
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermogalvanic bricks: optimising large dimension thermocells for air and water valorisation†
Rebecca
Haughton-James‡
a,
Sireenya
Mesawang‡
 a,
Mark A.
Buckingham
ab,
Robert
Taylor
c,
Patrick E.
Phelan
d and
Leigh
Aldous
*ae
a,
Mark A.
Buckingham
ab,
Robert
Taylor
c,
Patrick E.
Phelan
d and
Leigh
Aldous
*ae
aDepartment of Chemistry, King's College London, London, UK. E-mail: leighaldous@ntu.edu.tw
bDepartment of Materials, The University of Manchester, Manchester, UK
cMechanical and Manufacturing Engineering, UNSW Sydney, Sydney, Australia
dSchool for Engineering of Matter, Transport and Energy, Arizona State University, Tempe, USA
eDepartment of Chemical Engineering, National Taiwan University, Taipei, Taiwan
First published on 10th December 2024
Abstract
Thermogalvanic cells can potentially valorise the huge quantity of energy available as waste heat; using entropy-driven thermoelectrochemistry they can convert a thermal gradient into electricity. Most investigations exploit a thermal source (e.g. hot water, the human body, sunlight, electronics) via a heat exchanger (metal pipe, skin, housing, etc), combined with an unlimited heat sink (e.g. pumped cold water). Limited studies have used ambient air as the heat sink. This study is believed to be the first to explore using air as both the thermal source and heat sink. It compares thermogalvanic cell performance when using water–water and air–air as the thermal energy sources and sinks, respectively, for devices with relatively large physical dimensions (25 to 100 mm wide). Gelation improved power output under both scenarios, due to enhanced thermal isolation of the electrodes; power decreased with increasing width in the water–water setup, but power increased with increasing width for air–air harvesting. Water–water yielded higher power overall, yet the air–air system operated passively and could be further optimised for real-world applications, i.e. as thermogalvanic bricks or panels in building materials.
Introduction
Almost every activity undertaken by humanity produces low-grade (<100 °C) thermal energy. Industrial processes, refrigerators, air conditioners, data centres, and buildings create a vast source of continuous—yet unexploited—waste energy.1 Thermogalvanic cells (also known as thermocells) are two-electrode electrochemical devices which exploit entropy differences between two redox states in a redox couple (ΔSrc). These devices valorise a temperature gradient (ΔT e.g. from a waste heat source and a heat sink, Thot and Tcold, respectively) into electrical energy via a generated potential difference (ΔV, or VOCP), similar to the classic solid-state Seebeck effect.2 A cartoon of the process is shown in Fig. 1a. Fundamentally, these properties are intrinsically linked: | (1) |
 | ||
| Fig. 1 Annotated cartoons of the (top) water–water thermogalvanic set-up and (bottom) air–air thermogalvanic set-up. Photographs of the setup can be found in the ESI.† | ||
The Se directly correlates with the Vocp, and the Vocp represents the driving force behind the flow of current between the two electrodes. The highest voltage, Vocp, and the highest current generated (the short circuit current density, jsc) can be modelled via Butler–Volmer kinetics, as summarised by eqn (2);4
 | (2) |
The maximum electrical power, Pmax, produced by thermocells typically obeys the relationship Pmax = 0.25Vocpjsc.2 Under typical conditions, F, C, α and R can all be taken as constants. Removing these constants from eqn (2) (discussed in more detail in the ESI† section), then we can see that;
 | (3) |
For these reasons, during investigations ‘semi-infinite’ heat sources and heat sinks are typically used to heat exchange with the two thermocell electrodes. These precisely control the applied Thot and Tcold and therefore the ΔT, by using e.g. Peltier modules5 and water circulation.6 These mirror some limited ‘real life’ applications, such as in data centre cooling.7 However, unlimited quantities of pumped ‘hot’ and ‘cold’ water is only encountered in limited areas.
There are several examples where air forms one side of the heat exchange of a thermocell, e.g. during body heat harvesting8,9 (air = cold side), hot water pipe valorisation10 (cold side), solar irradiation11,12 (hot side), battery heat harvesting13 (cold side), and nighttime radiative cooling14 (cold side). Other cooling mechanisms can be applied, such as evaporation.13 However, we are unaware of prior reports of thermogalvanic cell heat exchange performed with both sides exposed to air.
Large scale (electro)chemical devices are being considered for incorporation into building structures, such as phase change materials15 and batteries.16 Other technologies have significant promise, such as electrochemical air conditioning,17 and electrochromic materials controlling UV/vis/IR-interactions.18 Building structures typically have a ΔT across dividers, reaching as high as ΔT = 70 K in loft compartments.19
Here we set out to compare a ‘model’ heat source/sink (i.e. water bath-regulated copper heat exchangers) vs. a ‘real’ scenario using air as both heat source and sink. This was undertaken using relatively large thermocells containing liquid or gelled electrolyte comprised of 0.2 M K3[Fe(CN)6] and 0.2 M K4[Fe(CN)6]. The cell width ranged from 25 mm to 100 mm. These were chosen because ceiling panels are typically 8–25 mm thick, pre-fab wall panels 25–100 mm thick, and the standard UK metric house brick is 102.5 mm wide; all represent real-life scenarios where two air bodies are separated by bricks or panels into different temperature areas, and thus could potentially be used for ambient temperature difference valorisation in to electricity.
Experimental
Electrolyte and gelation
The electrolyte consisted of 0.2 M K3[Fe(CN)6] and 0.2 M K4[Fe(CN)6] dissolved in ultrapure water. Gelation was achieved by adding sodium poly(acrylate) powder (supplied as ‘Instant Artificial Snow Magic Powder’, Amazon, UK) at fixed weight per volume values (wt/v%). Typically, only ca. 5 minutes was required for the system to equilibrate. Because the sodium poly(acrylate) powder was slightly acidic, for safety reasons20 0.05 M potassium carbonate (K2CO3) was also added to the electrolyte. This was not found to influence the performance of the electrolyte but was added as a precaution due to the risks associated with potential HCN evolution from heated, acidified ferri/ferrocyanide solutions.20The sodium poly(acrylate) dry powder rapidly expands and gels liquid media (within seconds for pure water, and within minutes for electrolyte solutions). Fig. S1† shows photographs of solutions containing 0.2 M K3[Fe(CN)6], 0.2 M K4[Fe(CN)6] and 0.05 M K2CO3, as a function of wt/v% of added sodium poly(acrylate) powder. After 5 minutes, the solutions were inverted, to determine whether they passed the inversion test.21 For 0.5 and 1 wt/v%, the swollen gel material tended to sink to the bottom of the electrolyte, causing a different phase. For 1.5 to 2.5 wt/v%, the swollen gel filled the electrolyte (e.g. was relatively homogeneously dispersed throughout the phase, but as a heterogeneous quasi-solid phase) but the mixture could still flow, making it a slurry. For 3.0 wt/v% or above, the mixture formed a soft material that could resist flow due to gravity, making it a genuinely gelled K3/4[Fe(CN)6] phase.
Thermogalvanic bricks
Four thermocells with electrode separations of 25, 50, 75 and 100 mm were custom cut and built from extruded poly(methyl methacrylate) (PMMA, PerspexSheet.uk, UK). Example photographs are shown in Fig. S2.† They comprised 4 pieces, with dimensions 5 mm × 50 mm × y mm (where y = 25–100 mm). Three pieces were secured into a u-shape with adhesive (EVO-STIK Tensol 70 Two Component Cement, RS Components, UK), with a fourth piece placed on top as a detachable lid. This fourth piece had holes drilled into it for introducing thermistors, and if not used were sealed with Blu-Tack. This arrangement resulted in an external cross-sectional area of 55 × 55 mm, with an internal area of 45 × 45 mm. Previously characterised4 graphite electrodes (amorphous graphite – 99.5% pure graphite gasket foil, 1 mm thick, Xiaochengshop, China) were cut to shape, and secured to the two faces of the thermocell using the same adhesive. These cells were reused for multiple measurements; between measurements the graphite electrode surfaces were resurfaced by rubbing with low-lint tissues (Kimtech Kimwipes).When filling the thermocell, some irreproducible results were initially observed due to capillary forces sucking up the electrolyte between the lid and the electrode, resulting in variable electrode surface areas. For this reason, the cells were filled with electrolyte which took the depth up to ca. 85% of the maximum depth. Due to slight differences in the cells, different filling proportions were required, but the volume added each time was kept constant. This then resulted in consistent electrode surface areas being wetted by the electrolyte, which was physically measured by callipers for the specified volumes of electrolyte. The volume used and resulting geometric surface area of the electrodes are summarised in the ESI in Table S1.†
Thermoelectrochemical setup using the water–water temperature-controlled apparatus
Water heating and cooling of the thermocells was undertaken using the setup shown by the schematic in Fig. 1a (photographs are shown in Fig. S3†). The graphite electrodes were placed in thermal contact with copper-based CPU heat exchangers (Floratek CPU Water Cooling Block with 50 mm Copper Base, Amazon, UK) using thermal paste (multicomp Silicone Heat Transfer Compound, Farnell, UK). The copper blocks had their temperature controlled by water flow from two thermostatic circulator baths (a Grant Optima refrigerated TX150-R2 circulating bath, and Grant Optima TX150-ST5 Heated Circulating Bath, respectively, both from Grant Instruments (Cambridge) Ltd, UK, both operating with <0.1 °C accuracy) with an applied temperature difference, ΔT, of 20 K (T of cold electrode = 20 °C).Thermogalvanic measurement parameters for the water–water apparatus
Thermogalvanic measurements were recorded using a source measure unit (SMU, Keysight B2901A Precision Source/Measure Unit, Keysight, UK). Measurements initially followed the sequence of potentials technique described in detail elsewhere2 to generate a 5-point power curve, with later measurements using 3-point (Vocp, current at 0.5Vocp for Pmax, and jsc). Each value was generated by continuously measuring for 600 s (0.1 s per data point), then averaging the 3000 data points from 300.1 to 600.0 seconds.Thermoelectrochemical setup using the air–air temperature-controlled apparatus
The air–air apparatus was constructed around an insulated box (34L HoCo Polystyrene Box, I.D. 420 mm × 320 mm × 250 mm, wall thickness 24 mm, Amazon, UK). A schematic is included above (Fig. 1b), and photographs are included in the ESI (Fig. S2†). A square hole was cut in the centre of the largest wall; the thermocell was balanced half-in and half-out of the box. The inside of the insulated box was heated to 45 °C using a heating element (Dimplex 1 foot Tubular Heater with Built in Thermostat, Argos, UK); because the hysteresis in the built-in thermostat was too large, the power to this heater was controlled via a temperature controller (Elitech Temperature Controller Thermostat with an NTC Probe, ebay.co.uk, UK). An electric fan (Pro-Elec 6” USB Mini Desk Fan, Farnell, UK) was used within the box for heat dispersal. The fan was positioned at the back wall, facing towards the insulated box wall holding the thermocell, with the face of the fan at a distance of 23.3 cm away from the thermocell wall. The tubular heater was positioned in between the fan and the thermocell, at an average distance of 11.1 cm from the wall housing the thermocell. The temperature controller thermistor was positioned equidistant between the heater and the thermocell.The ‘cold’ side in the air–air system was the ambient air in an air-conditioned laboratory. The air conditioning was set to 20 °C. For preliminary experiments no forced convection was applied this side of the thermocell; later experiments aimed to match the convection occurring inside the box by positioning a second, identical electric fan (Pro-Elec 6” USB Mini Desk Fan, Farnell, UK) facing towards the thermocell cold electrode. The face of this fan was at a distance of 20.0 cm away from the insulated wall holding the thermocell.
Thermogalvanic measurement parameters for the air–air apparatus
Thermogalvanic measurements for the air heating set-up were performed using a potentiostat (Autolab PGSTAT101, Metrohm, UK) in two-electrode mode, and using potentiostatic and amperometric procedures (using NOVA 2.0, Metrohm, UK). The instrument was set to measure a data point every 0.1 s, to match the measurements made on the water–water setup using a SMU. Because equilibration was relatively slow, and there were some repeating fluctuations in the temperature (due to a hysteresis in the temperature control unit) and some non-repeating fluctuations (e.g. laboratory door opening), the two-point sequence of potentials technique described in detail elsewhere2 was used, where the circuit potential (VOCP) and short circuit current density (jsc) were initially recorded for roughly 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s and 40500 s respectively (total run time ca. 23 h). This allowed averaging of data over extended periods of time (ca. 400000 data point each) and ensured that genuine ‘steady state’ measurements were performed.2 The maximum power (Pmax) was given by 0.25VOCPjsc. During subsequent repeat measurements, it was determined that shorter time period measurements (ca. 6000 seconds steady state data, or 60000 data points) produced data with equivalent quality. Exemplar raw data is shown in Fig. S4.†
000 s and 40500 s respectively (total run time ca. 23 h). This allowed averaging of data over extended periods of time (ca. 400000 data point each) and ensured that genuine ‘steady state’ measurements were performed.2 The maximum power (Pmax) was given by 0.25VOCPjsc. During subsequent repeat measurements, it was determined that shorter time period measurements (ca. 6000 seconds steady state data, or 60000 data points) produced data with equivalent quality. Exemplar raw data is shown in Fig. S4.†
Temperature monitoring and temperature profile generation using thermistors
Temperature was recorded during every air–air experiment using a battery-powered data logger (GP2 Data Logger and Controller, Delta-T Devices Ltd, UK) with six thermistors (MT3 sealed catheter-style 2K thermistor probes in flexible nylon tubes, Delta-T Devices Ltd, UK). These were used to record the air temperature inside and outside of the box, the air temperature ca. 1 mm away from the hot and cold electrodes, and the electrolyte temperature ca. 1 mm away from the hot and cold electrodes. They were also used to generate the temperature profiles across the cells shown later in Fig. 2e and 4e.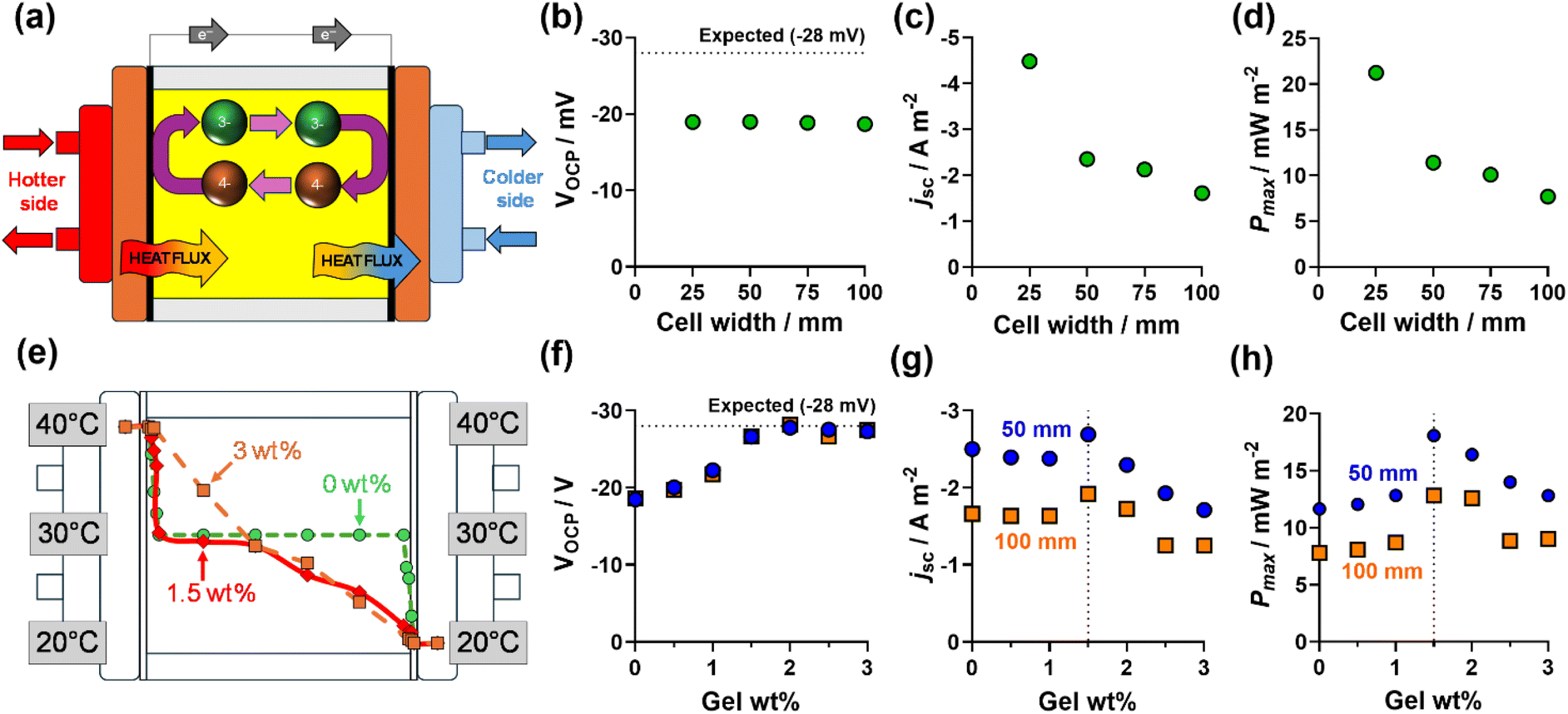 | ||
| Fig. 2 The top row shows (a) a cartoon of the water–water heat exchanged thermogalvanic cell; and the measured (b) open-circuit voltage (VOCP), (c) short-circuit current density (jSC) and (d) the maximum power density (Pmax) measured for K3/K4[Fe(CN)6] as a function of cell width when exposed to ΔT = 20 K (Tcold = 20 °C, Thot = 40 °C). The bottom row summarises the results from varying the wt/v% of polyacrylate gel added to K3/K4[Fe(CN)6], showing (e) the temperature gradient measured across the 100 mm cell for 0, 1.5, and 3 wt% (data tabulated in the ESI†), and the measured (f) VOCP, (g) jSC and (h) Pmax for 50 mm (blue circles) and 100 mm (orange squares) wide thermocells. | ||
For standard temperature monitoring, data logging was started for ca. 20 minutes before the heating was switched on, and left measuring for 1 h after the heating was switched off. The thermistors were connected to channels 1–6, with channel 6 = suspended in the centre of the thermostatic heated box; 5 = suspended in the ‘hot’ air ca. 1 mm away from the hot electrode; 4 = immersed in the electrolyte and ca. 1 mm away from the hot electrode; 3 = immersed in the electrolyte and ca. 1 mm away from the cold electrode; 2 = suspended in the ‘cold’ air and ca. 1 mm away from the hot electrode; and 1 = suspended in the air of the air conditioned laboratory, ca. 20 cm away from the heated box and thermocell. Exemplar data showing ca. 19 h recordings for two thermocells is shown in the ESI, Fig. S5.†
Results and discussion
The full experimental details of this study are included above in the Experimental section, but briefly the relatively large electrodes (ca. 30 cm2 each) were comprised of previously characterised amorphous graphite electrodes.4 The water heating/cooling apparatus has also been previously characterised22,23 and a cross-section schematic is shown in Fig. 2a. The air apparatus is novel to this study and comprised a tubular resistive heater and fan-convection, while the heat sink was the ambient air in an air-conditioned laboratory (schematic shown in Fig. 4a).First the water heat source/water heat sink system (henceforth referred to as water–water) was explored by; (i) characterising the effect of cell width; (ii) the effect of increasing gelation of the electrolyte, at various cell widths; and (iii) investigating the temperature gradient across the cell.
Fig. 2b shows the VOCP measured vs. cell width; the VOCP was largely constant, but also consistently below the expected value of ca. −28 mV (for a Seebeck coefficient of −1.4 mV K−1,4,5) corresponding to a lower ‘observed’ temperature difference (ca. 13.5 K) at the interior surface of the graphite electrodes than the applied one (20 K). The significant (ca. 33%) loss of temperature gradient is likely caused by significant parasitic convection though the liquid electrolyte in this physically large cell;24 under extreme circumstances thermal convection can exceed 1000 W m−2 K−1.25 The jSC decreased with increasing cell width (Fig. 2c) due to increasing mass transport resistance in the thermocell;4,26 this resulted in the Pmax following the same downward trend (Fig. 2d). One simple method of reducing natural convection and improving the temperature gradient in thermocells is by gelling the liquid.21,27–31
Gelation of the electrolyte was achieved by adding polyacrylate powder. The effect of polyacrylate powder on the electrolyte is shown visually in Fig. S1;† typically >0 wt% to 1.5 wt% (w/v) formed a heterogenous mixture, 1.5–2.5 wt% formed a homogenous slurry, and 3 wt% or more resulted in it passing the ‘inversion test’,21 confirming the electrolyte was fully gelled.
The VOCP was measured in the cells as a function of gel wt/v% (Fig. 2f) and increasing gelation increased VOCP (by suppressing the parasitic convection), such that ≥1.5 wt% resulted in the ΔT experienced between the two electrodes being equal to the applied ΔT. However, just as increasing cell width decreased jSC due to increased mass transport resistance, increasing gelation is known to do the same.32 Therefore, as shown in Fig. 2g, increased thermal resistance increased jSC to a maximum at 1.5 wt%, but beyond this point thermal resistance was already sufficient and increasing gel content only hindered mass transport, thus having a negative effect upon the current generated. As shown in Fig. 2h, Pmax clearly peaked at 1.5 wt% sodium poly(acrylate), and this was observed for all 4 cell widths. A similar optimum was previously observed when measuring the genuine thermogalvanic conversion efficiency (in a much smaller thermocell).33
Thermistors were next used to probe the temperature gradient across the 100 mm cell containing 0, 1.5, and 3.0 wt% polyacrylate, with the results plotted in Fig. 2e. For the liquid electrolyte, ‘hot’ and ‘cold’ electrolyte was only found immediately adjacent to the electrolyte surface, and 94% of the width of the electrolyte was homogenously 30 °C. For 3.0 wt%, the gelled electrolyte had a linear temperature gradient across the entire width of the cell. For the ‘slurry’ caused by 1.5 wt%, the ‘hot’ electrolyte was only within 2.5 mm of the electrode surface before dropping to ca. 30 °C (similar to the un-gelled electrolyte), while the colder half of the cell had a clear temperature gradient (similar to the fully gelled system), suggesting the majority of the polymeric slurry accumulated in the colder electrolyte and frustrated heat transfer in primarily just this half of the cell.
The above discussion related to thermistors submerged to half the depth. However, some temperature variation as a function of depth was observed (due to convection and radiation), so 2D IR imaging was utilised to investigate this. Fig. 3 shows photographs (top) and IR camera images (bottom) for side-on and top-down views of cells containing 0 and 3 wt% polyacrylate (see Fig. S7† for 1.5 wt% photographs). The liquid (0 wt%) electrolyte had a homogeneous temperature across the middle of the electrolyte (matching the thermistor results in Fig. 2e). Conversely, the gelled electrolyte showed a steady temperature gradient and no evidence of convection, with the side-on and top-down images having similar temperature profiles (again, matching the results in Fig. 2e).
 | ||
| Fig. 3 Photographs (top) and IR images (bottom) of (a and c) liquid electrolyte and (b and d) 3 wt% gelled electrolyte in (a and b) side-on profile and (c and d) top-down profile. For the IR images the temperature scales are colour-coded going from white (ca. 40 °C) to dark blue (ca. 20 °C). Note that the IR images only capture the surface values and not necessarily bulk values. Furthermore the angular emissivity was not calibrated for the wide variety of different surfaces, hence the values should be taken as semi-quantitative (up to ±5 °C). The IR images still accurately highlight relative temperature differences across surfaces composed of the same material. | ||
Having established the underlying trends when semi-infinite heat sources and sinks are employed, next, a novel air-based heat source and sink system (or ‘air–air’) was explored. This study followed the same trend, namely (i) the effect of cell width for liquid (0 wt/v% gel) electrolyte; (ii) the effect of polyacrylate wt/v% loading, and finally (iii) temperature gradient characterisation.
Fig. 4a displays a cartoon of the setup, whereby the hot air side was created using a heater and a fan; the cold side consisted of an air-conditioned laboratory without any forced convection (full details in Experimental and ESI†). Fig. 4b compares the measured VOCP for the air–air set-up (purple circles) with the water–water setup above (orange squares). The air–air system applied a greater ΔT of 25 K (Tambient air ≈ 20 °C, Thot air = 45 °C, due to enhanced stability) hence VOCP should be ca. −35 mV. However, due to the different thermal resistance between air and electrolyte, this could not be achieved. As cell width increased, the thermal resistance of the thermocell increased; therefore, the experienced ΔT increased, and VOCP increased (Fig. 4b). Consequently, jsc (Fig. 4c) and Pmax (Fig. 4d) also increased, but showed completely opposite trends to the same cells measured in the water–water setup. Furthermore, in the air–air setup, the experienced ΔT measured at the thermogalvanic brick electrodes (derived from the Se values) reached only ca. 20% of the applied ΔT, resulting in overall power ca. 50-fold smaller than the water–water system.
 | ||
| Fig. 4 The top row shows (a) a cartoon of the air–air heat exchange setup, and a comparison of the recorded (b) VOCP, (c) jSC and (d) Pmax for liquid K3/K4[Fe(CN)6]3−/4− in the 100 mm cell using the water–water setup from earlier (orange squares) vs. the air heating and cooling (purple circles). The bottom row shows (d) the temperature gradient measured across the 100 mm cell for the electrolyte containing 0 wt% and 2.5 wt% polyacrylate and an empty cell (quantitative data tabulated in the ESI†), and (f–h) a comparison of the outputs for the water–water vs. air–air setup as a function of wt/v% polyacrylate gel added to K3/K4[Fe(CN)6]. Please note the water–water setup used an applied ΔT = 20 K (Tcold = 20 °C, Thot = 40 °C), while the air–air used ΔT = 25 K (Tcold = 20 °C, Thot = 45 °C). In (f) and (g) the dotted lines are used to highlight peak performance areas. Also shown in (e) are the temperature gradients measured using thermistors for an empty (air-filled) 100 mm cell, and the same cell filled with electrolyte (containing 0 wt% or 2.5 wt% polyacrylate). Full data for these and polyacrylate loadings up to 4.5 wt% are tabulated in the ESI.† | ||
Since the air–air set-up was struggling to establish reasonable ΔT values, gelation of the electrolyte was once again explored. As shown in Fig. 4f, increasing gel content in the 100 mm cell increased VOCP to a plateau of ca. −28 mV (NB: ca. −35 mV was the ideal value). As seen in Fig. 4e, once again a peak in jsc is observed at the optimum compromise between frustrated heat transfer and frustrated mass transfer. However, because more gel was required before this was achieved (2.5 wt%), the system suffers from lower current by this optimum point. As seen in Fig. 4h, although Pmax has clearly significantly increased at the optimum gelation point, the highest air–air Pmax is only 40% of the highest in the water–water system, despite having a 20% higher applied ΔT value.
The measured temperature gradient across the cell is visualised in Fig. 4e; the electrolyte-filled cell (dashed green line) had a negligible temperature gradient across the width of the electrolyte, as expected due to extensive convective heat transfer, and slow heat exchange with the surroundings (especially at the colder electrode). As the gel wt% was increased, the steepness of this temperature gradient across the width of the electrolyte increased, reaching a maximum steepness at 2.5 wt% (shown as the red line); beyond 2.5 wt% the temperature gradient remained unchanged. As a comparison, the empty cell (i.e. filled with only air) was also measured. Here a temperature gradient also formed across the width of the air inside the empty cell, and interestingly the gradient was less steep than the ≥2.5 wt% gelled electrolyte, despite waiting for the systems to reach equilibrium. This demonstrates that the gelled electrolyte-containing ‘brick’ was a superior insulator than air.
Another factor that was clear from Fig. 4e was that the temperature gradient across the cold-side electrode was more significant than the hot-side; therefore, the colder electrode was dissipating the thermal energy to its surroundings slower than the heated side. This is to be somewhat expected, since the heated side employed a fan to improve thermal homogeneity throughout the heated box. To further probe this imbalance, a study was performed as a function of (i) cell width, and (ii) with and without forced air convection at the colder electrode. As shown in Fig. 5a, power increased as cell width increased (NB: the opposite trend of the water–water setup, showing the different cell design compromises required). Furthermore, air convection at the colder electrode successfully increased VOCP (to −33 mV) and ca. doubled the power (full data tabulated in the ESI). Fig. 5b highlights the contradictory trend observed in the water–water and air–air setups; power decreases with width in the water–water setup because mass transfer is the critical limiting factor, whereas in the air–air scenario, lack of thermal resistance through the electrolyte is the critical limiting factor, and is only overcome at a cell width of ca. 100 mm. In the future, other methods of thermal resistance should be explored, beyond gelation and electrolyte width between electrodes.
 | ||
| Fig. 5 Showing Pmax for various cells widths comparing (a) air–air setup with different convection at the colder electrode, and (b) comparing the air–air setup vs. water–water. All other conditions as per Fig. 2 and 4. | ||
The observations made in this study apply to thermogalvanic cells in horizontal arrangements, where the hot and cold electrodes are parallel to both each other and gravity (as might be found in a wall). Although it was not investigated herein, natural convection will play a larger role in vertical arrangements than horizontal arrangements due to temperature-induced density differences.34,35 For example, it is expected that if the cold electrode sits above the hot electrode, the gelation-based enhancements observed here should be more pronounced. Conversely, in a vertical arrangement where the hot electrode sits physically above the cold electrode, buoyancy should restrict flow with stagnant layers of higher temperature forming at the top.34 Thus, in a hot-over-cold arrangement, such gelation effects are expected to be less significant.
This study demonstrates generally higher power for the water–water setup and could seem negative towards the air–air system of thermogalvanic energy harvesting. However, the water–water system represents a far less common real-life scenario (especially the concept of having unlimited flowing ‘cold’ water), it requires heat exchangers, and it consumes energy via the water-pumping process. Conversely, the air–air system operates entirely passively, and represents a scenario found all over the world, such as the temperature difference across ceiling tiles and building walls. It is also still unoptimised, e.g. fins could be applied to the colder electrode surface to aid in heat dissipation.
The results demonstrate that for air–air ambient valorisation by thermogalvanic devices, large dimensions and gelation are essential. The application of optimised gels (e.g. directed ion transfer channels36) is expected to boost power further. Furthermore, these large-dimension insulating thermocells could be applied to air–water arrangements, such as on the outside of pipes which are passing through an environment with a dissimilar temperature to the interior. Sustaining a large temperature gradient across the thermogalvanic cell will ensure that the pipe contents are insulated, and electricity is generated via this temperature difference.
A final caveat is that more appropriate electrolytes are also required before application. Highly concentrated K3/4[Fe(CN)6] devices need to be handled with care under research conditions due to the risk of toxic HCN evolution.20 In large scale applications, fires, UV exposure, natural disasters etc. would increase the risk. This also extends to electrolytes based upon toxic and/or scarce metals (such as Co), and polyhalides (such as I−/I3−). Substitution with either abundant natural elements (e.g. Fe) and sustainable ligands,22 or sustainable organic redox couples37 may be required, but at present the performance metrics for these have not reached those of the ferri/ferrocyanide-based systems.
Conclusions
This study has demonstrated that thermogalvanic cells designed for ambient temperature valorisation, using air as both the thermal source and sink, can feasibly convert low-grade thermal energy into electricity. By comparing air–air and water–water setups, we confirmed that the water–water configuration generates higher power due to more efficient thermal transfer at the electrode-thermal source interfaces, resulting in a larger sustained temperature difference (ΔT). However, the air–air system operates passively, requiring no external pumping. Importantly, partial gelation (and not complete gelation) was found to be crucial for both configurations; partial gelation was able to minimise parasitic convective heat transfer within the electrolyte, thus improving the temperature gradient across electrodes and optimising power output, while minimising the negative mass transfer effects typical for fully gelled electrolytes.For real-world applications, building materials typically require good thermal resistance. For air–air thermogalvanic cells, increased cell dimensions and optimised gelation was also required to optimise the thermal resistance, and thus optimise valorising two temperature-differentiated air bodies into electricity. A thermogalvanic cell with the width of a house brick (100 mm) and partially gelled electrolyte (using 2.5 wt% sodium poly(acrylate)) demonstrated superior thermal resistance than air. Future studies should explore alternative or additional methods of increasing internal thermal resistance, combined with enhanced heat exploitation (e.g. solar irradiation) and especially heat dissipation (e.g. fins, radiative cooling) at the two electrodes.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualisation (RT, PEP, LA); funding acquisition (RT, PEP, LA); methodology (RHJ, SM, MAB, LA); investigation (RHJ, SM); validation (SM); supervision (MAB, LA); writing – original draft (RHJ, SM, MAB); writing – review & editing (all).Conflicts of interest
Three authors (R. T., P. E. P, L. A) hold a patent related to earlier prototypes of this air–air cell.38 There are no other conflicts of interest to declare.Acknowledgements
S. M. and L. A. acknowledge the Royal Society of Chemistry for an Undergraduate Research Bursary (U21-3197306810). M. A. B and L. A. acknowledge the EPSRC for a Standard Research Studentship (DTP; EP/N509498/1). R. T., P. E. P. and L. A. acknowledge the PLuS Alliance for a Seed Grant (PA17046).Notes and references
- R. O. Fitriani, B. D. Long, M. C. Barma, M. Riaz, M. F. M. Sabri, S. M. Said and R. Saidur, Renewable Sustainable Energy Rev., 2016, 64, 635–659 CrossRef CAS.
- M. A. Buckingham and L. Aldous, J. Electroanal. Chem., 2020, 872, 114280 CrossRef CAS.
- J. T. Hupp and M. J. Weaver, Inorg. Chem., 1984, 23, 3639–3644 CrossRef CAS.
- M. A. Buckingham, S. Hammoud, H. Li, C. J. Beale, J. T. Sengel and L. Aldous, Sustainable Energy Fuels, 2020, 4, 3388–3399 RSC.
- L. Zhang, T. Kim, N. Li, T. J. Kang, J. Chen, J. M. Pringle, M. Zhang, A. H. Kazim, S. Fang, C. Haines, D. Al-Masri, B. A. Cola, J. M. Razal, J. Di, S. Beirne, D. R. MacFarlane, A. Gonzalez-Martin, S. Mathew, Y. H. Kim, G. Wallace and R. H. Baughman, Adv. Mater., 2017, 29, 1605652 CrossRef PubMed.
- K. Laws, M. A. Buckingham and L. Aldous, Chem. Sci., 2024, 15, 6958–6964 RSC.
- A. H. Kazim, A. S. Booeshaghi, S. T. Stephens and B. A. Cola, Sustainable Energy Fuels, 2017, 1, 1381–1389 RSC.
- Y. Liu, S. Zhang, Y. Zhou, M. A. Buckingham, L. Aldous, P. C. Sherrel, G. G. Wallace, G. Ryder, S. Faisal, D. L. Officer, S. Beirne and J. Chen, Adv. Energy Mater., 2020, 10, 2002539 CrossRef CAS.
- P. Yang, K. Liu, Q. Chen, X. Mo, Y. Zhou, S. Li, G. Feng and J. Zhou, Angew. Chem., Int. Ed., 2016, 55, 12050–12053 CrossRef CAS.
- R. Hu, B. A. Cola, N. Haram, J. N. Barisci, S. Lee, S. Stoughton, G. Wallace, C. Too, M. Thomas, A. Gestos, M. E. Dela Cruz, J. P. Ferraris, A. A. Zakhidov and R. H. Baughman, Nano Lett., 2010, 10, 838–846 CrossRef CAS.
- B. Yu, J. Duan, J. Li, W. Xie, H. Jin, R. Liu, H. Wang, L. Huang, B. Hu and J. Zhou, Research, 2019, 1, 1–10 Search PubMed.
- J. Zhao, X. Wu, H. Yu, Y. Wang, P. Wu, X. Yang, D. Chu, G. Owens and H. Xu, EcoMat, 2023, 5, e12302 CrossRef CAS.
- S. Pu, Y. Liao, K. Chen, J. Fu, S. Zhang, L. Ge, G. Conta, S. Bouzarif, T. Cheng, X. Hu, K. Liu and J. Chen, Nano Lett., 2020, 20, 3791–3797 CrossRef CAS.
- M. Wang, J. Ruan and X. Zhang, Therm. Sci. Eng. Prog., 2023, 45, 102083 CrossRef.
- Z. Zhang, N. Zhang, Y. Yuan, P. E. Phelan and S. Attia, J. Energy Storage, 2023, 62, 106912 CrossRef.
- H. Wang, Y. Diao, Y. Lu, H. Yang, Q. Zhou, K. Chrulski and J. M. D'Arcy, Nat. Commun., 2020, 11, 3882 CrossRef CAS.
- H. Zhou, F. Matoba, R. Matsuno, Y. Wakayama and T. Yamada, Adv. Mater., 2023, 35, 2303341 CrossRef CAS.
- Z. Jia, Y. Sui and L. Qian, et al. , Nat. Commun., 2024, 15, 6110 CrossRef CAS.
- G. Mensah, A. Andrews, F. Davis, K. Mensah-Darkwa and P. Phelan, Therm. Sci. Eng. Prog., 2022, 34, 101448 CrossRef CAS.
- M. A. Buckingham, K. Laws, J. T. Sengel and L. Aldous, Green Chem., 2020, 22, 6062–6074 RSC.
- J. Wu, J. J. Black and L. Aldous, Electrochim. Acta, 2017, 225, 482–492 CrossRef CAS.
- M. A. Buckingham, K. Laws, E. Cross, A. J. Surman and L. Aldous, Green Chem., 2021, 23, 8901–8915 RSC.
- M. A. Buckingham, F. Marken and L. Aldous, Sustainable Energy Fuels, 2018, 2, 2717–2726 RSC.
- S. W. Hasan, S. M. Said, M. F. M. Sabri, A. S. A. Bakar, N. A. Hashim, M. M. I. M. Hasnan, J. M. Pringle and D. R. Macfarlane, Sci. Rep., 2016, 6, 29328–29339 CrossRef CAS.
- Y. Ikeda, Y. Cho and Y. Murakami, Sustainable Energy Fuels, 2021, 5, 5967–5974 RSC.
- Y. Mua and T. I. Quickenden, J. Electrochem. Soc., 1996, 143, 2558–2564 CrossRef CAS.
- H. A. H. Alzahrani, M. A. Buckingham, F. Marken and L. Aldous, Electrochem. Commun., 2019, 102, 41 CrossRef CAS.
- L. Jin, G. W. Greene, D. R. MacFarlane and J. M. Pringle, ACS Energy Lett., 2016, 1, 654–658 CrossRef CAS.
- Y. Zhou, S. Zhang, M. A. Buckingham, L. Aldous, S. Beirne, C. Wu, Y. Liu, G. Wallace and J. Chen, Chem. Eng. J., 2022, 449, 137775 CrossRef CAS.
- C. Han, X. Qian, Q. Li, B. Deng, Y. Zhu, Z. Han, W. Zhang, W. Wang, S. Feng, G. Chen and W. Liu, Science, 2020, 368, 1091–1098 CrossRef CAS.
- Z. Wu, B. Wang, J. Li, Y. Jia, S. Chen, H. Wang, L. Chen and L. Shuai, Nano Lett., 2023, 23, 10297–10304 CrossRef CAS PubMed.
- M. A. Buckingham, S. Zhang, Y. Liu, J. Chen, F. Marken and L. Aldous, ACS Appl. Energy Mater., 2021, 4, 11204–11214 CrossRef CAS.
- M. A. Trosheva, M. A. Buckingham and L. Aldous, Chem. Sci., 2022, 13, 4984–4998 RSC.
- A. Gunawan, H. Li, C. H. Lin, D. A. Buttry, V. Mujica, R. A. Taylor, R. S. Prasher and P. E. Phelan, Int. J. Heat Mass Transfer, 2014, 78, 423–434 CrossRef CAS.
- Y. Cengel and A. Ghajar, Heat and Mass Transfer: Fundamentals and Applications, McGraw-Hill, 5th edn, 2018 Search PubMed.
- S.-H. Hong, C.-C. Hsu, T.-H. Liu, T.-C. Lee, S.-H. Tung, H.-L. Chen, J. Yu and C.-L. Liu, Mater. Today Energy, 2024, 42, 101546 CrossRef CAS.
- B. Guo, Y. Hoshino, F. Gao, K. Hayashi, Y. Miura, N. Kimizuka and T. Yamada, J. Am. Chem. Soc., 2020, 142, 17318–17322 CrossRef CAS.
- P. Phelan, L. Aldous, R. Taylor and B. Obeng, WO Pat., 2020082028A1, 2020 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Exemplar raw data; temperature study; and tabulated results. See DOI: https://doi.org/10.1039/d4se01498g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |