
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLateral flow assay-based detection of nuclear fusion oncoprotein: implications for screening of acute promyelocytic leukemia†
Maede
Chabi
a,
Binh
Vu
 b,
Kristen
Brosamer
a,
Sophia
Song
b,
Vijay
Maranholkar
c,
Zihua
Zeng
d,
Youli
Zu
d,
Rashmi
Kanagal-Shamanna
e,
Jacinta C.
Conrad
*b,
Richard C.
Willson
*abcf and
Katerina
Kourentzi
*b
b,
Kristen
Brosamer
a,
Sophia
Song
b,
Vijay
Maranholkar
c,
Zihua
Zeng
d,
Youli
Zu
d,
Rashmi
Kanagal-Shamanna
e,
Jacinta C.
Conrad
*b,
Richard C.
Willson
*abcf and
Katerina
Kourentzi
*b
aDepartment of Biomedical Engineering, University of Houston, Houston, Texas 77204, USA. E-mail: willson@uh.edu
bDepartment of Chemical and Biomolecular Engineering, University of Houston, Houston, Texas 77204, USA. E-mail: jcconrad@central.uh.edu; ekourent@central.uh.edu
cDepartment of Biology and Biochemistry, University of Houston, Houston, Texas 77204, USA
dDepartment of Pathology and Genomic Medicine, Houston Methodist Hospital, USA
eDepartment of Hematopathology, The University of Texas MD Anderson Cancer Center, USA
fInstitute for Obesity Research, Tecnológico de Monterrey, Monterrey, Nuevo León 64710, Mexico
First published on 21st March 2025
Abstract
Due to the slow progression of most cancers, speed of diagnosis is not of primary concern. However, the diagnosis of acute promyelocytic leukemia (APL) is unusually urgent because its hemorrhagic complications can result in death within a few days. APL is highly treatable, but the turnaround time for standard molecular testing often exceeds the window for life-saving treatment, even in advanced medical centers. The hallmark of APL is the fusion of the PML and RARα genes (t(15;17)) resulting in the expression of a growth-promoting PML–RARα fusion protein. Toward timely screening for APL, we have developed a sensitive europium-based lateral flow immunoassay for direct detection of nuclear PML–RARα fusion oncoprotein. We demonstrated a limit of detection of 11% fusion protein positive NB4 cells spiked into healthy peripheral blood mononuclear cells and an integrated filter-based sample preparation workflow showcasing its potential for clinically actionable utility in prompt APL screening. With further validation with clinical human samples this lateral flow immunoassay has the potential to enable fusion-protein based cancer diagnostics at true point-of-care.
1 Introduction
Acute promyelocytic leukemia (APL) is a distinctive subtype of acute myeloid leukemia (AML) in which immature promyelocyte “blasts” accumulate in the bone marrow. APL presents with an abnormally high number of white blood cells and blasts, a low platelet count, coagulopathy, and pulmonary or cerebral hemorrhage.Early mortality within 30 days occurs in up to one in three patients, with the majority of deaths occurring within one week of diagnosis due to hemorrhagic complications.1–5 APL is highly curable if diagnosed in time. Prompt administration of all-trans retinoic acid (ATRA) therapy combined with arsenic trioxide (ATO) and/or anthracycline chemotherapy6 has transformed APL from the most rapidly-fatal to the most frequently curable form of acute leukemia, with long-term survival rates up to 90%.7,8 Thus timely diagnosis is crucial for emergency treatment to improve long-term survival.
The hallmark of APL is a translocation between chromosomes 15 and 17, t(15;17)(q24.1;q21.1), which leads to fusion of the promyelocytic leukemia (PML) and retinoic acid receptor alpha (RARα) genes. The breakpoint in the RARα gene is always located in intron 2, whereas breakpoints in the PML gene cluster are found in the breakpoint cluster region (bcr) of intron 6 (bcr1; 55% of cases), or in exon 6 (bcr2; 5% of cases) or intron 3 (bcr3; 40% of cases). In the presence of physiological levels of retinoic acid, the RARα/retinoid X receptor (RXR) heterodimers control the transcription of genes involved in myeloid differentiation by binding to retinoic acid response element. The expression of PML–RARα fusion protein in APL interferes with the normal function of wild-type RARα protein and represses the transcription of genes required for hematopoietic differentiation, causing the accumulation of immature promyelocytes in the bone marrow. Moreover, PML–RARα fusion protein results in the delocalization of PML from the nuclear body, which interferes with cell differentiation and inhibits apoptosis. Pharmacological concentrations of ATRA and ATO target RARα by restoring retinoid signaling and reorganization of PML to nuclear bodies, resulting in degradation of the fusion protein via various pathways and restoring myeloid differentiation.6,8–10
After complete blood count (CBC) and microscopy and standard flow cytometry for immunophenotyping, clinical diagnosis of APL is ultimately based on confirmation of the translocation by karyotyping, fluorescence in situ hybridization (FISH), and RT-PCR, especially for cytogenetically cryptic cases.11 Modern techniques like optical genome mapping and whole transcriptome sequencing for further work up and identification of alternate breakpoints or previously-unknown partners for RARα are available in MD Anderson Cancer Center. However, the standard cytogenetic methods have too-long turnaround times even in major medical centers (e.g. karyotyping: 5–7 days, FISH and RT-PCR: 1–2 days, STAT-FISH: 24 hours). Providing urgent results using these standard cytogenetic and molecular methods requires extensive laboratory resources and expertise and is associated with high cost.5,12–15 In-house developed immunofluorescence staining of PML protein16 provides quicker results (ca. 4 hours) but requires skilled interpretation, has accuracy dependent on pathologist experience, is prone to human error and is not broadly available17 (Table 1). Moreover, current best practice recommends that APL be excluded in each patient with newly-diagnosed AML. ATRA is not commonly given upon clinical suspicion, and early-stage APL may not have a classical clinical presentation or distinctive pathological findings.6
| Clinical test | Turnaround time | Accessibility | Ease of use | Cost |
|---|---|---|---|---|
| a Can be done STAT, within 24 hours for urgent cases. b Available only at MDACC. | ||||
| Morphology with cytochemical stain for myeloperoxidase | Few hours | High | Moderate | Low |
| Conventional karyotyping (G-banding) | 7–14 days | Moderate | Difficult | Moderate |
| FISH (fluorescence in situ hybridization) | 24–48 hoursa | Moderate | Moderate | Moderate/high |
| RT-PCR | 48 hours | Moderate | Moderate | Moderate/high |
| Flow cytometry (immunophenotyping) | 24–48 hours | Moderate | Moderate | Moderate |
| PML immunofluorescence staining (PML oncogenic domain/POD assay) | 4 hoursb | Moderate | Low/moderate | Moderate |
| Optical genome mapping | 4–5 days | Low | Moderate | High |
| Anchored multiplex PCR technology-based targeted NGS for fusions | <7 days | Moderate | Moderate | Moderate |
| Whole transcriptome sequencing | 2 weeks | Low | Difficult | High |
| APL LFA (this study) | <1 hour | High | Easy | Low |
Thus, while genetic confirmation of the APL gene fusion is ultimately required, there is great interest in accurate, rapid, and easily interpretable ancillary tests for the detection of the fusion PML–RARα oncoprotein to enable the earlier administration of differentiation therapy and thereby prevent early death due to delayed diagnosis.7,12,14,15,17,18 Recently described efforts include a flow cytometric immunoassay19 and a microfluidic chip-based magnetic immunoassay.20 Both assays are based on complex instrumentation or workflows that pose barriers to the widespread adoption of the technology.
Lateral flow assays (LFAs), brought into wide usage during the COVID-19 pandemic, enable easy, decentralized, and rapid diagnosis, especially in emergency settings without the need for sophisticated instrumentation. In an LFA, a porous nitrocellulose membrane provides a platform for the target-containing sample and reporter-labeled target-specific antibodies to move through capillary channels and interact with and be captured by antibodies immobilized on the test and the control lines. In the presence of the target, labeled antibodies and the target form a sandwich with antibodies immobilized on the test line, resulting in a signal line indicating a positive test. The control line antibodies bind to unbound labeled antibodies, resulting in a signal that confirms proper reagent flow and the validity of the test.
Given high-performance antibodies, LFA sensitivity depends greatly on the detectability of the reporters.21 Whereas commercial LFAs typically use gold nanoparticles as reporters, next-generation light-emitting reporters boost assay sensitivity to levels achieved with laboratory-based diagnostics such as ELISA.22 Specifically, europium chelate nanoparticles enable time-resolved fluorescence (TRF) measurements leading to reduced autofluorescence (background fluorescence is short-lived) and enhanced assay sensitivity, especially in biological samples.23
Application of LFA technology to cancer diagnostics is limited to serum or biomarkers contained in plasma,24 and not widely extended to the detection of intracellular oncoproteins. Here we demonstrate a rapid, sensitive, and easy-to-use LFA utilizing europium chelate nanoparticle reporters for the detection of intracellular PML–RARα oncoprotein using a compact time-resolved fluorescence LFA reader combined with point-of-care leukocyte isolation and on-filter cell lysis and showcase the potential of LFA for the detection of nuclear fusion proteins for cancer diagnostics at true point-of-care.
We initially evaluated the performance of eighty-six antibody pairs directed against different epitopes in PML–RARα fusion protein. We then demonstrated the LFA-based detection of PML–RARα fusion protein in leukemic cells spiked into normal peripheral blood mononuclear cells (PBMC) or in human AML (non-APL) HL-60 cells. We also demonstrated antibody specificity and compatibility with other cell lines and leftover, post-diagnosis leukemia blood samples negative for PML–RARα fusion protein.
2 Materials and methods
2.1 Materials
The following reagents were purchased from Thermo Fisher Scientific: 1× RIPA lysis and extraction buffer (89901; 25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS), 100× Halt protease inhibitor cocktail/EDTA (87786; 1×: AEBSF, 1 mM; aprotinin, 800 nM; bestatin, 50 μM; E64, 15 μM; leupeptin, 20 μM; pepstatin A 10 μM in dimethyl sulfoxide (DMSO) and EDTA 5 mM), EDC (A35391), M-PER mammalian protein extraction reagent (78501), and bicinchoninic acid (BCA) Protein Assay kit (23225). Additional cell lysis reagents tested were: 10× RIPA buffer (9806; Cell Signaling Technology), 1× cell lysis buffer (9803; Cell Signaling Technology). Phosphate-buffered saline (1× PBS) tablets were purchased from Takara. MES potassium salt (39946-25-3), NHS (130672), IGEPAL CA-630, bovine serum albumin (A7906, ≥98%), and polyvinylpyrrolidone (mol wt 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000; PVP-40) were from Sigma Aldrich. All chemicals were used as received unless otherwise indicated. Lymphoprep™ (07801), SepMate-50 tubes (395576), the EasySep Direct Human PBMC Isolation kit (19654) and the EasySep magnet stand (18001) were from StemCell Technologies. Acrodisc WBC filters (C4951) were from Pall/Cytiva Corporation.
000; PVP-40) were from Sigma Aldrich. All chemicals were used as received unless otherwise indicated. Lymphoprep™ (07801), SepMate-50 tubes (395576), the EasySep Direct Human PBMC Isolation kit (19654) and the EasySep magnet stand (18001) were from StemCell Technologies. Acrodisc WBC filters (C4951) were from Pall/Cytiva Corporation.
2.2 Cell culture and lysis
We employed two myeloid leukemic cell lines: HL-60 (non-APL AML; lacks t(15;17)), and NB4 (APL, PML–RARα fusion protein-positive) as well as other control non-AML cell lines: Jurkat T-cell, CA46 B-cell, and U-937 histiocytic lymphoma. All cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (Corning 35-011-CV), 100 unit mL−1 penicillin and 100 μg mL−1 streptomycin at 37 °C in a humidified atmosphere with 5% CO2. After two days' culture, cells were harvested, and viable cells were counted using trypan blue stain. After washing twice in PBS, cells were centrifuged, resuspended in RPMI 1640 medium with 10% DMSO, and stored at −80 °C for later use.The frozen cells were thawed in a water bath at 37 °C for 10 minutes. Next, the cells were centrifuged at 500 × g for 10 min at 4 °C and washed twice with cold 1× PBS. Halt protease inhibitor cocktail and EDTA solution were added to the RIPA lysis buffer at 10 μL mL−1 RIPA buffer immediately before use. Lysis buffer was then added to the cell pellet at 1 mL per 5 × 106 cells. The cells were incubated on ice for 15 min on a shaker and then centrifuged at 18000 × g for 15 min at 4 °C to remove the cell debris. The supernatant was stored at −20 °C for further analysis. Total protein in the lysates was measured using the BCA assay. A histone H3 quantification ELISA (Abcam; ab115091) was used to determine the total histone H3 proteins in the cell lysate and confirm nuclear extraction.
2.3 Point-of-care clinical sample preparation
Cryopreserved human peripheral blood mononuclear cells (PBMC) from healthy donors were purchased from AllCells, LLC. De-identified, non-coded peripheral whole blood samples derived from healthy volunteers were obtained from the Gulf Coast Regional Blood Center (Houston, TX) and used immediately or stored at 4 °C for up to 48 h until use. Leftover, de-identified peripheral blood (PB) or bone marrow (BM) samples obtained under IRB-approved protocols at Houston Methodist Hospital and MD Anderson Cancer Center were also used in assay development and validation.White blood cells (WBCs) were counted using a three-part differential hematology analyzer (Medonic M, Boule). White blood cells were collected from peripheral blood (PB) and bone marrow (BM) by filtering through an Acrodisc WBC filter (Pall Corporation, NY). The volume of whole blood containing 2.5 × 106 WBCs was calculated and applied to the Acrodisc filter to capture WBCs. Then the filter was washed with 20 mL 1× PBS to remove the red blood cells (RBCs). For lysis of the captured WBCs, 600 μL 1× RIPA buffer was added while the filter outlets were closed with Parafilm. The filter was incubated on ice on a shaker unconstrained for 20 min. The cell lysate was then pushed through the filter with 5 mL air using a syringe and the sample was immediately analyzed by LFA.
2.4 Nanoparticle conjugation
Carboxylated europium chelate polystyrene nanoparticles (0.2 μm, COOH surface titer 139 μeq. g−1; Bangs Laboratories, FCEU002) were mixed with 25 mM MES buffer, pH 6 at 0.5% solids concentration. The particles were centrifuged at 20000 × g for 10 min and re-suspended in 25 mM MES buffer, pH 6 at 0.5% solids concentration using a bath sonicator. This washing process was repeated three times. Next, 33 μL of 10 mg mL−1 EDC in DI water and 32 μL of 50 mg mL−1 NHS in DI water were added per 1 mL of particle suspension for a molar ratio of 2.5 EDC and 20 NHS per carboxyl group. The suspension was incubated for 30 min at room temperature on a rotator. Next, the particles were centrifuged and washed in 1× PBS. The particles were resuspended to 0.5% solids each time. An antibody solution in 1× PBS was added to a final concentration of 0.4 mg antibody per mL of the particle suspension (0.24 mg antibody per mL of the particle suspension for antibody screening). The solution was incubated for 2–3 h at room temperature on a rotator. The particles were passivated by overnight incubation with BSA at a final concentration of 40 mg mL−1 at 4 °C. The particle solution was then washed thrice with 10 mg mL−1 BSA in 1× PBS and finally stored in 10 mg mL−1 BSA in 1× PBS at 1% solids concentration at 4 °C until further use.
2.5 Lateral flow assay assembly
A UniSart CN95 nitrocellulose membrane (Sartorius Stedim), a Whatman standard 14 sample pad (Cytiva Life Sciences), and a ReliaFlow™ 440 absorbent pad (Ahlstrom-Munksjö) were assembled on a backing card (DCN, MIBA-020). Test line antibodies (Table 2) at 0.5 mg mL−1 and control line antibodies (a 1:1 mixture of anti-mouse IgG (Arista Biologicals; ABGAM-0500) and anti-rabbit IgG (Arista Biologicals; ABGAR-0500)) at 0.5 mg mL−1 in 1× PBS were dispensed onto the nitrocellulose membrane using a Biodot XYZ3060 system (1 μL cm−1, 30 μL per 30 cm card) to configure the test line and control line, respectively. After the initial antibody screening study, to obtain higher test line/control line (TL/CL) ratios we increased the concentration of the detection antibodies to 1 mg mL−1 and decreased the concentration of the capture antibodies to 0.25 mg mL−1. Antibodies were allowed to dry on the membrane in an incubator at 50 °C for 1 h and overnight at room temperature in a desiccator chamber (Totech; SuperDry Desiccant Cabinet; #SD-151-21) at 5% humidity. The cards were cut into 3 mm strips using a ZQ2000 Guillotine Cutter (Kinbio Tech) and stored in a desiccator at room temperature until use.
| # | Host species/Clonality | Target | Vendor | Cat # |
|---|---|---|---|---|
| R1 | Mouse monoclonal | 63-362 RARα protein | Santa Cruz Biotechnology | sc-515796 |
| R2 | Mouse monoclonal | 315-424 RARα protein | Santa Cruz Biotechnology | sc-293417 |
| R3 | Rabbit polyclonal | 322-349 RARα protein | Aviva Systems Biology | OAAB10147 |
| R4 | Rabbit polyclonal | RARα protein | Aviva Systems Biology | ARP97651_P050 |
| R5 | Rabbit polyclonal | RARα protein | Aviva Systems Biology | ARP42824_P050 |
| R6 | Rabbit polyclonal | RARα protein | Aviva Systems Biology | ARP40315_T100 |
| R7 | Rabbit monoclonal | 1-100 RARα protein | ABclonal Technology | A19551 |
| R8 | Rabbit polyclonal | 1-457 RARα protein | ABclonal Technology | A0370 |
| P1 | Mouse monoclonal | 37-51 PML protein | Santa Cruz Biotechnology | sc-966 |
| P2 | Mouse monoclonal | 157-394 PML protein | Santa Cruz Biotechnology | sc-377340 |
| P3 | Rabbit polyclonal | PML protein | Aviva Systems Biology | ARP38184_P050 |
| P4 | Rabbit polyclonal | PML protein | Aviva Systems Biology | ARP33048_P050 |
| P5 | Rabbit monoclonal | 1-100 PML protein | ABclonal Technology | A19646 |
| P6 | Rabbit polyclonal | 510 to C terminus PML protein | ABclonal Technology | A18134 |
| F1 | Rabbit polyclonal | 500-600 PML–RARα fusion junction | Abcam | ab43152 |
| F2 | Rabbit polyclonal | PML–RARα fusion protein | ABclonal Technology | A7525 |
2.6 Lateral flow assay
Cell lysates from cell lines (section 2.2) or clinical samples (section 2.3) were mixed with four volumes of LFA running buffer (1× PBS pH 7.4, 10 mg mL−1 BSA, 10 μL mL−1 IGEPAL CA-630, 5 mg mL−1 PVP-40). 45 μL of the sample was applied to the sample pad of the LFA strip. Next, 20 μL of europium chelate particles at approximately 3.3 × 1010 particles per mL was applied to the sample pad followed by 3 washes with 20 μL LFA buffer. After approximately 15 min the LFA strips were scanned with a compact time-resolved fluorescence LFA analyzer (LTRIC-600, Lumigenex) to read the signals of the test and control lines. The signals were extracted from the instrument intensity profiles and the area under each TL and CL peak was quantified using ImageJ or the software supplied with the reader. We defined the cutoff as the average plus 1.645 × standard deviation of blank strips plus 1.645 × standard deviation of the lowest positive strips (μblank + 1.645σblank + 1.645σlow).25 We determined the limit of detection by identifying the crossing point using linear interpolation between the two experimental data points that surround the cutoff.2.7 Antibody screening
Eighty-six antibody pairs, including two anti-PML–RARα fusion junction antibodies, eight anti-RARα protein antibodies, and six anti-PML protein antibodies, were screened on LFA using NB4 cells as the PML–RARα-bcr1 positive sample and U937 as the negative sample. For the first round of screening, NB4 and U937 cell lysates were diluted ten-fold in LFA running buffer to a concentration of approximately 3.7 × 107 lysed cells per mL. In the second round of screening, NB4 and U937 cell lysates were diluted twenty-fold in LFA buffer to approximately 1.8 × 107 cells per mL. The strips were run as described in section 2.6. Antibody pairs were ranked based on the difference between the test line (TL) intensities of positive and negative tests obtained from the LFA reader according to rank = TLNB4 − TLU937.2.8 Sensitivity and specificity testing of the APL-LFA
The sensitivity of the LFA was determined by analyzing serial dilutions of NB4 cell lysate spiked into normal PBMC lysate or of NB4 cells spiked into HL60 cells and lysed together, keeping the total cells at 33000 cells per sample. The specificity of the LFA was confirmed by analysis of several non-APL cell line lysates and leftover non-APL peripheral blood or bone marrow clinical samples.
3 Results and discussion
3.1 Europium-based LFA for cancer diagnostics
We developed a lateral flow assay (LFA) for the detection of the PML–RARα oncoprotein, the hallmark of APL (Fig. 1). Peripheral blood, bone marrow, and isolated mononuclear cells that include lymphocytes, monocytes, dendritic cells, and immature blasts (e.g. promyelocytes in the case of APL) are easily accessible samples since they are used in the patient's routine workup. Incorporating a white blood cell-selective syringe filter in the workflow allowed the direct use of blood or bone marrow samples without the need for lab equipment (e.g., a centrifuge) for cell isolation. We found that RIPA cell lysis buffer can successfully liberate intact PML–RARα fusion protein with nuclear localization and is compatible with the downstream LFA after lysate diluting/conditioning. We used europium chelate reporters that can be sensitively imaged with a portable, inexpensive LFA analyzer. | ||
| Fig. 1 Schematic illustration of APL-LFA and integrated point-of-care (POC) filter-based blood sample prep. Whole blood flows through a filter that captures white blood cells (WBCs). The red blood cells (RBCs) are washed away with PBS. Lysis buffer is applied to the filter to lyse the white blood cells, and the lysate sample is used to run LFA. In an APL-positive test, PML–RARα fusion protein is captured by anti-RARα antibodies on the test line and then detected by europium-conjugated anti-PML antibodies. Unbound europium-conjugated anti-PML antibodies are captured with the secondary anti-mouse antibodies on the control line. Created with https://BioRender.com. | ||
3.2 Toward point-of-care whole blood sample prep
The current laboratory standard for blood cell separation is density gradient centrifugation. Technical advances including new gradient media such as Lymphoprep (StemCell) and filter-based tubes such as SepMate (StemCell) have led to protocols that are faster, more efficient, and user-friendly but that still require a centrifuge. An alternative approach is filtration through leukocyte reduction filters such as the Acrodisc WBC syringe filter (Cytiva), which retain white blood cells (WBCs) due to their size, limited deformability, surface structure, and charge.26 Peripheral blood mononuclear cells (PBMC) also can be isolated from whole blood using immunomagnetic negative selection (EasySep Direct Human PBMC Isolation kit; StemCell), where granulocytes, platelets, and erythrocytes are tagged with antibody-functionalized magnetic particles that recognize specific cell surface markers and removed using a magnet. We tested (following the manufacturers' protocols) and evaluated these three commonly-used workflows for cell recovery, cost, and overall suitability for integration with LFA at point of care. All methods showed cell recoveries within their respective specifications (Fig. 2; density gradient centrifugation: 54.1%, WBC filter: 97.59%, magnetic immunoseparation: 34.7%). The density gradient protocol was time-consuming, laborious, and required significant technical skill. Moreover, it is centrifugation-based and thus not suitable for point of care. Magnetic-based immuno-separation is expensive (uses specific antibodies), requires several pipetting steps, and showed the highest variability. Leukocyte depletion filters require no significant training or equipment. Furthermore, depletion filter yield approached 100%, and in situ lysis of captured cells is straightforward and suitable for point of care. | ||
| Fig. 2 Average recovery/separation of white blood cells (WBCs) or peripheral blood mononuclear cells (PBMC) from whole blood with three commonly-used methods. Error bars are one standard deviation of measurements from five samples. WBC filters efficiently capture more than 90% WBCs from whole blood. | ||
3.3 Lysis buffer selection
The APL lateral flow assay requires the efficient liberation of PML–RARα from the cell nucleus using a lysis buffer compatible with downstream LFA immunoassay. We compared four commonly-used cell lysis buffers for mammalian cells (Fig. 3); 1× RIPA-T buffer (Thermo Scientific), 1× RIPA-CS buffer (Cell Signaling Technology), cell lysis buffer (Cell Signaling Technology), and M-PER (Thermo Scientific). 1× RIPA-T buffer lysate showed the highest extracted total protein concentration as measured by BCA assay (Fig. 3a). 1× RIPA-T also gave the highest liberated concentration of histone H3, commonly used as a normalizing control in nuclear extracts (Fig. 3b). The increased lysis efficiency of 1× RIPA-T may be due to the included sodium dodecyl sulfate (Fig. 3d). Most importantly, 1× RIPA-T showed the strongest LFA signal when used to treat NB4 cells (Fig. 3c). Thus, 1× RIPA-T was chosen for all subsequent experiments. | ||
| Fig. 3 Comparison of different extraction methods by total protein released, histone released, and signal on APL-LFA. (a) Bicinchoninic acid assay (BCA): absorbance at 562 nm corresponding to total protein extracted by each lysis buffer. The background signal from each lysis buffer is subtracted from the positive signal for the same lysis buffer. (b) Histone H3 quantification ELISA: absorbance at 450 nm corresponding to histone H3 nuclear extraction using each lysis buffer. To eliminate the potential interference of lysis buffers, the signal was normalized in each lysis buffer with the blank in the same buffer. (c) LFA performance: TL/CL intensity ratio for each cell lysate when run in LFA. Figures a–c show the highest signals for lysate extracted by RIPA-T compared to other lysis buffers (n = 3). (d) Composition of each lysis buffer tested as provided by the vendor. | ||
3.4 Antibody screening
We performed multiple rounds of antibody screening on LFA to identify the best-performing antibody pairs on APL-LFA. We screened commercially available antibodies raised against the amino acid sequence at the fusion junction of PML–RARα protein or, alternatively, against different epitopes on the N-terminus of PML or C-terminus of RARα protein expected to be present in the fusion protein (Table 2). In the screening, we used the human NB4 APL cell line as the PML–RARα protein-positive sample and the non-APL U937 histiocytic lymphoma cells as the negative control. Since the particles were conjugated with antibodies from different species (mouse or rabbit), we used a mixture of anti-mouse and anti-rabbit antibodies on the control line for screening strips. To minimize the effect of the variation of binding of different particles (conjugated with antibodies from different species) to the control line during screening, the antibodies were ranked based on the difference between the specific signal at the TL of the positive strip (NB4 cells) and the non-specific background at the TL of the negative strip (U937 cells). We do not expect significant interference of wild-type (not-fused) PML or RARα protein, as they are expressed in significantly lower quantities than the fusion protein in APL leukemic cells.27,28 Nevertheless, we did not pursue pairs of the same type of antibodies (i.e. two anti-PML antibodies or two anti-RARα antibodies) to avoid false signals in clinical testing.From the first round (Fig. 4) we selected fourteen antibody pairs that were then tested with a higher dilution (twenty-fold diluted) of cell lysate in the second round. Although the performance of the anti-PML–RARα antibody (F1; captures fusion junction of PML–RARα protein) was satisfactory, ultimately, we selected an anti-RARα antibody (sc-515796; R1) and an anti-PML antibody (sc-966; P1) as the capture and detection antibody, respectively because of the availability of the antibodies and to allow the capture of different isoforms of the PML–RARα fusion protein independent of the position of the breakpoint on the PML gene.
 | ||
| Fig. 4 Screening (first round) of different anti-PML protein (P), anti-RARα protein (R), and anti-PML–RARα fusion protein (F) antibody pairs with NB4 (positive) and U937 (negative) cell lysates on LFA. The values correspond to the difference of test line values of positive and negative strips (TLNB4 − TLU937). Darker blues correspond to larger differences. N/A indicates pairs that were not tested. The pair circled in yellow was selected for the subsequent experiments. | ||
It is worth mentioning that our in-house (recombinant protein expression in E. coli or in a cell-free expression system) and external Contract Research Organization (in baculovirus-insect system) efforts to produce recombinant protein did not yield satisfactory protein yields. We also set up a large-scale culture of NB4 APL cancer cells and purified PML–RARα fusion protein in-house with ion chromatography (IEX, UNO-Q, Biorad) followed by size exclusion chromatography (Superdex-200). However, the yield and purity of the purified protein were not sufficient to be used as a standard/positive control in immunoassay development. Given the extended efforts and concerns for authenticity, we decided to use lysates of NB4 cells as genuine standard, positive control in LFA development.
3.5 Performance of APL-LFA with human cancer cell lines
We evaluated the sensitivity of the APL-LFA by testing PML–RARα-positive NB4 cell lysate spiked into normal PBMC lysate (reflective of the true clinical sample background; Fig. 5) or NB4 cells spiked into HL60 cells and lysed together in a POC-compatible workflow (Fig. 6) while maintaining the same number of cells offered per test in all samples. The LFA signal at 11% NB4 cells lysate in PBMC background and 8.1% NB4 cells in HL60 APL-negative background (with 33000 total cells offered per test) exceeded the estimated cutoff and would be readily detectable (we attribute the difference in the background signal between different experiments to the different matrices used). It is worth noting that that the majority of APL patients have much higher blast counts (easily detectable by our method) by the time that symptoms appear and they present at the hospital.7,16,29 A rapid APL screening test is meant to be used in the clinic, at point-of-care, when a patient presents with symptoms that trigger APL suspicion. Blood collection and isolation of PBMCs as well as blood marrow aspiration are standard-of-care for any hematological neoplasia diagnosis and thus PBMC and blood marrow samples will be readily available for the LFA workflow. The demonstrated LFA sensitivity suggests the potential for clinically-actionable use, though the development of a diagnostic for a particular condition will require extensive study of clinical samples. In our experience, timely access to clinical samples has been challenging due to the rarity of the disease and the suspected interference of ATRA with the detectability of the fusion protein in an immunoassay.13 Prospective collection of samples from newly diagnosed patients before ATRA administration could alleviate this problem.
 | ||
| Fig. 5 Sensitive APL-LFA-based detection of PML–RARα fusion protein in NB4 cell lysate spiked into PBMC lysate. NB4 cell lysate was mixed with PBMC lysate in various percentages (in a total of 33000 cells). The LFA signals were measured using a portable LFA reader and quantified using ImageJ. Error bars are one standard deviation of three measurements. The cutoff (dashed line) is the average plus 1.645 times the standard deviation of the negative tests (100% healthy PBMC lysate) plus 1.645 times the standard deviation of the low positive tests (5% NB4 cell lysate) giving a limit of detection (LoD) of 11% NB4 cell lysate in a total of 33000 cells. | ||
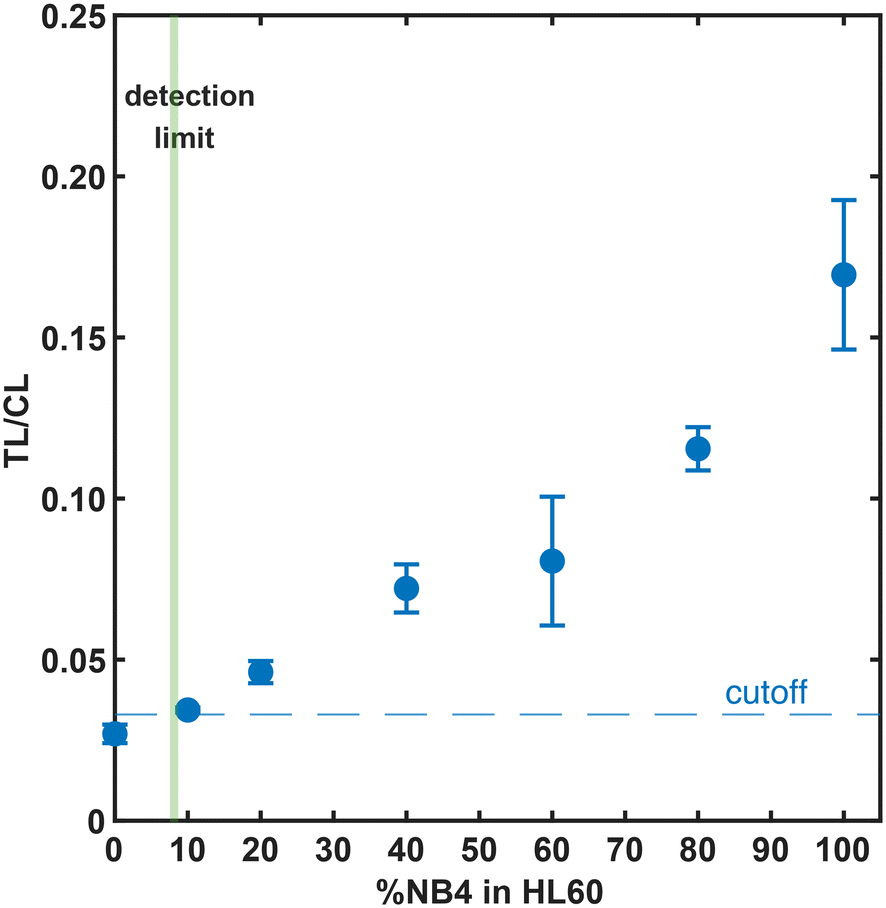 | ||
| Fig. 6 Sensitive APL-LFA-based detection of PML–RARα fusion protein in NB4 cells spiked into HL60 cells. NB4 cells were mixed with HL60 cells in various percentages and lysed together (in a total of 33000 cells). The LFA signals were measured using a portable LFA reader. Error bars are one standard deviation of three measurements. The cutoff (dashed line) is the average plus 1.645 times the standard deviation of the negative tests (100% HL60 cell lysate) plus 1.645 times the standard deviation of the low positive tests (10% NB4 cell lysate), giving a limit of detection (LoD) of 8.1% NB4 cells in a total of 33000 cells. | ||
We also evaluated the specificity of the antibodies and assay compatibility with cell lysates from different hematopoietic cancer cell lines: HL60; acute myeloid leukemia, U937; histiocytic lymphoma, JeKo-1; mantle cell lymphoma, KBM7; chronic myeloid leukemia (positive for BCR–ABL fusion oncoprotein), and SUP-B15; acute lymphoblastic leukemia (positive for BCR–ABL fusion oncoprotein). The APL-LFA showed a specific high positive signal for APL-positive NB4 cell lysate (Fig. 7). The background signal from cancer cell lines differed but was similar to the signal of realistic clinical sample background, blood or PBMC samples, and not significant; a similar number of NB4 cells had a signal 15–50 times higher than those of the negative samples. Nevertheless, ROC analysis is typically required to determine the clinically-relevant final cutoff for the targeted levels of sensitivity and specificity.
 | ||
| Fig. 7 Detection of PML–RARα fusion protein in various clinical samples and hematopoietic cancer cell lines. White blood cells (WBCs) were isolated using a point-of-care workflow from healthy donors' peripheral blood (PB, n = 5) and non-APL bone marrow samples (BM, including AML, B-cell lymphoma, CHL, MDS, and CML, and other patient bone marrow samples negative for myeloid or lymphoid malignancy; n = 11). Cell lysates from various cell lines known to be negative for PML–RARα fusion protein: HL60, U937, JeKo-1, KBM7, and SUP-B15 cell lines were also tested (n = 3). Healthy PB or APL fusion-protein negative samples gave significantly lower LFA signal than PML–RARα positive NB4 cell lysate. Error bars are one standard deviation of replicate measurements. | ||
4 Conclusion
Lateral flow assay is the most popular point-of-care (POC) immunoassay technology but its use in cancer diagnostics has been limited to the detection of serum biomarkers.24 Here, we demonstrate an LFA combined with point-of-care leukocyte isolation and on-filter lysis workflow that is, to the best of our knowledge, the first rapid assay workflow with the potential for prompt detection of PML–RARα nuclear fusion oncoprotein. We evaluated the performance of 86 pairs of commercially available antibodies recognizing different epitopes of the PML–RARα fusion protein, determined a sensitivity of ca. 11% NB4 APL leukemic cells spiked into normal peripheral blood mononuclear cells (PBMC) or in human AML (non-APL) HL-60 cells and confirmed its potential for clinically actionable use. This LFA is not intended to replace the gold standard tests for genetic confirmation of APL but rather complement the standard diagnostic workflow and facilitate the prompt initiation of therapy. We anticipate the LFA workflow to require less than 1 hour starting from bone marrow samples. LFA strips and blood filters are stable for at least one year, can be stored on site and are available to use as needed by minimally-trained personnel. Implementation of this proof-of-concept assay will require extensive study of clinical samples as well as the development of standardized sample handling and storage, which must be developed during clinical testing.Data availability
The data supporting the findings of this work are available within the article. Source data underlying the graphs presented in the main figures are included in the ESI.†Author contributions
M. C.: conceptualization, investigation, formal analysis, visualization, writing – original draft, review and editing; B. V.: resources, writing – review and editing; K. B.: resources; S. S.: resources, investigation; V. M.: resources, investigation; Z. Z.: resources, investigation; Y. Z.: resources, project administration; R. K. S.: resources, project administration; J. C. C.: conceptualization, supervision, visualization, writing – review and editing; R. C. W.: conceptualization, supervision, funding acquisition, writing – review and editing; K. K.: conceptualization, resources, supervision, visualization, project administration, funding acquisition, writing – original draft, review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Prof. Sergey Shevkoplyas for access to the hematology counter. We thank Dr. Rongwei Lei for initial investigations of the leukocyte-depletion filters, Anton Mukhamedshin for helpful discussions and Omar Rezk at StemCell Technologies. We gratefully acknowledge financial support from University of Houston (GEAR Program), Welch Foundation (Grant #E-1869-2024-0404), Ladies Leukemia League, DOD CDMRP (Grant #W81XWH-21-1-0975) and NIH/NIAID (Grant #R61AI174294).References
- S. Lehmann, A. Ravn, L. Carlsson, P. Antunovic, S. Deneberg, L. Möllgård, Å. Rangert Derolf, D. Stockelberg, U. Tidefelt, A. Wahlin, L. Wennström, M. Höglund and G. Juliusson, Leukemia, 2011, 25, 1128–1134 CrossRef CAS.
- A. P. Jillella and V. K. Kota, Blood Rev., 2018, 32, 89–95 CrossRef CAS.
- M. Yilmaz, H. Kantarjian and F. Ravandi, Blood Cancer J., 2021, 11, 123 CrossRef PubMed.
- J. K. Altman, A. Rademaker, E. Cull, B. B. Weitner, Y. Ofran, T. L. Rosenblat, A. Haidau, J. H. Park, S. L. Ram, J. M. Orsini, S. Sandhu, R. Catchatourian, S. M. Trifilio, N. G. Adel, O. Frankfurt, E. M. Stein, G. Mallios, T. Deblasio, J. G. Jurcic, S. Nimer, L. C. Peterson, H. C. Kwaan, J. M. Rowe, D. Douer and M. S. Tallman, Leuk. Res., 2013, 37, 1004–1009 CrossRef CAS PubMed.
- H. Gill, Y. Yung, H.-T. Chu, W.-Y. Au, P.-K. Yip, E. Lee, R. Yim, P. Lee, D. Cheuk, S.-Y. Ha, R. Y. Y. Leung, E. S. K. Ma, C. R. Kumana and Y.-L. Kwong, Blood Adv., 2021, 5, 2829–2838 CrossRef CAS PubMed.
- G.-B. Zhou, W.-L. Zhao, Z.-Y. Wang, S.-J. Chen and Z. Chen, PLoS Med., 2005, 2, e3–e12 CrossRef.
- M. M. Ryan, J. Adv. Pract. Oncol., 2018, 9, 178–187 Search PubMed.
- L. Cicconi and F. Lo-Coco, Ann. Oncol., 2016, 27, 1474–1481 CrossRef CAS PubMed.
- C. Nervi, F. F. Ferrara, M. Fanelli, M. R. Rippo, B. Tomassini, P. F. Ferrucci, M. Ruthardt, V. Gelmetti, C. Gambacorti-Passerini, D. Diverio, F. Grignani, P. G. Pelicci and R. Testi, Blood, 1998, 92, 2244–2251 CAS.
- A. Liquori, M. Ibañez, C. Sargas, M. Á. Sanz, E. Barragán and J. Cervera, Cancers, 2020, 12, 624 CAS.
- A. Rashidi and S. I. Fisher, Blood Cancer J., 2015, 5, e320 CAS.
- J. A. Dyck, R. P. Warrell, R. M. Evans and W. H. Miller, Blood, 1995, 86, 862–867 CAS.
- N. Chien, M. Petrasich, G. Chan, E. Theakston, A. Ruskova, N. Eaddy, T. Hawkins, L. Berkahn, R. Doocey, P. J. Browett, T. N. Green and M. L. Kalev-Zylinska, Pathology, 2019, 51, 412–420 Search PubMed.
- B. Kárai, M. Habók, G. Reményi, L. Rejtő, A. Ujfalusi, J. Kappelmayer and Z. Hevessy, Ann. Hematol., 2019, 98, 1413–1420 Search PubMed.
- O. Spinelli, A. Rambaldi, F. Rigo, P. Zanghì, E. D'Agostini, G. Amicarelli, F. Colotta, M. Divona, C. Ciardi, F. Lo Coco and G. Minnucci, Onco Targets Ther, 2014, 2, 50–58 Search PubMed.
- N. D. Dimov, L. J. Medeiros, H. M. Kantarjian, J. E. Cortes, K. S. Chang, C. E. Bueso-Ramos and F. Ravandi, Cancer, 2010, 116, 369–376 CAS.
- P. Manescu, P. Narayanan, C. Bendkowski, M. Elmi, R. Claveau, V. Pawar, B. J. Brown, M. Shaw, A. Rao and D. Fernandez-Reyes, Sci. Rep., 2023, 13, 2562 CAS.
- M. Breccia, R. Latagliata, L. Cannella, C. Minotti, G. Meloni and F. Lo-Coco, Haematologica, 2010, 95, 853–854 Search PubMed.
- E. H. Dekking, V. H. van der Velden, R. Varro, H. Wai, S. Böttcher, M. Kneba, E. Sonneveld, A. Koning, N. Boeckx, N. Van Poecke, P. Lucio, A. Mendonça, L. Sedek, T. Szczepański, T. Kalina, V. Kanderová, P. Hoogeveen, J. Flores-Montero, M. C. Chillón, A. Orfao, J. Almeida, P. Evans, M. Cullen, A. L. Noordijk, P. M. Vermeulen, M. T. de Man, E. P. Dixon, W. M. Comans-Bitter and J. J. van Dongen, Leukemia, 2012, 26, 1976–1985 CAS.
- B. Emde, H. Kreher, N. Bäumer, S. Bäumer, D. Bouwes and L. Tickenbrock, Int. J. Mol. Sci., 2020, 21, 8942 CAS.
- Y. Deng, H. Jiang, X. Li and X. Lv, Microchim. Acta, 2021, 188, 379 CAS.
- A. N. Danthanarayana, J. Brgoch and R. C. Willson, ACS Appl. Bio Mater., 2022, 5, 82–96 CAS.
- S. Natarajan, E. Saatçi and J. Joseph, J. Fluoresc., 2022, 32, 419–426 CAS.
- T. Mahmoudi, M. de la Guardia and B. Baradaran, TrAC, Trends Anal. Chem., 2020, 125, 115842 CrossRef CAS.
- NCCLS, Protocols for Determination of Limits of Detection and Limits of Quantitation; Approved Guideline, NCCLS document EP17-A [ISBN 1-56238-551-8], 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2004.
- R. Henschler, B. Rüster, A. Steimle, H. L. Hansmann, W. Walker, T. Montag and E. Seifried, Ann. Hematol., 2005, 84, 538–544 CrossRef CAS PubMed.
- M. T. Daniel, M. Koken, O. Romagne, S. Barbey, A. Bazarbachi, M. Stadler, M. C. Guillemin, L. Degos, C. Chomienne and H. de Thé, Blood, 1993, 82, 1858–1867 CrossRef CAS.
- H.-M. Mu, K.-V. Chin, J.-H. Liu, G. Lozano and K.-S. Chang, Mol. Cell. Biol., 1994, 14, 6858–6867 Search PubMed.
- J.-N. Eckardt, T. Schmittmann, S. Riechert, M. Kramer, A. S. Sulaiman, K. Sockel, F. Kroschinsky, J. Schetelig, L. Wagenführ and U. Schuler, et al. , BMC Cancer, 2022, 22, 201 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sd00357h |
| This journal is © The Royal Society of Chemistry 2025 |