
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical signal amplification for pathogen nucleic acid detection utilizing a cobalt-based DNA-binding metallo-intercalator†
Joshua
Rainbow‡
 *ab,
Emily P.
Judd-Cooper
c,
Simon J. A.
Pope
d,
Niklaas J.
Buurma
c and
Pedro
Estrela
*ab
*ab,
Emily P.
Judd-Cooper
c,
Simon J. A.
Pope
d,
Niklaas J.
Buurma
c and
Pedro
Estrela
*ab
aCentre for Bioengineering & Biomedical Technologies (CBio), University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: Joshua.rainbow@wyss.harvard.edu; P.Estrela@bath.ac.uk
bDepartment of Electronic and Electrical Engineering, University of Bath, Claverton Down, Bath BA2 7AY, UK
cPhysical Organic Chemistry Centre, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, UK
dSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, UK
First published on 27th March 2025
Abstract
This paper reports the development of a highly sensitive and rapid electrochemical biosensor for the detection of pathogen nucleic acids. The primary objective was to enhance the detection sensitivity of DNA biosensors for pathogen nucleic acids commonly found in fresh and wastewaters, the food industry, and clinical samples. This enhanced sensitivity was achieved through the addition of a [Co(GA)2(aqphen)]Cl intercalating complex to increase the electrostatic field at the sensor surface/solution interface. Voltammetric and impedance-based detection techniques were employed to characterize the intercalation and redox-active properties of the compound. Additionally, non-faradaic impedance and voltammetric methods were characterized as appropriate techniques for electrochemical detection. Implementing the [Co(GA)2(aqphen)]Cl intercalator led to increased voltammetric signal output using DPV, facilitating the rapid and sensitive detection of target DNA sequences. Notably, the [Co(GA)2(aqphen)]Cl permitted detection using non-faradaic impedance in the absence of [Fe(CN)6]3−/4−. Characterization by cyclic voltammetric measurements revealed a surface-controlled redox mechanism and reversible electrochemistry of the compound intercalated with double-stranded DNA (dsDNA). Upon binding of 1 μM target DNA and 200 μM [Co(GA)2(aqphen)]Cl, a 2250% current peak increase was achieved. This increase enabled the sensitive detection of a target DNA sequence representative of E. coli DNA in buffer with an LOD of 67.5 pM, 100-fold more sensitive than the standard unlabeled assay while maintaining assay simplicity, low cost, and quick response. The use of [Co(GA)2(aqphen)]Cl among similar compounds in DNA biosensors offers a cost-effective and sensitive method for detecting waterborne pathogens such as E. coli. This approach could significantly improve environmental monitoring and pollution control by enabling more reliable and rapid monitoring of pathogens in water sources. Additionally, it has the potential to be of great use within the food industry and in point-of-care clinical settings.
Introduction
The detection of nucleic acids is an important tool for the diagnosis of disease, and prognosis of disorders as well as applications in food safety and monitoring of pathogenic microbes within the environment. Simple and rapid detection of nucleic acid by handheld biosensor devices is currently an attractive area of research that holds many advantages over traditional techniques.1,2 At present, techniques such as polymerase chain reaction (PCR), fluorescence, mass spectrometry, microbial culturing, and micro-arrays are traditionally used for the detection of nucleic acids for a multitude of applications.Unfortunately, these techniques suffer several drawbacks that make them impractical for real-time, point-of-care (PoC), or in situ monitoring purposes.3 These disadvantages include long sample-to-answer times, expensive machinery, high reagent use, user expertise, and complex data processing. Thus, the development of PoC electrochemical biosensors for the detection of nucleic acids has been seen as an attractive alternative to solve these limitations. Recent advances in biosensor devices offer several benefits. These include quick sample analysis, lower costs due to the use of standard components, reduced need for reagents and samples, user-friendly interfaces with easy-to-understand data outputs, and increased sensitivity.4 Detection of signature DNA sequences of pathogens through monitoring duplex formation with an immobilized capture strand takes advantage of the intrinsic sequence selectivity of DNA duplex formation and should therefore be compatible with programmable detection of a wide range of pathogens without requirement for detection optimization steps for each pathogen. Similarly, the modularity of this approach also underpins potential multiplexing of detection. These features make biosensors suitable for point-of-care and on-site applications, such as in doctors' offices and public waterways.5
Electrochemical biosensors that detect the presence of nucleic acids, also known as genosensors, are devices that convert molecular nucleic acid hybridization events through a transducer into an electrical signal output. These devices work by functionalizing a transducer surface with a biological probe, e.g. DNA, and measuring the current, potential, or impedance between the electrolyte solution and the functionalized transducer surface.6 Upon binding of a target, the observable signal will increase or decrease due to changes in the biolayer and electrochemical double layer, changes in the redox behavior for the layer, charge, and size of the captured molecule, or conformational change of the DNA probe. Signal changes can be measured by both voltammetric techniques, e.g. cyclic voltammetry (CV), square wave voltammetry (SWV), or differential pulse voltammetry (DPV) as well as potentiometric or impedimetric techniques such as electrochemical impedance spectroscopy (EIS). Labeled methods are often employed with DNA-based biosensors to boost the observable signal as a result of the small size of the target and resulting small signal changes at low concentrations.7 Two labeling methods are predominantly used with DNA biosensors; direct and indirect labeling. Direct labeling involves modifying the probe or target nucleic acids by covalently attaching redox-active functional groups or nanoparticles. Alternatively, indirect labels, such as intercalators, can be utilized through their characteristic binding to nucleic acid molecules.8
DNA-binding molecules increase the observable signal by binding to nucleic acids between the base pairs (intercalators), to the charged phosphate backbone (electrostatic binders), or in the grooves of the double-stranded helix (groove binders) (Scheme 1).9,10 Intercalating molecules have been explored and developed within the pharmaceutical industry as potential therapeutics for disorders caused by genetic mutations, e.g., cancers and neurodegenerative disorders.11,12 Similarly, in sensing, non-sequence-selective intercalating molecules and complexes can be used as signal amplifiers in pathogen nucleic acid detection. The resulting pathogen detection approach uses the intrinsic sequence selectivity of DNA duplex formation and is thus, in principle, compatible with the detection of any pathogen species for which a DNA signature can be identified and does not require optimization of individual sensitizers for different pathogens. In this study, an intercalating cobalt-based compound, that inserts itself between the nitrogenous base pairs of the double-stranded DNA helix structure, was utilized. This compound is based upon a previous study on a mixed-ligand complex, [Co(GA)2(phen)], containing 1,10-phenanthroline (phen) and glycolic acid (GA).13 The structure was subsequently modified by Regan et al. to contain an extended planar ligand (aqphen = naphtho[2,3-a]dipyrido[3,2-h:2′,3′-f]phenazine-5,18-dione) with a highly conjugated anthraquinone unit to improve binding affinity using a protocol by López et al. (Scheme 2).14,15
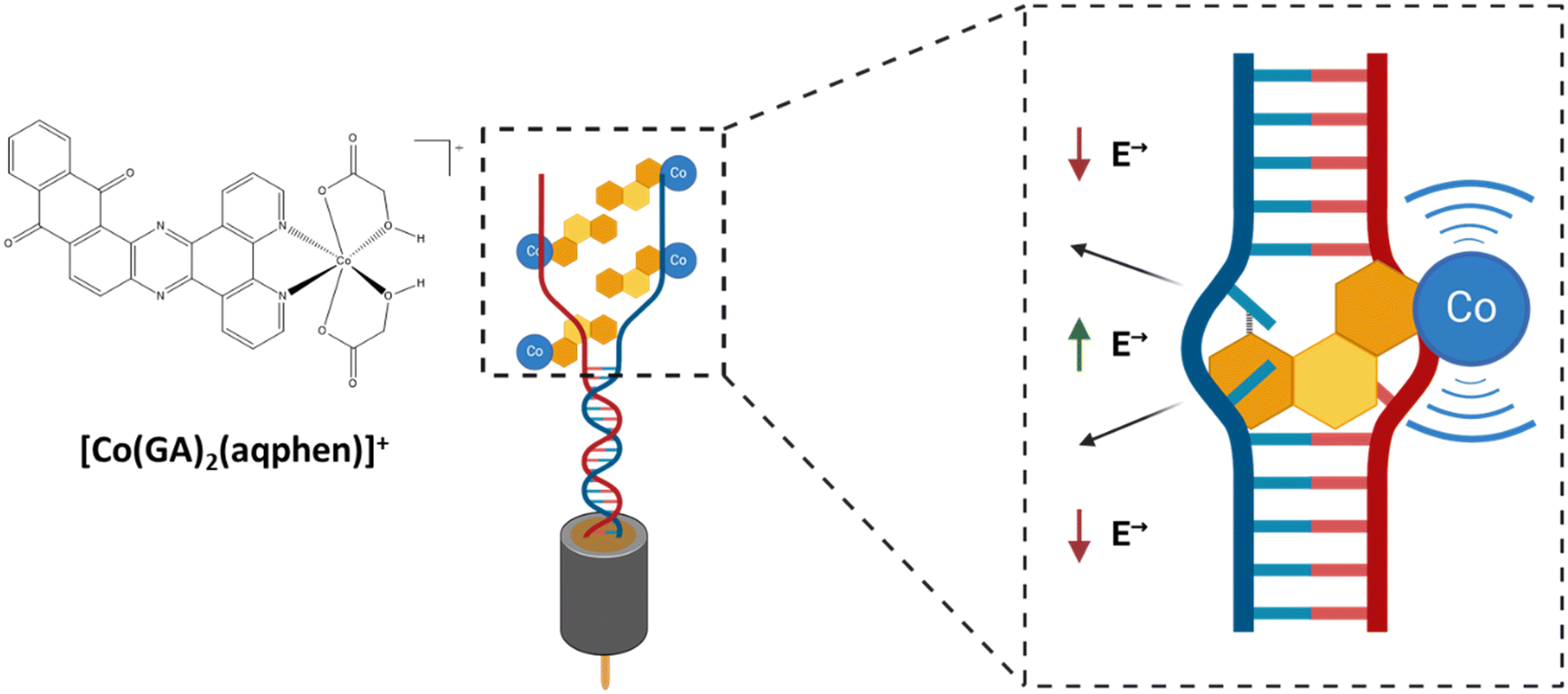 | ||
| Scheme 1 Docking of [Co(GA)2(aqphen)]Cl interacting with dsDNA between the base pairs, causing unwinding and an increase in the electrostatic field in-between DNA strands as well as adding redox-signal through the presence of the cobalt ligand. | ||
 | ||
| Scheme 2 Synthesis of the cobalt aqphen complex. (i) EtOH, heat; (ii) EtOH, 1 eq. CoCl2·6H2O, 2 eq. glycolic acid/KOH(aq). | ||
The resulting [Co(GA)2(aqphen)]Cl compound has a high potential for being effective as an intercalating compound for the electrochemical detection of nucleic acids due to its intercalative binding mode with the dsDNA helix structure as well as the presence of the redox-active cobalt ion as well as a redox-active ligand. Firstly, we have previously shown that [Co(GA)2(aqphen)]Cl binds to duplex DNA and that binding is highly likely to be between the nucleotide base pairs of the dsDNA helix causing the helix structure to unwind, increasing the associated electrical field. [Co(GA)2(aqphen)]Cl also contains the aqphen ligand which acts as a redox probe through its ability to exist in multiple oxidation states. Moreover, cobalt can transition between Co2+ and Co3+ oxidation states acting as a reducing or oxidizing agent. In electrochemical setups, cobalt can therefore mediate electron transfer between the bulk electrolyte and the transducer surface. These two mechanisms work together to modify the interfacial properties between the bulk electrolyte and transducer surface to amplify observable electrochemical signal changes. [Co(GA)2(aqphen)]Cl was previously explored to demonstrate its intercalating properties for amplification of both voltammetric and Faradaic impedance signals for the detection of a 21-base TCT-repeat oligonucleotide sequence.14 Herein, we have used an environmentally relevant sequence from E. coli which could be implemented for the detection of pathogenic species in water, clinical, and food safety samples. This study also provides insights on the improvement of compound dissolution using organic solvents to improve the long-term stability of stock solutions as well as the short-term stability of aqueous sensitizer solutions of the compound when in use. We have also characterized the electrochemical properties of the compound using additional voltammetric and non-faradaic techniques to explore further potential uses of the compound in biological field-effect transistor (BioFET) and point-of-care (PoC) devices. Finally, we have explored the effect of co-incubating the compound with target ssDNA to determine whether primary binding of the compound to ssDNA target molecules may increase overall target binding affinity and reduce assay complexity.
Experimental
Materials
HPLC-purified unmodified and thiol C6-modified DNA oligonucleotides (Table 1) were procured from Sigma-Aldrich (UK). 6-Mercapto-1-hexanol (451088), potassium phosphate monobasic solution (P8709), potassium phosphate dibasic solution (P8584), potassium sulfate (P0772), potassium hexacyanoferrate(III) (P8131), potassium hexacyanoferrate(II) trihydrate (P3289), magnesium chloride (M8266), ethylenediaminetetraacetic acid (324506), dimethyl sulfoxide (D8418), sulfuric acid (258105), hydrogen peroxide solution (H1009) and nuclease-free water (3098) were all purchased from Sigma-Aldrich (UK). Ethanol (12337163) was purchased from Fisher Scientific (UK). Gold 1.6 mm diameter working electrodes (MF-2014) were purchased from BASi Research Products (USA). Polishing pads (50318-05) and 0.05 μm alumina slurry (50368-10) were purchased from Electron Microscopy Sciences (USA). All aqueous solutions were prepared using 18.2 MΩ cm ultra-pure water (Millipore, USA) unless otherwise stated. All data values were recorded using PSTrace 5.9 (PalmSens BV, The Netherlands) with data plotted using GraphPad Prism 10 (GraphPad Software, USA). The cobalt complex [Co(GA)2(aqphen)]Cl was synthesized as previously described.14| Name | Sequence |
|---|---|
| Probe DNA (22-base) | 5′-HS-(CH2)6-TTT TTG GTC CGC TTG CTC TCG C-3′ |
| Target DNA (17-base) | 5′-GCG AGA GCA AGC GGA CC-3′ |
| Non-complementary DNA (20-base) | 5′-GCG TGA ACG TTG TAC CGC TA-3′ |
Sensor fabrication
Gold working electrodes with a radius of 1.6 mm were cleaned using a previously optimized protocol.16 Briefly, working electrodes were stripped of any previously functionalized thiols by cyclic voltammetry (CV) pre-treatment between −1.5 V and −0.5 V vs. Ag/AgCl at a scan rate of 1.0 V s−1 and a step potential of 0.01 V for 200 cycles in 0.1 M NaOH. This was followed by mechanically polishing each WE with an alumina slurry (0.05 μm) on a polishing pad for 3 minutes. Electrodes were ultrasonically cleaned in MilliQ water for 2 minutes to remove alumina residue. The electrodes were then chemically cleaned by dipping them in a hot piranha solution containing 30% hydrogen peroxide solution and concentrated sulfuric acid (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) for 15 minutes. The electrodes were then rinsed and ultrasonicated with MilliQ water for another minute. Electrochemical polishing was performed by cycling working electrodes between −0.2 V and 1.5 V vs. Ag/AgCl at a scan rate of 0.1 V s−1 and step potential of 0.01 V for 25 cycles in 0.1 M H2SO4 aqueous solution. The residual gold oxide layer was then immediately electrochemically stripped by running 10 cycles from 0.2 V to 0.8 V vs. Ag/AgCl at the same scan rate and step potential in 0.1 M H2SO4. Electrodes were then placed in absolute ethanol for 20 minutes to chemically reduce any remaining gold oxide formed during the cleaning process.16
:3) for 15 minutes. The electrodes were then rinsed and ultrasonicated with MilliQ water for another minute. Electrochemical polishing was performed by cycling working electrodes between −0.2 V and 1.5 V vs. Ag/AgCl at a scan rate of 0.1 V s−1 and step potential of 0.01 V for 25 cycles in 0.1 M H2SO4 aqueous solution. The residual gold oxide layer was then immediately electrochemically stripped by running 10 cycles from 0.2 V to 0.8 V vs. Ag/AgCl at the same scan rate and step potential in 0.1 M H2SO4. Electrodes were then placed in absolute ethanol for 20 minutes to chemically reduce any remaining gold oxide formed during the cleaning process.16
Each working electrode was then dried under a stream of nitrogen and exposed to 150 μL of a solution containing 1 M thiol-terminated probe DNA and 6-mercapto-1-hexanol (MCH) (1:10) in a humidity chamber for ≥16 hours at 4 °C. Probe ssDNA was modified with a thiol-C6 group on the 5′ end and had a 22-base sequence of 5′-TTT TTG GTC CGC TTG CTC TCG C-3′ from the genome of E. coli O157:H7 serotype. Five thymine bases were added at the 5′ end to increase the distance of the probe DNA from the electrode surface. The immobilization solution contained 0.8 M phosphate buffer (PB) + 1.0 M NaCl + 5 mM MgCl2 + 1 mM ethylene diamine tetra acetic acid (EDTA), pH 7.0. After initial self-assembled monolayer (SAM) formation, electrodes were rinsed with a wash buffer containing 50 mM PB + 100 mM K2SO4 + 10 mM EDTA, pH 7.0, to remove residual Mg2+ ions. To ensure complete coverage of the gold electrode surface and reduce the chance of pinholes, electrodes were backfilled with a solution of 1 mM MCH in MilliQ water for 1 hour (Scheme 3). Finally, electrodes were rinsed with MilliQ water and placed in 50 mM PB + 100 mM K2SO4, pH 7.0, for 1.5 hours to ensure SAM stability.
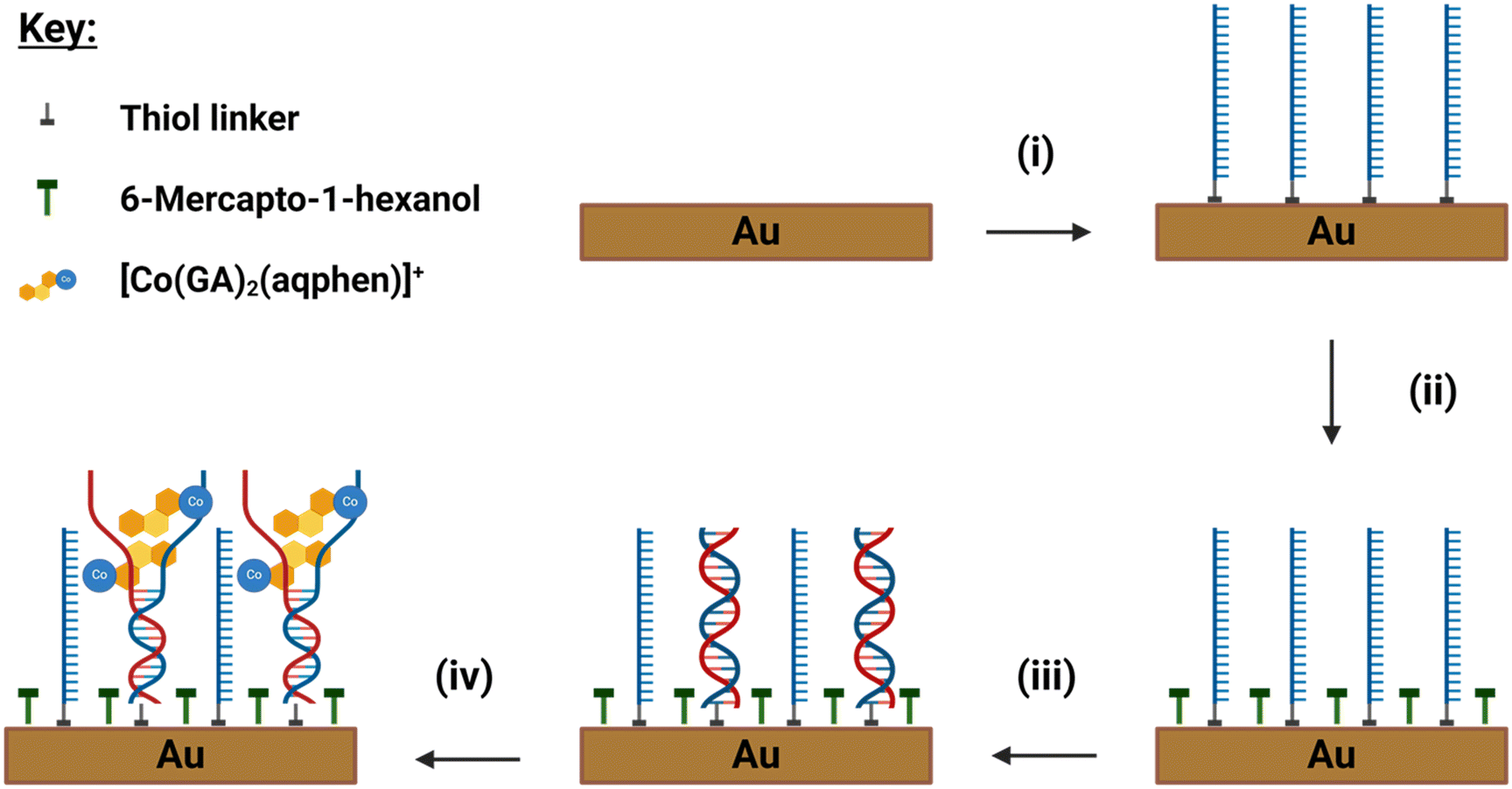 | ||
| Scheme 3 Design and fabrication of the DNA sensor and assay steps for detection of E. coli ssDNA. (i) Incubation in a 1:10 solution of HS-(CH)6-ssDNA and MCH overnight. (ii) Backfilling with MCH to fill pinholes. (iii) Hybridization with target ssDNA to form surface-bound dsDNA. (iv) [Co(GA)2(aqphen)]Cl was incubated and intercalated with dsDNA, causing unwinding of the dsDNA helix structure. | ||
Sample preparation and incubation
Complementary target ssDNA was incubated on the functionalized electrode surface with a 17-base sequence of 5′-GCG AGA GCA AGC GGA CC-3′. Target DNA was prepared as aliquots from a lyophilized state in nuclease-free water (Sigma-Aldrich, UK) at a stock concentration of 100 μM and serial diluted to working concentrations in 50 mM PB + 100 mM K2SO4, pH 7.0. Once SAM stability was achieved, electrodes were incubated with 100 μL of target ssDNA solution (100 pM–1 μM) for 1 hour at ambient room temperature (RT). Electrodes were then rinsed with 50 mM PB + 100 mM K2SO4, pH 7.0 for 2–3 s.Once target DNA had hybridized with the surface-bound probe DNA, electrodes were incubated with 100 μL of 200 μM [Co(GA)2(aqphen)]Cl at ambient RT. For sequential incubation of the cobalt compound after DNA hybridization, a stock of [Co(GA)2(aqphen)]Cl was dissolved in DMSO to a concentration of 1 mM by ultrasonication for 2 hours at 70 °C. Stock [Co(GA)2(aqphen)]Cl was then diluted to 200 μM in 50 mM PB + 100 mM K2SO4, pH 7.0, and vortexed thoroughly followed by incubation on the electrode for 30 minutes. For co-incubation of the ssDNA target and [Co(GA)2(aqphen)]Cl, 100 μL of solution containing the required concentration of ssDNA and 200 μM of [Co(GA)2(aqphen)]Cl was prepared in 50 mM PB and 100 mM K2SO4, pH 7.0, and incubated on electrodes for 1 hour at ambient RT (Scheme 3).
Measurement setup
Electrochemical impedance spectroscopy (EIS), differential pulse voltammetry (DPV), and cyclic voltammetry (CV) were performed on a Palmsens4 potentiostat using the PSTrace 5.9 software (PalmSens BV, The Netherlands). A three-electrode setup was used with a 1.6 mm gold working electrode (WE) (BASi, USA), a platinum wire counter electrode (CE) (ALS Instruments, Japan), and an Ag/AgCl (3 M KCl) reference electrode (RE) (BASi, USA) placed into a salt bridge containing 50 mM PB + 100 mM K2SO4, pH 7.0. All electrodes were placed into 8 mL of buffer solution within a 10 mL beaker with a Teflon cap containing holes for the electrode to fit firmly within.For faradaic measurements, the electrochemical impedance spectrum was measured in a solution of 2 mM K[Fe(CN)6]4− + 2 mM K[Fe(CN)6]3− in 50 mM PB + 100 mM K2SO4, pH 7.0. The impedance spectrum was measured over the frequency range of 100 kHz to 100 MHz, with a 10 mV AC voltage superimposed on a DC bias of 0.2 V, corresponding to the bias potential of the redox couple. For non-faradaic measurements determining capacitance and open circuit potential (OCP), experiments were carried out in 100 mM PB. The impedance spectrum was measured over a frequency range of 100 kHz to 100 MHz, with a 10 mV AC voltage superimposed on a DC bias of 0.0 V vs. OCP.
For DPV measurements, electrodes were placed in 100 mM PB, and DPV scanned between −0.257 V and 0.143 V vs. Ag/AgCl with a scan rate of 0.05 V s−1, step potential of 0.005 V, pulse potential of 0.05 V, and pulse time of 0.05 s. During CV, electrodes were scanned between −0.5 V and 0.5 V in 100 mM PB with a scan rate of 0.2 V s−1 and step potential of 0.01 V.
Results & discussion
Optimization of [Co(GA)2(aqphen)]Cl dissolution
Due to the hydrophobic, aromatic structure of the aqphen ligand, [Co(GA)2(aqphen)]Cl does not dissolve well in water-based buffers. These characteristics cause the compound to form a suspension rather than a fully dissolved solute within these buffers (Fig. S1†). When preparing a solution of [Co(GA)2(aqphen)]Cl in MilliQ water, it was observed that the compound only partially dissolved. Only upon dilution to concentrations lower than the apparent solubility limit of 600 μM did the compound appear to fully dissolve. Unfortunately, however, the compound aggregated immediately when this stock solution in MilliQ water was further diluted into a high ionic strength buffer. The observation of aggregation in high ionic strength buffers is in line with the expectation that this compound will have a solubility product, meaning that the apparent solubility limit strongly depends on the nature and concentration of the anions in the buffer. Whereas the thermodynamic solubility limit of the complex in different buffers depends on the structure of the complex and the formulation of the buffer, the kinetics of precipitation may be slowed down by the judicious selection of a method for the preparation of the solution. We selected DMSO as a cosolvent to explore because its low volatility allows solution composition to remain the same over extended periods of time. In addition, a small amount of DMSO is known to not significantly affect base pairing. To increase solution stability, [Co(GA)2(aqphen)]Cl was dissolved first in dimethyl sulfoxide (DMSO) before diluting in the high ionic strength buffers used for DNA hybridization. [Co(GA)2(aqphen)]Cl was therefore dissolved in a 1 mM stock concentration in DMSO (Fig. S2†). Two dilution pathways were tested, the first being dilution in DMSO to 600 μM followed by a final dilution step to 200 μM into the high ionic strength buffer (50 mM PB + 100 mM K2SO4.) In this dilution pathway, [Co(GA)2(aqphen)]Cl diluted well to 600 μM in DMSO but then aggregated when diluted to 200 μM in 50 mM PB + 100 mM K2SO4. After only 2 minutes the compound had completely aggregated and settled. It was observed that by diluting the stock solution of 1 mM [Co(GA)2(aqphen)]Cl directly to 200 μM in a high ionic strength buffer, the compound was more readily dissolved and formed a stable solution for ≥4 hours. It is important to note that the stock solution of 1 mM [Co(GA)2(aqphen)]Cl in DMSO remained stable for ≥3 months.Impedance response to [Co(GA)2(aqphen)]Cl intercalation
To determine the limit of detection (LOD) of the developed sensor without the assistance of the cobalt compound, electrodes were immobilized with probe DNA, and the electrochemical impedance spectra were measured upon binding of five concentrations of fully complementary target DNA from 0 pM to 1 μM (Fig. S3†). The LOD for the detection of E. coli target ssDNA was calculated to be LOD = 3.3σ/slope = 9.73 nM. This higher detection limit is expected when utilizing faradaic EIS in a label-free approach without the use of pre-amplification steps or any modification of the transducer surface such as with conductive nanoparticles.A typical Nyquist plot is shown in Fig. 1 demonstrating the percentage change in charge transfer resistance (Rct) observed from the hybridization of the probe and target ssDNA as well as the change observed from the intercalation of the [Co(GA)2(aqphen)]Cl compound. An increase of 19.2% was observed upon binding of 1 μM complementary ssDNA. The increase in Rct observed after incubation with [Co(GA)2(aqphen)]Cl is a result of the unwinding of the DNA double helix causing an increase in electrostatic resistance to [Fe(CN)6]3−/4−. An increase of 9.66% in Rct was seen upon binding of 200 μM [Co(GA)2(aqphen)]Cl to the dsDNA. This change upon binding of [Co(GA)2(aqphen)]Cl was relatively small, likely due to the charge screening effect of the negatively charged dsDNA with the positively charged [Co(GA)2(aqphen)]+. Thus, it was expected that measuring the binding of [Co(GA)2(aqphen)]Cl to the dsDNA using non-faradaic EIS measurements, i.e. capacitance or OCP in the absence of [Fe(CN)6]3−/4−, may be beneficial to specifically measure the presence of [Co(GA)2(aqphen)]Cl.
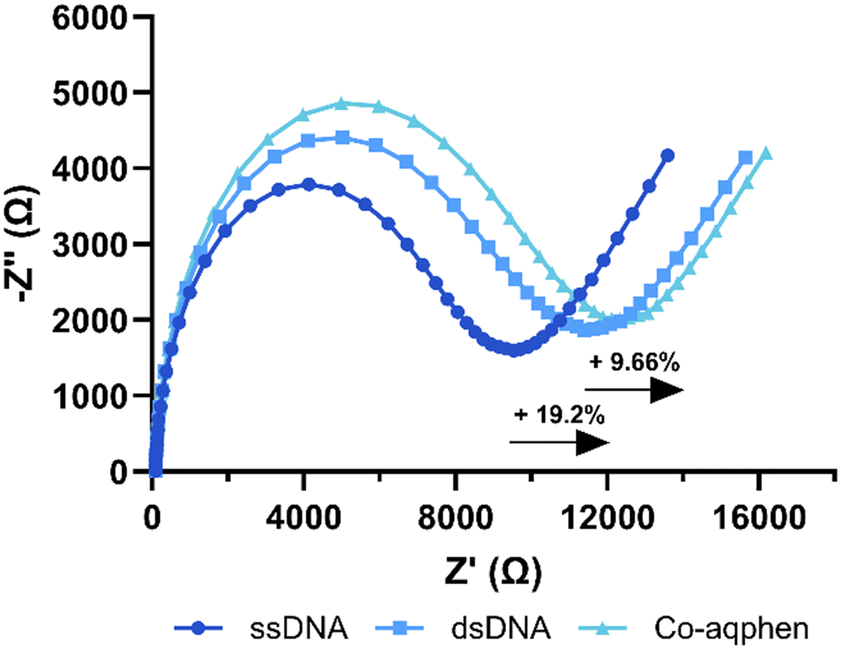 | ||
| Fig. 1 A typical Nyquist plot showing faradaic impedance response to DNA hybridization and [Co(GA)2(aqphen)]Cl intercalation in 50 mM PB + 100 mM K2SO4 + 2 mM [Fe(CN)6]3−/4−. | ||
To measure the redox-active properties of [Co(GA)2(aqphen)]Cl, measurements using EIS were carried out in 100 mM phosphate buffer electrolyte without [Fe(CN)6]3−/4−. Due to the absence of [Fe(CN)6]3−/4−, measurements were carried out using a DC bias of 0 V versus the open circuit potential (OCP). This was done so that effects on the capacitance of the electrical double-layer (Cdl) and OCP could be assessed. OCP values were derived from the PSTrace 5.9 software and Cdl values were obtained by fitting the EIS spectra with a Randles circuit. Using the same surface chemistry mentioned previously (Scheme 3), measurements were taken after incubating with 1 μM complementary target DNA for 1 hour and again after 30 minutes of [Co(GA)2(aqphen)]Cl binding. After incubation with the target DNA, a small increase in Cdl of 0.06 nF (SD: ±7) was observed (Fig. 2), compared to a large increase of 45 nF (SD: ±1) with binding of [Co(GA)2(aqphen)]Cl. Fig. 2 also displays the effect of [Co(GA)2(aqphen)]Cl binding to dsDNA on the OCP. After the binding of target DNA, a decrease of 15 mV (SD ± 42) occurred. Upon measuring the impedance in the absence of a surface-bound intercalator or redox solution e.g. [Fe(CN)6]3−/4−, the OCP was observed to fluctuate significantly. This is due to the lack of well-defined equilibrium at the solution-electrode interface in the absence of redox-active molecules. After incubating with the redox-active [Co(GA)2(aqphen)]Cl, a large decrease of 154 mV (SD: ±8) was observed, which corresponded to the binding of the intercalator. The large reduction in variability suggests that the redox-active properties of [Co(GA)2(aqphen)]Cl increased the stability of the potential in the circuit.
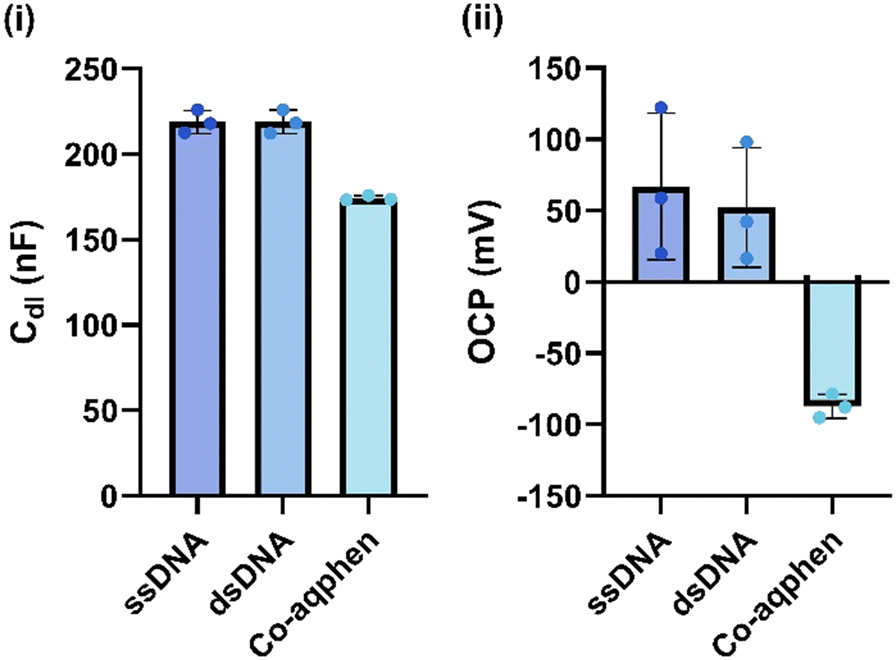 | ||
| Fig. 2 The responses of DNA hybridization and [Co(GA)2(aqphen)]Cl intercalation on non-faradaic EIS measurements in 100 mM PB. (i) Double-layer capacitance response. (ii) Response of the open circuit potential (OCP) (n ≥ 3). | ||
Voltammetric response to [Co(GA)2(aqphen)]Cl intercalation
The compound [Co(GA)2(aqphen)]Cl displays clear redox activity, allowing for electrochemical investigation using techniques such as cyclic voltammetry (CV), differential pulse voltammetry (DPV), or square wave voltammetry (SWV). Here we have utilized CV to assess the reversibility of this compound's redox reaction and DPV to measure the binding response to dsDNA. Fig. 3 shows a typical cyclic voltammogram of probe ssDNA immobilized onto the electrode surface with 1 μM target DNA hybridized and 200 μM [Co(GA)2(aqphen)]Cl bound to the dsDNA helix. Voltammetric scans of the surface-bound [Co(GA)2(aqphen)]Cl in 100 mM PB at a scan rate of 0.2 V s−1 gave peak values of 0.04 V and −0.16 V, for the anodic peak potential (Epa) and cathodic peak potential (Epc), respectively, which might be attributed to the Co(II)/Co(III) redox couple. Using these values, peak-to-peak separation (ΔEp) was calculated as 200 mV, which suggests that the redox reaction of [Co(GA)2(aqphen)]Cl is likely to be a quasi-reversible reaction with a complex mechanism that may involve steps such as proton transfer.17,18 The half-wave potential (E½) was then calculated as E½ = (Epa + Epc) /2 = −0.06 V. This half-wave potential value can be used as the bias potential (E0′) in impedance experiments.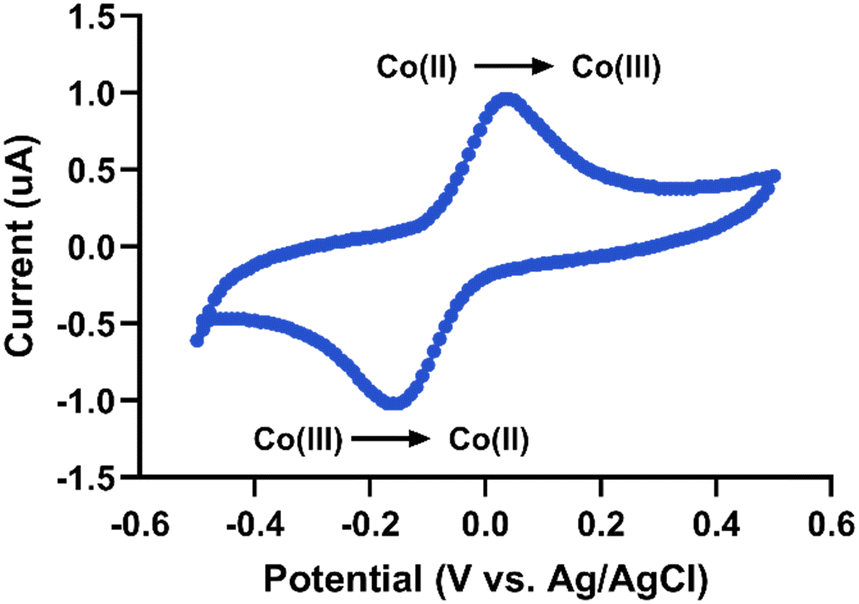 | ||
| Fig. 3 Cyclic voltammogram of dsDNA-bound [Co(GA)2(aqphen)]Cl in 100 mM PB at a scan rate of 0.2 V s−1vs. Ag/AgCl. | ||
To assess the reversibility of the electrochemically generated products of [Co(GA)2(aqphen)]Cl, the current ratio was calculated using the current peak values for both oxidation (0.96 μA) and reduction (−1.02 μA) of the compound:
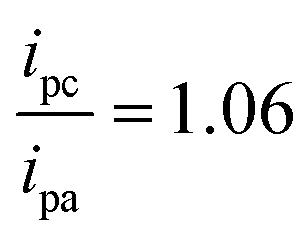
Fig. S5† shows anodic and cathodic peak current data for CV measurements at varying scan rates (10 mV s−1–1 V s−1) in the absence or presence of the redox couple [Fe(CN)6]3−/4−. By comparing the peak current data to the scan rate or the square root of the scan rate, it is possible to determine the type of redox process occurring at the electrode surface. When the peak current is linearly proportional to the scan rate, the redox process is surface-controlled. However, if the peak current is linearly proportional to the square root of the scan rate, then the redox process is diffusion-based. Fig. S5i and ii† show cyclic voltammetry data where [Co(GA)2(aqphen)]Cl has intercalated with surface-bound dsDNA. The peak current for surface-bound [Co(GA)2(aqphen)]Cl displays a linear relationship with scan rate (R2 = 0.99 (Ipa), 0.99 (Ipc)) over the square root of scan rate (R2 = 0.97 (Ipa), 0.97 (Ipc)). This suggests that the redox process associated with the surface-bound [Co(GA)2(aqphen)]Cl follows a surface-controlled process as expected. However, when the same measurements were carried out in the presence of [Fe(CN)6]3−/4−, the redox process followed a diffusion-based process (Fig. S4†).
When [Co(GA)2(aqphen)]Cl was incubated with dsDNA, a clear peak was observed at −0.056 V versus an Ag/AgCl reference electrode (Fig. 5). The current peak values were 0.017 μA for ssDNA, 0.175 μA for ssDNA + 200 μM [Co(GA)2(aqphen)]+, and 0.462 μA for 1 μM dsDNA + 200 μM [Co(GA)2(aqphen)]+. The current value increase of 0.461 μA for target DNA + [Co(GA)2(aqphen)]Cl was significantly larger than the addition of [Co(GA)2(aqphen)]Cl to ssDNA of 0.174 μA. No peak was observed in the absence of [Co(GA)2(aqphen)]Cl.
Co-incubation of target DNA with [Co(GA)2(aqphen)]Cl
In the previous study by Regan et al., it was hypothesized that the positive charge of [Co(GA)2(aqphen)]Cl may screen the negative charges of the phosphates on the DNA backbone.14 If so, masking the negative charges may increase the binding affinity of the target DNA to the surface-functionalized probe DNA by reducing the electrostatic repulsion between DNA strands caused by the crowding of surface-bound DNA. To test this hypothesis the same surface chemistry with ssDNA and MCH was utilized, however, instead of incubating the target DNA and compound separately, measurements were taken after co-incubation with 1 μM complementary target DNA and 200 μM of [Co(GA)2(aqphen)]Cl in 100 mM PB for 1 hour. Co-incubation further reduced the assay time by 30 minutes and reduced the assay complexity for future microfluidic and automated integration. After co-incubation, an increase in Cdl of 54 nF (SD: ±8 nF) was observed, 20.88% higher than incubating the target DNA and [Co(GA)2(aqphen)]Cl separately (Fig. 4i). A decrease in OCP of 153 mV (SD: ±3 mV) was likewise observed (Fig. 4ii), comparable with the change detected when incubating separately. | ||
| Fig. 4 Data for co-incubation of target DNA and [Co(GA)2(aqphen)]Cl intercalator showing the response of simultaneous hybridization and intercalation on (i) capacitance, and (ii) OCP in 100 mM PB (n ≥ 3). | ||
 | ||
| Fig. 5 Differential pulse voltammograms after co-incubation with target DNA and [Co(GA)2(aqphen)]Cl in 100 mM PB at a scan rate of 50 mV s−1vs. Ag/AgCl. | ||
The current values observed for the DPV measurements were 0.02 μA (SD: ±0.03) for ssDNA and 0.5 μA (SD: ±0.04) for dsDNA + [Co(GA)2(aqphen)]Cl (Fig. 5). A current value increase of 0.45 μA was comparable to the incubation of target complementary DNA and [Co(GA)2(aqphen)]Cl separately, suggesting co-incubation was a viable alternative to incubating separately. DPV was determined to be the best technique for measuring the intercalation of [Co(GA)2(aqphen)]Cl for a dose–response of target DNA. This is due to the larger potential dynamic signal change observed between the absence and presence of the compound. While both OCP and capacitance showed a large change upon going from absence to binding of the compound, there was no significant difference observed between different concentrations of target DNA.
A dose–response experiment was then carried out to determine the detection limit of the assay; simultaneous incubation of the target DNA with [Co(GA)2(aqphen)]Cl was combined with DPV detection due to the larger potential dynamic signal change. Target DNA was serially diluted in 50 mM PB containing 100 mM K2SO4 (0.1–1000 nM) with 200 μM [Co(GA)2(aqphen)]Cl and incubated onto functionalized electrodes for 1 hour at ambient RT. The dose–response curve revealed that 100 pM target DNA gave a significantly larger current response than the negative control of 0 nM + [Co(GA)2(aqphen)]Cl (P < 0.0001, Table S3†). However, the assay exhibited a saturation effect at around 10 nM. The calibration curve follows a sigmoidal (4PL) logarithmic curve (Fig. 6):

 | ||
| Fig. 6 Differential pulse voltammetry current response to the binding of ssDNA and [Co(GA)2(aqphen)]Cl. (i) Dose–response curve for detection between 0.1–1000 nM E. coli ssDNA in 100 mM PB (n ≥ 3). (ii) The results of an ordinary one-way ANOVA related to the dose–response of E. coli ssDNA between 0.1–1000 nM (n ≥ 3). | ||
Conclusions
In this study, the improvement of dissolution was explored for the redox-active intercalator, [Co(GA)2(aqphen)]Cl. It was observed that by dissolving the compound in water, a low dissolution capacity was achieved. However, upon dissolving the complex first in the organic solvent DMSO, a higher dissolution rate and higher solubility were achieved, allowing usefully concentrated stock solutions with long-term stability. A kinetically stable aqueous solution was attained in the high ionic strength buffer required for surface in vitro DNA hybridization. To test the efficacy of [Co(GA)2(aqphen)]Cl to increase the sensitivity of DNA detection, a biosensor was fabricated using simple thiol-based surface chemistry. To measure the inherent redox-active properties of [Co(GA)2(aqphen)]Cl, non-faradaic EIS was used to observe changes in Cdl and OCP. Upon binding of [Co(GA)2(aqphen)]Cl, with 1 μM of target DNA, an increase of 44.53 nF ±1.48 was achieved compared with 0.06 nF ±6.87 after binding of 1 μM of target DNA alone. Additionally, a decrease in OCP of 14.7 mV ±41.9 was observed compared to 154.1 mV ±8.3 after binding of [Co(GA)2(aqphen)]Cl. The standard deviation values suggest in both cases that upon binding of the redox-active intercalator, stability in both capacitance and OCP values was increased due to the strong redox characteristics of the compound.To characterize the intercalating properties of [Co(GA)2(aqphen)]Cl, CV and DPV were utilized to determine the electrochemical stability and binding kinetics to the surface-bound dsDNA. The cyclic voltammogram for the surface-bound dsDNA after intercalation of [Co(GA)2(aqphen)]Cl gave a ΔEp value of 200 mV, suggesting that the redox reaction is quasi-reversible, likely due to the complexity of the redox mechanism.
A current ratio of 1.06 was achieved, suggesting a highly stable electrochemically generated product with highly reversible electron transfer. DPV measurements demonstrated a significant 257% increase in the current peak upon binding of [Co(GA)2(aqphen)]Cl, compared with only a 7.69% increase after target DNA hybridization. Finally, we have shown that by incubating target DNA simultaneously with [Co(GA)2(aqphen)]Cl, signal changes are 20.88% higher with capacitance and comparable within OCP and DPV measurements. This implies that co-incubation of the compound with the target DNA sample is a viable method for reducing the complexity, and time required for labeled assays in electrochemical DNA-sensing platforms. Finally, a dose–response curve of the target DNA co-incubated with [Co(GA)2(aqphen)]Cl demonstrated a LOD of 67.5 pM for the detection of E. coli in buffer samples, i.e. an over 100-fold increase in assay sensitivity.
We hypothesize that the screening effect of [Co(GA)2(aqphen)]Cl with negatively charged DNA provides the high sensitivity observed. However, it is as yet unexplained why the upper dynamic range of the assay becomes affected by increasing concentrations of compound binding. It is our current theory that the positive charge of [Co(GA)2(aqphen)]Cl may work to negate the binding of further compounds with increasing DNA concentrations. One way of negating this effect may involve adding a mild biological detergent such as TWEEN-20 to the binding and washing buffers to mitigate non-specific binding of [Co(GA)2(aqphen)]Cl to reduce the saturation of signal at higher target concentrations. Further modifications of [Co(GA)2(aqphen)]Cl can be explored to increase dissolution further within biologically compatible buffers as well as to increase the strength of the redox signal from the Co-based ligand. Further study of [Co(GA)2(aqphen)]Cl would be combined in a sensor that integrates isothermal amplification of target nucleic acid samples within a microfluidic device. This could enable ultrasensitive detection of nucleic acids in complex clinical and environmental samples such as blood, saliva, and both fresh and wastewater.
Data availability
Data supporting this article have been included as part of the ESI.† Further data are available upon request from the authors.Author contributions
JR: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft; EJC: resources; SP: resources, writing – review & editing; NB: conceptualization, resources, supervision, writing – review & editing; PE: conceptualization, funding acquisition, supervision, writing – review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
JR acknowledges funding through the UK Natural Environment Research Council (NERC) GW4 FRESH CDT. EJC acknowledges funding through the UK Engineering and Physical Science Research Council (EPSRC) DTP interdisciplinary doctoral training hub in Biosensors and Diagnostics (EP/T517951/1).References
- N. Kumar, et al., Emerging biosensor platforms for the assessment of water-borne pathogens, Analyst, 2018, 143(2), 359–373 RSC.
- J. Vidic and M. Manzano, Electrochemical biosensors for rapid pathogen detection, Curr. Opin. Electrochem., 2021, 29, 100750 CrossRef.
- K. Koo, et al., Enabling miniaturised personalised diagnostics: from lab-on-a-chip to lab-in-a-drop, Lab Chip, 2017, 17(19), 3200–3220 RSC.
- S. Schumacher, et al., Highly-integrated lab-on-chip system for point-of-care multiparameter analysis, Lab Chip, 2012, 12(3), 464–473 Search PubMed.
- S. Arshavsky-Graham and E. Segal, Lab-on-a-chip devices for point-of-care medical diagnostics, Microfluidics in Biotechnology, 2020, pp. 247–265 Search PubMed.
- N. Bhalla, et al., Introduction to biosensors, Essays Biochem., 2016, 60(1), 1–8 Search PubMed.
- X. Zhou, et al., Recent advances in signal amplification to improve electrochemical biosensing for infectious diseases, Front. Chem., 2022, 10, 911678 CrossRef PubMed.
- A. A. Almaqwashi, et al., Mechanisms of small molecule–DNA interactions probed by single-molecule force spectroscopy, Nucleic Acids Res., 2016, 44(9), 3971–3988 Search PubMed.
- N. Buurma and I. Haq, Calorimetric and spectroscopic studies of Hoechst 33258: self-association and binding to non-cognate DNA, J. Mol. Biol., 2008, 381(3), 607–621 CAS.
- H. Liu and P. Sadler, Metal complexes as DNA intercalators, Acc. Chem. Res., 2011, 44(5), 349–359 CAS.
- M. F. Brana, et al., Intercalators as anticancer drugs, Curr. Pharm. Des., 2001, 7(17), 1745–1780 CrossRef CAS PubMed.
- M. Khalifa, et al., Topo II inhibition and DNA intercalation by new phthalazine-based derivatives as potent anticancer agents: design, synthesis, anti-proliferative, docking, and in vivo studies, J. Enzyme Inhib. Med. Chem., 2022, 37(1), 299–314 CrossRef CAS PubMed.
- H. Lin, et al., Crystal structure and DNA binding studies of a cobalt (II) complex containing mixed-ligands of 1, 10-phenanthroline and glycollic acid, Chin. Chem. Lett., 2011, 22(8), 969–972 CAS.
- E. Regan, et al., A novel cobalt complex for enhancing amperometric and impedimetric DNA detection, Electrochim. Acta, 2014, 128, 10–15 CrossRef CAS.
- B. R. López, et al., Synthesis of a new phenanthroline derived ligand with acceptor properties, Tetrahedron Lett., 1996, 37(31), 5437–5440 Search PubMed.
- J. Tkac and J. Davis, An optimised electrode pre-treatment for SAM formation on polycrystalline gold, J. Electroanal. Chem., 2008, 621(1), 117–120 Search PubMed.
- N. Elgrishi, et al., A practical beginner's guide to cyclic voltammetry, J. Chem. Educ., 2018, 95(2), 197–206 CrossRef.
- D. Guziejewski, et al., Reversible and quasireversible electron transfer under conditions of differential square-wave voltammetry, J. Phys. Chem. C, 2022, 126(12), 5584–5591 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sd00322e |
| ‡ Current address: Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, MA, USA. |
| This journal is © The Royal Society of Chemistry 2025 |