
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper(I) bis(diimine) sensitized titania nanotube array photoelectrodes for photoelectrochemical water oxidation†
Joseph D.
Chiong
 ab,
Zujhar
Singh
a,
Joseph F.
Ricardo-Noordberg
a,
Nhat Truong
Nguyen
*b and
Marek B.
Majewski
*a
ab,
Zujhar
Singh
a,
Joseph F.
Ricardo-Noordberg
a,
Nhat Truong
Nguyen
*b and
Marek B.
Majewski
*a
aDepartment of Chemistry and Biochemistry and Centre for NanoScience Research, Concordia University, 7141 Sherbrooke Street West, Montreal, Quebec, Canada H4B 1R6. E-mail: marek.majewski@concordia.ca
bDepartment of Chemical and Materials Engineering, Concordia University, 1515 Saint-Catherine Street West, Montreal, Quebec, Canada H3G 2WI. E-mail: truong.nguyen@concordia.ca
First published on 4th February 2025
Abstract
Dye-sensitized photoelectrochemical cells (DS-PECs), devices inspired by photosynthesis, are being developed to advance the goal of using the sun as the sole source of energy for converting abundant resources to fuel and valuable chemicals. Herein, we report compact and vertically aligned titanium dioxide nanotubes grown through self-organized electrochemical anodization as semiconducting materials functionalized with a molecular copper(I) bis(diimine)-based acceptor–chromophore–donor to yield a photoanode capable of carrying out oxidative processes. The ability of these dye-sensitized photoanodes to drive oxidative processes is further confirmed photoelectrochemically through activation of a molecular iridium(III) water oxidation pre-catalyst where ultimately a Faradaic efficiency of 84% is found for O2 production.
Introduction
Creating devices that replicate intricate systems found in nature has been a research point of interest towards achieving current sustainability goals. Such devices, inspired by natural photosynthesis, are dye-sensitized photoelectrochemical cells (DS-PECs).1,2 DS-PECs store energy by conversion of sunlight to chemical bonds allowing for the generation of “solar fuels” at the point of deployment.3,4 In these devices, semiconductor photoelectrodes generate excitons from light absorption, leading to free charge carriers that can drive redox reactions like the direct reduction/oxidation of adsorbed species on the photocathode/anode.5–8 Previously, copper(I) bis(diimine) complexes have been used to sensitize wide bandgap semiconductors for visible light absorption to construct photoelectrodes for DS-PEC applications.9–14 In these systems, the fast recombination of electron–hole pairs was mitigated through the use of acceptor–chromophore–donor triads to generate long-living charge separated states. These molecular copper(I)-based photosensitizers are of interest when considering the low-cost and analogous photophysical properties compared to ruthenium(II) polypyridine photosensitizers typically used in these types of cells.15–19Titania, TiO2, is one of the most broadly studied wide bandgap semiconductors (often as a photocatalyst where the anatase polymorph has a bandgap of 3.2 V and, as a result, can only carry out photocatalysis under UV-light irradiation).1,20–24 TiO2 is considered a sustainable material, is relatively inexpensive, chemically stable, and is the prototypical photocatalyst for water oxidation.22,25–27 One-dimensional (1D) semiconductor nanostructures of TiO2 have been shown to grant added benefits such as two-dimensional confinement, directional charge transport, and orthogonal charge separation.23,24,28,29 Previous reports have highlighted self-organized electrochemical anodization (SOA) as a method to yield organized vertically oriented arrays of TiO2 nanotubes.23,25,26,28,30–32
Herein, we describe the use of TiO2 nanotubes (NTs) as a semiconducting surface for photoelectrodes sensitized with a Cu(I) bis(diimine)-based acceptor–chromophore–donor A–Cu(I)–D complex (Fig. 1). Through characterization and photoelectrochemical studies of the supposedly undecorated TiO2 NTs, we find that they are intrinsically sensitized with carbon impurities (e.g., carbon nitride). As-prepared TiO2 NTs are photosensitized with a previously reported acceptor–chromophore–donor triad A–Cu(I)–D where the electron acceptor A is 3,6-dimethyldipyrido[3,2-a:2′,3′-c]-phenazine-11-carboxylic acid (dppz) and electron donor D is 4-(6,9-dimesityl-1H-imidazo[4,5-f][1,10]-phenanthrolin-2-yl)-N,N-diphenylaniline (TPA) (Fig. 2).10 To validate the resulting photoelectrode architectures, we report photocatalytic water oxidation by incorporating two water oxidation catalysts (WOCs): molecular [Cp*Ir(pyalc)OH] (Cp* = pentamethyl-cyclopentadienyl, pyalc = 2-(2′-pyridyl)-2-propanoate) pre-catalyst and surface deposited cobalt oxide CoOx.33,34 In all cases, the as-prepared photoanodes can drive oxidative processes. A significant increase of photocurrent generated by the photoanode is observed when functionalizing TiO2 NTs with A–Cu(I)–D (Δj = 58 μA cm−2 at 0.2 V vs. SCE), attributed to the photoinduced spatial charge separation found in acceptor–chromophore–donor systems.
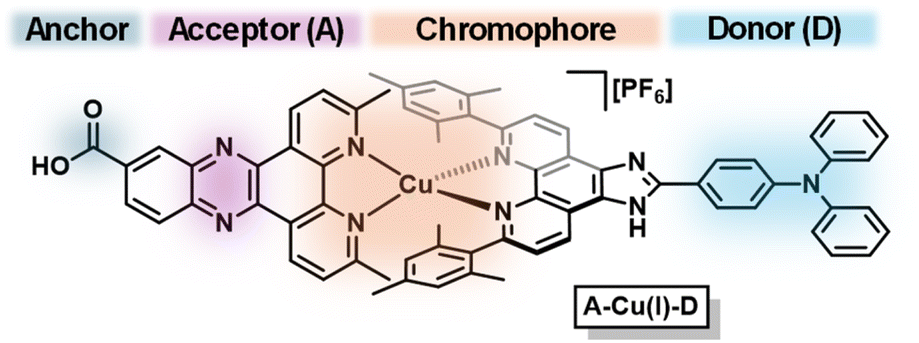 | ||
| Fig. 1 Proposed structure of acceptor–chromophore–donor triad A–Cu(I)–D. | ||
 | ||
| Fig. 2 Fabrication protocol for the TiO2 NTs|A–Cu(I)–D photoanode described in this work along with the proposed light-driven oxidation scheme of [Cp*Ir(pyalc)OH] (pre-catalyst) into [IrIV(pyalc)(H2O)2(μ-O)]22+ (water oxidation catalyst) and subsequent water oxidation. | ||
Experimental
Materials
All chemicals were used as received from commercial sources. Dichloromethane, ethanol, methanol, sodium sulfate, ammonium fluoride, cobalt nitrate hexahydrate, potassium phosphate monobasic, ethylene glycol, and triethanolamine (TEOA) were purchased from Fisher Scientific. Titanium foil (0.127 mm thick, 99.7% purity) was purchased from Strem Chemicals. Potassium phosphate tribasic monohydrate was purchased from Acros Organics. Triethylamine (TEA) was purchased from TCI. Fluorine doped tin oxide (10 Ω) coated glass substrates (FTO) were purchased from Sigma-Aldrich.General methods
All moisture sensitive reactions were carried out using standard Schlenk techniques under an inert atmosphere of nitrogen. 1H NMR spectra were recorded on a 500 MHz Varian spectrometer and a 300 MHz Bruker spectrometer with chemical shifts referenced to the residual solvent peaks.UV-Vis spectra of A–Cu(I)–D and its component ligands were collected in CH2Cl2 using quartz cuvettes with a 1 cm path length on a dual beam Varion Cary 5000 UV-Vis near-infrared (NIR) spectrophotometer with wavelength changeover at 350 nm and a scan rate of 10 nm s−1. Diffuse reflectance UV-Vis spectra of photoanodes were collected using the same instrument with an EasiDiff diffuse reflectance accessory (PIKE Technologies) with Ti foil and TiO2 NTs as the baseline for Ti|TiO2 NTs and Ti|TiO2 NTs|A–Cu(I)–D, respectively.
Electrochemical measurements on molecular species were performed using a WaveDriver 20 Integrated Bipotentiostat/Galvanostat workstation (Pine Research Instrumentation, Inc.) in a conventional three-electrode cell using a glassy carbon button (2 mm) as the working electrode, platinum mesh as the counter electrode and saturated calomel electrode (SCE) as the reference electrode. Photoelectrochemical experiments (deposition of CoOx and subsequent characterization) were conducted using the same instrument in a three-electrode cell configuration with a TiO2 NT photoelectrode as the working electrode, platinum mesh as the counter electrode, and a saturated calomel electrode (SCE) as the reference electrode. The working electrode was illuminated with white light from a single Cree CXB3590 LED on a 140 mm pin heatsink. The diode was equipped with a 400 nm long pass filter and has a power output of 540 mW cm−2 as measured with a PM16-405 thermal sensor power meter from ThorLabs.
Scanning electron microscopy (SEM) images were recorded on a Phenom ProX Desktop instrument and on a Hitachi Regulus 8230 FE-SEM instrument. The films were not sputter-coated. Transmission electron microscopy (TEM) images were recorded on a Talos F200X STEM G2. Samples were analyzed in scanning transmission electron microscopy mode with images obtained using a high-angle annular dark-field (HAADF) detector. Energy-dispersive X-ray spectroscopy (EDS) analyses were done using four SDD Super-X detectors. Sample preparation consists of scratching TiO2 NTs samples into a vial containing 5 mL of isopropanol, sonicating the resulting suspension for 15 min and drop casting twice on a TEM grid using a micropipette.
Powder X-ray diffraction (PXRD) patterns were obtained using a Rikagu Miniflex 600/600C benchtop diffractometer (Applied Rikagu Technologies Inc., Austin, TX, USA) equipped with a CuKα X-ray source (wavelength, λ = 1.54 Å, radiation operating at 40 kV and 15 mA), a D/teX Ultra 2 high-speed 1D detector or HyPix-400 MF 2D sensitive detector (Applied Rigaku Technologies Inc., Austin, TX, USA) and a nickel filter. Sample data were collected using a continuous coupled θ/2θ scan in the 2θ – range of 20–80° with a step size of 0.01°, scan speed of 10° per minute, and spin speed of 20 rpm. X-ray photoelectron spectroscopy (XPS) was done using a Nexus G2 Surface Analysis System.
Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was done using a Thermo Scientific Nicolet iS50 FTIR spectrometer equipped with a MCT detector with a resolution of 1 cm−1 in the range of 4000–650 cm−1.
Self-organizing anodization of titanium dioxide nanotube arrays
Growth of TiO2 NTs was carried out in a custom-made electrochemical cell (Scheme S1†) using an adapted previously reported self-organized anodization method.28 Titanium foils (ca. 2 cm × 1.5 cm) were degreased by sonication in acetone, ethanol, and deionized water and dried in air. Titanium foil, acting as a working electrode, was pressed against a copper contact in the electrochemical cell, leaving a circular area (ca. 1 cm in diameter) exposed to a fluoride-containing electrolyte solution (0.15 M NH4F and 3 wt% H2O in ethylene glycol). A platinum mesh (12.5 mm × 12.5 mm) was submerged in the electrolyte solution above the titanium foil, serving as a counter electrode. Anodization took place when a potential of 60 V was applied for 15–180 minutes using a Xantrex XFR 100-12 DC power supply. Following the anodization period, the sample was rinsed, immersed in ethanol, and dried in air. The TiO2 NTs were then annealed at 450 °C for 1 hour in air using a Thermolyne FB1315M muffle furnace. The resulting photoelectrodes were characterized by SEM, PXRD, XPS, diffuse reflectance UV-Vis and DRIFTS.Ligand and complex synthesis
Photoanode assembly
TiO2 NTs electrodes were left to soak in 0.0125 mM solution of A–Cu(I)–D in CH2Cl2 for 12 hours. The photoelectrodes were removed from the solution, rinsed with CH3OH and CH2Cl2, and then dried in an oven for 2 hours at 90 °C. The photoelectrodes were characterized by diffuse reflectance UV-Vis, DRIFTS and TEM. Loading of A–Cu(I)–D on working electrode surface was obtained by determining the concentration, using UV-Vis of a soaking solution of photosensitizer before and after decoration of the TiO2 NTs. The concentration was calculated using the molar absorptivity constant (ε) 7900 L mol−1 cm−1 for the 1MLCT transition in A–Cu(I)–D (centered at ca. 460 nm).10 With the known volume of soaking solution used, the amount (in mol) can be calculated and divided by the area of TiO2 NTs to yield the loading per surface area of the device.Deposition of CoOx water oxidation catalyst on photoanodes
The CoOx water oxidation catalyst was deposited on photoanodes by photoelectrochemical deposition adapted from previously reported experimental conditions.34,42 A representative procedure is as follows: photoanodes were immersed in cobalt precursor-containing aqueous electrolyte solution of 0.5 mM Co(NO3)2 in 0.1 M pH 7 phosphate buffer (KPi) and held at +0.2 V versus SCE under illumination from a single 540 mW cm−2 UV-filtered white LED for 180 seconds. Following the deposition, the samples were immersed in and rinsed with deionized H2O and dried in air.Photoelectrochemical testing
Photoelectrochemical testing of photoanodes was performed using an adapted method outlined by Jewell et al.34 It was carried out in a single compartment Pyrex® cell with a circular quartz window filled with aqueous 0.1 M Na2SO4 pH 8.9 as electrolyte solution. The photoanode was illuminated using the same LED used for photoelectrochemical deposition. Before carrying out the measurements the cell was allowed to rest in the dark for 2 minutes to disperse any prior charge buildup. First, the open-circuit potential, Voc, was measured against SCE for 90 seconds in the dark and measured again under illumination. Second, chopped-light chronoamperometry was performed, where the photoanode was held at 0 V versus SCE for 300 seconds with 20 seconds light/dark transient, and then the process was carried out a second time at 0.2 V. Third, the circuit was allowed to rest again for 2 minutes before re-measuring Voc in the dark and under illumination again. Fourth and finally, a current–voltage (i–V) photocurrent transient experiment (linear sweep voltammetry) was carried out with a scan region from −0.2 to +1.0 V versus SCE with a 10 mV s−1 scan rate and 5 seconds light/dark transient.Oxygen detection
Oxygen detection was carried out using the two-electrode collector-generator method. This technique is used in DS-PECs for in situ quantification of photoelectrochemically produced O2 where the two electrodes, the collector and the generator, were assembled together with a 1 mm glass spacer (Fig. S2†).43 The procedure for O2 detection and quantification was derived from work carrying out similar studies.2,9,34,43–47 The generator Ti|TiO2 NTs|A–Cu(I)–D and collector (unfunctionalized FTO) were sandwiched and used as working electrodes in an H-cell with a porous glass separator. The collector-generator assembly and SCE reference electrode were placed together in one compartment and the counter electrode alone was placed in the opposite compartment. The H-cell was filled with aqueous 0.1 M Na2SO4 pH 8.9 electrolyte solution which was deaerated by bubbling with N2 for 1 hour prior to use and further deaerated with Ar in both compartments for an additional 10 minutes prior to current measurements. Photocurrent generated at the TiO2 NTs photoanode and current generated at the FTO collector from reduction of O2 to H2O was measured with a potential held at 0.2 V and −0.6 V versus SCE respectively, which begins with a dark period of 300 seconds, followed by 300 seconds with light illumination and ending with 300 seconds in the dark ensuring residual O2 diffusion is detected. The resulting data were used to calculate the Faradaic efficiency for oxygen generation (ηO2) for the photoanodes following where Qcol and Qgen are the total charge passed at the collector and generator electrode respectively and ηeff is the collection efficiency of the collector-generator assembly (70%).43
where Qcol and Qgen are the total charge passed at the collector and generator electrode respectively and ηeff is the collection efficiency of the collector-generator assembly (70%).43
Results and discussion
Preparation of TiO2 nanotube arrays
The formation of the TiO2 NTs by self-organizing electrochemical anodization was carried out using modifications of previously reported methods.28,31,32 The obtained morphology was observed through SEM and consists of an ordered array of nanotubes where the tubes are aligned in a compact manner, resulting in a uniform surface (Fig. 3A). The lack of spacing between the individual tubes results from the initiation layer, which is described as the collar at the mouth of the tubes.28 This initiation layer provides the surface for photosensitizer deposition and will likely act as the primary site of photoactivity (Scheme S2†). The length of the tubes is directly influenced by the time of the anodization period up to the extent of reaching equilibrium between the formation of the tubes and their dissolution in the fluoride-containing electrolyte. The tube length is obtained by taking cross-sectional SEM of the NTs by physical slicing at the surface using a snap-blade knife (Fig. 3B). The tube length for an anodization period of 15 minutes is observed to be 6.0 ± 0.1 μm with a tube aperture of 73.0 ± 0.5 nm in diameter (Fig. S3†). After the growth process, the films were annealed at 450 °C for 1 hour resulting in an observable change to the color of the surface (Fig. S4†). EDS elemental mapping confirms the presence of TiO2 and furthermore that it is contacted on Ti metal (Fig. 3C and S5†). PXRD confirms the presence of the desired anatase phase with the appearance of a characteristic (101) reflection at 25.3° 2θ.48 The post-anodization product is amorphous with reflections associated primarily with the base Ti foil (Fig. 3D). | ||
| Fig. 3 (A) SEM micrograph of a TiO2 NT surface, (B) cross-sectional SEM micrograph of TiO2 NTs, (C) EDS elemental analysis of TiO2 NTs, (D) PXRD diffractogram of TiO2 NTs (simulated anatase AMCSD 0019093 and simulated rutile AMCSD 0001735) and (E) XPS survey of TiO2 NTs. | ||
XPS was used to further examine the surface composition of the TiO2 NTs. XPS survey confirms the surface composition showing binding energies associated with TiO2 NTs and of additional interest, surface composition also includes 14.36% carbon and 1.48% nitrogen (Fig. 3E). In the high-resolution scan of Ti 2p, a pair of well-separated binding energies is present, 459.45 and 465.14 eV, associated with Ti 2p3/2 and Ti 2p1/2, respectively, generally associated with Ti4+ and further confirming the anatase phase (Fig. S6†).49 In the high-resolutions scan of O 1 s, the main component of the signal is the Ti–O bond at 532.04 eV associated with TiO2 and another component at 530.69 eV is attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O resulting from the incorporation of carbon into the inner walls of the nanotubes (Fig. S7†).50 Since ethylene glycol electrolyte-based TiO2 NTs are known to have a carbon rich inner shell that fuses into the inner tube walls during the annealing process, the nature of these carbon impurities is of interest.32 Presumably, these carbon impurities can exist as carbon nitrides incorporated into the nanotubular structure of TiO2. XPS helps support this presumption as the observed binding energy of N 1s at 401 eV is attributed to carbon nitride (Fig. S8†).51–53 The high-resolution scan of C 1s yields peaks at 287.33 and 289.43 eV that can be associated to C–O and CO, respectively (Fig. S9†).54 The peaks at 285.70 and 286.64 eV are associated with C–NHx (x = 1, 2) and –CN, respectively, and hint at the carbon nitride nature of the carbon impurities.55–59 The presence of these impurities should result in a TiO2 surface that is intrinsically sensitized to visible light (as opposed to just UV as implied by the bandgap energy of anatase phase TiO2).
O resulting from the incorporation of carbon into the inner walls of the nanotubes (Fig. S7†).50 Since ethylene glycol electrolyte-based TiO2 NTs are known to have a carbon rich inner shell that fuses into the inner tube walls during the annealing process, the nature of these carbon impurities is of interest.32 Presumably, these carbon impurities can exist as carbon nitrides incorporated into the nanotubular structure of TiO2. XPS helps support this presumption as the observed binding energy of N 1s at 401 eV is attributed to carbon nitride (Fig. S8†).51–53 The high-resolution scan of C 1s yields peaks at 287.33 and 289.43 eV that can be associated to C–O and CO, respectively (Fig. S9†).54 The peaks at 285.70 and 286.64 eV are associated with C–NHx (x = 1, 2) and –CN, respectively, and hint at the carbon nitride nature of the carbon impurities.55–59 The presence of these impurities should result in a TiO2 surface that is intrinsically sensitized to visible light (as opposed to just UV as implied by the bandgap energy of anatase phase TiO2).
Photoanode assembly and characterization
TiO2 NTs samples were grown for periods of 15, 30, 45, 60, 90, 120, 180 minutes and the corresponding photocurrents (without decorating with a photosensitizer) were found to increase in a linear fashion up to 90 minutes (Fig. S10A and B†). From these data alone it is difficult to determine why this may be the case; however, we speculate that there are three possible scenarios: (i) after a 90 minutes anodization period, an equilibrium between the formation of TiO2 NTs and dissolution has been reached, (ii) the nanotube length associated with the growth up to 90 minutes represent the limit for light absorption (i.e., any light-absorbing sites found deeper within the structure are no longer being illuminated) and (iii) the nanotube length becomes too long to the extent that there is slower charge transfer between the surface and the conductive Ti foil in addition to increasing recombination thus becomes less effective than shorter nanotube lengths. In the regime where nanotube growth corresponds to a linear increase in photocurrent, the respective nanotube lengths were measured (through the cross-sectional SEM micrograph of nanotube array fragments) and used to normalize photocurrent data. From this analysis, photocurrent density per μm of TiO2 NTs averages resulted in constant values of 0.64 μA cm−2 μm−1 at 0 V of applied bias and 1.74 μA cm−2 μm−1 at 0.2 V of applied bias (Fig. S10C and D†). The photocurrent density of TiO2 NTs photoelectrode can be maximized up to a plateau photocurrent of ≈20 μA cm−2 for an anodization period of 90 minutes, yielding ≈12 μm long TiO2 NTs (Fig. S10C†).The acceptor ligand (A), donor ligand (D) and triad A–Cu(I)–D were synthesized and characterized as previously reported by our lab and confirmed by 1H NMR (Fig. S11†).9,10,35–37 The sensitized photoanodes were constructed by soaking TiO2 NT films for 15 min in a solution of A–Cu(I)–D to yield a Ti|TiO2 NTs|A–Cu(I)–D film (Fig. 2). This differs from previously reported surface-grafting procedures as the semiconducting TiO2 NTs are grown using a top-down method, resulting in them being directly attached to the Ti foil.34,42,46,60 The resulting films were characterized using diffuse reflectance UV-Vis spectroscopy, where the TiO2 NTs show absorbance associated with the carbon impurities in the visible part of the electromagnetic spectrum extending from the blue-violet region (Fig. 4A).61,62 These data were used as the baseline for films decorated with A–Cu(I)–D, and as such, any additional absorbance will originate from the addition of the photosensitizer. TiO2 NTs|A–Cu(I)–D films show an 1MLCT band (centered at ca. 480 nm) that can be directly compared with the 1MLCT observed in the A–Cu(I)–D solution UV-Vis spectrum (Fig. 4B) with a λmax red-shift (Δλ = 10 nm) associated with increased conjugation of the ligands through planarization caused by rigidification of the aromatic groups when the complexes are anchored onto the surface of TiO2 (Fig. 4C).63 By comparing the DRIFTS spectra of the A–Cu(I)–D complex in a KBr pellet with the TiO2 NTs films decorated with the complex, we can confirm its attachment through the observation of a mode centered at 786 cm−1 associated with Ti–O (Fig. 4D).64,65 Additional modes centered around 3000 to 3100 cm−1 indicate the sp2 stretching vibrations associated with the presence of aromatic rings in A–Cu(I)–D while modes centered around 2850 to 3000 cm−1 observed are associated with the sp3 stretching vibrations of the methyl groups present on the acceptor ligand. Additionally, when A–Cu(I)–D is on the surface of TiO2 NTs, the spectra also show pairs of bands at 1585 cm−1 and 1604 cm−1 and 1356 cm−1 and 1371 cm−1 associated with asymmetric stretches and vibrations of carboxylates, respectively.10,66 A characteristic feature of A–Cu(I)–D in KBr, the carbonyl stretching mode at 1733 cm−1, is lost on TiO2 NTs upon conversion of carboxylic acid to carboxylate when anchored onto the surface.10,67,68 Additionally, we note that on the unfunctionalized TiO2 NTs films, we observe possible CN and C–N aromatic stretching modes in the ranges of 1400 to 1600 cm−1 further hinting at the presence of carbon impurities (Fig. S12†).57,69,70 Further supporting evidence of carbon impurities on the surface of unfunctionalized TiO2 NTs and its decoration with A–Cu(I)–D is obtained by TEM and presented in the HAADF micrographs and corresponding elemental maps (Fig. S13 and 14†).
 | ||
| Fig. 4 (A) Diffuse reflectance UV-Vis spectrum of TiO2 NTs. (B) UV-Vis spectrum of A–Cu(I)–D in CH2Cl2 at room temperature (black arrow indicates the position of the proposed 1MLCT transition). (C) Diffuse reflectance UV-Vis spectrum of TiO2 NTs|A–Cu(I)–D (black arrow indicates the position of the proposed 1MLCT transition mirroring that of the triad in solution). (D) DRIFTS spectrum of TiO2 NTs|A–Cu(I)–D (red) and A–Cu(I)–D in a KBr pellet (black). | ||
Photoelectrochemistry of unfunctionalized TiO2 NTs
For comparison purposes, an amorphous sample of TiO2 NTs and an anatase sample of TiO2 NTs were examined. Unfunctionalized TiO2 NTs, after undergoing heat treatment at 450 °C to yield the anatase phase, are capable of visible-light absorption and photocurrent generation (Fig. 5A and S15†) from carbon impurities introduced during annealing.32 TiO2 nanotubes resulting from anodization of Ti in ethylene glycol-containing electrolyte are known to be composed of an outer shell that consists of pure TiO2 and an inner shell composed of carbon-rich titanium oxide or oxy-hydroxide following the electrochemical anodization.71–74 In addition to anatase generally exhibiting higher photoactivity, crystallization into this phase provides high electron mobility compared to its rutile or amorphous counterparts.75 Thermal treatment of as-grown TiO2 NTs at 450 °C in a furnace for 1 hour yields the desired anatase phase. It has been reported that thermal treatments with rates above 50 °C s−1 result in the inner and outer shells to be fused together.71 Thus carbon-derived impurities such as carbon nitride are likely incorporated into the inner surface of the TiO2 NTs allowing for photocurrents to be generated upon illumination with light (as supported by XPS analysis, vide supra). In our hands, this was also partially confirmed by the fact that CoOx was deposited on the surface of unfunctionalized TiO2 NTs under white light illumination—a process that requires the oxidation of Co2+ at the electrode surface through the photogeneration of oxidizing equivalents (Fig. S16A†). Moreover, transient photocurrent measurements of unfunctionalized TiO2 NTs show plateau photocurrents of ≈8–12 μA cm−2 under white light illumination (Fig. 5A). By comparison, as-grown TiO2 NTs generate negligible amounts of photocurrent without a thermal treatment step (Fig. 5A and S15†). Oxidative charge build-up is observed at the surface of the TiO2 NTs photoelectrode as indicated by the local capacitance (photocurrent “spiking”) upon initial illumination with light. | ||
| Fig. 5 (A) Chopped-light chronoamperometry experiments of amorphous TiO2 NTs (yellow), anatase TiO2 NTs (blue) and anatase TiO2 NTs with triethylamine as a sacrificial electron donor (red) at 0.2 V vs. SCE. Transient photocurrent resulting from the addition of [Cp*Ir(pyalc)OH] to (B) TiO2 NTs|A–Cu(I)–D and (C) TiO2 NTs. (D) Photocurrent resulting from electrodes with photoelectrochemically deposited CoOx on TiO2 NTs and TiO2|A–Cu(I)–D. All experiments were conducted in aqueous 0.1 M Na2SO4 pH 8.9 solution with the photoelectrode as working electrode, platinum mesh as counter electrode and a saturated calomel electrode as reference electrode. | ||
With the addition of triethylamine (TEA) as a sacrificial electron donor to the 0.1 M Na2SO4 pH 8.9 electrolyte solution, a maximum plateau photocurrent of ≈22 μA cm−2 is observed due to quenching the oxidative charge build-up at the surface of the photoanode and further injection of electrons into the conduction band of the TiO2 NTs (Fig. 5A).
Photoelectrochemical water oxidation
To examine the oxidative capabilities of the fabricated photoanodes, we investigated their ability to activate water oxidation catalysts (WOC) either in an ensemble with the catalyst in solution or with the catalyst bound to the surface. Here, we use a champion molecular electrocatalyst [Cp*Ir(pyalc)OH], to probe the performance of the photoanodes by transforming this precursor into a dimeric form and carrying out water oxidation. From a preliminary analysis it appears there exists a misalignment of redox potentials across this system but we note that it has been shown in other work that it is possible to drive such oxidative processes despite the mismatch (Scheme S3†).9 UV-Vis data show that [Cp*Ir(pyalc)OH] has no absorbance in the visible region, but when oxidized into the dimer [IrIV(pyalc)(H2O)2(μ-O)]22+ a broad absorbance band centered around 600 nm is observed (Fig. S17†) and associated with the IV–IV state of [IrIV(pyalc)(H2O)2(μ-O)]22+ that acts as the active water oxidation catalyst.33In parallel, a CoOx WOC was successfully deposited on TiO2 and TiO2|A–Cu(I)–D, yielding TiO2|CoOx and TiO2|A–Cu(I)–D|CoOx photoelectrodes. CoOx photoelectrodeposition was initiated through bulk electrolysis where chronoamperograms show a stable current after turning the light on at 3 seconds and throughout the 180 seconds of photoelectrochemical deposition indicating a steady injection of electrons originating from the oxidation of the Co2+ precursor into higher oxidation states and depositing at the surface as CoOx (Fig. S16†). Photoelectrochemical deposition of CoOx results in a monolayer at the surface of TiO2 NTs localized on areas where there are high concentrations of photogenerated holes.42,76,77 This is confirmed by TEM and presented in the HAADF micrograph and elemental maps in which cobalt is present on the surface of the TiO2 NTs (Fig. S18†).
The sequence of photoelectrochemical testing follows both the light/dark transient at 0 V and 0.2 V of applied bias. All the studied photoanodes present the ability to generate photocurrent under illumination with visible light. The addition of the A–Cu(I)–D triad leads to a drastic increase of the plateau photocurrent from typical ≈8–12 μA cm−2 at 0.2 V observed for bare TiO2 NTs to ≈70 μA cm−2 by the end of the 5 minutes light on/off cycling (Fig. 5B). For the presented photoanode, the addition of ≈50 μL of triethanolamine (TEOA) to the electrolyte solution results in enhanced photocurrents with decreased local capacitance at the onset of illumination resulting from faster regeneration kinetics yielding increased electron injection into the TiO2 NTs (Fig. S19†). The photoanodes were evaluated for their ability to activate the [Cp*Ir(pyalc)OH] WOC pre-catalyst.
In the presence of the pre-catalyst, unfunctionalized TiO2 NTs show a decrease in the photocurrent (Fig. 5C). This can be attributed to the previously reported “anti-catalyst” effect where carbon impurities present at the surface of the TiO2 NTs can act as trap states for recombination from the WOC.34,78–80 For the TiO2 NTs|A–Cu(I)–D photoelectrode, a minimal decrease in photocurrent is observed (Fig. 5B). This is expected from the delay in forming the active form of the WOC and photosensitizer desorption over time (Fig. S20†). Despite this minimal decrease in photocurrent, close observations before and after the addition of [Cp*Ir(pyalc)OH] indicate that the ratio of capacitance from the moment the light is turned on and the photocurrent plateau prior to turning the light off is consistently lower after the addition of the pre-catalyst which may indicate a redox process taking place on the surface of the photoanode (Fig. S21†). The addition A–Cu(I)–D on the surface of the TiO2 NTs may passivate surface localized impurities negating the “anti-catalyst” effect. Although it has been reported previously that despite having a photosensitizer-decorated metal oxide, the “anti-catalyst” effect expresses itself upon the addition of the WOC,34,78–80 however, in the present work, the siting of the carbon impurities may be restricted to the surface. In other semiconductor preparations, owing to the use of organic stabilizers and binders for templating the assembly layers of metal oxide nanoparticles, carbon impurities are incorporated throughout the bulk of the material compared rather than only at the surface of TiO2 NTs. Where addition of CoOx to Ti|TiO2 NTs resulted in a net photocurrent increase, no increase in photocurrent in the TiO2 NTs|A–Cu(I)–D|CoOx photoanode was observed (Fig. 5D) where some desorption of A–Cu(I)–D during photoelectrochemical deposition is likely (Fig. S22†).
Photocurrent origins
To further elucidate the basis of the photocurrent being observed on unfunctionalized and functionalized TiO2 NTs films, photoaction spectra were plotted. High power density LEDs with specific wavelengths were used for unfunctionalized TiO2, and a variable wavelength light source with low power density was used for Ti|TiO2 NTs|A–Cu(I)–D (Scheme S4†). Using different light sources with different power densities should elucidate the contribution to the photocurrent based on light penetration depth and the surface composition of the TiO2 NTs. When examining the photocurrent plateau of Ti|TiO2 NTs|A–Cu(I)–D, we observe that the highest photocurrent can be found at excitation wavelengths around 480 nm (Fig. S23†), and in general the photoaction spectrum matches that of the diffuse reflectance UV-Vis where the photocurrent maxima match the absorbance of the 1MLCT associated with the A–Cu(I)–D complex (Fig. S24†). In unfunctionalized TiO2 NTs, the photocurrent maximum is found around 475 nm (Fig. S25†). We note that the controls for the respective photoaction spectra do not generate any significant photocurrent at any given wavelength, further confirming that the photocurrent plateau observed can be attributed to the carbon impurities in the inner walls and A–Cu(I)–D on the initiation layer using high power and low power density lights sources respectively (Fig. S23–25†). From these data, the incident photon-to-current efficiency (IPCE) was determined to be 4% and 1% for maximum photocurrent at 0.2 V associated with the absorption maxima for TiO2 NTs and Ti|TiO2 NTs|A–Cu(I)–D respectively (Tables S1 and 2†). IPCE for the TiO2 NTs is highly dependent on morphology, and illumination intensity plays a role in what surface (i.e., initiation layer or inner tube walls) of the TiO2 NTs will generate the photocurrent.81Collector-generator Faradaic efficiency
The collector-generator method was used to probe the efficacy of the Ti|TiO2 NTs|A–Cu(I)–D architecture for generating O2 from H2O. Faradaic efficiency of O2 generation based on the photoanode systems TiO2 NTs|A–Cu(I)–D|CoOx and TiO2NTs|A–Cu(I)–D + [Cp*Ir(pyalc)OH] versus TiO2 NTs|A–Cu(I)–D and bare TiO2 NTs as controls was calculated from their respective current response. The two-electrode collector-generator method has been widely used to evaluate O2 evolution in photoelectrochemical water oxidation and validated with Clark-type O2 sensors.82–84 The Faradaic efficiencies of the controls TiO2 NTs and Ti|TiO2 NTs|A–Cu(I)–D are 15% and 27% respectively, both able to carry out water oxidation without the presence of a WOC, however with low Faradaic efficiencies, and the addition of the acceptor–chromophore–donor A–Cu(I)–D system represent a significant increase almost doubling the Faradaic efficiency of unfunctionalized TiO2 NTs (Fig. S26†). It is important to note that the Faradaic efficiencies mentioned are calculated based on the ratio of total charges (Qgen and Qcol) for the period between 100 s and 400 s which represents an overall underestimation of the true Faradaic efficiency given that there is still O2 diffusing to the collector after the light has been turned off (Fig. S27†). When quantifying oxygen production with the addition of [Cp*Ir(pyalc)OH] to the TiO2 NTs|A–Cu(I)–D photoanode, stoichiometric ratio of the acting oxidant and pre-catalyst need to be considered. This is shown when using a stoichiometric ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :14 (0.1134 mM [Cp*Ir(pyalc)OH]) and 1:1 (0.0081 mM [Cp*Ir(pyalc)OH]) of A–Cu(I)–D:[Cp*Ir(pyalc)OH] to run the collector-generator experiment resulting in 64% and 84% Faradaic efficiencies, respectively (Fig. 6). The ratios used are estimates based on loading (3.24 × 10−8 mol cm−2) of A–Cu(I)–D on TiO2 NTs (Fig. S28†). The pre-catalyst needs to be converted to the catalytically active dimer through oxidation at the photoanode surface. In other words, one excited A–Cu(I)–D can transfer one oxidizing equivalent to an approaching pre-catalyst molecule to yield one equivalent of active catalyst. In a solution where the concentration of [Cp*Ir(pyalc)OH] is lower, the current at the collector electrode increases and plateaus more quickly, hinting that conversion of the pre-catalyst is completed rapidly (Fig. 6B). Furthermore, when 0.1134 mM [Cp*Ir(pyalc)OH] is added to the electrolyte when using a bare TiO2 NTs photoanode, a Faradaic efficiency of 29% is obtained which represents a 14% increase upon addition of [Cp*Ir(pyalc)OH] compared to a 36% increase when using the TiO2 NTs|A–Cu(I)–D photoanode with the same concentration of [Cp*Ir(pyalc)OH] (Fig. S26 and 27†). This highlights the effect of the A–Cu(I)–D photosensitizer on the surface of the TiO2 NTs which allows visible light absorption and the photoinduced charge transfer that enables the oxidation of [Cp*Ir(pyalc)OH]. With TiO2 NTs|A–Cu(I)–D|CoOx as the generator electrode, a Faradaic efficiency of 11% (close to the TiO2 NTs control) was found, attributed to the “anti-catalyst” effect, photosensitizer desorption, or a potential mismatch between the excited state of A–Cu(I)–D and CoOx (Fig. S27†).
:14 (0.1134 mM [Cp*Ir(pyalc)OH]) and 1:1 (0.0081 mM [Cp*Ir(pyalc)OH]) of A–Cu(I)–D:[Cp*Ir(pyalc)OH] to run the collector-generator experiment resulting in 64% and 84% Faradaic efficiencies, respectively (Fig. 6). The ratios used are estimates based on loading (3.24 × 10−8 mol cm−2) of A–Cu(I)–D on TiO2 NTs (Fig. S28†). The pre-catalyst needs to be converted to the catalytically active dimer through oxidation at the photoanode surface. In other words, one excited A–Cu(I)–D can transfer one oxidizing equivalent to an approaching pre-catalyst molecule to yield one equivalent of active catalyst. In a solution where the concentration of [Cp*Ir(pyalc)OH] is lower, the current at the collector electrode increases and plateaus more quickly, hinting that conversion of the pre-catalyst is completed rapidly (Fig. 6B). Furthermore, when 0.1134 mM [Cp*Ir(pyalc)OH] is added to the electrolyte when using a bare TiO2 NTs photoanode, a Faradaic efficiency of 29% is obtained which represents a 14% increase upon addition of [Cp*Ir(pyalc)OH] compared to a 36% increase when using the TiO2 NTs|A–Cu(I)–D photoanode with the same concentration of [Cp*Ir(pyalc)OH] (Fig. S26 and 27†). This highlights the effect of the A–Cu(I)–D photosensitizer on the surface of the TiO2 NTs which allows visible light absorption and the photoinduced charge transfer that enables the oxidation of [Cp*Ir(pyalc)OH]. With TiO2 NTs|A–Cu(I)–D|CoOx as the generator electrode, a Faradaic efficiency of 11% (close to the TiO2 NTs control) was found, attributed to the “anti-catalyst” effect, photosensitizer desorption, or a potential mismatch between the excited state of A–Cu(I)–D and CoOx (Fig. S27†).
 | ||
| Fig. 6 Collector-generator dual working electrode experiment on TiO2 NTs|A–Cu(I)–D with the addition of (A) 0.1134 mM and (B) 0.0081 mM [Cp*Ir(pyalc)OH] in aqueous 0.1 M Na2SO4 pH 8.9. The generator electrode was held at 0.2 V and illuminated with white light. The collector electrode was held at −0.6 V. reference electrode: saturated calomel electrode; counter electrode: platinum mesh. The experiment is carried out in an H-cell in which the collector-generator assembly and reference electrode are placed together in one compartment and the counter electrode alone is placed in the opposite compartment across a porous glass frit. | ||
Conclusion
The present work represents a study of TiO2 NTs as a potential nanostructured semiconducting material for applications in DS-PECs. The nanostructure morphology of vertically oriented arrays of TiO2 nanotubes yields desired directional charge transport properties in addition to somewhat unexpected optical properties that stem from the incorporation of carbon impurities. Photoelectrochemical studies show that the TiO2 nanotubes can generate photocurrents yielding oxidative potential at the surface and that is further enhanced when sensitized with the acceptor–chromophore–donor system where in the latter we observe contributions primarily from the surface installed triad. We have shown that the TiO2 NTs can be optimized (maximizing the base photocurrent) for the given application by carefully tuning the anodization period considering the length of the nanotubes by increasing surface area and mitigating the limitation of light absorption associated with the depth of the nanotubes. Photoelectrochemical studies show that the resulting photoanodes can oxidize a [Cp*Ir(pyalc)OH] pre-catalyst into [IrIV(pyalc)(H2O)2(μ-O)]22+ active water oxidation catalyst and proceed to carry out water oxidation with a Faradaic efficiency as high as 84% for the TiO2 NTs|A–Cu(I)–D photoanode.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Natural Sciences and Engineering Research Council of Canada (NSERC) [M. B. M funding reference number RGPIN-2018-04391; N. T. N. funding reference number RGPIN-2022-04678], the Fonds de Recherche du Québec – Nature et technologies (FRQNT) and the Quebec Centre for Advanced Materials (QCAM) for financial support. The authors thank Tyler Rutherford for his assistance in acquiring NMR spectra. Mass spectrometry was carried out at the Centre for Biological Applications of Mass Spectrometry (CBAMS). The authors thank Dr Melanie Hazlett for allowing us to use the IR spectrometer in her lab and Chris Copeman for assistance in acquiring DRIFTS spectra. TEM imaging and EDS analyses were carried out at the Facility for Electron Microscopy Research (FEMR) – McGill with the assistance of Dr Jesus Angel Valdez Aguilar. XPS analysis was carried out at the McGill Institute for Advanced Materials (MIAM) with the assistance of Dr Lihong Shang. FE-SEM imaging was carried out at the Concordia Materials Characterization Platform (CMCP) with the assistance of Dr Dmytro Kevorkov.References
- J. Willkomm, K. L. Orchard, A. Reynal, E. Pastor, J. R. Durrant and E. Reisner, Chem. Soc. Rev., 2016, 45, 9–23 RSC.
- W. J. Youngblood, S.-H. A. Lee, Y. Kobayashi, E. A. Hernandez-Pagan, P. G. Hoertz, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131, 926–927 CrossRef CAS PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- Y. Gao, X. Ding, J. Liu, L. Wang, Z. Lu, L. Li and L. Sun, J. Am. Chem. Soc., 2013, 135, 4219–4222 CrossRef CAS PubMed.
- W. Song, A. Ito, R. A. Binstead, K. Hanson, H. Luo, M. K. Brennaman, J. J. Concepcion and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 11587–11594 CrossRef CAS PubMed.
- A. Magnuson, H. Berglund, P. Korall, L. Hammarström, B. Åkermark, S. Styring and L. Sun, J. Am. Chem. Soc., 1997, 119, 10720–10725 CrossRef CAS.
- O. S. Wenger, Coord. Chem. Rev., 2009, 253, 1439–1457 CrossRef CAS.
- Z. Singh, P. R. Donnarumma and M. B. Majewski, Inorg. Chem., 2020, 59, 12994–12999 CrossRef CAS PubMed.
- Z. Singh, S. Kamal and M. B. Majewski, J. Phys. Chem. C, 2022, 126, 16732–16743 CrossRef CAS.
- J.-H. Yum, I. Jung, C. Baik, J. Ko, M. K. Nazeeruddin and M. Grätzel, Energy Environ. Sci., 2009, 2, 100–102 RSC.
- J. C. Freys, J. M. Gardner, L. D'Amario, A. M. Brown and L. Hammarström, Dalton Trans., 2012, 41, 13105 RSC.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- M. Sandroni, A. Maufroy, M. Rebarz, Y. Pellegrin, E. Blart, C. Ruckebusch, O. Poizat, M. Sliwa and F. Odobel, J. Phys. Chem. C, 2014, 118, 28388–28400 CrossRef CAS.
- C. Dragonetti, M. Magni, A. Colombo, F. Melchiorre, P. Biagini and D. Roberto, ACS Appl. Energy Mater., 2018, 1, 751–756 CrossRef CAS.
- A. Rosas-Hernández, C. Steinlechner, H. Junge and M. Beller, Green Chem., 2017, 19, 2356–2360 RSC.
- O. S. Wenger, J. Am. Chem. Soc., 2018, 140, 13522–13533 CrossRef CAS PubMed.
- D. V. Scaltrito, D. W. Thompson, J. A. O'Callaghan and G. J. Meyer, Coord. Chem. Rev., 2000, 208, 243–266 CrossRef CAS.
- T. Bessho, E. C. Constable, M. Graetzel, A. Hernandez Redondo, C. E. Housecroft, W. Kylberg, Md. K. Nazeeruddin, M. Neuburger and S. Schaffner, Chem. Commun., 2008, 3717 RSC.
- J. T. Carneiro, T. J. Savenije, J. A. Moulijn and G. Mul, J. Phys. Chem. C, 2011, 115, 2211–2217 CrossRef CAS.
- Y. Park, W. Kim, D. Monllor-Satoca, T. Tachikawa, T. Majima and W. Choi, J. Phys. Chem. Lett., 2013, 4, 189–194 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- X. Feng, K. Shankar, O. K. Varghese, M. Paulose, T. J. Latempa and C. A. Grimes, Nano Lett., 2008, 8, 3781–3786 CrossRef CAS PubMed.
- P. Roy, S. Berger and P. Schmuki, Angew. Chem., Int. Ed., 2011, 50, 2904–2939 CrossRef CAS PubMed.
- K. Lee, A. Mazare and P. Schmuki, Chem. Rev., 2014, 114, 9385–9454 CrossRef CAS PubMed.
- S. R. Sitaaraman, A. N. Grace, J. Zhu and R. Sellappan, Nanoscale Adv., 2024, 6, 2407–2418 RSC.
- N. T. Nguyen, M. Altomare, J. E. Yoo, N. Taccardi and P. Schmuki, Adv. Energy Mater., 2016, 6, 1501926 CrossRef.
- Z. Sun, J. H. Kim, Y. Zhao, F. Bijarbooneh, V. Malgras, Y. Lee, Y.-M. Kang and S. X. Dou, J. Am. Chem. Soc., 2011, 133, 19314–19317 CrossRef CAS PubMed.
- N. T. Nguyen, S. Ozkan, O. Tomanec, R. Zboril and P. Schmuki, ChemistryOpen, 2018, 7, 131–135 CrossRef CAS PubMed.
- J. M. Macák, H. Tsuchiya and P. Schmuki, Angew. Chem., Int. Ed., 2005, 44, 2100–2102 CrossRef PubMed.
- F. Riboni, N. T. Nguyen, S. So and P. Schmuki, Nanoscale Horiz., 2016, 1, 445–466 RSC.
- U. Hintermair, S. W. Sheehan, A. R. Parent, D. H. Ess, D. T. Richens, P. H. Vaccaro, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2013, 135, 10837–10851 CrossRef CAS PubMed.
- C. F. Jewell, A. Subramanian, C.-Y. Nam and R. G. Finke, ACS Appl. Mater. Interfaces, 2022, 14, 25326–25336 CrossRef CAS PubMed.
- Y. Pellegrin, M. Sandroni, E. Blart, A. Planchat, M. Evain, N. C. Bera, M. Kayanuma, M. Sliwa, M. Rebarz, O. Poizat, C. Daniel and F. Odobel, Inorg. Chem., 2011, 50, 11309–11322 CrossRef CAS PubMed.
- S. O. Fürer, B. Bozic-Weber, M. Neuburger, E. C. Constable and C. E. Housecroft, RSC Adv., 2015, 5, 69430–69440 RSC.
- C. Dragonetti, M. Magni, A. Colombo, F. Fagnani, D. Roberto, F. Melchiorre, P. Biagini and S. Fantacci, Dalton Trans., 2019, 48, 9703–9711 RSC.
- M. Sandroni, M. Kayanuma, A. Planchat, N. Szuwarski, E. Blart, Y. Pellegrin, C. Daniel, M. Boujtita and F. Odobel, Dalton Trans., 2013, 42, 10818 RSC.
- M. Schmittel and A. Ganz, Chem. Commun., 1997, 999–1000 RSC.
- J. F. Hull, D. Balcells, J. D. Blakemore, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2009, 131, 8730–8731 CrossRef CAS PubMed.
- U. Hintermair, S. M. Hashmi, M. Elimelech and R. H. Crabtree, J. Am. Chem. Soc., 2012, 134, 9785–9795 CrossRef CAS PubMed.
- J. T. Kirner, J. J. Stracke, B. A. Gregg and R. G. Finke, ACS Appl. Mater. Interfaces, 2014, 6, 13367–13377 CrossRef CAS PubMed.
- B. D. Sherman, M. V. Sheridan, C. J. Dares and T. J. Meyer, Anal. Chem., 2016, 88, 7076–7082 CrossRef CAS PubMed.
- B. D. Sherman, D. L. Ashford, A. M. Lapides, M. V. Sheridan, K.-R. Wee and T. J. Meyer, J. Phys. Chem. Lett., 2015, 6, 3213–3217 CrossRef CAS.
- A. M. Lapides, B. D. Sherman, M. K. Brennaman, C. J. Dares, K. R. Skinner, J. L. Templeton and T. J. Meyer, Chem. Sci., 2015, 6, 6398–6406 RSC.
- K.-R. Wee, B. D. Sherman, M. K. Brennaman, M. V. Sheridan, A. Nayak, L. Alibabaei and T. J. Meyer, J. Mater. Chem. A, 2016, 4, 2969–2975 RSC.
- J. R. Swierk, D. D. Méndez-Hernández, N. S. McCool, P. Liddell, Y. Terazono, I. Pahk, J. J. Tomlin, N. V. Oster, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U.S.A., 2015, 112, 1681–1686 CrossRef CAS PubMed.
- C. J. Howard, T. M. Sabine and F. Dickson, Acta Crystallogr., Sect. B:Struct. Sci., 1991, 47, 462–468 CrossRef.
- L. Zhu, Q. Lu, L. Lv, Y. Wang, Y. Hu, Z. Deng, Z. Lou, Y. Hou and F. Teng, RSC Adv., 2017, 7, 20084–20092 RSC.
- H. Nohira, W. Tsai, W. Besling, E. Young, J. Petry, T. Conard, W. Vandervorst, S. De Gendt, M. Heyns, J. Maes and M. Tuominen, J. Non-Cryst. Solids, 2002, 303, 83–87 CrossRef CAS.
- P. Lazar, R. Mach and M. Otyepka, J. Phys. Chem. C, 2019, 123, 10695–10702 CrossRef CAS.
- M. Kim, D.-H. Nam, H.-Y. Park, C. Kwon, K. Eom, S. Yoo, J. Jang, H.-J. Kim, E. Cho and H. Kwon, J. Mater. Chem. A, 2015, 3, 14284–14290 RSC.
- G.-M. Rignanese, A. Pasquarello, J.-C. Charlier, X. Gonze and R. Car, Phys. Rev. Lett., 1997, 79, 5174–5177 CrossRef CAS.
- A. Shchukarev and D. Korolkov, Open Chem., 2004, 2, 347–362 CrossRef CAS.
- M. Salahvarzi, A. Setaro, K. Ludwig, P. Amsalem, T. Schultz, E. Mehdipour, M. Nemati, C. Chong, S. Reich and M. Adeli, Environ. Res., 2023, 238, 117078 CrossRef CAS PubMed.
- Y. M. Shulga, S. A. Baskakov, E. I. Knerelman, G. I. Davidova, E. R. Badamshina, N. Yu. Shulga, E. A. Skryleva, A. L. Agapov, D. N. Voylov, A. P. Sokolov and V. M. Martynenko, RSC Adv., 2014, 4, 587–592 RSC.
- Y. Zhou, L. Zhang, W. Huang, Q. Kong, X. Fan, M. Wang and J. Shi, Carbon, 2016, 99, 111–117 CrossRef CAS.
- C. Fettkenhauer, J. Weber, M. Antonietti and D. Dontsova, RSC Adv., 2014, 4, 40803–40811 RSC.
- P. Tiong, H. O. Lintang, S. Endud and L. Yuliati, RSC Adv., 2015, 5, 94029–94039 RSC.
- W. Song, C. R. K. Glasson, H. Luo, K. Hanson, M. K. Brennaman, J. J. Concepcion and T. J. Meyer, J. Phys. Chem. Lett., 2011, 2, 1808–1813 CrossRef CAS.
- Y. Zhang, J. Liu, G. Wu and W. Chen, Nanoscale, 2012, 4, 5300 RSC.
- H. Lin and L. Zhao, J. Mater. Sci.: Mater. Electron., 2019, 30, 18191–18199 CrossRef CAS.
- Z. Wei, Z.-Y. Gu, R. K. Arvapally, Y.-P. Chen, R. N. McDougald, J. F. Ivy, A. A. Yakovenko, D. Feng, M. A. Omary and H.-C. Zhou, J. Am. Chem. Soc., 2014, 136, 8269–8276 CrossRef CAS PubMed.
- Z. Liu, Z. Jian, J. Fang, X. Xu, X. Zhu and S. Wu, Int. J. Photoenergy, 2012, 2012, 1–8 Search PubMed.
- D. C. L. Vasconcelos, V. C. Costa, E. H. M. Nunes, A. C. S. Sabioni, M. Gasparon and W. L. Vasconcelos, Mater. Sci. Appl., 2011, 2, 1375–1382 CAS.
- M. H. El-Newehy, S. M. Osman, M. S. Refat, S. S. Al-Deyab and A. El-Faham, J. Macromol. Sci., Part A:Pure Appl. Chem., 2015, 52, 561–576 CrossRef CAS.
- X. Jia, J. Ma, F. Xia, Y. Xu, J. Gao and J. Xu, Nat. Commun., 2018, 9, 933 CrossRef PubMed.
- F. Herold, O. Leubner, K. Jeschonek, C. Hess, A. Drochner, W. Qi and B. J. M. Etzold, Carbon Trends, 2021, 3, 100020 CrossRef CAS.
- Q. Hao, X. Niu, C. Nie, S. Hao, W. Zou, J. Ge, D. Chen and W. Yao, Phys. Chem. Chem. Phys., 2016, 18, 31410–31418 RSC.
- S. Sunasee, K. H. Leong, K. T. Wong, G. Lee, S. Pichiah, I. Nah, B.-H. Jeon, Y. Yoon and M. Jang, Environ. Sci. Pollut. Res., 2019, 26, 1082–1093 CrossRef CAS PubMed.
- S. P. Albu, A. Ghicov, S. Aldabergenova, P. Drechsel, D. LeClere, G. E. Thompson, J. M. Macak and P. Schmuki, Adv. Mater., 2008, 20, 4135–4139 CrossRef CAS.
- N. Liu, H. Mirabolghasemi, K. Lee, S. P. Albu, A. Tighineanu, M. Altomare and P. Schmuki, Faraday Discuss., 2013, 164, 107 RSC.
- S. So, I. Hwang and P. Schmuki, Energy Environ. Sci., 2015, 8, 849–854 RSC.
- I. Hwang, S. So, M. Mokhtar, A. Alshehri, S. A. Al-Thabaiti, A. Mazare and P. Schmuki, Chem.–Eur. J., 2015, 21, 9204–9208 CrossRef CAS PubMed.
- H. Tang, K. Prasad, R. Sanjinès, P. E. Schmid and F. Lévy, J. Appl. Phys., 1994, 75, 2042–2047 CrossRef CAS.
- M. Bledowski, L. Wang, A. Ramakrishnan, A. Bétard, O. V. Khavryuchenko and R. Beranek, ChemPhysChem, 2012, 13, 3018–3024 CrossRef CAS PubMed.
- E. M. P. Steinmiller and K.-S. Choi, Proc. Natl. Acad. Sci. U.S.A., 2009, 106, 20633–20636 CrossRef CAS PubMed.
- C. F. Jewell, A. Subramanian, C.-Y. Nam and R. G. Finke, Sustainable Energy Fuels, 2021, 5, 5257–5269 RSC.
- C. F. Jewell, A. Subramanian, W.-I. Lee, C.-Y. Nam and R. G. Finke, Sustainable Energy Fuels, 2023, 7, 4567–4579 RSC.
- R. J. Kamire, K. L. Materna, W. L. Hoffeditz, B. T. Phelan, J. M. Thomsen, O. K. Farha, J. T. Hupp, G. W. Brudvig and M. R. Wasielewski, J. Phys. Chem. C, 2017, 121, 3752–3764 CrossRef CAS.
- N. Denisov, X. Zhou, G. Cha and P. Schmuki, Electrochim. Acta, 2021, 377, 137988 CrossRef CAS.
- D. Wang, R. N. Sampaio, L. Troian-Gautier, S. L. Marquard, B. H. Farnum, B. D. Sherman, M. V. Sheridan, C. J. Dares, G. J. Meyer and T. J. Meyer, J. Am. Chem. Soc., 2019, 141, 7926–7933 CrossRef CAS PubMed.
- B. D. Sherman, Y. Xie, M. V. Sheridan, D. Wang, D. W. Shaffer, T. J. Meyer and J. J. Concepcion, ACS Energy Lett., 2017, 2, 124–128 CrossRef CAS.
- M. S. Eberhart, D. Wang, R. N. Sampaio, S. L. Marquard, B. Shan, M. K. Brennaman, G. J. Meyer, C. Dares and T. J. Meyer, J. Am. Chem. Soc., 2017, 139, 16248–16255 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se01768d |
| This journal is © The Royal Society of Chemistry 2025 |