
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diffusive evaporation dynamics in polymer solutions is ubiquitous†
Max
Huisman
 a,
Wilson C. K.
Poon
a,
Patrick B.
Warren
ab,
Simon
Titmuss
a and
Davide
Marenduzzo
*a
a,
Wilson C. K.
Poon
a,
Patrick B.
Warren
ab,
Simon
Titmuss
a and
Davide
Marenduzzo
*a
aSUPA and School of Physics and Astronomy, The University of Edinburgh, Peter Guthrie Tait Road, Edinburgh EH9 3FD, UK. E-mail: davide.marenduzzo@ed.ac.uk
bThe Hartree Centre, STFC Daresbury Laboratory, Warrington, WA4 4AD, UK
First published on 22nd January 2025
Abstract
Recent theory and experiments have shown how the buildup of a high-concentration polymer layer at a one-dimensional solvent–air interface can lead to an evaporation rate that scales with time as t−1/2 and that is insensitive to the ambient humidity. Using phase field modelling we show that this scaling law constitutes a naturally emerging robust regime, diffusion-limited evaporation (DLE). This regime dominates the dynamical state diagram of the system, which also contains regions of constant and arrested evaporation, confirming and extending understanding of recent experimental observations and theoretical predictions. We provide a theoretical argument to show that the scaling observed in the DLE regime occurs for a wide range of parameters, and our simulations predict that it can occur in two-dimensional geometries as well. Finally, we discuss possible extensions to more complex systems.
1 Introduction
Solvent evaporation from concentrated solutions or suspensions is an omnipresent phenomenon. The apparent simplicity of this process is deceptive: it is in fact ridden with complications due to the interaction between multiple components and the environment, leading to complex and fascinating dynamics. Such dynamics can in turn fundamentally alter the evaporative behaviour. Its understanding is therefore important for designing or controlling any process that involves drying. For instance, evaporative dynamics controls the application of paints and inks, where the formation of a defect-free skin upon drying is desired.1 It is also important in food preservation, where moisture content reduction increases shelf-life, but may also adversely affect flavour or texture.2 Due to such practical applications and fundamental interest in the dynamics of multi-component complex systems, the physics of evaporating polymer solutions and colloidal suspensions has inspired numerous investigations.3–7A number of these pertain to quasi-one-dimensional evaporation from the open end of a long capillary. In this geometry, the phase behaviour of aqueous lipid solutions can respond to varying ambient water activity ae (equivalently, relative humidity) in such a way as to render the evaporation rate practically independent of ae.5 It is suggested that in this regime, the mass flux is diffusion-controlled, Jevap ∼ t−1/2. A similar regime was predicted theoretically by Salmon et al.6 They argue that the evaporation-induced advective flux causes the growth of a concentrated ‘polarization layer’ at the interface, leading to mass loss increasing with the square root of time, hereafter referred to as diffusion-limited evaporation (DLE). However, the stability range of this DLE regime in parameter space was not explored because current calculations assume diffusional dynamics.
We have recently tested the predictions of Salmon et al., and observed a regime of ae -independent evaporation in which the rate decreases as t−1/2.8 We also found a transition from constant evaporation rate at early times to this t−1/2 regime, consistent with the building up of a polymeric ‘polarisation layer’. The inclusion of elastic effects from the formation of a very thin ‘gelled skin’ right at the air–solution interface9,10 improves the agreement between theory and experiments.
On general grounds, we expect that Jevap ∼ t−1/2 should only be one of at least three dynamical regimes of mass loss in an evaporating polymer solution. At very low polymer concentration, we should approach pure solvent evaporation, where the mass loss m(t) ∼ t, giving a constant evaporation rate. In the absence of any evaporative driving force, for instance when the air is saturated with solvent, we expect m(t) ∼ t0. So, we expect that DLE, where Jevap ∼ t−1/2 or m(t) ∼ t1/2, represents an intermediate regime. Surprisingly, however, this is the only behaviour observed experimentally to date at long times.5,8,11 Why this is so is currently a puzzle.
Here we set up and investigate a one-dimensional continuum phase-field model for evaporation of a polymer solution, where the evaporation is hindered by the polymer. We indeed find three main evaporative regimes that transition into one another, with predictions for the diffusive regime that agree with other studies to date. Importantly, the DLE regime dominates the predicted state diagram of the system. We characterise the generic nature of this regime and propose an argument to explain why it is so pervasive. The physical picture that emerges is a simple one. A polymer layer grows from the interface into the bulk solution. When this layer becomes concentrated enough to act as a ‘porous plug’, Darcy solvent flow through this layer is the rate limiting step, so that the evaporative dynamics becomes diffusive. Finally, we show how the model can be extended to higher dimensions, or to study more complex systems such as aerosol droplets, important in respiratory virus transmission, or multilayered paints and coatings.
2 Methods
2.1 A phase field model for evaporation
Our phenomenological model consists of two phases, inside the drop and outside (= the atmosphere), connected through a continuous interface, with periodic boundary conditions (see ESI† for details). Two continuum fields are considered. The first of these is the order parameter ϕ. In phase field modelling, the field ϕ is used to distinguish between two phases, for instance indicating a change in orientational order.12 In our model ϕ represents the total volume density of the droplet, that differentiates the droplet phase with a higher value of ϕ, from the surrounding air, with a lower value of ϕ. The second field, c, represents the concentration of polymer, and is in practice non-zero only in the droplet phase. Note that ϕ and c have dimensionless units. Inside the droplet ϕ ≃ ϕ1, and c has some finite value, with c = c0 < ϕ1 initially. We follow the phase field convention that ϕ1 = 1 inside the drop, and note that therefore our model is fundamentally different from a two-fluid model, as ϕ + c ≠ 1. Outside the drop, there is no polymer (c ≃ 0), and the droplet phase field has some finite value ϕ ≃ ϕ0 < ϕ1: this phase represents ambient air, with ϕ0 the equivalent of the relative humidity.The dynamic evolution of these fields is governed by chemical potential gradients around the interfaces, stemming from a coupled free energy density f(ϕ,c). The two fields themselves are coupled through a convective term v(ϕ,c). Each phase field is described by a modified Cahn–Hilliard equation with an additional evaporative term:13
 | (1a) |
 | (1b) |
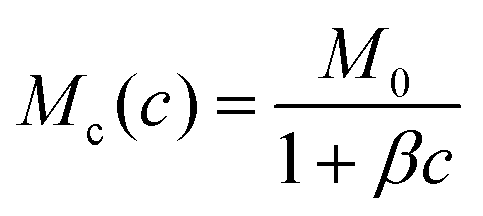 . We note that using a constant polymer mobility leads to qualitatively similar evaporation dynamics, which was also observed in ref. 6.
. We note that using a constant polymer mobility leads to qualitatively similar evaporation dynamics, which was also observed in ref. 6.
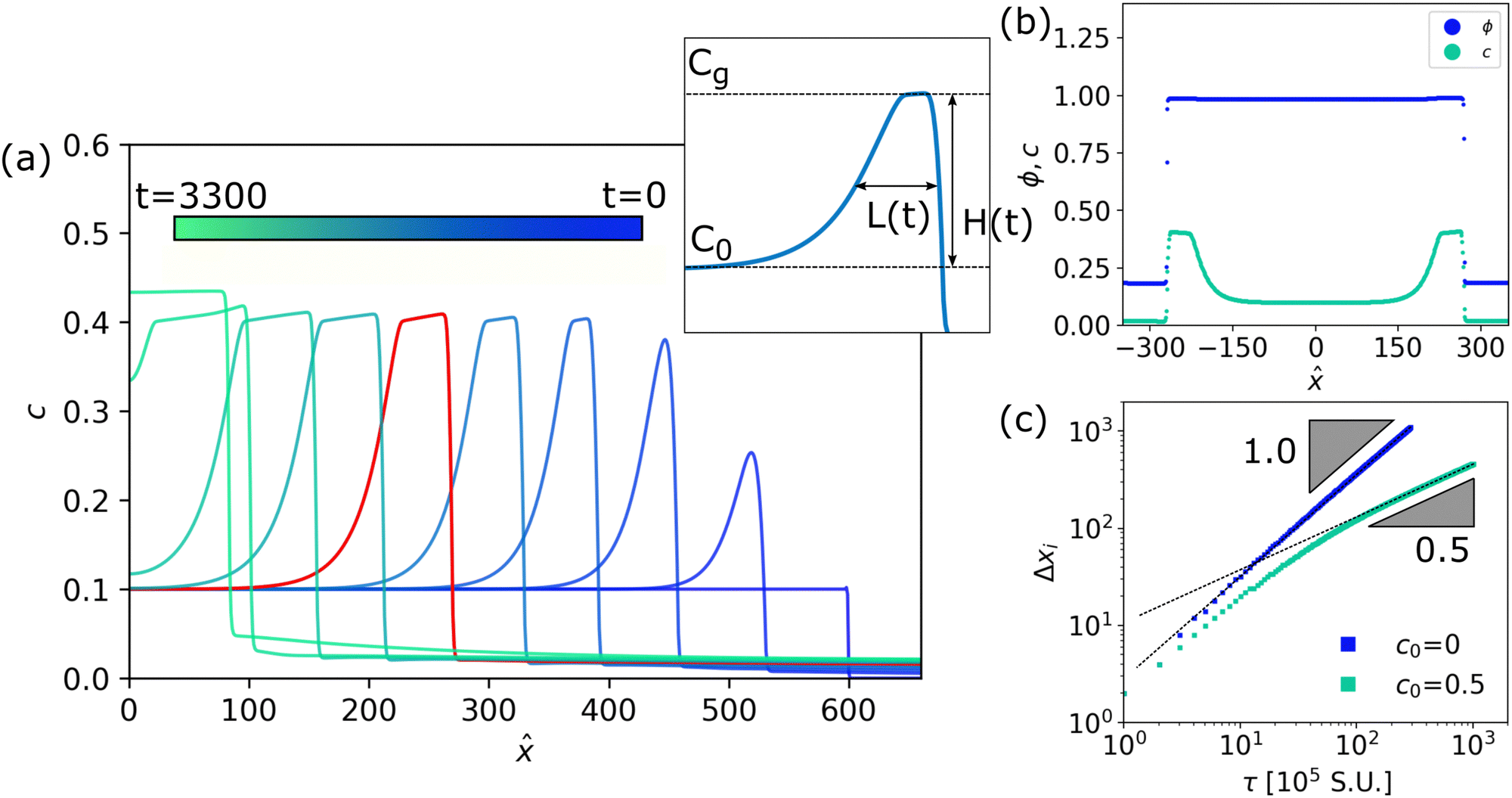 | ||
| Fig. 1 Dynamic evolution of a unidirectional drying polymer-solvent system in 1D. (a) Time evolution of polymer concentration profiles c in a 1D polymer-solvent slab during evaporation. The colorbar indicates the time corresponding to the concentration profiles. The x-coordinates are shifted from the simulation coordinates such that the middle of the droplet is located at x = 0. Inset: Schematic of the initial concentration c0, gelation concentration cg, width of the polymer layer L(t) (taken as the Full Width Half Maximum) and the peak height of the polymer layer H(t). (b) Snapshot of a typical simulation profile for ϕ and c, at time t = 1800, which corresponds to the red profile of c in panel (a). The x-coordinates are shifted so that the middle of the droplet is located at x = 0. (c) Evolution of the interface Δxi plotted over time, comparing a system with added polymer to a system of pure solvent. A log–log scaling is applied to highlight the different long time power-law behaviour. Simulation units (S.U.) for space Δxi and time t can be converted to physical units by applying the scalings L = 2.5 × 10−10 m and T = 6.3 × 10−9 s, respectively, as presented in the main manuscript. | ||
The quantity vi is the interfacial velocity of the evaporating droplet. Physically, the driving force for evaporation is the interfacial water activity difference.14 In the absence of polymer, it was suggested that this can be represented by a gradient in ϕ,13 leading to vi ∼ ∇ϕ. In our model, as the water activity inside the droplet reduces with increasing presence of polymer, an increasing polymer concentration c should reduce evaporation. Therefore we take the phenomenological expression vi = γ∇(ϕ − (γ′/γ)c), with the parameters γ determining the relative importance of evaporation to the phase fields and γ′ the contribution of c to reducing evaporation. In the droplet with binary composition, ∇(ϕ − (γ′/γ)c) is the effective solvent gradient leading to evaporation. We note that this expression is only valid for evaporation if ϕ ≥ (γ′/γ)c, and that the expression is similar to those used in other models in terms of water activity6 or partial pressure.15 Using an alternative expression, where the interface velocity also depends on the local phase field as vi = ϕ × γ∇(ϕ − (γ′/γ)c), gives similar results in the same parameter range, with a slight renormalization, as we show in the ESI.† Finally, v = −vi in eqn (1b) is the water velocity which advects the polymer towards the interface. Inside the droplet ∇c ≠ 0, so in eqn (1b) there exists a convective flux of polymer towards the interface, which is the evaporating solvent flux to the interface, compressing the polymer.16 This is not the case in eqn (1a), as inside the droplet ∇ϕ = 0, hence the dynamics there is purely diffusive.
The term driving droplet evaporation is therefore vi·∇ϕ at the droplet–air interface, which leads to droplet shrinking. Interestingly, in the absence of c, this has the form of a square gradient term, similar to the key nonlinearity in the Kardar–Parisi–Zhang (KPZ)17 equation, but with the opposite sign with respect to the one normally considered for growing interfaces.
The local chemical potential μ is derived from the free energy density f as  for the solvent and
for the solvent and 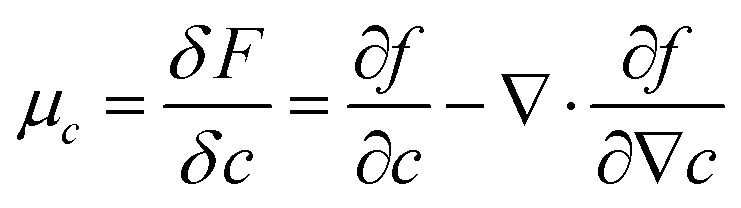 for the polymer. We note that in phase field modelling, it is common practice to use a free energy landscape f(ϕ,c) of the phase fields ϕ and c to generate physically appropriate chemical potential terms that describe the evolution of the phase fields in eqn (1a) and (1b).12 This definition of ϕ and c as phase fields is different compared to theoretical treatments such as Landau–Ginzburg theory, that use thermodynamic density fields. We use a Landau-like free energy density for the phase fields ϕ and c as
for the polymer. We note that in phase field modelling, it is common practice to use a free energy landscape f(ϕ,c) of the phase fields ϕ and c to generate physically appropriate chemical potential terms that describe the evolution of the phase fields in eqn (1a) and (1b).12 This definition of ϕ and c as phase fields is different compared to theoretical treatments such as Landau–Ginzburg theory, that use thermodynamic density fields. We use a Landau-like free energy density for the phase fields ϕ and c as
 | (2) |
The first term ensures that the system separates into a solvent-rich droplet ϕ1 with polymer (c > 0) and a surrounding vapor phase ϕ0 that contains no polymer (c = 0), so that ϕ0 represents the solvent concentration. κϕ and κc determines the bare surface tension of the droplet and polymer. The phenomenological term  is necessary to confine the polymer into the droplet interior, and its form is chosen in analogy to other types of phase field modelling;18 similar to those types of models, we expect that different forms favouring polymer confinement should lead to qualitatively similar results. The term
is necessary to confine the polymer into the droplet interior, and its form is chosen in analogy to other types of phase field modelling;18 similar to those types of models, we expect that different forms favouring polymer confinement should lead to qualitatively similar results. The term  penalises the transfer of polymer across the interface, where
penalises the transfer of polymer across the interface, where 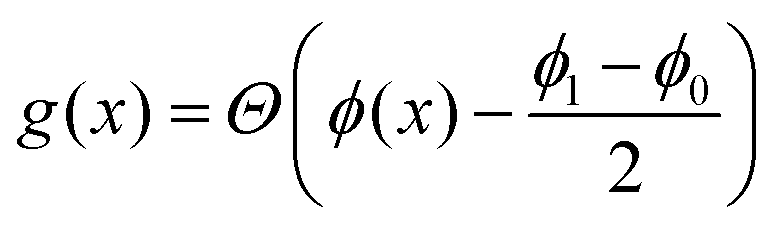 is an indicator function defined in terms of the Heaviside Θ such that g = 0 if
is an indicator function defined in terms of the Heaviside Θ such that g = 0 if  and g = 1 otherwise. When c is high enough to induce gelation, a permanent elastic stress develops, increasing the osmotic pressure and thereby the chemical potential.19 We approximate the bulk osmotic modulus contribution with
and g = 1 otherwise. When c is high enough to induce gelation, a permanent elastic stress develops, increasing the osmotic pressure and thereby the chemical potential.19 We approximate the bulk osmotic modulus contribution with  ,10 where Kg is a (constant) bulk osmotic modulus, cg is the gelation concentration of the polymer, and G(x) = Θ(c(x) − cg) is another indicator function defined again in terms of the Heaviside Θ. The remaining terms represent the virial coefficient for polymer diffusion (b0) and excluded volume effects of the polymer (b1). We note that eqn (2) should be accordingly adjusted to be used for modelling other systems than evaporating polymer–water droplets.
,10 where Kg is a (constant) bulk osmotic modulus, cg is the gelation concentration of the polymer, and G(x) = Θ(c(x) − cg) is another indicator function defined again in terms of the Heaviside Θ. The remaining terms represent the virial coefficient for polymer diffusion (b0) and excluded volume effects of the polymer (b1). We note that eqn (2) should be accordingly adjusted to be used for modelling other systems than evaporating polymer–water droplets.
From eqn (2), the system spontaneously phase-separates into a droplet phase and an environment phase, independent of initial conditions. We tested this by initializing the system with a sinusoidal variation of ϕ and c with x, see ESI,† and found that this system quickly phase-separates and stabilizes into the general profiles seen in Fig. 1b.
Finally, we note that another way to approach the evaporation problem could be to have a two-field (or Landau–Ginzburg) model, with one field for the interior of the droplet and another for the exterior, which are joined by a moving boundary condition. However, the implementation of such a model is non-trivial. In our implementation of the single phase-field model the moving boundary, i.e. the interface, is an emergent feature from the underlying physics. A numerical drawback of our implementation is that a fine-grained grid is required to resolve the interface structure. For one-dimensional and radially symmetric problems this is not an issue, but such limitations might become more relevant for systems with increased spatial heterogeneity. Another numerical cost is the need to construct a sufficiently elaborate phase-field model that captures all the relevant physics, which leads to a relatively large amount of numerical parameters in the governing equations. This is, however, expected to be similar compared to moving boundary problems, where multiple numerical parameters are also likely to be required to correctly describe the physics in the system. For our phase-field model, we have thoroughly explored the relevant parameter spaces, for which our findings are robust.
3 Results and discussion
3.1 Unidirectional drying in 1D
We solve a 1D version of our model with periodic boundary conditions using the computational procedure provided in the ESI,† and defining the origin of the x coordinate to be in the middle of the droplet. For the case of ϕ1 = 1, c0 = 0.1, ϕ0 = 0.2 and γ′/γ = 1.50, Fig. 1a shows the polymer c(x,t) in the right half (x > 0) of the droplet at a series of time points, while Fig. 1b plots the profiles of ϕ and c in the full droplet at time t = 1800. These results agree qualitatively with previous work.9 As the drop shrinks, a peak in c(x,t) develops just within the interface – a ‘polarisation layer’. When the peak height, H(t), reaches cg, the peak stops growing and flattens into a plateau of increasing width, L(t) (Fig. 1a inset), until the concentration in the droplet is homogeneous and the droplet continues to shrink slowly. We note that outside the droplet the polymer concentration is not exactly c = 0, indicating minor leakage of polymer into the environment. Such minor leakage does not significantly affect the overall evaporation dynamics.To quantify evaporative dynamics, consider the position of the interface xi, taken to be the position of the peak in c(x,t). Fig. 1c shows a log–log plot of Δxi(t) = xi(t) − xi(0) = m(t) by mass conservation if only solvent leaves the interface. For pure solvent (c0 = 0), Δxi ∼ t, so that J ∼ d(Δxi)/dt is constant, which is a well-known result.14 A solvent-polymer mixture (c0 = 0.5, with ϕ0 = 0.35 and γ′/γ = 1.50) behaves differently. After an initial linear regime, the evaporation slows down and approaches a steady state where Δxi ∼ t1/2 and J ∼ t−1/2, as found by experiments5,8 and theory.6
3.2 Stability of the DLE regime
To assess the relative stability of the Δxi ∼ t and ∼t1/2 regimes and explore the possibility of other forms of scaling, we scan two parameters. The first, γ′/γ, regulates the extent to which c reduces the convective evaporation speed – recall vi = γ∇(ϕ − (γ′/γ)c). The second is the phase field value outside the droplet, ϕ0, which is equivalent to relative humidity and governs the evaporative driving force.For the exponent α in m(t) ∼ tα, the state diagram in Fig. 2a displays three dynamical regimes separated by relatively sharp boundaries, as indicated by the white contour lines. We identify the pure solvent-like regime (α → 1) in the bottom left corner of the state diagram and the arrested evaporation regime (α → 0) in the top right corner of the state diagram. The DLE regime where α ≈ 0.5 occupies the largest region in the state diagram, and is therefore the most robust. This is consistent with the fact that diffusive dynamics is the behaviour typically reported in experiments to date.
 | ||
Fig. 2 State diagrams of the α exponent in m(t) ∼ tα with varying ϕ0 and γ′/γ, in a system with c0 = 0.5, using (a) a Landau expansion for the polymer free energy 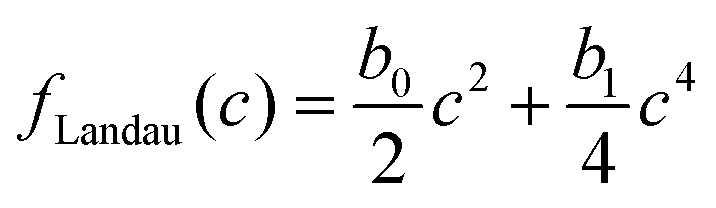 and (b) a Flory–Huggins free energy fFH(c) = b0(1 − c)ln(1 − c) +χc(1 − c). Countour lines (white) are added to highlight the transition between the different evaporation regimes. and (b) a Flory–Huggins free energy fFH(c) = b0(1 − c)ln(1 − c) +χc(1 − c). Countour lines (white) are added to highlight the transition between the different evaporation regimes. | ||
To understand the stability of the Δxi ∼ t1/2 regime, note that eqn (1) become diffusion equations in the limit vi → 0.6 The system cannot start in this regime, but can only approach it asymptotically: having γ′/γ ∼ ∇iϕ/∇ic to give vi → 0 (where ∇i ≡ gradient at the interface) means no evaporation in the first place. We therefore need γ′/γ ≲ ∇iϕ/∇ic to confer a finite initial evaporation rate, which then decreases with time as interfacial polymer accumulates and the system approaches the diffusive regime (α = 0.5) asymptotically, Fig. 3a. How fast this happens depends on the effectiveness of interfacial polymer in reducing evaporation, which is controlled by γ′.
 | ||
Fig. 3 Diffusion-limited evaporation (DLE) after long times that is independent of the external driving force. (a) Evolution of the exponent α in m(t) ∼ tα over time steps τ in a system with ϕ0 = 0.35 and γ′/γ = 1.5, settling on a time exponent α = 0.5. α is found through power law fitting of m(t) = Btα using α = d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnm(t)/dlnt. (b) Evolution of Δxi over the square root of time τ1/2, showing independence of the external driving force ϕ0. Systems shown are ϕ0 = 0.7 with γ′/γ = 0.415, ϕ0 = 0.5 with γ′/γ = 1.00, ϕ0 = 0.3 with γ′/γ = 1.85 and ϕ0 = 0.1 with γ′/γ = 2.75. In (a) and (b) simulation units (S.U.) for space and time are L = 2.5 × 10−10 m and T = 6.3 × 10−9 s, respectively. (c). Evolution of the interface in dimensional units, scaled to experimental data as ΔXi = [X]Δxi, with [X] = 4.5 × 10−5 m, and T = [T]t, with [T] = 9 × 10−4 s. Simulation dataset is from a system with ϕ0 = 0.5 with γ′/γ = 1.00. Experimental dataset at 50% relative humidity is reproduced from ref. 8. lnm(t)/dlnt. (b) Evolution of Δxi over the square root of time τ1/2, showing independence of the external driving force ϕ0. Systems shown are ϕ0 = 0.7 with γ′/γ = 0.415, ϕ0 = 0.5 with γ′/γ = 1.00, ϕ0 = 0.3 with γ′/γ = 1.85 and ϕ0 = 0.1 with γ′/γ = 2.75. In (a) and (b) simulation units (S.U.) for space and time are L = 2.5 × 10−10 m and T = 6.3 × 10−9 s, respectively. (c). Evolution of the interface in dimensional units, scaled to experimental data as ΔXi = [X]Δxi, with [X] = 4.5 × 10−5 m, and T = [T]t, with [T] = 9 × 10−4 s. Simulation dataset is from a system with ϕ0 = 0.5 with γ′/γ = 1.00. Experimental dataset at 50% relative humidity is reproduced from ref. 8. | ||
As γ′/γ drops, this effectiveness decreases, requiring a larger polarisation layer that takes longer to establish to approach the diffusive regime. So, for finite system size and observation time, there exists a (γ′/γ)min = ε below which the system will not cross over to Δxi ∼ t1/2 behaviour. We expect ε to increase with the polymer mobility, Mc: more mobile polymers require a longer time to build up a large enough polarisation layer to slow evaporation. On the other hand, a stronger driving force for evaporation due to lower external solvent concentration, ϕ0, requires the polymer to be more effective in reducing evaporation for diffusive behaviour to emerge; so, ε should decrease with increasing ϕ0, as observed, Fig. 2.
We establish that the scaling results from our modelling approach are insensitive to the exact implementation of the polymer free energy, by replacing the polymeric contribution from a Landau expansion 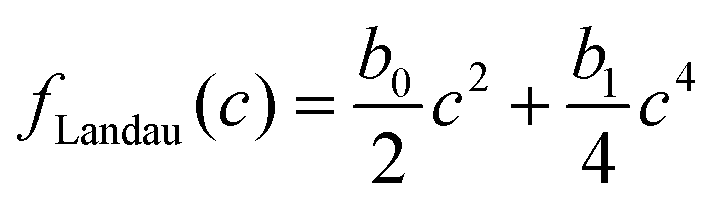 to the free energy density (eqn (2)) with the Flory–Huggins mean field expression fFH(c) = b0(1 − c)ln(1 − c) + χc(1 − c), assuming a large degree of polymerization N ≫ 1, see ESI† for more details. The resulting state diagram for varying γ′/γ and ϕ0 in Fig. 2b is dominated by a large region where m(t) ∼ t0.5, indicating DLE, and the transition of long-times exponents from α = 1 to α = 0.5 and α = 0 in m(t) ∼ tα is qualitatively similar between Fig. 2a and b upon increasing γ′/γ and ϕ0. We note that the boundaries between the evaporation regimes in Fig. 2b are less well-defined, which is shown by comparing a line in the state diagrams in the ESI.† We attribute this effect to the divergence of the term ∼ln(1 − c) in fFH(c) for c → 1, which leads to increased spreading and therewith slight variations in the settling interface concentration ci < 1, that are likely to depend on the evaporation driving force, set by ϕ0.
to the free energy density (eqn (2)) with the Flory–Huggins mean field expression fFH(c) = b0(1 − c)ln(1 − c) + χc(1 − c), assuming a large degree of polymerization N ≫ 1, see ESI† for more details. The resulting state diagram for varying γ′/γ and ϕ0 in Fig. 2b is dominated by a large region where m(t) ∼ t0.5, indicating DLE, and the transition of long-times exponents from α = 1 to α = 0.5 and α = 0 in m(t) ∼ tα is qualitatively similar between Fig. 2a and b upon increasing γ′/γ and ϕ0. We note that the boundaries between the evaporation regimes in Fig. 2b are less well-defined, which is shown by comparing a line in the state diagrams in the ESI.† We attribute this effect to the divergence of the term ∼ln(1 − c) in fFH(c) for c → 1, which leads to increased spreading and therewith slight variations in the settling interface concentration ci < 1, that are likely to depend on the evaporation driving force, set by ϕ0.
To understand the physics underpinning the late-stage, diffusive regime, note that by this stage, the chemical potential of the solvent (at partial pressure p) just inside the interface (where the polarisation layer is at its most concentrated), μ(p)|xi, has nearly equilibrated with that of the solvent vapour outside, and so is nearly constant. At the same time, the solvent chemical potential in the middle of the droplet, μ(p)|x=0, is also constant. So, there is a constant osmotic pressure difference driving solvent flow through the polarisation layer towards the interface, Δp = p|xi−px=0 < 0 (because μ(p)|xi < μ(p)|x=0). Treating the growing polarisation of thickness L(t) as a porous medium of Darcy permeability k implies the solvent flux  , with η the solvent viscosity. In the DLE regime in steady state, this flux is exactly balanced by the evaporative flux, which mass conservation requires to be dL/dt, so that we have dL/dt ∼ L−1, or L(t) ∼ t1/2, as also found in a recent theory20 as well in our numerics (see ESI†). Darcy's law then implies J ∼ t−1/2, consistent with our finding that Δxi ∼ t1/2 at long times (recall that J ∼ d(Δxi)/dt), Fig. 1c.
, with η the solvent viscosity. In the DLE regime in steady state, this flux is exactly balanced by the evaporative flux, which mass conservation requires to be dL/dt, so that we have dL/dt ∼ L−1, or L(t) ∼ t1/2, as also found in a recent theory20 as well in our numerics (see ESI†). Darcy's law then implies J ∼ t−1/2, consistent with our finding that Δxi ∼ t1/2 at long times (recall that J ∼ d(Δxi)/dt), Fig. 1c.
For this physical argument to hold, we require that the rate of advective polymer accumulation, giving rise to the (growing) polarisation layer, dominates polymer diffusion which counteracts this buildup, i.e. the Péclet number  . Equivalently, this means that the diffusion is slow compared to the evaporation rate. While this is true at short times, it seems at least at first sight that this condition will be broken as late times where navely one might expect Pe → 0 as vi → 0. However, in the DLE regime, our argument suggests vi ∼ ṁ(t) ∼ t−1/2, L(t) ∼ t1/2. So in fact at late times Pe ∼ t0, provided that
. Equivalently, this means that the diffusion is slow compared to the evaporation rate. While this is true at short times, it seems at least at first sight that this condition will be broken as late times where navely one might expect Pe → 0 as vi → 0. However, in the DLE regime, our argument suggests vi ∼ ṁ(t) ∼ t−1/2, L(t) ∼ t1/2. So in fact at late times Pe ∼ t0, provided that 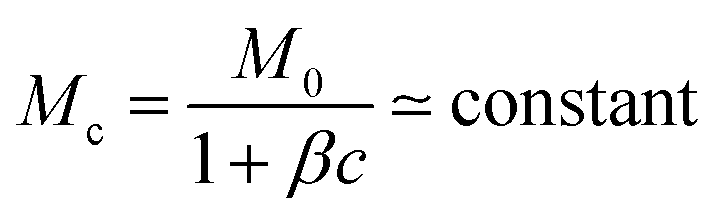 . The latter is a fair approximation for systems with β ∼ 0.1–1.0 and c ∼ 0.1–1.0. Our physical argument for the diffusive regime at long times is therefore self consistent, because the balance between advection and diffusion remains constant even as vi → 0. For a final check, we find Pe ≈ 102 ≫ 1 for a typical system in the DLE regime (γ′/γ = 1.50, ϕ0 = 0.30), so that, indeed, advection near the interface dominates over diffusion.
. The latter is a fair approximation for systems with β ∼ 0.1–1.0 and c ∼ 0.1–1.0. Our physical argument for the diffusive regime at long times is therefore self consistent, because the balance between advection and diffusion remains constant even as vi → 0. For a final check, we find Pe ≈ 102 ≫ 1 for a typical system in the DLE regime (γ′/γ = 1.50, ϕ0 = 0.30), so that, indeed, advection near the interface dominates over diffusion.
Considering the concentration dependence of our results, we identify the limiting evaporation regimes of m(t) ∼ tα for a varying initial concentration c0. For a system where c0 → 0, we expect that DLE never occurs and the system evaporates at a constant rate (α → 1). On the other hand, if c0 is sufficiently high that the system is already at thermodynamic equilibrium with the environment, evaporation never occurs (α → 0). For any other value of c0, we expect to observe the behaviour where 0 < α < 1, which approaches α → 0.5 as long as Pe ≫ 1 and the system is sufficiently large that a polarization layer can form at the solution–air interface.
In the diffusive regime, it was previously predicted6 that the mass loss rate should be independent of the external driving force. For water evaporation into air, the driving force is the relative humidity ae,14 which for us is ‘tuned’ by ϕ0. Plotting m vs. t1/2 whilst varying ϕ0 and γ′/γ, Fig. 3b, shows that this is indeed the case in our model.
This is a direct consequence of the fact that interfacial polymer concentration has reached a constant value, cg, so that it is the polymer concentration gradient in the polarisation layer rather than the external humidity that drives water transport. In the theory of Salmon et al.,6 the same physics emerges due to the sharp fall in water activity at high polymer concentrations, so that the late stage interfacial polymer concentration varies very little over a broad range of external water activities. In both cases, the humidity independence is necessarily correlated with the emergence of DLE. However, such correlation is not logically necessary. In our earlier experiments, the formation of a thin polymer skin at the solution–air interface due to rapid adsorption also gives rise to humidity-independent evaporation, but without a porous polarisation layer, the dynamics is not diffusive.8
3.3 Simulation units and comparison to experimental systems
We find the simulation units in time (T) and space (L) of our system, by comparing the simulation parameters in eqn (2) to measured physical quantities of a water-PVA solution, for which measurements have been performed in experiments where a solution evaporates unidirectionally.8 Using the units of the simulation parameters from Table S1 in the ESI,† we obtain a set of equations for (1) the surface tension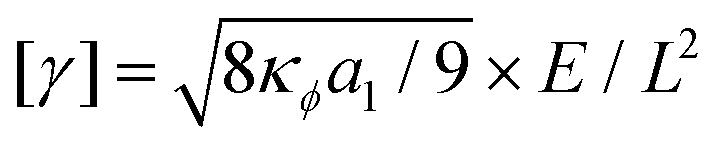 ,21 (2) the diffusion coefficient [D] = M0b0 × L2/T and (3) the osmotic pressure [π] = b0 × E/L3, where E is the unit of energy. Using [γ] ≈ 0.07, [D] ≈ 10−11 (ref. 15) and [π] ≈ 107 Pa,22 we find L ≈ 2.5 × 10−10 m, T ≈ 6.3 × 10−9 s and energy unit E ≈ 1.56 × 10−20 J. Finally, we calculate a dimensional evaporation rate from the slope of a simulation without polymer (ϕ0 = 0.5, c0 = 0) as Vev ≈ 2 × 10−5L/T ≈ 0.8 μm s−1, which is comparable to measurements in ref. 8.
,21 (2) the diffusion coefficient [D] = M0b0 × L2/T and (3) the osmotic pressure [π] = b0 × E/L3, where E is the unit of energy. Using [γ] ≈ 0.07, [D] ≈ 10−11 (ref. 15) and [π] ≈ 107 Pa,22 we find L ≈ 2.5 × 10−10 m, T ≈ 6.3 × 10−9 s and energy unit E ≈ 1.56 × 10−20 J. Finally, we calculate a dimensional evaporation rate from the slope of a simulation without polymer (ϕ0 = 0.5, c0 = 0) as Vev ≈ 2 × 10−5L/T ≈ 0.8 μm s−1, which is comparable to measurements in ref. 8.
To establish that our simulations are representative of experimental data, we map our results to the data from ref. 8 in Fig. 3c. We find excellent agreement between these two systems by scaling our simulation data to dimensional units as ΔXi = [X]Δxi in meters [m], where [X] = 4.5 × 10−5 m, and T = [T]t in seconds [s], where [T] = 9 × 10−4 s. It should be noted that these scalings are much higher than the simulation units in the system, which can be attributed to the experimental system being much larger than we can reasonably simulate, but that the ratios L/T ≈ 0.05 and [X]/[T] ≈ 0.043 are consistent.
3.4 Extensions of the model to more complex systems
One of the major advantages of our phase field model is the ease with which it can be adapted to more complex systems and used to explore higher dimensions. Fig. 4a shows the evolution of a 2D evaporating drop geometry for a system with c0 = 0.2, ϕ0 = 0.35 and γ′/γ = 3.0. As in 1D, a concentrated polymer layer forms at the droplet–air interface over time. | ||
| Fig. 4 Dynamic evolution of evaporating droplets in 2D. (a) Snapshots of the time evolution of polymer concentration c in a 2D evaporating droplet, with c0 = 0.2, ϕ0 = 0.35 and γ′/γ = 3.0. (b) Evolution of the log-corrected area D2ln[(Lx/D) + 0.5]/G of the system in (a) plotted over time, comparing a system with added polymer to a system of pure solvent. (c) Evolution of the exponent ζ in D2ln[(Lx/D) + 0.5] ∼ tζ over timesteps τ in a system with ϕ0 = 0.35 and γ′/γ = 3.0, settling on a time exponent ζ = 0.5. ζ is found through power law fitting using ζ = dln[1 − (D2)ln[(Lx/D) + 0.5]/G]/dlnt. In (b) and (c) simulation units (S.U.) for space and time are L = 2.5 × 10−10 m and T = 6.3 × 10−9 s, respectively. | ||
For 2D or 3D droplets of diameter D evaporating in an unconfined environment, D2 ∼ t, so that the evaporation rate is constant.23 However, solving the same problem in a confined system with a constant solvent chemical potential imposed at the system's boundary leads to deviations from this ‘D2 law’. If viscous and buoyancy effects can be neglected, theory23 predicts that in a 2D finite system of this kind,
| D2ln[(Lx/D) + 0.5]/G = 1 − Ct, | (3) |
ln[(Lx/D0) + 0.5]. Over the course of the simulations that give the results shown in Fig. 4, the value of ϕ at the (periodic) boundaries increases by ≲ 2%, so that we may expect eqn (3) to hold to a good approximation.
Our data for the evaporation of a pure solvent droplet, Fig. 4b, (blue; c0 = 0) indeed agree with eqn (3), as apparent from the approximately linear evolution of D2ln[(Lx/D) + 0.5]/G with t. However, in a system with added polymer, deviations are observed, Fig. 4b (green, c0 = 0.2), which recall Fig. 1c. We therefore fit these data to a power law with a running exponent ζ, D2ln[(Lx/D) + 0.5] ∼ tζ. The resulting ζ, Fig. 4c, clearly recalls Fig. 3a for the 1D case.
So, while a full study of the 2D case is beyond our scope, it seems reasonable to surmise that the state diagram in this case should also display a robust diffusive regime, provided that Pe is high enough. Previous experiments have demonstrated ambient humidity independent evaporation of droplets containing large glycoproteins,24 which is consistent with our surmise.
4 Conclusions and outlook
In summary, we have applied a phase field modelling approach to study the evaporative dynamics of a polymer–solvent mixture. Whilst our approach is phenomenological, rather than being derived from rigorously coarse-graining a microscopic theory, we expect it to describe the system in a qualitatively accurate way, in line with previous work on phase fields. Our key result is that the DLE regime, where evaporation rate decays with time as t−1/2, is a robust dynamical regime found over a range of parameter values. We rationalise this scaling with a simple mathematical and physical argument, according to which the −1/2 exponent is due to a diffusive growth of the polymer layer, and a Darcy flow of the solvent due to the ensuing pressure difference close to the droplet–air interface. For this argument to be self-consistent, the Péclet number Pe should remain high and nearly constant, to achieve a non-equilibrium steady state where advection dominates over diffusion at all times. We show that this requirement is, perhaps surprisingly, indeed met.Our model is in quantitative agreement with previous theoretical and experimental results, including a near-independence of evaporation rates on relative humidity in the DLE regime. Such agreement gives confidence for applying this phase field model to study solvent and solute transfer in more complex systems and geometries. We have shown preliminary results for the evaporation of a 2D droplet to demonstrate this potential. Possible future applications include dissolution processes (e.g., making instant coffee) and the drying of multilayers involving multiple solvents and solutes (e.g., oil paintings25). In the latter case, our approach has the added advantage that no assumptions need to be made on the phase of layers and/or the location of the interface over time.
Data availability
All data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding was provided by the University of Edinburgh. For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising from this submission.Notes and references
- B. J. D. Gans and U. S. Schubert, Langmuir, 2004, 20, 7789–7793 CrossRef PubMed.
- Z. Erbay and F. Icier, CRC Crit. Rev. Food Sci. Nutr., 2010, 50, 441–464 CrossRef PubMed.
- M. G. Hennessy, G. L. Ferretti, J. T. Cabral and O. K. Matar, J. Colloid Interface Sci., 2017, 488, 61–71 CrossRef CAS PubMed.
- P. G. de Gennes, Eur. Phys. J. E, 2002, 7, 31–34 Search PubMed.
- K. Roger, M. Liebi, J. Heimdal, Q. D. Pham and E. Sparr, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 10275–10280 CrossRef CAS PubMed.
- J. B. Salmon, F. Doumenc and B. Guerrier, Phys. Rev. E, 2017, 96, 032612 CrossRef PubMed.
- M. Rezaei and R. R. Netz, Phys. Fluids, 2021, 33, 091901 CrossRef CAS PubMed.
- M. Huisman, P. Digard, W. C. Poon and S. Titmuss, Phys. Rev. Lett., 2023, 131, 248102 CrossRef CAS PubMed.
- K. Ozawa, T. Okuzono and M. Doi, Japan. J. Appl. Phys., 2006, 45, 8817–8822 CrossRef CAS.
- T. Okuzono and M. Doi, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 77, 030501(R) CrossRef PubMed.
- K. Roger and J. J. Crassous, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2105530118 CrossRef CAS PubMed.
- N. Provatas and K. Elder, Phase-field methods in materials science and engineering, John Wiley & Sons, 2011 Search PubMed.
- H. G. Lee, J. Yang, S. Kim and J. Kim, Appl. Math. Comput., 2021, 390, 125591 CrossRef.
- E. L. Cussler, Diffusion: Mass Transfer in Fluid Systems, CUP, Cambridge, UK, 1997 Search PubMed.
- M. Okazaki, K. Shioda, K. Masuda and R. Toei, J. Chem. Eng. Jpn., 1974, 7, 99–105 CrossRef CAS.
- F. Meng, L. Luo, M. Doi and Z. Ouyang, Eur. Phys. J. E, 2016, 39, 1–10 CrossRef CAS PubMed.
- M. Kardar, G. Parisi and Y.-C. Zhang, Phys. Rev. Lett., 1986, 56, 889–892 CrossRef CAS PubMed.
- R. Mueller, J. M. Yeomans and A. Doostmohammadi, Phys. Rev. Lett., 2019, 122, 048004 CrossRef CAS PubMed.
- L. Leibler and K. Sekimoto, Macromolecules, 1993, 26, 6937–6939 CrossRef CAS.
- L. Talini and F. Lequeux, Soft Matter, 2023, 19, 5835–5845 RSC.
- M. E. Cates and E. Tjhung, J. Fluid Mech., 2018, 836, P1 CrossRef.
- P. Bacchin, J. Leng and J.-B. Salmon, Chem. Rev., 2021, 122, 6938–6985 CrossRef PubMed.
- L. Fei, F. Qin, G. Wang, K. H. Luo, D. Derome and J. Carmeliet, Phys. Rev. E, 2022, 105, 025101 CrossRef CAS PubMed.
- E. P. Vejerano and L. C. Marr, J. R. Soc. Interface, 2018, 15, 20170939 CrossRef PubMed.
- J. R. Duivenvoorden, R. P. Kramer, M. H. van Eikema Hommes, P. D. Iedema, J. J. Hermans and K. Keune, Int. J. Heat Mass Tran., 2023, 202, 123682 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm01215a |
| This journal is © The Royal Society of Chemistry 2025 |