
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Liquid crystalline self-assembly of mixtures of rod- and wedge-shaped MIDA boronates†
Christopher
Schilling
a,
Soeren M.
Bauch
 a,
Eugen
Wuckert
a,
Anna
Zens
a,
Johanna R.
Bruckner
b and
Sabine
Laschat
*a
a,
Eugen
Wuckert
a,
Anna
Zens
a,
Johanna R.
Bruckner
b and
Sabine
Laschat
*a
aInstitut für Organische Chemie, Universität Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany. E-mail: sabine.laschat@oc.uni-stuttgart.de
bInstitut für Physikalische Chemie, Universität Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany
First published on 31st January 2025
Abstract
Rod-like MIDA boronates form smectic mesophases, while wedge-shaped MIDA boronates self-assemble into columnar mesophases. However, the phase behavior of mixtures is less understood. In order to obtain further insight on the molecular self-assembly of MIDA boronate mixtures two series of binary mixtures of rod-like and wedge-shaped mesogens were prepared. The phase behavior was studied using differential scanning calorimetry (DSC), polarizing optical microscopy (POM) and X-ray diffraction (XRD). The study revealed a strong dependency of the mesophase structure on the mesogen composition. Usage of a less bulky columnar mesogen suppressed the formation of columnar mesophases in the mixture and led to a decrease in melting and clearing temperatures. The phase behavior is discussed in terms of the packing parameter model typically applied for lyotropic liquid crystals.
Introduction
Liquid crystalline mixtures of rod-like mesogens are very important for applications such as liquid crystal displays (LCDs). This is due to the fact that the temperature range and phase stability of the mixture is significantly expanded as compared to the single components. Moreover, in such mixtures physical properties such as viscosity, optical refractive indices, elastic constants, electric permittivities etc. can be efficiently tailored in a desired fashion.1 Mixtures of rod-like mesogens can also lead to new mesophase types, which were not observed in the pure compounds as was recently reported by Mandle for the splay nematic phase NS.2Also for other functional liquid crystals the importance of mixtures to obtain new properties has been highlighted.3–7 On the one hand, mixtures of discotic liquid crystals have been examined in several theoretical studies.8–36 Most recently Sambritzki investigated binary mixtures of discotic liquid crystals differing in their molecular thickness by molecular dynamics (MD) simulations.37 Various experimental studies on discotic mixtures have been performed, dealing for example with bent-core mesogens38–41 forming columnar banana phases, chiral dopants forming chiral nematic discotic  phases in discotic host materials,42–45 donor–acceptor systems consisting of electron-rich discotic molecules and non-mesogenic electron acceptors such as trinitrofluorenone (TNF).46–49 In addition, mixtures of structurally closely related discotic compounds were also studied with respect to phase behaviour, electric conductivity or magnetic properties.50–60 On the other hand, mixtures of rod- and disk-shaped molecules,61 rod- and rod-shaped molecules62 and ILC compounds63 were much less explored.
phases in discotic host materials,42–45 donor–acceptor systems consisting of electron-rich discotic molecules and non-mesogenic electron acceptors such as trinitrofluorenone (TNF).46–49 In addition, mixtures of structurally closely related discotic compounds were also studied with respect to phase behaviour, electric conductivity or magnetic properties.50–60 On the other hand, mixtures of rod- and disk-shaped molecules,61 rod- and rod-shaped molecules62 and ILC compounds63 were much less explored.
Previous work by us has shown that the N-methyl-iminodiacetic acid (MIDA) boronate unit is a strongly beneficial mesophase promoter both from a fundamental point of view64–66 but also for applications such as electrooptical birefringence (Kerr effect).67 This is due to the high polarity of the MIDA unit (μ = 7.6 D) as compared to boronic acids (μ = 2.8 D) and borolanes (μ = 1.4 D), resulting in a pronounced tendency to form polar subdomains within the mesophase.64,68–70 Depending on the structure of the (hetero)aryl core and the number of alkoxy side chains, the phase type and stability of the corresponding rod-like or wedge-shaped MIDA boronates was controlled,64–66i.e., rod-like MIDA boronates formed lamellar smectic A (SmA) phases while wedge-shaped derivatives formed columnar hexagonal (Colh) phases (Scheme 1). With respect to mixtures between these components we were curious whether the rod-like or the wedge-shaped MIDA boronate would control the self-assembly of the mixture. The results towards this goal are reported below.
 | ||
| Scheme 1 Overview of preliminary work on rod-like and wedge-shaped MIDA boronates.64 | ||
Results and discussion
For a better overview and sake of clarity the following nomenclature will be used for the mixtures: the first series of mixtures consisting of MIDA boronates 1 and 2 carrying 1 and 2 dodecyloxy side chains respectively will be abbreviated 1/2 [X1] with the molar fraction X1 of 1. The second series of mixtures consisting of MIDA boronates 1 and 3 carrying 1 and 3 dodecyloxy side chains respectively will be abbreviated 1/3 [X1] with the molar fraction X1 of 1.Mixtures with molar fractions of X1 = 0.2, 0.4, 0.5, 0.6, 0.8 were prepared initially and studied by polarizing optical microscopy (POM), differential scanning calorimetry (DSC) regarding their phase behaviour. Selected mixtures were investigated by X-ray diffraction (XRD) to assign the phase structure. The results were compared with the known behaviour of the pure MIDA compounds 1, 2 and 3 respectively.64
First the series 1/2 [X1] with the molar fraction X1 of 1 was examined by POM. The pure mono-substituted boronate 1 (1/2 [1.0]), displayed at 180 °C upon cooling from the isotropic phase large areas of homeotropic alignment with a few Maltese cross defects, which are characteristic for a SmA phase (Fig. 1a) as reported earlier.64 For the pure disubstituted MIDA boronate 2 (1/2 [0.0]), fan-shaped textures were observed (Fig. 1g) in agreement with the previously assigned Colh phase for this MIDA boronate 2. Upon increasing the amount of bis-dodecyloxy MIDA boronate 2 the tendency towards homeotropic alignment decreased with decreasing molar fraction of the mono-dodecyloxy MIDA boronate 1. Textures changed from Maltese crosses and fan textures (e.g.Fig. 1b for X1 = 0.8) to uncharacteristic textures for X1 = 0.2 (Fig. 1f). For all studied mixtures 1/2 [X1] with X1 = 0.8–0.2 textures were easily shearable and highly fluid, suggesting smectic phases with low order were favoured as compared to columnar phase.71
 | ||
| Fig. 1 POM textures of mixture series 1/2 [X1] with molar fractions X1 = (a) 1.0 (180 °C), (b) 0.8 (125 °C), (c) 0.6 (128 °C), (d) 0.5 (124 °C), (e) 0.4 (120 °C), (f) 0.2 (123 °C), (g) 0.0 (160 °C). Taken during cooling from the isotropic liquid (cooling rate 5 K min−1). | ||
Next the series 1/3 [X1] with the molar fraction X1 of 1 was examined by POM (Fig. 2). For the pure tri-substituted MIDA boronate 3 (1/3 [0.0]), large fan-shaped textures were visible under the POM (Fig. 2g), which are characteristic for columnar phases in agreement with recent work.64 The mixtures 1/3 [X1] with X1 = 0.8 and X1 = 0.6 were easily shearable and possessed a high fluidity in analogy to the 1/2 [X1] mixtures of the other series. In contrast, 1/3 [X1] mixtures with X1 = 0.5–0.2 displayed a much higher viscosity as compared to the mixtures with higher molar fractions (X1 = 0.6–0.8). In comparison with the pure trisubstituted MIDA compound 3 (1/3 [0.0]), mixtures 1/3 [X1] with X1 = 0.5–0.2 showed a lower viscosity and better shearability.
 | ||
| Fig. 2 POM textures of mixture series 1/3 [X1] with molar fractions X1 = (a) 1.0 (180 °C), (b) 0.8 (121 °C), (c) 0.6 (82 °C), (d) 0.5 (119 °C), (e) 0.4 (120 °C), (f) 0.2 (125 °C), (g) 0.0 (82 °C). Taken during cooling from the isotropic liquid (cooling rate 5 K min−1). | ||
In order to assign the phase transition temperatures and enthalpies differential scanning calorimetry (DSC) experiments were performed. All DSC data of mixtures 1/2 [X1] are shown in Fig. S3 (ESI†) and are summarized in Table S3 (ESI†). The DSC curves of exemplary mixtures 1/2 [0.15], 1/2 [0.4] and 1/2 [0.5] are shown in Fig. 3. In the DSC curve of 1/2 [0.5] weak endothermal melting and clearing transitions at 55 °C and 145 °C, respectively, are visible (Fig. 3a). In the cooling cycle the isotropic to mesophase transition appeared at 146 °C, while no crystallization could be observed. Instead, slow vitrification of the sample occurred upon cooling in agreement with the DSC results reported for the MIDA boronates 1, 2 and 364 and Pyr(n)MIDA66 (for details of the molecular structure, see Fig. S1, ESI†). The DSC curves of 1/2 [0.4] (Fig. 3b) displayed endothermal melting and clearing at 57 °C and 134 °C upon heating, and in the cooling cycle the exothermal phase transitions appeared at 136 °C and 72 °C. In comparison with the pure mono- and bis-substituted MIDA boronates 1 (1/2 [1.0]), and 2 (1/2 [0.0]), melting and clearing transitions decreased considerably for the mixtures 1/2 [X1] with X1 = 0.5, 0.4. A phase diagram summarizes the results of the various mixtures 1/2 [X1] (Fig. 4a). When comparing the whole series of mixtures 1/2 [X1] with X1 = 0.8–0.2 with the pure compounds 1 (1/2 [1.0]), and 2 (1/2 [0.0]), clearing transitions decreased with decreasing X1, i.e., decreasing amount of 1 from 194 °C (X1 = 1.0) until the minimum value 137 °C for X1 = 0.4 and then increased again up to 169 °C for X1 = 0.0. On the other hand, melting transitions of the mixtures (X1 = 0.8–0.2) were significantly lower as compared to the values for the pure compounds 1 (1/2 [1.0]) and 2 (1/2 [0.0]), but for all mixtures (X1 = 0.8–0.2) the melting transitions remained relatively constant around 54–62 °C irrespective of the molar fraction.
 | ||
| Fig. 3 DSC traces of 1/2 [X1] mixture series: (a) 1/2 [0.5], (b) 1/2 [0.4] and (c) 1/2 [0.15] (2nd heating (bottom) and 2nd cooling (top) cycle, heating/cooling rate 10 K min−1). | ||
 | ||
| Fig. 4 Phase diagram of the binary systems (a) 1/2 [X1] and (b) 1/3 [X1] from DSC measurements (2nd heating cycle; rate 10 K min−1; red: SmA phase; blue: Colh phase; grey: crystalline; crosshatched grey: not investigated, but added for enhanced comparability; note that the SmA to Colh transition temperatures of X1 = 0.2 in (a) and X1 = 0.5 and 0.45 in (b) were determined from the 2nd cooling cycle). | ||
While initial POM investigation of the 1/2 [X1] mixtures suggested solely the presence of a SmA phase based on homeotropic alignment, bâtonnets and focal-conic fan texture (Fig. 2b–f), we were curious about the transition between the SmA and the Colh phases which has to occur somewhere between X1 = 0 and 0.2. Due to the symmetry change from D∞h symmetry with only one-dimensional long-range translational order in the SmA phase to the p6/mmm symmetry in the Colh phase, the phase transition has to be of first order according to Landau theory.72 Thus, a biphasic region is necessary to occur between the two phases. Therefore, the mixtures 1/2 [X1] with X1 = 0.15, 0.2 and 0.25 were examined more closely by DSC and POM. The DSC curve of mixture 1/2 [0.15] displayed an endothermal melting transition at 54 °C, a very weak mesophase to mesophase transition at 96 °C and a weak clearing transition at 125 °C in the heating cycle (Fig. 3c). In the cooling cycle the isotropic to mesophase transition occurred at 131 °C, the mesophase to mesophase transition at 104 °C and the crystallization at 60 °C, respectively. The change of the texture was also visible by POM upon cooling (Fig. S6a–c, ESI†) supporting the presence of Colh and SmA phases at higher and lower temperatures, respectively. At high temperatures the typical fan texture73 of the Colh phase prevails (Fig. S6a, ESI†) which turns into a less distinct, grainy texture at lower temperatures (Fig. S6c, ESI†). The latter uncharacteristic texture is associated with the uncommon rearrangement of the mesogens from the columnar to the lamellar structure of the SmA phase. Indeed, a biphasic region is observed in between the two phases, as predicted by symmetry arguments, in which the fan texture is spotted with darker patches (Fig. S6b, ESI†). DSC curves of 1/2 [0.20] (Fig. S3b, ESI†) again suggested the presence of both SmA and Colh phases. However, the transition was too weak to be analysed from the heating cycle, and thus could only be determined from the cooling cycle, which leaves some doubts whether or not the Colh phase at this composition is enantiotropic or only monotropic. In contrast, in the mixture 1/2 [0.25] the higher temperature mesophase totally disappeared (Fig. S6d, ESI†). Complementary POM studies showed Maltese crosses and homeotropic alignment (Fig. S6d, ESI†) suggesting that in this mixture only an enantiotropic SmA phase prevails. The phase diagram constructed from this data is shown in Fig. 4a. The presence of biphasic regions was neglected in the diagram due to their narrow temperature ranges and the associated difficulties in determining them.
Next, the mixtures 1/3 [X1] were investigated. All DSC data of 1/3 [X1] are shown in Fig. S4 (ESI†) and summarized in Table S4 (ESI†). The DSC curve of mixture of 1/3 [0.6] displayed an endothermal melting transition at 45 °C and an endothermal clearing transition at 96 °C upon heating, while the corresponding exothermal transition appeared at 102 °C and 55 °C in the cooling cycle (Fig. 5a). As the POM texture showed Maltese crosses with homeotropic alignment (Fig. 2a–c), an enantiotropic SmA phase was assigned. On the other hand, for the mixture 1/3 [0.4] melting and clearing transitions at 39 °C and 142 °C were both visible in the heating and cooling cycle (47 °C, 143 °C) (Fig. 5b). For this mixture the POM fan-like textures (Fig. 2e) strongly suggested a Colh phase. In order to characterise the SmA to Colh transition of the phase diagram shown in Fig. 4b, 1/3 [X1] mixtures with X1 = 0.55, 0.5 and 0.45 were examined additionally. For 1/3 [0.55] a single enantiotropic mesophase was detected (Fig. S6f, ESI†), which was assigned as a SmA phase based on the Maltese cross textures and homeotropic alignment observed by POM. In the DSC curve of 1/3 [0.45] a weak endothermal melting at 37 °C and a clearing transition at 126 °C were detected together with an extremely small hump which hints at the existence of two enantiotropic mesophases, but is too small to evaluate (Fig. 5c).
 | ||
| Fig. 5 Exemplary DSC traces of 1/3 [X1] mixture series: (a) 1/3 [0.6], (b) 1/3 [0.4] (c) 1/3 [0.45] and (2nd heating (bottom) and 2nd cooling (top) cycle, heating/cooling rate 10 K min−1). | ||
Upon subsequent cooling a small isotropic to mesophase transition at 130 °C, a weak exothermal mesophase to mesophase transition at 61 °C and a weak exothermal crystallization peak at 53 °C were detected. By POM fan-shaped textures at 100 °C indicated the presence of the Colh phase as a higher temperature phase (Fig. S6e, ESI†) and the SmA phase as lower temperature phase. Likewise, the DSC curve of the mixture 1/3 [0.5] displayed a lower temperature SmA phase and a higher temperature Colh phase (Fig. S4d, ESI†). Whether or not the Colh phase in these two mixtures is monotropic or if the transition enthalpy is just extremely small could not be fully elucidated at this point.
While Fig. 4a demonstrated the strong influence of the rod-like mesogen 1 on the phase type and the temperature-modulating effect of 2 in the 1/2 [X1] series, Fig. 4b revealed a much more pronounced effect of the wedge-shaped mesogen 3 on both phase type and transition temperatures in the 1/3 [X1] mixtures. Upon increasing the amount of trisubstituted MIDA boronate 3 a decrease of melting and particularly clearing temperatures were observed for 1/3 [0.8] and 1/3 [0.6] resulting in a decrease of the stability and temperature range of the SmA phase from 75 K (X1 = 1.0) to 31 K (X1 = 0.55) phase widths (Fig. S5, ESI†). Upon further increasing the amount of wedge-shaped mesogen 3 in the mixture, the SmA phase was disfavoured against the Colh phase. Both mesophases were present for X1 = 0.5, 0.45 until X1 = 0.4, where the SmA phase completely disappeared and only the Colh phase remained. Regarding clearing transitions of the Colh phase, increasing the relative amount of 3 (i.e., decrease of X1) resulted in increasing clearing temperatures with a maximum at X1 = 0.4 and then decrease until X1 = 0 (pure wedge-shaped mesogen 3). Similarly, to the 1/2 [X1] series, melting points were relatively little affected by the change of X1.
It appears that the additional side chain of 3 in comparison with 2 introduces a stronger effect on the molecular self-assembly. Whereas in the 1/2 [X1] system a high amount of 2 of up to X1 = 0.25 only resulted in the formation of a smectic layer structure rather than the formation of a columnar structure, while the 1/3 [X1] system showed a columnar phase at molar X1 fractions as high as X1 = 0.5. This may be rationalized by the increasing steric demand of 3 in comparison to 2 resulting in larger effective wedge size and increased space filling favouring columnar over layered phase structures.
For selected mixtures of both series X-ray diffraction experiments (WAXS, SAXS) were carried out. The equimolar mixture 1/2 [0.5] displayed a sharp small angle reflection at 36.0 Å, which was assigned as the (001) layer reflection of the SmA phase and a broad halo in the wide angle region around 4.8 Å caused by the liquid-like order of the flexible side chains (Fig. 6a, b and Table S5, ESI†). Temperature-dependent XRD measurements of the layer reflection showed a decrease of the d values with increasing temperature, which is characteristic for the SmA phase.74
 | ||
| Fig. 6 (a) XRD results of 1/2 [X1] mixtures; (a) WAXS profile of X1 = 0.5 at 108 °C and corresponding 2D patterns of the SAXS (left image, at 91 °C) and WAXS measurement (left image, at 108 °C); (b) temperature dependent layer spacing of X1 = 0.5; (c) comparison of temperature dependent layer spacing of the mixture of X1 = 0.5 with X1 = 1.0, i.e., the neat 1. | ||
Upon comparison of temperature-dependent layer distances of the mixture 1/2 [0.5] and the pure mono-substituted MIDA boronate 1 surprisingly the d values of 1/2 [0.5] increased much stronger with decreasing temperature as compared to the pure compound 1, i.e., the slope was 4 times larger as compared to the former case (Fig. 6c). This result might be rationalized by the different layer structures in the mixture vs. pure MIDA boronate. The smectic layer of pure 1 already possesses a high degree of order, which increases only slightly upon cooling and thus the d value also increases only slightly. In contrast, the mixture 1/2 [0.5] presumably possesses a lower order parameter close to the clearing temperature, resulting in a strong increase of the d value due to the strong increase of the order parameter upon cooling. Due to the observed thermal decomposition for all kind of different MIDA boronates,64–66 the temperature dependent XRD measurements of 1/2 [0.5] were only conducted at temperatures closer to the melting point, since the set of data points was considered sufficient to determine the phase order of the lamellar phase, i.e., SmA or SmC.
Presumably within the smectic layers a statistic distribution of both components 1 and 2 seems to be reasonable. From the temperature-dependent layer distance the reduced layer distance dred = 32.5 Å was obtained by extrapolation at the reduced temperature Tred.74 For MIDA boronates 1 and 2 the molecular lengths were calculated to 24.3 Å and 24.4 Å respectively via the Avogadro software (Version Avogadro 1.2.0) using an Universal Force Drive (OFF) for geometry optimization.75 In analogy to the known pyridine and pyrimidine MIDA boronates Pyr(n)MIDA, Pym(n)MIDA66 and phenylester MIDA derivatives Ph(n)EsterMIDA65 (for details, see Scheme S1, ESI†) the reduced layer distance dred is larger than the molecular length Lcalc and much smaller than the twofold molecular lengths 2 Lcalc, i.e., 2 Lcalc < dred > Lcalc, which can be explained by a smectic bilayer arrangement with interdigitated side chains (Fig. 7).
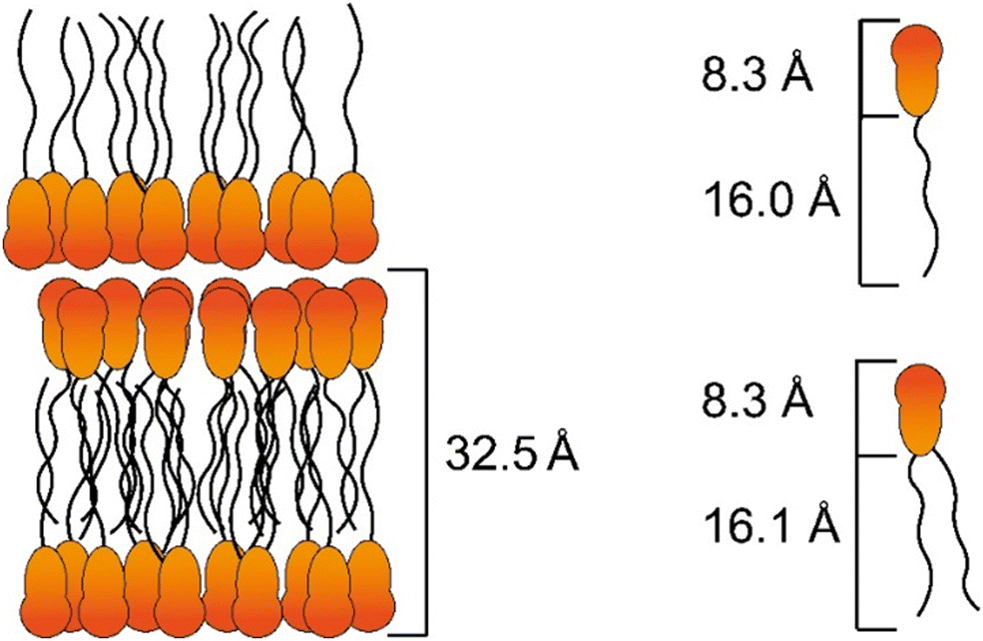 | ||
| Fig. 7 Proposed packing model of the mixture series 1/2 [X1] with calculated reduced layer spacing dred and molecular lengths. | ||
To confirm the correct assignment of both phases in the 1/2 phase diagram, the mixture 1/2 [0.15] was studied by SAXS and WAXS at 120 °C in the Colh phase and at 85 °C in the SmA phase (Fig. S7, ESI†). Both diffraction patterns look rather similar, again featuring one sharp scattering maximum in the small-angle region and a broad halo in the wide-angle region. Unfortunately, elongating the measurement time did not lead to the appearance of further reflexes. However, by plotting the small-angle scattering maxima vs. the azimuthal angle χ (Fig. S8, ESI†), several individual maxima turn up in the scattering experiment obtained at 120 °C. Measuring the angle between these maxima reveals that they originate from two differently aligned domains with hexagonal symmetry, since the angle between maxima belonging together amounts to approximately 60° (Table S6, ESI†). Cooling down to 85 °C leads to the emergence of two strong scattering peaks at χ = 0 and 180 °C as typically found for aligned layered structures, with the other maxima present at higher temperature dwindling down to soft undulations. These findings strongly suggest that the assignment of phases in the 1/2 system, i.e., the Colh phase at high temperatures and the SmA phase at low temperatures, is correct.
XRD experiments at 117 °C of the equimolar mixture 1/3 [0.5] showed a sharp reflection at 32.4 Å in the SAXS and a broad halo around 4.8 Å in the wide-angle section (Fig. 8 and Table 1). Further scattering patterns of the mixtures 1/3 [X1] with X1 = 0.45, 0.55 are shown in Fig. S10 and S12 (ESI†). Extensive efforts were conducted to obtain aligned samples in order to clarify the phase geometry. However, attempts to prepare aligned samples by fiber extrusion, alignment in a magnetic field and slow cooling from the isotopic phase failed to result in monodomain samples. Nevertheless, when plotting the small-angle diffraction pattern of X1 = 0.45 at 100 °C vs. the azimuthal angle χ (Fig. S11, ESI†), a multitude of clear-cut scattering maxima emerges, some of which may be attributed to individual hexagonal domains (Table S7, ESI†). By cooling down to 57 °C the peaks become less distinct and even vanish in some cases, but still reflect a multi-domain alignment. In total, evidence from XRD experiments for the correct assignment of the Colh phase at high temperatures and the SmA phase at low temperatures is weaker for the 1/3 mixtures than the 1/2 mixtures, but considering the structural similarities of the molecules, characteristic textures observed by POM, the high viscosity and low shearability which hints at the presence of a more ordered phase, as compared to the rather fluid SmA phase, the assignment seems more than likely. Thus, the distinct reflection at higher temperatures was assigned as (001) reflection of a Colh phase.
 | ||
| Fig. 8 (a) SAXS profile of 1/3 [0.5] at 107 °C and corresponding 2D pattern; (b) WAXS profile of 1/3 [0.5] at 117 °C and corresponding 2D pattern. | ||
| Compound | Mesophase | a/Å | d exp/Å | Miller indices | Z |
|---|---|---|---|---|---|
| 1/3 [0.5] | Colh at 117 °C | a = 37.3 | 32.4 | (001) | 6 |
| 4.8 | Halo | ||||
| 2 | Colh at 120 °C | a = 37.7 | 32.6 | (001) | 5 |
| 16.3 | (002) | ||||
| 4.1 | Halo | ||||
| 3 | Colh at 70 °C | a = 36.3 | 31.5 | (001) | 4 |
| 4.7 | Halo |
For the molecular mass the median of the masses from 1 and 3 were taken and the number Z of molecules per unit cell was calculated according to the procedure described in ref. 76 Thus, a Colh (p6mm) phase with lattice parameter a = 37.3 Å and Z = 6 was calculated (Table 1) for the equimolar mixture 1/3 [0.5]. When comparing these values with the known lattice parameters of 364 (Colh (p6mm) at 70 °C, a = 36.3 Å, halo 4.7 Å, Z = 4), the optimum space filling can be obtained for the mixture 1/3 [0.5] if 3 rod-like MIDA boronates 1 and 3 wedge-shaped MIDA boronates 3 co-assemble in a supramolecular disc, i.e., a slice through the columnar aggregate, which has a slightly larger diameter (i.e., a value) than the corresponding disk consisting of only 4 wedge-shaped molecules 3 (Table 1). These results correlate well with a recent study by Williams on the coexistence of folded and unfolded shape-incompatible, conformers of discotic dimers resulting in modest perturbations of the phase behaviour.77–79 Moreover, Shimizu reported for triphenylens with attached peripheral alkoxyazobenzenes calamitic discotic mesomorphism and a dynamic change of molecular conformation generating a variation of the liquid crystalline phases.80
By studying the phase behaviour of two different series of binary mixtures containing mono-dodecyloxy-substituted MIDA boronates 1 and either bis-dodecyloxy-substituted MIDA boronates 2, i.e., 1/2 [X1] or tri-dodecyloxy-substituted MIDA boronates 3, i.e., 1/3 [X1], we obtained new insights into the liquid crystalline self-assembly of mixtures of rod-like and wedge-shaped mesogens. The results revealed that contrary to the common observation that shape incompatible mesogens often phase separate quite readily unless they are covalently linked (e.g. in dendritic molecules ternary block-copolymers, rod-coil molecules, polyphilic low molecular weight block molecules, amphiphiles81,82 and disc-rod mesogens83,84) rods and wedges were fully miscible at all concentrations. Presumably, the highly polar boronate groups interact strongly via dipole–dipole interactions, which overcomes the tendency to separate.85 This would be in good agreement with our previous experimental oberservation that related mesogens with less polar “head goups”, such as boronates, pinacol borolanes, or iminoboronates, do not self-assemble into mesophases regardless of the number and lengths of the alkoxy side chains at the aryl units.64 Thus, most likely due to the polar MIDA head groups the boronates tend to aggregate in layers or columns, forming a polar sublayer or polar core, in order to avoid charge separation.
A second important finding of our study is that in 1/2 [X1] mixtures the monosubsituted boronate 1 drives the phase behaviour leading to the SmA phase dominating the phase diagram, while in 1/3 [X1] mixtures both boronates 1 and 3 contribute, which results in a well-balanced proportion of the SmA and the Colh phase. This finding agrees well with studies by Tschierske et al.86 who investigated mixtures of amphiphilic N-benzoyl-1-amino-1-deoxy-D-glucitol derivatives with one, two or three aliphatic chains as well as by Finkelmann et al.87–89 who studied lyotropic liquid crystals composed of amphiphile mixtures with one or two hydrophobic or hydrophilic chains. Both groups discuss their results in terms of the average molecular geometry and the packing parameter model by Israelachvili.90 Thus, it seems reasonable to apply the same approach and draw analogies with mixed lyotropic systems. The model, which we adjusted to our thermotropic case, defines the packing parameter P as
 | (1) |
In addition, these considerations may explain the rather uncommon phase sequence of the Colh phase at high temperatures and the SmA phase at lower temperatures, which is typically observed in reversed order due to the dimensionality of long-range order in the two mesophases. Increasing the temperature will lead to more thermal fluctuations of the dodecyloxy chains, which results in an increase of its volume Vc. Hence, the packing parameter is shifted to larger values at higher temperature, favouring the formation of inverse columnar aggregates over bilayers.
Conclusion
In order to understand the phase behaviour of mixtures of rod- and wedge-shaped MIDA boronates two binary series were prepared, which differed in the number and relative amount of dodecyloxy side chains. According to POM, DSC and XRD studies of the mixtures, rods and wedges were fully miscible at all concentrations due to the strong dipole–dipole interactions of the MIDA headgroups. The observed preference of either SmA or Colh phase could be rationalized by the chain volume Vc in agreement with the packing parameter model adapted from lyotropic liquid crystals. Future work needs to demonstrate, whether the design principle to control liquid crystalline self-assembly presented in this study can be transferred to other mixtures of rod- and wedge-shaped mesogens forming supramolecular liquid crystals as well and if the analogies to the formation of lyotropic liquid crystal phases hold true.Data availability
The data supporting this article have been included in the main manuscript and as part of the ESI.†Conflicts of interest
The authors declare no competing interest.Acknowledgements
Generous financial support by the Ministerium für Wissenschaft, Forschung und Kunst des Landes Baden-Württemberg, the Fonds der Chemischen Industrie, the Deutsche Forschungsgemeinschaft and the Carl-Schneider-Stiftung Aalen (shared instrumentation grant) is gratefully acknowledged. We would like to thank the referees for valuable suggestions and comments.Notes and references
- M. Klasen-Memmer and H. Hirschmann, in Handbook of liquid crystals, ed. T. Kato, H. Gleeson, P. Raynes, J. W. Goodby, P. J. Collings and C. Tschierske, Wiley-VCH, Weinheim, 2nd edn, 2014, pp. 1–25 Search PubMed.
- P. L. M. Connor and R. J. Mandle, Soft Matter, 2020, 16, 324–329 RSC.
- T. Kato, J. Uchida, T. Ichikawa and T. Sakamoto, Angew. Chem., Int. Ed., 2018, 57, 4355–4371 CrossRef CAS PubMed.
- H. Shimura, M. Yoshio, K. Hoshino, T. Mukai, H. Ohno and T. Kato, J. Am. Chem. Soc., 2008, 130, 1759–1765 CrossRef CAS PubMed.
- T. Kato, M. Yoshio, T. Ichikawa, B. Soberats, H. Ohno and M. Funahashi, Nat. Rev. Mater., 2017, 2, 17001 CrossRef.
- M. K. Sonali, P. Bhagavath, M. Srinivasulu, R. K. Sinha and K. Swamynathan, J. Fluorine Chem., 2022, 259–260, 110002 Search PubMed.
- F. S. Alamro, H. A. Ahmed, M. S. Khushaim, N. S. Bedowr and N. S. Al-Kadhi, Crystals, 2023, 13, 899 CrossRef CAS.
- T. Coussaert and M. Baus, J. Chem. Phys., 2002, 116, 7744–7751 CrossRef CAS.
- D. De Las Heras and M. Schmidt, Philos. Trans. R. Soc., A, 2013, 371, 20120259 CrossRef PubMed.
- A. Galindo, G. Jackson and D. J. Photinos, Chem. Phys. Lett., 2000, 325, 631–638 CrossRef CAS.
- A. Galindo, A. J. Haslam, S. Varga, G. Jackson, A. G. Vanakaras, D. J. Photinos and D. A. Dunmur, J. Chem. Phys., 2003, 119, 5216–5225 CrossRef CAS.
- F. Gámez, R. D. Acemel and A. Cuetos, Mol. Phys., 2013, 111, 3136–3146 CrossRef.
- J. Landman, E. Paineau, P. Davidson, I. Bihannic, L. J. Michot, A.-M. Philippe, A. V. Petukhov and H. N. W. Lekkerkerker, J. Phys. Chem. B, 2014, 118, 4913–4919 CrossRef CAS PubMed.
- M. Chen, H. Li, Y. Chen, A. F. Mejia, X. Wang and Z. Cheng, Soft Matter, 2015, 11, 5775–5779 RSC.
- N. Doshi, G. Cinacchi, J. S. Van Duijneveldt, T. Cosgrove, S. W. Prescott, I. Grillo, J. Phipps and D. I. Gittins, J. Phys.: Condens. Matter, 2011, 23, 194109 CrossRef CAS.
- J. Phillips and M. Schmidt, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2010, 81, 041401 CrossRef PubMed.
- H. N. W. Lekkerkerker, R. Tuinier and H. H. Wensink, Mol. Phys., 2015, 113, 2666–2673 CrossRef CAS.
- T. Nakato, Y. Yamashita, E. Mouri and K. Kuroda, Soft Matter, 2014, 10, 3161 RSC.
- M. Golmohammadi and A. D. Rey, Entropy, 2008, 10, 183–199 CrossRef CAS.
- M. Golmohammadi and A. D. Rey, Liq. Cryst., 2009, 36, 75–92 CrossRef CAS.
- G. Cinacchi and A. Tani, J. Chem. Phys., 2002, 117, 11388–11395 CrossRef CAS.
- M. A. Bates and G. R. Luckhurst, Liq. Cryst., 1998, 24, 229–241 CrossRef CAS.
- M. A. Bates, Liq. Cryst., 2003, 30, 181–190 CrossRef CAS.
- J. Idé, R. Méreau, L. Ducasse, F. Castet, H. Bock, Y. Olivier, J. Cornil, D. Beljonne, G. D’Avino, O. M. Roscioni, L. Muccioli and C. Zannoni, J. Am. Chem. Soc., 2014, 136, 2911–2920 CrossRef PubMed.
- O. Cienega-Cacerez, J. A. Moreno-Razo, E. Díaz-Herrera and E. J. Sambriski, Soft Matter, 2014, 10, 3171 RSC.
- R. Berardi and C. Zannoni, Soft Matter, 2012, 8, 2017 RSC.
- S. Belli, A. Patti, R. Van Roij and M. Dijkstra, J. Chem. Phys., 2010, 133, 154514 CrossRef PubMed.
- S. Varga, E. Velasco, L. Mederos and F. J. Vesely, Mol. Phys., 2009, 107, 2481–2492 CrossRef CAS.
- A. A. Verhoeff, H. H. Wensink, M. Vis, G. Jackson and H. N. W. Lekkerkerker, J. Phys. Chem. B, 2009, 113, 13476–13484 CrossRef CAS PubMed.
- Y. Martínez-Ratón, E. Velasco and L. Mederos, J. Chem. Phys., 2005, 123, 104906 CrossRef.
- T. Koda, A. Nishioka and S. Ikeda, J. Phys.: Condens. Matter, 2005, 17, S2875–S2878 CrossRef CAS.
- S. Lago, A. Cuetos, B. Martínez-Haya and L. F. Rull, J. Mol. Recognit., 2004, 17, 417–425 CrossRef CAS.
- G. Cinacchi, L. Mederos and E. Velasco, J. Chem. Phys., 2004, 121, 3854–3863 CrossRef CAS.
- Y. Martínez-Ratón and J. A. Cuesta, J. Chem. Phys., 2003, 118, 10164–10173 CrossRef.
- S.-M. Cui and Z. Y. Chen, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1994, 50, 3747–3754 CrossRef CAS.
- R. A. Wheeler and G. R. Van Hecke, in Liquid Crystals and Ordered Fluids, ed. A. C. Griffin and J. F. Johnson, Springer US, Boston, MA, 1984, pp. 283–297 Search PubMed.
- O. Cienega-Cacerez, C. García-Alcántara, J. A. Moreno-Razo, E. Díaz-Herrera and E. J. Sambriski, Soft Matter, 2016, 12, 1295–1312 RSC.
- A. Juríková, K. Csach, J. Miškuf, N. Tomašovičová, Z. Mitróová, P. Kopčanský, N. Éber, K. Fodor-Csorba and A. Vajda, Mol. Cryst. Liq. Cryst., 2015, 610, 187–192 CrossRef.
- D. S. S. Rao, M. V. Kumar, S. K. Prasad, U. S. Hiremath, M. Sarvamangala and S. Basavaraja, J. Mater. Chem. C, 2013, 1, 7488 RSC.
- M. Kohout, J. Tůma, J. Svoboda, V. Novotná, E. Gorecka and D. Pociecha, J. Mater. Chem. C, 2013, 1, 4962 RSC.
- Y. Yamazaki, Y. Takanishi and J. Yamamoto, EPL, 2009, 88, 56004 CrossRef.
- H. Taing, J. G. Rothera, J. F. Binder, C. L. B. Macdonald and S. H. Eichhorn, Liq. Cryst., 2018, 45, 1147–1154 CrossRef CAS.
- A. Hauser, M. Thieme, A. Saupe, G. Heppke and D. Krüerke, J. Mater. Chem., 1997, 7, 2223–2229 RSC.
- C. J. Booth, D. Krüerke and G. Heppke, J. Mater. Chem., 1996, 6, 927–934 RSC.
- P. Le Barny, J. Billard and J.-C. Dubois, in Liquid Crystals and Ordered Fluids, ed. A. C. Griffin and J. F. Johnson, Springer US, Boston, MA, 1984, pp. 57–74 Search PubMed.
- L. An, M. Jing, B. Xiao, X.-Y. Bai, Q.-D. Zeng and K.-Q. Zhao, Chin. Phys. B, 2016, 25, 096402 CrossRef.
- A. Grafe, D. Janietz, T. Frese and J. H. Wendorff, Chem. Mater., 2005, 17, 4979–4984 CrossRef CAS.
- J. R. Hughes, G. R. Luckhurst, K. Praefcke, D. Singer and W. M. Tearle, Mol. Cryst. Liq. Cryst., 2003, 396, 187–225 CrossRef CAS.
- N. Usoltseva, V. Bykova, E. Kudrik, G. Shaposhnikov, A. Smirnova, G. Ananjeva and I. Nikolaev, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 2001, 367, 509–516 CrossRef CAS.
- S.-H. Wu and H.-H. Chen, Tetrahedron, 2019, 75, 220–229 CrossRef CAS.
- S. Iqbal and A. A. Khan, RSC Adv., 2019, 9, 6335–6345 RSC.
- M. Jasiński, K. Szymańska, A. Gardias, D. Pociecha, H. Monobe, J. Szczytko and P. Kaszyński, ChemPhysChem, 2019, 20, 636–644 CrossRef PubMed.
- A. A. Khan, G. Rughoobur, M. A. Kamarudin, A. Sepe, J. A. Dolan, A. J. Flewitt, M. M. Qasim and T. D. Wilkinson, Org. Electron., 2016, 36, 35–44 CrossRef CAS.
- Y. Shimizu, Y. Matsuda, F. Nekelson, Y. Miyake, H. Yoshida, A. Fujii and M. Ozaki, in Emerging Liquid Crystal Technologies VII, ed. L.-C. Chien, SPIE, 2012, 82790G Search PubMed.
- S. Sergeyev, O. Debever, E. Pouzet and Y. H. Geerts, J. Mater. Chem., 2007, 17, 3002 RSC.
- A. Pecchia, O. R. Lozman, B. Movaghar, N. Boden, R. J. Bushby, K. J. Donovan and T. Kreouzis, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 104204 CrossRef.
- T. Kreouzis, K. Scott, K. J. Donovan, N. Boden, R. J. Bushby, O. R. Lozman and Q. Liu, Chem. Phys., 2000, 262, 489–497 CrossRef CAS.
- W. K. Lee, B. A. Wintner, E. Fontes, P. A. Heiney, M. Ohba, J. N. Haseltine and A. B. Smith, Liq. Cryst., 1989, 4, 87–102 CrossRef CAS.
- C. Destrade, P. Foucher, J. Malthete and N. Huu Tinh, Phys. Lett. A, 1982, 88, 187–190 CrossRef.
- C. Vauchier, A. Zann, P. L. Barny, J. C. Dubois and J. Billard, Mol. Cryst. Liq. Cryst., 1981, 66, 103–114 CrossRef.
- W.-R. Chen, C.-M. Chen, J.-C. Hwang and B.-R. Liaw, Jpn. J. Appl. Phys., 2005, 44, 3126 CrossRef CAS.
- N. Lindner, M. Kölbel, C. Sauer, S. Diele, J. Jokiranta and C. Tschierske, J. Phys. Chem. B, 1998, 102, 5261–5273 CrossRef CAS.
- M. Inb-Elhaj, D. Guillon, A. Skoulios, P. Maldivi, A. M. Giroud-Godquin and J.-C. Marchon, J. Phys. II, 1992, 2, 2237–2253 CrossRef.
- T. Wöhrle, R. Gündemir, W. Frey, F. Knecht, A. Köhn and S. Laschat, Chem. – Eur. J., 2017, 23, 4149–4159 CrossRef PubMed.
- C. Schilling, F. Schulz, A. Köhn and S. Laschat, Org. Mater., 2020, 02, 288–299 CrossRef.
- C. Schilling, A. Bauer, J. A. Knöller, F. Schulz, A. Zens and S. Laschat, J. Mol. Liq., 2022, 367, 120519 CrossRef CAS.
- M. C. Schlick, N. Kapernaum, M. M. Neidhardt, T. Wöhrle, Y. Stöckl, S. Laschat and F. Giesselmann, ChemPhysChem, 2018, 19, 2305–2312 CrossRef CAS PubMed.
- For other polar boron containing liquid crystals see ref. 69 and 70.
- L. Jacob, E. Rzeszotarska, M. Koyioni, R. Jakubowski, D. Pociecha, A. Pietrzak and P. Kaszynski, Chem. Mater., 2022, 34, 6476–6491 CrossRef CAS.
- A. Jankowiak, A. Baliński, J. E. Harvey, K. Mason, A. Januszko, P. Kaszyński, V. G. Young and A. Persoons, J. Mater. Chem. C, 2013, 1, 1144–1159 RSC.
- Y. Shimizu, H. Monobe, M. Heya and K. Awazu, Mol. Cryst. Liq. Cryst., 2005, 441, 287–295 CrossRef CAS.
- L. D. Landau, Zh. Eksp. Teor. Fiz., 1937, 7, 926 Search PubMed.
- S. J. Cowling, in Handbook of liquid crystals, ed. J. W. G. Goodby, P. J. Collings, T. Kato, C. Tschierske, H. F. Gleeson and E. P. Raynes, Wiley-VCH, Weinheim, 2014, pp. 1–38 Search PubMed.
- P. H. J. Kouwer and T. M. Swager, J. Am. Chem. Soc., 2007, 129, 14042–14052 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- M. Lehmann, C. Köhn, H. Meier, S. Renker and A. Oehlhof, J. Mater. Chem., 2006, 16, 441–451 RSC.
- C. O. Zellman and V. E. Williams, J. Org. Chem., 2021, 86, 15076–15084 CrossRef CAS PubMed.
- C. O. Zellman and V. E. Williams, Phys. Chem. Chem. Phys., 2023, 25, 1363–1371 RSC.
- C. O. Zellmann-Parrotta and V. E. Williams, Soft Matter, 2024, 20, 4504–4514 RSC.
- Y. Shimizu, H. Sanada, K. Kitagawa, A. Oka, N. Ohta, T. Kawai and K. Uchida, Liq. Cryst., 2024, 1–8 CrossRef.
- C. Tschierske, J. Mater. Chem., 2001, 11, 2647–2671 RSC.
- C. Tschierske, Curr. Opin. Colloid Interface Sci., 2002, 7, 355–370 CrossRef CAS.
- P. H. Kouwer, C. J. Welch, G. McRobbie, B. J. Dodds, L. Priest and G. H. Mehl, J. Mater. Chem., 2004, 14, 1798–1803 RSC.
- P. H. Kouwer and G. H. Mehl, J. Mater. Chem., 2009, 19, 1564–1575 RSC.
- We would like to thank one of the referees for suggesting this rationalization.
- K. Borisch, S. Diele, P. Goring, H. Muller and C. Tschierske, Liq. Cryst., 1997, 22, 427–443 CrossRef CAS.
- K. Kratzat, F. Guittard, E. Taffin de Givenchy and A. Cambon, Langmuir, 1996, 12, 6346–6350 CrossRef CAS.
- K. Kratzat, C. Stubenrauch and H. Finkelmann, Colloid Polym. Sci., 1995, 273, 257–262 CrossRef CAS.
- J. M. Walsh and G. J. Tiddy, Langmuir, 2003, 19, 5586–5594 CrossRef CAS.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm01132e |
| This journal is © The Royal Society of Chemistry 2025 |