
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-enzymatic electrochemical assay of N-acetyl-D-neuraminic acid through competitive chemoreceptor binding with (thiophen-3-yl)boronic acid†
Athira
Tomy
,
Saurav K.
Guin
,
Conor
Cassidy
and
Eithne
Dempsey
 *
*
Department of Chemistry, Kathleen Lonsdale Institute for Human Health Research, Maynooth University, Maynooth, County Kildare W23 DD4R, Ireland. E-mail: eithne.dempsey@mu.ie; Tel: +353 4747172
First published on 8th May 2025
Abstract
Electroanalytical methods which can aid in the selective quantitation of saccharides such as the sialic acid N-acetyl-D-neuraminic acid (Neu5Ac) are very attractive due to their significance in a wealth of human diseases and food/nutritional products. Using cyclic voltammetry, boronic acid–diol recognition based on a redox indicator displacement assay (RIDA) strategy was exploited for non-enzymatic comparative electroanalysis of Neu5Ac vs. fructose using the redox active reporter Alizarin Red S (ARS). The concept has its foundation in the classical competition between an analyte and an indicator (ARS) for the same binding site on a host (boronic acid) molecule. The pH dependent assay employed first-time use of (thiophen-3-yl)boronic acid (TBA) as heterocyclic chemoreceptor. Electrochemistry of ARS in equilibrium with TBA resulted in proton coupled redox processes at −0.48 V (free ARS), −0.29 V (ARS–TBA boronate ester) and +0.51 V vs. Ag|AgCl (free ARS) correlating with ARS concentration in the TBA–ARS equilibrium or in competition equilibrium with a sugar species. Saccharide driven boronic acid displacement resulted in the reinstatement of the free ARS redox processes, forming the basis for the analytical signal. Voltammetry and optical investigations established the optimum conditions for Neu5Ac measurement relative to competing species such as fructose, enabling pH tunable ratiometric quantitation over the range 1–10 mM Neu5Ac (0.1 M sodium acetate buffer pH 5.6) with sensitivity 0.119 ± 0.009 μA mM−1 and LOD 0.63 mM (using differential pulse voltammetry). The homogeneous studies paved the way for film formation and preliminary displacement testing when ARS was surface confined within a chitosan biopolymer layer on a glassy carbon electrode.
1 Introduction
Bioinspired chemically modified electrodes build upon bioelectroanalytical sensing approaches which rely on entrapped recognition elements e.g., enzymes, antibodies etc., suitably responsive for the analyte.1 The exploitation of molecular recognition principles which overcome some of the limitations of natural biomolecules include the use of synthetic materials as sorbents in molecularly imprinted polymers1 and exploitation of boronic acids as chemosensors for polyols/saccharides.Ever since the establishment of covalent and reversible formation of cyclic boronate ester linkages between boronic acids and diols, boronic acids have been employed in drug delivery, separation science2 and as detection tools for chemosensing of diol-containing biomolecules and sugars.3,2 The sp2 hybridised boron atom has a vacant p orbital4 making them a mild class of organic Lewis acids capable of coordinating basic molecules. They have pKa values ranging 4–10 and result in conversion from a trigonal sp2 hybridised (neutral) form to a tetrahedral sp3 hybridised (anionic) boronate when pH > pKa of the boron centre.5 Boronic acids, having established affinity primarily for syn-periplanar (cis) 1,2 diols6 result in reversible boronate ester formation,7 whose stability is pH and solvent dependent, enabling construction of saccharide sensors. The boronate–saccharide interaction is dependent on the pKa of both the boronic acid and cis-diol containing compound and thus the solution pH. Furthermore, boronic acids can be regarded as “green” and environmentally friendly compounds owing to their low toxicity and degradation into boric acid.4
Carbohydrates are involved in many physiological processes from the immune response to viral infections and are widely used in the food industry. The use of colorimetric sugar probes based on boronic acid substituted azobenzenes have been reported.8 The successful deployment of boronic acids as fluorescent carbohydrate sensors was recently reviewed by Fang et al.9 The use of boronic acids in colorimetric competitive binding in dye displacement assays have also been investigated, where the analyte displaces a dye from its receptor, resulting in a measurable colour/optical response which is directly related to the amount of analyte present.3 Dye displacement assays rely on a specific receptor binding event which is followed by addition of a competitive analyte with production of an optical/electrochemical signal.10 Indicator displacement assays have advantages of speed, selectivity and sensitivity for indirect measurement11 and such approaches have employed 1,2 diol containing dihydroxyanthraquinone dyes12 such as 3,4-dihydroxy-9,10-dioxo-anthracene-2-sulfonate (ARS)13,14 and dopamine.3,15 Having electron donor (phenolic) and electron acceptor (quinone) moieties,13 the catechol derivative ARS forms covalent adducts with boronic acids and this equilibrium has been examined optically as an assay for qualitative and quantitative saccharide binding.4
Much of the activity in this area has focused on glucose sensing and suitable probes tend to be generally indiscriminate in their binding. Hence, explorations into new boronic acid species and binding strategies which target alternative sugars and vitamins (e.g. ascorbic acid, riboflavin) of clinical and nutritional significance remain a current need. Vitamin B2 (riboflavin) plays a role in the conversion of carbohydrates into glucose and Fernandes et al.16 recently explored electrodeposited riboflavin–boric acid interactions using square wave voltammetry. In addition, poly(pyridine-3-boronic acid) modified glassy carbon electrode was used for simultaneous determination of ascorbic acid in the presence of co-existing species.17 Boronic acid binding to ligands with functional groups such as α-hydroxycarboxylates extends biorecognition beyond vicinal diols with pKa, buffer pH, ionic strength and steric factors playing a role.6
The glycosylation of a protein influences stability and activity with abnormal changes associated with a number of pathogenic states including cancers.18 Sialic acids make up a significant proportion of glycan structures, and N-acetyl-D-neuraminic acid (Neu5Ac) acts as a terminal sugar moiety in many glycoproteins. These sugar residues are linked with tumour growth and increased expression of sialylated glycans is a common manifestation of cancer progression and metastatic potential.19 Previously, we reviewed the emerging biomedical and nutritional importance of sialic acids18 with those distributed in free, polymeric and conjugated forms playing a role in brain development, the central nervous and immune system, lactation and infant cognition.18 Rajan et al. reported that total sialic acid levels in human plasma increased for pregnant women during the 3rd trimester and further increased in the post-partum period.20 Neu5Ac in human milk is responsible for the neurocognitive development of the human brain and alteration of the gut-microbiota in early stages of life.21 The total concentration of sialic acid in commercial infant formulas usually lies in the range 0.05–0.63 mM which is ca. 25% less than that in human breast milk. The European Commission have recently approved the inclusion of Neu5Ac·2H2O as a nutritional additive (COMMISSION IMPLEMENTING DECISION (EU) 2017/2375 of 15 December 2017).‡
Monosaccharide analysis is also significant in food chemistry in relation to quality and control of illegal addition/adulterant into fruit juices/honey. Fructose has been extensively employed as a sweetener in the food and beverages industry, being present in honey, juices and energy drinks11 and as residual sugar in wine following fermentation.22 Excessive consumption can result in obesity and disorders such as nonalcoholic fatty liver disease hyperlipidaemia, hypertension and hyperuricemia. The primary carbohydrate in human and milk based infant formulas is lactose and non-lactose carbohydrates are increasingly in use,23 driven by concerns surrounding infant lactose intolerance. Corn syrup/maltodextrin are the most common carbohydrates replacing lactose in these formulas, while in the case of follow-on formulas, the addition of sucrose and fructose may be considered acceptable.24 Early nutrition has been shown to impact the infant's immune system, brain and behavioural development and can shape immune function, gut microbiota and metabolism.25 Slupsky et al.26 have compared serum insulin and plasma metabolites in 3-month infants upon consumption of human milk, lactose or corn syrup solids-based formulas and Jones et al. have shown that lactose reduced infant formula with added corn syrup is associated with altered nutritional profiles inducing changes in glycaemic index.25
Given their significance in human and maternal health, the immune system, pathogenicity and disease6 and as a nutritional supplement, it is imperative that rapid and reliable analytical methodologies be designed with respect to selective measurement of sialic acids and co-existing saccharides (e.g. fructose, lactose). The redox silence of these analytes limits their direct electrochemical detection and typically chromatographic, enzyme-based spectrophotometry techniques have been employed for their measurement. In relation to Neu5Ac assays, recent years have seen an increase in the use of photoluminescence, voltammetry, electrochemical impedance spectroscopy, photoelectrochemistry and electrochemiluminescence approaches.18 Potentiometric analysis of sialic acid based on boronate affinity was reported by Zhou et al.27 using molecularly imprinted polymers electropolymerised with 3-aminophenylboronic acid. A non-enzymatic approach to sialic acid reported by Liu et al.15 involved dopamine coupled to a tetra(4-carboxylphenyl)porphine-graphene oxide material exploiting the surface binding competition of 2-fluorophenylboronic acid to realise electrochemical determination of sialic acid by competition assay upon monitoring the dopamine redox signal. Recently, there have been reports of heterocyclic classes of boronic acids based on the pyridine structure,19 however there have been little to no investigations of sulfur containing species such as (thiophen-3-yl)boronic acid (TBA) with the exception of a study involving isothermal titration calorimetry.28 This work reported polymer films of TBA and their binding to fructose and sorbitol28 with binding constant of ∼3400 M−1 for fructose with more alkaline conditions leading to an increase in binding enthalpy. Non-enzymatic electrochemical sialic acid measurement has been under the spotlight lately and our previous work21 reported an enzyme free electrochemical assay for Neu5Ac exploiting the ferrocene boronic acid chemoreceptor, confirming thermodynamically stable geometry of the FcBA–Neu5Ac complex relative to the corresponding complexes of glucose and α-hydroxy acids such as lactic acids. Enzyme free approaches to quantitation have advantages of lower cost, less complexity and longer-term stability for device deployment while advantages of electroanalytical assays include ease of miniaturisation, suitability for quality control environments, with design for facile operation.
In relation to surface confinement of the redox indicator ARS, this has been achieved in a polyelectrolyte layer-by-layer film of phenyl boronic acid modified poly(ethyleneimine) and carboxymethylcellulose29 with testing of glucose, L-DOPA and sorbitol.30 The polymerisation of ARS has been reported in acidic media by potential sweeping, resulting in diffusion controlled (poly)alizarin films31 while an overoxidised poly(alizarin red S) at a graphite electrode was employed for the simultaneous determination of hydroquinone and catechol.32 Additionally, adsorption of ARS on glassy carbon electrode was investigated electrochemically by Dadpou et al.13 and ARS/multiwalled carbon nanotube nanocomposites based on π–π stacking non-covalent interactions were employed for hydrogen peroxide reduction.33
Given the attractive optical and electrochemical properties of ARS and its green credentials, it was employed here for the first time in combination with (thiophen-3-yl)boronic acid (TBA) in both free and immobilised form in a displacement assay format. The boronic acid bound ARS can be displaced by fructose or Neu5Ac, reinstating the ARS redox behaviour in a manner related to the saccaride level. Our findings confirm that preferential binding interaction of Neu5Ac with TBA under acidic conditions contrasts with that of fructose at neutral pH. To our knowledge, no such electrochemical study of ARS with Neu5Ac has been reported and the work presented here is an important proof of concept with use of ratiometric signalling for indirect quantitation of the redox silent saccharide species, encouraging transfer of this single step electroanalytical assay into an immobilised sensing strategy for Neu5Ac. An ARS–biopolymer encapsulation approach resulted in films which retained redox properties and resulted in analogous solution phase behaviour (as a proof of principle study), albeit with the challenge of pH driven leaching limitations under optimal assay conditions.
2 Materials and methods
2.1 Reagents
Analytical grade chemicals (>95% purity) purchased from Sigma Aldrich included Alizarin Red S, (C14H7NaO7S) (purity >95%), chitosan, (thiophen-3-yl)boronic acid (TBA), monobasic dihydrogen phosphate, dibasic monohydrogen phosphate, sodium acetate D(−) fructose (β-D-fructofuranose), xylitol, D-mannitol, D-lactose monohydrate, L-lactic acid, sucrose, galactose. Acetic acid and D(+)glucose (α-D-glucopyranose) were from Fischer, hydrochloric acid (HCl) and sodium hydroxide (NaOH) from Fluka, potassium chloride from Merck, and N-acetyl neuraminic acid (Neu5Ac) from Fluorochem. All solutions were purged with nitrogen (N2) gas prior to measurements and all aqueous solutions were prepared in ultrapure water.2.2 Instrumentation and software
Electrochemical measurements (cyclic voltammetry and differential pulse voltammetry) were conducted using either a Metrohm i-STAT-400S or a CHI660 workstation, CH Instruments Inc. CH660C electrochemical potentiostat, 3700 Tennison Hill Drive Austin, TX 78738-5012, USA. Voltammetric studies involved a three-electrode electrochemical cell consisting of glassy carbon electrode (GCE, Ø = 3 mm, 0.0706 cm2 (geometric area)) as working electrode, an Ag|AgCl (3 M KCl) as the reference electrode, and platinum wire as the counter electrode. Ag|AgCl reference electrodes were stored in a 3 M KCl solution when not in use. All electrochemical synthesis and measurements were performed at room temperature. The pH of the supporting electrolyte was measured using a Model 8000, pH Meter (Jenway model 570). The UV/vis and reflectance probe studies employed a Lambda 365 UV/vis Spectrophotometer and reflectance probe/spectrophotometer was an Ocean Insight (brand of Ocean Optics BV) USB650 miniature spectrometer. Before each measurement GCEs were subjected to mechanical polishing in 1 μm diamond slurry (Aka-Mono) on a polishing cloth (Aka Napal 250 mm) for 5 min followed by sonication for 2 min in DI water. The electrode was polished before each experiment to ensure consistent results. Solutions were prepared freshly for each experiment to avoid any degradation and were stored at 4 °C in the dark if reused. All data were plotted using Origin2022b software.2.3 Procedures
 | ||
| Scheme 1 [A] Solution phase homogeneous displacement assay. [B] Preparation of ARS–chitosan modified electrode. | ||
3 Results and discussion
3.1 Electrochemical and optical studies of Alizarin Red S in solution phase
Fig. 1[A] presents the UV-vis spectrum of 0.15 mM ARS solution in 0.1 M phosphate buffer saline (50 mM KCl) at pH 7.4 (black trace) and 4.9 (red trace). The resulting λmax at 335 and 515 nm at pH 7.4 are due to the n → π* electronic transition at the carbonyl units and HOMO (centered on the aromatic ring linked to the phenolate and sulphonic acid groups) → LUMO (centered on two adjacent rings) electronic transition, respectively.34 At acidic pH i.e., 4.9, the phenolate group is protonated resulting in shorter electronic conjugation and thus the HOMO–LUMO electronic transition shifted to lower wavelength at λmax 426 nm. Two pKas have been reported for ARS at 5.5 and 10.85 (ref. 35) corresponding to the removal of the first and second phenol proton, respectively.7 The colour evident at pH 4.9 corresponds to alizarin molecules existing in the undissociated form (yellow, ARSH2) while at pH 7.4 deprotonation occurs resulting in a monoanion structure (red, ARSH−)36 as shown in the inset of Fig. 1[A]. Confirmation of the influence of pH on the optical properties of ARS were relevant in the context of boronic acid interactions and associated proton release which also influenced the redox activity of ARS. | ||
| Fig. 1 [A] UV-vis spectra for 0.15 mM ARS in 0.1 M phosphate buffer saline at pH 7.4 (black trace) and 4.94 (red trace). [B] Cyclic voltammogram of 0.15 mM ARS in 0.1 M phosphate buffer at pH 4.9 and 7.4 at a GCE at ν = 0.1 V s−1 with ARS redox processes indicated. | ||
Fig. 1[B] presents the cyclic voltammogram (CV) of 0.15 mM ARS solution in 0.1 M phosphate buffer saline (50 mM KCl) at pH 7.4 and 4.9 at a polished GCE with insert showing the corresponding redox processes involved, as labelled in the figure. The voltametric peaks 1′ and 1 correspond to reduction and oxidation of the hydroquinone type unit of ARS, respectively, whereas peak 3 corresponds to the irreversible oxidation of the catechol type unit of ARS resulting in 3,4,9,10-tetraoxo-3,4,9,10-tetrahydroanthracene-2-sulfonate (THS).32,37,38 Due to an inherent reactivity of the ortho-quinone structure, THS can undergo a further chemical transformation to a polymeric product randomly adhering on the electrode surface (evident from an anodic process >0.60 V, see ESI† S1[B]). This process was not evident in the first cycle of the CV (cathodic direction, initial potential −0.2 V) and in order to avoid this complication, this remained the experimental protocol throughout the cyclic voltammetry studies.
Peak 1/1′ correspond to the proton coupled electron transfer reaction of ARS to its reduced form (3,4,9,10-tetrahydroxyanthracene-2-sulfonate) (THA) followed by reoxidation. Peak 1/1′ formal potential values were E°′ = −0.515 V and −0.392 V with peak to peak separation ΔEp = 101 mV and 68 mV at pH 7.4 and 4.9, respectively, in agreement with literature values at pH 7.4.14 This indicates that less energy for the reduction process and higher energy for the oxidation process are required in the acidic medium compared to the neutral medium. On the other hand, peak 3 was found to experience a 104 mV anodic shift as pH decreased from 7.5 to 4.9 owing to the protonation of the catechol oxygen below pKa1 (5.5).
The scan rate (υ) of CV over the potential range −1.0 V to +1.0 V was varied over 0.01–2.00 V s−1 for 1 mM ARS in 0.1 M acetate buffer at pH 5.6 (ESI† S1). The use of higher (1 mM) concentration of ARS resulted in clearer redox behaviour for this study with improved solubility of ARS in acetate buffer (pH 5.6) while correlating with the conditions employed for differential pulse voltammetry experiments below. Excellent linearity was observed between log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Iapvs. logν plots with slope values of 0.48 (r2 = 0.9999), 0.49 (r2 = 0.9996) and 0.51 (r2 = 0.9975), for peaks 3, 1 and 1′ respectively, indicating the diffusion-controlled electrochemical behaviour of the process. This was also deduced from the linearity of the peak current vs. square root of scan rate (Ipvs. υ1/2) plot using peak currents from the second cycle of CVs. The Randles Sevcik equation was employed to estimate the diffusion coefficient (Dox) at 0.94 × 10−6 ± 2.41 × 10−11 and 0.19 × 10−6 ± 1.20 × 10−11 cm2 s−1 for peak 3 and peak 1 respectively. The emergence of a cathodic peak at −0.066 V in the second cycle of the voltammogram (ESI† S1[B]) confirmed the oxidation process of 3,4,9,10-tetraoxo-3,4,9,10-tetrahydroanthracene-2-sulfonate, THS, as per inset of Fig. 1[B].37 This process was also evident from a study of the influence of the anodic potential limit with emergence of the follow-on oxidation at more anodic potentials, being dependent on the upper return voltage limit (Efinal). Values >0.60 V resulted in the appearance of the cathodic process (peak 2) at Ecp = −0.06 V vs. Ag|AgCl (ESI† S2[A]).
Iapvs. logν plots with slope values of 0.48 (r2 = 0.9999), 0.49 (r2 = 0.9996) and 0.51 (r2 = 0.9975), for peaks 3, 1 and 1′ respectively, indicating the diffusion-controlled electrochemical behaviour of the process. This was also deduced from the linearity of the peak current vs. square root of scan rate (Ipvs. υ1/2) plot using peak currents from the second cycle of CVs. The Randles Sevcik equation was employed to estimate the diffusion coefficient (Dox) at 0.94 × 10−6 ± 2.41 × 10−11 and 0.19 × 10−6 ± 1.20 × 10−11 cm2 s−1 for peak 3 and peak 1 respectively. The emergence of a cathodic peak at −0.066 V in the second cycle of the voltammogram (ESI† S1[B]) confirmed the oxidation process of 3,4,9,10-tetraoxo-3,4,9,10-tetrahydroanthracene-2-sulfonate, THS, as per inset of Fig. 1[B].37 This process was also evident from a study of the influence of the anodic potential limit with emergence of the follow-on oxidation at more anodic potentials, being dependent on the upper return voltage limit (Efinal). Values >0.60 V resulted in the appearance of the cathodic process (peak 2) at Ecp = −0.06 V vs. Ag|AgCl (ESI† S2[A]).
The proton dependent nature of ARS (process peak 1/1′) over the pH range 5.2–7.4 at 100 mV s−1 was also confirmed with E°′ vs. pH resulting in a slope of −0.054 V dec−1 (ESI† S2[B–D]), being close to the theoretical value and confirming equal numbers of protons and electrons (2 electrons–2 protons) in the process. The maximum current response for peak 3 was observed in the pH range 5.25–5.75, which is the pKa1 value, after which it decreased as the solution approached neutral pH and the associated proton is dissociated from the catechol oxygen atom.
An investigation of optical and cyclic voltammetric studies of the relative binding of fructose and Neu5Ac with respect to TBA formed the next step. These target saccharides may coexist in certain paediatric nutritional products, hence their comparative TBA binding ability was of interest prior to follow on Neu5Ac quantitative studies. Hence, exploration of the electrochemical (CV) changes, correspondent to the ARS–TBA interaction and reinstatement of free ARS upon competition with a suitable saccharide analyte followed.
3.2 (Thiophen-3-yl)boronic acid initial binding study and fructose displacement assay by cyclic voltammetry
TBA is known to be hydrolysed to the anionic tetrahedral boronate species (due to its Lewis acidity) and subsequently into the cyclic boronate ester upon addition of fructose (see Fig. 2). The formation of the hydroxyboronate complexes of 1,2 diols is accompanied by a release of angle strain from rehybridisation of boron from sp2 to sp3 where the trivalent neutral form is in equilibrium with the anionic tetrahedral form. The formation of a boronate ester involves donation of an electron pair from the diol to the empty p-orbital on the boronic acid, resulting in a stable 5 or 6 membered ring and it has been shown that ligand binding increases the acidity of the boron resulting in cyclic tetrahedral anionic boronate esters.6 Given the strong tendency for fructose to bind to boronic acids in its fructofuranose form (pKa 12.1) and that the pKa of TBA and ARS are 8.25 and 5.5 (pKa1) respectively, physiological pH was therefore selected for fructose driven redox displacement of the ARS–TBA boronate ester (Scheme 2). | ||
| Fig. 2 [A] UV-vis spectra of 0.15 mM ARS (phosphate buffer pH 7.4) (black curve) followed by addition of 3 mM TBA (red curve) with λmax shifted from 516 to 459 nm confirming altered optical properties upon formation of TBA–ARS boronate ester species (visible red to yellow colour change evident). [B] Cyclic voltammograms showing 1–5 mM additions of TBA to a solution containing 0.15 mM ARS (phosphate buffer pH 7.4) t = 5 min incubation time. [C] UV-vis spectroscopy confirmation of the indicator displacement ARS homogeneous assay following TBA–ARS boronate ester formation with bathochromic effect on λmax corresponding to fructose additions (10–150 mM) which reinstate the original ARS solution colour at pH 7.4 (phosphate buffer). [D] Cyclic voltammetry data response to 100 mM fructose addition, v = 100 mV s−1Ei = 0.2 V, cathodic sweep, t = 10–60 min. [E] Corresponding ratiometric plots (n = 3) of relative ARS–TBA boronate ester (peak 4) and free ARS peak signals (peak 1 and peak 3). [F] Signal increase in peak 3 (free ARS) and decrease in peak 4 (TBA–ARS boronate ester process) over time upon addition of 100 mM fructose. | ||
 | ||
| Scheme 2 Square scheme for TBA (trigonal and tetrahedral) equilibria showing fructose binding to the TBA molecule. | ||
Initially, the binding of the catechol groups of ARS with TBA was examined optically and electrochemically. Optical studies (Fig. 2[A]) resulted in a blue shift by 58 nm due to boronate esterification and corresponding visible change in colour of the ARS solution from red to yellow upon addition of TBA. This indicates the decrease in electronic conjugation of the catecholic unit to the aromatic ring owing to its involvement in the esterification reaction. The released proton during this esterification reaction decreases the pH of the solution resulting in a slight blue shift in the UV region peak corresponding to n → π* electronic transitions of the anthraquinone unit. Upon formation of the boronate ester (see chemical structure insert Fig. 2[A]), the pKa of the ester decreases relative to the boronic acid, resulting in an increased acidity of the solution. The addition of the diol induces a decrease in the pKa of the boronic acid, due to consumption of the boronate form. In order to maintain the acid–base equilibrium, the boronic acid changes to the boronate form and accordingly, diol interaction leads to a decrease in apparent pKa.8Fig. 2[B] shows the equivalent electrochemical impact of TBA additions on the “free” ARS redox processes and confirmed the appearance of a specific redox signal reflecting the ARS–TBA boronate ester formation, labelled as peak 4 herein. The redox behaviour of TBA alone was examined over the same potential range with no evidence of inherent electroactivity under similar conditions.
The addition of fructose was found to restore the optical and electrochemical properties of the ARS due to the higher affinity of the TBA for the sugar molecule relative to ARS. To demonstrate this, Fig. 2[C and D] show the optical and electrochemical impact of additions of fructose into a solution containing 0.15 mM ARS and 3 mM TBA. In relation to the absorbance changes, a visible reinstatement of the ARS colour (yellow to red) indicative of ARS release10 was evident, with a red shift of absorbance values approaching that of the free ARS. A plot of absorbance ratio Ao/A for 458 nm and A/Ao for 520 nm respectively relative to fructose additions over the range 10–150 mM for samples taken at 4 and 24 hours confirmed the solution stability over this period (ESI† S3). Corresponding cyclic voltammetry studies of the “free” ARS oxidation peak 3 response (Fig. 2[D]) reached a maximum within 10 min while the peak 4 (ARS–TBA ester) signal decreased gradually over the incubation time period examined. These changes reflect the competition between TBA and fructose with subsequent release of ARS and lowering of the redox active peak 4 response as an indirect measure of fructose–boronic acid binding. The TBA–fructose boronate ester is electro-inactive, hence the ARS redox signal is the key response being monitored. Scheme 3 gives an overview of the various chemical equilibria involved upon addition of the saccharide molecule.
 | ||
| Scheme 3 Equilibria for fructose–TBA displacement assay showing release of free ARS upon displacement from the TBA bound form, driven by the preferential saccharide interaction. | ||
A normalisation approach was employed as an aid to make data more comparable and to compensate for any signal variations. Hence a ratiometric representation of the peak 4 signal relative to peaks 1 and 3 are shown in Fig. 2[E] with sigmoidal responses evident and enhanced signal with the use of peak 3. The so formed ARS–TBA boronate ester showed a concentration dependence up to 4 mM and the peak 4 process relates to the reoxidation of the hydroquinone moieties of the ARS–TBA boronate ester species formed as a product of the initial cathodic sweep. This finding advances the prior work of Schumacher14 who relied on “free” ARS peak 1 and peak 3 signals in their fructose binding studies using phenyl boronic acid. Incubation times from 2–45 min were examined (data not shown) and the peak current magnitude as measured by cyclic voltammetry for peak 4 (ARS–TBA ester at Eap = −0.353 V) increased to a maximum value at t = 5 min and that of peak 1 and peak 3 decreased to a steady minimum.
3.3 N-Acetyl neuraminic acid-(thiophen-3-yl)boronic acid binding and displacement assay by cyclic voltammetry
Having established competitive saccharide binding at neutral pH with the fructose analyte, optical and electrochemical studies for the preferential binding of Neu5Ac followed. This was informed by our prior work involving the electrochemical assay of Neu5Ac using a ferrocene boronic acid probe21 where acidic conditions provided optimum conditions for selective interaction. Neu5Ac has diol functionality at C7/8,9 and an α-hydroxy carboxylic acid at C1,2 as shown in Scheme 4 and insert of Fig. 3[A] with pKa of 2.6 and in physiological condition exists in an anionic pyranose form.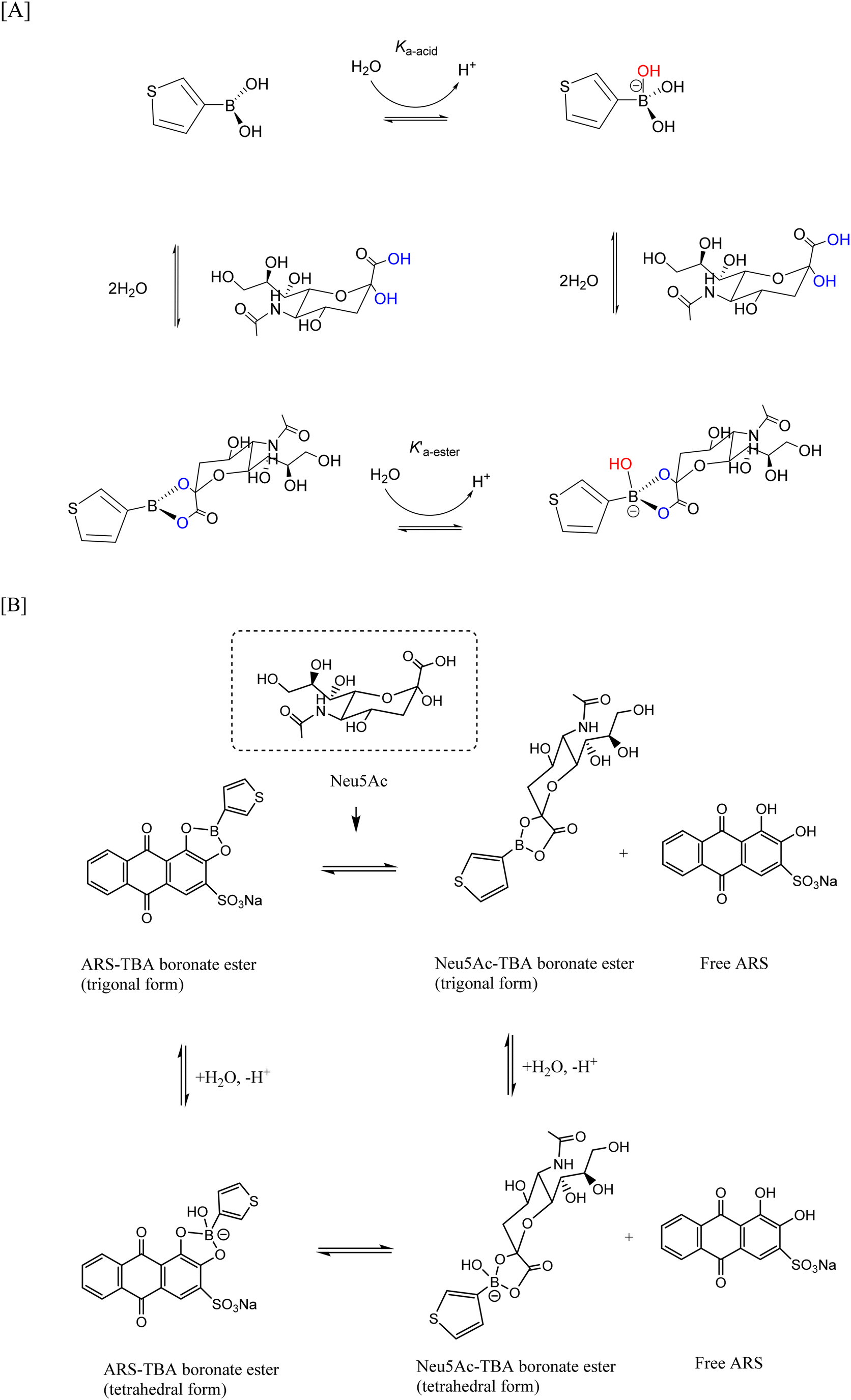 | ||
| Scheme 4 [A] Equilibria for TBA–Neu5Ac interaction and [B] displacement of the ARS–TBA boronate ester upon competition with Neu5Ac and release of free ARS. | ||
 | ||
| Fig. 3 Comparative UV-vis indicator displacement behaviour of 0.15 mM ARS and 3 mM TBA upon addition of [A] Neu5Ac (10 mM addition) relative to [B] fructose (10 mM addition) at pH 4.9 (0.1 M phosphate buffer) with t = 5 min incubation. [C] Cyclic voltammetry showing Neu5Ac (10 mM) response (incubation time t = 10 min, ν = 100 mV s−1) upon addition to a solution containing 0.15 mM ARS and 3 mM TBA pH 4.9 (0.1 M phosphate buffer) with evident current increase at peak 3 including a small anodic shift in Ep reflecting a local pH change [D] cyclic voltammetry showing Neu5Ac (10 mM) response (incubation time t = 10 min) upon addition to a solution containing 0.15 mM ARS and 3 mM TBA at pH 7.4 (0.1 M phosphate buffer). | ||
Hence, UV-vis spectra were performed at pH 4.9 comparing the optical displacement assay for Neu5Ac relative to fructose by monitoring the expected free ARS and ARS–TBA absorbances (Fig. 3[A and B]). The experiment involved addition of 10 mM of each analyte following the initial ARS–TBA incubation for 10 min. UV-vis spectra showed that the Neu5Ac species successfully displaced the TBA–ARS as evident from the hypsochromic effect with shift of λmax from 441 nm back to the original free ARS having λmax at 426 nm (Fig. 3[A]). Addition of fructose meanwhile was not competitive at this pH having little impact on the λmax of the TBA–ARS species, reflecting the relative stability of the ARS–TBA boronate species (Fig. 3[B]).
This interaction was also confirmed electrochemically under identical conditions (at pH 4.9) via cyclic voltammetry (Fig. 3[C]) where the more anodic ARS oxidation process (peak 3) was reinstated. This was accompanied by an initial anodic shift in Eap together with a fluctuation in peak height response with increased incubation time (following a 10 mM Neu5Ac addition) with CVs recorded every 10 minutes (maximum changes occurred at incubation time longer than 10 min). The same experiment carried out under neutral conditions resulted in no Neu5Ac induced peak 3 or 4 alterations Fig. 3[D], reflecting the absence of Neu5Ac binding under neutral conditions and the promising use of pH control for monosaccharide–boronic acid selectivity studies.
The first equilibrium between the boronic acid and redox active reporter molecule ARS can be measured electrochemically and the addition of Neu5Ac sets up a second equilibrium resulting in a Neu5Ac–TBA complex which realises a change in the redox properties of the ARS, reinstating the free form. The extent to which the Neu5Ac addition perturbs the redox behaviour depends on the affinity of the TBA chemoreceptor and the Neu5Ac (see Scheme 4[A]) and is expected to involve the α-hydroxy carboxylic acid at C1,2 under these conditions. Preference for acidic conditions in the case of Neu5Ac relative to fructose is consistent with our previous reports on ferrocene boronic acid interactions21 and previous work on phenylboronic acid and 4-isoquinoline boronic acid.6 The increased Neu5Ac binding at acidic pH values being lower than the pKa of TBA (8.25 ± 0.1) implies that a fraction of the boronic acid (in contrast to the boronate) is involved, which is consistent with previous sialic acid binding profiles for nitrogen containing heterocyclic molecules.19
In order to ensure sufficient binding time, an incubation time of 30 min was employed for the next cyclic voltammetry study which involved additions of Neu5Ac (10–40 mM) to a solution containing 0.15 mM ARS and 3 mM TBA at pH 4.9 with typical traces shown in Fig. 4[A]. The evident anodic Eap shift together with the increase in current for peak 3 (free ARS process) enabled comparative Iap responses for the key processes as per Fig. 4[B and C] with ratiometric plot (peak 1 and 4 relative signal) shown in [D].
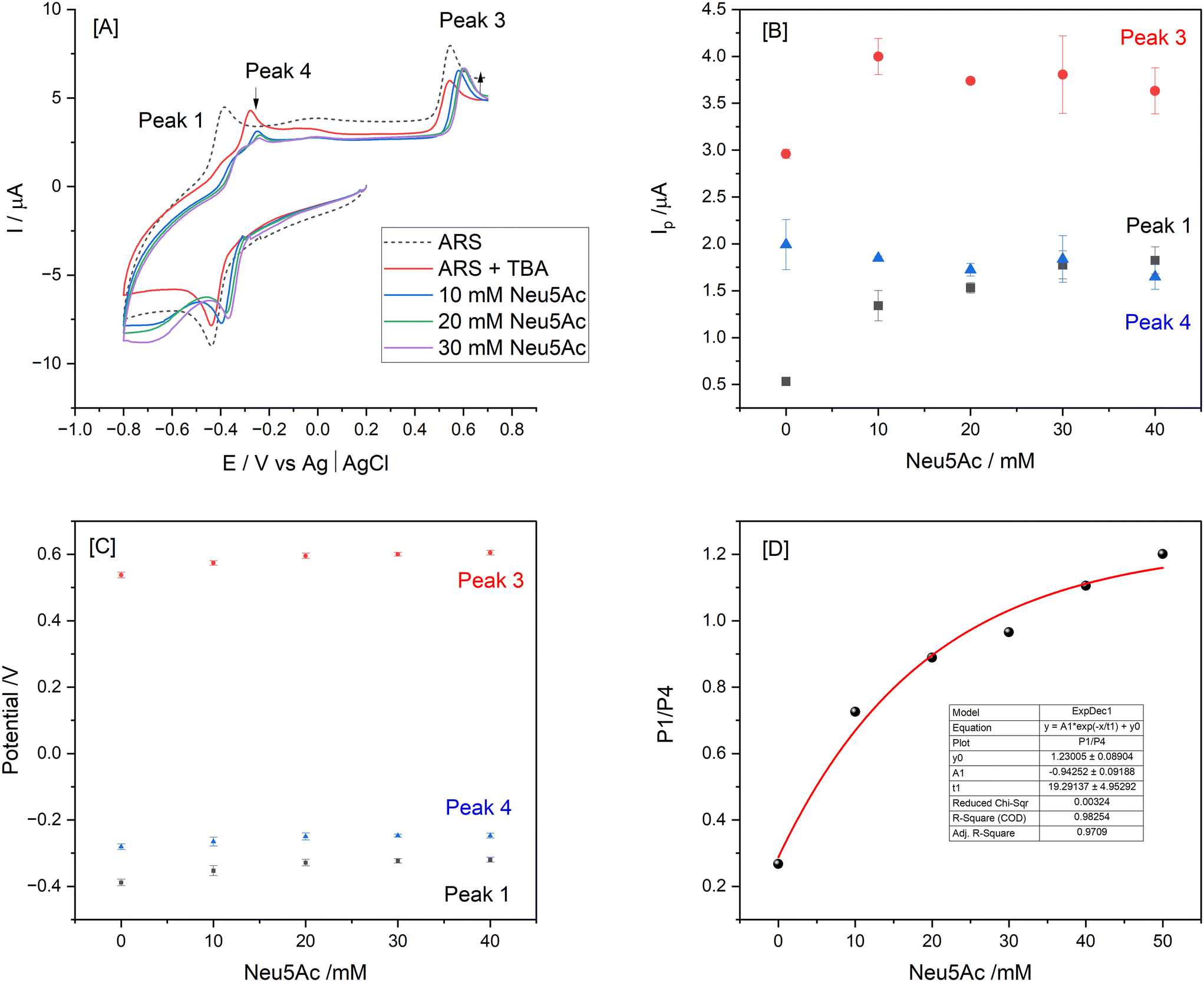 | ||
| Fig. 4 [A] Cyclic voltammograms for 0.15 mM ARS alone and following addition of 3 mM TBA followed by 10–40 mM Neu5Ac additions (pH 4.9, 0.1 M phosphate buffer). Effect of Neu5Ac additions on change in [B] peak current and [C] Ep values over the range 10–40 mM Neu5Ac with incubation time t = 30 min [D] corresponding ratiometric plot of P1/P4. | ||
In order to improve peak resolution and to enable lower Neu5Ac concentration ranges to be examined, differential pulse voltammetry (DPV) was employed for further quantitative studies. To approach more physiological conditions and for more accurate pH control, the buffer system for the next part of the work employed 0.1 M acetate buffer at pH 5.6. The use of the acetate system was also informed by the potential complication of the acidic phosphate moiety to co-ordinate with Neu5Ac, forming ternary complexes with the boronate ester.6,39 In addition, the solubility of ARS improved under these conditions and a comparison of 0.1 mM and 1 mM responses for Neu5Ac justified the use of the higher ARS concentration (see ESI† S4). In the case of 1.0 mM ARS there was a 30%-fold increase in the peak 1 free ARS, reinstating the free form and the noise level was improved with a clearer ARS–TBA ester peak which also responded to Neu5Ac, encouraging use of these conditions for subsequent studies. The use of DPV also resulted in the emergence of a new process at −0.815 V which decreased following Neu5Ac addition (10 mM). This was interesting and suggestive of a secondary ARS boronate complex which was easier to oxidise relative to the main peak 4 signal and warranted further investigations as an analytical signal (labelled peak 5 herein).
3.4 N-Acetyl neuraminic acid-(thiophen-3-yl)boronic acid binding and displacement assay by differential pulse voltammetry
An ARS–TBA binding study followed, as monitored using DPV (Fig. 5[A and B]) with increasing additions of TBA up to >6 fold excess of ARS. Considering ARS as the host molecule that forms a complex with the guest boronic acid TBA, the binding constant βs can be described as per the expression below.40,41The following relationship has been employed by Gu et al. and others40,41 for electrochemical determination of apparent binding constants where in this instance the anodic oxidation of ARS acts like a reporter and the current signal decrease is proportional to TBA concentration, being attributed to the binding of ARS to TBA as described below,

where m (slope) is the stoichiometry and βs represents the apparent binding constant, ΔI is the difference in peak currents in the presence and absence of TBA, ΔImax represents the maximum change in peak 3 (free ARS) current when the concentration of TBA in solution is much higher than that of ARS.42 The formation of an adduct between ARS and TBA can be examined using this approach43 and a plot of

 as a function of the log of concentration of TBA was nonlinear (Fig. 5[D]) indicative of multiple complex formations.42 The curve was divided into two parts with linear portions corresponding to m1 = 1.5 (0.2–2.6 mM TBA) and m2 = 3.6 (3.0–6.3 mM TBA), with values of βs = 2.5 × 104 and 3.9 × 109 M−1 respectively. Fig. 5[C] shows the decay of the peak 1 ARS oxidation with insert giving the mole ratio plot for both components. A crossover at approx. 1 mM corresponded to the emergence of the peak 5 process (>1 mM) TBA and the levelling off of the response for the initial boronate ester formed at concentrations of TBA < 0.8 mM, i.e. the TBA level at which the new boronate ester peak arises (peak 5 (Eap) = −0.808 V) and peak 4 ceases to increase.
as a function of the log of concentration of TBA was nonlinear (Fig. 5[D]) indicative of multiple complex formations.42 The curve was divided into two parts with linear portions corresponding to m1 = 1.5 (0.2–2.6 mM TBA) and m2 = 3.6 (3.0–6.3 mM TBA), with values of βs = 2.5 × 104 and 3.9 × 109 M−1 respectively. Fig. 5[C] shows the decay of the peak 1 ARS oxidation with insert giving the mole ratio plot for both components. A crossover at approx. 1 mM corresponded to the emergence of the peak 5 process (>1 mM) TBA and the levelling off of the response for the initial boronate ester formed at concentrations of TBA < 0.8 mM, i.e. the TBA level at which the new boronate ester peak arises (peak 5 (Eap) = −0.808 V) and peak 4 ceases to increase.
 | ||
| Fig. 5 [A] and [B] ARS–TBA binding study using 1 mM ARS with additions of 0.2–8 mM TBA (shown up to 6.3 mM) until saturation in signal. [C] Free ARS current decrease upon TBA additions over range 0.2–8 mM and [D] log(ΔI/ΔImax − ΔI) vs. log TBA plot with two linear regions at low and higher TBA ranges respectively. | ||
The emergence of this cathodic process (peak 5) reflects a new type of redox active molecule independent of the free ARS. Solution acidity upon proton release via ARS–TBA ester formation at TBA concentration higher than 0.8 mM may play a role, where promotion of a more than 1:1 interaction with the reduced form of the ARS (THA – see Fig. 1[B] insert) is possible. This might be electrochemically driven upon application of the initial −1.0 V potential, followed by its oxidation to the ARS–TBA ester (ox) form. A detailed separate study will be valuable in order to establish more insight into this species.
The  plot was also generated using peak 4 data resulting in slope (m) of 3.4 (in 1.0–4.5 mM region) and a slope (m) of 3.9 was obtained in the case of peak 5, reflecting the higher stoichiometry of this species and aligning with the stoichiometry above for m2.
plot was also generated using peak 4 data resulting in slope (m) of 3.4 (in 1.0–4.5 mM region) and a slope (m) of 3.9 was obtained in the case of peak 5, reflecting the higher stoichiometry of this species and aligning with the stoichiometry above for m2.
The equilibrium constant of the chemical reaction of TBA with ARS in the oxidised and reduced state can be represented by the following expression, as employed in the case of the square scheme for ferrocene boronic acids.21,44

When the Neu5Ac interaction at lower concentrations of TBA (0.8 mM) was examined there was no evidence of this new process peak 5, confirming the above (see ESI† S5). Relative free and bound ARS responses were enhanced at higher boronic acid levels, with the aid of ratiometric responses estimated using the peak 5 process, thus justifying the continued use of 3 mM TBA in the assay conditions.
Fig. 6[A] shows a typical DPV trace for the homogeneous assay with impact of incubation time assessed. There is confirmation that Neu5Ac displacement of the ARS–TBA complex occurred within 5 min based on relative changes in peak currents for the various redox processes (Fig. 6[B] for n = 3). The expected reinstatement of free ARS peaks (i.e., peak 1 and peak 3) took place in addition to the emergence of peak 4, representing the ARS–TBA complex with the more cathodic redox process emerging as identified above (peak 5). The relative peak responses prior to and following 10 mM Neu5Ac addition are shown in Fig. 6[C] with the ratiometric peak 3/5 signal dominating. Anodic shifts in Ep for peak 3 (from 0.504 V to 0.515 V), peak 1 (from −0.596 V to −0.491 V) and peak 5 (from −0.819 V to −0.773 V) occurred within 5 min of the Neu5Ac addition to the solution, reflecting increased acidity upon boronate ester formation (due to the expected change in pKa of this species) following which there was no further change in values.
 | ||
| Fig. 6 Neu5Ac incubation time study (5–30 min) using differential pulse voltammetry upon addition of 10 mM Neu5Ac in 0.1 M acetate buffer (pH 5.6). Showing [A] DPV trace and [B] peak current trend vs. time for all relevant signals (n = 3) and [C] bar chart showing relative peak ratios in the presence and absence of 10 mM Neu5Ac for 5 min incubation time. | ||
Neu5Ac calibration studies (Fig. 7) show a typical DPV anodic scan from −1.0 V to 0.8 V which resulted in the expected processes as labelled (Fig. 7[A]) with corresponding plots for absolute current [B] and ratiometric responses [C]. The peak 3 ratio plot results in regression equation I (μA) = 0.119 ± 0.009 Neu5Ac (mM) + 6.31 ± 0.05 and peak ratio 1/5 gives slope of 0.114 ± 0.005 with r2 = 0.9793. The limit of detection (LOD) was estimated as 1.5 mM (based on peak 3 response) and 0.63 mM based on peak ratio 3/5 data.
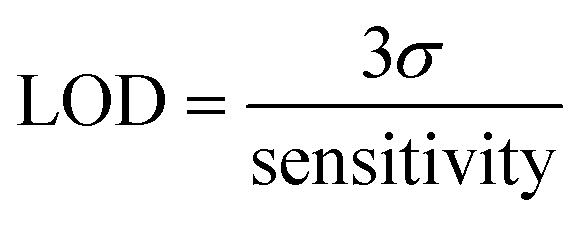
 | ||
| Fig. 7 [A] DPV showing calibration of Neu5Ac from 0.5–10 mM in the presence of 1 mM ARS with 3 mM TBA incubated for 10 min at pH 5.6. [B] Corresponding calibration showing current response vs. concentration of Neu5Ac and peak ratios. [C] Impact on peak potential Ep with increasing Neu5Ac additions. | ||
3.5 pH and selectivity studies by differential pulse voltammetry
To evaluate the specificity of co-existing species which can also displace the ARS–TBA adduct, a number of mono and disaccharides (galactose, lactose, D-mannitol, sucrose, xylitol and glucose) together with L-lactic acid were examined at 5 mM level and their response relative to that of Neu5Ac presented (ESI† S6). The closest competitors based on the peak 3 response were L-lactic acid followed by fructose and D-mannitol with the disaccharide lactose resulting in the least interference. Overall, the TBA interaction was Neu5Ac > L-lactic acid > fructose ∼ D-mannitol > xylitol > glucose > sucrose > lactose. Boronate ester formation with α-hydroxycarboxylates can depend on relative acidities with L-lactic acid (pKa 3.96) < Neu5Ac (pKa 2.60) and dynamic displacement of L-lactic acid using a polymer indicator displacement assay has been reported,45 being enhanced under acidic conditions where the reactive trigonal boron form exists.Given the interest in and future potential for this methodology in quantitative analysis or quality control of infant nutrition/food samples, further studies as presented below (Fig. 8) focused on equimolar additions of each of fructose and L-lactic acid as competing species together with Neu5Ac. Comparative responses were estimated for each redox process using the formula 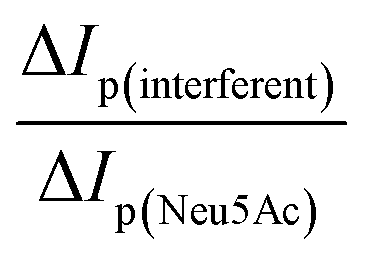 (n = 3) with corresponding data summarised in Table 1.
(n = 3) with corresponding data summarised in Table 1.
 | ||
| Fig. 8 Effect of pH of 0.1 M acetate buffer solution on the change in Neu5Ac signal for all redox peaks relative to signal following addition of 3 mM TBA to a 1 mM ARS solution. Effect of pH on relative interferent response (all peaks) [A], pH 5.4 [B], 5.6 [C], and 5.8 [D] upon addition of 10 mM Neu5Ac for 5 min incubation time followed by addition of 10 mM L-lactic acid or fructose for 5 min incubation time. Neu5Ac(1) and (2) data relates to individual experimental data prior to addition of either L-lactic acid (LA) or fructose (FRU). | ||
 values presented with average of n = 3 for each redox process
values presented with average of n = 3 for each redox process
| L-Lactic acid | ||||
|---|---|---|---|---|
| pH | Peak 5 | Peak 1 | Peak 4 | Peak 3 |
| 5.4 | 1.24 | 1.34 | — | 1.00 |
| 5.6 | 1.29 | 1.22 | 0.38 | 0.89 |
| 5.8 | 1.10 | 1.07 | 0.38 | 0.86 |
| Fructose | ||||
|---|---|---|---|---|
| pH | Peak 5 | Peak 1 | Peak 4 | Peak 3 |
| 5.4 | 1.15 | 0.59 | 0.21 | 0.83 |
| 5.6 | 1.16 | 0.81 | 0.55 | 0.85 |
| 5.8 | 1.05 | 0.84 | 0.34 | 0.79 |
The relative responses for Neu5Ac over this pH range is also depicted in Fig. 8[A] and while the slightly higher pH (i.e., 5.8) results in greater signal changes for Neu5Ac alone, overall, pH 5.4 was deemed to be slightly more favourable in relation to the relative L-lactic acid and fructose responses as shown at pH 5.4, 5.6 and 5.8 [B–D] respectively.
In order to advance this homogeneous assay investigation and to examine the potential for binding to surface confirmed ARS, ARS–Chit films were prepared as described in section 2.3.8 followed by optical and electrochemical film studies.
3.6 Optical and electrochemical studies of Alizarin Red S–chitosan biopolymer film
Electrostatic interactions between amino-containing polycations such as chitosan and ARS were previously exploited by Dadpou et al.13 The composite film showed good stability in acidic media, reflecting favourable electrostatic interaction between the positively charged chitosan (pKa 6.3) and partially deprotonated ARS species. This approach has not been explored previously and provides new exploratory receptor surfaces for more robust sensing formats. As mentioned previously, ARS turns from pale yellow to yellow-orange to red as pH changes from 2.0 to 4.9 to 6.2, respectively and has two ionisable functional groups. Absorbance at 261 nm can be assigned to the π → π* electronic transition of the quinonic structure, peaks at 326 nm and 520 nm can be assigned to the n → σ* electronic transition of the protonated and deprotonated forms of the hydroxy groups respectively.46 Dadpou et al.13 have reported ARS layer by layer assembly with branched poly(ethyleneimine) BPEI and reported reflectance probe measurements resulting in absorbance bands at 252 nm, 288 nm and 325 nm reflecting π → π* electronic transition of benzenoid, quinonoid and benzenoid units, respectively. Alizarin species are considered as n-type photosensitive organic semiconductors47 and the optical properties of ARS thin films were examined by Awed et al., having behaviour characteristic of direct transition with optical band energy Eg = 2.93–2.98 eV.Here, optical studies employed a reflectance probe with ARS–Chit film modified GCE immersed in buffers of pH 4.4 and 7.4 (Fig. 9[A and B]) with parallel solution studies (Fig. 9[C]). The absorption edge is indicative of the optical transition from the valence to conduction band in the material and was found to be 471 nm (pH 4.4), shifting to lower energy (625 nm) at pH 7.4 (Fig. 9[A and B]). The direct optical band gap value (Eg) of the film was estimated at 2.15 eV using the optical absorption method48 (Fig. 9[D]) from the plot of (αhν)2 as a function of photon energy with extrapolation to (αhν)2 = 0.
| (αhν)1/n = A(hυ − Eg) |
 | ||
| Fig. 9 ARS–Chit film pH [A] 4.4 and [B] 7.4 reflectance probe investigations, [C] 0.1 mM ARS pH 4.4 solution phase absorbance and reflectance probe signals. [D] Plot of (αhν)2vs. photon energy for absorbance data. Optical band gap determined by extrapolating the linear portion of the plot to (αhν)2 = 0 with hv = 2.15 eV. | ||
Surface immobilisation provides advantages of convenience and enhanced performance due to interactions at the electrode/solution interface.2 The electrochemical evaluation of the so formed films followed (Fig. 10), resulting in symmetric shapes of the voltammograms. The central quinone functionality of the adsorbed ARS underwent a quasi-reversible process with E°′ = −0.402 V for the peak 1/1′ process with ΔEp = 120 mV. This confirmed the surface confirmed redox system with the aid of the chitosan biopolymer and proton coupled electron transfer facilitated balance of charge in the ARS–Chit adsorbed film. This correlated with previous findings using layer by layered approaches13 (see inset of Fig. 10[A] for the proposed structure with electrostatic interactions).
 | ||
| Fig. 10 [A] Voltammogram of ARS–Chit film following stabilisation in pH 4.9 buffer with insert showing proposed chemical structure. [B] Scan rate study over the range 0.01–1.0 V s−1. [C] Corresponding Ipvs. ν and ν1/2 [D] plots and logIpvs. logv [E] and Epvs. logv [F] for peaks 3, 1, 1′. | ||
Voltammograms at different scan rates (0.01 to 1 V s−1) were recorded in 0.1 M acetate buffer (pH 4.9) for consistency with respect to the earlier optical studies. Fig. 10[B] shows typical CVs where anodic and cathodic peak current increased in proportion to  indicating diffusion controlled redox behaviour of the thick films where anion and cation diffusion into and out of the film upon oxidation and reduction are involved. The diffusion-controlled nature was deduced from the linearity of the Ipvs. v or v1/2 plots using peak currents from the final cycle (Fig. 10[C and D]). This was confirmed by the logarithm of oxidation peak currents and logarithm of scan rate plot (Fig. 10[E]) with correlation coefficient of 0.9972 and average slope of 0.59 ± 0.038. The surface coverage (Γ) was estimated from the slope of the linear Ipvs. ν plots resulting in 2.544 × 10−10 and 2.37 × 10−10 mol cm−2 based on peak 3 and peak 1′ data, respectively. With increasing scan rates, the oxidation peaks (peak 1 and peak 3) shifted anodically and the reduction (peak 1′) shifted cathodically. Intercepts γa and γc gave the transfer coefficient α (see expression below) and the number of electrons (n) involved was estimated from the slope of the Eapvs. logν graph (Fig. 10[F]).32 These values were found to be α = 0.5 and n = 2 (1.9 and 1.86) based on slope values of 6.08 × 10−2 and 6.03 × 10−2 (γa and γc), respectively.
indicating diffusion controlled redox behaviour of the thick films where anion and cation diffusion into and out of the film upon oxidation and reduction are involved. The diffusion-controlled nature was deduced from the linearity of the Ipvs. v or v1/2 plots using peak currents from the final cycle (Fig. 10[C and D]). This was confirmed by the logarithm of oxidation peak currents and logarithm of scan rate plot (Fig. 10[E]) with correlation coefficient of 0.9972 and average slope of 0.59 ± 0.038. The surface coverage (Γ) was estimated from the slope of the linear Ipvs. ν plots resulting in 2.544 × 10−10 and 2.37 × 10−10 mol cm−2 based on peak 3 and peak 1′ data, respectively. With increasing scan rates, the oxidation peaks (peak 1 and peak 3) shifted anodically and the reduction (peak 1′) shifted cathodically. Intercepts γa and γc gave the transfer coefficient α (see expression below) and the number of electrons (n) involved was estimated from the slope of the Eapvs. logν graph (Fig. 10[F]).32 These values were found to be α = 0.5 and n = 2 (1.9 and 1.86) based on slope values of 6.08 × 10−2 and 6.03 × 10−2 (γa and γc), respectively.

Finally, the average area σ occupied by a molecule of ARS in the adsorbed layer can be estimated by the following expression,

where NA is Avogadro's constant, and Γo is the amount of adsorbed ARS (mol cm−2) resulting in a value of 6.53 × 10−15 cm2, an order of magnitude higher than the literature value of 4.54 × 10−16 cm2 for adsorbed ARS.13

As the films were assembled mainly via electrostatic interactions, they were sensitive to pH and became unstable, which is also encountered in the literature.29 Film stability was examined with ARS film control studies in the absence of TBA providing evidence of a 3.3% decrease in peak 3 current over a period of 20 min (selected to match the binding time employed). A similar study was performed upon addition of 10 mM Neu5Ac with incubation of 20 min resulting in a 6% increase in peak 3, possibly due to some film swelling/buffer interaction. Despite these effects which partially cancel each other out, Neu5Ac and fructose ARS film test interactions involved addition of 10 mM of the saccharide and performing cyclic voltammetry at t = 2–50 min intervals to monitor redox signals associated with the Neu5Ac driven competition between ARS–TBA boronate ester and Neu5Ac–TBA, reinstating the ARS film behaviour. CVs which capture the signal changes for the various redox peaks are shown in ESI† S7 with graphical data in Fig. 11. These data follow incubation of the ARS–Chit films in 3 mM TBA where the expected peak 4 emerged and then lowered upon displacement with the saccharide (analogous to the homogeneous studies). Despite the fructose (as comparative saccharide) response with respect to reinstatement of peak 3, there was a more significant signal increase in the case of Neu5Ac with a more evident Ep anodic shift (Fig. 11[A vs. B]), following 20 min incubation time. Overall, the requirement for operation at a pH favorable for Neu5Ac interaction may require a compromise in relation to maintenance of film retention via electrostatic interactions.
 | ||
| Fig. 11 Plot of peak 3 current and potential for 10 mM [A] Neu5Ac and [B] fructose at ARS–Chit film immobilised on a GCE, pH 4.9 buffer with incubation time from 0–50 min. | ||
This preliminary study of Neu5Ac interaction at ARS–chitosan films on GCE represents a new approach with promising insights into the potential for further optimisation for Neu5Ac vs. co-existing saccharide signals. Design of next generation boronic acid-based sensors requires knowledge of intrinsic binding affinities between boronic acid and diol moieties and this ARS system could be used to rapidly compare the affinities of many boronic acid bearing compounds.
This aspect of the work was informed by the extensive homogeneous non-enzymatic Neu5Ac studies which involved optical, cyclic voltammetric and differential pulse voltammetric binding and quantitative studies, which progressed knowledge beyond the current state of the art, exploiting pH control and realising an understanding of the ARS–TBA equilibria at play in competition with saccharide driven displacement studies.
4 Conclusions
The significant biological relevance of sialic acids in human health and nutrition has accelerated interest in design of sensing methodologies as alternatives to existing analytical assays. As knowledge of the biological role of these important group of monosaccharides grows, extended quantitation approaches are paramount to underpin further explorations and understanding of their significance in human health and disease. The regulatory approval of Neu5Ac and 3′- and 6′-sialyllactose sialic acids as nutrition additives in milk-based products together with the increased use of lactose free carbohydrate substitutes require analytical methodologies that can discriminate Neu5Ac relative to fructose and other added saccharides.The relatively unexplored boronic acid chemoreceptor (thiophen-3-yl)boronic acid (TBA) formed the focus of this work for non-electroactive saccharide binding studies. This diol–boronic acid equilibrium was exploited as a first time competitive colorimetric and redox indicator displacement assay for binding investigations and quantitative determination of saccharides containing α hydroxy carboxylates (Neu5Ac vs.L-lactic acid) vs. fructose with binding under pH control. Comparative UV-vis and cyclic voltammetry was employed for fructose vs. Neu5Ac homogeneous investigations with respect to the influence of pH, incubation time, concentration of ARS and TBA. This enabled DPV monitoring of the emerging redox active boronate ester (ARS–TBA) signal(s) being suggestive of higher binding constants >3.0 mM TBA as reflected by estimates of 2.5 × 104 and 3.9 × 109 M−1 corresponding to stoichiometries of 1.5 (for 0.2–2.6 mM TBA) and 3.6 (for 3.0–6.3 mM TBA). Subsequent addition of the model sialic acid (Neu5Ac) and its impact on current magnitude and shift in redox potentials of the key processes was examined with reinstatement of the free ARS anodic signal. Ratiometric responses to Neu5Ac concentrations using DPV realised quantitation over the range 1–10 mM (pH 5.6) with sensitivity 0.119 ± 0.009 mA mM−1 and LOD 0.63 mM.
Neu5Ac(α2,6) lactose (i.e., 6′-sialyllactose) and Neu5Ac(α2,3) lactose (i.e., 3′-sialyllactose) acidic milk oligosaccharides exist in the range 0.3–0.5 and 0.1–0.3 g L−1, respectively in human milk18 whereas only 6′-sialyllactose is found in bovine milk in lower concentration (0.03–0.06 g L−1) compared to human milk. The free Neu5Ac LOD achieved in this work lies in the mid human milk range and is an order of magnitude lower than that required for quantitation in the case of bovine milk. However, sialic acid supplemented infant nutritional products may include 0.4 g L−1 (6.3 × 10−4 M) of 6′-sialyllactose in reconstituted products which is in line with the analytical performance achieved in this work.
This enzyme free homogeneous electrochemical investigation paved the way to evaluate surface entrapped electrochemically active Neu5Ac selective probes (ARS confined within a chitosan biopolymer layer) with the aid of a first time electrochemical and reflectance probe spectroscopy investigation. The proof of principle findings for this part of the study provides an excellent platform for follow on testing.
Overall, this electroanalytical methodology holds promise for extension to real sample matrices and this forms the next phase of the work. Dairy and food matrices have inherent operational challenges and require sample preparation e.g. acetic acid hydrolysis for Neu5Ac release from 3 or 6-sialyllactose and implementation of this approach is underway in our group.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
A. Tomy: methodology, investigation, data curation and visualisation; Saurav K. Guin: data curation and visualisation, review and editing; C. Cassidy: investigation, methodology; E. Dempsey: writing, conceptualisation, methodology, resources and supervision, funding acquisition, project administration, writing, review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Research Ireland [grant number GOIPG/2023/3535]; Enterprise Ireland and the European Union's Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie Career FIT PLUS [grant agreement no. 847402].References
- NewdevelopmentsinAnalyticalChemistryResearchNova2015.pdf.
- M. Li, W. Zhu, F. Marken and T. D. James, Electrochemical sensing using boronic acids, Chem. Commun., 2015, 51(78), 14562–14573 RSC.
- A. Varyambath, C. H. Tran, W. L. Song and I. Kim, Hyper-Cross-Linked Polypyrene Spheres Functionalized with 3-Aminophenylboronic Acid for the Electrochemical Detection of Diols, ACS Omega, 2017, 2(11), 7506–7514 CrossRef CAS PubMed.
- D. G. Hall, Structure, Properties, and Preparation of Boronic Acid Derivatives. Overview of Their Reactions and Applications, Boronic Acids, 2005, pp. 1–99 Search PubMed.
- H. S. Geethanjali, R. M. Melavanki, D. Nagaraja, P. Bhavya and R. A. Kusanur, Binding of boronic acids with sugars in aqueous solution at physiological pH – Estimation of association and dissociation constants using spectroscopic method, J. Mol. Liq., 2017, 227, 37–43 CrossRef.
- N. Wellington, S. Macklai and P. Britz-McKibbin, Elucidating the Anomalous Binding Enhancement of Isoquinoline Boronic Acid for Sialic Acid Under Acidic Conditions: Expanding Biorecognition Beyond Vicinal Diols, Chemistry, 2019, 25(67), 15277–15280 CrossRef CAS PubMed.
- G. Springsteen and B. Wang, A detailed examination of boronic acid–diol complexation, Tetrahedron, 2002, 58(26), 5291–5300 CrossRef CAS.
- Y. Egawa, R. Miki and T. Seki, Colorimetric Sugar Sensing Using Boronic Acid-Substituted Azobenzenes, Materials, 2014, 7(2), 1201–1220 CrossRef.
- G. Fang, H. Wang, Z. Bian, J. Sun, A. Liu and H. Fang, et al. Recent development of boronic acid-based fluorescent sensors, RSC Adv., 2018, 8(51), 294–29427 RSC.
- E. V. Lampard, A. C. Sedgwick, T. Sombuttan, G. T. Williams, B. Wannalerse and A. T. A. Jenkins, et al. Dye Displacement Assay for Saccharides using Benzoxaborole Hydrogels, ChemistryOpen, 2018, 7(3), 266–268 CrossRef CAS.
- F. Liu and X. Kan, Dual-analyte electrochemical sensor for fructose and alizarin red S specifically sensitive detection based on indicator displacement assay, Electrochim. Acta, 2019, 319, 286–292 CrossRef CAS.
- Y. Jiang, T. Niu, Z. Wang, W. Tan, F. Liu and Y. Kong, Electrochemical polymerization of alizarin and the electrochemical properties of poly(alizarin), Ionics, 2017, 24(5), 1391–1397 CrossRef.
- B. Dadpou and D. Nematollahi, Electrochemical Oxidation of Alizarin Red-S on Glassy Carbon Electrode: Mechanistic Study, Surface Adsorption and Preferential Surface Orientation, J. Electrochem. Soc., 2016, 163(7), H559–H565 CrossRef CAS.
- S. Schumacher, T. Nagel, F. W. Scheller and N. Gajovic-Eichelmann, Alizarin Red S as an electrochemical indicator for saccharide recognition, Electrochim. Acta, 2011, 56(19), 6607–6611 CrossRef CAS.
- T. Liu, B. Fu, J. Chen, Z. Yan and K. Li, A non-enzymatic electrochemical sensor for detection of sialic acid based on a porphine/graphene oxide modified electrode via indicator displacement assay, Electrochim. Acta, 2018, 269, 136–143 CrossRef CAS.
- Evaluation of Different Substrates for Electrodeposition of Riboflavin Films for Determination of Boric Acid in Eye Drops, Developments and Advances in Defense and Security, ed. J. O. Fernandes, R. S. de Lima Garcia, A. S. Araújo, F. G. Lepri, W. F. Pacheco and R. M. Dornellas, et al., Springer Nature Singapore, Singapore, 2024 Search PubMed.
- Z. Wu, H. Zhao, Y. Xue, Y. He, X. Li and Z. Yuan, Poly(pyridine-3-boronic acid)/Multiwalled Carbon Nanotubes Modified Glassy Carbon Electrodes for Simultaneous Determination of Ascorbic Acid, 3,4-Dihydroxyphenylacetic Acid and Uric Acid, Electroanalysis, 2010, 22(19), 2196–2201 CrossRef CAS.
- S. K. Guin, T. Velasco-Torrijos and E. Dempsey, Explorations in a galaxy of sialic acids: a review of sensing horizons, motivated by emerging biomedical and nutritional relevance, Sens. Diagn., 2022, 1(1), 10–70 RSC.
- A. Matsumoto, A. J. Stephenson-Brown, T. Khan, T. Miyazawa, H. Cabral and K. Kataoka, et al. Heterocyclic boronic acids display sialic acid selective binding in a hypoxic tumor relevant acidic environment, Chem. Sci., 2017, 8(9), 6165–6170 RSC.
- R. Rajan, A. R. Sheth and S. S. Rao, Sialic acid, sialyltransferase and neuraminidase levels in maternal plasma, urine and lymphocytes during pregnancy and post-partum period--a longitudinal study in women, Eur. J. Obstet. Gynecol. Reprod. Biol., 1983, 16(1), 37–46 CrossRef CAS.
- S. K. Guin, T. Krämer and E. Dempsey, A single-step enzyme-free electrochemical assay of N-acetyl-D-neuraminic acid, Electrochim. Acta, 2023, 460, 142618 CrossRef CAS.
- R. Yaa'ri, E. Schneiderman, V. B. Aharon, M. Stanevsky and E. Drori, Development of a Novel Approach for Controlling and Predicting Residual Sugars in Wines, Fermentation, 2024, 10(3), 125 CrossRef.
- P. Mokhtari, K. A. Schmidt, M. Babaei and M. I. Goran, Altered Nutrient Composition of Lactose-Reduced Infant Formula, Nutrients, 2024, 16(2), 276 CrossRef CAS.
- N. Fidler Mis, C. Braegger, J. Bronsky, C. Campoy, M. Domellöf and N. D. Embleton, et al. Sugar in Infants, Children and Adolescents, J. Pediatr. Gastroenterol. Nutr., 2017, 65(6), 681–696 CrossRef CAS PubMed.
- R. B. Jones, P. K. Bpintoerger, J. F. Plows, T. L. Alderete, J. Millstein and J. Fogel, et al. Lactose-reduced infant formula with added corn syrup solids is associated with a distinct gut microbiota in Hispanic infants, Gut Microbes, 2020, 12(1), 1813534 CrossRef.
- C. M. Slupsky, X. He, O. Hernell, Y. Andersson, C. Rudolph and B. Lönnerdal, et al. Postprandial metabolic response of breast-fed infants and infants fed lactose-free vs regular infant formula: A randomized controlled trial, Sci. Rep., 2017, 7(1), 3640 CrossRef.
- Y. Zhou, H. Huangfu, J. Yang, H. Dong, L. Liu and M. Xu, Potentiometric analysis of sialic acid with a flexible carbon cloth based on boronate affinity and molecularly imprinted polymers, Analyst, 2019, 144(21), 6432–6437 RSC.
- Y. Efremenko and V. M. Mirsky, 3-Thienylboronic Acid as a Receptor for Diol-Containing Compounds: A Study by Isothermal Titration Calorimetry, Chemosensors, 2022, 10(7), 251 CrossRef CAS.
- S. Takahashi, I. Suzuki, T. Ojima, D. Minaki and J. I. Anzai, Voltammetric Response of Alizarin Red S-Confined Film-Coated Electrodes to Diol and Polyol Compounds: Use of Phenylboronic Acid-Modified Poly(ethyleneimine) as Film Component, Sensors, 2018, 18(1), 317 CrossRef PubMed.
- W. Ma, Y. Zhang, F. Li, D. Kou and J. L. Lutkenhaus, Layer-by-Layer Assembly and Electrochemical Study of Alizarin Red S-Based Thin Films, Polymer, 2019, 11(1), 165 Search PubMed.
- H. Filik, A. A. Avan and Y. Mümin, Simultaneous Electrochemical Determination of Caffeine and Vanillin by Using Poly(Alizarin Red S) Modified Glassy Carbon Electrode, Food Anal. Methods, 2016, 10(1), 31–40 CrossRef.
- N. Aravindan and M. V. Sangaranarayanan, Differential pulse voltammetry as an alternate technique for over oxidation of polymers: Application of electrochemically synthesized over oxidized poly (Alizarin Red S) modified disposable pencil graphite electrodes for simultaneous detection of hydroquinone and catechol, J. Electroanal. Chem., 2017, 789, 148–159 CrossRef CAS.
- H. Han, X. Wu, S. Wu, Q. Zhang, W. Lu and H. Zhang, et al. Fabrication of alizarin red S/multi-walled carbon nanotube nanocomposites and their application in hydrogen peroxide detection, J. Mater. Sci., 2013, 48(9), 3422–3427 CrossRef CAS.
- S. Ranjitha, V. Aroulmoji, T. Mohr, P. Anbarasan and G. Rajarajan, Structural and Spectral Properties of 1,2-dihydroxy-9,10-anthraquinone Dye Sensitizer for Solar Cell Applications, Acta Phys. Pol. A, 2014, 126(3), 833–839 CrossRef CAS.
- W. Lemlikchi, P. Sharrock, M. Fiallo, A. Nzihou and M. O. Mecherri, Hydroxyapatite and Alizarin Sulfonate ARS Modeling Interactions for Textile Dyes Removal from Wastewaters, Procedia Eng., 2014, 83, 378–385 CrossRef CAS.
- A. Turcanu and T. Bechtold, pH Dependent redox behaviour of Alizarin Red S (1,2-dihydroxy-9,10-anthraquinone-3-sulfonate) – Cyclic voltammetry in presence of dispersed vat dye, Dyes Pigm., 2011, 91(3), 324–331 CrossRef CAS.
- G. Deffo, R. C. T. Temgoua, S. F. Mbokou, E. Njanja, I. Kenfack Tonlé and E. Ngameni, A sensitive voltammetric analysis and detection of Alizarin Red S onto a glassy carbon electrode modified by an organosmectite, Sens. Int., 2021, 2, 100126 CrossRef.
- K. Chetankumar and B. E. K. Swamy, Electrochemically nitric acid pre-treated glassy carbon electrode sensor for catechol and hydroquinone: A voltammetric study, Sens. Int., 2020, 1, 100001 CrossRef.
- J. A. Peters and K. Djanashvili, Exploration and exploitation of the uncommon pH profile of the dynamic covalent interactions between boronic acids and N-acetylneuraminic acids, Coord. Chem. Rev., 2023, 491, 215254 CrossRef CAS.
- W. Gu, Y. Wang, T. Wu, W. Li, H. Wang and W. Xia, Linear sweep voltammetric studies on the complex of alizarin red s with aloe polysaccharide and determination of aloe polysaccharide, Carbohydr. Res., 2012, 349, 82–85 CrossRef CAS.
- M. Dilshad, A. Shah and S. Munir, Electroanalysis of Ibuprofen and Its Interaction with Bovine Serum Albumin, Molecules, 2022, 28(1), 49 CrossRef PubMed.
- D. Ravelli, P. Isernia, A. Acquarulo, A. Profumo and D. Merli, Voltammetric Determination of Binding Constant and Stoichiometry of Albumin (Human, Bovine, Ovine)–Drug Complexes, Anal. Chem., 2019, 91(15), 10110–10115 CrossRef CAS PubMed.
- A. Esokkiya, S. Sudalaimani, K. Sanjeev Kumar, P. Sampathkumar, C. Suresh and K. Giribabu, Poly(methylene blue)-Based Electrochemical Platform for Label-Free Sensing of Acrylamide, ACS Omega, 2021, 6(14), 9528–9536 CrossRef CAS PubMed.
- M. Li, S.-Y. Xu, A. J. Gross, J. L. Hammond, P. Estrela and J. Weber, et al. Ferrocene-Boronic Acid–Fructose Binding Based on Dual-Plate Generator–Collector Voltammetry and Square-Wave Voltammetry, ChemElectroChem, 2015, 2(6), 867–871 CrossRef CAS PubMed.
- S. M. Wikeley, P. Lozano-Sanchez, M. Caffio, T. D. James and F. Marken, Polymer indicator displacement assay (PIDA) with boronic acid receptors on graphene foam electrodes for self-optimised impedimetric lactic acid determination, Sens. Actuators, B, 2023, 377, 133089 CrossRef CAS.
- O. Thomas and M. Brogat, Organic constituents, in UV-Visible Spectrophotometry of Waters and Soils, ed. O. Thomas and C. Burgess, Elsevier, 3rd edn, 2022, ch. 4, pp. 95–160 Search PubMed.
- A. S. Awed, N. A. El-Ghamaz, M. M. El-Nahass and H. M. Zeyada, Linear and nonlinear optical properties of alizarin red S thin films, Indian J. Phys., 2019, 93(7), 861–868 CrossRef CAS.
- F. Yakuphanoglu and B. F. Şenkal, Electrical conductivity and optical properties of poly(3-thiophene boronic acid) organic semiconductor, Polym. Eng. Sci., 2009, 49(4), 722–726 CrossRef CAS.
- H. Cao, D. P. Yang, D. Ye, X. Zhang, X. Fang and S. Zhang, et al. Protein-inorganic hybrid nanoflowers as ultrasensitive electrochemical cytosensing interfaces for evaluation of cell surface sialic acid, Biosens. Bioelectron., 2015, 68, 329–335 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sd00034c |
| ‡ COMMISSION IMPLEMENTING DECISION (EU) 2017/2375 of 15 December 2017 authorizing the placing on the market of N-acetyl-D-neuraminic acid as a novel food ingredient under Regulation (EC) No 258/97 of the European Parliament and of the Council (notified under document C(2017) 8431) https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=OJ:L:2017:337:TOC. |
| This journal is © The Royal Society of Chemistry 2025 |