DOI:
10.1039/D5SC06640A
(Edge Article)
Chem. Sci., 2025,
16, 22147-22159
Self-doped and planar nitrogen-bay-functionalized perylene diimides: unravelling structure–property relationships
Received
28th August 2025
, Accepted 15th October 2025
First published on 16th October 2025
Abstract
Bay-functionalization of perylene diimides (PDIs) offers a strategy for modulating electronic properties to achieve optimal n-type charge transport character, but can result in core twisting and reduced planarity, leading to compromised π-conjugation and disruption of tight π–π stacking in the solid state. Herein, we report the synthesis and characterization of two new nitrogen-bay-functionalized N-annulated PDIs, where N-annulation effectively enforces planarity of the polycyclic aromatic core thus preserving π-conjugation even with bulky bay substituents present. Both compounds exhibit intramolecular charge transfer character, tunable optical and electrochemical properties, and strong paramagnetic behaviour without external dopants present, as confirmed by UV-visible spectroscopy, cyclic voltammetry, and solid-state electron paramagnetic resonance, respectively. Guided by Hansen solubility parameters, we formulated green solvent blends that enabled concentrated ink formulations (10 mg mL−1) for large-area film deposition via slot-die coating. These findings highlight the promise of bay-substituted, N-annulated PDIs as self-doped, solution-processable materials ripe for applications in organic electronics.
Introduction
Perylene diimides (PDIs) are π-conjugated chromophores that are commonly used to derive n-type charge transport in thin-film form, with established applications in organic light-emitting diodes,1,2 photovoltaics,3–5 field-effect transistors,6–8 and are now emerging in photocatalysis.9–11 The rigid perylene core promotes tight π-stacking in the film, enabling electron mobilities comparable to or exceeding fullerene benchmarks.12 Numerous PDI derivatives have been synthesized with functional modifications aimed at improving solubility, electronic properties, and film morphology. In particular, imide substitution with various derivatives is often used to enhance solubility and tune film morphology,8,13,14 whereas bay-position functionalization allows for modulation of the frontier molecular orbital (FMO) energies and control over solid-state packing.2,15,16 Furthermore, owing to the formation of highly stable radical anions upon reduction, the incorporation of extrinsic dopants into PDI-based films is a well-established strategy that enhances device performance and is widely employed across organic electronic systems.17–19
To impart enhanced performance in organic electronic devices as a whole, a growing body of work now leverages the concept of “self-doping” in PDIs, which has enabled enhanced carrier concentrations without the miscibility and morphology issues associated with extrinsic dopants in the solid state.20,21 Notable compounds, such as PDINO and PDINN (Fig. 1a), exploit pendant amino or nitroxide motifs on the imide position to generate strong paramagnetic signatures detectable by electron paramagnetic resonance (EPR) spectroscopy, and when employed as cathode interlayers in single-junction organic solar cells device efficiencies exceeded 17%.22 These self-doped systems work by inducing electron transfer from the nitrogen-based functional groups to the perylene core thereby increasing electron density in the π-conjugated backbone that is responsible for charge transport, a process which is enhanced upon photo-excitation.23 Most reports confine self-doping functional groups to the imide position while bay-functionalized analogues remain scarce, which could be because bay-substituents twist the perylene core and attenuate intermolecular π-overlap in the solid state.24 The study of two bis(n-octylamino) PDIs, one being (C8H17NH)2-PDI (Fig. 1b), by Ahrens et al. illustrates this trade-off, observing stable radical cations upon electrochemical oxidation and strong intramolecular transfer (ICT) character in optoelectronic spectra of the neutral compounds, but with compromised planarity as the “core twist” of these compounds about the two fused central naphthalene units are as high as 21°.25 More recently, a bis-PDI macrocycle, termed “the pink box”, reported core twisting of 21° when bay-substituted with triazoles linkers.26 Therefore, a balance exists between introducing effective bay-dopant functional groups and preserving molecular planarity, which is essential for maintaining core π-conjugation and enabling efficient charge transport.
 |
| | Fig. 1 Design strategies for introducing nitrogen-functionalities onto perylene diimide (PDI) frameworks: (a) self-doping via pendant electron-donating groups (e.g., nitroxide or amine) on the imide position as seen in PDINN and PDINO,22 (b) bay-functionalization with electron-rich amines to afford (C8H17NH)2-PDI and PDIN-H, (c) this work: bay-substituted, N-annulated PDIs (NO2-PDIN-H and NH2-PDIN-H) combine structural rigidity and acidic pyrrolic N–H handles. | |
To circumvent torsion and loss of intermolecular π-overlap caused by bay-functionalization of nitrogen-based dopants, we have identified N-annulation as a structural strategy for enforcing planarity while supplying a mildly acidic pyrrolic N–H handle ripe for further derivatization. Although N-annulated PDIs, such as PDIN-H (Fig. 1b) & other closely related derivatives, have been explored as non-fullerene acceptors,27 cathode interlayers,28–30 light emitters,31,32 and as photocatalysts,33,34 to the best of our knowledge investigation into self-doped bay derivatives have not been considered. Herein, we present a nitro-bay-substituted N-annulated perylene diimide (NO2-PDIN-H; Fig. 1c) and an amino-bay-substituted N-annulated perylene diimide (NH2-PDIN-H; Fig. 1c) as new PDIs that (1) retain near-planar cores (twist 1.9° for NO2-PDIN-H, 7.3° for NH2-PDIN-H), (2) display characteristics of strong ICT in UV-visible & photoluminescence spectroscopy, and (3) exhibit strong paramagnetic behaviour in the solid-state, as identified by EPR, without external dopants present. We report the new syntheses, optoelectronic properties, electrochemical properties, EPR spectra, and density functional theory (DFT)/time-dependent DFT (TDDFT) calculations to support experimental findings for NO2-PDIN-H and NH2-PDIN-H. Notably, green-solvent ink formulations guided by Hansen solubility parameters were achieved to afford uniform coatings at high concentrations (10 mg mL−1) on flexible substrates, highlighting practical advantages for printed electronics.
Results & discussion
Synthesis & characterization
The synthesis of NO2-PDIN-H and NH2-PDIN-H (Scheme 1) are reported for the first time, which is accomplished by starting from PDIN-H, which has been previously reported.27 First, PDIN-H is nitrated with nitric acid in dichloromethane at room temperature, providing NO2-PDIN-H as the sole regio-isomer. The reaction mixture is washed with water to quench residual acid, the organic layer is concentrated under reduced pressure, and solid slurried with methanol with subsequent collection by vacuum filtration to afford analytically pure NO2-PDIN-H (90% yield). The nitro group in NO2-PDIN-H is reduced by refluxing in a THF/H2O solution (pH 8) containing L-ascorbic acid and NaOH. After completion, the mixture is poured into water and stirred overnight. The precipitate is filtered and recrystallized from isopropanol & water to give NH2-PDIN-H as a dark purple solid (90% yield). The synthesis of NO2-PDIN-H followed a traditional nitration using nitric acid, and was optimized by using dilute nitric acid to suppress rapid di-substitution that would otherwise yield the known (NO2)2-PDIN-H compound.35 Nitration of aromatic compounds is a well-known strategy for tuning electrochemical and optoelectronic properties through the introduction of highly electron-withdrawing nitro groups.36 For PDIs, nitration is emerging as a versatile route to provide building blocks that can be further functionalized, including π-conjugation extension.37 Notably, nitro-substituted PDIs and N-annulated PDIs have been employed as electrophilic building blocks in Suzuki–Miyaura couplings, providing efficient alternatives to brominated PDIs for the synthesis of PDI multimers.38 We adopted a green-chemistry strategy in preparing NH2-PDIN-H by substituting our traditional method of using tin(II) reductants with L-ascorbic acid (vitamin C). This reagent is a mild, non-toxic, organic reductant and aligns with sustainable chemistry principles.39 In basic conditions, L-ascorbic acid deprotonates to form ascorbate mono- and dianions which act as potent reducing species to donate electrons and protons in consecutive two-electron steps, reducing nitro (–NO2) to nitroso (–NO), then hydroxylamine (–NHOH), and finally the amine (–NH2).40 Similar to nitro-aromatics, amino-aromatic compounds are experiencing renewed interest as modular building blocks for constructing extended π-conjugated architectures via condensation reactions, providing Schiff-base (imine) or imide linkages.41–43 Recent examples include the synthesis of semiconducting polymers through imine condensation,44 as well as the reaction of a diamino-PDI with aromatic aldehydes to form polyimines that undergo oxidative photocyclization, yielding fused, electron-deficient polymers with enhanced stability.45 In the context of NH2-PDIN-H, the PDI core bearing a primary amine offers a reactive site for condensation with complementary aldehyde or anhydride partners. Structural characterizations via proton nuclear magnetic spectroscopy (1H-NMR), high-resolution mass spectrometry (HRMS), and elemental analysis (EA) of NO2-PDIN-H and NH2-PDIN-H are provided in the SI (Fig. S1–S8). Note, 13C-NMR spectroscopy was attempted, but due to the low solubility in common deuterated solvents, the spectra were not well resolved and therefore not reported. The materials were found to be stable to light and air when stored under ambient conditions over the course of several months; 1H-NMR spectroscopy experiments indicated no change in structure after one year of storage. Additionally, the thermal stability of the N-annulated PDIs were probed by thermal gravitational analysis (TGA; Fig. S9–S11), which revealed decomposition onsets of (Tonset) 389 °C, 363 °C, and 375 °C with maximum decomposition (Tmax) at 417 °C, 378 °C, and 409 °C for PDIN-H, NO2-PDIN-H, and NH2-PDIN-H, respectively. This demonstrates the high thermal stability known to PDIs and relevant π-conjugated organic compounds.46,47
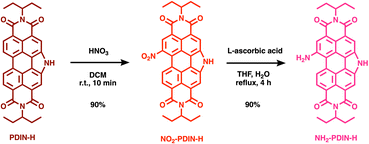 |
| | Scheme 1 Synthetic pathway for NO2-PDIN-H and NH2-PDIN-H starting from PDIN-H. | |
Molecular conformation
DFT calculations were performed on PDIN-H, NO2-PDIN-H, NH2-PDI, and NH2-PDIN-H to determine the gas-phase optimized geometries and understand the effect of bay functionalization with nitrogen moieties on molecular conformation (Fig. 2). All geometry optimizations were performed using the LC-ωHPBE functional with the 6-31G(d) basis set (note that the ω parameter was gap tuned for each molecule; see SI for details).48,49 The results demonstrate that PDIN-H maintains a planar perylene core, whereas NO2-PDIN-H introduces a slight distortion (dihedral angle of 1.9°) due to steric repulsion from the nitro group. Note, the single crystal X-ray diffraction of a di-nitro N-annulated PDI also showed minimal twisting at the core even in the presence of two sterically demanding substituents.35 NH2-PDIN-H exhibits the largest deviation from planarity for the N-annulated PDIs, with a calculated dihedral angle of 7.3° between the central perylene plane and the amino-substituted bay region, which disrupts π-conjugation across the core. However, when considering the non-N-annulated analogue (NH2-PDI; Fig. 2c), the core twists an additional 10° (to 17.3°) about the amino-bay-substituted site and an additional 10.5° (to 11.1°) about the pyrrolic site. Pictorial representations of select FMOs (Fig. S13) show that the wavefunctions tend to be symmetrically distributed across the molecular core, highlighting N-annulation as a strategy to retain core planarity in the presence of bay-functional groups. Note, attempts to obtain single crystals of NO2-PDIN-H and NH2-PDIN-H suitable for X-ray diffraction were unsuccessful, likely due to the unsymmetric nature of these compounds. Among the multitude of procedures used to attempt single crystal growth, two were analogous to that for PDIN-H and (NO2)2-PDIN-H,35,50 where all attempts resulted in small needle like microstructures with poor diffraction. Collectively, the calculated geometries suggest that bay-substitution is likely to drive distinct solid-state packing motifs. The core planarity in PDIN-H enables close π–π stacking,50 which is expected to be largely maintained in the nitrogen-bay-substituted N-annulated PDIs due to the preserved planarity. However, the addition of nitrogen substituents at the open bay positions is hypothesized to distort the packing as follows: (1) nitro groups on aromatics are known to induce strong dipole moments, promoting dipole–dipole interactions that favour slipped π-stacked arrangements,51 and (2) aromatic amino groups are known to engage in hydrogen bonding, which can drive the formation of columnar or chain-like supramolecular assemblies.52 Thus, while the electronic delocalization across the perylene core remains intact, the nature of the bay-substituent is expected to direct packing geometry.
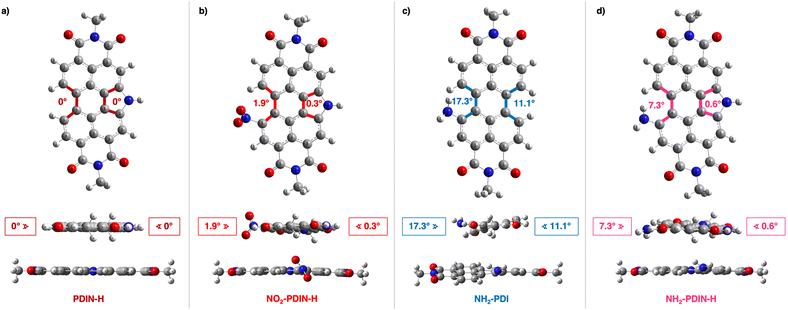 |
| | Fig. 2 DFT-optimized chemical structures as derived at the LC-ωHPBE/6-31G(d) level of theory. Top-down and in-plane views with relevant dihedral angles for (a) PDIN-H, (b) NO2-PDIN-H, (c) NH2-PDI, and (d) NH2-PDIN-H are provided. | |
Optoelectronics
Experimental solution & simulated gas-phase UV-visible absorptions.
The optoelectronic properties of NO2-PDIN-H and NH2-PDIN-H were evaluated experimentally in solution, with PDIN-H included as a reference. The solution UV-visible absorption and photoluminescence spectra were collected using tetrahydrofuran (THF) as the solvent, where additional experiments to probe quantum yield & solvatochromism were performed in toluene and dimethyl sulfoxide (DMSO). Additionally, TDDFT calculations at the LC-ωHPBE/6-31G(d) level of theory were performed to investigate the gas-phase electronic excitations for PDIN-H, NO2-PDIN-H, and NH2-PDIN-H.53 In THF, PDIN-H shows an onset of 2.32 eV with a λmax of 520 nm, NO2-PDIN-H an onset of 2.20 eV with a λmax of 585 nm, and NH2-PDIN-H shows an onset of 2.08 eV with a λmax of 564 nm (Table 1 and Fig. 3). TDDFT calculations in the gas phase (Table 1 and Fig. S12) revealed a λmax of 443 nm for PDIN-H, a λmax of 464 nm for NO2-PDIN-H, and a λmax of 465 nm for NH2-PDIN-H; the TDDFT results generally follow the measured values. The classical 0 → 0, 0 → 1, and 0 → 2 vibronic transitions of the perylene core are present for PDIN-H and NO2-PDIN-H in the experimental spectra, however, NH2-PDIN-H exhibits broadening and a pronounced redshift of the primary transition. Considering the DFT-derived geometries, PDIN-H shows no significant twisting and thus retains planarity and symmetric HOMO and LUMO distribution. This results in the well-resolved vibronic progression observed in the absorption spectrum, which is indicative of delocalized π–π* transitions with minimal ICT character or conformational asymmetry. NO2-PDIN-H shows a red-shifted absorption relative to PDIN-H owing to FMO energy stabilization with functionalization. Additionally, the bulky substituent does not significantly distort the core structure (dihedral angle near 0°), allowing for similar wavefunction delocalization. However, the presence of the secondary and tertiary blue shifted peaks at 311 nm (3.99 eV) and 402 nm (3.08 eV), respectively, arise from electronic asymmetry introduced by the nitro substituent. NH2-PDIN-H exhibits both a red-shifted primary absorption and the appearance of a secondary, blue-shifted peak. This spectral splitting arises from two competing effects: (1) electron donation & delocalization, and (2) steric-induced twisting. The amino group donates electron density into the perylene core through resonance leading to HOMO energy destabilization and a reduction in the HOMO–LUMO energy gap. As seen in HOMO isosurface plots (Fig. S13), the amino group participates significantly in the HOMO (and HOMO-1), thus causing the energy destabilization and subsequent red-shift in the primary optical transition. Additionally, amino functionalization introduces moderate dihedral twisting of the perylene core (7.3°) which somewhat disrupts the delocalization of the wavefunction across the molecule and causes localization of electronic transitions and a break in symmetry. As a result, a secondary, blue-shifted peak appears due to a transition involving a more localized excitation (HOMO-1 → LUMO) with reduced ICT character. Amino-bay-functionalized PDIs without N-annulation also exhibit primary and secondary optical transitions, however, the primary band is markedly broadened and is significantly bathochromically shifted.25,54 The bathochromic shift of these primary transitions increases in the order of tertiary > secondary > primary amino substituents, consistent with the inductive effects of the alkyl groups. Moreover, introducing a second amino group (di-substitution) further distorts the PDI core resulting in core twists of up to 20°,54 and increases destabilization of the FMOs.
Table 1 Optical properties for PDIN-H, NO2-PDIN-H, and NH2-PDIN-H in THF and in a thin film (on quartz post annealing at 100 °C for 15 minutes); results from the TDDFT calculations are provided for reference
| Compound |
Solution λmax (nm) |
TDDFT λmax (nm) |
ε (M−1 cm−1) |
Solution Eoptgap (eV) |
Film λmax (nm) |
Film Eoptgap (eV) |
| PDIN-H |
520 |
443 |
98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 479 479 |
2.32 |
502 |
2.06 |
| NO2-PDIN-H |
526 |
464 |
44726 |
2.20 |
510 |
2.01 |
| NH2-PDIN-H |
564 |
465 |
50191 |
2.08 |
545 |
1.92 |
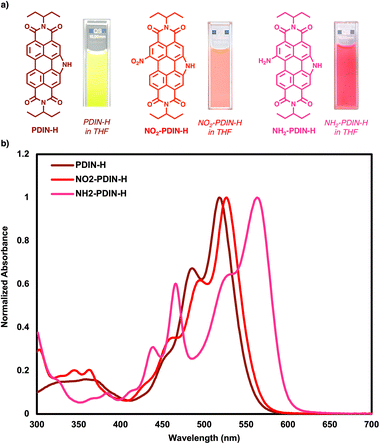 |
| | Fig. 3 a) Molecular structures and photographs of the solutions of PDIN-H, NO2-PDIN-H, and NH2-PDIN-H dissolved in THF with (b) corresponding UV-visible absorption spectra. | |
These optical features are also observed in secondary cyclic amino groups at the bay position of PDIs.55–57 In short, the spectral behaviour of these nitrogen functionalized PDIs is a combined outcome of electronic substituent effects and geometric distortion induced by bay functionalization.
Solvatochromism & quantum yield.
Solvent-stabilization of an ICT state in organic compounds can be probed by comparing optical signatures of a compound in solvents with variable polarity indices. Increasing solvent polarity is known to stabilize the dipole moment of the excited state for push–pull systems with strong ICT, which results in increased bathochromic shifts and Stokes shifts but decreased quantum yield (Φ).58,59 PDIN-H, NO2-PDIN-H, and NH2-PDIN-H were probed for solvatochromism and Φ in toluene (polarity index: 2.4), THF (polarity index: 4.0), and dimethyl sulfoxide (DMSO; polarity index: 7.2) (Fig. 4, S21–S30 and Table 2).60 The quantum yield (Φ) of each were calculated using Fluorescein as a standard (Fig. S21),61 see SI for full experimental details. All data are summarized in Table 2. First, PDIN-H maintains a high Φ across the three solvents (74–99%), owing to the rigid and planar core which facilitates radiative relaxation. Although Φ decreases in DMSO to 74%, this drop is likely due to non-radiative decay pathways enabled by intermolecular interactions, such as hydrogen bonding, with the polar solvent.62 PDIN-H exhibits minimal solvatochromism, suggesting a low ground-state dipole moment and negligible ICT character. Given the low concentrations used in the quantum yield experiments, PDIN-H behaves similarly to other non-aggregated PDI derivatives, which are known to exhibit high Φ values.2 Second, NO2-PDIN-H displays the widest range of Φ values across the three solvents (33–68%), along with the largest Stokes shifts (0.19–0.30 eV). Solvatochromism remains minimal, with only a 10 nm bathochromic shift in DMSO, which is nearly identical to that observed for PDIN-H and indicates a small change in dipole moment between the ground and excited states. Nonetheless, the pronounced Stokes shifts suggest stabilization of an excited ICT state by polar solvents which is a feature of fluorosolvatochromism.63 These characteristics have been observed in nitroperylene, where strong hydrogen bonding character and the solvent dependent torsion angle of the nitro group can lead to ultrafast nonradiative decay upon excitation.64 Lastly, NH2-PDIN-H exhibits the lowest Φ in toluene and THF, and is a mere 1% higher in DMSO compared to NO2-PDIN-H, suggesting strong ICT and nonradiative pathways stabilized by polar solvents. Similar to other amino-substituted PDIs, NH2-PDIN-H exhibits strong solvatochromism with bathochromic shifts of 11 nm from toluene to THF and 17 nm from THF to DMSO, indicative of significant ICT character in both ground and excited states and a significant change from ground to excited state dipole moments.25 While the Stokes shifts are not as large as those of NO2-PDIN-H, they are at least double those of PDIN-H, further suggesting that an excited ICT state is stabilized by polar environments.
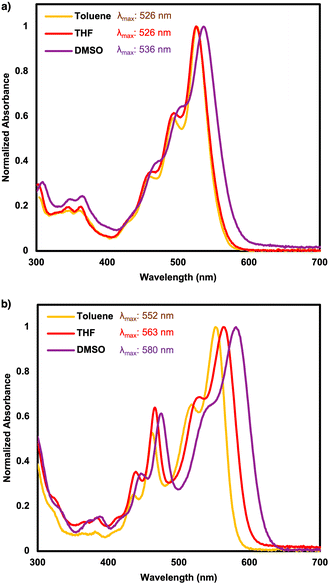 |
| | Fig. 4 UV-visible absorption spectra for (a) NO2-PDIN-H and (b) NH2-PDIN-H in toluene, THF, and DMSO with associated λmax values. | |
Table 2 Solvatochromism & quantum yield optical data for PDIN-H, NO2-PDIN-H, and NH2-PDIN-H
| Compound |
Abs λmax (nm) |
PL λmax (nm) |
Φ
|
Stokes shift (eV) |
Abs λmax (nm) |
PL λmax (nm) |
Φ
|
Stokes shift (eV) |
Abs λmax (nm) |
PL λmax (nm) |
Φ
|
Stokes shift (eV) |
|
Solvent = toluene.
Solvent = THF.
Solvent = DMSO.
|
| PDIN-H |
521 |
530 |
0.99 |
0.04 |
519 |
530 |
0.90 |
0.05 |
528 |
546 |
0.74 |
0.08 |
| NO2-PDIN-H |
526 |
572 |
0.68 |
0.19 |
526 |
582 |
0.57 |
0.23 |
536 |
616 |
0.33 |
0.30 |
| NH2-PDIN-H |
552 |
578 |
0.62 |
0.10 |
563 |
598 |
0.47 |
0.13 |
580 |
626 |
0.34 |
0.16 |
Base treatments in solution.
Previously, PDIN-H was shown to undergo reversible reactivity with base in both solution and solid states, wherein the pyrrolic N–H moiety can be deprotonated to form stable ion pairs in solution (Fig. 6a).50 This deprotonation results in distinct changes to the optical properties due to the lone pair on the pyrrolic nitrogen aligning parallel to the π-conjugated core, thereby increasing electron density and altering the electronic structure. Specifically, a bathochromic shift of 76 nm was observed upon deprotonation. Herein, PDIN-H, NO2-PDIN-H and NH2-PDIN-H were treated with an organic base, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), in DMSO to compare the deprotonated optical spectra for each (Fig. 5b and c). PDIN-H exhibited a greater bathochromic shift when deprotonated by DBU in DMSO (82 nm) compared to NaOH in isopropanol (76 nm), requiring 5 equivalents of DBU for complete deprotonation, as further additions produced no change in the absorption spectrum. For NO2-PDIN-H, 4 equivalents of DBU were sufficient for complete deprotonation, and resulted in a bathochromic shift of 88 nm, indicative of a strong push–pull system upon deprotonation. In contrast, NH2-PDIN-H required 100 equivalents of DBU for complete deprotonation, yielding a more modest bathochromic shift of 41 nm. Furthermore, the pKa of each molecule in DMSO were evaluated at the LC-ωHPBE/6-31G(d) level of theory. The DFT-derived pKa were 22.9, 20.9, and 23.7 for PDIN-H, NO2-PDIN-H, and NH2-PDIN-H, respectively; see SI for details. Additionally, pKa was determined for the molecules in THF and toluene, with the DFT-derived pKa being higher (36–40 in THF and 59–65 in toluene; Table S3) than those for DMSO. This trend reflects the relative solvent polarity (DMSO > THF > toluene), where DMSO, being highly polar, most effectively stabilizes the deprotonated/ionic species for the lowest pKa values. These findings are consistent with our experimental results, which reveal the trend of acidity being NO2-PDIN-H > PDIN-H > NH2-PDIN-H, and underscores the influence of bay substituents on pyrrolic N–H deprotonation. Similar trends have been observed for N-annulated PDIs bearing sulfide and sulfone substituents, where the electron-donating sulfide reduced acidity and the electron-withdrawing sulfone increased acidity.65 Consistently, the electron-withdrawing nitro group in NO2-PDIN-H enhances acidity (requiring fewer equivalents for deprotonation), while the electron-donating amino group in NH2-PDIN-H reduces acidity (requiring more equivalents) relative to PDIN-H. In solution, PDIN-H appears orange, while in a deprotonated form it is purple. For NO2-PDIN-H, the neutral form presents pink, and deprotonated is blue. Lastly, NH2-PDIN-H appears pink in its neutral state and shifts to a bluish-purple upon deprotonation. All compounds exhibit two isosbestic points during the neutral-to-anionic transition, indicating that no intermediate species are formed. These pronounced colour changes highlight the potential of these derivatives as colorimetric pH probes. Note, it was observed that when treated with the strong inorganic base, NaOH, NH2-PDIN-H will continue to react with increasing equivalences of NaOH producing a new spectral signature with a bathochromically shifted λmax (790 nm; Fig. S31). The origins of the change are unknown at this time; however, a proposed mechanism is provided in Fig. S31a. Here, we hypothesize the amino moiety is deprotonated at high equivalences of NaOH, as this would reduce sterics at the bay area.
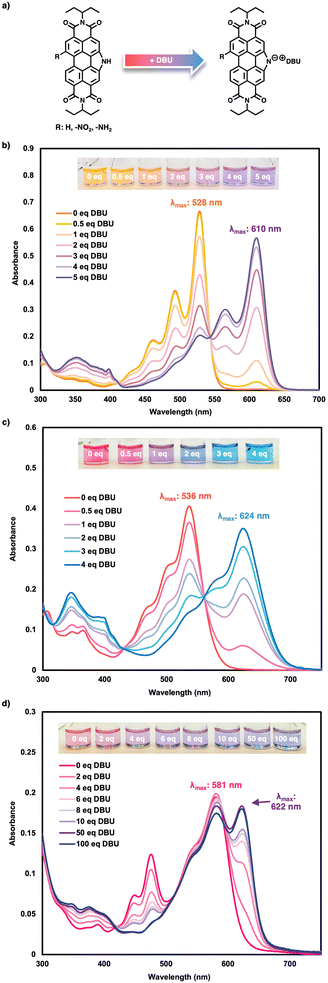 |
| | Fig. 5 Treating PDIN-H, NO2-PDIN-H and NH2-PDIN-H with increasing equivalences of DBU; (a) proposed deprotonation mechanism, and UV-visible spectra of (b) PDIN-H, (c) NO2-PDIN-H, and (d) NH2-PDIN-H at 10 μM each with increasing equivalences of DBU & respective inset photographs of solutions used in the experiment. | |
Electrochemical properties
The electrochemical properties of PDIN-H, NO2-PDIN-H, and NH2-PDIN-H were probed using solution cyclic voltammetry (CV; Fig. 6 and Table 3) and differential pulse voltammetry (DPV; SI, Fig. S32–34), using dichloromethane (DCM) as the solvent. For PDIN-H and NH2-PDIN-H, the oxidation and reduction waves collected using CV were used to estimate the HOMO and LUMO energies via E1/2 values, respectively, with Fc/Fc+ as the internal standard. For NO2-PDIN-H, the reduction wave collected by CV was used to determine the LUMO energy via E1/2 values, while DPV was used to determine the oxidation potential and thus the HOMO energy level, in both cases using Fc/Fc+ as the internal standard. Here, DPV was used to determine the HOMO energy as the oxidation wave for NO2-PDIN-H overlaps with the solvent oxidation event in CV. DPV was used to confirm all other energy levels determined by CV, to which all values were in good agreement. PDIN-H and NH2-PDIN-H exhibited two quasi-reversible reduction waves and one quasi-reversible oxidation wave, while NO2-PDIN-H exhibits two quasi-reversible reduction waves. Relative to PDIN-H, the addition of the electron withdrawing nitro group on an open bay position stabilizes the HOMO and LUMO energies by 0.2 eV. For NH2-PDIN-H relative to PDIN-H, the addition of the electron donating amine group on an open bay position destabilized the HOMO energy level by 0.3 eV and the LUMO by 0.2 eV. This results in PDIN-H and NO2-PDIN-H exhibiting the same optical gap of 2.2 eV, and an optical gap of 2.1 eV for NH2-PDIN-H. This trend showcases the predictable structure–property relationships of N-annulated perylene diimides as it is typical for electron withdrawing and donating groups to stabilize and destabilize the FMO energies, respectively.
 |
| | Fig. 6 Electrochemical properties of NO2-PDIN-H, PDIN-H, and NH2-PDIN-H; (a) HOMO/LUMO energy levels determined from CV E1/2 values, except the HOMO energy level for NO2-PDIN-H was estimated using DPV, and (b) corresponding CV traces for NO2-PDIN-H, PDIN-H, and NH2-PDIN-H using Fc/Fc+ as the internal standard. | |
Table 3 Electrochemical properties for PDIN-H, NO2-PDIN-H, and NH2-PDIN-H in DCM
| Compound |
Solution HOMO (eV) |
Solution LUMO (eV) |
Solution Egap (eV) |
E
1/2_ox (V) |
E
1/2_red (V) |
E
onset_ox (V) |
E
onset_red (V) |
| NO2-PDIN-H |
−6.0 |
−3.8 |
2.2 |
— |
−1.02 |
— |
−0.96 |
| PDIN-H |
−5.8 |
−3.6 |
2.2 |
1.06 |
−1.24 |
0.99 |
−1.18 |
| NH2-PDIN-H |
−5.5 |
−3.4 |
2.1 |
0.65 |
−1.37 |
0.59 |
−1.30 |
Ionization energies (IE) and electron affinities (EA) were determined by DFT calculations (Table S2) at the LC-ωHPBE/6-31G(d) level of theory. FMO energies were also extracted from the neutral ground-state structures for comparison with experimental electrochemical data. The HOMO and LUMO energies of PDIN-H reflect a baseline for comparison. Substitution at the bay position with an electron-withdrawing nitro group (NO2-PDIN-H) results in stabilization of both frontier orbitals: the HOMO energy is lowered by 0.25 eV, and the LUMO energy by 0.36 eV relative to PDIN-H. This stabilization results in a slightly reduced HOMO–LUMO gap (5.34 eV vs. 5.45 eV), and notably increases the electron affinity (2.51 eV), supporting the enhanced electron-accepting character of the nitro substituent. The ionization energy also increases (7.81 eV), consistent with a more stabilized HOMO energy level. Conversely, NH2-PDIN-H displays destabilization of both the HOMO and LUMO energies relative to PDIN-H, with the HOMO energy level raised by 0.34 eV and the LUMO energy level by 0.18 eV. This reflects the electron-donating nature of the amino group, leading to a narrowed optical gap (5.30 eV), reduced EA (1.94 eV), and decreased IE (7.12 eV), indicating enhanced electron richness. While the gas-phase energy level calculations differ from experimental CV/DPV measurements performed in solution, these results align well with experimental trends and affirm the tunability of electronic structure via bay substitution in N-annulated PDI systems, as seen for cyanated N-annulated PDIs, which have HOMO and LUMO energies of 6.1 eV and 3.8 eV, respectively.30 Notably, the experimentally derived FMO energies for NO2-PDIN-H and NH2-PDIN-H are comparable to widely recognized molecular components used in organic electronics. For instance, Y6 (BTP-4F) is a benchmark non-fullerene acceptor, OPV devices constructed using it in the bulk heterojunction achieve power conversion efficiencies over 19%, and exhibits an estimated HOMO energy level of −5.7 eV and a LUMO energy level of −4.1 eV.66,67 On the interlayer front, PDINN is commonly used as a cathode interlayer with a measured HOMO energy level of −6.0 eV and LUMO energy level of −3.8 eV.22 Within this landscape, NO2-PDIN-H, featuring stabilized FMOs comparable to PDINN, closely mimics the alignment of industry-standard electron-selective interlayers. Conversely, NH2-PDIN-H, with relatively destabilized HOMO and LUMO energy levels, aligns more with donor-type or ambipolar semiconductors, suggesting suitability for OFETs or sensor applications where electron-rich behaviour is beneficial. However, the amino functionality in NH2-PDIN-H induces strong paramagnetic character (vide infra) consistent with self-doping motifs in amino-modified PDIs, a feature that may compensate for destabilized FMOs and support electron transport via radical-anion conduction pathways.20,23
Electron paramagnetic resonance
To investigate potential self-doping effects via paramagnetic signatures, EPR spectroscopy was performed on PDIN-H, NO2-PDIN-H, and NH2-PDIN-H in the solid state (Fig. 7). The parent compound, PDIN-H, served as a reference to assess the impact of electron-donating and electron-withdrawing substituents. PDIN-H exhibited a near negligible EPR signal, and thus highlights the absence of self-doping effects in the unsubstituted system. NO2-PDIN-H exhibits a moderate signal centred at g = 2.0060, and NH2-PDIN-H has the most intense resonance centred at g = 2.0057. The increased signal intensity in NH2-PDIN-H implies increased self-doping character, consistent with spontaneous electron donation into the π-system. The g-value of 2.0057 is slightly lower than that of NO2-PDIN-H, placing it closer to the free electron value (2.0023).68 These results underscore the substituent-dependent tuning of paramagnetic character in N-annulated PDI derivatives; the nitro and amino groups not only modulate electronic structure but also influence geometric character, where disruption of π-orbital conjugation can lead to stabilized ICT states and thus increases self-doping characteristics. These results are supported by the strong electron donating and electron withdrawing nature of the amino- and nitro-bay substituents, respectively, as seen in the partial charges determined via DFT at the LC-ωHPBE/6-31G(d) level of theory (Fig. S14), derived from CM5-corrected Mulliken population analysis (CM5) for the radical cation, radical anion, and neutral forms.69 Note, in the case of NO2-PDIN-H, this data suggests ICT occurs from the PDI core to the nitro group, whereas for NH2-PDIN-H electron density is more likely to be donated into the core. This charge redistribution could give rise to the paramagnetic signatures observed, however, further investigation is required to validate this hypothesis. While we cannot compare our data directly to known self-doped PDIs, we acknowledge the intensity of the EPR signals of the NO2-PDIN-H and NH2-PDIN-H derivatives are lower than that of PDINN,22 for example. This indicates a lower degree of paramagnetic character for our derivatives, yet the presence of discernible paramagnetic signatures nonetheless suggests that bay-functionalization with nitrogen functional groups does impart measurable self-doping behaviour, providing a promising foundation for further molecular tuning that could bring the associated charge transport properties closer to that of benchmark systems such as PDINN. Furthermore, while our EPR measurements offer valuable insights into the self-doping nature of NO2-PDIN-H, and NH2-PDIN-H, they do not assess the stability or lifetime of radical species under varying illumination, thermal, or electrochemical conditions. Both nitro- and amino-substituted aromatics may generate radical anions or cations, and the persistence of such depends critically on π-delocalization and molecular packing. To further evaluate radical character and stability in these N-annulated PDI derivatives, it would be beneficial in future work to employ spectroelectrochemical and chemical doping experiments.
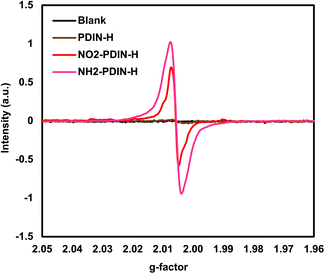 |
| | Fig. 7 Electron paramagnetic resonance spectra for PDIN-H, NO2-PDIN-H, and NH2-PDIN-H in the solid-state. | |
Green solvent processing for large area coatings
Hansen solubility parameters.
The coating of thin films is a widely pursued fabrication method for organic electronic devices owing to low-cost, good energy efficiency, and high-throughput potential.70–72 The choice of solvent for ink formulation critically impacts both the economic and environmental aspects of the process. As such, the development of formulations using low-cost, non-toxic “green” solvents is a growing priority in sustainable materials research.73 One predictive method for evaluating solubility in various solvents and blends without extensive experimental screening is the use of Hansen solubility parameters (HSPs). By leveraging HSPs, researchers can formulate green inks that are compatible with specific substrates, streamlining solvent selection and minimizing trial-and-error. Here, we applied this method to evaluate green ink formulations and the resulting large area processing of PDIN-H, NO2-PDIN-H, and NH2-PDIN-H thin-films (vide infra). The HSPs for PDIN-H, NO2-PDIN-H, and NH2-PDIN-H (Table 4 and Fig. S35–S37) were calculated using the approach originally developed by Steven Abbott in 2013 and subsequently improved by Díaz de los Ríos and Hernández Ramos.74,75 Solubility testing was simulated across 25 different solvents, each at a concentration of 1 mg mL−1 (Table S4). The resulting HSP values are: for PDIN-H δD = 17.0, δP = 6.1, δH = 8.7, R = 4.6; for NO2-PDIN-H, δD = 17.3, δP = 6.8, δH = 8.8, R = 5.0; and for NH2-PDIN-H, δD = 16.8, δP = 12.1, δH = 6.6, R = 6.5. This data is visualized as individual parameter plots (Fig. S35–S37), which highlight the narrow solvent compatibility for each compound. The lack of solubility at moderate concentrations is indicative of strong intermolecular forces, where the pyrrolic N–H, and amino groups participate in hydrogen bonding as seen in variable temperature NMR spectroscopy experiments for NH2-PDIN-H (Fig. S4) and within the solid-state packing for parent PDIN-H as per single crystal X-ray diffraction.50 These strong intermolecular interactions result in ‘solvent-resistant’ thin films of PDIN-H, an important property for the deposition of subsequent layers and one that is typically difficult to achieve. Furthermore, the planarity of these compounds enables tight π–π stacking about the aromatic core, thus promoting aggregation over miscibility with common solvents. Despite this, we have utilized HSPs to enable green solvent processing (vide infra).
Table 4 Hansen solubility parameters (at concentrations of 1 mg mL−1) for PDIN-H, NO2-PDIN-H, and NH2-PDIN-H
| Compound |
δ
D
|
δ
P
|
δ
H
|
R
|
| PDIN-H |
17.0 |
6.1 |
8.7 |
4.6 |
| NO2-PDIN-H |
17.3 |
6.8 |
8.8 |
5.0 |
| NH2-PDIN-H |
16.8 |
12.1 |
6.6 |
6.5 |
Solid state optical properties.
Thin films of PDIN-H, NO2-PDIN-H, and NH2-PDIN-H were fabricated on 1 inch quartz substrates via spin coating using 5 mg mL−1 THF solutions (Fig. 8). THF was selected due to suitable wettability on quartz and optimal miscibility with all three dyes. The optical gaps of the resulting films before annealing are 1.99 eV (PDIN-H), 1.97 eV (NO2-PDIN-H), and 1.88 eV (NH2-PDIN-H), with λmax values of 506 nm, 509 nm, and 546 nm, respectively. Each film exhibited clear vibronic features corresponding to the 0 → 0, 0 → 1, and 0 → 2 transitions, with the 0 → 1 transition dominating each spectrum prior to annealing. After thermal annealing at 100 °C for 15 minutes, all films exhibited an increase in absorbance intensity. Annealing typically improves molecular packing and enhances π–π interactions in PDI thin films,76 however, further studies are needed to elucidate the precise structural changes responsible for this behaviour. The optical gaps of the resulting films post annealing become 2.07 eV (PDIN-H), 2.02 eV (NO2-PDIN-H), and 1.92 eV (NH2-PDIN-H) with λmax values at 539 nm, 509 nm, and 541 nm, respectively. For PDIN-H, the 0 → 0 transition became the most prominent after annealing, reshaping the spectrum, while NO2-PDIN-H and NH2-PDIN-H mostly retained their spectral profiles, with the 0 → 1 transition remaining the most intense. Comparing the solution UV-visible spectra of PDIN-H, NO2-PDIN-H, and NH2-PDIN-H in THF with film spectra, the signatures are broadened and red-shifted (Fig. S38). This film-state behaviour parallels general trends observed in PDI derivatives, where transition to thin films leads to red-shifts and supressed vibronic fine structure due to aggregation.24,77 Within this series, NH2-PDIN-H demonstrates the largest red-shift and most pronounced vibronic suppression, whereas PDIN-H retains greater vibronic resolution and exhibits only moderate red-shifting. NO2-PDIN-H exhibits intermediate behaviour with a modest red-shift and more spectral broadening than PDIN-H, yet less than NH2-PDIN-H. These observations align with literature reports showing that bay-substituents in PDI frameworks influence aggregation geometry and optical signatures in films.78,79 The strong visible-light absorption, tunable optical gaps, and enhanced molecular ordering upon thermal annealing for these N-annulated PDI derivatives support the potential for thin-film optoelectronic applications. For indoor organic photovoltaics (iOPVs), in particular, the broad absorption profiles in the visible light spectrum are ideal for efficient low-light harvesting. The absorption range (400–650 nm) closely matches the emission spectra of artificial light sources such as warm and cool LEDs (400–700 nm), aligning well with iOPV operational conditions. Additionally, the relatively higher-lying LUMO energies, compared to fullerenes or ring-fused non-fullerene acceptors, can yield wider optical gaps which can contribute to higher open-circuit voltages.80,81 Moreover, the high absorption coefficients and paramagnetic character of NO2-PDIN-H and NH2-PDIN-H in the solid-state suggest strong potential for organic photodetectors by enabling efficient visible-to-near-IR light harvesting and stable radical anions for photo-driven ultra-sensitive detection.82 Lastly, these same properties make NO2-PDIN-H and NH2-PDIN-H promising candidates for photocatalysis by enabling efficient photoinduced charge separation and electron–transfer processes. For example, Koenig and coworkers have shown that N-annulated PDI tethered rhenium bipyridine dyads can achieve efficient CO2-to-CO photocatalysis, with electron-donating substituents such as –NH2 yielding turnover numbers (TONCO) as high as ∼234, and –NO2 derivatives achieving ∼137 TONCOvia in situ conversion to –NH2 during catalysis.33,34
 |
| | Fig. 8 UV-visible absorption spectra of thin films of PDIN-H, NO2-PDIN-H, and NH2-PDIN-H spin-coated from 5 mg mL−1 THF solutions onto 1 inch quartz substrates. (a) Spectra of all films after annealing at 100 °C for 15 min, (b) spectra of PDIN-H, (c) NO2-PDIN-H, and (d) NH2-PDIN-H before and after annealing with corresponding film images shown in the insets. | |
Green ink formulations & large area coatings.
Green-solvent inks incorporating biobased or biodegradable components minimize volatile emissions and worker exposure while enabling sustainable printed electronics with lower life-cycle impact.73,83,84 Using HSP screening, we formulated green solvent blends that fully dissolved PDIN-H, NO2-PDIN-H, and NH2-PDIN-H at 1 mg mL−1 and 10 mg mL−1. Inks for large-area, high-throughput fabrication of solution processed organic electronics must meet both solubility and coating requirements, particularly for slot-die coating, which is a pre-metered, non-contact process that delivers uniform films with high material efficiency and translates seamlessly to roll-to-roll production.85 Achieving stable wetting and drying within a controlled coating window is therefore critical for commercial deployment. All three compounds were soluble at 1 mg mL−1 in a 1:1 blend of ethyl lactate (EL) and 2-methyltetrahydrofuran (2-MeTHF) (Fig. 9). Although EL is underutilized, likely due to having a relatively high boiling point (∼154 °C), it has recently been employed in perovskite processing, valued for low cost, bio-derived origin, excellent wettability, low toxicity, and strong film-forming capability.86 In contrast to EL, 2-MeTHF has been successfully used as a processing solvent in high-performance OPVs, offering bio-derived origin, low toxicity, favourable evaporation kinetics, and excellent compatibility with non-fullerene acceptor blends.87,88 In this blend, EL delivers ideal substrate wetting and film formation, while 2-MeTHF (boiling point ≈ 78 °C) accelerates drying for high-quality coating dynamics. At higher concentrations (10 mg mL−1), NO2-PDIN-H was soluble in a 1:1 ethyl lactate to anisole blend, demonstrating the practical utility of HSP-guided selection, though substrate wettability was less consistent. To further improve solubility, as these compounds are prone to strong intermolecular interactions, n-butylamine (BA) was introduced. For N-annulated PDIs like PDIN-H, deprotonation by BA has been observed as a means to ionize the compound and bring into polar solvents, such as alcohols.50 Therefore, a ternary 1:1:1 mixture of EL, 2-MeTHF, and BA enabled 10 mg mL−1 solubility for all three derivatives (Fig. 9). For NO2-PDIN-H, the resulting deep blue ink is consistent with deprotonation, as seen for the base treatments. Note, BA evaporates during coating, therefore the films revert to their intrinsic colours observed in the solid state which is red for PDIN-H and NO2-PDIN-H, and purple for NH2-PDIN-H. Slot-die coating of the 10 mg mL−1 inks onto vinyl substrates yielded brightly coloured, large-area films (Fig. 9), demonstrating the compatibility of these materials with scalable processing techniques and potential use in organic electronic applications requiring ink-based deposition. Building on the solvent-optimized inks, we successfully coated 10 mg mL−1 films of PDIN-H, NO2-PDIN-H, and NH2-PDIN-H onto PET substrates using the ternary 1:1:1 EL:2-MeTHF:BA blend, yielding uniform, coloured films (Fig. S39). Notably, water-based processing, a critical target for fully sustainable printed electronics, was achieved by formulating 10 mg mL−1 inks in 1:1 and 3:2 BA/water mixtures for PDIN-H/NO2-PDIN-H and NH2-PDIN-H, respectively, enabling slot-die coating onto PET/ITO substrates (Fig. S40). We acknowledge that the films coated from the BA/water solvent blends are visibly thinner than those from the organic solvent blends, which highlights the influence of meniscus dynamics and solvent viscosity during slot-die coating. However, further optimization of coating parameters can be pursued to obtain thicker films when warranted. Regardless, achieving stable aqueous coatings is inherently difficult due to the hydrophobic nature of PDIs alongside the strong tendency to aggregate, and the poor wetting of ITO with water. Indeed, diverse chemical strategies have been employed to develop aqueous-processable organic semiconductors, including modulation of side-chain polarity, introduction of hydrophilic functionalities, and the use of cleavable nanoparticle dispersions.89–91 Here, BA serves a dual role: (1) deprotonating the pyrrolic N–H groups to generate soluble ionized species compatible with polar media, and (2) enhancing substrate wetting to promote uniform spreading during coating. These coating designs demonstrate that with thoughtful engineering of solvent blends and interfacial interactions N-annulated PDI films can be reliably coated across diverse substrates, including low-energy plastics and conductive PET/ITO from both organic and water-based inks. The successful deposition of adherent, uniform thin films from water represents a meaningful strategy to afford green and high-throughput solution processing of N-annulated PDIs onto relevant substrates for organic electronic applications.
 |
| | Fig. 9 Green solvent ink formulations and slot-die coated films of (a) PDIN-H, (b) NO2-PDIN-H, and (c) NH2-PDIN-H. Initial 1 mg mL−1 solutions in a 1:1 EL to 2-MeTHF blend were converted into 10 mg mL−1 concentrated inks via addition of n-butylamine. The resulting deep-coloured inks were slot-die coated onto white vinyl substrates to yield uniform, large-area films (15 cm × 1.5 cm), showcasing their compatibility with scalable, solution-processing techniques. | |
Conclusion
In summary, we demonstrated N-annulation as a powerful design strategy for retaining planarity in nitrogen-bay-functionalized PDIs, enabling electronic fine-tuning without vastly compromising core planarity and thus π-conjugation. The synthesized NO2-PDIN-H and NH2-PDIN-H derivatives exhibit strong ICT, measurable paramagnetic character, and clear substituent-driven modulation of the HOMO and LUMO energies. A combination of DFT and TDDFT calculations, optical spectroscopy, electrochemical measurements, and EPR analysis collectively support the presence of substituent-induced self-doping effects, which can be leveraged for enhanced electron transport. Using HSP-guided ink formulation, we developed concentrated (10 mg mL−1), green solvent inks capable of forming uniform coatings on diverse flexible substrates, including vinyl, PET, and PET/ITO, through scalable slot-die deposition. Achieving uniform coatings from green solvent-based inks, particularly water-based inks, highlights the value of solvent engineering and molecular design in overcoming the challenges of poor wettability and strong aggregation in hydrophobic PDIs. This substrate-versatile, sustainable processing underscores the practical potential of N-annulated PDIs in high-throughput, roll-to-roll-compatible organic electronics. These findings provide new insight into how nitrogen bay substitution, when paired with N-annulation, enables self-doped, tuneable, and sustainable electron-transport materials. Future work exploring alternative annulation motifs, diverse bay dopants, and pyrrolic N-functionalization could further expand the design space, opening avenues toward highly efficient and environmentally responsible organic electronic devices.
Author contributions
K. M. W. conducted the majority of the experiments and was the main author of the manuscript under the supervision of G. C. W., alongside written contributions from Z. T. G. and edits from all authors. Z. T. G. performed all DFT and TDDFT calculations under the guidance of C. R. Synthesis optimization and some structural characterization was done by Z. M. S. All EPR experiments were carried out by A. H.
Conflicts of interest
The authors have no conflict of interest to declare.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: this data includes detailed synthetic procedures, structural characterizations (NMR spectra, mass spectra, and elemental analysis), DFT and TD-DFT experimental procedures and data, additional electrochemical data (DPV), Hansen solubility parameter data and procedures, and coating of films data and procedures. See DOI: https://doi.org/10.1039/d5sc06640a.
Acknowledgements
KMW is grateful for the scholarships that enabled this work, and thanks the Natural Sciences and Engineering Research Council of Canada (NSERC) and Alberta Innovates. The University of Calgary, NSERC Disocvery Grants program (RGPIN-2025-04443) and the NSERC Green Electronic Network are thanked for providing resources and funding. The work at the University of Kentucky was supported by the Office of Naval Research (ONR) through award number N00014-22-1-2179. Supercomputing resources were provided by the University of Kentucky Information Technology Department and Center for Computational Sciences (CCS).
References
- G. Li, Y. Zhao, J. Li, J. Cao, J. Zhu, X. W. Sun and Q. Zhang, J. Org. Chem., 2015, 80, 196–203 CrossRef CAS PubMed.
- F. Zhang, Y. Ma, Y. Chi, H. Yu, Y. Li, T. Jiang, X. Wei and J. Shi, Sci. Rep., 2018, 8, 8208 CrossRef PubMed.
- P. Murugan, E. Ravindran, V. Sangeetha, S.-Y. Liu and J. W. Jung, J. Mater. Chem. A, 2023, 11, 26393–26425 RSC.
- P. Cheng, X. Zhao and X. Zhan, Acc. Mater. Res., 2022, 3, 309–318 CrossRef CAS.
- E. Kozma and M. Catellani, Dyes Pigm., 2013, 98, 160–179 CrossRef CAS.
- M. Barra, F. V. D. Girolamo, F. Chiarella, M. Salluzzo, Z. Chen, A. Facchetti, L. Anderson and A. Cassinese, J. Phys. Chem. C, 2010, 114, 20387–20393 CrossRef CAS.
- A. L. Briseno, S. C. B. Mannsfeld, C. Reese, J. M. Hancock, Y. Xiong, S. A. Jenekhe, Z. Bao and Y. Xia, Nano Lett., 2007, 7, 2847–2853 CrossRef CAS PubMed.
- L. I. Kuznetsova, A. A. Piryazev, D. V. Anokhin, A. V. Mumyatov, D. K. Susarova, D. A. Ivanov and P. A. Troshin, Org. Electron., 2018, 58, 257–262 CrossRef CAS.
- F. Zhang, W. Li, T. Jiang, X. Li, Y. Shao, Y. Ma and J. Wu, RSC Adv., 2020, 10, 23024–23037 RSC.
- H. Li and O. S. Wenger, Angew. Chem., Int. Ed., 2022, 61, e202110491 CrossRef CAS PubMed.
- C. Rosso, G. Filippini and M. Prato, Eur. J. Org Chem., 2021, 2021, 1193–1200 CrossRef CAS.
- G. S. Perez, S. Dasgupta, W. Żuraw, R. F. Pineda, K. Wojciechowski, L. K. Jagadamma, I. Samuel and N. Robertson, J. Mater. Chem. A, 2022, 10, 11046–11053 RSC.
- J.-P. Sun, A. D. Hendsbee, A. J. Dobson, G. C. Welch and I. G. Hill, Org. Electron., 2016, 35, 151–157 CrossRef CAS.
- R. Muñoz-Mármol, P. G. Boj, J. M. Villalvilla, J. A. Quintana, N. Zink-Lorre, Á. Sastre-Santos, J. Aragó, E. Ortí, P. Baronas, D. Litvinas, S. Juršėnas, F. Fenández-Lázaro and M. A. Díaz-García, J. Phys. Chem. C, 2021, 125, 12277–12288 CrossRef.
- R. Ganesamoorthy, R. Vijayaraghavan, K. Ramki and P. Sakthivel, J. Sci.:Adv. Mater. Devices, 2018, 3, 99–106 Search PubMed.
- X. Shang, J. Ahn, J. H. Lee, J. C. Kim, H. Ohtsu, W. Choi, I. Song, S. K. Kwak and J. H. Oh, ACS Appl. Mater. Interfaces, 2021, 13, 12278–12285 CrossRef CAS PubMed.
- A. Arulkashmir, B. Jain, J. C. John, K. Roy and K. Krishnamoorthy, Chem. Commun., 2014, 50, 326–328 RSC.
- C. Che, S. Tong, Y. Jia, J. Yang, X. He, S. Han, Q. Jiang and Y. Ma, Front. Chem., 2023, 11, 1–9 Search PubMed.
- Y. Kim, H. W. Lee, M. J. Jung, S. Lee, J. H. Oh and E. K. Lee, Adv. Mater. Technol., 2025, e00681 CrossRef CAS.
- D. Powell and L. Whittaker-Brooks, Mater. Horiz., 2022, 9, 2026–2052 RSC.
- D. Powell, X. Zhang, C. I. Nwachukwu, E. J. Miller, K. R. Hansen, L. Flannery, J. Ogle, A. Berzansky, J. G. Labram, A. G. Roberts and L. Whittaker-Brooks, Adv. Mater., 2022, 34, 2204656 CrossRef CAS PubMed.
- J. Yao, B. Qiu, Z.-G. Zhang, L. Xue, R. Wang, C. Zhang, S. Chen, Q. Zhou, C. Sun, C. Yang, M. Xiao, L. Meng and Y. Li, Nat. Commun., 2020, 11, 2726 CrossRef CAS PubMed.
- D. Powell, Z. Rhodes, X. Zhang, E. J. Miller, M. Jonely, K. R. Hansen, C. I. Nwachukwu, A. G. Roberts, H. Wang, R. Noriega, S. D. Minteer and L. Whittaker-Brooks, ACS Mater. Au, 2022, 2, 482–488 CrossRef CAS PubMed.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- M. J. Ahrens, M. J. Tauber and M. R. Wasielewski, J. Org. Chem., 2006, 71, 2107–2114 CrossRef CAS PubMed.
- S. E. Penty, M. A. Zwijnenburg, G. R. F. Orton, P. Stachelek, R. Pal, Y. Xie, S. L. Griffin and T. A. Barendt, J. Am. Chem. Soc., 2022, 144, 12290–12298 CrossRef CAS PubMed.
- A. D. Hendsbee, J.-P. Sun, W. K. Law, H. Yan, I. G. Hill, D. M. Spasyuk and G. C. Welch, Chem. Mater., 2016, 28, 7098–7109 CrossRef CAS.
- M. Sadeghianlemraski, C. R. Harding, G. C. Welch and H. Aziz, ACS Appl. Energy Mater., 2020, 3, 11655–11665 CrossRef CAS.
- M. E. Farahat, M. A. Anderson, M. Martell, E. L. Ratcliff and G. C. Welch, ACS Appl. Mater. Interfaces, 2022, 14, 43558–43567 CrossRef CAS PubMed.
- K. M. Wolfe, S. Alam, E. German, F. N. Alduayji, M. Alqurashi, F. Laquai and G. C. Welch, Beilstein J. Org. Chem., 2023, 19, 1620–1629 CrossRef CAS PubMed.
- S. V. Dayneko, E. Cieplechowicz, S. S. Bhojgude, J. F. Van Humbeck, M. Pahlevani and G. C. Welch, Mater. Adv., 2021, 2, 933–936 RSC.
- P. Kumar Behera, S. Lenka, F.-R. Chen, M. Roy, I. Mondal, D. S. Shankar Rao, S. P. Senanayak, J.-H. Jou and A. S. Achalkumar, Chem. Eng. J., 2024, 497, 154719 CrossRef CAS.
- J. D. B. Koenig, Z. S. Dubrawski, K. R. Rao, J. Willkomm, B. S. Gelfand, C. Risko, W. E. Piers and G. C. Welch, J. Am. Chem. Soc., 2021, 143, 16849–16864 CrossRef CAS PubMed.
- J. D. B. Koenig, W. E. Piers and G. C. Welch, Chem. Sci., 2022, 13, 1049–1059 RSC.
- J. Cann, B. S. Gelfand and G. C. Welch, Mol. Syst. Des. Eng., 2020, 5, 1181–1185 RSC.
- M. R. Niazi, E. Hamzehpoor, P. Ghamari, I. F. Perepichka and D. F. Perepichka, Chem. Commun., 2020, 56, 6432–6435 RSC.
- L. Rocard, A. Goujon and P. Hudhomme, Molecules, 2020, 25, 1402 CrossRef CAS PubMed.
- L. Rocard, D. Hatych, T. Chartier, T. Cauchy and P. Hudhomme, Eur. J. Org Chem., 2019, 2019, 7635–7643 CrossRef CAS.
- M. Palomba, G. Carotenuto and A. Longo, Materials, 2022, 15, 6456 CrossRef CAS PubMed.
- C. Liang, Y.-T. Lin and J.-W. Shiu, J. Hazard. Mater., 2016, 302, 137–143 CrossRef CAS PubMed.
- S. Barik, T. Bletzacker and W. G. Skene, Macromolecules, 2012, 45, 1165–1173 CrossRef CAS.
- A. Bolduc, C. Mallet and W. G. Skene, Sci. China:Chem., 2013, 56, 3–23 CrossRef CAS.
- M. Bourgeaux and W. G. Skene, Macromolecules, 2007, 40, 1792–1795 CrossRef CAS.
- T. Lei, M. Guan, J. Liu, H.-C. Lin, R. Pfattner, L. Shaw, A. F. McGuire, T.-C. Huang, L. Shao, K.-T. Cheng, J. B.-H. Tok and Z. Bao, Proc. Natl. Acad. Sci. U.S.A., 2017, 114, 5107–5112 CrossRef CAS PubMed.
- A. Gapin, E. Chatir, O. Alévêque, C. Pasgrimaud, A. H. G. David, A. De Maria, M. Legros, L. Le Bras, E. Levillain and A. Goujon, J. Am. Chem. Soc., 2025, 147, 12218–12227 CrossRef CAS PubMed.
- W. Yan, Z. He, J. Jiang, D. Lu, Y. Gong, W. Yang, R. Xia, W. Huang and H. Xin, J. Mater. Chem. C, 2020, 8, 14773–14781 RSC.
- Z. B. Henson, G. C. Welch, T. Van Der Poll and G. C. Bazan, J. Am. Chem. Soc., 2012, 134, 3766–3779 CrossRef CAS PubMed.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- C. R. Harding, J. Cann, A. Laventure, M. Sadeghianlemraski, M. Abd-Ellah, K. R. Rao, B. S. Gelfand, H. Aziz, L. Kaake, C. Risko and G. C. Welch, Mater. Horiz., 2020, 7, 2959–2969 RSC.
- R. Mathew, A. Mazumder, P. Kumar, J. Matula, S. Mohamed, P. Brazda, M. Hariharan and B. Thomas, Chem. Sci., 2024, 15, 490–499 RSC.
- S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, J. Am. Chem. Soc., 2015, 137, 3300–3307 CrossRef CAS PubMed.
-
M. E. Casida, in Recent Advances in Computational Chemistry, World Scientific, 1995, vol. 1, pp. 155–192 Search PubMed.
- H.-Y. Tsai and K.-Y. Chen, Dyes Pigm., 2013, 96, 319–327 CrossRef CAS.
- D. H. Harris, S. Brixi, B. S. Gelfand, B. H. Lessard and G. C. Welch, J. Mater. Chem. C, 2020, 8, 9811–9815 RSC.
- I. E. Park, S. Brixi, M. Martell, M. U. Ocheje, R. D. Pettipas, D. H. Harris, B. S. Gelfand, S. Rondeau-Gagné, B. H. Lessard and G. C. Welch, J. Mater. Chem. C, 2021, 9, 13630–13634 RSC.
- R. D. Pettipas, A. Hoff, B. S. Gelfand and G. C. Welch, ACS Appl. Mater. Interfaces, 2022, 14, 3103–3110 CrossRef CAS PubMed.
- F. Bureš, RSC Adv., 2014, 4, 58826–58851 RSC.
- H. Zhu, M. Li, J. Hu, X. Wang, J. Jie, Q. Guo, C. Chen and A. Xia, Sci. Rep., 2016, 6, 24313 CrossRef CAS PubMed.
- L. R. Snyder, J. Chromatogr. Sci., 1978, 16, 223–234 CAS.
- L. Porrès, A. Holland, L.-O. Pålsson, A. P. Monkman, C. Kemp and A. Beeby, J. Fluoresc., 2006, 16, 267–273 CrossRef PubMed.
- O. F. Mohammed, O. Kwon, C. M. Othon and A. H. Zewail, Angew. Chem., Int. Ed., 2009, 48, 6251–6256 CrossRef CAS PubMed.
- H. A. Z. Sabek, A. M. M. Alazaly, D. Salah, H. S. Abdel-Samad, M. A. Ismail and A. A. Abdel-Shafi, RSC Adv., 2020, 10, 43459–43471 RSC.
- O. F. Mohammed and E. Vauthey, J. Phys. Chem. A, 2008, 112, 3823–3830 CrossRef CAS PubMed.
- M. J. Grant, A. Hoff, L. G. Kaake and G. C. Welch, Sens. Diagn., 2024, 3, 817–821 RSC.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- Z. Chen, X. Li, S. Qin, Y. Gong, Z. Liu, M. Yuan, T. Liang, L. Meng and Y. Li, CCS Chem., 2025, 7, 507–518 CrossRef CAS.
- P. J. Mohr, B. N. Taylor and D. B. Newell, Rev. Mod. Phys., 2012, 84, 1527–1605 CrossRef CAS.
- A. V. Marenich, S. V. Jerome, C. J. Cramer and D. G. Truhlar, J. Chem. Theory Comput., 2012, 8, 527–541 CrossRef CAS PubMed.
- K. Fukuda, Y. Takeda, Y. Yoshimura, R. Shiwaku, L. T. Tran, T. Sekine, M. Mizukami, D. Kumaki and S. Tokito, Nat. Commun., 2014, 5, 4147 CrossRef CAS PubMed.
- V. Vohra, N. T. Razali, R. Wahi, L. Ganzer and T. Virgili, Opt. Mater.:X, 2022, 13, 100127 CAS.
- M. A. Butt, Coatings, 2022, 12, 1115 CrossRef CAS.
- C. Larsen, P. Lundberg, S. Tang, J. Ràfols-Ribé, A. Sandström, E. Mattias Lindh, J. Wang and L. Edman, Nat. Commun., 2021, 12, 4510 CrossRef CAS PubMed.
-
S. Abbott and H. Yamamoto, HSPiP Software, 5th edn, (version 5.2.05), 2015 Search PubMed.
- M. Díaz De Los Ríos and E. Hernández Ramos, SN Appl. Sci., 2020, 2, 676 CrossRef.
- P. Ślęczkowski, Y. Xiao, J. W. Wu, C. Adachi, L. S. Vargas, D. Kreher, B. Heinrich, J.-C. Ribierre and F. Mathevet, J. Phys. Chem. C, 2024, 128, 21826–21835 CrossRef PubMed.
- C. Shao, M. Grüne, M. Stolte and F. Würthner, Chem. - Eur. J., 2012, 18, 13665–13677 CrossRef CAS PubMed.
- N. Zink-Lorre, M. G. Ramírez, S. Pla, P. G. Boj, J. A. Quintana, J. M. Villalvilla, Á. Sastre-Santos, F. Fernández-Lázaro and M. A. Díaz-García, Molecules, 2023, 28, 6776 CrossRef CAS PubMed.
- A. Hoff, M. Martell, A. Gasonoo, J. D. B. Koenig, P. Simón Marqués, E. Cieplechowicz, M. Pahlevani and G. C. Welch, Adv. Eng. Mater., 2023, 25, 2201437 CrossRef CAS.
- S. V. Dayneko, M. Pahlevani and G. C. Welch, ACS Appl. Mater. Interfaces, 2019, 11, 46017–46025 CrossRef CAS PubMed.
- Y. Cui, Y. Wang, J. Bergqvist, H. Yao, Y. Xu, B. Gao, C. Yang, S. Zhang, O. Inganäs, F. Gao and J. Hou, Nat. Energy, 2019, 4, 768–775 CrossRef CAS.
- H. Ren, J. Chen, Y. Li and J. Tang, Adv. Sci., 2021, 8, 2002418 CrossRef CAS PubMed.
- C. J. Clarke, W.-C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed.
- L. Sanchez-Duenas, E. Gomez, M. Larrañaga, M. Blanco, A. M. Goitandia, E. Aranzabe and J. L. Vilas-Vilela, Materials, 2023, 16, 3940 CrossRef CAS PubMed.
- R. Abbel, Y. Galagan and P. Groen, Adv. Eng. Mater., 2018, 20, 1701190 CrossRef.
- S. M. Majeed, M. K. A. Mohammed and D. S. Ahmed, J. Mater. Chem. C, 2022, 10, 16480–16491 RSC.
- J. Panidi, E. Mazzolini, F. Eisner, Y. Fu, F. Furlan, Z. Qiao, M. Rimmele, Z. Li, X. Lu, J. Nelson, J. R. Durrant, M. Heeney and N. Gasparini, ACS Energy Lett., 2023, 8, 3038–3047 CrossRef CAS PubMed.
- C. Liao, M. Zhang, X. Xu, F. Liu, Y. Li and Q. Peng, J. Mater. Chem. A, 2019, 7, 716–726 RSC.
- S. J. Jeon, N. Zhao, Y. Yuan and Y. Li, Adv. Mater. Technol., 2024, 9, 2301542 CrossRef CAS.
- A. Holmes, E. Deniau, C. Lartigau-Dagron, A. Bousquet, S. Chambon and N. P. Holmes, ACS Nano, 2021, 15, 3927–3959 CrossRef CAS PubMed.
- C. Xie, X. Zeng, C. Li, X. Sun, S. Liang, H. Huang, B. Deng, X. Wen, G. Zhang, P. You, C. Yang, Y. Han, S. Li, G. Lu, H. Hu, N. Li and Y. Chen, Energy Environ. Sci., 2024, 17, 2441–2452 RSC.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Zachary T.
Gardner
a,
Zachary T.
Gardner









