
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational molecular engineering of porphyrins for enhanced performance in dye-sensitized solar cells†
Faraghally A.
Faraghally‡
a,
Yu-Hsuan
Chen‡
 b,
Tsung-Zu
Lee
b,
Yan-Da
Chen
b,
Tzu-Chien
Wei
*a and
Chen-Yu
Yeh
*b
b,
Tsung-Zu
Lee
b,
Yan-Da
Chen
b,
Tzu-Chien
Wei
*a and
Chen-Yu
Yeh
*b
aDepartment of Chemical Engineering, National Tsing Hua University, Hsinchu City 30044, Taiwan. E-mail: tcwei@mx.nthu.edu.tw
bDepartment of Chemistry, i-Center for Advanced Science and Technology (i-CAST), Innovation and Development Center of Sustainable Agriculture (IDCSA), National Chung Hsing University, Taichung City 402, Taiwan. E-mail: cyyeh@dragon.nchu.edu.tw
First published on 17th March 2025
Abstract
Porphyrin dyes are promising sensitizers for high-efficiency dye-sensitized solar cells (DSSCs) due to their remarkable light-harvesting capabilities. However, realizing their full potential in photovoltaic devices necessitates meticulous optimization of their molecular structure. This study explores the structure–performance relationship in nine novel porphyrin dyes (TZ1–TZ9), aiming to enhance the overall power conversion efficiency (PCE). The impact of donor ability and its bulkiness is investigated using bis(4-hexylphenyl)amine and a bulky modified Hagfeldt donor (bis(2′,4′,6′-tris(hexyloxy)-[1,1′-biphenyl]-4-yl)amine). Indacenodithiophene IDT and thiophene groups are also incorporated as π-spacers to improve the light-harvesting efficiency and increase the overall molecular bulkiness. Furthermore, diverse acceptors (benzothiadiazole, benzotriazole, and cyanoacrylic acid) are introduced to fine-tune photophysical and electrochemical properties. Among these porphyrin dyes, TZ1 exhibits a remarkable PCE of 9.90% (short circuit photocurrent (JSC) = 15.675 mA cm−2, open circuit photovoltage (VOC) = 0.834 V, and fill factor (FF) = 0.758), surpassing the PCE of 9.20% (JSC = 14.737 mA cm−2, VOC = 0.817 V, and FF = 0.764) achieved by GY50 under similar fabrication conditions.
1. Introduction
Driven by escalating fossil fuel depletion and intensifying environmental concerns, the quest for sustainable energy solutions has become a paramount global challenge. Among renewable energy technologies, solar energy stands out as a frontrunner.1–4 In this context, dye-sensitized solar cells (DSSCs) have attracted significant interest within the scientific community as a promising technology for future renewable energy systems. This heightened interest stems from their advantageous attributes, encompassing a cost-effective, solution-based fabrication process, a demonstrably minimal environmental impact, and the remarkable ability to show excellent efficiency under low-light conditions.5–12Crucially, the sensitizer within a DSSC plays a significant role in capturing solar energy and facilitating its conversion into an electric current. Among the sensitizer types employed in DSSCs (Organic dyes, ruthenium complexes, porphyrins, and natural dyes),8,13,14 porphyrin dyes hold significant promise due to their unique properties. These include intense light absorption across the visible spectrum, readily available constituent elements, robust thermal stability, and the ability to tailor their electrochemical properties through molecular engineering.15,16
The meticulous design of porphyrin sensitizers plays a vital role in maximizing the power conversion efficiency (PCE) of DSSCs. To achieve this objective, strategic manipulation of the donor, π-conjugated spacer, and acceptor moieties within the porphyrin scaffold, particularly at the meso and β-positions, is essential. These modifications enable better energy level alignment, enhancing the efficient transfer of charge within the molecule. Consequently, improving the light-harvesting efficiency (LHE) and optimized charge dynamics are facilitated within the DSSC architecture.15–22 Additionally, incorporating sterically hindered substituents strategically suppresses dye aggregation and reduces charge recombination, thus contributing significantly to the long-term stability and sustained performance of DSSCs.21,23–31
However, early porphyrin-based exhibited low device performance until the YD series of porphyrins was introduced.32–36 The YD-series of porphyrins, featuring D–π–A molecular design, boosted both the electron injection efficiency and the light-harvesting, enabling YD2 to achieve a PCE of 11%.37 This advancement paved the way for the synthesis of YD2-o-C8, an ingeniously designed derivative incorporated with judiciously tailored alkoxy chains. These modifications demonstrably suppressed dye aggregation and mitigated interfacial back electron transfer, resulting in a noteworthy PCE of 11.9% under AM 1.5G irradiation.25 Furthermore, GY50 dye was obtained by incorporating an electron-withdrawing benzothiadiazole (BTD) moiety between the core of porphyrin and the anchoring group. This strategic enhancement extended conjugation and heightened intramolecular charge transfer (ICT), resulting in an impressive PCE of 12.8%.17 Subsequently, the development of SM315 involved the integration of a bulky donor moiety, bis(20,40-bis(hexyloxy)-[1,10-biphenyl]-4-yl)amino group, culminating in a remarkable PCE of 13%.26 A recently developed double-fence porphyrin dye incorporated strategically positioned alkoxyl groups at the ortho-positions of its phenyl rings mitigated charge recombination and diminished the dye aggregation.38 Continuing this successful design strategy, further research efforts have been directed towards incorporating the Indacenodithiophene (IDT) group within the porphyrin dyes as a donor moiety to achieve enhanced photovoltaic performance. As a result, YS7-based devices exhibited an exceptional PCE of 11.4%.39 Despite the demonstrably successful application of porphyrin dyes in dye-sensitized solar cells (DSSCs), a disparity remains between their achieved efficiencies and the theoretical efficiency of 20% PCE. This disparity underscores the imperative for further investigation into targeted molecular engineering to realize highly efficient and cost-competitive solar cell technologies.
In this respect, we systematically modified the donor, π-spacer, and acceptor moieties within the D–π–A porphyrin framework. This strategic approach enables the investigation of the structure–performance relationship governing the sensitizers, thus enhancing the overall PCE. As illustrated in Fig. 1, nine novel porphyrins (TZ1–TZ9) were synthesized and subsequently employed as DSSC sensitizers. To examine the influence of donor ability and its steric bulk on the performance of DSSCs, we employed two distinct donor types: an alkyl arylamine and a bulky modified Hagfeldt donor incorporating six alkoxy chains.40 Moreover, to fine-tune the photophysical and electrochemical properties of the dyes, three distinct acceptor units were strategically incorporated: benzothiadiazole (BTD), benzotriazole (BTA), and cyanoacrylic acid. Finally, the influence of the π-spacer on photovoltaic performance was investigated through two strategies. The first strategy involved incorporating an IDT unit, enhancing π-conjugation and overall molecular bulkiness. The second strategy focused on extending the dye structure by introducing an additional thiophene unit coupled with BTA moiety. This modification aims to enhance the light-harvesting capability, mitigate charge recombination processes, and optimize the dye loading capacity within the DSSC. Among these newly synthesized porphyrin dyes, TZ1 exhibited a remarkable PCE of 9.90%% (JSC = 15.675 mA cm−2, VOC = 0.834 V, and FF = 0.758), surpassing the PCE of 9.20% (JSC = 14.737 mA cm−2, VOC = 0.817 V, and FF = 0.764) achieved by the benchmark GY50 under identical fabrication conditions. This improvement highlights the crucial role of molecular design in optimizing DSSC performance.
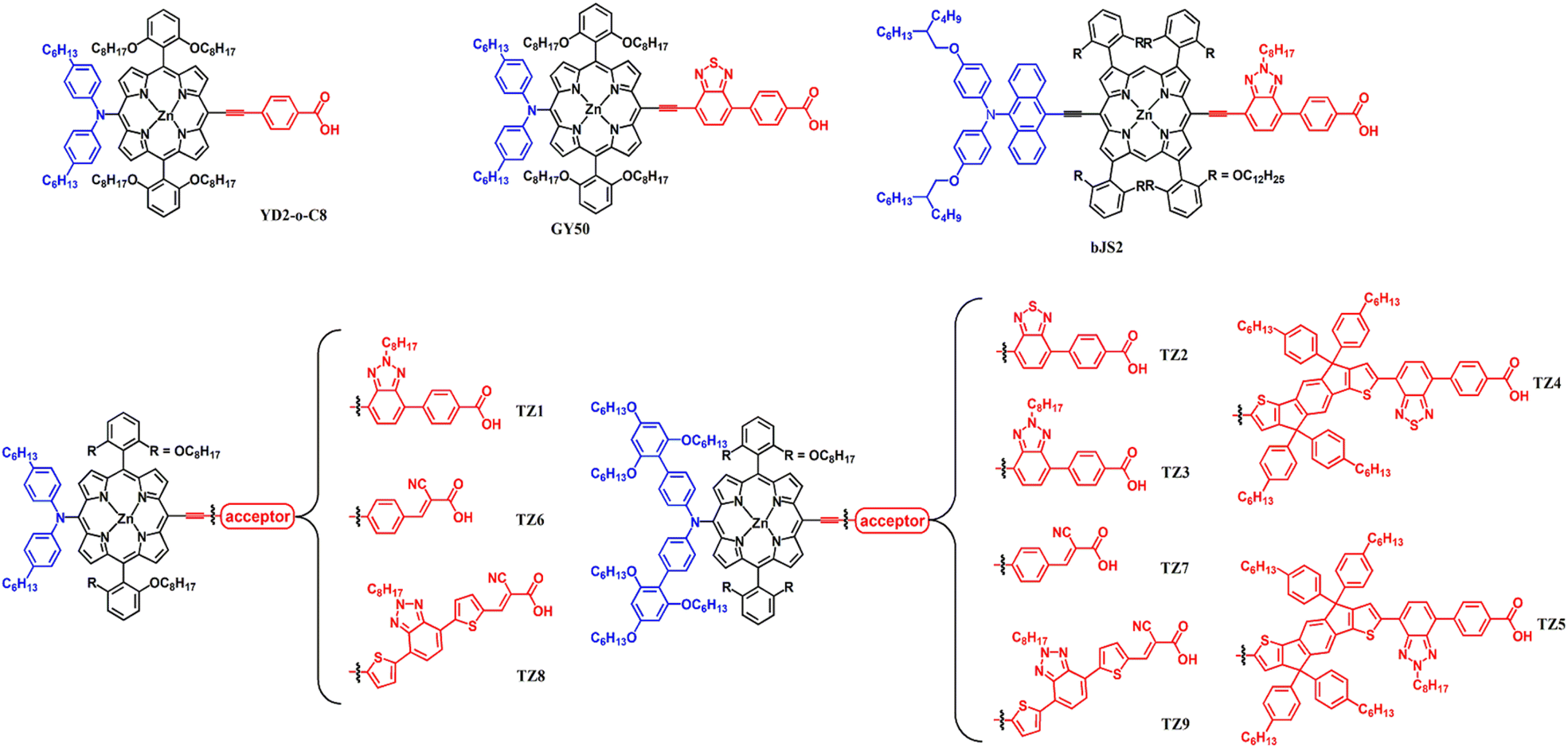 | ||
| Fig. 1 Chemical structures of YD2-o-C8, GY50, bJS2, and TZ porphyrin dyes. | ||
2. Experimental section
2.1 Experimental procedures
The syntheses of precursors (4, 9–14) are described in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as eluent to give TZ8 (41 mg, 78%) as a brown solid. 1H NMR (400 MHz, CDCl3): δ 9.57 (d, J = 4.5 Hz, 2H), 9.15 (d, J = 4.5 Hz, 2H), 8.88 (d, J = 4.5 Hz, 2H), 8.66 (d, J = 4.5 Hz, 2H), 8.25 (d, J = 3.8 Hz, 1H), 8.09–8.05 (m, 1H), 7.70–7.66 (m, 3H), 7.57 (dd, J = 32.0, 3.6 Hz, 2H), 7.45 (d, J = 7.6 Hz, 1H), 7.21 (d, J = 8.7 Hz, 4H), 7.00 (d, J = 8.5 Hz, 5H), 6.94 (d, J = 8.7 Hz, 5H), 4.91 (t, J = 7.1 Hz, 2H), 3.92–3.81 (m, 8H), 2.46 (t, J = 7.8 Hz, 4H), 2.34–2.27 (m, 3H), 1.70–1.44 (m, 18H), 1.43–1.14 (m, 116H), 1.12–1.10 (m, 20H), 1.08–0.93 (m, 20H), 0.93–0.77 (m, 69H), 0.74–0.51 (m, 53H). 13C NMR (151 MHz, CDCl3): δ 159.8, 159.7, 152.0, 151.7, 150.5, 150.4, 150.1, 134.6, 132.2, 131.9, 130.5, 130.3, 129.8, 128.7, 121.9, 120.9, 114.2, 105.4, 77.2, 77.0, 76.8, 68.8, 68.6, 38.7, 37.1, 35.2, 34.4, 32.7, 31.9, 31.8, 31.7, 31.6, 31.5, 31.4, 31.3, 30.4, 30.2, 30.0, 29.7, 29.5, 29.4, 29.2, 29.1, 29.0, 28.9, 28.7, 28.6, 28.4, 28.2, 28.1, 28.0, 27.1, 26.7, 25.1, 25.0, 24.8, 23.8, 23.0, 22.7, 22.6, 22.4, 22.3, 22.2, 19.7, 14.1, 13.9, 13.8, 13.7, 11.0. MALDI-TOF (HRMS): m/z calcd. for C116H141N9O6S2Zn: [M]+ 1883.9732; found 1883.9983.
:1 as eluent to give TZ9 (23 mg, 47%) as a brown solid. 1H NMR (400 MHz, CDCl3): δ 9.62 (d, J = 4.3 Hz, 2H), 9.29–9.23 (m, 2H), 8.86 (d, J = 4.5 Hz, 2H), 8.65 (dd, J = 15.5, 4.5 Hz, 2H), 8.16–8.09 (m, 2H), 7.81–7.78 (m, 1H), 7.72–7.66 (m, 2H), 7.61–7.75 (m, 3H), 7.51–7.46 (m, 2H), 7.30 (d, J = 8.5 Hz, 5H), 7.13 (d, J = 8.5 Hz, 4H), 6.98–6.92 (m, 4H), 6.11 (s, 4H), 4.81 (d, J = 8.4 Hz, 2H), 3.91–3.76 (m, 26H), 2.22–2.16 (m, 1H), 1.90–1.71 (m, 54H), 1.59–1.53 (m, 18H), 1.48–1.36 (m, 17H), 1.34–1.18 (m, 122H), 1.07–0.92 (m, 19H), 0.89–0.85 (m, 24H), 0.82–0.73 (m, 40H), 0.66–0.48 (m, 40H), 0.45–0.32 (m, 23H),. 13C NMR (151 MHz, CDCl3): δ 159.8, 159.2, 157.9, 152.2, 151.6, 150.8, 150.3, 150.2, 132.0, 131.7, 130.9, 130.1, 129.7, 125.8, 121.2, 120.9, 114.4, 113.3, 105.1, 92.9, 77.2, 77.0, 76.8, 68.7, 68.6, 67.9, 45.3, 37.1, 34.4, 31.9, 31.8, 31.6, 31.3, 30.1, 30.0, 29.7, 29.4, 29.3, 29.1, 29.0, 28.6, 28.5, 28.4, 27.1, 26.6, 25.7, 25.5, 25.1, 25.0, 23.0, 22.7, 22.6, 22.5, 22.2, 22.1, 19.7, 14.1, 14.0, 13.8, 8.6. MALDI-TOF (HRMS): m/z calcd. for C152H197N9O12S2Zn: [M]+ 2468.3809; found 2468.3563.
:1 as eluent to give TZ8 (41 mg, 78%) as a brown solid. 1H NMR (400 MHz, CDCl3): δ 9.57 (d, J = 4.5 Hz, 2H), 9.15 (d, J = 4.5 Hz, 2H), 8.88 (d, J = 4.5 Hz, 2H), 8.66 (d, J = 4.5 Hz, 2H), 8.25 (d, J = 3.8 Hz, 1H), 8.09–8.05 (m, 1H), 7.70–7.66 (m, 3H), 7.57 (dd, J = 32.0, 3.6 Hz, 2H), 7.45 (d, J = 7.6 Hz, 1H), 7.21 (d, J = 8.7 Hz, 4H), 7.00 (d, J = 8.5 Hz, 5H), 6.94 (d, J = 8.7 Hz, 5H), 4.91 (t, J = 7.1 Hz, 2H), 3.92–3.81 (m, 8H), 2.46 (t, J = 7.8 Hz, 4H), 2.34–2.27 (m, 3H), 1.70–1.44 (m, 18H), 1.43–1.14 (m, 116H), 1.12–1.10 (m, 20H), 1.08–0.93 (m, 20H), 0.93–0.77 (m, 69H), 0.74–0.51 (m, 53H). 13C NMR (151 MHz, CDCl3): δ 159.8, 159.7, 152.0, 151.7, 150.5, 150.4, 150.1, 134.6, 132.2, 131.9, 130.5, 130.3, 129.8, 128.7, 121.9, 120.9, 114.2, 105.4, 77.2, 77.0, 76.8, 68.8, 68.6, 38.7, 37.1, 35.2, 34.4, 32.7, 31.9, 31.8, 31.7, 31.6, 31.5, 31.4, 31.3, 30.4, 30.2, 30.0, 29.7, 29.5, 29.4, 29.2, 29.1, 29.0, 28.9, 28.7, 28.6, 28.4, 28.2, 28.1, 28.0, 27.1, 26.7, 25.1, 25.0, 24.8, 23.8, 23.0, 22.7, 22.6, 22.4, 22.3, 22.2, 19.7, 14.1, 13.9, 13.8, 13.7, 11.0. MALDI-TOF (HRMS): m/z calcd. for C116H141N9O6S2Zn: [M]+ 1883.9732; found 1883.9983.
:1 as eluent to give TZ9 (23 mg, 47%) as a brown solid. 1H NMR (400 MHz, CDCl3): δ 9.62 (d, J = 4.3 Hz, 2H), 9.29–9.23 (m, 2H), 8.86 (d, J = 4.5 Hz, 2H), 8.65 (dd, J = 15.5, 4.5 Hz, 2H), 8.16–8.09 (m, 2H), 7.81–7.78 (m, 1H), 7.72–7.66 (m, 2H), 7.61–7.75 (m, 3H), 7.51–7.46 (m, 2H), 7.30 (d, J = 8.5 Hz, 5H), 7.13 (d, J = 8.5 Hz, 4H), 6.98–6.92 (m, 4H), 6.11 (s, 4H), 4.81 (d, J = 8.4 Hz, 2H), 3.91–3.76 (m, 26H), 2.22–2.16 (m, 1H), 1.90–1.71 (m, 54H), 1.59–1.53 (m, 18H), 1.48–1.36 (m, 17H), 1.34–1.18 (m, 122H), 1.07–0.92 (m, 19H), 0.89–0.85 (m, 24H), 0.82–0.73 (m, 40H), 0.66–0.48 (m, 40H), 0.45–0.32 (m, 23H),. 13C NMR (151 MHz, CDCl3): δ 159.8, 159.2, 157.9, 152.2, 151.6, 150.8, 150.3, 150.2, 132.0, 131.7, 130.9, 130.1, 129.7, 125.8, 121.2, 120.9, 114.4, 113.3, 105.1, 92.9, 77.2, 77.0, 76.8, 68.7, 68.6, 67.9, 45.3, 37.1, 34.4, 31.9, 31.8, 31.6, 31.3, 30.1, 30.0, 29.7, 29.4, 29.3, 29.1, 29.0, 28.6, 28.5, 28.4, 27.1, 26.6, 25.7, 25.5, 25.1, 25.0, 23.0, 22.7, 22.6, 22.5, 22.2, 22.1, 19.7, 14.1, 14.0, 13.8, 8.6. MALDI-TOF (HRMS): m/z calcd. for C152H197N9O12S2Zn: [M]+ 2468.3809; found 2468.3563.
3. Results and discussions
3.1 Synthesis and characterization
The targeted TZ porphyrin dyes were synthesized following the route outlined in Scheme 1. The detailed procedures for each step are provided in the ESI.† Briefly, intermediates 1 and acceptor moieties with benzotriazole (5), cyanoacrylic acid derivatives (6 and 7), and benzothiadiazole (8) were prepared according to previously reported literature.5,17,26,38–41 The synthetic approach involved desilylation of intermediate 1, followed by Sonogashira coupling with acceptors 5 and 6 to afford final dyes TZ1 and TZ6, respectively. For TZ8 synthesis, the desilylated intermediate 1 was first reacted with compound 7, followed by subsequent condensation with cyanoacetic acid. A modified bulky Hagfeldt donor 3 was prepared using established methods.40 Key intermediate 4 was constructed via Buchwald–Hartwig amination of precursors 2 and 3. Similar to the previously described approach, desilylation of compound 4 was followed by Sonogashira coupling with the corresponding acceptors (5–8, 11 and 14) to furnish the target TZ dye series(TZ2–TZ5, TZ7, and TZ9) respectively This methodology enables systematic variation of both donor and acceptor units within the porphyrin framework, providing a versatile approach for tailoring the final dyes' properties. | ||
| Scheme 1 Synthetic routes of TZ porphyrin dyes. (i) Pd (OAc)2, NaOtBu, P(tBu)3, toluene. (ii) tetra-n-Butylammonium fluoride (TBAF), THF. (iii) Pd2(dba)3, AsPh3, Et3N, THF. (iv) 2-cyanoacetic acid, piperidine, CHCl3. | ||
3.2 Optical properties
The influence of various moieties on the light-harvesting properties of TZ-dyes was investigated through UV-vis spectroscopy in a tetrahydrofuran (THF) solution. As shown in Fig. 2, the UV-vis absorption spectra of TZ dyes reveal characteristic porphyrin Soret bands within the 400–550 nm range, accompanied by moderate-intensity Q bands spanning the 600–700 nm region. Among the TZ dyes series, TZ1, featuring an alkylamine donor and BTA acceptor, displays the highest extinction coefficient. TZ2 incorporating BTD unit exhibits a broadened absorption spectrum with a bathochromic shift (400–540 and 620–720 nm) in the sorb and Q bands, respectively. This red-shifted absorption could be attributed to the reduced HOMO–LUMO energy gap due to the electron-withdrawing nature of the BTD moiety, which facilitated the absorption of lower-energy photons, consequently broadening the light-harvesting spectrum of the dye. Incorporation of the IDT unit in TZ4 and TZ5 results in the broadest absorption profile observed among TZ dyes series, encompassing a wide spectral range of the Soret band (390–588 nm) and Q band (622–694 nm). This extended light absorption arises from the well-established ability of IDT units to promote extended π-conjugation and enhance π-electron delocalization throughout the molecule. Moreover, the high extinction coefficient suggests the presence of strong ICT processes within these dyes.42–44 Furthermore, the strategic placement of thiophene groups coupled with BTA in TZ8 and TZ9 allowed for the modulation of π-conjugation, offering valuable insights into fine-tuning light-harvesting properties. Consequently, these dyes exhibit a red-shifted and broadened absorption range, highlighting the influence of the thiophene group as a spacer on their light-capture profiles. | ||
| Fig. 2 UV-vis absorption spectra of TZ dyes in THF. | ||
Steady-state fluorescence spectra were recorded in THF (Fig. S35†). A slight redshift in the fluorescence maximum of TZ2 was observed, consistent with the trend evident in the absorption spectra. Furthermore, time-resolved fluorescence measurements were conducted using time-correlated single-photon counting (TCSPC) (Fig. S36†), and the average fluorescence lifetimes (τ) are summarized in Table 1. Notably, TZ4 and TZ5 exhibited longer lifetimes, suggesting their effectiveness in mitigating dye aggregation in dilute solutions.45
| Dye | Absorptionaλmax (nm) [ε/104 M−1 cm−1] | Emission λmax (nm) | τ AV (ns) | E ox [V] | E 0−0 [eV] | E S1 V | ΔGinjf (eV) | ΔGregj (eV) |
|---|---|---|---|---|---|---|---|---|
| a Wavelength maximum for absorption and emission are recorded in THF at 25 °C. b Average fluorescence decays lifetimes of dyes in THF solution (1 × 10−6 M). c The oxidation potentials were determined by cyclic CV in THF containing 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as electrolyte and were calibrated with ferrocene/ferrocenium (Fc/Fc+) as internal reference at 25 °C and corrected to NHE by adding +0.63 V. d E 0−0 were determined from the intersection of normalized absorption and emission spectra in THF. e Calculated by the following equation: ES1 = EHOMO − E0−0. f Driving force for electron injection from the S+/S* to the CB of TiO2 (−0.5 V vs. NHE). j Driving force for dye regeneration (cobalt redox electrolyte) (0.56 V vs. NHE). | ||||||||
| TZ1 | 466 (2.0), 654 (0.5) | 678 | 1.56 | +0.80 | 1.86 | −1.06 | 0.56 | 0.24 |
| TZ2 | 435 (0.6), 454 (1.0), 772 (0.4) | 731 | 1.54 | +0.66 | 1.77 | −1.11 | 0.61 | 0.10 |
| TZ3 | 466 (1.7), 660 (0.4) | 691 | 1.50 | +0.67 | 1.84 | −1.17 | 0.67 | 0.11 |
| TZ4 | 474 (1.7), 666 (0.7) | 702 | 2.00 | +0.66 | 1.81 | −1.15 | 0.65 | 0.10 |
| TZ5 | 478 (1.6), 495 (1.7), 666 (0.6) | 690 | 2.05 | +0.68 | 1.83 | −1.15 | 0.65 | 0.12 |
| TZ6 | 442 (1.16), 659 (0.28) | 677 | 1.26 | +0.85 | 1.86 | −1.01 | 0.51 | 0.29 |
| TZ7 | 444 (1.13), 662 (0.34) | 694 | 1.09 | +0.75 | 1.83 | −1.08 | 0.58 | 0.19 |
| TZ8 | 465 (0.89), 664 (0.35) | 684 | 1.15 | +0.80 | 1.83 | −1.03 | 0.52 | 0.24 |
| TZ9 | 471 (0.98), 666 (0.37) | 696 | 1.00 | +0.70 | 1.81 | −1.11 | 0.61 | 0.14 |
3.3 Electrochemical properties
Cyclic voltammetry was employed in a three-electrode configuration to investigate the electrochemical behaviours of TZ dyes. The electrochemical cell encompassed dry THF, TBAPF6 as a supporting electrolyte, and ferrocene/ferrocenium as an internal reference electrode (Fig. S37†). Potentials obtained versus Fc+/Fc redox couple were meticulously converted to the NHE scale by adding +0.63 V. EHOMO was estimated using the redox potential (E1/2(ox)) of the first oxidation, while the first excited states of TZ dyes were calculated by eqn (1):| ELUMO = EHOMO − E0−0 | (1) |
As shown in Fig. 3, incorporating alkoxy chains into the donor (modified Hagfeldt donor) upshifts the oxidation potential. It resulted in a narrowed band gap for TZ2, TZ3, TZ4, TZ5, TZ7, and TZ9 compared to TZ1, TZ6, and TZ8 (alkylamine-based donor). This could be ascribed to the augmented electron-donating propensity of the modified Hagfeldt donor, which promotes a more stable oxidized state and leads to a narrower band gap. Notably, TZ2, equipped with the strong electron-withdrawing BTD unit, exhibits the narrowest energy band gap among all dyes (1.77 eV). This suggests that the combined influence of the modified Hagfeldt and BTD units optimizes the electronic landscape for efficient light harvesting. Furthermore, employing thiophene groups coupled with BTA as a spacer in TZ8 and TZ9 results in narrower band gaps than their counterparts TZ6 and TZ7. As demonstrated by UV-vis and CV measurements, the targeted manipulation of the molecular architecture emerges as a critical strategy in fine-tuning the optoelectronic properties of TZ dyes.
 | ||
| Fig. 3 Energy levels diagram of TZ dyes based on their optical and electrochemical data. | ||
3.4 Photovoltaic performance
The photovoltaic performance of DSSCs incorporating TZ dyes was examined under simulated AM 1.5G illumination. The resulting current density–voltage (J–V) curves and incident photon-to-current efficiency (IPCE) spectra are presented in Fig. 4. Detailed descriptions of device fabrication and measurement procedures are included in the ESI.† As shown in Table 2, devices incorporating TZ2–TZ5 dyes exhibited reduced dye-loading capacities compared to TZ1. This disparity could be attributed to the steric hindrance imposed by the bulkier molecular structures of TZ2–TZ5, which impeded the efficient adsorption of dye molecules onto the TiO2 surface. Consequently, the devices incorporating TZ2–TZ5 exhibited lower JSC than the TZ1-based device, following the TZ1 > TZ2 > TZ3 > TZ4 > TZ5 trend. Moreover, the observed superiority in JSC for TZ1 could be ascribed to its enhanced driving force for dye regeneration (0.24 eV) compared to TZ2–TZ5 (≃0.11 eV), as elucidated by CV measurements and shown in Table 1.46–50 Notably, TZ4 and TZ5, incorporating the IDT moiety, displayed significantly higher VOC of 0.863 V and 0.883 V, respectively, exceeding all other TZ dyes, despite lower dye-loading capacities. This superior ability to suppress charge recombination at the TiO2 photoanode–electrolyte interface is attributed to the steric hindrance effect caused by the bulky IDT unit. On the other hand, although TZ8 and TZ9 demonstrated the highest dye-loading capacities, they showed low PCEs of 2.1% and 3.5%, respectively. This relatively low performance could be attributed to their elongated molecular structures, which promote intermolecular π–π interactions and increase dye aggregation. As shown in Fig. S38†, upon application to a 2 μm thick TiO2 electrode, TZ8 and TZ9 exhibited significant blue shifts of 31 nm and 34 nm, respectively, whereas TZ1 displayed a comparatively smaller blue shift of 12 nm. Furthermore, the elongated molecular structures of TZ8 and TZ9 may facilitate a larger tilt angle, potentially enhancing charge recombination (low VOC), thereby negating the potential benefits of increased light harvesting achieved by the extended structures.6,51–53 | ||
| Fig. 4 (a) J–V curves under 1 sun illumination. (b) IPCE spectra of TZ dyes and GY50-based devices. | ||
| Dyea | J SC [mA cm−2] | V OC [V] | FF | PCE % | J IPCESC [mA cm−2] | Dye loadingb [10−8 mol cm−2] |
|---|---|---|---|---|---|---|
| a Photovoltaic parameters were obtained from the champion cells. Averaged values of four independent cells with standard errors are presented in parentheses. b The values were obtained by averaging four independent cells. | ||||||
| TZ1 | 15.675 (15.512 ± 0.163) | 0.834 (0.826 ± 0.008) | 0.758 (0.738 ± 0.014) | 9.909 (9.456 ± 0.439) | 14.872 | 6.70 |
| TZ2 | 12.351 (12.078 ± 0.272) | 0.844 (0.846 ± 0.003) | 0.759 (0.756 ± 0.003) | 7.921 (7.738 ± 0.182) | 11.655 | 4.52 |
| TZ3 | 9.774 (9.702 ± 0.072) | 0.845 (0.845 ± 0.00) | 0.785 (0.775 ± 0.010) | 6.480 (6.356 ± 0.124) | 8.976 | 3.50 |
| TZ4 | 9.001 (8.885 ± 0.116) | 0.862 (0.859 ± 0.003) | 0.746 (0.728 ± 0.018) | 5.789 (5.556 ± 0.233) | 8.495 | 3.10 |
| TZ5 | 5.997 (5.638 ± 0.358) | 0.883 (0.872 ± 0.011) | 0.736 (0.743 ± 0.007) | 3.901 (3.650 ± 0.251) | 5.412 | 2.94 |
| TZ6 | 11.031 (10.825 ± 0.206) | 0.826 (0.824 ± 0.002) | 0.770 (0.772 ± 0.002) | 7.022 (6.901 ± 0.121) | 10.331 | 8.25 |
| TZ7 | 5.086 (4.911 ± 0.175) | 0.776 (0.777 ± 0.001) | 0.773 (0.771 ± 0.002) | 3.055 (2.943 ± 0.112) | 4.484 | 7.55 |
| TZ8 | 2.344 (2.168 ± 0.176) | 0.783 (0.786 ± 0.003) | 0.753 (0.738 ± 0.015) | 1.384 (1.258 ± 0.126) | 1.976 | 10.10 |
| TZ9 | 5.513 (5.251 ± 0.262) | 0.772 (0.770 ± 0.002) | 0.732 (0.745 ± 0.014) | 3.116 (3.015 ± 0.101) | 4.895 | 8.15 |
| GY50 | 14.737 (14.779 ± 0.042) | 0.817 (0.815 ± 0.002) | 0.764 (0.757 ± 0.007) | 9.207 (9.133 ± 0.074) | 13.812 | 6.91 |
IPCE spectra were employed to further elucidate the light-harvesting capability of the TZ dyes, as presented in Fig. 4b. All investigated dyes displayed characteristic IPCE profiles with two maxima around 480 nm and 700 nm, corresponding to the Soret and Q-band absorption transitions, respectively, with a minor dip observed at approximately 600 nm. The plateau heights in the IPCE spectra follow the trend: TZ1 > TZ2 > TZ6 > TZ3 > TZ4 > TZ5 > TZ7 ≈ TZ9 > TZ8. This trend is mirrored in the integrated Jsc values derived from the IPCE spectra, which closely correlate with the Jsc values obtained from the J–V curves, as summarized in Table 2. TZ1 displays a superior IPCE value of 65%, surpassing its counterparts and the benchmark GY50 dye (60%). The superiority of TZ1 could be ascribed to the combination of dye loading capacity, the larger driving force for dye regeneration, and the higher extinction coefficient. On the other hand, TZ7, TZ8, and TZ9 showed lower IPCE values due to the dye aggregation and limitations in charge collection efficiency, as will be investigated using electrochemical impedance spectroscopy.
3.5 Electrochemical impedance spectroscopy study (EIS)
EIS under varying applied potential biases in darkness was employed to investigate the charge collection efficiency (ηcoll), charge recombination resistance (RCT) at the dye/TiO2/electrolyte interface, and charge transport resistance (RT). As shown in Fig. 5a, TZ2–TZ5-based devices exhibit comparable RCT to TZ1 despite a lower dye loading, suggesting that strategic bulking of the dye structures suppresses charge recombination through steric hindrance effects. Conversely, TZ8 and TZ9 display the lowest RCT despite having the highest dye loading, which can be attributed to dye aggregation.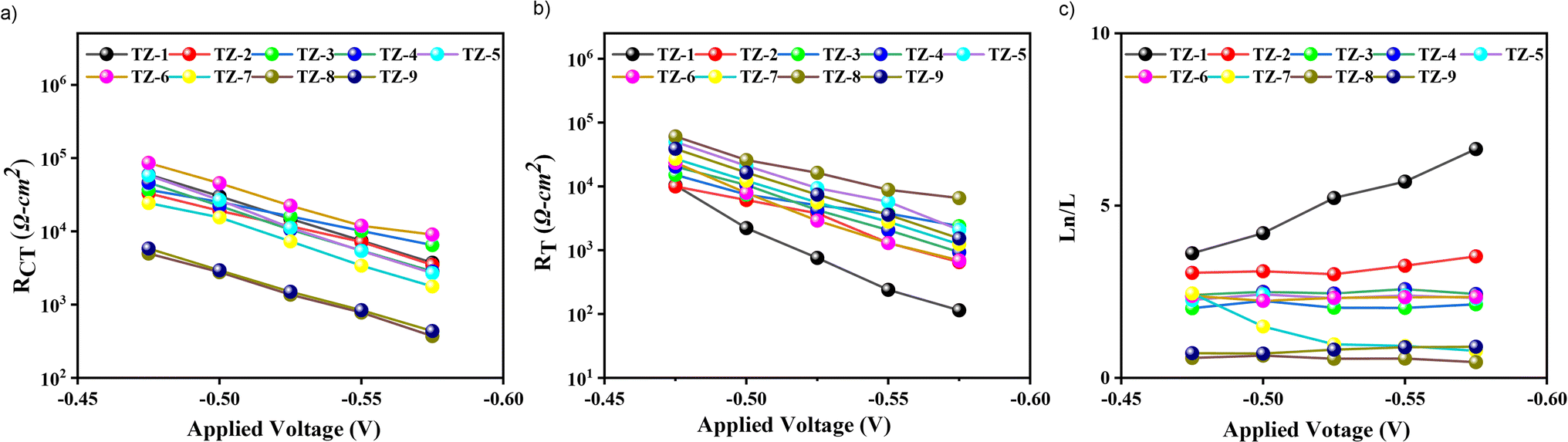 | ||
| Fig. 5 (a) Logarithmic dependence of charge-recombination resistance. (b) Logarithmic dependence of charge-transport resistance. (c) Charge collection efficiency was calculated by Ln/L ratio. | ||
Consequently, these devices exhibited lower VOC values (0.78–0.79 V, respectively). Notably, the DSSCs-based TZ1 dye shows the lowest RT values, as illustrated in Fig. 5b, consistent with its high JSC. Conversely, the elevated RT observed in the TZ8-based DSSC may be attributed to dye aggregation phenomena. This aggregation could impede efficient charge injection, which is consistent with the observed low JSC. Finally, ηcoll can be quantified using the equation Ln/L = (RCT/RT)1/2, where L denotes TiO2 thickness, and Ln represents the electron diffusion length. For efficient charge collection, the Ln/L ratio should exceed unity. As shown in Fig. 5c, TZ7, TZ8, and TZ9 exhibit lower charge collection efficiencies, providing further evidence for their lower JSC, VOC, and, consequently, lower PCE values. Overall, among the investigated dyes, TZ1-based DSSC exhibited the highest PCE of 9.90%, outperforming both the other TZ dyes and GY50 (PCE of 9.20%) under similar fabrication conditions. Conversely, TZ7–TZ9 displayed lower PCEs (3.98%, 1.29%, and 3.52%, respectively) due to insufficient ηcoll and increased dye aggregation.
3.6 Stability studies of TZ dyes
The photostability of TZ dyes was investigated by subjecting dye-coated nanocrystalline TiO2 films to 1 sun illumination for a day. As illustrated in Fig. S39,† the UV-vis absorption spectra of the dyes showed a slight decrease, demonstrating their photostability.54 Moreover, 1H NMR analysis of powdered dyes after 24 hours of continuous illumination (Fig. S40 and S41† for TZ4 and TZ5 as representative examples) confirmed their structural integrity under prolonged light exposure.To assess thermal stability, thermogravimetric analysis (TGA) was employed. As presented in Fig. S42,† all TZ derivatives exhibited remarkable thermal stability, remaining stable up to 300 °C. To evaluate long-term device performance, DSSCs based on TZ1 were subjected to aging tests at 60 °C for 30 days, utilizing both cobalt- and iodide-based electrolytes in 3-methoxypropionitrile (MPN). As depicted in Fig. 6, devices employing the iodide-based electrolyte outperformed cobalt-based devices, retaining 92% of their initial PCE compared to only 36%. This disparity could be attributed to the lower volatility of the iodide-based DSSCs.6
 | ||
| Fig. 6 Stability trail TZ1-based DSSCs using cobalt and iodine-based electrolytes at 60 °C. | ||
Furthermore, under continuous indoor illumination at 6000 lux, TZ1-based DSSCs maintained 93% and 76% of their initial PCE for iodine and cobalt-based electrolytes, respectively (Fig. S43 and Table S1†). The combination of high thermal and photostability, coupled with acceptable durability, particularly under dim light conditions, positions TZ dyes as promising candidates for practical solar energy conversion applications.
4. Conclusion
This study meticulously described the design, synthesis, and performance evaluation of nine novel porphyrin sensitizers, designated as TZ dyes, for the application in DSSCs. A systematic investigation was undertaken to explore the structure–performance relationship of these sensitizers. By strategically modifying the donor, π-spacer, and acceptor units, we aimed to understand the critical interplay between molecular design and photovoltaic efficiency. Incorporating a bulky modified Hagfeldt donor resulted in a red-shifted absorption spectrum compared to their counterparts containing alkylamine-based donors. Furthermore, introducing IDT units as π-spacer nTZ4 and TZ5 led to enhanced LHE and demonstrably suppressed the charge recombination through steric hindrance effects. These combined effects translated to high VOC values for TZ4 and TZ5. However, this approach comes at the cost of a lower dye loading capacity, consequently leading to a diminished JSC. Further extension of the π-spacer with thiophene and benzothiadiazole (BTA) moieties in TZ8 and TZ9 further red-shifted the absorption spectrum however, it increased the dye aggregation, limiting their photovoltaic performance. All TZ dye derivatives exhibited remarkable thermal stability, demonstrating robust structural integrity up to 300 °C. Moreover, these dyes displayed excellent photochemical stability.Among the investigated dyes, DSSCs based on TZ1 exhibited a remarkable PCE of 9.90%% (JSC = 15.675 mA cm−2, VOC = 0.834 V, and FF = 0.758), surpassing the PCE of 9.20% (JSC = 14.737 mA cm−2, VOC = 0.817 V, and FF = 0.764) achieved by the benchmark GY50 under identical fabrication. Furthermore, the exceptional thermal and photochemical stability of TZ1, coupled with its promising long-term operational stability, underscores its potential for practical application in sustainable solar energy conversion technologies.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Ministry of Science and Technology, Taiwan (MOST 108-2221-E-007-102-MY3 and MOST 110-2113-M-005-023-MY3) and “Innovation and Development Center of Sustainable Agriculture” from the Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan. The authors also thank the Instrument Center at National Chung Hsing University for providing valuable assistance in MS measurements.References
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed.
- P. Szuromi, B. Jasny, D. Clery, J. Austin and B. Hanson, Science, 2007, 315, 781 CAS.
- A. Costello, M. Abbas, A. Allen, S. Ball, S. Bell, R. Bellamy, S. Friel, N. Groce, A. Johnson, M. Kett, M. Lee, C. Levy, M. Maslin, D. McCoy, B. McGuire, H. Montgomery, D. Napier, C. Pagel, J. Patel, J. A. P. deOliveira, N. Redclift, H. Rees, D. Rogger, J. Scott, J. Stephenson, J. Twigg, J. Wolff and C. Patterson, Lancet, 2009, 373, 1693–1733 Search PubMed.
- J. Rogelj, M. DenElzen, N. Höhne, T. Fransen, H. Fekete, H. Winkler, R. Schaeffer, F. Sha, K. Riahi and M. Meinshausen, Nature, 2016, 534, 631–639 CAS.
- Y. S. Tingare, N. S. N. Vinh, H.-H. Chou, Y.-C. Liu, Y.-S. Long, T.-C. Wu, T.-C. Wei and C.-Y. Yeh, Adv. Energy Mater., 2017, 7, 1700032 Search PubMed.
- C.-C. Chen, V. S. Nguyen, H.-C. Chiu, Y.-D. Chen, T.-C. Wei and C.-Y. Yeh, Adv. Energy Mater., 2022, 12, 2104051 CAS.
- C. Teng, X. Yang, C. Yang, S. Li, M. Cheng, A. Hagfeldt and L. Sun, J. Phys. Chem. C, 2010, 114, 9101–9110 Search PubMed.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CAS.
- Y. Xie, Y. Tang, W. Wu, Y. Wang, J. Liu, X. Li, H. Tian and W. H. Zhu, J. Am. Chem. Soc., 2015, 137, 14055–14058 Search PubMed.
- M. Grätzel, Acc. Chem. Res., 2009, 42, 1788–1798 Search PubMed.
- S. Venkatesan, T.-H. Hsu, H. Teng and Y.-L. Lee, Sol. RRL, 2023, 7, 2300220 Search PubMed.
- F. A. Faraghally, A. F. Musa, C.-C. Chen, Y.-H. Chen, Y.-D. Chen, C.-Y. Yeh and T.-C. Wei, Small Struct., 2024, 5, 2400236 Search PubMed.
- M. Grätzel, in Pure and Applied Chemistry, Walter de Gruyter GmbH, 2001, vol. 73, pp. 459–467 Search PubMed.
- M. Grätzel, J. Photochem. Photobiol. C Photochem. Rev., 2003, 4, 145–153 Search PubMed.
- S. Huang, Q. Li, S. Li, C. Li, H. Tan and Y. Xie, Chem. Commun., 2024, 60, 4521–4536 CAS.
- J. M. Ji, H. Zhou and H. K. Kim, J. Mater. Chem. A, 2018, 6, 14518–14545 CAS.
- A. Yella, C.-L. Mai, S. M. Zakeeruddin, S. N. Chang, C.-H. Hsieh, C.-Y. Yeh and M. Grätzel, Angew. Chem. Int. Ed., 2014, 53, 2973–2977 CAS.
- S. H. Kang, I. T. Choi, M. S. Kang, Y. K. Eom, M. J. Ju, J. Y. Hong, H.-S. Kang and H. K. Kim, J. Mater. Chem. A, 2013, 1, 3977–3982 CAS.
- C. Yi, F. Giordano, N. LeCevey-Ha, H. N. Tsao, S. M. Zakeeruddin and M. Grätzel, ChemSusChem, 2014, 7, 1107–1113 CAS.
- M. Ishida, D. Hwang, Z. Zhang, Y. J. Choi, J. Oh, V. M. Lynch, D. Y. Kim, J. L. Sessler and D. Kim, ChemSusChem, 2015, 8, 2967–2977 CAS.
- S. H. Kang, M. J. Jeong, Y. K. Eom, I. T. Choi, S. M. Kwon, Y. Yoo, J. Kim, J. Kwon, J. H. Park and H. K. Kim, Adv. Energy Mater., 2017, 7, 1602117 CrossRef.
- Z. Li, Q. Li, C. Li and Y. Xie, Mater. Chem. Front., 2023, 8, 652–680 RSC.
- J. E. Kroeze, N. Hirata, S. Koops, M. K. Nazeeruddin, L. Schmidt-Mende, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2006, 128, 16376–16383 CrossRef CAS PubMed.
- N. Koumura, Z. S. Wang, S. Mori, M. Miyashita, E. Suzuki and K. Hara, J. Am. Chem. Soc., 2006, 128, 14256–14257 CrossRef CAS.
- A. Yella, H.-W. W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CAS.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CAS.
- T. Ripolles-Sanchis, B. C. Guo, H. P. Wu, T. Y. Pan, H. W. Lee, S. R. Raga, F. Fabregat-Santiago, J. Bisquert, C.-Y. Yeh and E. W.-G. Diau, Chem. Commun., 2012, 48, 4368–4370 CAS.
- H.-Y. Hsu, H.-C. Chiang, J.-Y. Hu, K. Awasthi, C.-L. Mai, C.-Y. Yeh, N. Ohta and E. W.-G. Diau, J. Phys. Chem. C, 2013, 117, 24761–24766 CAS.
- M. R. DiNunzio, B. Cohen, S. Pandey, S. Hayse, G. Piani and A. Douhal, J. Phys. Chem. C, 2014, 118, 11365–11376 CAS.
- J. Luo, Z. Xie, J. Zou, X. Wu, X. Gong, C. Li and Y. Xie, Chin. Chem. Lett., 2022, 33, 4313–4316 CAS.
- J. Zou, Y. Wang, G. Baryshnikov, J. Luo, X. Wang, H. Ågren, C. Li and Y. Xie, ACS Appl. Mater. Interfaces, 2022, 14, 33274–33284 CAS.
- L. Schmidt-Mende, W. M. Campbell, Q. Wang, K. W. Jolley, D. L. Officer, M. K. Nazeeruddin and M. Grätzel, ChemPhysChem, 2005, 6, 1253–1258 CrossRef.
- Q. Wang, W. M. Campbell, E. E. Bonfantani, K. W. Jolley, D. L. Officer, P. J. Walsh, K. Gordon, R. Humphry-Baker, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. B, 2005, 109, 15397–15409 Search PubMed.
- W. M. Campbell, K. W. Jolley, P. Wagner, K. Wagner, P. J. Walsh, K. C. Gordon, L. Schmidt-Mende, M. K. Nazeeruddin, Q. Wang, M. Grätzel and D. L. Officer, J. Phys. Chem. C, 2007, 111, 11760–11762 CrossRef CAS.
- S. L. Wu, H. P. Lu, H. T. Yu, S. H. Chuang, C. L. Chiu, C. W. Lee, E. W.-G. Diau and C. Y. Yeh, Energy Environ. Sci., 2010, 3, 949–955 RSC.
- C.-W. Lee, H.-P. Lu, C.-M. Lan, Y.-L. Huang, Y.-R. Liang, W.-N. Yen, Y.-C. Liu, Y.-S. Lin, E. W.-G. Diau and C.-Y. Yeh, Chem.–Eur. J., 2009, 15, 1403–1412 CrossRef CAS.
- T. Bessho, S. M. Zakeeruddin, C.-Y. Yeh, E. W.-G. Diau and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6646–6649 CrossRef CAS PubMed.
- C.-C. Chen, J.-S. Chen, V. S. Nguyen, T.-C. Wei and C.-Y. Yeh, Angew. Chem., 2021, 133, 4936–4943 CrossRef.
- C.-C. Chen, Y.-H. Chen, V. S. Nguyen, S.-Y. Chen, M.-C. Tsai, J.-S. Chen, S.-Y. Lin, T.-C. Wei and C.-Y. Yeh, Adv. Energy Mater., 2023, 13, 2300353 CrossRef CAS.
- Y.-H. Chen, C.-C. Chen, V. S. Nguyen, M.-N. Lu, Y.-D. Chen, Y.-C. Lin, T.-C. Wei and C.-Y. Yeh, ACS Appl. Energy Mater., 2022, 5, 13544–13553 CrossRef CAS.
- J. M. Ji, H. Zhou, Y. K. Eom, C. H. Kim and H. K. Kim, Adv. Energy Mater., 2020, 10, 2000124 Search PubMed.
- Y. Li, K. Yao, H. L. Yip, F. Z. Ding, Y. X. Xu, X. Li, Y. Chen and A. K. Y. Jen, Adv. Funct. Mater., 2014, 24, 3631–3638 CrossRef CAS.
- Y.-X. Xu, C.-C. Chueh, H.-L. Yip, F.-Z. Ding, Y.-X. Li, C.-Z. Li, X. -Li, W.-C. Chen and A. K.-Y. Jen, Adv. Mater., 2012, 24, 6356–6361 CrossRef CAS PubMed.
- Y. Liu, Y. Cao, W. Zhang, M. Stojanovic, M. I. Dar, P. Péchy, Y. Saygili, A. Hagfeldt, S. M. Zakeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2018, 57, 14125–14128 CrossRef CAS PubMed.
- Y. Zhang, T. Higashino, I. Nishimura and H. Imahori, ACS Appl. Mater. Interfaces, 2024, 16, 67761–67770 CrossRef CAS PubMed.
- S. Hattori, Y. Wada, S. Yanagida and S. Fukuzumi, J. Am. Chem. Soc., 2005, 127, 9648–9654 CAS.
- S. M. Feldt, G. Wang, G. Boschloo and A. Hagfeldt, J. Phys. Chem. C, 2011, 115, 21500–21507 Search PubMed.
- M. Hussain, A. Islam, I. Bedja, R. K. Gupta, L. Han and A. El-Shafei, Phys. Chem. Chem. Phys., 2014, 16, 14874–14881 Search PubMed.
- Y. Saygili, M. Söderberg, N. Pellet, F. Giordano, Y. Cao, A. B. Munoz-García, S. M. Zakeeruddin, N. Vlachopoulos, M. Pavone, G. Boschloo, L. Kavan, J. E. Moser, M. Grätzel, A. Hagfeldt and M. Freitag, J. Am. Chem. Soc., 2016, 138, 15087–15096 CAS.
- M. Hu, J. Shen, Z. Yu, R. Z. Liao, G. G. Gurzadyan, X. Yang, A. Hagfeldt, M. Wang and L. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 30409–30416 Search PubMed.
- H. Imahori, S. Kang, H. Hayashi, M. Haruta, H. Kurata, S. Isoda, S. E. Canton, Y. Infahsaeng, A. Kathiravan, T. Pascher, P. Chábera, A. P. Yartsev and V. Sundström, J. Phys. Chem. A, 2011, 115, 3679–3690 CrossRef CAS PubMed.
- S. Ye, A. Kathiravan, H. Hayashi, Y. Tong, Y. Infahsaeng, P. Chabera, T. Pascher, A. P. Yartsev, S. Isoda, H. Imahori and V. Sundström, J. Phys. Chem. C, 2013, 117, 6066–6080 CAS.
- C.-C. Chen, J.-S. Chen, V. S. Nguyen, T.-C. Wei and C.-Y. Yeh, Angew. Chem. Int. Ed., 2021, 60, 4886–4893 CrossRef CAS PubMed.
- Y. Wang, B. Chen, W. Wu, X. Li, W. Zhu, H. Tian and Y. Xie, Angew. Chem., Int. Ed., 2014, 126, 10955–10959 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: It includes the synthesis procedures, 1H NMR spectra, 13C NMR spectra, mass spectra, details of theoretical calculations and all necessary figures and tables of the new compounds. See DOI: https://doi.org/10.1039/d5se00043b |
| ‡ F. A. Faraghally and Y.-H. Chen contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |