
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceClick-assembled N-graphene–C60 hybrids for ultrafast electron transfer
Luis M.
Arellano
 a,
Habtom B.
Gobeze
b,
Youngwoo
Jang
b,
María J.
Gómez-Escalonilla
*a,
Paul A.
Karr
c,
Francis
D'Souza
*b and
Fernando
Langa
*a
a,
Habtom B.
Gobeze
b,
Youngwoo
Jang
b,
María J.
Gómez-Escalonilla
*a,
Paul A.
Karr
c,
Francis
D'Souza
*b and
Fernando
Langa
*a
aUniversidad de Castilla-La Mancha, Instituto de Nanociencia, Nanotecnología y Materiales Moleculares (INAMOL), 45071-Toledo, Spain. E-mail: Fernando.Langa@uclm.es
bChemistry and Materials Science and Engineering, University of North Texas, 76203-5017 Denton, TX, USA. E-mail: francis.dsouza@unt.edu
cDepartment of Physical Sciences and Mathematics, Wayne State College, 1111 Main Street, Wayne, Nebraska 68787, USA
First published on 6th October 2025
Abstract
A novel donor–acceptor hybrid derived from N-doped graphene (NG) and an electron acceptor, C60, has been newly synthesized using click chemistry and characterized by a suite of physico–chemical techniques. The usage of click chemistry resulted in a relatively high degree of functionalization. Due to the presence of two C12 alkyl chains on the fulleropyrrolidine moiety, the NG-C60 hybrid was found to be relatively soluble in most organic solvents, facilitating both spectroscopic and electrochemical characterization. Fluorescence studies revealed quenching of the fulleropyrrolidine emission, indicating the occurrence of excited-state events. While DFT studies provided insights into the geometry and localization of the frontier orbitals, the TD-DFT studies performed at the B3LYP/6-311G(d,p) level suggested the possibility of excited-state charge transfer from several excited states. Subsequent femtosecond transient absorption studies performed in DMF confirmed electron transfer, wherein the material could be characterized. The charge transfer state persisted for approximately 12 ps before populating the low-lying 3C60*, highlighting the material's potential for light energy harvesting applications.
Introduction
The increasing global demand for sustainable energy solutions has intensified research into materials capable of harvesting and converting solar energy with high efficiency. Among the various strategies explored, the design of donor–acceptor (D–A) systems that facilitate photoinduced electron transfer (ET) has emerged as a particularly promising approach.1,2 These systems emulate key aspects of natural photosynthesis, where efficient charge separation and migration are essential for energy conversion.3 Fullerenes, especially C60, have long been recognized as prototypical zero-dimensional electron acceptors due to their high electron affinity, low reorganization energy, and excellent photostability.4 Their integration into artificial photosynthetic assemblies has led to significant advances in the development of photovoltaic and photocatalytic devices.5–12In parallel, graphene and its derivatives have acquired considerable attention as potential electron donors in D–A systems.5,13–20 While pristine graphene is a zero-band-gap material with limited reactivity, heteroatom doping—particularly with nitrogen—introduces localized electronic states and reactive sites that enhance both its chemical versatility and electronic properties.21–23 Nitrogen-doped graphene (NG) exhibits improved conductivity, tunable band structure, and enhanced interaction with electron-accepting species, making it an ideal platform for constructing hybrid nanomaterials.24,25
A few years ago, we reported the first example of nitrogen-doped graphene (NG) functionalization via derivatization at the heteroatom.26 Building on this strategy, we subsequently achieved the covalent attachment of C60 to NG, thereby constructing a novel donor–acceptor hybrid.27 However, the synthetic approach employed in that study was relatively inefficient, yielding only moderate product quantities and requiring extended reaction times. To overcome these limitations, the present work introduces a new and more effective method for linking C60 to NG, employing the well-established copper-catalyzed azide–alkyne cycloaddition (click chemistry). This approach enables higher functionalization efficiency, improved yields, and reduced reaction times, facilitating the development of advanced carbon-based donor–acceptor systems.28 As demonstrated in this study, the use of click chemistry enabled significantly improved reaction yields, reduced reaction times, and a higher degree of functionalization. Additionally, femtosecond transient spectroscopy revealed ultrafast charge separation, highlighting the novelty of this work. Following the successful synthesis of the NG-C60hybrid, comprehensive characterization was carried out using a range of physico–chemical techniques, including UV-vis absorption and fluorescence spectroscopy, FT-IR, Raman spectroscopy, X-ray photoelectron spectroscopy (XPS), and electrochemical analysis. The potential of the hybrid to undergo excited-state electron transfer was subsequently investigated through density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations. Finally, femtosecond transient absorption spectroscopy provided direct evidence of photoinduced charge transfer, revealing the formation of a charge-separated state with broad spatial and temporal characteristics. The key findings of this work are detailed in the following sections.
Results and discussion
The synthetic route for the preparation of the target material, NG-C60, is outlined in Scheme 1. Previously, the fulleropyrrolidine derivative 3 was obtained in moderate yield (45%) by 1,3-dipolar cycloaddition reaction between N-(3,5-didodecyloxybenzyl)glycine 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29 and 4-((trimethylsilyl)ethynyl)benzaldehyde 230 (see Experimental section in SI for further details), following the procedure reported by Prato et al.31 To enhance the solubility of the fullerene derivative, dodecyloxy chains were incorporated. Compound 3 was characterized using standard spectroscopic techniques (see Fig. S1–S3 in SI). The typical pyrrolidine fingerprint appears in the 1H-NMR spectrum between 5.19 and 4.17 ppm, along with the TMS signal from the protected triple bond (0.24 ppm). The structure of 3 was unequivocally confirmed by MALDI-TOF-MS spectrometry (see Fig. S3).
29 and 4-((trimethylsilyl)ethynyl)benzaldehyde 230 (see Experimental section in SI for further details), following the procedure reported by Prato et al.31 To enhance the solubility of the fullerene derivative, dodecyloxy chains were incorporated. Compound 3 was characterized using standard spectroscopic techniques (see Fig. S1–S3 in SI). The typical pyrrolidine fingerprint appears in the 1H-NMR spectrum between 5.19 and 4.17 ppm, along with the TMS signal from the protected triple bond (0.24 ppm). The structure of 3 was unequivocally confirmed by MALDI-TOF-MS spectrometry (see Fig. S3).
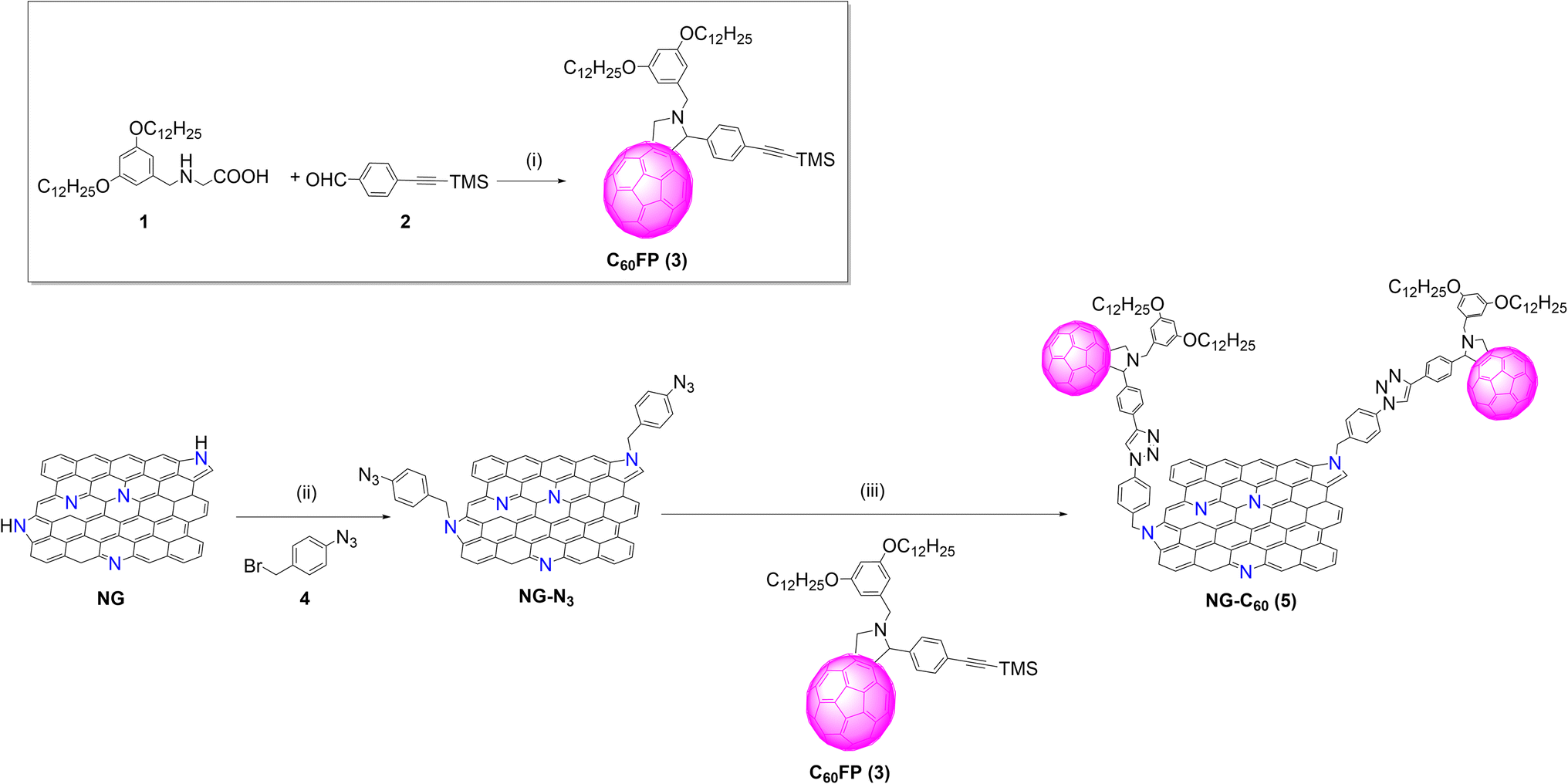 | ||
| Scheme 1 Synthetic route for the preparation of NG-C60 (5) hybrid. Reagents and conditions: (i) C60, toluene, 6 h, reflux, 45%; (ii) K2CO3, NMP, 70 °C, 48 h, (iii) TBAF, THF, NMP, rt or 0 °C, 3 h; then, CuSO4·5H2O, sodium ascorbate, NMP, 70 °C, 48 h. | ||
Next, commercial nitrogen-doped graphene, NG (http://www.timesnano.com), was first exfoliated using N-methyl-2-pyrrolidone (NMP);32 this resulted in a dispersion of few-layer graphene sheets (3 or 4 layers) (see Fig. S4a–c). The Raman spectrum (laser 532 nm) of exfoliated NG displays the characteristic D and G bands for graphitic materials at 1353 and 1586 cm−1 respectively (Fig. S4a). As previously noted for nitrogen-doped graphene,37 the presence of significant defects due to nitrogen within the graphitic network increased the intensity of the D band and broadened the Raman bands. Also, atomic force microscopy (AFM) analysis revealed that the material exhibits an average thickness of approximately 3 nm. Additionally, thinner nanosheets with an average height of ∼1.4 nm, corresponding to single-layer graphene, were also identified (Fig. S4b). Finally, transmission electron microscopy (TEM) further confirmed the presence of agglomerated few-layer graphene domains, displaying lateral dimensions of several hundred nanometers and high crystallinity (Fig. S4c).
The reaction of exfoliated NG with 1-azido-4-(bromomethyl) benzene 433 in NMP, in the presence of potassium carbonate (K2CO3) yielded the functionalized NG-N3 (see further details in SI); the triple bond of fullerene derivative 3 was deprotected with tetra-n-butylammonium fluoride (TBAF), followed by its reaction with NG-N3 in NMP via copper-catalysed alkyne–azide cycloaddition (click chemistry).12,20,34,35 This process afforded the target donor–acceptor hybrid material NG-C60 (5).
Various techniques were employed to investigate the structural and thermal properties of the new NG-fullerene hybrid. Thermogravimetric analysis (TGA) of the starting NG material showed a weight loss of approximately 10.3%, attributed to intrinsic defects; on the other hand, NG-N3 exhibited a gradual decrease in weight of 32.5%, a higher value attributed to the covalent incorporation of the azide group. After the “click” reaction, the hybrid material 5 showed an additional weight loss of 9.6% (after subtracting the 32.5% loss from the NG-N3 precursor), which is attributed to the decomposition of the fulleropyrrolidine unit 3 (Fig. S5).
The FT-IR spectrum of NG-C60 (5) (Fig. 1a) displays the aliphatic C–H stretching modes between 2850 and 2950 cm−1 (from the alkyl chains), along with the characteristic vibrational peak of C60 at 529 cm−1, confirming effective covalent functionalization of the N-doped graphene. Additionally, the bands associated with the alkyne group in 3 and the azide moiety in NG-N3 disappeared, further verifying the success of the functionalization.20,36
 | ||
Fig. 1 (a) FTIR spectra of NG-C60 (5) ( ) compared with the corresponding spectra of NG ( ) compared with the corresponding spectra of NG ( ), NG-N3 ( ), NG-N3 ( ), and 3 ( ), and 3 ( ). The coloured area highlights the typical region for ν(N3) and ν(alkyne) vibrations; (b) Superimposed Raman spectra (514 nm) for (b) starting NG ( ). The coloured area highlights the typical region for ν(N3) and ν(alkyne) vibrations; (b) Superimposed Raman spectra (514 nm) for (b) starting NG ( ), 5 ( ), 5 ( ) and 3 ( ) and 3 ( ); (c) Expanded G band region for NG ( ); (c) Expanded G band region for NG ( ) and functionalized NG-C60 (5) ( ) and functionalized NG-C60 (5) ( ). The spectra are normalized to the intensity of the G-mode to facilitate observation of the peak shift (7 cm−1). ). The spectra are normalized to the intensity of the G-mode to facilitate observation of the peak shift (7 cm−1). | ||
The Raman spectrum of NG-C60 (5) is shown in Fig. 1b, alongside the spectra of the starting NG material and derivative 3. The spectrum of 5 displays the two most intense peaks typical for carbon materials: the D band (1348 cm−1) and the G band (1590 cm−1) bands. Evidence for the presence of the C60 cage is provided by the emergence of a Raman mode at 1456 cm−1, assigned to the C60 Ag (2) pentagonal pinch mode, which is red-shifted by 4 cm−1 compared to that of fulleropyrrolidine precursor.7,20,27 Furthermore, the G band in 5 is upshifted by 7 cm−1 relative to NG (Fig. 1c), attributed to the p-doping effect of C60, consistent with previous reports on similar fullerene-containing hybrids.7,20,27 Finally, deformation of the 2D band was also observed, likely caused by nitrogen atom intercalation.27,37 It is important to emphasize that the hybrid material investigated in this study is nitrogen-doped graphene, and therefore not composed exclusively of carbon atoms. The incorporation of nitrogen modifies both the atomic structure and the electronic properties of the material, rendering conventional methods for estimating the degree of functionalization inapplicable. Likewise, the ID/IG ratio obtained from Raman spectroscopy (vide infra) cannot be reliably used as an indicator of functionalization, due to defect-induced band broadening associated with nitrogen doping.
X-ray photoelectron spectroscopy (XPS) analysis of NG, 3, and 5 was performed (Tables S1, S2 and Fig. S6–S8). The high-resolution N 1s XPS spectrum of the starting NG material shows three distinct components located at 398.2, 400.2 and 401.8 eV, corresponding to pyridinic-N, pyrrolic-N, and quaternary (or “graphitic) nitrogen atoms, respectively27 (see Fig. S6b in the SI). After the first functionalization, the N 1s spectrum of NG-N3 revealed two additional bands at binding energies of 401.5 and 404.4 eV (Fig. S7b), which are reasonably attributed to the differently charged nitrogen atoms in the azide groups.38 Finally, XPS analysis of the C 1s and N 1s core regions of the final NG-C60 (5) material (Fig. 2 and Table S1) provided definitive proof that the click reaction had occurred: (1) no peak corresponding to the free azide group at 405 eV was observed in N 1s XPS region, confirming the formation of the triazole ring; (2) the presence of sp3 carbon atoms (285.7 eV) from the alkyl chains of the fullerene derivative 3 was detected in the C 1s region, consistent with previous works.28
 | ||
| Fig. 2 (a) C 1s and (b) N 1s core level XPS regions of NG-C60 (5) and their relative fits. | ||
All these data agree with the FTIR results discussed above, confirming the successful formation of material 5via click chemistry.
To further characterize the newly prepared hybrid 5, electrochemical redox potentials were investigated by Osteryoung Square Wave Voltammetry (OSWV) in o-dichlorobenzene/acetonitrile (o-DCB/AcCN, 4:1 v/v) containing 0.1 M tetra-n-butylammonium hexafluorophosphate (TBAPF6) under an argon atmosphere and at room temperature (Table 1). The NG-C60 (5) hybrid exhibits redox activity in the cathodic region, attributed to the reduction of the attached fullerene derivative. It displays the characteristic peaks of C60 cage at −1.23, −1.63, −2.14 and −2.54 V (Table 1 and Fig. S9). These peaks are broader and shifted by 50–90 mV compared to those of fulleropyrrolidine precursor 3 (−1.14, −1.55, −2.09 and −2.55 V respectively). These shifts are attributed to electronic interactions between the NG surface and the fullerene cage.7,27
| Sample | E red 1/V | E red 2/V | E red 3/V | E red 4/V |
|---|---|---|---|---|
| a o-DCB/ACN solution containing 0.1 M TBAPF6 and using Ag/AgNO3 as a reference electrode, glassy carbon as a working electrode, and a Pt wire counter electrode. Scan rate: 100 mV s−1. Potentials are referenced to Fc/Fc+. | ||||
| 3 | −1.14 | −1.55 | −2.09 | −2.55 |
| NG-C60 (5) | −1.23 | −1.63 | −2.14 | −2.54 |
The spectral properties of these materials were also investigated. The UV-vis absorption spectra of the pristine NG material revealed broad absorption across the entire spectral range, with a lower energy shoulder at 328 nm; upon attachment of the fullerene derivative, a new broad band appeared with a maximum at 429 nm, which is blue-shifted (∼5 nm) compared to the characteristic absorption peak of the precursor fullerene derivative 3 (433 nm) (Fig. 3a). Similarly, in the emission spectra, precursor fulleropyrrolidine 3 displays a weak emission band at 711 nm upon excitation at 433 nm. This band is markedly quenched in N-graphene-fullerene hybrid 5 (Fig. 3b), suggesting the occurrence of excited-state processes such as electron transfer or energy transfer.7,20,27
 | ||
Fig. 3 (a) UV-Visible spectra of NG-C60 (5) hybrid material ( ) compared to NG ( ) compared to NG ( ) and derivative 3 ( ) and derivative 3 ( ) recorded in o-DCB. (b) Fluorescence spectra of NG-C60 (5) hybrid material () compared to that of 3 ( ) recorded in o-DCB. (b) Fluorescence spectra of NG-C60 (5) hybrid material () compared to that of 3 ( ) in o-DCB as solvent, upon excitation at 433 nm. ) in o-DCB as solvent, upon excitation at 433 nm. | ||
DFT and TD-DFT studies
DFT and time-dependent DFT studies play a significant role in establishing the geometry, electronic structure, and excited state properties of hybrid systems composed of donor and acceptor entities. In the present study, the NG-C60 structure was built on a local pc using GaussView 6.0. The structure was then uploaded to a supercomputer and optimized to a stationary point on the Born-Oppenheimer surface in solution at the CAM-B3LYP/6-311G(d,p) level utilizing the Self-Consistent Reaction Field (SCRF) Polarizable Continuum Model (PCM) with o-dichlorobenzene (κ = 9.9949) as parametrized in the Gaussian 16 software suite. The coordinates of the optimized structure are given in Table S3 in the SI. For this purpose, a reasonably large NG with N-atoms embedded in the graphene structure and functionalized through the terminal five-membered ring containing a doped N (the most reactive site for chemical functionalization) is utilized. Fig. 4 shows the optimized structure, frontier HOMO and LUMO orbitals (Fig. 4a–c). The doped N to the center of C60 was about 16.7 Å, revealing a lack of geometrical crowding. However, due to limited solubility, they likely exist in some aggregated form, as discussed earlier from optical studies. Notably, the HOMO on NG and LUMO on C60, establishing their role of electron donor and electron acceptor, was borne out. This could also be envisioned from the ground-state molecular electrostatic potential map (MEP, see Fig. 4d). This trend persisted when a second C60 unit on the NG was added. Further, time-dependent DFT (TD-DFT) studies were performed for the first 25 excited states. The excitation energies ranged between 1.48 and 2.97 eV, most revealing positive oscillator strength values. The computed spectrum is bathoscopic from ∼420 to ∼840 nm. However, of the twenty-five computed excitations, only two indicate charge transfer. The calculated charge transfer band is at ∼490 nm. The band is comprised of two absorptions. A weak absorption at ∼493 nm (2.52 eV), which is the 11th excited state (HOMO-3 → LUMO+1). A strong absorption at ∼490 nm (2.53 eV), which is the 13th excited state (HOMO-2 → LUMO+3). The charge difference maps from the ground state to the assigned one, and the excited state MEPs are shown in Fig. 4e and f for the first excited state, while Fig. S10 and S11 in the SI show the first six excited states. Clear segregation of charges on the fullerene (negative charge) and NG (positive charge) parts was witnessed for the first five excited states (the 6th is a locally excited state), indicating the occurrence of excited-state charge separation in this hybrid. | ||
| Fig. 4 (a) Optimized geometry, (b) HOMO, (c) LUMO, (d) ground state MEP, (e) first excited state MEP, and (f) charge difference map for the first excited state of NG-C60 hybrid calculated at the CAM-B3LYP/6-311G(d,p) level with the SCRF-PCM solvation model in o-dichlorobenzene. | ||
Femtosecond transient absorption studies
Encouraged by these theoretical predictions, femtosecond transient absorption (fs-TA) spectral studies were performed in DMF by exciting the samples at 440 nm to seek evidence of excited-state charge separation in this hybrid. First, the fs-TA spectra of the C60 derivative used in the click reaction were investigated. As shown in Fig. 5a, the instantaneously formed 1C60* revealed excited state absorption (ESA) peaks in the 640–720 nm range, and at 772 nm. Weaker ground state bleaching (GSB) peaks at 483 and 511 nm, and a stimulated emission (SE) type peak at 745 nm was observed. Decay and recovery of the ESA and GSB/SE peaks revealed a new broad peak in the 550–650 nm range corresponding to 3C60*, a product of intersystem crossing (ISC). The fs-TA spectra of pristine NG shown in Fig. 5b revealed no measurable signal (the sharp peak at 510 nm is due to instrument scattering); however, in the near-IR region, weak transient features corresponding to an exciton were observed (Fig. 5c). Kinetic analysis of this signal (Fig. 5d) resulted in a lifetime of 0.18 ps, which agreed with the earlier reported value. The photo response of both NG and C60 used in building the donor–acceptor hybrid is borne out by this study. | ||
| Fig. 5 Fs-TA spectra at the indicated delay times of C60 used in the click reaction, (b and c) of NG covering the visible and near-IR region, and (d) the NG exciton time profile. The samples were excited using a 100-fs pulsed laser tuned to 433 nm for C60 and 440 nm for NG in DMF. | ||
Fig. 6a and b show the fs-TA spectra at the indicated delay times for NG-C60 in DMF. At earlier delay times, the spectral features were what was observed for the C60 control in Fig. 5a. That is, the formation of 1C60* and later, undergoing ISC to populate 3C60*. Interestingly, in the near-IR region, the excitonic peak of NG also had a clear peak in the 1000 nm region (see spectrum at 890 fs in Fig. 6c), expected for C60− that lasted beyond the excitonic peak recovery time (see spectrum at 3.44 ps in Fig. 6c), indicating charge transfer in the NG-C60 hybrid. The broad nature of this peak could be attributed to the low solubility (aggregation) of the hybrid. Global data analysis was subsequently performed, which resulted in a lifetime of ∼12 ps for the charge transfer state (see Fig. 6d). These results provide clear evidence that NG-1C60* undergoes a charge transfer state and subsequently populates the 3C60* before returning to the ground state, as evidenced by the data in Fig. 6b.
 | ||
| Fig. 6 Fs-TA spectra at the indicated delay times of the NG-C60 hybrid (a and b covering the visible region and c covering the near-IR region). The result of global target analysis is shown in Fig. 6d. The sample was excited using a 100-fs pulsed laser tuned to 433 nm, corresponding to C60 in DMF. | ||
Summary and conclusions
In this study, we successfully synthesized a novel donor–acceptor hybrid material composed of nitrogen-doped graphene (NG) covalently linked to a fullerene (C60) derivative via copper-catalyzed azide–alkyne cycloaddition (click chemistry). This synthetic strategy enabled a higher degree of functionalization, improved reaction yields, and reduced reaction times compared to previously reported methods. The resulting NG-C60 hybrid exhibited good solubility in organic solvents, facilitating comprehensive structural and spectroscopic characterization.Theoretical investigations using DFT and TD-DFT calculations supported the formation of a charge-separated state, with the HOMO localized on the NG donor and the LUMO on the C60 acceptor. These predictions were experimentally validated by femtosecond transient absorption spectroscopy, which revealed ultrafast charge separation and the formation of a charge transfer state with a lifetime of approximately 12 ps, followed by population of the triplet C60* state.
Overall, the NG-C60 hybrid developed in this work represents a promising platform for light-harvesting applications, offering a robust and tunable architecture for efficient photoinduced charge separation. These findings contribute to the broader development of carbon-based donor–acceptor systems for energy conversion technologies.
Author contributions
All the authors approved the final version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Data availability
The data supporting the findings of this study are included in the main text and in the supplementary information (SI). Additionally, they are available from the corresponding author upon reasonable request. The data have also been deposited in Zenodo and can be accessed via the following link: https://zenodo.org/records/17293193. See DOI: https://doi.org/10.1039/d5sc06142c.Acknowledgements
This work was financially supported by MICINN of Spain (PID2022-141687OB-I00 and TED2021-131255B-C42) and the Junta de Comunidades de Castilla-La Mancha, together with European FEDER funds (SBPLY/21/180501/000142) and by the US-National Science Foundation (2345836 to FD). M. A. thanks MINECO (CTQ2016-79189-R) for a doctoral FPI grant.References
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890 CrossRef CAS PubMed.
- F. D'Souza and O. Ito, Chem. Soc. Rev., 2012, 41, 86 RSC.
- M. R. Wasielewski, Chem. Rev., 1992, 92, 435 CrossRef CAS.
- G. B. Daniel, K. Nachimuthu and J. L. Nallasivam, Chem Asian J., 2025, 20, e202401800 CrossRef CAS PubMed.
- M. Barrejón, L. M. Arellano, F. D'Souza and F. Langa, Nanoscale, 2019, 11, 14978 RSC.
- M. Vizuete, M. Barrejón, M. J. Gómez-Escalonilla and F. Langa, Nanoscale, 2012, 4, 4370 RSC.
- M. Barrejón, H. B. Gobeze, M. J. Gómez-Escalonilla, J. L. G. Fierro, M. Zhang, M. Yudasaka, S. Iijima, F. D'Souza and F. Langa, Nanoscale, 2016, 8, 14716 RSC.
- T. Umeyama and H. Imahori, J. Phys. Chem. C, 2013, 117, 3195 CrossRef CAS.
- T. Umeyama and H. Imahori, Nanoscale Horiz., 2018, 3, 352 RSC.
- M. Chen, R. Guan and S. Yang, Adv. Sci., 2019, 6, 1800941 CrossRef PubMed.
- M. Vizuete, M. J. Gómez-Escalonilla, J. L. G. Fierro, K. Ohkubo, S. Fukuzumi, M. Yudasaka, S. Iijima, J. F. Nierengarten and F. Langa, Chem. Sci., 2014, 5, 2072 RSC.
- M. Vizuete, M. J. Gómez-Escalonilla, J. L. G. Fierro, M. Yudasaka, S. Iijima, M. Vartanian, J. Iehl, J.-F. Nierengarten and F. Langa, Chem. Commun., 2011, 47, 12771 RSC.
- G. Bottari, M. Á. Herranz, L. Wibmer, M. Volland, L. Rodríguez-Pérez, D. M. Guldi, A. Hirsch, N. Martín, F. D'Souza and T. Torres, Chem. Soc. Rev., 2017, 46, 4464 RSC.
- K. Jayaramulu, S. Mukherjee, D. M. Morales, D. P. Dubal, A. K. Nanjundan, A. Schneemann, J. Masa, S. Kment, W. Schuhmann, M. Otyepka, R. Zbořil and R. A. Fischer, Chem. Rev., 2022, 122, 17241 CrossRef CAS PubMed.
- A. Criado, M. Melchionna, S. Marchesan and M. Prato, Angew. Chem., Int. Ed., 2015, 54, 10734 CrossRef CAS PubMed.
- Z. Li, Z. Liu, H. Sun and C. Gao, Chem. Rev., 2015, 115, 7046 CrossRef CAS PubMed.
- M. Barrejón, M. J. Gómez-Escalonilla, J. L. G. Fierro, P. Prieto, J. R. Carrillo, A. M. Rodríguez, G. Abellán, M. C. López-Escalante, M. Gabás, J. T. López-Navarrete and F. Langa, Phys. Chem. Chem. Phys., 2016, 18, 29582 RSC.
- A. Stergiou, R. Cantón-Vitoria, M. N. Psarrou, S. P. Economopoulos and N. Tagmatarchis, Prog. Mater. Sci., 2020, 114, 100683 CrossRef CAS.
- R. Canton-Vitoria, A. Z. Alsaleh, G. Rotas, Y. Nakanishi, H. Shinohara, F. D' Souza and N. Tagmatarchis, Nanoscale, 2022, 14, 15060 RSC.
- M. Barrejón, M. Vizuete, M. J. Gómez-Escalonilla, J. L. G. Fierro, I. Berlanga, F. Zamora, G. Abellán, P. Atienzar, J.-F. Nierengarten, H. García and F. Langa, Chem. Commun., 2014, 50, 9053 RSC.
- H. Xu, L. Ma and Z. Jin, J. Energy Chem., 2018, 27, 146 CrossRef.
- R. Yadav and C. K. Dixit, J. Sci. Adv. Mater. Devices, 2017, 2, 141 CrossRef.
- P. Lazar, R. Mach and M. Otyepka, J. Phys. Chem. C, 2019, 123, 10695 CrossRef CAS.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781 CrossRef CAS.
- C. Hu, D. Liu, Y. Xiao and L. Dai, Prog. Nat. Sci., 2018, 28, 121 CrossRef CAS.
- M. Barrejón, A. Primo, M. J. Gómez-Escalonilla, J. L. G. Fierro, H. García and F. Langa, Chem. Commun., 2015, 51, 16916 RSC.
- M. Barrejón, L. M. Arellano, H. B. Gobeze, M. J. Gómez-Escalonilla, J. L. G. Fierro, F. D'Souza and F. Langa, Chem. Sci., 2018, 9, 8221 RSC.
- L. M. Arellano, H. B. Gobeze, Y. Jang, M. Barrejón, C. Parejo, J. C. Álvarez, M. J. Gómez-Escalonilla, Á. Sastre-Santos, F. D'Souza and F. Langa, Chem.–Eur. J., 2022, 28, e202200254 CrossRef CAS PubMed.
- F. Ajamaa, T. M. Figueira Duarte, C. Bourgogne, M. Holler, P. W. Fowler and J.-F. Nierengarten, Eur. J. Org Chem., 2005, 3766 CrossRef CAS.
- F. Auras, L. Ascherl, A. H. Hakimioun, J. T. Margraf, F. C. Hanusch, S. Reuter, D. Bessinger, M. Döblinger, C. Hettstedt, K. Karaghiosoff, S. Herbert, P. Knochel, T. Clark and T. Bein, J. Am. Chem. Soc., 2016, 138, 16703 CrossRef CAS PubMed.
- M. Prato and M. Maggini, Acc. Chem. Res., 1998, 31, 519 CrossRef CAS.
- H. B. Gobeze, L. M. Arellano, A. M. Gutiérrez-Vílchez, M. J. Gómez-Escalonilla, Á. Sastre-Santos, F. Fernández-Lázaro, F. Langa and F. D'Souza, Nanoscale Adv., 2019, 1, 4009 RSC.
- M. Belkheira, D. El Abed, J.-M. Pons and C. Bressy, Chem.–Eur. J., 2011, 17, 12917 CrossRef CAS PubMed.
- M. Vizuete, M. J. Gómez-Escalonilla, J. L. G. Fierro, A. S. D. Sandanayaka, T. Hasobe, M. Yudasaka, S. Iijima, O. Ito and F. Langa, Chem.–Eur. J., 2010, 16, 10752 CrossRef CAS PubMed.
- J. Mateos-Gil, L. Rodríguez-Pérez, M. Moreno Oliva, G. Katsukis, C. Romero-Nieto, M. Á. Herranz, D. M. Guldi and N. Martín, Nanoscale, 2015, 7, 1193 RSC.
- L. Rodríguez-Pérez, J. Ramos-Soriano, A. Pérez-Sánchez, B. M. Illescas, A. Muñoz, J. Luczkowiak, F. Lasala, J. Rojo, R. Delgado and N. Martín, J. Am. Chem. Soc., 2018, 140, 9891 CrossRef PubMed.
- N. Mahmood, C. Zhang, H. Yin and Y. Hou, J. Mater. Chem. A, 2014, 2, 15 RSC.
- G. Tuci, D. Mosconi, A. Rossin, L. Luconi, S. Agnoli, M. Righetto, C. Pham-Huu, H. Ba, S. Cicchi, G. Granozzi and G. Giambastiani, Chem. Mater., 2018, 30, 8257 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |