
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctional assessment of migration and adhesion to quantify cancer cell aggression†
Lauren E.
Mehanna
 a,
James D.
Boyd
b,
Chloe G.
Walker
a,
Adrianna R.
Osborne
a,
Martha E.
Grady
*b and
Brad J.
Berron
*a
a,
James D.
Boyd
b,
Chloe G.
Walker
a,
Adrianna R.
Osborne
a,
Martha E.
Grady
*b and
Brad J.
Berron
*a
aDepartment of Chemical and Materials Engineering, University of Kentucky, 177 F. Paul Anderson Tower, Lexington, KY 40506, USA. E-mail: brad.berron@uky.edu; Tel: +1 (859) 257-2791
bDepartment of Mechanical and Aerospace Engineering, University of Kentucky, 151 Ralph G. Anderson Building, Lexington, KY 40506, USA. E-mail: m.grady@uky.edu; Tel: +1 (859) 323-4107
First published on 17th March 2025
Abstract
During epithelial-to-mesenchymal transition (EMT), cancer cells lose their cell–cell adhesion junctions as they become more metastatic, altering cell motility and focal adhesion disassembly associated with increased detachment from the primary tumor and a migratory response into nearby tissue and vasculature. Current in vitro strategies characterizing a cell's metastatic potential heavily favor quantifying the presence of cell adhesion biomarkers through biochemical analysis; however, mechanical cues such as adhesion and motility directly relate to cell metastatic potential without needing to first identify a cell specific biomarker for a particular type of cancer. This paper presents a comprehensive comparison of two functional metrics of cancer aggression, wound closure migration velocity and cell detachment from a culture surface, for three pairs of epithelial cancer cell lines (breast, endometrium, tongue tissue origins). It was found that one functional metric alone was not sufficient to categorize the cancer cell lines; instead, both metrics were necessary to identify functional trends and accurately place cells on the spectrum of metastasis. On average, cell lines with low metastatic potential (MCF-7, Ishikawa, and Cal-27) were more aggressive through wound closure migration compared to loss of cell adhesion. On the other hand, cell lines with high metastatic potential (MDA-MB-231, KLE, and SCC-25) were on average more aggressive through loss of cell adhesion compared to wound closure migration. This trend was true independent of the tissue type where the cells originated, indicating that there is a relationship between metastatic potential and the predominate type of cancer aggression. Our work presents one of the first combined studies relating cell metastatic potential to functional migration and adhesion metrics across cancer cell lines from selected tissue origins, without needing to identify tissue-specific biomarkers to achieve success. Using functional metrics provides powerful clinical relevancy for future predictive tools of cancer metastasis.
1. Introduction
Cancer is the second leading global cause of death, contributing to nearly 10 million deaths per year in 2022.1 In the United States, cancer is the leading cause of death among people less than 85 years of age, with over 2 million new cancer diagnoses per year.2 Cancer metastasis is the multi-step process in which cancer cells detach and travel from the primary tumor site and deposit in secondary locations in the body, contributing upwards of 90% of these cancer deaths.3 The metastatic cascade encompasses 5 key steps: local invasion of tumor cells into adjacent tissue, intravasation into local vasculature, survival in circulation, extravasation at distant organs, and proliferation at these sites leading to colonization.4,5 Local invasion, the initial stage of metastasis, involves dysregulated cell proliferation and upregulated migration through the stroma towards blood or lymphatic vessels.4 Metastatic cells are known to lose their adhesion to other cells (cell–cell adhesion) and their environment (cell–matrix adhesion), detaching and circulating through the bloodstream to secondary organs in a process called intravasation.6–8Key contributions have been made in cancer metastasis research in vitro through identifying cell adhesion biomarkers, such as cadherins, integrins, selectins, and proteoglycans, and studying their migratory behavior.9–14 The quantification of these biomarkers in relation to cell metastatic potential can be complex as they are tissue-specific and require knowledge about the tissue origin to be successful. This work develops a label-free tissue-agnostic quantification method that relates cell metastatic potential to functional metrics rather than tissue-specific biomarker expression. Functional metrics of interest include quantifying the differences in cell adhesion between cancer cells from a culture surface at various metastatic levels and comparing these differences in cell adhesion to their migratory capabilities. We seek to understand the in vitro characteristics of various epithelial cancer cell lines by studying these two metrics of cancer aggression that relate to the first two steps of the metastatic cascade: wound closure migration velocity (local invasion) and cell detachment from a culture surface (intravasation) (Fig. 1).
 | ||
| Fig. 1 Schematic representation of two metrics of cancer aggression– 1) migration into nearby tissue and 2) detachment of cancerous cells from the primary tumor site. | ||
We have chosen to study three pairs of human epithelial cell lines from various tumor tissue locations (breast, endometrium, tongue). Each pair contains a cell line with high and low metastatic potential, to directly compare their cell adhesion and migratory characteristics. While breast cancer cell lines are commonly researched for specific biomarkers and functional metrics related to metastatic potential, there is far less research available about cell lines originating from other tissues in the body. Therefore, we specifically chose to include a well-studied cell line pairing as well as additional cell lines from other tissues to create a comprehensive comparison of specific functional metrics.
Two commonly studied human breast cancer cell lines in regards to cell metastatic potential are MCF-7 and MDA-MB-231. MCF-7 is hormone dependent, with functional estrogen, progesterone, and glucocorticoid receptors. They are considered poorly aggressive and non-invasive, having low metastatic potential.15 MDA-MB-231 is hormone independent, also called a triple negative cell line, without any hormone receptors that can be targeted for treatment. MDA-MB-231 is highly aggressive and considered to have high metastatic potential compared to MCF-7.16–18
Endometrial cancer is classified by type – with type 1 representing a cell line with a high expression of hormone receptors (less aggressive) and type 2 having a low expression of hormone receptors (more aggressive). Ishikawa cells are a common type 1 endometrial cancer with low metastatic potential while KLE cells are a type 2 endometrial cancer with high metastatic potential.19,20
Oral squamous cell carcinomas (OSCCs) are a subset of head and neck squamous cell carcinoma and are often studied for invasion and metastasis into surrounding lymph tissue. Cal-27 and SCC-25 are both oral squamous cell carcinomas isolated from tongue tissue. Cal-27 is considered a non-metastatic cell line with low metastatic potential, while SCC-25 is a OSCC cell line with high metastatic potential.21,22 This study of three pairs of cancer cell lines provides a more robust relationship between cell migration and adhesion than current literature and identifies any dependency between these metrics and tissue origin.
A common baseline for observing cell invasiveness via 2-dimensional cell migration is a wound healing assay, also called a scratch assay, in which a confluent monolayer of cells is intentionally scratched to create a region free of cells.23,24 This region is then studied over time to observe how the remaining cells respond and migrate into the wound area to reestablish cell–cell connections. In comparison to transwell migration assays, in which cells are seeded on a culture insert and migrate through a semi-permeable membrane to a separate culture compartment, scratch assays are a simple, quick, and inexpensive means of measuring cell migration and wound healing in vitro.25,26 Scratch assays also allow for time-lapse imaging rather than strictly endpoint imaging, which is a major limitation in transwell assays.27,28 In scratch assays, cells that migrate faster into the scratched region have a greater wound closure velocity compared to cells that migrate slower.
Metastatic cancers are often associated with the epithelial-to-mesenchymal transition (EMT), in which tightly connected, immobile epithelial cells differentiate to a more migratory mesenchymal phenotype.29 This transition increases cell invasiveness, enhances cell resistance to apoptosis, and promotes reorganization of the cell cytoskeleton.30–32 A hallmark of EMT is the loss of cell polarity and cell adhesion junctions, contributing to tumor aggression through increased cell migration and detachment from the primary tumor site.33 We contribute to the current knowledge of metastatic cell adhesion beyond gene or protein expression by probing adhesion function through direct comparisons of cell detachment in vitro. Previous literature supports using differences in non-specific cell adhesion from tissue biopsies on untreated cell culture surfaces as a simplistic model for cell selection.34–37 We propose applying this type of adhesion-based system with cancerous cells of varying metastatic potential to determine quantitative differences in adhesive function. While cancer invasiveness has been explored using adhesion-based assays,38 direct comparisons of cell lines with varying metastatic potential across multiple tissue origins has yet to be fully developed. A microfluidic device is frequently employed to discern the varying strengths of cell adhesion, achieved by introducing fluid flow to cells attached to a culture surface.39 A parallel plate flow chamber is a widely accepted and well-developed microfluidic device that creates a unidirectional, single channel of fluid flow between two plates. A lateral shear force is induced on cells adhered to one of the plates, called the wall shear stress, and mimics the forces a cell experiences from blood flow in vivo.40,41 The cells detach from the surface when the shear force from fluid flow exceeds the adhesive force of the cells to the culture surface. Due to the loss of cell adhesion junctions during metastasis, we hypothesize that more metastatic cancer cell lines will have greater detachment from the surface compared to the less metastatic cancer cell lines.
The purpose of this study is to determine how the migration velocity and adhesion function of each individual cancer cell line relates to metastatic potential in head-to-head comparisons across cells of various tissue origins without biomarker identification. This purpose is motivated by the need to identify physical (rather than molecular), phenotypic (rather than genotypic) differences of cancer cell aggression.42 Thus, we provide quantitative metrics of physical processes across established cell lines from multiple tissue origins. According to EMT, as cancerous cells become increasingly metastatic, they transition to a more mesenchymal phenotype and lose their adhesion junctions; therefore, we hypothesize that cell lines traditionally described as having higher metastatic potential will have enhanced migratory and reduced adhesion characteristics when cultured as homogenous populations.29,33 We then expand our analysis to heterogenous populations consisting of high and low metastatic potential cell lines, as would be present in a given tumor.43 Since the EMT process has been confirmed in vivo, which contains variability in cell types, we predict that adhesion differences in the homogenous populations of low and high metastatic potential cell lines will also be present when the cells are co-cultured as heterogeneous populations.44,45
This work presents novel head-to-head comparisons of functional metrics among different cancer cell lines that have not been previously combined in this way. By comparing phenotypic differences between cell lines from different tissue origins, we can identify general trends between cell migration, cell adhesion, and metastatic potential. Our work provides a pathway for future predictive tools of metastatic potential based on functional metrics alone, with applications in additional tissues of interest to better categorize cells on the spectrum of metastasis.
2. Materials and methods
2.1 Cell culture
Three pairs of human cancerous cell lines were studied, with each pair containing a less invasive and more invasive cancer cell line. Breast cancer (mammary adenocarcinoma) cell lines studied were MCF-7 and MDA-MB-231. Tongue cancer (squamous cell carcinoma) cell lines studied were SCC-25 and Cal-27. Endometrial cancer (endometrium adenocarcinoma) cell lines studies were Ishikawa and KLE. MCF-7, MDA-MB-231, SCC-25, Cal-27, and KLE cell lines were obtained from ATCC. Ishikawa cell line was obtained from the laboratory of Dr Brittany E. Givens at the University of Kentucky and authenticated using ATCC STR Profiling services. The profiling results indicated a similar match to the STR profile for cell line Ishikawa 3-H-4 listed in Expasy. KLE cells were cultured in DMEM/F-12 (Thermo Fisher) media supplemented with 10% FBS (Corning) and 1% penicillin–streptomycin (VWR) while all other cell lines were cultured in DMEM (Millipore Sigma) media with 10% FBS and 1% penicillin–streptomycin. All cell lines were cultured at 37 °C with 5% CO2.2.2 Wound healing assay
Each cell line was seeded at 2.08 × 104 cells per cm2 in a 6-well culture plate (VWR) in their respective cell media and grown to confluence. Once confluent, a 20 μL pipette tip was used to scratch a vertical line down the middle of each well. The wells were washed once with 1× PBS (HyClone) to remove any floating cells, then replaced with the appropriate (serum-containing or serum-free) cell media. Images of the scratch were taken every 12 h on an Eclipse Ti-E (Nikon) inverted microscope for the first 48 h to determine the initial wound closure velocity of each cell type. The scratch was then imaged every 24 h thereafter until the wound was closed. Cell media exchange occurred at 48 h after plating, and every 24 h thereafter. ImageJ (NIH) MRI Wound Healing Tool was used to determine the scratch area at each timepoint for n = 5 representative images for each cell type. For images that did not have enough contrast between the cells and the background for the tool alone to recognize the scratch area, the following ImageJ commands were used to threshold the image prior to inputting into the MRI Wound Healing Tool: Find Edges, Sharpen, Threshold, Find Edges, and Invert LUT. The thresholding protocol was verified for n = 5 images with clear contrast and produced results within 5% of the output of the MRI Wound Healing Tool, indicating that the thresholding protocol was appropriate for analysis of images with poor contrast between the cells and the background (Fig. S1, ESI†).The wound closure migration velocity was calculated for each cell line in the first 12 h following the scratch using established quantification methods.23,46 The wound closure migration velocity was calculated using the following equation,
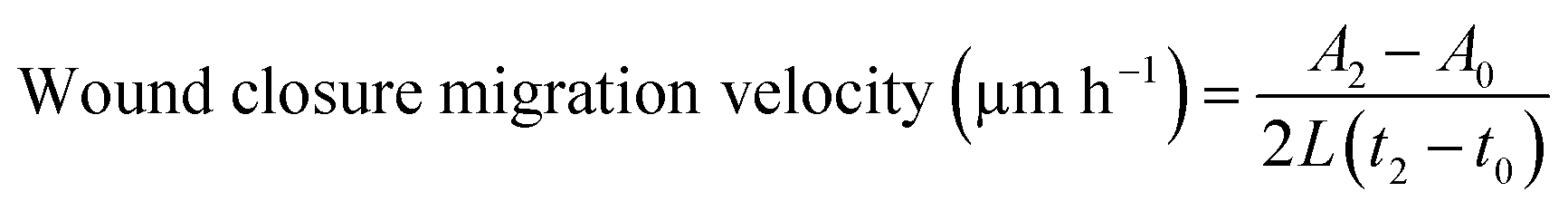 | (1) |
2.3 Cell adhesion of homogenous populations
Glass microscope slides (VWR) were marked with a hydrophobic barrier pen (Vector Laboratories) to outline the rectangular 1 cm × 5.85 cm microfluidic flow path. Each cell population was seeded at 5000 cells per cm2 in their respective media in the outlined area of the glass microscope slide. Cells were seeded on a surface with no pretreatment or ECM protein deposition, which has been justified in other fluid flow assays in literature.47–51 Cells were incubated overnight at 37 °C and 5% CO2 to adhere to the slide surface. The cell media was removed, and the slides were gently washed with 1× PBS. The cells were stained with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 Hoechst 33342 (Thermo Fisher) nuclear stain solution for 20 min, then gently rinsed three times with 1× PBS. Cell adhesion to the slide was tested with shear flow through a parallel plate flow chamber microfluidics device having a 1.0 cm flow path width and 0.0254 cm gasket thickness (Glycotech #31-010). The flow chamber was sealed to the microscope slide using a vacuum pump (Gast). A syringe pump (New Era, NE-4000) with two 60 mL syringes was used to control the flow of 1× PBS through the flow path. Each cell line was exposed to a volumetric flow rate of 50 mL min−1, which corresponds to a wall shear stress of 77.5 dynes per cm2 (7.75 Pa), for 60 s. The volumetric flow rate of PBS was related to the wall shear stress within the chamber using the equation,
:1000 Hoechst 33342 (Thermo Fisher) nuclear stain solution for 20 min, then gently rinsed three times with 1× PBS. Cell adhesion to the slide was tested with shear flow through a parallel plate flow chamber microfluidics device having a 1.0 cm flow path width and 0.0254 cm gasket thickness (Glycotech #31-010). The flow chamber was sealed to the microscope slide using a vacuum pump (Gast). A syringe pump (New Era, NE-4000) with two 60 mL syringes was used to control the flow of 1× PBS through the flow path. Each cell line was exposed to a volumetric flow rate of 50 mL min−1, which corresponds to a wall shear stress of 77.5 dynes per cm2 (7.75 Pa), for 60 s. The volumetric flow rate of PBS was related to the wall shear stress within the chamber using the equation, | (2) |
Image analysis was completed using ImageJ for n = 3 independent microscope slide replicates of each cell type and flow rate. ImageJ ‘Threshold’ and ‘Analyze Particles’ features were used to count the initial number of cells in the image frame and the number of cells remaining on the slide every 15 s after being exposed to shear flow. Cell counting using ImageJ was determined to be within 5% error of manual cell counting. Fluorescent images of Hoechst-stained cells were used for cell counting to prevent incorrect counting of any cell aggregates. Hoechst staining of cell nuclei created easier distinctions between individual cells when aggregated, which ImageJ counted as one large cell in brightfield images.
2.4 Cell adhesion in co-cultures
Co-culture studies assessed adhesion differences between MCF-7/MDA-MB-231, Ishikawa/KLE, and SCC-25/Cal-27 cell pairings. For each cell pair, the less metastatic cell line (MCF-7, Ishikawa, and Cal-27) was stained with 1:1000 Cell Tracker Green CMFTA in DMEM (Invitrogen). The more metastatic cell line (MDA-MB-231, KLE, and SCC-25) was stained with 3:1000 Cell Tracker Blue CMAC (Invitrogen) in DMEM. Each nuclear stain was incubated with the appropriate cell population for 30 minutes. The cell lines were centrifuged for 3 min at 400 × g, then resuspended in fresh DMEM. Glass microscope slides were marked with a hydrophobic barrier pen (Vector Laboratories) to outline the 5.85 cm2 area of the microfluidic flow path as described above. A 50/50 cell mixture of each cell pair was created in DMEM and seeded in the outlined area of the glass microscope slides at a total seeding density of 5000 cells per cm2 (2500 cells per cm2 per cell line). The slides were incubated overnight at 37 °C and 5% CO2 to allow the cells to adhere to the slide surface.
Shear flow was introduced to the sample using the same parallel plate microfluidic system used in the homogenous cell population studies. Each sample was exposed to a volumetric flow rate of 50 mL min−1, which corresponds to a wall shear stress of 77.5 dynes per cm2 (7.75 Pa), for 60 s. Initial and final images of the samples were taken for both fluorescent molecules to quantify the number of cells from each cell line before and after exposure to shear flow. ImageJ was used to analyze the number of cells in each fluorescence channel before and after shear flow for n = 3 independent replicates of each cell type and flow rate.
2.5 MTT calibration curves
Each cell line was seeded at specific concentrations in individual 96 well plates with n = 3 independent replicates per seeding concentration. The seeding concentrations tested were 0, 1 × 103, 5 × 103, 1 × 104, 5 × 104, 1 × 105, 5 × 105, and 1 × 106 cells per mL in each cell lines appropriate cell media. The cells were incubated overnight at 37 °C and 5% CO2 to adhere to the plates. An MTT stock solution was prepared at 5 mg mL−1 in 1× PBS. A working solution was then created by diluting the stock solution in DMEM media to a final concentration of 0.5 mg mL−1. The cells were washed twice with 1× PBS, then incubated with the MTT working solution for 4 h in the dark at 37 °C and 5% CO2. The MTT working solution was removed and a 10% DMSO and 90% isopropanol solution was added to the wells. A plate reader was used to measure the absorbance of the wells at 570 nm. The average of the triplicate absorbance measurements was plotted against the initial cell seeding concentration to create a calibration curve for each cell line.2.6 MTT cell proliferation assay
Each cell line was seeded at a concentration of 1 × 105 cells per mL in their respective cell media in a 96 well plate with n = 3 independent replicates per cell line. The plates were incubated at 37 °C and 5% CO2 for 72 h, with the media being changed at 48 h. At 72 h, the cell media was removed, and the wells were washed twice with 1× PBS. The MTT working solution described in Section 2.5 was added to the wells and the plates were incubated in the dark for 4 h at 37 °C and 5% CO2. The MTT solution was removed, and the DMSO/isopropanol mixture was added to the wells. The absorbance of each well was measured using a plate reader at 570 nm. The calibration curves created in Section 2.5 were used to relate the absorbance to the cell concentration at the 72 h timepoint (Fig. S2, ESI†). Each cell line's doubling time was then calculated by the following equation: | (3) |
2.7 Statistical methods
For each test, data was plotted as the average ± standard deviation of n ≥ 3 independent replicates. Statistical analysis using one-way ANOVA was performed to compare each cell line's 12 h wound closure migration velocity in the presence or absence of serum. Additionally, one-way ANOVA was performed to compare the percentage of cell detachment after exposure to 60 s of shear flow for each cell line cultured in homogenous or co-culture populations. For all ANOVA testing, Tukey–Kramer HSD post-hoc testing was used to adjust the p-value for multiple comparisons. An independent two-group Student's t-test was used to compare each cell line's percentage of cell retention after exposure to shear flow in homogenous and co-culture populations. For all statistical testing, significance was noted as * p < 0.05, ** p < 0.01, *** p < 0.001.3. Results and discussion
Local invasion of cancer cells into surrounding tissue and intravasation into vasculature are the first two steps in cancer metastasis. Our goal is to study function-based quantifiable in vitro metrics (wound closure migration velocity and cell detachment from a culture surface) to place cancer cell lines within the spectrum of metastatic invasiveness. By studying cell lines from a variety of tissue types (breast, endometrium, tongue), we determined overall trends relating cell migration and adhesion properties to cell metastatic potential.3.1 Cell migration through wound closure analysis
Tumor invasion is driven by changes in cell motility, which enables cells to migrate to nearby vasculature, enter the bloodstream, and metastasize to secondary sites.55 Cancer cells are known to have genetic mutations and altered gene expression that contribute to dysregulated cell motility and altered migratory behavior.56 Many researchers currently study therapeutic options that restrict cancer cell motility, hypothesizing that cells with higher metastatic potential are more migratory.57–59 We studied this hypothesis linking cell migration and metastatic potential through wound healing assays of each cancer cell line, which produced a quantitative measure for cell migration velocity. We used this assay to determine the migration characteristics of cancer cell lines with varying levels of reported metastatic potential. The tumor microenvironment is more complex than depicted in in vitro assays due to variability in cell types and the surrounding extracellular matrix.60,61 We chose to study the components of the cell media and their influence on cell migration as one example of this complexity. Cell migration can be influenced by cell proliferation, in which cell division can result in tumor growth and infiltration in surrounding healthy tissue. In vitro, cell proliferation is controlled through the addition of serum in the cell culture media as a source of growth, attachment, and hormonal factors.62 We compared this traditional approach using serum in the culture media to serum-free culture media, which does not provide the factors necessary for cell growth and proliferation. Representative images of wound closure are provided in Fig. 2.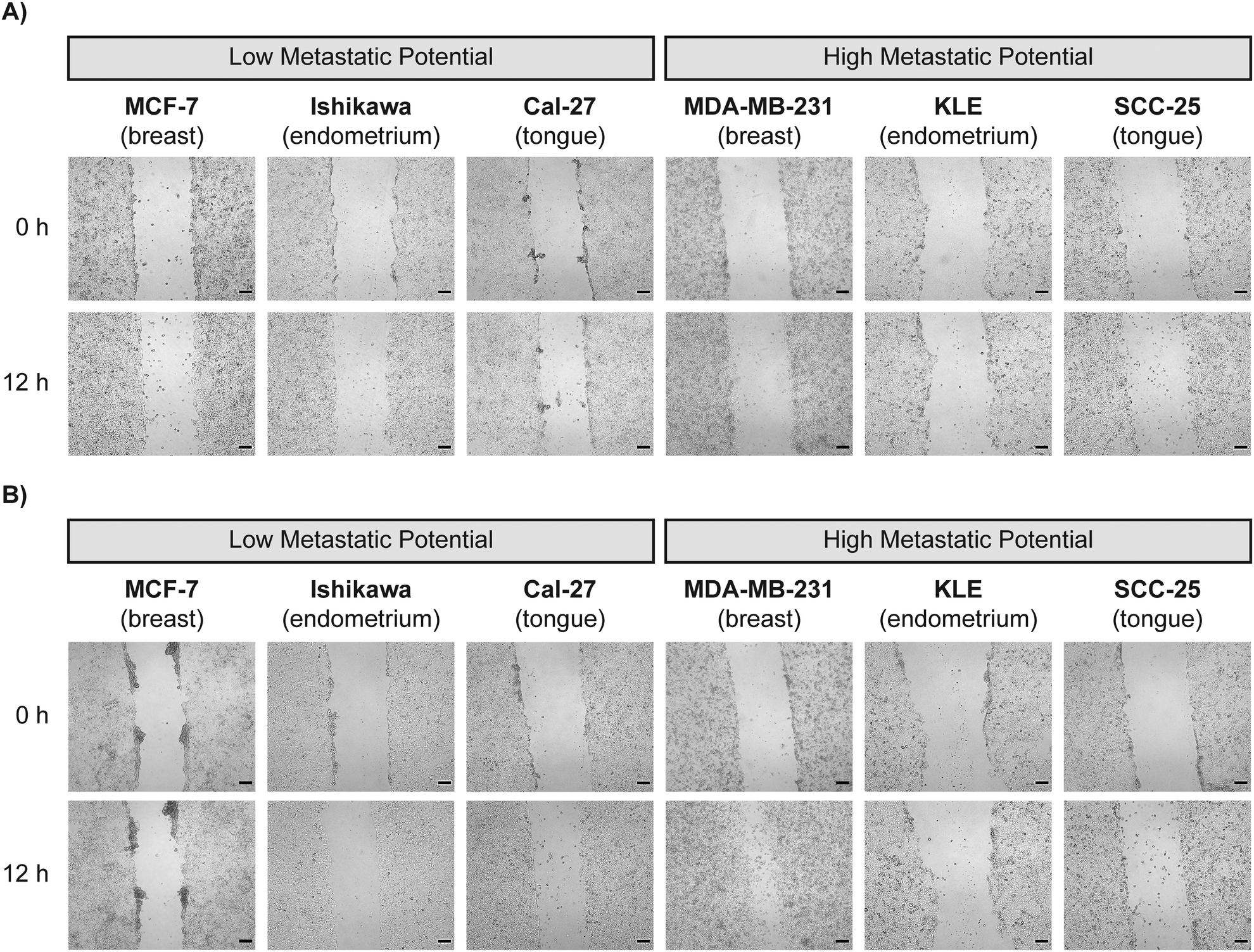 | ||
| Fig. 2 Wound closure imaging of cancer cell lines based on serum in culture media. (A) Representative images of the 6 cancer cell lines in serum-free media immediately after (0 h) and 12 h after the scratch wound was created. (B) Representative images of the 6 cancer cell lines in serum-containing media immediately after (0 h) and 12 h after the scratch wound was created. (C) All wound areas were visualized using brightfield microscopy at 4× magnification. Scale bars represent 200 μm. | ||
There was a visible decrease in wound area for all cell lines within the first 12 h of the wound being established, regardless of the presence of serum. The most noticeable decrease in wound area was in the MDA-MB-231 cell line when serum was present in the media (Fig. 2B). Wound area was determined using the ImageJ MRI Wound Healing Tool every 24 h for 144 h (Fig. 3).
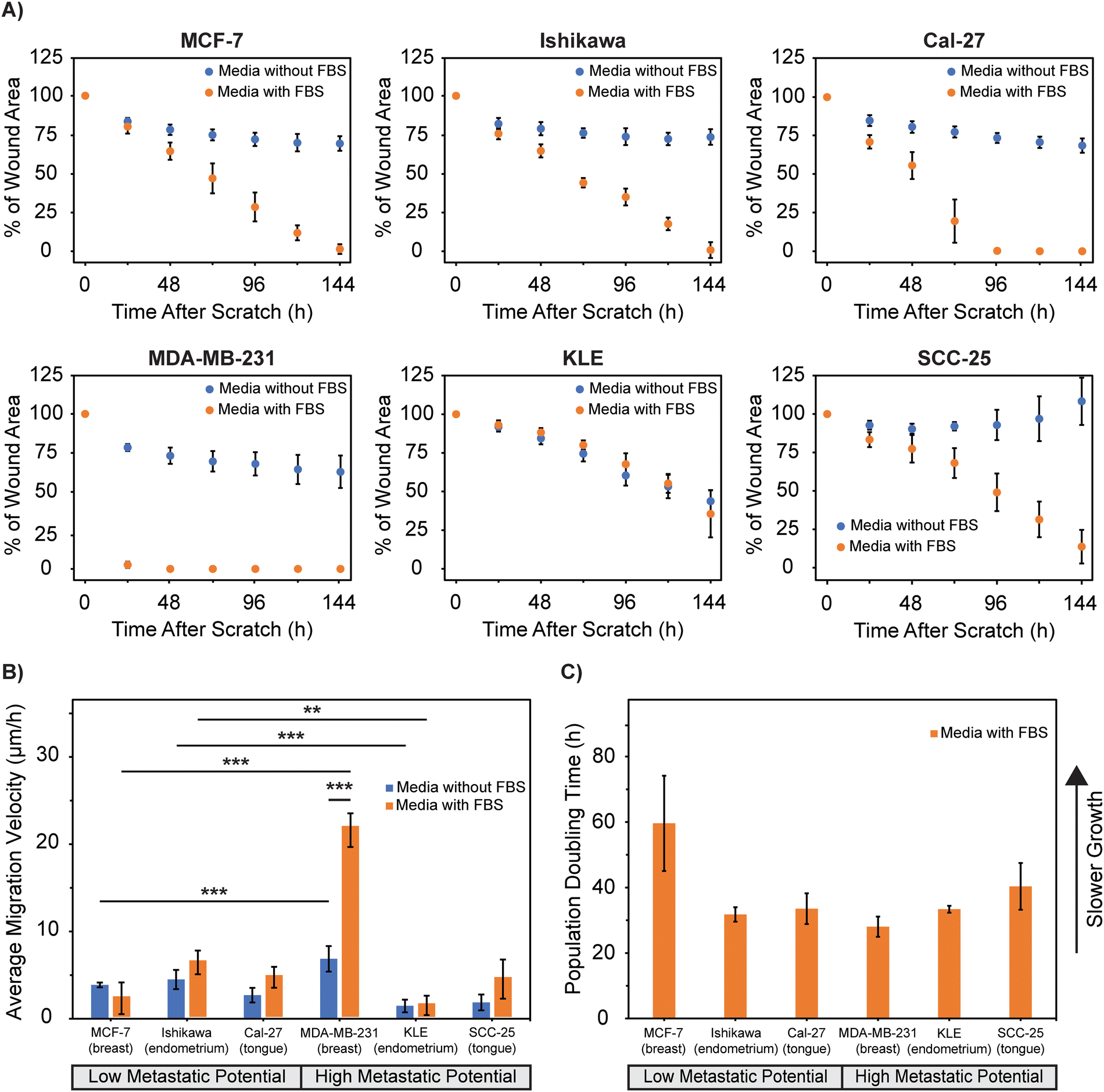 | ||
| Fig. 3 Wound closure quantification of cancer cell lines. (A) Wound area expressed as a percentage of the original wound area when cultured in serum-containing or serum-free media. (B) Wound closure average velocity of each cell line in the first 12 h after the wound was created when cultured in serum-containing or serum-free media. Significance testing compared the migration velocity across cell lines and influence of serum in the media using one-way ANOVA with Tukey–Kramer HSD post hoc testing to adjust the p-value for multiple comparisons (* p < 0.05, ** p < 0.01, *** p < 0.001). The absence of * is considered not significant. (C) Population doubling times of each cell line as determined from the MTT assay. Data is plotted as the mean ± standard deviation of n = 5 representative images for each cell line. | ||
For all cell lines, the presence of serum in the cell media enhanced wound closure, allowing each wound area to be fully closed over time (Fig. S3, ESI†). The time for complete wound closure to occur varied, with MDA-MB-231 and Cal-27 being the only cell lines to close the wound area prior to 144 h (48 h and 96 h respectively). Overall, MDA-MB-231 cells closed the area the fastest (48 h) and KLE cells closed the area the slowest (216 h). When serum was absent in the media, cell proliferation was inhibited and complete wound closure did not occur for any of the cell lines except KLE (384 h, Fig. S3, ESI†). For the SCC-25 cell line, once the cells stopped migrating, they began to aggregate on the culture surface, contributing to an increase in wound area output by the ImageJ MRI Wound Healing Tool at time points beyond 72 h (108% of wound area at 144 h). Although these controlled comparisons have yet to be performed across all of the tested cell lines, similar wound healing has been achieved in literature for individual cell lines, validating these results (Tables S1 and S2, ESI†). Additionally, similar results have been achieved in other studies showing that increasing the concentration of serum in the media increases the percentage of wound closure by the cell line.63 These results indicate that cell growth and proliferation are necessary to wound closure and prolonged cell migration.
The wound closure migration velocity for the first 12 h after the scratch was calculated for each cell line (eqn (1)). MDA-MB-231 cells migrated the quickest into the gap region, with an average migration velocity of 21.8 μm h−1, over two-fold higher than any of the other cell lines studied (Fig. 3B). For the majority of cell lines, the presence of serum in the media significantly increased the wound closure velocity (Fig. 3B). The wound closure velocity was significantly greater for MDA-MB-231 (p < 1.0 × 10−4) when serum was present in the media compared to when the serum was absent. However, there was not a significant difference in the closure velocity for MCF-7 (p = 0.79), Ishikawa (p = 0.63), KLE (p = 1.0), Cal-27 (p = 0.26), or SCC-25 (p = 0.088) cell lines when serum was present in the media compared to when the serum was absent.
The population doubling time of each cell line was studied to see if cell proliferation, driven by serum in the cell media, played a role in the migration velocity. We expected that the increase in migration velocity between cells exposed to serum was due to the cell's population doubling time, as metastatic cells are known to have greater uncontrolled cell proliferation.64,65 However, the cell doubling times determined experimentally did not support this hypothesis. MDA-MB-231 (28 h), Ishikawa (31 h), KLE (34 h), Cal-27 (34 h), and SCC-25 (40 h) cell lines all had similar population doubling times (Fig. 3C), supported by other studies in literature, but not all of these cell lines exhibited migration velocities enhanced with the presence of serum.66–71
When serum was present in the media, the average migration velocity was significantly greater for MDA-MB-231 compared to MCF-7 breast lines (p < 1.0 × 10−4) and Ishikawa compared to KLE endometrial lines (p = 1.2 × 10−3). Similarly, the average migration velocity was also significantly greater when serum was absent from the media for MDA-MB-231 compared to MCF-7 (p = 6.0 × 10−4) and Ishikawa compared to KLE (p = 5.0 × 10−4). There were no significant differences between the average migration velocity between the tongue tissue Cal-27 and SCC-25 lines when serum was present (p = 1.0) or absent (p = 0.74) from the media. These results indicate that there is not a clear relationship between migration velocity and cell metastatic potential. Since there were no apparent trends between cell migration velocity and cell doubling times, additional factors relating to cell motility may be contributing to the differences in migration velocity, rather than proliferation. There is strong evidence suggesting that cancer cells exhibit highly altered focal adhesions, which are protein complexes that link the cell cytoskeleton to the extracellular matrix through integrins.72,73 During cell migration, there is continuous formation of focal adhesions at the leading edge of the cell while there is simultaneous disassembly of focal adhesions at the lagging edge.74 It has been observed that cell lines with quicker focal adhesion disassembly are more migratory.52 The disassembly of these focal adhesions likely varies based on metastatic potential and is an additional factor involved in cell migration velocity rather than proliferation alone.52,75
3.2 Cell adhesion through shear flow analysis
Cell detachment from the primary tumor site is another component involved in local invasion and intravasation within the metastatic cascade. According to observations associated with EMT, cells will lose their cell–cell adhesion junctions and rearrange their cytoskeleton as they become more metastatic, altering cell motility, focal adhesion disassembly, and interactions with their microenvironment.33,52,76 Based on this description of EMT, we expected the cell lines commonly described as having a higher metastatic potential to be less adherent to a culture surface under shear flow compared to lower metastatic potential lines.Our direct analysis of adhesive function supports this hypothesis, as purified populations of cell lines with a higher metastatic potential (MDA-MB-231, KLE, and SCC-25) are more susceptible to detachment from a cell culture surface compared to their lower metastatic cell counterparts (MCF-7, Ishikawa, Cal-27) (Fig. 4).
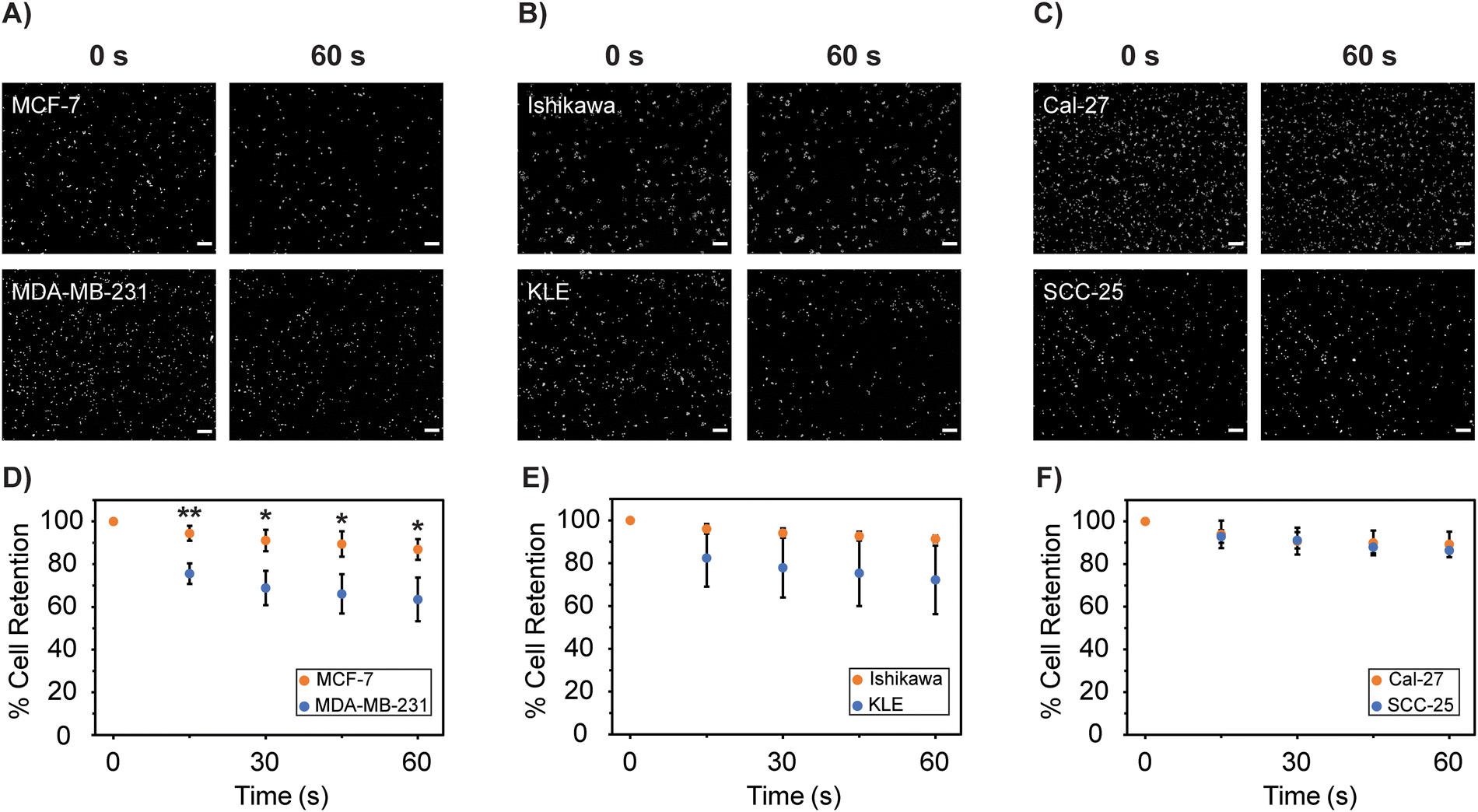 | ||
| Fig. 4 Cell adhesion of homogeneous cell populations. (A)–(C) Representative images of Hoechst-stained cells before (0 s) and after (60 s) exposure to shear flow at 50 mL min−1. (D)–(F) Percentage of initial cells retained after exposure to shear flow at 50 mL min−1 over 60 s. Data is plotted as the mean ± standard deviation of n = 3 independent replicates for each cell line. Significance testing compared the two cell lines in each pair using a two-group independent Student's t-test (* p < 0.05, ** p < 0.01, *** p < 0.001). Absence of * is considered not significant. Scale bars represent 200 μm. | ||
At 60 s, MCF-7 breast lines had a significantly greater percentage of cell retention at 86.9 ± 4.86% compared to MDA-MB-231 cells at 63.5 ± 10.2% (p = 0.023). For endometrial tissues, the apparent increase in cell retention of Ishikawa at 91.3 ± 1.56% compared to KLE cells at 72.2 ± 16.0% was not significant (p = 0.11). Similarly, the apparent increase in cell retention of the Cal-27 at 89.2 ± 5.99% over SCC-25 cells at 85.6 ± 1.15% was not significant (p = 0.35).
Functional cell behavior, such as cell adhesion, are downstream outcomes resulting from molecular changes associated with EMT, and therefore should follow similar trends. Our adhesive function findings for each of these six cancer cell lines provide novel information that is not currently available in literature for comparison. To validate our results, we have chosen to compare our results on adhesive function to previous molecular biomarker data, which is supported by current literature (Table S3, ESI†). Our lab has previously investigated the presence of adhesion biomarkers, including cell-to-cell adhesion proteins called cadherins, on the cell surface of MDA-MB-231 and MCF-7 cell lines using flow cytometry.77 We specifically studied the expression of E-cadherin, an epithelial biomarker that is downregulated in EMT, and N-cadherin, a mesenchymal biomarker that is upregulated during EMT. This inverse expression is called the cadherin switch and is a hallmark of EMT.78 As expected, MDA-MB-231, with higher metastatic potential, has a decreased cell surface expression of E-cadherin and increased expression of N-cadherin compared to MCF-7, with lower metastatic potential.77,79 Further western blot analysis from literature confirm this cadherin switch in between E- and N-cadherin in MDA-MB-231 and MCF-7 cell lines.80–83 Our study expands these works further by relating a cell line's metastatic phenotype, based on the presence of cell adhesion biomarkers, to their adhesive function under shear flow. MDA-MB-231, with surface biomarkers that are indicative of a more metastatic phenotype, is less adhesive to a culture surface under fluid flow, compared to MCF-7.
Quantifying cell surface expression of adhesion biomarkers using flow cytometry has yet to be widely performed on all cancer cell lines, but the relative expression of these markers has been determined using western blot analysis. These studies also support the cadherin switch, where more highly metastatic KLE has a greater relative expression of N-cadherin, while less metastatic Ishikawa has a greater relative expression of E-cadherin.84–87 Similarly, more metastatic SCC-25 has a greater relative expression of N-cadherin, while less metastatic Cal-27 has a greater relative expression of E-cadherin.88–91 When relating the relative presence of these biomarkers to our shear flow study, cells with downregulated E-cadherin and upregulated N-cadherin expression (KLE, SCC-25) had greater cell detachment under shear flow after 60 s, indicative of their more metastatic phenotype. In comparison to western blot analysis for MCF-7/MDA-MB-231, there are less drastic differences in the expression of E-cadherin and N-cadherin between the Ishikawa/KLE and Cal-27/SCC-25 cell lines. These less drastic differences translate similarly into our adhesion assays, where the differences in cell adhesion between the Ishikawa/KLE and Cal-27/SCC-25 cell lines were not significant at 60 s. These results also indicate that adhesive function alone may not be a definitive measurement of cell metastatic potential for all cancer cell lines.
Cancerous tumors are comprised of heterogenous populations of cells with varying metastatic potential. To expand the adhesion platform to reflect this heterogeneity, we applied our shear flow system to co-cultures (50/50 mixture) of each cell pairing (Fig. 5).
 | ||
| Fig. 5 Cell adhesion of cancer cells in co-culture. (A)–(C) Representative images of cell tracker green (less metastatic) and cell tracker blue (more metastatic) stained cells cultured together before (0 s) and after (60 s) exposure to shear flow at 50 mL min−1. (D)–(F) Percentage of initial cells retained after exposure to shear flow at 50 mL min−1 for 60 s. Data is plotted as the mean ± standard deviation of n = 3 independent replicates for each cell line pair. Significance testing compared the two cell lines in each pair using a two-group independent Student's t-test (* p < 0.05, ** p < 0.01, *** p < 0.001). Absence of * is considered not significant. Scale bars represent 200 μm. | ||
There were less drastic adhesion differences when the cells were co-cultured and exposed to shear flow as opposed to being cultured as homogenous cell populations. On average, all cell lines except KLE had a larger percentage of cell detachment from the glass culture surface after 60 s of shear flow when co-cultured compared to when cultured as homogenous cell populations. For example, there was a significantly greater percentage of MCF-7 (p < 1.0 × 10−4) and MDA-MB-231 (p = 1.0 × 10−4) cells that detached in co-culture compared to when cultured as homogenous populations.
Only the cell pairings with significant adhesion differentials when studied as homogenous cell populations, had significant differences in cell adhesion when co-cultured. MCF-7 breast cells had a significantly greater percentage of cell retention (43.6 ± 4.72%) compared to MDA-MB-231 (28.1 ± 2.91%) breast cells (p = 8.3 × 10−3). However, there were no significant differences in the percentage of cell retention between homogenous cell pairings with less drastic differences in cell adhesion, such as endometrial cell lines (Ishikawa = 78.8 ± 9.92% and KLE = 78.5 ± 4.52%, p = 0.96), or tongue cell lines (Cal-27 = 78.9 ± 0.586% and SCC-25 = 77.7 ± 5.14%, p = 0.72). Cells in culture are known to secrete and remodel extracellular matrix (ECM) components and small soluble molecules over time.92 These components play a large role in modulating cell adhesion, proliferation, and migration.60 Studies also suggest mutations in proteins secreted by cancer cell lines, potentially leading to differences in cell adhesion.93–95 The discrepancies in cell adhesion between the homogenous and co-culture cell populations could be driven by differences in these cell secretions, as cells in co-culture could have more similar adhesion properties when faced with similar secreted molecules on the culture surface.
Normalizing the data provides a powerful visual display that highlights general trends between in vitro wound closure migration velocity, cell detachment, and metastatic potential. Normalization reorganizes the data by removing the units from each axis and scaling the data between 0 and 1, focusing more on trends between data points. This process allows for simplistic comparisons that have greater translational potential into clinical applications. Specifically in this work, normalization aids in developing future predictive tools for cell metastatic potential based on functional properties. Wound closure migration velocity and cell detachment were normalized to the highest value among the cell lines for each data set (MDA-MB-231), where a value of 1 corresponds to most aggressive, while a value of 0 corresponds to least aggressive for each metric within the set of cell lines (Fig. 6).
 | ||
| Fig. 6 Comparison of the average wound closure migration velocity and average percentage of cell detachment after 60 s of shear flow for each cell line normalized to the cell line with the highest value of each metric. | ||
There were clear trends that related cell migration and detachment to metastatic potential. On average, cell lines with low metastatic potential (MCF-7, Ishikawa, and Cal-27) were more aggressive through wound closure migration compared to cell detachment from a culture surface. On the other hand, cell lines with high metastatic potential (MDA-MB-231, KLE, and SCC-25) were on average more aggressive through cell detachment compared to wound closure migration. This trend was true independent of the tissue type where the cells originated, indicating that there is a relationship between metastatic potential and the predominate type of cancer aggression.
The goal of this work was to develop a comprehensive comparison of functional metrics in relation to cell metastatic potential. Displaying the data in this way revealed that one functional metric was not sufficient to categorize cell lines according to their metastatic potential, as both adhesion and migration needed to be accounted for to accurately see any trends. While this work provides a baseline for comparison, it is recommended that future work expand these comparisons with additional molecular assays, such as those specific to a tissue origin, to relate changes in the metastatic cancerous genotype to the overall trends found in this work.
4. Conclusions
This study examined wound healing migration velocity and cell detachment from a culture surface as quantifiable label-free function-based metrics of cancer aggression in vitro linked to local invasion and intravasation in the metastatic cascade. We give a comprehensive comparison of these function-based metrics across epithelial cancer cell lines from a variety of tissue origins (breast, endometrium, tongue) to help draw broader conclusions between cell migration and adhesion to metastatic potential than what is currently available. Our data suggests that cancer cells with low metastatic potential are more aggressive through migration rather than detachment while high metastatic potential cells are more aggressive through detachment rather than migration. Wound healing studies measured cell migratory behavior, but found no relationships between cell migration velocity and cell metastatic potential. The presence of serum in the culture media influenced whether cells could migrate in combination with proliferation; however, we found that the presence of serum did not necessary enhance the cell's migration velocity for all cell types and that any increase in velocity was not due to the population doubling time. Additionally, we demonstrated that when cultured as individual populations, cells with high metastatic potential are more likely to have poor adhesion properties and detach from a culture surface, as determined in our shear flow system and consistent with cell adhesion characteristics related to EMT. Co-cultures of high and low metastatic potential cells revealed less drastic differences in adhesion function between the cell types, as there are likely excreted proteins that influence their adhesion properties. Through our shear flow system, we have highlighted trends between cell metastatic potential and adhesive function, expanding on work that has traditionally relied on gene or protein expression alone. Through our observations, and with comparable literature, we demonstrate that functional cell behavior through migration and adhesion provides information on cell metastatic potential independent of the tissue of origin.This work reveals that one functional metric alone is not enough to draw accurate conclusions about cell metastatic potential. Instead, multiple functional metrics, such as wound healing and adhesion properties, are necessary to categorize each cell population. These results have major clinical relevancy, with potential to develop a tissue-agnostic predictive tool for cell metastatic potential based solely on functional behavior.
Data availability
The raw data required to reproduce these findings are available to download from https://doi.org/10.18126/2zmk-n291via the Materials Data Facility.Conflicts of interest
The authors have no competing interests to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. 1839289 and CAREER Award Grant No. 2045853. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation. We would also like to thank Dr. Brittany E. Givens and Dr. Claire E. Rowlands at the University of Kentucky for providing the Ishikawa and KLE cells used in these studies.References
- F. Bray, M. Laversanne, H. Sung, J. Ferlay, R. L. Siegel, I. Soerjomataram and A. Jemal, Ca-Cancer J. Clin., 2024, 74, 229–263 CrossRef.
- R. L. Siegel, T. B. Kratzer, A. N. Giaquinto, H. Sung and A. Jemal, Ca-Cancer J. Clin., 2025, 75, 10–45 CrossRef PubMed.
- X. Guan, Acta Pharm. Sin. B, 2015, 5, 402–418 Search PubMed.
- F. van Zijl, G. Krupitza and W. Mikulits, Mutat. Res., Rev. Mutat. Res., 2011, 728, 23–34 CAS.
- E. Pachmayr, C. Treese and U. Stein, Visc. Med., 2017, 33, 11–20 Search PubMed.
- M. C. Moh and S. Shen, Cell Adhes. Migr., 2009, 3, 334–336 CrossRef PubMed.
- T. Okegawa, Y. Li, R.-C. Pong and J.-T. Hsieh, J. Urol., 2002, 167, 1836–1843 CrossRef CAS PubMed.
- A. K. Shenoy and J. Lu, Cancer Lett., 2016, 380, 534–544 CrossRef CAS PubMed.
- J. Yayan, K.-J. Franke, M. Berger, W. Windisch and K. Rasche, Mol. Biol. Rep., 2024, 51, 165 CrossRef CAS PubMed.
- G. Lee, C. Wong, A. Cho, J. J. West, A. J. Crawford, G. C. Russo, B. R. Si, J. Kim, L. Hoffner and C. Jang, Cancer Res., 2024, 84, 2820–2835 CrossRef CAS PubMed.
- M. R. Chastney, J. Kaivola, V.-M. Leppänen and J. Ivaska, Nat. Rev. Mol. Cell Biol., 2025, 26, 147–167 CrossRef CAS PubMed.
- L. Depau, J. Brunetti, C. Falciani, M. Garfì, M. F. Paolocci, A. Pini and L. Bracci, Cancer Res., 2024, 84, 1508 CrossRef.
- M. Janiszewska, M. C. Primi and T. Izard, J. Biol. Chem., 2020, 295, 2495–2505 CrossRef CAS PubMed.
- Z. Karagiorgou, P. N. Fountas, D. Manou, E. Knutsen and A. D. Theocharis, Cancers, 2022, 14, 5328 CrossRef CAS PubMed.
- Ş. Comşa, A. M. Cimpean and M. Raica, Anticancer Res., 2015, 35, 3147–3154 Search PubMed.
- C. Gest, U. Joimel, L. Huang, L.-L. Pritchard, A. Petit, C. Dulong, C. Buquet, C.-Q. Hu, P. Mirshahi and M. Laurent, BMC Cancer, 2013, 13, 1–14 CrossRef PubMed.
- F. Tang, R. Zhang, Y. He, M. Zou, L. Guo and T. Xi, PLoS One, 2012, 7, e35435 CrossRef CAS PubMed.
- E. W. Thompson, S. Paik, N. Brünner, C. L. Sommers, G. Zugmaier, R. Clarke, T. B. Shima, J. Torri, S. Donahue and M. E. Lippman, J. Cell. Physiol., 1992, 150, 534–544 CrossRef CAS PubMed.
- J. Kozak, P. Wdowiak, R. Maciejewski and A. Torres, Cytotechnology, 2018, 70, 339–350 CrossRef PubMed.
- T. Van Nyen, C. P. Moiola, E. Colas, D. Annibali and F. Amant, Int. J. Mol. Sci., 2018, 19, 2348 CrossRef PubMed.
- M. V. Bais, M. Kukuruzinska and P. C. Trackman, Oral Oncol., 2015, 51, 476–482 CrossRef PubMed.
- A. Bugshan and I. Farooq, F1000Research, 2020, 9, 229 CAS.
- J. E. Jonkman, J. A. Cathcart, F. Xu, M. E. Bartolini, J. E. Amon, K. M. Stevens and P. Colarusso, Cell Adhes. Migr., 2014, 8, 440–451 CrossRef PubMed.
- G. Cory, Cell Migration: developmental methods and protocols, 2011, pp. 25–30 Search PubMed.
- L. G. Rodriguez, X. Wu and J.-L. Guan, Cell Migration: Developmental Methods and Protocols, 2005, pp. 23–29 Search PubMed.
- A. Grada, M. Otero-Vinas, F. Prieto-Castrillo, Z. Obagi and V. Falanga, J. Invest. Dermatol., 2017, 137, e11–e16 CrossRef CAS PubMed.
- N. Kramer, A. Walzl, C. Unger, M. Rosner, G. Krupitza, M. Hengstschläger and H. Dolznig, Mutat. Res., Rev. Mutat. Res., 2013, 752, 10–24 CrossRef CAS PubMed.
- J. Pijuan, C. Barceló, D. F. Moreno, O. Maiques, P. Sisó, R. M. Marti, A. Macià and A. Panosa, Front. Cell Dev. Biol., 2019, 7, 107 CrossRef PubMed.
- F. Martin-Belmonte and M. Perez-Moreno, Nat. Rev. Cancer, 2012, 12, 23–38 CrossRef CAS PubMed.
- R. Kalluri and R. A. Weinberg, J. Clin. Invest., 2009, 119, 1420–1428 CAS.
- P. Nisticò, M. J. Bissell and D. C. Radisky, Cold Spring Harbor Perspect. Biol., 2012, 4, a011908 Search PubMed.
- X. Lai, Q. Li, F. Wu, J. Lin, J. Chen, H. Zheng and L. Guo, Front. Cell Dev. Biol., 2020, 8, 760 Search PubMed.
- M. W. Klymkowsky and P. Savagner, Am. J. Pathol., 2009, 174, 1588–1593 CAS.
- S. A. Fleit, H. B. Fleit and S. Zolla-Pazner, J. Immunol. Methods, 1984, 68, 119–129 CAS.
- N. Delirezh, E. Shojaeefar, P. Parvin and B. Asadi, Cell J., 2013, 15, 218 CAS.
- M. J. Tomlinson, S. Tomlinson, X. B. Yang and J. Kirkham, J. Tissue Eng., 2013, 4, 2041731412472690 Search PubMed.
- P. Bouchalova and P. Bouchal, Cancer Cell Int., 2022, 22, 394 Search PubMed.
- N. Paddillaya, K. Ingale, C. Gaikwad, D. K. Saini, P. Pullarkat, P. Kondaiah, G. I. Menon and N. Gundiah, Soft Matter, 2022, 18, 4378–4388 CAS.
- S. Kumar and P. LeDuc, Exp. Mech., 2009, 49, 11–23 CAS.
- C. Zhang and S. Neelamegham, Platelets, 2017, 28, 434–440 CAS.
- Y. Zhang and S. Neelamegham, J. Immunol. Methods, 2003, 278, 305–317 CAS.
- Z. Wan and R. D. Kamm, Nat. Biomed. Eng., 2019, 3, 423–424 Search PubMed.
- P. T. Winnard, A. P. Pathak, S. Dhara, S. Y. Cho, V. Raman and M. G. Pomper, J. Nucl. Med., 2008, 49, 96S–112S Search PubMed.
- Z. Zhao, X. Zhu, K. Cui, J. Mancuso, R. Federley, K. Fischer, G.-J. Teng, V. Mittal, D. Gao and H. Zhao, Cancer Res., 2016, 76, 2094–2104 CrossRef CAS PubMed.
- P. Savagner, Ann. Oncol., 2010, 21, vii89–vii92 CrossRef PubMed.
- A. V. P. Bobadilla, J. Arévalo, E. Sarró, H. M. Byrne, P. K. Maini, T. Carraro, S. Balocco, A. Meseguer and T. Alarcón, J. R. Soc., Interface, 2019, 16, 20180709 CrossRef CAS PubMed.
- B. D. Riehl, E. Kim, J. S. Lee, B. Duan, R. Yang, H. J. Donahue and J. Y. Lim, J. Biomech. Eng., 2020, 142, 101001 CrossRef PubMed.
- T. Van Kooten, J. Schakenraad, H. Van der Mei and H. Busscher, J. Biomed. Mater. Res., 1992, 26, 725–738 CrossRef CAS PubMed.
- J. Hu, Y. Zhou, J. D. Obayemi, J. Du and W. O. Soboyejo, J. Mech. Behav. Biomed. Mater., 2018, 86, 1–13 CrossRef CAS PubMed.
- D. J. Modulevsky, D. Tremblay, C. Gullekson, N. V. Bukoresthliev and A. E. Pelling, PLoS ONE, 2012, 7, e45329 CrossRef CAS PubMed.
- T. F. Didar and M. Tabrizian, Lab Chip, 2010, 10, 3043–3053 RSC.
- P. Beri, A. Popravko, B. Yeoman, A. Kumar, K. Chen, E. Hodzic, A. Chiang, A. Banisadr, J. K. Placone and H. Carter, Cancer Res., 2020, 80, 901–911 CrossRef CAS PubMed.
- W. O. Lane, A. E. Jantzen, T. A. Carlon, R. M. Jamiolkowski, J. E. Grenet, M. M. Ley, J. M. Haseltine, L. J. Galinat, F.-H. Lin and J. D. Allen, J. Visualized Exp., 2012, e3349 Search PubMed.
- C. K. Zarins, D. P. Giddens, B. Bharadvaj, V. S. Sottiurai, R. F. Mabon and S. Glagov, Circ. Res., 1983, 53, 502–514 CrossRef CAS PubMed.
- C. H. Stuelten, C. A. Parent and D. J. Montell, Nat. Rev. Cancer, 2018, 18, 296–312 CrossRef CAS PubMed.
- J. Kassis, D. A. Lauffenburger, T. Turner and A. Wells, Semin. Cancer Biol., 2001, 11, 105–117 CrossRef CAS PubMed.
- B. Kortüm, H. Radhakrishnan, F. Zincke, C. Sachse, S. Burock, U. Keilholz, M. Dahlmann, W. Walther, G. Dittmar and D. Kobelt, Oncogene, 2022, 41, 4446–4458 CrossRef PubMed.
- M. Raudenská, K. Petrláková, T. Juriňáková, J. L. Fialová, M. Fojtů, M. Jakubek, D. Rösel, J. Brábek and M. Masařík, Trends Cancer, 2023, 9, 293–308 CrossRef PubMed.
- T. D. Palmer, W. J. Ashby, J. D. Lewis and A. Zijlstra, Adv. Drug Delivery Rev., 2011, 63, 568–581 CrossRef CAS PubMed.
- J. Nicolas, S. Magli, L. Rabbachin, S. Sampaolesi, F. Nicotra and L. Russo, Biomacromolecules, 2020, 21, 1968–1994 CrossRef CAS PubMed.
- R. Baghban, L. Roshangar, R. Jahanban-Esfahlan, K. Seidi, A. Ebrahimi-Kalan, M. Jaymand, S. Kolahian, T. Javaheri and P. Zare, Cell Commun. Signaling, 2020, 18, 1–19 CrossRef PubMed.
- D. Barnes and G. Sato, Cell, 1980, 22, 649–655 CAS.
- P. Y. Yue, E. P. Leung, N. Mak and R. N. Wong, J. Biomol. Screening, 2010, 15, 427–433 CAS.
- C. J. Sherr, Science, 1996, 274, 1672–1677 CAS.
- L. Li and W. B. Neaves, Cancer Res., 2006, 66, 4553–4557 CAS.
- K. J. Sweeney, A. Swarbrick, R. L. Sutherland and E. A. Musgrove, Oncogene, 1998, 16, 2865–2878 CAS.
- K. Wosikowski, D. Schuurhuis, G. Kops, M. Saceda and S. E. Bates, Clin. Cancer Res., 1997, 3, 2405–2414 CAS.
- M. Nishida, Cell and Molecular Biology of Endometrial Carcinoma, Springer, 2003, pp. 35–58 Search PubMed.
- W. Qu, Y. Zhao, X. Wang, Y. Qi, C. Zhou, Y. Hua, J. Hou and S.-W. Jiang, Clin. Chim. Acta, 2019, 489, 225–232 CAS.
- J. Gioanni, J.-L. Fischel, J.-C. Lambert, F. Demard, C. Mazeau, E. Zanghellini, F. Ettore, P. Formento, P. Chauvel and C.-M. Lalanne, Eur. J. Cancer Clin. Oncol., 1988, 24, 1445–1455 CAS.
- T. B. Steinbichler, A. Alshaimaa, M. V. Maria, D. Daniel, R. Herbert, D. Jozsef and S. Ira-Ida, Oncotarget, 2018, 9, 3641 Search PubMed.
- M. Maziveyi and S. K. Alahari, Oncotarget, 2017, 8, 48471 Search PubMed.
- T. A. Martin, L. Ye, A. J. Sanders, J. Lane and W. G. Jiang, Madame Curie Bioscience Database [Internet], Landes Bioscience, 2013 Search PubMed.
- M. Nagano, D. Hoshino, N. Koshikawa, T. Akizawa and M. Seiki, Int. J. Cell Biol., 2012, 2012, 310616 Search PubMed.
- K. Bijian, C. Lougheed, J. Su, B. Xu, H. Yu, J. Wu, K. Riccio and M. Alaoui-Jamali, Br. J. Cancer, 2013, 109, 2810–2818 CrossRef CAS PubMed.
- S. E. Leggett, A. M. Hruska, M. Guo and I. Y. Wong, Cell Commun. Signaling, 2021, 19, 32 CrossRef CAS PubMed.
- C. F. Cahall, J. L. Lilly, E. A. Hirschowitz and B. J. Berron, Breast Cancer: Basic Clin. Res., 2015, 9, BCBCR.S25461 CrossRef PubMed.
- M. Yilmaz and G. Christofori, Cancer Metastasis Rev., 2009, 28, 15–33 CrossRef PubMed.
- N. Singh, R. Chakraborty, R. P. Bhullar and P. Chelikani, Biochem. Biophys. Res. Commun., 2014, 446, 499–503 CAS.
- X. Wan, J. Hou, S. Liu, Y. Zhang, W. Li, Y. Zhang and Y. Ding, Front. Cell Dev. Biol., 2021, 9, 583572 Search PubMed.
- A. H. Klopp, L. Lacerda, A. Gupta, B. G. Debeb, T. Solley, L. Li, E. Spaeth, W. Xu, X. Zhang and M. T. Lewis, PLoS One, 2010, 5, e12180 Search PubMed.
- X. Zhang, Z. Gao, K. Chen, Q. Zhuo, M. Chen, J. Wang, X. Lai and L. Wang, Food Funct., 2022, 13, 4967–4976 CAS.
- Z. Colín-Val, N. E. López-Díazguerrero and R. López-Marure, J. Steroid Biochem. Mol. Biol., 2021, 208, 105818 CrossRef PubMed.
- H.-Y. Chen, Y.-F. Chiang, J.-S. Huang, T.-C. Huang, Y.-H. Shih, K.-L. Wang, M. Ali, Y.-H. Hong, T.-M. Shieh and S.-M. Hsia, Cancers, 2021, 13, 1236 CAS.
- W.-N. Yang, Z.-H. Ai, J. Wang, Y.-L. Xu and Y.-C. Teng, J. Gynecol. Oncol., 2014, 25, 36–42 CAS.
- S. Sheng, W. Liu, Y. Xue, Z. Pan, L. Zhao, F. Wang and X. Qi, Int. J. Environ. Res. Public Health, 2022, 19, 15344 CrossRef CAS PubMed.
- X. Luan, C. Ma, P. Wang and F. Lou, OncoTargets Ther., 2017, 1389–1402 CAS.
- B. F. Matte, A. Kumar, J. K. Placone, V. G. Zanella, M. D. Martins, A. J. Engler and M. L. Lamers, J. Cell Sci., 2019, 132, jcs224360 Search PubMed.
- S. Y. Moon, P. S. de Campos, B. F. Matte, J. K. Placone, V. L. G. Zanella, M. D. Martins, M. L. Lamers and A. J. Engler, Mol. Biol. Cell, 2023, 34, ar89 CAS.
- S. Wang, H. Fan and J. Bai, J. Cell. Mol. Med., 2025, 29, e70342 CAS.
- X. Liu, L. Meng, X. Li, D. Li, Q. Liu, Y. Chen, X. Li, W. Bu and H. Sun, Int. J. Oral Sci., 2020, 12, 1–11 CAS.
- L. A. Sawicki, L. H. Choe, K. L. Wiley, K. H. Lee and A. M. Kloxin, ACS Biomater. Sci. Eng., 2018, 4, 836–845 CrossRef CAS PubMed.
- S. Mathivanan, H. Ji, B. J. Tauro, Y.-S. Chen and R. J. Simpson, J. Proteomics, 2012, 76, 141–149 CrossRef CAS PubMed.
- O. K. Kwon, J. M. Jeon, E. Sung, A.-Y. Na, S. J. Kim and S. Lee, Cancer Genomics Proteomics, 2018, 15, 279–290 CrossRef CAS PubMed.
- N. M. Novikov, S. Y. Zolotaryova, A. M. Gautreau and E. V. Denisov, Br. J. Cancer, 2021, 124, 102–114 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm01351d |
| This journal is © The Royal Society of Chemistry 2025 |