy, and single-molecule volume, all calculated using DFT, were incorporated as molecular properties.
Based on this training set, we evaluated the ability to quantitatively predict (1) the existence of solid–solid transitions into a plastic crystal state, and (2) the related thermodynamic properties of the solid–solid transition.23 To tackle the problem of high dimensionality from the molecular descriptors where only limited data are available, we utilized the sure-independence screening and sparsifying operator (SISSO) approach to explore the large combinatorial spaces that use operators (addition, subtraction, and multiplication) and all available features. In this study, we applied a feature selection strategy to tackle this low-data problem.24–27 The results show that the strategy of using a correlation matrix and SISSO is effective in extracting information from the original descriptors and in improving the predictive accuracy. Furthermore, interpreting the operation of the original descriptors could potentially guide us to the relationship between the descriptors.
2 Material and methods
In this work, the term descriptor is used when an individual parameter that is obtained from the chemical species or environment. The term feature is used when referring to the direct input to the machine learning model.2.1 Dataset parameters
The possible chemical space investigated in this study consists of small globular carbon-centered tetrahedral molecules, only a small number of which are observed to exhibit a rotationally disordered plastic crystalline state. We conducted an extensive literature search to identify the appropriate training and test sets for these systems. The resulting database, TetraPlastC5,6,10–13,28–46 consists of globular and tetrahedral molecules wherein the enthalpy of the solid–solid plastic crystalline transition is either comparable to or larger than the latent heat of melting. While there are other globular molecules that are proven plastic crystals (e.g., adamantane), these do not exhibit tetrahedral configurations, and thus, are considered out of the scope of this study. Additionally, we only considered relatively small functional groups (shown in Table 1) in the tetrahedral structure to reduce the steric variability.| Functional group | Donor element and strength | Acceptor element and strength |
|---|---|---|
| –CH3 | C: 0.5 | No acceptor |
| –C2H5 | C: 0.5 | No acceptor |
| –CH2OH | O: 4.1 | O: 6.3 |
| –COOH | O: 5.5 | O: 6.3 |
| –NH2 | N: 2.0 | N: 8.6 |
| –CH2NH2 | N: 2.0 | N: 8.6 |
| –NO2 | No donor | O: 5.5 |
| –X (X = F, Cl, Br, I) | No donor | F: 3.9, Cl: 3.10, Br: 2.90, I: 2.61 |
| –CH2X (X = F, Cl, Br, I) | C: 1.3 | F: 3.9, Cl: 3.10, Br: 2.90, I: 2.61 |
| –SH | S: 2.0 | S: 3.5 |
| –CHO | O: 0.9 | O: 5.5 |
| –C4H9 | C: 0.5 | No acceptor |
| –CONH2 | N: 4.5 | O: 8.3 |
Reviewing the identified plastic crystalline compounds, it was observed that the dataset collected is biased toward compounds that contain hydroxyl, methoxy, and alkyl groups. This occurrence is likely due to the wide utilization of these compounds in synthesis, for their stability in ambient environments and their presence in the most well-studied plastic crystalline compounds such as NPG. 86 molecules with tetrahedral-like structures were collected from the literature, including compounds which had only a single type of functional group (e.g., neopentane), and increasingly complex compounds which had two different functional groups (e.g., NPG), and three or even four different functional groups (e.g., 2-methyl-2-butanethiol, which is observed to exhibit a plastic crystal transition47).
The plastic crystals (49) in the dataset include (1) phases that have direct experimental evidence of rotational disorder (e.g., electrical impedance spectroscopy), as well as (2) phases that are calorimetrically determined to have large changes in configurational entropy at this solid–solid transformation relative to the melting transformation (as defined by dSs–s − dSs–l). In contrast, molecules that are determined to “not exhibit” a plastic crystal state (37) included (1) compounds that present data (e.g., temperature-dependent heat capacity data) which conclusively demonstrate the lack of an additional solid–solid phase transformation over the relevant temperature range of interest, (2) phases which exhibit low-entropy transitions from one solid crystalline state to another solid crystalline state, and (3) studies which present thermophysical data (e.g., enthalpies of fusion) that would have made it more likely than not to observe a solid–solid plastic crystal transition if one exist near the melting transition. Compounds with limited reported information e.g., only low-temperature single crystal structure determination, or room-temperature spectroscopy information) were labeled as “inconclusive” (14) and were not included as part of our known dataset. 84 molecules were identified to have no calorimetric data and thus, were not included into our dataset. The summary of the dataset, classification labels, and sources are provided in our GitHub repository (link provided in the ESI†).
2.2 Features
Five categories of descriptors of small molecules are considered, as follows: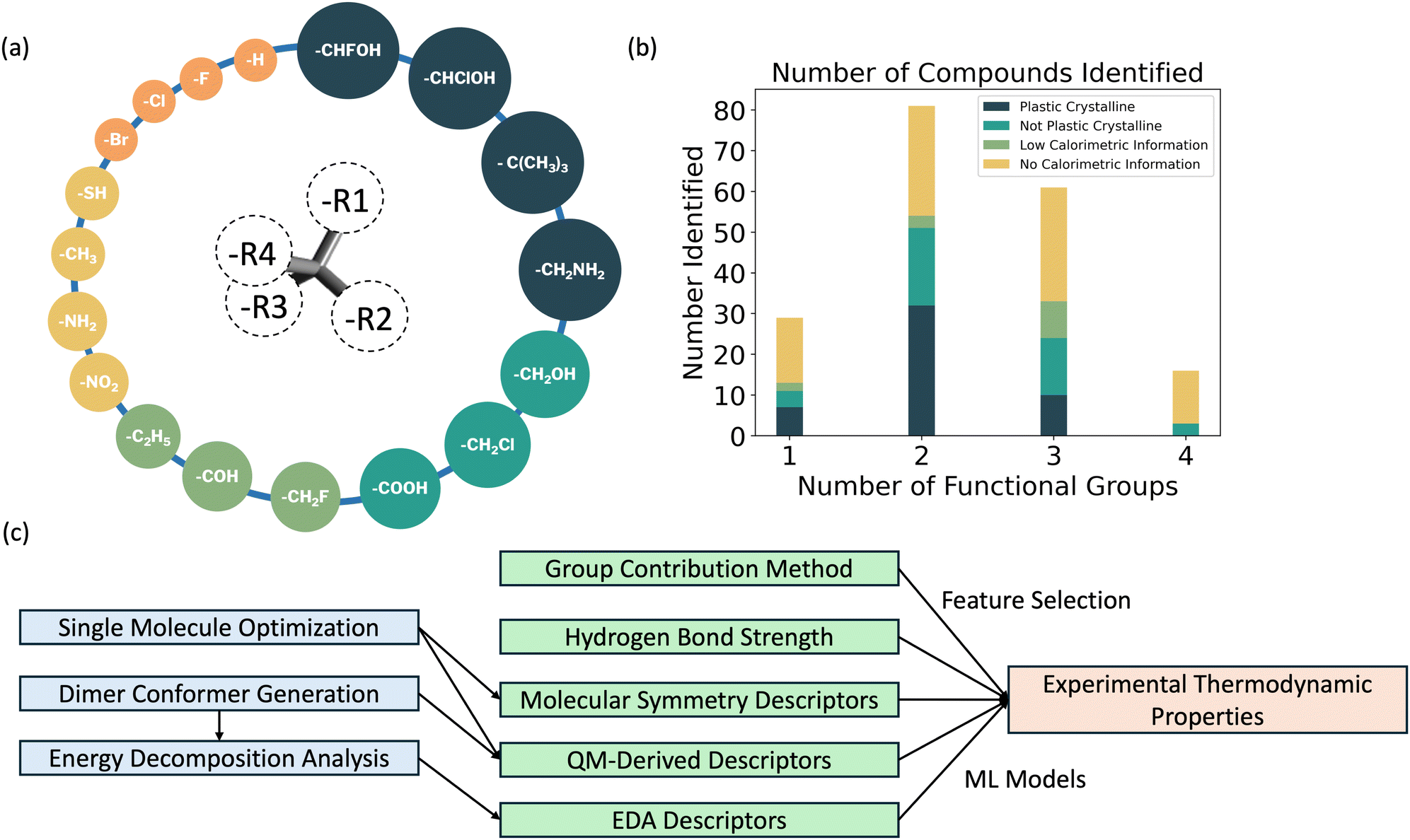 | ||
| Fig. 1 (a) Illustration of an idealized tetrahedral-like molecule (b) database sparsity (c) calculation workflow. | ||
Group contribution methods have been shown in other applications to predict the melting point and boiling point of aromatic compounds,48 as well as enthalpies and entropies of fusion of broad sets of low molecular weight organic compounds.49–51 They also form the basis for many cheminformatics-based models on organic molecule solubility, synthesizability, and other applications.52,53
 ,
,  where M = mass and n = 4). All bonds and angles considered are described in Fig. 1(a).
where M = mass and n = 4). All bonds and angles considered are described in Fig. 1(a).
| Identifiers | Sampling | |||||
|---|---|---|---|---|---|---|
| ΔS | ΔH | T tr | T m | |||
| a | –H | Hydrogen | 1.9 | −0.5 | 10.9 | 19.4 |
| b | –CH3 | Methyl | 6.7 | 1.0 | 44.6 | 64.0 |
| c | –C2H5 | Ethyl | 0.3 | −4.6 | 14.4 | 2.3 |
| d | –CH2OH | Hydroxymethyl | 19.1 | 7.6 | 100.9 | 117.9 |
| e | –CHFOH | Hydroxyfluoromethyl | −0.7 | −3.4 | 38.4 | 124.4 |
| f | –CHClOH | Hydroxychloromethyl | 11.8 | 0.4 | 31.4 | 56.1 |
| g | –COOH | Carboxyl | 20.3 | 10.8 | 153.2 | 162.6 |
| h | –NH2 | Amino | 26.7 | 8.7 | 97.2 | 80.6 |
| i | –CH2NH2 | Methylamine | 19.1 | 5.5 | 66.5 | 67.8 |
| j | –NO2 | Nitro | 10.6 | 3.6 | 81.9 | 87.4 |
| k | –F | Fluoro | 5.1 | 0.4 | 19.1 | 22.4 |
| l | –Cl | Chloro | 7.2 | 1.6 | 56.9 | 59.3 |
| m | –Br | Bromo | 5.7 | 1.8 | 78.5 | 88.6 |
| o | –CH2F | Fluoromethyl | 11.1 | 2.4 | 58.8 | 89.8 |
| p | –CH2Cl | Chloromethyl | 6.1 | 1.7 | 61.1 | 69.9 |
| r | –SH | Thiol | 21.7 | 5.7 | 36.6 | 60.6 |
| v | –CHO | Aldehyde | 6.3 | 1.7 | 50.0 | 79.9 |
| z | –C4H9 | Butyl | 113.2 | 2.9 | 70.8 | 133.0 |
2.3 Downselection of features
Once all descriptors were evaluated for the molecules in this database, we wanted to investigate the limit of informative linear regressions with discrete descriptors. To achieve this, an F-test was conducted on two linear regression models to select the top five descriptors. The first model utilized the group contribution method as input to predict transformational entropy, while the second model relied on all other descriptors. The downselection in the group contribution model aimed to identify the functional groups most critical for prediction. Meanwhile, the other descriptors, being structure-related, were analyzed to determine the key factors influencing the transformation. Reducing the number of features to five in both models also helped to prevent overfitting of the training data.3 Results and discussion
3.1 Classification of plastic crystals
We first consider the classification problem. Classifying whether a particular compound exhibits a plastic crystalline phase can help screen candidate molecules, and prioritize certain compounds over others. Therefore, we collected other non-plastic crystal tetrahedral-like molecules into our dataset for classification. The final set of tetrahedral molecules that do not contain a plastic crystal phase contains 37 molecules. Subsequently, we constructed a correlation matrix and then employed SISSO to obtain sets of two features from combinations of the descriptors. In order to avoid overfitting, we conducted tests using different numbers of allowed operators and descriptors. The results indicated that reducing the number of operations to three (seven was used in the regression) could be a sweet spot that balances between underfitting and overfitting (testing accuracy improves from 50% to 75%). Using a limited set of descriptors, we obtained a classification model with 70 percent accuracy (Fig. 2(b)–(e)). We also mapped the two features obtained from this model to see if a boundary exists (in feature space) that effectively separates molecules that do or do not exhibit plastic crystal states. From Fig. 2(b) and (c), it can be determined that plastic and non-plastic crystals can be separated using this set of features. The results above indicate that the SISSO-reducing technique helps with (1) identifying the key descriptors and (2) improving the performance compared to models that take all the descriptors as inputs. Within this low data regime, fewer inputs can avoid overfitting. | ||
| Fig. 2 (a) Distribution that shows descriptors that are obtained from SISSO for classification. The scores are plotted to show which descriptors tend to give better performance. Maximum hydrogen acceptor strength, charge transfer and dispersion are the top three descriptors. (b) and (c) By mapping the two features and labeled with types (blue for plastic crystal and red for non-plastic crystal). The overall mapping shows that there is a non-linear separation between two types of data. (d) and (e) One random seed train/test split classification result. The overall accuracy is around 0.7. | ||
3.2 Multiple linear regression analysis
:2, which is quite high for most machine learning models. As previously mentioned, the data set is highly biased towards hydroxymethyl and methyl compounds.
 | ||
| Fig. 3 (a) Tailored group contribution model of the entropy of transition for the TetraPlastC database. (b) The results of linear regression with all descriptors in the database. (c) The results for the downselected descriptor linear regressions of the entropy of the transition. | ||
Through consideration of the weighing strength of each functional group, this dataset is capable of determining functional groups that have a greater impact on the plastic crystal transition (Table 2). The larger the weighting factor, the more influential that specific functional group on that thermodynamic property. The more influential functional groups tend to be those that have strong hydrogen bonds. The results for using all 30 features (types of functional groups) from group contribution methods have high correlation factors. Additionally, the tailored group contribution method is inflexible for prediction as with this method, there are no ways to predict the properties of compounds containing functional groups that are not present or that are poorly sampled in the testing database. The present model provides reasonable predictions for key thermodynamic properties related to the functional groups tested, but cannot be extended to those outside the testing dataset, since the number of features is still considered too high compared to the size of the existing dataset.
3.3 Low data machine learning techniques: employing SISSO
Based on the results presented in the previous sections, it remains difficult to accurately predict the classification and thermodynamic properties of unknown molecules. In the following section, we assess the performance using the distribution of 100 randomly generated train/test split sets to avoid the risk of obtaining a good performance from a “lucky” set. Additionally, due to the limited amount of available data, we employed two separate dimensionality reduction techniques: (1) removing high correlation descriptors within each group of descriptors and (2) applying sure-independence screening and sparsifying operator (SISSO) techniques. By limiting the number of descriptors in the machine learning model, we can avoid the overfitting problem for testing set molecules. In order to maintain an appropriate number for each group, we used different criteria to screen out unnecessary descriptors. The highly correlated descriptors were determined by a value of the correlation coefficient, r ≥ 0.5 (r ≥ 0.1 for group contribution method threshold of 0.5 (0.1 for group contribution method to ensure that not too many descriptors are included). After screening out the highly correlated descriptors, we combined these descriptors into the SISSO task and selected two features as our final inputs for the regression (or classification model). To evaluate the validity of this strategy, we examined a subset of 30 molecules out of the 49 plastic crystal molecules contained in the full data set and checked the testing score distribution. By choosing 30 data points, we are able to conduct “every-set” testing as the number of combinations of hypothetical train/test split is computationally feasible (30!/(24!6!) = 593775). Plotting the testing score distribution can indicate the overall performance in case the sampled train/test split is occasionally the “easy” set to predict. We compared the testing score distribution between the original descriptors and the two features that were down-selected after the two screening steps (Fig. 4(a)–(e)). Additionally, the comparison is done on five groups of descriptors since these have been shown to have a high correlation with transformational entropy. The results of all five testing groups indicate that the dimensionality reduction strategy here improves the overall performance significantly (the peak shifts to the right for all five cases).
 | ||
| Fig. 4 To show the effectiveness of SISSO, we plotted testing distributions using the following categories of descriptors: (a) group contribution method, (b) molecular symmetry, (c) hydrogen bond strength, (d) Svib, volume, interaction energy, (e) energy decomposition analysis. The green bars use all descriptors, while the yellow bars are SISSO-reduced features. These are trained and tested on the sampled training and sets, and the distribution is based on every possible train/test split. (f) and (g) 70000 scores obtained from 100 train per test splits sampled from the whole dataset. The scores are calculated among 100 × 7 (7-fold cross-validation) × 100 (top 100 models from SISSO) sets. | ||
3.4 Predicting transformational entropy and enthalpy
As the subset of 30 sampled molecules described in the previous section demonstrated the validity of the strategy, we further applied this strategy to the complete dataset of 49 plastic crystal molecules. We excluded the division operation in the SISSO regression task, as the division operation could possibly lead to infinity if one of the descriptors in the data is 0, which will results in untrainable features. Since the number of potential train/test splits is very large, an every-set testing approach (where every possible train/test split is tested) is not feasible. We sampled 100 train per test splits to approximate the distribution of the scores over these possible splits. We performed a 7-fold cross-validation (because of the dataset size of 49). Furthermore, by using SISSO, we identified the top 100 combinations of descriptors from the original set. To summarize, we obtained 100 feature sets from one train/test split and an overall 70000 scores from the sampling (Fig. 4(f) and (g)). Here we plotted the distribution for all 100 splits. The peak in the distribution is around R2 0.5, which is already an improvement within the data size we have. In Fig. 5(c)–(f), predictions on two different splits are shown to show some edge cases in the distribution. The selected descriptors are listed below each panel. This result demonstrates that the performance is strongly dependent on which specific train/test split is initially selected. This strategy highlights that examining the overall correlation across the dataset is insufficient to determine the model's suitability for future screening. The down-selection approach can help pinpoint key descriptors, but its predictive power remains uncertain since it relies solely on fitting the entire dataset. Furthermore, for small and sparse datasets, evaluating models across various data splits is crucial to assess their overall predictive performance.
 | ||
| Fig. 5 One train/test split prediction obtained from the regression task. Selected features are described below the plot. (a) and (b) Distribution that shows features that are obtained from SISSO for regression. The scores are plotted to show which descriptors tend to give better performance. Interaction energy, maximum hydrogen acceptor strength, dispersion, and average distances between central carbon and four functional groups are the top four descriptors. (c) and (d) Examples showing a good prediction of both entropy and enthalpy. (e) and (f) Examples showing a poor prediction of both entropy and enthalpy. | ||
3.5 Chronological assessment
Over the past few decades, the total sum of knowledge on tetrahedral plastic crystal molecules has steadily increased. In this section, we considered this accumulation of knowledge and posed the question of, “How has our ability to accurately predict the properties of solid–solid phase transformations between rotationally ordered and disordered states evolved over time?” (if these models had existed throughout time). To answer this question, we re-trained the SISSO model at different points in time, including the cumulative body of knowledge that was known at that point in time, and evaluated the validity of that model on data that was collected after that point in time. This test evaluates whether certain periods of time introduced new functional groups that complicate the overall prediction or if the measured experimental values are relatively inaccurate. On the training side, we can observe the evolution of the relative performance of the model and whether a particular data set deteriorated the performance of the model. On the testing side, we can estimate the difficulty of a certain functional group not being within the training data, which we expect makes the overall training harder. We tested on both regression and classification tasks. For regression (Fig. 6(a) and (b)), the molecules associated with higher prediction losses in 1999 are highlighted above the plot. Specifically, 1,3-dichloro-2,2-dimethylpropane and 2,2-dichloropropane are identified as problematic molecules for these models. For classification, a significant drop in accuracy occurred in 2008, coinciding with the addition of two molecules containing carbonyl groups. In 2019, the accuracy declined again, primarily due to the very small size of the test set. The trends in entropy and enthalpy predictions indicate that the discovery of new plastic crystals and corresponding experimental data generally improved the models predictive performance. From Fig. 6, we can tell that predictions after 1994 and 2005 started to show a decreasing trend in the prediction loss, which corresponds to a training data size of 20 and 34. Additionally, after the chloro-containing compounds are added to the training set, the prediction loss decreases. | ||
| Fig. 6 Chronological plots for regression (a) and (b) and classification (c) that the data before a certain year was trained and tested on all the other. The molecules labeled are the ones that were added in that year, which decreased the overall performance. The yellow triangle, square, and circle dots correspond to training, testing, and total dataset size. | ||
3.6 Correlation between selected features and model performance
From the top 100 models and 100 sampled train/test splits, we generated 10000 combinations of features selected using SISSO. We then counted the occurrences of certain descriptors and color-coded them based on the corresponding model scores. The results are shown in the Fig. 5(a) and (b). Among these descriptors, maximum acceptor strength, charge transfer, and dispersion energies are the top-performing descriptors. All these terms are related to either hydrogen bond strength or molecular interactions between two molecules. As secondary ranking descriptors, geometry-related descriptors are also commonly selected, particularly those used for understanding the shape of the molecule.
We conclude that electronic interactions and molecular shapes are the top descriptors that correlate with transformational enthalpy and entropy, aligning with our observations using an f-test down-selection method. For commonly seen operating correlations, we identified two frequently observed feature operations:
(1) Maximum acceptor strength – center of mass and
(2) Center of mass × Edispersion
Based on these observations, we can conclude both molecular rotation and interaction between molecules are important to transformational entropy. Although the relationship between mass and dispersion energy is not entirely clear, their contribution to transformational entropy is significant enough to warrant inclusion in future training. Overall, by running 100 models, we identified the top candidates for future model applications. Compared to other thermodynamic property models, such as a predictive model for Tg,60–65 our model demonstrates several advantages. First, we present the performance distribution across multiple train/test splits, ensuring that the results are not biased by datasets that are easier to learn. Additionally, our model achieves moderate performance without incorporating melting temperature as a descriptor. According to the literature, melting points are highly correlated with the glass transition temperature, yet they are not easily accessible features. In contrast, our model benefits from using descriptors that can be computed theoretically, making it more suitable for candidate screening. Finally, while some previous models achieve R2 > 0.863,65 (with melting point as one of their features), our model shows a peak performance around 0.7 (70% accuracy), which is an improvement over earlier Tg prediction models.60–65
4 Conclusions
In conclusion, we have aggregated previously reported experimental ΔS data for all previously reported carbon-centered tetrahedral-like plastic crystal molecules. Using five types of molecular descriptors, including DFT calculations, we have demonstrated that both correlation matrix and SISSO approaches can effectively reduce feature dimensions and improve overall model performance. To show the effectiveness of this approach, with sampled set testing, the performance distribution is consistently right-shifted, regardless of which descriptors are used. We used the same strategy for classification, which resulted in an average of 0.7 accuracy. By plotting the score and selecting features from each model, we were able to obtain top performance descriptors. We have quantified the impact of structural descriptors that are essential for developing predictive models and identified key mechanisms underlying the plastic crystalline transition: (1) hydrogen bond strength and center of mass, and (2) center of mass and dispersion energy in the dimer configuration. These descriptors can be categorized into molecular interactions and molecular shape. Both hydrogen bonding and dispersion forces contribute to intermolecular interactions and are the dominant forces in plastic crystalline materials, as demonstrated in previous studies.16,17 Regarding the center of mass, previous research has explored how variations in the moment of inertia influence plastic crystal transitions, and our findings align with these studies. These simple regression models provide valuable insights for designing new plastic crystalline compounds with tailored thermal properties, expanding the scope of materials suitable for practical applications.Conflicts of interest
There are no conflicts to declare.Data availability
All code and data are available in the following Github repository: https://github.com/Tabor-Research-Group/TetraPlastiC.Acknowledgements
T.-H. Chao and D. P. Tabor acknowledge support from the Robert A. Welch Foundation, grant no. A-2049-20230405. P. J. Shamberger and A. Foncerrada acknowledge support from the Department of the Navy, Office of Naval Research under award no. N00014-22-1-2050. Portions of this research were conducted with high-performance research computing resources provided by Texas A&M University HPRC. The authors thank Chase Somodi for feedback and helpful discussions on the manuscript.Notes and references
- E. Molenbroek, M. Smith, N. Surmeli, S. Schimschar, P. Waide, J. Tait and C. McAllister, Saving and benefits of global regulations for energy efficient products. A “cost of non-world” study, European Commission, Directorate-General for Energy, 2015. Accessed 2025-06-14, https://energy.ec.europa.eu/publications/savings-and-benefits-global-regulations-energy-efficient-products-cost-non-world-study_en.
- S. Fähler and V. K. Pecharsky, MRS Bull., 2018, 43, 264–268 CrossRef.
- B. Shen, J. Sun, F. Hu, H. Zhang and Z. Cheng, Adv. Mater., 2009, 21, 4545–4564 CrossRef CAS.
- X. Moya, S. Kar-Narayan and N. D. Mathur, Nat. Mater., 2014, 13, 439–450 CrossRef CAS PubMed.
- B. Li, Y. Kawakita, S. Ohira-Kawamura, T. Sugahara, H. Wang, J. Wang, Y. Chen, S. I. Kawaguchi, S. Kawaguchi and K. Ohara, et al. , Nature, 2019, 567, 506–510 CrossRef CAS PubMed.
- P. Lloveras, A. Aznar, M. Barrio, P. Negrier, C. Popescu, A. Planes, L. Mañosa, E. Stern-Taulats, A. Avramenko and N. D. Mathur, et al. , Nat. Commun., 2019, 10, 1803 CrossRef CAS PubMed.
- W. Press, Science, 1980, 207, 880–881 CrossRef CAS PubMed.
- L. Staveley, Annu. Rev. Phys. Chem., 1962, 13, 351–368 CrossRef CAS.
- D. Bansal, J. Hong, C. W. Li, A. F. May, W. Porter, M. Y. Hu, D. L. Abernathy and O. Delaire, Phys. Rev. B, 2016, 94, 054307 CrossRef.
- E. F. Westrum Jr, Pure Appl. Chem., 1961, 2, 241–250 CrossRef.
- J. Font, J. Muntasell and E. Cesari, Mater. Res. Bull., 1995, 30, 839–844 CrossRef CAS.
- J. Li, D. Dunstan, X. Lou, A. Planes, L. Mañosa, M. Barrio, J.-L. Tamarit and P. Lloveras, J. Mater. Chem. A, 2020, 8, 20354–20362 RSC.
- Q. Ren, J. Qi, D. Yu, Z. Zhang, R. Song, W. Song, B. Yuan, T. Wang, W. Ren and Z. Zhang, et al. , Nat. Commun., 2022, 13, 2293 CrossRef CAS PubMed.
- C. Escorihuela-Sayalero, L. C. Pardo, M. Romanini, N. Obrecht, S. Loehlé, P. Lloveras, J.-L. Tamarit and C. Cazorla, npj Comput. Mater., 2024, 10, 13 CrossRef CAS.
- L. Schmidt, D. Van der Spoel and M.-M. Walz, ACS Phys. Chem. Au, 2022, 3, 84–93 CrossRef.
- F. Li, M. Li, X. Xu, Z. Yang, H. Xu, C. Jia, K. Li, J. He, B. Li and H. Wang, Nat. Commun., 2020, 11, 4190 CrossRef CAS PubMed.
- A. H.-T. Li, S.-C. Huang and S. D. Chao, J. Chem. Phys., 2010, 132, 024506 CrossRef PubMed.
- S. Das, A. Mondal and C. M. Reddy, Chem. Soc. Rev., 2020, 49, 8878–8896 RSC.
- W. Shen, J. Antonaglia, J. A. Anderson, M. Engel, G. van Anders and S. C. Glotzer, Soft Matter, 2019, 15, 2571–2579 RSC.
- R.-M. Dannenfelser and S. H. Yalkowsky, Ind. Eng. Chem. Res., 1996, 35, 1483–1486 CrossRef CAS.
- M. Wirth, A. Volkamer, V. Zoete, F. Rippmann, O. Michielin, M. Rarey and W. H. Sauer, J. Comput.-Aided Mol. Des., 2013, 27, 511–524 CrossRef CAS PubMed.
- D. Santos-Martins and S. Forli, J. Chem. Theory Comput., 2020, 16, 2846–2856 CrossRef CAS PubMed.
- F. Musil, S. De, J. Yang, J. E. Campbell, G. M. Day and M. Ceriotti, Chem. Sci., 2018, 9, 1289–1300 RSC.
- Y. Zhang and C. Ling, npj Comput. Mater., 2018, 4, 25 CrossRef.
- P. Xu, X. Ji, M. Li and W. Lu, npj Comput. Mater., 2023, 9, 42 CrossRef.
- R. Ouyang, E. Ahmetcik, C. Carbogno, M. Scheffler and L. M. Ghiringhelli, J. Phys.: Mater., 2019, 2, 024002 Search PubMed.
- A. L. Liu, R. Venkatesh, M. McBride, E. Reichmanis, J. C. Meredith and M. A. Grover, ACS Appl. Polym. Mater., 2020, 2, 5592–5601 CrossRef CAS.
- N. I. of Standards and Technology, NIST Computational Chemistry Comparison and Benchmark Databases, U.S. Department of Commerce Technical Report NIST Standard Reference Database Number 101 Release 22, May 2022, 2022.
- A. Aznar, P. Lloveras, M. Barrio and J. L. Tamarit, Eur. Phys. J.-Spec. Top., 2017, 226, 1017–1029 CrossRef CAS.
- A. Aznar, P. Lloveras, M. Barrio, P. Negrier, A. Planes, L. Mañosa, N. D. Mathur, X. Moya and J.-L. Tamarit, J. Mater. Chem. A, 2020, 8, 639–647 RSC.
- D. Chandra, W. Ding, R. A. Lynch and J. J. Tomilinson, J. Less-Common Met., 1991, 168, 159–167 CrossRef CAS.
- K. Kobashi and M. Oguni, J. Phys. Chem. B, 1999, 103, 7687–7694 CrossRef CAS.
- H. Enokido, T. Shinoda and Y.-i Mashiko, Bull. Chem. Soc. Jpn., 1969, 42, 84–91 CrossRef CAS.
- J. Hicks, J. Hooley and C. Stephenson, J. Am. Chem. Soc., 1944, 66, 1064–1067 CrossRef CAS.
- R. Andon, J. Counsell, D. Lee and J. Martin, J. Chem. Soc., Faraday Trans. 1, 1973, 69, 1721–1726 RSC.
- F. Meersman, B. Geukens, M. Wübbenhorst, J. Leys, S. Napolitano, Y. Filinchuk, G. Van Assche, B. Van Mele and E. Nies, J. Phys. Chem. B, 2010, 114, 13944–13949 CrossRef CAS PubMed.
- S. Hore, R. Dinnebier, W. Wen, J. Hanson and J. Maier, Z. Anorg. Allg. Chem., 2009, 635, 88–93 CrossRef CAS.
- A. B. Bazyleva, G. J. Kabo and A. V. Blokhin, Phys. B, 2006, 383, 243–252 CrossRef CAS.
- J. Font and J. Muntasell, J. Mater. Chem., 1995, 5, 1137–1140 RSC.
- D. Sake Gowda and R. Rudman, J. Chem. Phys., 1982, 77, 4666–4670 CrossRef.
- D. Sake Gowda and R. Rudman, J. Chem. Phys., 1982, 77, 4671–4677 CrossRef.
- D. Sake Gowda, N. Federlein and R. Rudman, J. Chem. Phys., 1982, 77, 4659–4665 CrossRef.
- N. Doshi, M. Furman and R. Rudman, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1973, 29, 143–144 CrossRef.
- P. Hall, Trans. Faraday Soc., 1971, 67, 556–562 RSC.
- J. N. Sherwood, The Plastically crystalline state: orientationally disordered crystals, John Wiley & Sons, 1979 Search PubMed.
- L. Silver and R. Rudman, J. Phys. Chem., 1970, 74, 3134–3139 CrossRef CAS.
- S. Kondo, Bull. Chem. Soc. Jpn., 1965, 38, 527–529 CrossRef CAS.
- P. Simamora and S. H. Yalkowsky, Ind. Eng. Chem. Res., 1994, 33, 1405–1409 CrossRef CAS.
- J. S. Chickos, C. M. Braton, D. G. Hesse and J. F. Liebman, J. Org. Chem., 1991, 56, 927–938 CrossRef CAS.
- F. Gharagheizi and G. R. Salehi, Thermochim. Acta, 2011, 521, 37–40 CrossRef CAS.
- K. Mansour and M. Korichi, Computer Aided Chemical Engineering, Elsevier, 2023, vol. 52, pp. 661–666 Search PubMed.
- F. Gharagheizi, A. Eslamimanesh, A. H. Mohammadi and D. Richon, Ind. Eng. Chem. Res., 2011, 50, 10344–10349 CrossRef CAS.
- E. Stefanis and C. Panayiotou, Int. J. Thermophys., 2008, 29, 568–585 CrossRef CAS.
- J. Wei, Ind. Eng. Chem. Res., 1999, 38, 5019–5027 CrossRef CAS.
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminf., 2011, 3, 1–14 Search PubMed.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng and X. Feng, et al. , Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- P. R. Horn and M. Head-Gordon, J. Chem. Phys., 2015, 143, 114111 CrossRef PubMed.
- J. McDonagh, T. van Mourik and J. B. Mitchell, Mol. Inf., 2015, 34, 715–724 CrossRef CAS PubMed.
- J. Zhang, M. Zhao, C. Zhong, J. Liu, K. Hu and X. Lin, Nanoscale, 2023, 15, 18511–18522 RSC.
- L. Tao, V. Varshney and Y. Li, J. Chem. Inf. Model., 2021, 61, 5395–5413 CrossRef CAS PubMed.
- T. Galeazzo and M. Shiraiwa, Environ. Sci.: Atmos., 2022, 2, 362–374 CAS.
- G. M. Casanola-Martin, A. Karuth, H. Pham-The, H. González-Daz, D. C. Webster and B. Rasulev, Commun. Chem., 2024, 7, 226 CrossRef CAS PubMed.
- G. Armeli, J.-H. Peters and T. Koop, ACS Omega, 2023, 8, 12298–12309 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sm00353a |
| This journal is © The Royal Society of Chemistry 2025 |