
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-entropy sulfide nanoflowers with multi-atomic catalytic sites for efficient nitrate-to-ammonia conversion
Yuanting
Lei
ab,
Lili
Zhang
b,
Xiaochen
Wang
b,
Dan
Wang
b,
Yafei
Zhao
 b,
Bing
Zhang
b,
Ning
Zhang
*ab and
Huishan
Shang
*ab
b,
Bing
Zhang
b,
Ning
Zhang
*ab and
Huishan
Shang
*ab
aState Key Laboratory of Critical Metals Beneficiation, Metallurgy and Purification, Zhengzhou University, Zhengzhou 450001, P. R. China. E-mail: shanghs@zzu.edu.cn; nzhang@zzu.edu.cn
bSchool of Chemical Engineering, Zhengzhou Key Laboratory of Advanced Separation Technology, Zhengzhou University, Zhengzhou 450001, P. R. China
First published on 2nd September 2025
Abstract
Electrocatalytic nitrate-to-ammonia conversion (NO3RR) offers a sustainable alternative to the energy-intensive Haber–Bosch process. High-entropy materials (HEMs), which exploit compositional diversity, lattice distortion, and d-band modulation, demonstrate remarkable electrocatalytic potential. However, they encounter significant synthesis challenges in achieving structural control and elemental homogeneity. Herein, a hollow spherical-flower NiCoFeV–S high-entropy sulfide is prepared via a mild hydrothermal method. After optimizing the metal compositions and their respective proportions, the hollow spherical-flower NiCoFeV–S exhibits exceptional bifunctional performance. It requires only 267 mV of overpotential for the oxygen evolution reaction (OER) at 100 mA cm−2 while simultaneously achieving remarkable performance in the NO3RR, with an ammonia yield of 16.6 mg h−1 mgcat.−1 and a faradaic efficiency of 93.2%. Theoretical investigations identify three enhancement mechanisms: (1) hierarchical nanoarchitecture enabling maximized active site accessibility, (2) multi-metal synergy fine-tuning charge transfer dynamics, and (3) an upshifted d-band center synergistically accelerating water dissociation and hydrogenation kinetics. This work develops a simple synthesis strategy for HEMs, offering insights into their electronic structure modulation and holding significant promise for energy applications.
Introduction
Ammonia (NH3) serves as a critical chemical raw material in industrial applications.1,2 Globally, the predominant method for ammonia production is the energy-intensive Haber–Bosch process, which consumes approximately 2% of the world's annual energy and accounts for roughly 1% of global greenhouse gas emissions.3,4 Therefore, it is imperative to explore clean and renewable ammonia synthesis technologies that align with sustainable development goals.5 Currently, there has been a growing emphasis on the utilization of electrochemical techniques for the conversion of nitrogen-containing small molecules into ammonia under ambient conditions. However, due to the low solubility of N2 and the difficulty in breaking the strong N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds, the electrocatalytic N2 reduction reaction exhibits relatively low production activity.6,7 By contrast, the electrocatalytic nitrate reduction reaction (NO3RR) holds greater promise due to the higher water solubility of nitrates and the lower bond energy of N
N bonds, the electrocatalytic N2 reduction reaction exhibits relatively low production activity.6,7 By contrast, the electrocatalytic nitrate reduction reaction (NO3RR) holds greater promise due to the higher water solubility of nitrates and the lower bond energy of N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O.8,9 Moreover, the excessive use of nitrogen-based fertilizers and inadequate treatment of industrial wastewater have resulted in severe nitrate pollution, posing significant risks to human health.10,11 Consequently, the NO3RR has been attracting significant research interest in recent years,12–16 as employing nitrate pollutants from wastewater as a nitrogen source for synthesizing high-value-added NH3 represents not only a green process consistent with sustainable development principles but also an effective strategy to mitigate nitrate-induced environmental pollution.17,18
O.8,9 Moreover, the excessive use of nitrogen-based fertilizers and inadequate treatment of industrial wastewater have resulted in severe nitrate pollution, posing significant risks to human health.10,11 Consequently, the NO3RR has been attracting significant research interest in recent years,12–16 as employing nitrate pollutants from wastewater as a nitrogen source for synthesizing high-value-added NH3 represents not only a green process consistent with sustainable development principles but also an effective strategy to mitigate nitrate-induced environmental pollution.17,18
The NO3RR proceeds through a complex, multi-step electron–proton transfer mechanism, requiring the sequential involvement of eight electrons and nine protons, as well as the formation of multiple reactive intermediates.19 This intricate pathway encounters two major challenges: first, the competing hydrogen evolution reaction becomes a dominant side process, especially under low overpotential conditions;20 second, the inherent limitations in nitrate adsorption capacity and slow electron transfer kinetics collectively impair the selectivity of NH3 production and Faraday efficiency, fundamentally constraining the NO3RR performance.21 Transition metal phthalocyanines (TMPcs) have emerged as highly promising electrocatalysts for the NO3RR due to the transition metal center serving as the primary active site.22,23 The catalytic performance is heavily influenced by the central metal ion in its M–N4 coordination environment.24–26 However, the M–N4 coordination provides single metal sites with limited electron transfer efficiency. Consequently, the development of multi-site catalytic systems is of paramount importance for the NO3RR. Notably, practical implementation of the NO3RR requires integration with the anodic oxygen evolution reaction (OER) in a dual-electrode configuration. However, the intrinsic thermodynamics and kinetic barriers of the OER significantly limit the overall system efficiency.27,28 Consequently, it is imperative to develop efficient catalysts to simultaneously optimize both the cathodic NO3RR and anodic OER processes.
High-entropy materials (HEMs) represent an innovative class of multi-component alloy systems, distinguished by their composition of five or more metallic elements in near-equal molar ratios (5–35%).29,30 This unique structural disorder imparts significant lattice distortion effects, d-band ligand modulation phenomena, and elevated configurational entropy.31,32 In HEMs, the presence of various adjacent atoms facilitates the emergence of a series of active sites that yield nearly continuous distributions of associated adsorption energies.33–35 By optimizing the free energy associated with intermediate adsorption and regulating the reaction barriers, it is possible to achieve precise control over electrocatalytic reaction pathways.36 Consequently, the application of compositional engineering strategies to establish electronic synergistic mechanisms among multiple active sites has emerged as a pivotal approach for enhancing the intrinsic catalytic activity of HEMs.15 Notably, the interplay between compositional regulation in HEMs and the morphological effects from nanosheet structures holds significant potential for substantially improving the electrocatalytic performance. This enhancement can be achieved through electronic structure optimization coupled with increased exposure of active sites.37,38 The HEMs synthesized under thermodynamically non-equilibrium conditions exhibit marked differences in heterogeneous nucleation and growth kinetics due to their multi-component nature.39,40 The synthesis of these HEMs typically necessitates stringent conditions, including elevated temperatures and pressures. In particular, with regard to nanoscale control, achieving the precise fabrication of nanosheet-like high-entropy materials under mild reaction conditions while ensuring a uniform distribution of elements remains a significant technical challenge currently encountered in this field.41–45
Herein, we constructed a high-entropy sulfide (NiCoFeV–S) with a hollow spherical-flower structure via a mild hydrothermal method. NiCoFeV–S exhibits exceptional bi-functional electrocatalytic performance in the OER and NO3RR. The optimized NiCoFeV–S catalyst achieved an impressively low OER overpotential of 267 mV at 100 mA cm−2, concurrently delivering remarkable NO3RR performance characterized by a high ammonia production rate of 16.6 mg h−1 mgcat.−1 and a faradaic efficiency of 93.2%. Mechanistic investigations that integrate experimental and computational analyses have identified three critical factors contributing to catalytic enhancement: (1) the unique hierarchical architecture providing abundant exposed active sites, (2) optimized electronic configurations through multi-metallic synergistic interactions, and (3) an elevated d-band center that facilitates water dissociation kinetics and promotes hydrogenation pathways for NH3 synthesis. This work establishes a facile synthesis strategy for HEMs while providing fundamental insights into their electronic structure modulation, paving the way for their broader application in sustainable energy conversion and environmental remediation technologies.
Results and discussion
The NiCoFeV–OH precursor was synthesized via a hydrothermal reaction in a mixed solution containing Ni, Co, Fe, V, urea, and NH4F at 120 °C for 8 hours. Multi-metal coprecipitation is achieved relying on the pH slow-release effect of urea, and ammonium fluoride assists in regulating the crystallinity, ultimately forming a uniform hydroxide solid solution. The NiCoFeV–S was subsequently produced through anion exchange between the S2− at 120 °C for an additional 8 hours (Fig. 1a). Ion exchange occurs between S2− provided by sodium sulfide and hydroxides, and lattice reconstruction is completed under high temperature and high pressure, resulting in the formation of a multi-metal sulfide solid solution. Similarly, the NiCoFe–S, NiCo–S, and Ni–S compounds were fabricated by varying the metal elements used during the hydrothermal step to synthesize their respective M–OH precursors. The morphology of NiCoFeV–S was investigated through scanning electron microscopy (SEM) and transmission electron microscopy (TEM) analyses. Fig. 1b–d and S1 illustrate well-defined hollow spherical-flower structures self-assembled from nanosheets for the NiCoFeV–S material. The notable presence of voids among the nanosheet units enhances active site exposure and facilitates mass-charge transport. Notably, the nanosheet units at the edges are visible in Fig. S2a, while a distinct lattice spacing of 0.288 nm is observed in Fig. S2b, which corresponds to the (311) planes of CoNi2S4 (PDF #24-0334).46 Elemental mapping images presented in Fig. S3, S4 and Table S1 demonstrate a uniform distribution and coexistence of Ni, Co, Fe, V, and S elements within the structure. These TEM results preliminarily indicate a homogeneous high-entropy phase for NiCoFeV–S. The morphological evolution of NiCoFe–S, NiCo–S, and Ni–S catalysts with varying metal compositions was systematically investigated through comprehensive SEM and TEM analyses (Fig. S5–S13) with quantitative elemental distributions detailed in Tables S2–S4. Notably, this structural reorganization appears to stem from fundamental differences in the distinct ionic radii and charge densities of the constituent metal ions. These critical physicochemical parameters directly influence their hydrolysis kinetics and coordination modes with hydroxide ions during catalyst formation.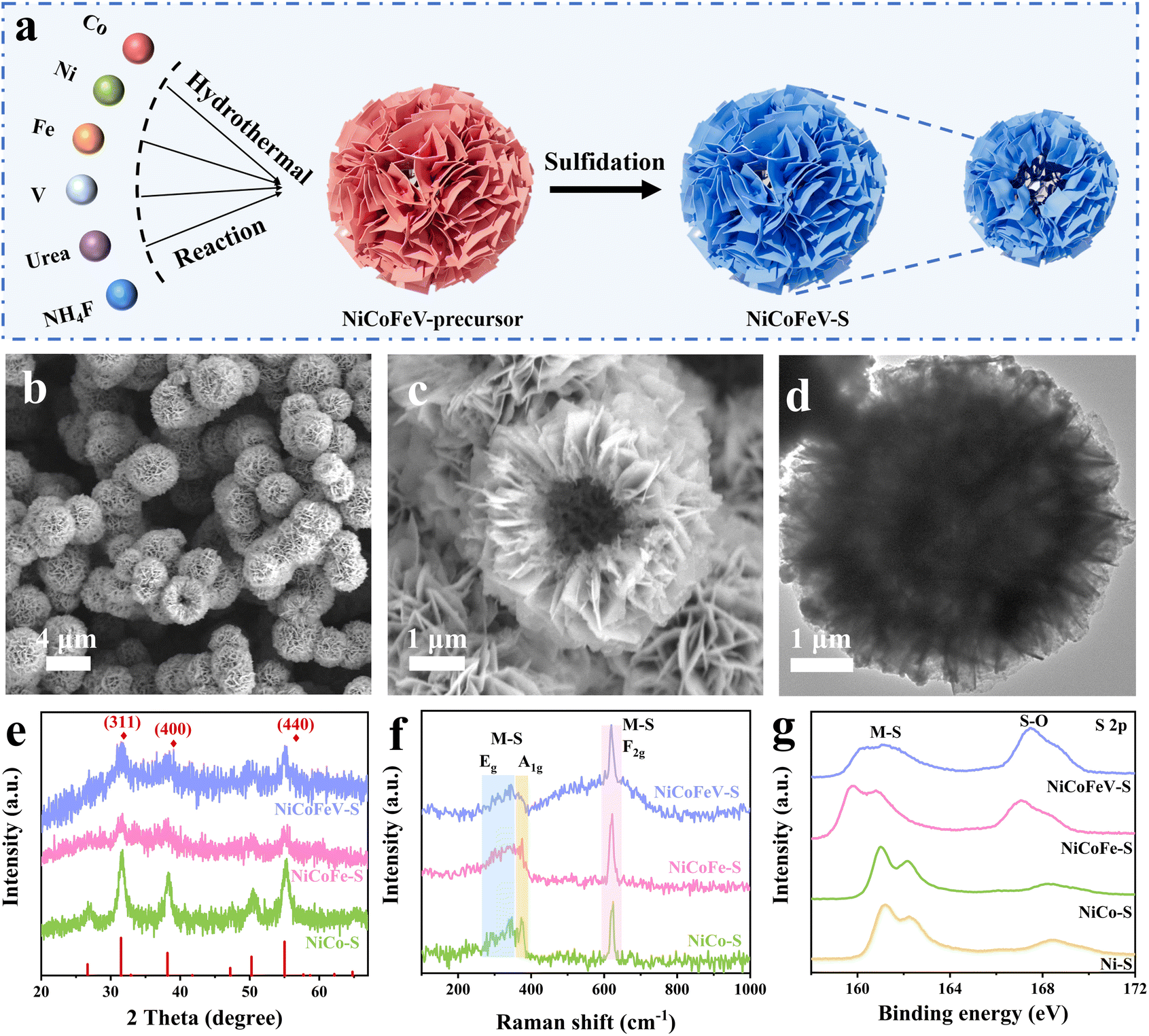 | ||
| Fig. 1 (a) A schematic illustration depicting the formation process of the hollow spherical-flower-like NiCoFeV–S. (b and c) SEM and (d) TEM images of NiCoFeV–S. (e) XRD patterns, (f) Raman spectra, and (g) S 2p XPS spectra of NiCoFeV–S, NiCoFe–S, and NiCo–S. | ||
The structural and compositional characteristics were analyzed using X-ray diffraction (XRD). As illustrated in Fig. S14, the XRD pattern of the NiCoFeV–OH precursor confirmed the formation of a single-phase high-entropy hydroxide precursor. Furthermore, both NiCoFe–OH and NiCo–OH exhibit diffraction peaks that are characteristic of typical hydroxides and consistent with those of NiCoFeV–OH (Fig. S15), thereby confirming the reliability of the synthesis method for producing a single-phase high-entropy hydroxide precursor. After the sulfidation process, the characteristic peaks observed in the XRD spectra of NiCo–S, NiCoFe–S, and NiCoFeV–S exhibited strong alignment with the standard CoNi2S4 (PDF #24-0334) (Fig. 1e).47,48 Specifically, peaks at 31.5°, 38.2°, 41.7°, 50.3°, and 55.3° were attributed to the (311), (400), (331), (511), and (440) planes, respectively, indicating that NiCoFeV–S is a single phase without any detectable impurities. This sulfide structure enables the disorderly and uniform distribution of each metal element. Moreover, the crystalline structure and chemical component of NiCoFeV–S were further studied by Raman spectra with NiCoFe–S and NiCo–S as a comparison. The spectra of NiCoFeV–S, NiCoFe–S, and NiCo–S displayed similar peaks, suggesting the synthesized catalysts were composed of identical single-phase sulfide structures (Fig. 1f). In particular, the peaks at ∼280–340 and ∼380 cm−1 were attributed to Eg and A1g modes, respectively, while the band around 620 cm−1 was associated with the F2g mode of M–S.49 The observation that the peak at 619.8 cm−1 of NiCoFeV–S exhibited a left shift compared to those of NiCoFe–S (622.2 cm−1) and NiCo–S (624.7 cm−1), consistent with the hypothesis that interstitial atoms are present after metal doping, as evidenced by the phenomenon of lattice expansion and in line with the TEM results.49,50 The Raman analysis further validated the single-phase structure of NiCoFeV–S, showing consistency with the TEM and XRD results.
Furthermore, the porous structure and specific surface area of hollow spherical-flower NiCoFeV–S were determined by the N2 adsorption–desorption test. The results are illustrated in Fig. S16; the NiCoFeV–OH precursor and NiCoFeV–S were found to deliver typical type IV isotherms with pronounced hysteresis loops, underscoring that the hollow-spherical-flower features a mesoporous structure.51 The specific surface areas of NiCoFeV–OH and NiCoFeV–S were determined to be 186.8 and 31.8 m2 g−1, respectively, using the Brunauer–Emmett–Teller (BET) method. The corresponding average pore diameter distribution curves revealed a mesoporous distribution for NiCoFeV–OH and NiCoFeV–S. The pore sizes were determined to be 3.8 nm and 12.3 nm, respectively, using the Barrett–Joyner–Halenda (BJH) method, with corresponding pore volumes of 0.815 and 0.309 cm3 g−1, respectively (Table S5). The decrease in the specific surface area and pore volume, coupled with an increase in the pore size of NiCoFeV–S, can principally be ascribed to defective structures resulting from sulfur replacement during sulfidation.52 The hollow spherical-flower-like morphology and mesoporous structure of NiCoFeV–S are expected to facilitate electrolyte diffusion, reduce the diffusion distance of reactants and electrons, and consequently enhance the electrocatalytic activity.
X-ray photoelectron spectroscopy (XPS) was employed to elucidate the chemical states of the elements present in NiCoFeV–S. First, as shown in Fig. S17, the survey spectra corroborated the co-existence of the Ni, Co, Fe, V, and S elements in NiCoFeV–S. The Ni 2p XPS spectra exhibited four peaks located at binding energies of 852.08, 855.08, 871.88, and 872.96 eV, which correspond to Ni0 and Ni2+ for Ni 2p3/2 and 2p1/2 (Fig. S18a), respectively.53,54 As shown in Fig. S18b, the Co 2p spectrum displayed Co 2p3/2 peaks at 777.49 and 780.02 eV and Co 2p1/2 peaks at 792.56 and 796.19 eV. Among them, the peaks at 777.49 and 792.56 eV were attributed to Co3+, and the peaks at 780.02 and 796.19 eV belonged to Co2+.55 The valence state of Fe was further demonstrated by the Fe 2p spectrum (Fig. S18c), the peaks at 710.50 and 712.90 eV were attributed to Fe2+ and Fe3+ of Fe 2p3/2, while the peaks at 723.60 and 726.30 eV were assigned to Fe2+ and Fe3+ of Fe 2p1/2.56 The V 2p spectra were fitted to two peaks of 515.74 and 522.82 eV assigned to V 2p3/2 and V 2p1/2, respectively, which are the characteristic peaks of V4+ (Fig. S18d).57,58 For the comparison samples Ni–S, NiCo–S, and NiCoFe–S (Fig. S19–S21), the valence characteristics were similar to those of NiCoFeV–S. Therefore, according to the Fe and V 2p spectra analysis, the Fe and V elements may be mixed in the tetrahedral/octahedral site and the octahedral site, respectively. It is noteworthy that the intensity of S–O differs for S 2p, as a consequence of different elemental compositions (Fig. 1g). Specifically, compared with Ni–S and NiCo–S, the increased peak intensity assigned to S–O bonds in NiCoFe–S and NiCoFeV–S suggests a robust interaction between the O 2p and S 2p orbitals, which will resist intense self-oxidation of catalysts in the subsequent OER process.59 When considered in conjunction with the XRD results, it can be concluded that the oxygen atom in the S–O bond partially replaces the lattice sulfur atoms, and the phase structure remains unaltered. The sulfur–oxygen bond is a “dual-functional regulatory factor” that takes into account both the OER and NO3RR. It demonstrates considerable potential to enhance the catalytic performance through inducing charge rearrangement and optimizing the adsorption energy of the active site for the OER/NO3RR intermediate; introducing O-related sites and high-valent metal ions to increase the number of effective active centers; the high bond energy S–O bond inhibits sulfur loss, enhance the stability, and adapts to different environments of the two reactions (oxidation/reduction). The XPS results demonstrated that incorporating different elements altered the electronic structure, thereby exerting an influence on the catalytic properties of the catalysts.
The catalytic activity towards the OER was investigated in a typical three-electrode system on a CHI660E in 1.0 M KOH. NiCoFe–S, NiCo–S, Ni–S, NiCoFeV–OH, and commercial RuO2 were also tested under the same experimental conditions for comparison. As illustrated in Fig. 2a, the linear sweep voltammetry (LSV) of NiCoFeV–S exhibited a lower overpotential of 267 mV at 100 mA cm−2, demonstrating the most favorable OER performance than NiCoFe–S (291 mV), NiCo–S (348 mV), and Ni–S (299 mV), as well as NiCoFeV–OH (297 mV) and commercial RuO2 (289 mV) (Fig. S22). It was demonstrated that the presence of both the sulfide phase and a high-entropy structure is essential for enhancing the OER activity. Moreover, the OER performance of NiCoFeV–S was better than that of some reported catalysts in 1.0 KOH at 100 mA cm−2 (Table S7), confirming the significant advantages of the high-entropy sulfide NiCoFeV–S. The enhanced OER activity is associated with the favorable reaction kinetics, and the OER kinetics were evaluated by the Tafel plots originating from LSV. As revealed in the Tafel plots in Fig. 2b, the Tafel value of 72.5 mV dec−1 for NiCoFeV–S was lower than those of NiCoFe–S (78.9 mV dec−1), NiCo–S (92.7 mV dec−1), Ni–S (86.0 mV dec−1), and commercial RuO2 (82.6 mV dec−1). The smaller Tafel slope observed for NiCoFeV–S reflects accelerated kinetics and enhanced catalytic activity.60 The enhanced performance of NiCoFeV–S could be attributed to the synergistic interactions between the constituent elements and sulfidation, in comparison to NiCoFe–S, NiCo–S, Ni–S, and the NiCoFeV–OH precursor. Furthermore, the electrochemical active surface area (ECSA) was evaluated through the electrochemical double-layer capacitance (Cdl), which provided a measurement of the number of available active sites over electrocatalysts (Fig. S23). As illustrated in Fig. S24a, the NiCoFeV–S showed a notable enhancement in Cdl reaching 6.87 mF cm−2, in comparison to NiCoFe–S (5.88 mF cm−2), NiCo–S (1.36 mF cm−2), and Ni–S (2.46 mF cm−2). The results emphasize that NiCoFeV–S exposes more effective active sites due to the hollow spherical-flower porous structure and multi-metallic components. Apart from this, the electrochemical impedance (EIS) curves were obtained to investigate the charge transfer of the catalysts, which is pertinent to the accelerated reaction rate. The EIS measurement presented that NiCoFeV–S possessed a smaller Nyquist semicircle diameter than the control sample, ensuring favorable kinetics and a rapid charge-transfer rate during the OER process (Fig. S24b and S25).61 The OER durability of NiCoFeV–S was evidenced by a stable 88 hour CP curve (Fig. 2c). Moreover, the morphology and composition were preserved without alteration after the durability test (Fig. S26 and S27), which indicates the exceptional structural stability of NiCoFeV–S.
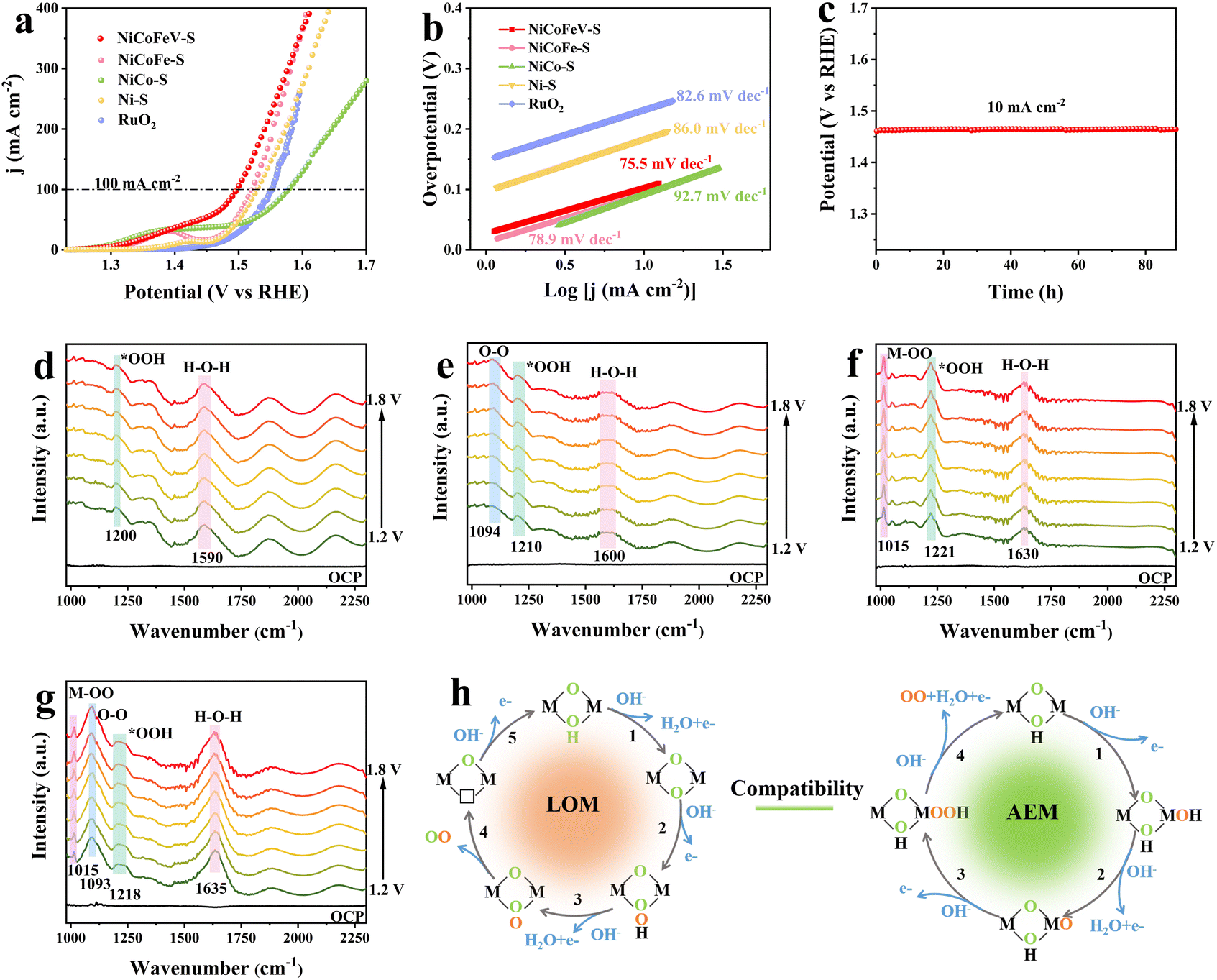 | ||
| Fig. 2 (a) OER polarization curves and (b) Tafel slopes of samples. (c) Chronopotentiometric response at 10 mA cm−2 of NiCoFeV–S. In situ electrochemical ATR-FTIR spectra of (d) Ni–S, (e) NiCo–S, (f) NiCoFe–S, and (g) NiCoFeV–S under different potential infrared signals. (h) Schematic illustration of the OER pathway over NiCoFeV–S. | ||
In situ attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR) was conducted to investigate the reaction pathways for the OER. In Fig. 2d and e, the characteristic peak at ∼1200 cm−1 was attributed to *OOH produced by the adsorbate evolution mechanism (AEM) pathway for Ni–S and NiCo–S.62 Noteworthily, NiCo–S exhibited a weak peak located at ∼1094 cm−1 belonging to the –O–O– formed during the lattice oxygen mechanism (LOM) pathway,63,64 suggesting the involvement of a weak LOM induced by the incorporation of Co. Moreover, NiCoFe–S exhibited a significantly enhanced intensity of the peaks corresponding to M–OO (∼1015 cm−1) and *OOH (∼1221 cm−1) in Fig. 2f.65,66 This demonstrates that the incorporation of Fe substantially enhances the AEM pathway and is conducive to the formation of the active phase, thereby improving the catalytic activity of the material. As shown in Fig. 2g, the characteristic peak of –O–O– (∼1093 cm−1) was markedly enhanced upon the incorporation of vanadium into NiCoFeV–S, thereby indicating that V facilitates the LOM pathway. Additionally, the presence of M–OO (∼1015 cm−1) and *OOH (∼1218 cm−1) further corroborates that the synergistic coupling of the AEM and LOM contributes to the improved activity and stability of NiCoFeV–S.62 Furthermore, the pronounced enhancement of the peak at 1635 cm−1 (H–O–H) is attributed to water,67 indicating the enhanced water adsorption and dissociation capabilities of NiCoFeV–S. In light of the aforementioned results, the in situ ATR-FTIR analysis further substantiated that the OER process was synergistically optimized through the compatibility of AEM and LOM, which was attributed to the synergistic effect within NiCoFeV–S (Fig. 2h). The high entropy sulfide NiCoFeV–S is employed as an electrocatalyst for the OER. Undoubtedly, the metal sites function as the primary active centers, facilitating the adsorption of OH− ions and optimizing the binding of key reaction intermediates (including O*, HO*, HOO*, and OO*) while simultaneously inducing lattice oxygen activation to synergistically enhance the OER performance.
Tetramethylammonium hydroxide (TMAOH) serves as a chemical probe that is commonly employed to detect the formation of active oxygen species (O22−) during the deprotonation process in the LOM pathway. This phenomenon can be ascribed to the strong electrostatic interaction between TMA+ and O22−, which adversely affects the activity of LOM-based electrocatalysts. A comparison of the data obtained in 1.0 M KOH and TMAOH electrolytes demonstrated that NiCoFeV–S exhibited larger current density differences compared to NiCoFe–S, NiCo–S, and Ni–S (Fig. S28). These results indicate that the OER activity of NiCoFeV–S was substantially suppressed in TMAOH, likely due to significant interference by TMA+ with the deprotonation step in the LOM pathway. It can be deduced that the rate-determining step (RDS) of the LOM pathway on NiCoFeV–S has been optimized, thereby enhancing the contribution of LOM to the OER process.68 As discussed earlier, the RDS of LOM exclusively involves proton transfer, which accounts for its significant pH dependence.69 Consequently, to further explore the underlying reasons for the enhanced lattice oxygen activity in NiCoFeV–S, LSV measurements were conducted in electrolytes with different pH values. As shown in Fig. S29a–d, the OER activity of NiCoFeV–S significantly improved with increasing pH, while NiCoFe–S, NiCo–S, and Ni–S demonstrated only a relatively slight pH dependency. Simultaneously, the order of the proton reaction was calculated on the reversible hydrogen electrode (RHE) scale (ρRHE) to assess the dependency of reaction kinetics on proton activity, providing deeper insight into the effect of pH on activity.70 A parameter closer to 1.0 indicates a higher sensitivity of the catalyst to pH changes.63 The ρRHE values of Ni–S, NiCo–S, and NiCoFe–S were 0.76, 0.65, and 0.97, respectively, significantly lower than that of NiCoFeV–S of 1.04 (Fig. S29e). This result suggests that the LOM pathway of NiCoFeV–S was achieved by increasing the proton transfer kinetics at the catalyst surface. Consequently, NiCoFeV–S demonstrated faster OER kinetics in comparison to NiCoFe–S, NiCo–S, and Ni–S.
The catalytic performance of the catalysts was investigated in a three-electrode H-type cell toward NO3− electroreduction at 25 °C. The LSV curves were sequentially compared with and without KNO3 with a scan rate of 5 mV s−1 to preliminarily study the NO3RR catalytic activity. NiCoFeV–S with NO3− exhibits significantly enhanced cathodic current compared to that in the absence of NO3−, indicating the high NO3RR activity of NiCoFeV–S. Moreover, the current increases with the incorporation of metal (Fig. 3a and S30), which demonstrates that the synergistic effect in NiCoFeV–S effectively contributes to the improved NO3RR performance. The electrolysis test was carried out at constant potentials from −0.4 to −1.0 V versus RHE for 2 hours in 1.0 M KOH solution containing 0.1 M KNO3. Following measurements, colorimetric techniques were used to determine the concentrations of products in the electrolyte. All the possible liquid products, including NO3−, NH3, and NO2−, were quantified by various colorimetric methods after plotting their standard calibration curves (Fig. S31–S33). The supplementary UV-vis spectra of NH4+ and NO2− after electrolysis at various potentials for the NiCoFeV–S, NiCoFe–S, NiCo–S, and Ni–S catalysts are provided in Fig. S34 and S35. Remarkably, NiCoFeV–S delivered an excellent NH3 yield of 16.6 mg h−1 mgcat.−1 at −0.8 V vs. RHE, compared to NiCoFe–S (9.2 mg h−1 mgcat.−1), NiCo–S (11.2 mg h−1 mgcat.−1), and Ni–S (5.9 mg h−1 mgcat.−1) (Fig. 3b). A correspondingly high NH3 FE of 93.2% was achieved by NiCoFeV–S at −0.8 V vs. RHE (Fig. 3c), which is higher than those of NiCoFe–S (82.7%), NiCo–S (67.4%), and Ni–S (34.6%). Moreover, the NH3 FE of NiCoFeV–S was the highest across various applied potentials among the prepared catalysts, indicating that NiCoFeV–S exhibits superior NH3 selectivity (Fig. 3d and S36). The H2 quantitative experimental results via gas chromatography revealed a positive correlation between the applied potential and both the hydrogen evolution yield and faradaic efficiency across all catalysts, except for NiCoFeV–S. Notably, NiCo–S and Ni–S demonstrated superior hydrogen evolution rates (Fig. S37a) and faradaic efficiency (Fig. S37b), while NiCoFe–S exhibited intermediate performance at elevated applied potentials. In contrast, NiCoFeV–S displayed comparatively lower H2 rates and faradaic efficiencies, particularly at an applied potential of −0.8 V. These findings provide further evidence for the enhanced selectivity of NiCoFeV–S toward the nitrate-to-ammonia electroreduction reaction under the investigated potential conditions.
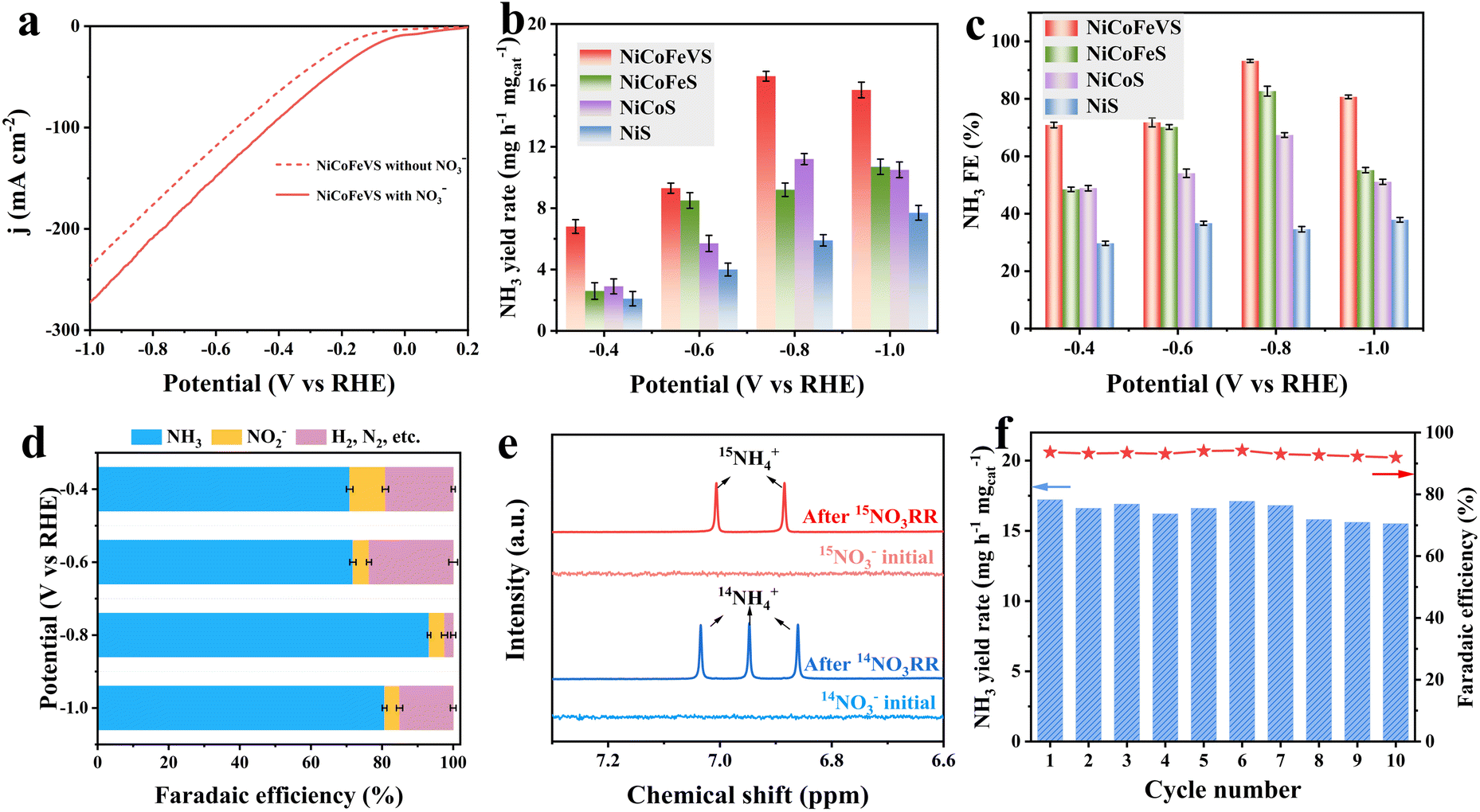 | ||
| Fig. 3 (a) LSV curves of NiCoFeV–S with and without 0.1 M NO3− in 1.0 M KOH. (b) NH3 yield and (c) NH3 FE of NiCoFeV–S, NiCoFe–S, NiCo–S, and Ni–S at different potentials. (d) NH3 FE of NiCoFeV–S at different potentials. (e) 1H NMR spectra of the electrolytes obtained before and after the NO3RR using 14NO3− and 15NO3− as the isotopic NO3− sources. (f) NH3 yield and FE of ten recycle tests. | ||
The ammonia generation performance of NiCoFeV–S was comparable to that of other noble metal catalysts and markedly superior to several transition metal sulfide catalysts (Table S8). Isotopic labeling using 1H NMR confirmed that ammonia was produced from nitrate electroreduction on NiCoFeV–S, thereby excluding the possibility of contamination (Fig. 3e). The presence of double peaks at 7.01 and 6.88 ppm has been observed when utilizing 15NO3− as the reactant. The triple peaks emerge at 7.03, 6.95, and 6.86 ppm when employing 14NO3− as the reactant.71 In contrast, no analogous signals indicative of double or triple distributions were observed in the electrolyte prior to the reaction. These observations provided compelling evidence that the detected generated NH3 originates from an electrocatalytic NO3RR process catalyzed by NiCoFeV–S.
The durability of NiCoFeV–S was evaluated by 10 consecutive electrolysis in an H-cell reactor under −0.8 V (Fig. 3f). It is clearly evident that both the yield and FE demonstrate a high level of stability, thereby providing compelling evidence for the exceptional stability of NiCoFeV–S. The multi-valence synergistic effect of nickel, cobalt, iron, and vanadium plays a pivotal role in enhancing structural stability and promoting the NO3RR. This multi-valence synergistic effect represents a distinctive property that is rarely observed in conventional single-metal or bimetallic sulfide systems. Furthermore, neither the morphology nor the composition undergoes any significant alterations after the durability test (Fig. S38 and S39), which reveals the superior structural stability of NiCoFeV–S when utilized as NO3RR catalysts. The XPS analysis revealed minor oxidation phenomena, whereas the predominant valence states of the constituent elements in the NiCoFeV–S catalyst remained largely unchanged, thereby providing further evidence for the structural stability of the high-entropy sulfide (Fig. S40). Moreover, the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image and the corresponding elemental mapping analysis of NiCoFeV–S following the NO3RR test reveal a homogeneous distribution of all constituent elements throughout the entire spherical nanoflower architecture (Fig. S41), providing further compelling evidence for the structural robustness of NiCoFeV–S. Quantitative elemental analysis demonstrated minimal variation in compositional ratios before and after the electrochemical process (Table S6), with negligible deviations in elemental content. These findings collectively substantiate the exceptional structural stability of NiCoFeV–S under electrochemical reduction reaction conditions.
To gain more insight into the intrinsic reaction pathway of the NO3RR over catalysts, in situ ATR-FTIR spectroscopy was conducted. As shown in Fig. S42 and 4a, Ni–S and NiCo–S exhibited low peak intensity of both *NH2OH and NH4+,72,73 suggesting that these samples exhibited slow reaction rates for the NO3RR. In contrast, NiCoFe–S demonstrated significantly enhanced *NH2OH peak intensities (Fig. 4b), indicating the incorporation of Fe promoted the production of *NH2OH, which is an important intermediate for ammonia generation. Obviously, NiCoFeV–S has been demonstrated to significantly enhance the peak intensity of NH4+ (Fig. 4c), indicating that the addition of V is conducive to the subsequent hydrogenation to generate NH3. Furthermore, the significantly increased H–O–H at 1636 cm−1 of NiCoFeV–S indicated that the water adsorption and dissociation were capable of supplying *H to hydrogenate NH3. A sharp comparison is provided by the vibrational bands of all reaction intermediates (*NH2OH, NH4+, and H–O–H) over NiCoFeV–S, which exhibit a significant enhancement in signal intensity with increasing potential. This indicates a significant enhancement in H2O absorption and hydrogenation, which can be ascribed to the synergistic effect of multiple elements. In situ infrared spectroscopy analysis of all samples demonstrated that the introduction of Fe and V catalyzed the enhanced formation of *NH2OH and the subsequent hydrogenation to generate NH3.
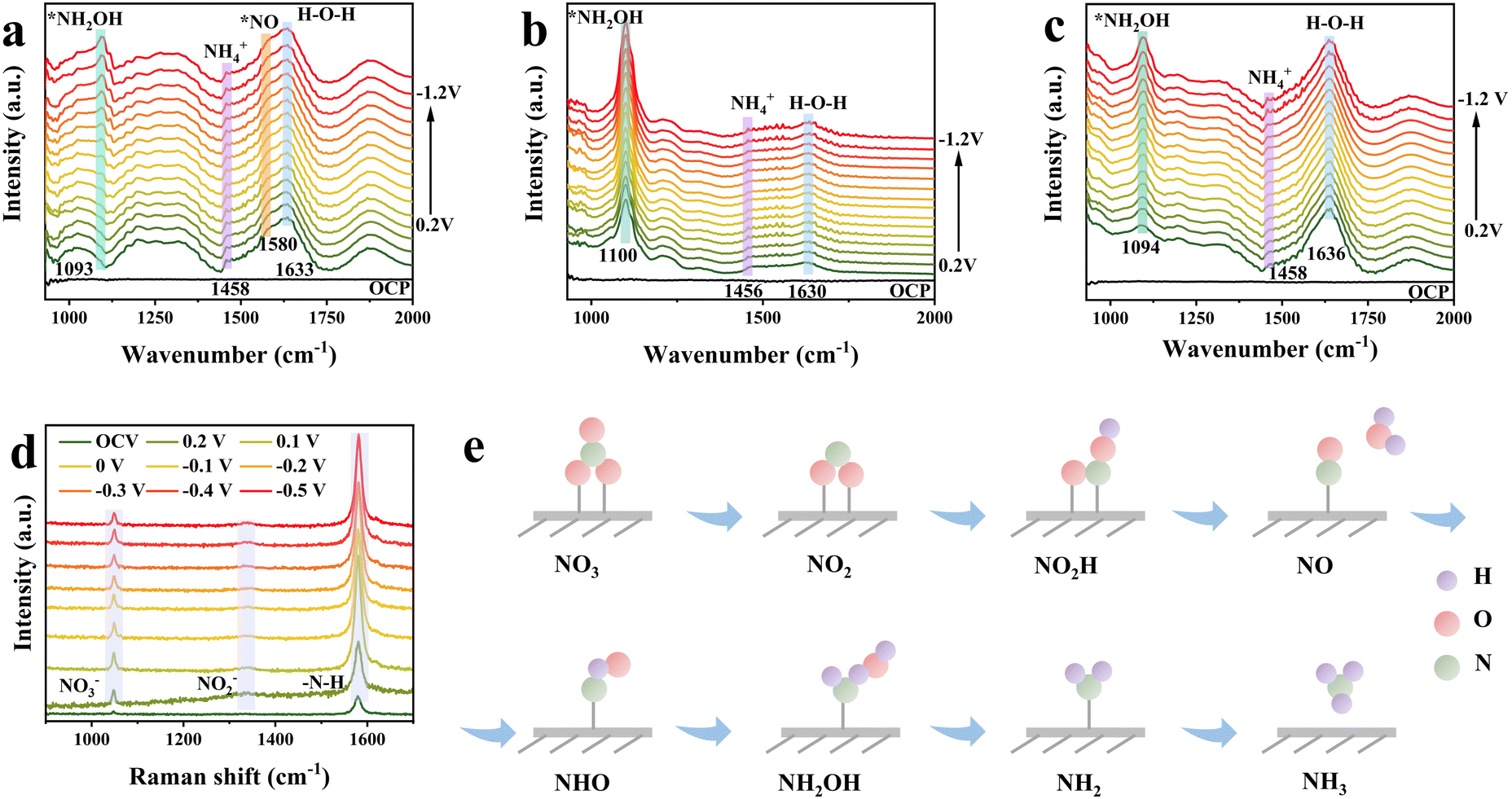 | ||
| Fig. 4 In situ electrochemical ATR-FTIR spectra of (a) NiCo–S, (b) NiCoFe–S, and (c) NiCoFeV–S under different potential infrared signals. (d) In situ Raman spectra of NiCoFeV–S at different operation potentials. (e) Schematic illustration of the NO3RR pathway. | ||
The in situ Raman spectroscopy of NiCoFeV–S was also performed to further understand the NO3RR process. The Raman spectra collected at different potentials from open-circuit voltage (OCV) to −0.5 V are shown in Fig. 4d. A characteristic Raman peak corresponding to the symmetric stretching mode of NO3− is observed at 1047 cm−1,6 indicating strong adsorption of nitrate ions. Upon application of a negative potential, the intensity of the NO3− peak diminishes concurrently with the emergence of a new peak at 1337 cm−1 attributed to NO2−, signifying rapid reduction of nitrate to nitrite. At potentials of −0.3 V and more negative values, a pronounced Raman band emerges at 1580 cm−1,9 assigned to the –N–H stretching vibration of NH3. Further reduction of the applied potential leads to a gradual attenuation of both NO3− and NO2− peaks, accompanied by significant enhancement of the –N–H stretching band. This spectral evolution suggests continuous and accelerated hydrogenation of intermediate, correlating with an increased rate of ammonia generation.74 Consequently, the formation of the critical intermediate *NH2OH was detected, thereby substantiating that NiCoFeV–S predominantly promotes the NO3RR process through the NHO pathway (Fig. 4e).75,76
To highlight the practical significance of a bifunctional NiCoFeV–S catalyst with low overpotentials for both the OER and NO3RR, the NiCoFeV–S catalyst was incorporated into an electrolyzer as both cathode and anode materials. As depicted in Fig. S43, the LSV curves of the electrocatalytic cathodic NO3RR coupled with the anodic OER over NiCoFeV–S were measured (Fig. 5a). The LSV presented in Fig. 5b showed the actual performance comparison of the HER/OER and NO3RR/OER electrolyzer. As expected, the NO3RR/OER required a lower cell voltage of 1.97 V to reach 10 mA cm−2, dropping approximately to 0.29 V to HER/OER and reflecting the accelerated kinetic reaction rates for the NO3RR/OER. Furthermore, the electrolyzer exhibited no discernible degradation during the 25 hours of observation (Fig. 5c), thereby suggesting that the electrochemical activity remained stable. Consequently, NiCoFeV–S enabled a high-performing electrolyzer towards the cathodic NO3RR and the anodic OER with excellent bifunctional electrocatalytic activity and durability. To alleviate the slow mass transfer process and further evaluate the potential of NiCoFeV–S in practical applications, a two-electrode flow cell coupling the NO3RR‖OER system with a NO3− reduction cathode (0.1 M KNO3 and 1.0 M KOH) and an OER anode (1.0 M KOH) was further assembled (Fig. S44). The LSV curves depicted in Fig. S45a indicated that the addition of 0.1 M KNO3 reduced the voltage required to achieve a high current density of 400 mA cm−2 at 2.42 V, which is substantially superior to the HER‖OER. Stability tests of the NiCoFeV–S‖NiCoFeV–S system were carried out at a current density of 100 mA cm−2. The system exhibited excellent long-term durability (Fig. S45b), maintaining stable performance over a 25 hour test at a constant current of 100 mA cm−2. Thus, the aforementioned results suggested that NiCoFeV–S exhibited excellent electrochemical stability and potential for practical applications in the NO3RR‖OER system.
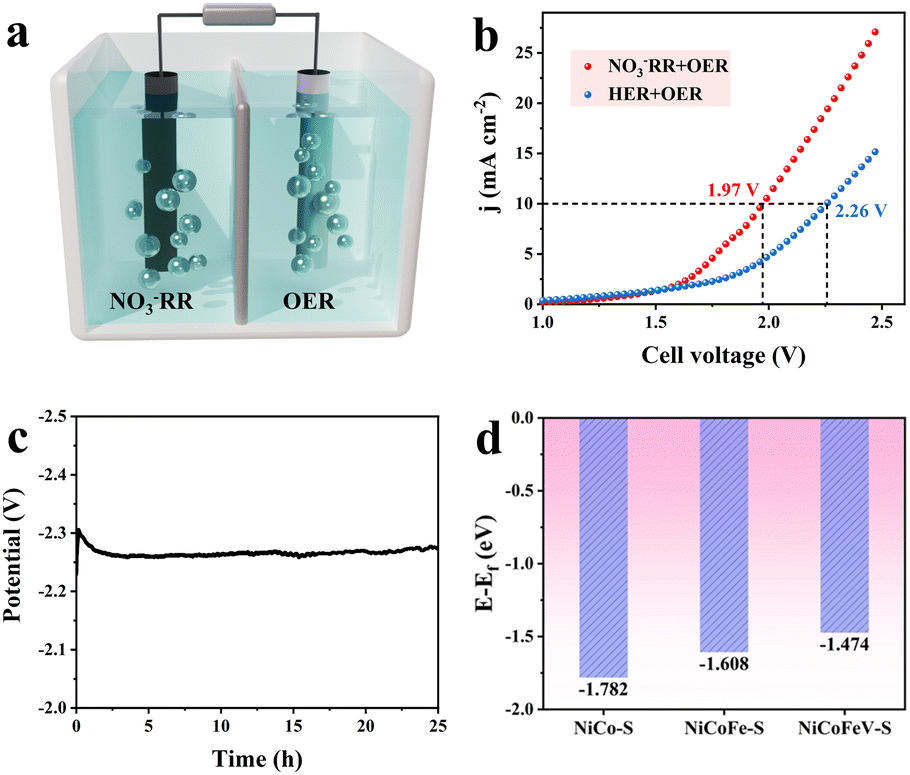 | ||
| Fig. 5 (a) Two-electrode configuration schematic illustrating simultaneous OER (anode) and nitrate reduction (NO3RR, cathode) processes. (b) LSV curves of the electrolyzer under varied conditions: anolyte (1.0 M KOH) and catholyte (1.0 M KOH + 0.1 M KNO3). (c) Chronopotentiometric test of the electrolyzer at 10 mA cm−2. (d) The statistics of d-band centers for NiCoFeV–S, NiCoFe–S, and NiCo–S. | ||
The potential benefits of NiCoFeV–S on catalytic reactions can be elucidated through density functional theory (DFT) calculations. The predicted models of NiCoFeV–S, NiCoFe–S, and NiCo–S are displayed in Fig. S46–S48. As illustrated by the density of states (DOS) in Fig. S49a, NiCoFeV–S exhibited a more continuous electronic structure in comparison to both NiCoFe–S (Fig. S49b) and NiCo–S (Fig. S49c). It facilitated the formation of highly active sites and broadened the range of adsorption energies.77,78 In addition, the significant overlap observed among the orbitals of all elements is indicative of robust bonding, which not only facilitates electron transfer between various metal sites but also provides multiple active sites for catalytic reactions.79 In accordance with the d-band theory, the adsorption energy of reaction intermediates can be optimized by modifying the d-band center with other metal dopants.80 As illustrated in Fig. 5d, NiCoFeV–S featured an upshifted d-band center at −1.474 eV, in contrast to NiCoFe–S (−1.608 eV) and NiCo–S (−1.728 eV). This observation indicates an enhancement in the adsorption strength of reactants (H2O and NO3−) on the catalyst surface, which consequently promotes the OER and NO3RR kinetic processes.81 In the complex multi-step NO3RR process, an upward shift of the d-band center can modulate the adsorption strength of intermediates (e.g., *NO, *NH2, etc.), thereby preventing the active site from being blocked by overly strong adsorption or failing to desorb due to excessively weak adsorption.82 An upward shift of the d-band center can also preferentially facilitate specific reaction pathways. As reported, the HER can be effectively suppressed and the NH3 Faraday efficiency enhanced through d-band center modulation during the NO3RR.82 In light of the aforementioned results, it can be concluded that the exceptional performance exhibited by NiCoFeV–S is primarily attributable to the synergistic effect of multimetallic sites, wherein the up-shifted d-band centers modulate the adsorption energies of reactants and intermediates.
Furthermore, we performed systematic elemental doping and removal studies through the synthesis of NiCoV–S and CoFeV–S samples to elucidate the specific contributions of individual elements. The microstructural morphology of NiCoV–S and CoFeV–S is depicted in Fig. S50. The NiCoV–S sample exhibits well-defined spherical nanostructures, whereas the CoFeV–S material demonstrates an irregular morphological configuration. Furthermore, the phase composition from XRD of NiCoV–S and CoFeV–S is illustrated in Fig. S51. NiCoV–S displays identical diffraction peaks to CoNi2S4 (PDF #24-0334), confirming that NiCoV–S adopts the characteristic spinel sulfide crystal structure. However, CoFeV–S did not display the characteristic diffraction peaks expected for a crystalline phase, suggesting an amorphous structural tendency. The electrocatalytic OER performance of NiCoV–S and CoFeV–S was systematically investigated. As illustrated in Fig. S52a, LSV reveals that NiCoV–S exhibits a significantly reduced overpotential of 283 mV at a current density of 100 mA cm−2, demonstrating superior OER performance compared to NiCoFe–S (291 mV), NiCo–S (348 mV), and CoFeV–S (304 mV). This finding further underscores the pivotal role of Fe/V incorporation and the synergistic interactions among these elements in enhancing the OER. Moreover, the catalytic performance of NiCoV–S and CoFeV–S for nitrate electroreduction was also systematically evaluated. As displayed in Fig. S52b, NiCoV–S delivered the NH3 yield of 7.5 mg h−1 mgcat.−1 at −0.6 V vs. RHE, higher than that of NiCo–S (5.7 mg h−1 mgcat.−1). CoFeV–S delivered the highest NH3 yield of 6.6 mg h−1 mgcat.−1 at −0.8 V vs. RHE. A correspondingly high NH3 FE of 75.6% was achieved by NiCoV–S at −0.8 V vs. RHE, which is higher than those of CoFeV–S (49.3%) and NiCo–S (67.4%) at −0.8 V vs. RHE. Moreover, among the prepared catalysts, NiCoFeV–S demonstrated the highest NH3 yield and faradaic efficiency (FE) across various applied potentials, underscoring its exceptional NH3 selectivity. This superior performance originates from the synergistic interactions among the constituent elements, with the Fe and V components playing particularly pivotal roles. The comprehensive analysis presented herein unequivocally demonstrates the pivotal role of Fe/V dopants and elucidates the pronounced synergistic enhancement effect exhibited by multi-metallic components in high-entropy sulfide systems.
Conclusions
In conclusion, we developed a mild hydrothermal synthesis method for fabricating hollow spherical-flower NiCoFeV–S high-entropy sulfide electrocatalysts. It was found that NiCoFeV–S exhibits exceptional performance in both the OER and NO3RR. Notably, the NiCoFeV–S catalyst achieved a low overpotential of 267 mV at a current density of 100 mA cm−2 for the OER, as well as a high ammonia FE of 93.2% and a yield rate of 16.6 mg h−1 mgcat.−1 for the NO3RR. The superior bifunctional activity was attributed to three key enhancement mechanisms: (1) the hierarchical nanoarchitecture facilitating maximized accessibility of active sites, (2) multi-metal synergy optimizing charge transfer dynamics, and (3) an upshifted d-band center synergistically enhancing water dissociation and hydrogenation kinetics. This work highlights the substantial potential of adopting a mild strategy for the development of advanced HEM electrocatalysts and offers profound insights into the modulation of their electronic structures, thereby holding great promise for practical energy applications.Author contributions
H. S. conceived the study, designed the methodology, and wrote the manuscript. Y. L. synthesized the samples, performed most of the reactions, collected and analyzed the data, and wrote the paper. Y. L., L. Z., and X. W. conducted the performance measurements. H. S., N. Z., Y. Z., D. W., and B. Z. helped to check and revise the paper.Conflicts of interest
There are no conflicts to declare.Data availability
All the data supporting this article have been included in the main text and the SI. Supplementary information: The supplementary characteristics and test results are included there. See DOI: https://doi.org/10.1039/d5sc04536c.Acknowledgements
This work was sponsored by the Natural Science Foundation of Henan (252300421175) and the National Natural Science Foundation of China (Grant No. U22A20143 and 22201262). The authors thank the Center for Advanced Analysis and Gene Sequencing of Zhengzhou University and the High Energy Photon Source for help with characterization studies. The authors thank Caini Ma for performing the electrochemical in situ Raman spectroscopy measurements.Notes and references
- X. Shi, W.-H. Huang, J. Rong, M. Xie, Q. Wa, P. Zhang, H. Wei, H. Zhou, M.-H. Yeh, C.-W. Pao, J. Wang, Z. Hu, X. Yu, J. Ma and H. Cheng, Nat. Commun., 2025, 16, 6161 CrossRef CAS.
- S. Sun, C. Dai, P. Zhao, S. Xi, Y. Ren, H. R. Tan, P. C. Lim, M. Lin, C. Diao, D. Zhang, C. Wu, A. Yu, J. C. J. Koh, W. Y. Lieu, D. H. L. Seng, L. Sun, Y. Li, T. L. Tan, J. Zhang, Z. J. Xu and Z. W. Seh, Nat. Commun., 2024, 15, 260 CrossRef CAS PubMed.
- S. Tian, R. Wu, H. Liu, C. Yan, Z. Qi, P. Song, W.-J. Chen, L. Song, Z. Wang and C. Lv, Angew. Chem., Int. Ed., 2025, e202510665 CAS.
- E. Murphy, B. Sun, M. Rüscher, Y. Liu, W. Zang, S. Guo, Y.-H. Chen, U. Hejral, Y. Huang, A. Ly, I. V. Zenyuk, X. Pan, J. Timoshenko, B. R. Cuenya, E. D. Spoerke and P. Atanassov, Adv. Mater., 2024, 36, 2401133 CrossRef CAS PubMed.
- X. Gu, J. Zhang, S. Guo, Y. Zhang, L. Xu, R. Jin and G. Li, J. Am. Chem. Soc., 2025, 147, 22785–22795 CrossRef CAS.
- C. Zhang, Y. Zhang, R. Deng, L. Yuan, Y. Zou, T. Bao, X. Zhang, G. Wei, C. Yu and C. Liu, Adv. Mater., 2024, 36, 2313844 CrossRef CAS.
- Z. Shen, G. Chen, X. Cheng, F. Xu, H. Huang, X. Wang, L. Yang, Q. Wu and Z. Hu, Sci. Adv., 2024, 10, eadm9325 CrossRef CAS PubMed.
- X. Shi, M. Xie, K. Yang, Y. Niu, H. Ma, Y. Zhu, J. Li, T. Pan, X. Zhou, Y. Cui, Z. Li, Y. Yu, X. Yu, J. Ma and H. Cheng, Angew. Chem., Int. Ed., 2024, 63, e202406750 CrossRef CAS.
- Y. Zou, Y. Yan, Q. Xue, C. Zhang, T. Bao, X. Zhang, L. Yuan, S. Qiao, L. Song, J. Zou, C. Yu and C. Liu, Angew. Chem., Int. Ed., 2024, 63, e202409799 CrossRef CAS.
- K. Zhang, P. Sun, Y. Huang, M. Tang, X. Zou, Z. Pan, X. Huo, J. Wu, C. Lin, Z. Sun, Y. Wan, X. Zhang and L. An, Adv. Funct. Mater., 2024, 34, 2405179 CrossRef CAS.
- H. Luo, S. Li, Z. Wu, M. Jiang, M. Kuang, Y. Liu, W. Luo, D. Zhang and J. Yang, Adv. Funct. Mater., 2024, 34, 2403838 CrossRef CAS.
- H. Fu, S. Lu, Y. Xin, S. Xiao, L. Chen, Y. Li and K. Shen, Energy Environ. Sci., 2025, 18, 818–830 RSC.
- L. Zheng, Y. Zhang, W. Chen, X. Xu, R. Zhang, X. Ren, X. Liu, W. Wang, J. Qi, G. Wang, C. Ma, L. Xu, P. Han, Q. He, D. Ma, J. Wang, C. Ling, D. Su, M. Shao and Y. Chen, Angew. Chem., Int. Ed., 2025, 64, e202500985 CrossRef CAS.
- M. J. Liu, C. A. F. Otero, D. U. Patino, H. Gong, M. D. Hossain, J. E. Matthews, K. S. Williams, A. Vargas, M. J. Zachman, A. S. Hoffman, D. Nordlund, M. Bajdich, S. R. Bare, M. B. Stevens, T. F. Jaramillo, Z. Bao and W. A. Tarpeh, J. Am. Chem. Soc., 2025, 147, 29026–29041 Search PubMed.
- J. Hao, T. Wang, R. Yu, J. Cai, G. Gao, Z. Zhuang, Q. Kang, S. Lu, Z. Liu, J. Wu, G. Wu, M. Du, D. Wang and H. Zhu, Nat. Commun., 2024, 15, 9020 CrossRef CAS PubMed.
- Z. Xiang, Y.-R. Lu, L. Meng, J. Lan, F. Xie, S. Gao, J. Li, M. Luo, M. Peng and Y. Tan, Adv. Mater., 2025, 37, 2501886 CrossRef CAS.
- M. Wang, S. Li, Y. Gu, W. Xu, H. Wang, J. Sun, S. Chen, Z. Tie, J.-L. Zuo, J. Ma, J. Su and Z. Jin, J. Am. Chem. Soc., 2024, 146, 20439–20448 CrossRef CAS PubMed.
- X. Wang, L. R. Winter, X. Wu, Y. Fan, Y. Zhao, J.-H. Kim and M. Elimelech, Sci. Adv., 2025, 11, eads6943 CrossRef CAS.
- F. Xie, X. Cui, X. Zhi, D. Yao, B. Johannessen, T. Lin, J. Tang, T. B. F. Woodfield, L. Gu and S.-Z. Qiao, Nat. Synth., 2023, 2, 129–139 CrossRef CAS.
- L. Wu, L. Zhang, J. Feng, S. Jia, R. Wang, X. Song, X. Ma, Q. Zhu, X. Kang, Q. Qian, X. Sun and B. Han, Chem, 2025, 102591 Search PubMed.
- W. Liu, M. Xia, C. Zhao, B. Chong, J. Chen, H. Li, H. Ou and G. Yang, Nat. Commun., 2024, 15, 3524 CrossRef CAS.
- N. Mukherjee, A. Adalder, N. Barman, R. Thapa, R. Urkude, B. Ghosh and U. K. Ghorai, J. Mater. Chem. A, 2024, 12, 3352–3361 RSC.
- A. Adalder, S. Paul, N. Barman, A. Bera, S. Sarkar, N. Mukherjee, R. Thapa and U. K. Ghorai, ACS Catal., 2023, 13, 13516–13527 CrossRef CAS.
- J. Mukherjee, A. Adalder, N. Mukherjee and U. K. Ghorai, Catal. Today, 2023, 423, 113905 CrossRef CAS.
- S. Sarkar, A. Adalder, S. Paul, S. Kapse, R. Thapa and U. K. Ghorai, Appl. Catal., B, 2024, 343, 123580 CrossRef CAS.
- S. Bhowmick, A. Adalder, A. Maiti, S. Kapse, R. Thapa, S. Mondal and U. K. Ghorai, Chem. Sci., 2025, 16, 4806–4814 RSC.
- D. Liu, L. Qiao, S. Peng, H. Bai, C. Liu, W. F. Ip, K. H. Lo, H. Liu, K. W. Ng, S. Wang, X. Yang and H. Pan, Adv. Funct. Mater., 2023, 33, 2303480 CrossRef CAS.
- L. Quan, H. Jiang, G. Mei, Y. Sun and B. You, Chem. Rev., 2024, 124, 3694–3812 CrossRef CAS.
- Y. Sun, M. Li, J. Duan, M. Antonietti and S. Chen, Angew. Chem., Int. Ed., 2024, 63, e202402678 CrossRef CAS.
- Y. Yao, Z. Huang, P. Xie, S. D. Lacey, R. J. Jacob, H. Xie, F. Chen, A. Nie, T. Pu, M. Rehwoldt, D. Yu, M. R. Zachariah, C. Wang, R. Shahbazian-Yassar, J. Li and L. Hu, Science, 2018, 359, 1489–1494 CrossRef CAS PubMed.
- M. Cui, C. Yang, B. Li, Q. Dong, M. Wu, S. Hwang, H. Xie, X. Wang, G. Wang and L. Hu, Adv. Energy Mater., 2020, 11, 2002887 CrossRef.
- H. Li, J. Lai, Z. Li and L. Wang, Adv. Funct. Mater., 2021, 31, 2106715 CrossRef CAS.
- J. Hao, Z. Zhuang, K. Cao, G. Gao, C. Wang, F. Lai, S. Lu, P. Ma, W. Dong, T. Liu, M. Du and H. Zhu, Nat. Commun., 2022, 13, 2662 CrossRef CAS.
- T. Löffler, A. Ludwig, J. Rossmeisl and W. Schuhmann, Angew. Chem., Int. Ed., 2021, 60, 26894–26903 CrossRef PubMed.
- H. Zhu, S. Sun, J. Hao, Z. Zhuang, S. Zhang, T. Wang, Q. Kang, S. Lu, X. Wang, F. Lai, T. Liu, G. Gao, M. Du and D. Wang, Energy Environ. Sci., 2023, 16, 619–628 RSC.
- J. Baek, M. D. Hossain, P. Mukherjee, J. Lee, K. T. Winther, J. Leem, Y. Jiang, W. C. Chueh, M. Bajdich and X. Zheng, Nat. Commun., 2023, 14, 5936 CrossRef CAS.
- K. Gu, D. Wang, C. Xie, T. Wang, G. Huang, Y. Liu, Y. Zou, L. Tao and S. Wang, Angew. Chem., Int. Ed., 2021, 60, 20253–20258 CrossRef CAS.
- H. Ge, L. Zheng, G. Yuan, W. Shi, J. Liu, Y. Zhang and X. Wang, J. Am. Chem. Soc., 2024, 146, 10735–10744 CrossRef CAS PubMed.
- Y. Kang, O. Cretu, J. Kikkawa, K. Kimoto, H. Nara, A. S. Nugraha, H. Kawamoto, M. Eguchi, T. Liao, Z. Sun, T. Asahi and Y. Yamauchi, Nat. Commun., 2023, 14, 4182 CrossRef CAS PubMed.
- Y. Sun, W. Zhang, Q. Zhang, Y. Li, L. Gu and S. Guo, Matter, 2023, 6, 193–205 CrossRef CAS.
- X. Liu, Y. Yu, K. Li, Y. Li, X. Li, Z. Yuan, H. Li, H. Zhang, M. Gong, W. Xia, Y. Deng and W. Lei, Adv. Mater., 2024, 2312583 CrossRef CAS.
- C. R. McCormick and R. E. Schaak, J. Am. Chem. Soc., 2021, 143, 1017–1023 CrossRef CAS PubMed.
- M. Cui, C. Yang, B. Li, Q. Dong, M. Wu, S. Hwang, H. Xie, X. Wang, G. Wang and L. Hu, Adv. Energy Mater., 2021, 11, 2002887 CrossRef CAS.
- T. Tian, M. Zheng, J. Lin, X. Meng and Y. Ding, Chem. Commun., 2019, 55, 1044–1047 RSC.
- Y. Peng, Q. Liu, B. Lu, T. He, F. Nichols, X. Hu, T. Huang, G. Huang, L. Guzman, Y. Ping and S. Chen, ACS Catal., 2021, 11, 1179–1188 CrossRef CAS.
- B. Yin, L. Hao, X. Li and Q. Yang, Chem. Eng. J., 2023, 476, 146569 CrossRef CAS.
- C. Hao, C. Ni, X. Wang, Y. Pan, Q. Wu, J. Wu and X. Wang, Chem. Eng. J., 2023, 465, 143024 CrossRef CAS.
- X. F. Lu, S. L. Zhang, W. L. Sim, S. Gao and X. W. Lou, Angew. Chem., Int. Ed., 2021, 60, 22885–22891 CrossRef CAS.
- D. Ma, B. Hu, W. Wu, X. Liu, J. Zai, C. Shu, T. T. Tsega, L. Chen, X. Qian and T. L. Liu, Nat. Commun., 2019, 10, 3367 CrossRef PubMed.
- G. Wang, B. Liang, C. Lin, C. Gao, X. Shen, Y. Liu and Q. Jiao, Appl. Mater. Today, 2022, 29, 101692 CrossRef.
- S. Yang, L. Peng, P. Huang, X. Wang, Y. Sun, C. Cao and W. Song, Angew. Chem., Int. Ed., 2016, 55, 4016–4020 CrossRef CAS PubMed.
- T. Zhou, H. Shan, H. Yu, C. a. Zhong, J. Ge, N. Zhang, W. Chu, W. Yan, Q. Xu, H. a. Wu, C. Wu and Y. Xie, Adv. Mater., 2020, 32, 2003251 CrossRef CAS.
- M. Chen, Y. Zhang, J. Chen, R. Wang, B. Zhang, B. Song and P. Xu, Small, 2024, 2309371 CrossRef CAS.
- J. Pan, K. Shi, H. Wu, J. Li, R. Zhang, Q. Liu and Z. Liang, Adv. Energy Mater., 2024, 14, 2302862 CrossRef CAS.
- Y. Liu, Z. Zhang, J. Tan, B. Chen, B. Lu, R. Mao, B. Liu, D. Wang, G. Zhou and H.-M. Cheng, Nat. Commun., 2024, 15, 2167 CrossRef CAS PubMed.
- T. X. Nguyen, Y. H. Su, C. C. Lin and J. M. Ting, Adv. Funct. Mater., 2021, 31, 2106229 CrossRef CAS.
- C. S. Rout, B.-H. Kim, X. Xu, J. Yang, H. Y. Jeong, D. Odkhuu, N. Park, J. Cho and H. S. Shin, J. Am. Chem. Soc., 2013, 135, 8720–8725 CrossRef CAS.
- S. Wang, F. Gong, S. Yang, J. Liao, M. Wu, Z. Xu, C. Chen, X. Yang, F. Zhao, B. Wang, Y. Wang and X. Sun, Adv. Funct. Mater., 2018, 28, 1801806 CrossRef.
- N. Zhang, Y. Hu, L. An, Q. Li, J. Yin, J. Li, R. Yang, M. Lu, S. Zhang, P. Xi and C.-H. Yan, Angew. Chem., Int. Ed., 2022, 61, e202207217 CrossRef CAS PubMed.
- Y. H. Wang, S. Zheng, W. M. Yang, R. Y. Zhou, Q. F. He, P. Radjenovic, J. C. Dong, S. Li, J. Zheng, Z. L. Yang, G. Attard, F. Pan, Z. Q. Tian and J. F. Li, Nature, 2021, 600, 81–85 CrossRef CAS PubMed.
- K. Yeom, J. Jo, H. Shin, H. Ji, S. Moon, J. E. Park, S. Lee, J. Shim, D. H. Mok, M. S. Bootharaju, S. Back, T. Hyeon and Y.-E. Sung, Adv. Funct. Mater., 2024, 2401095 CrossRef CAS.
- Y. Hu, Y. Zheng, J. Jin, Y. Wang, Y. Peng, J. Yin, W. Shen, Y. Hou, L. Zhu, L. An, M. Lu, P. Xi and C.-H. Yan, Nat. Commun., 2023, 14, 1949 CrossRef CAS PubMed.
- X. Ren, Y. Zhai, N. Yang, B. Wang and S. Liu, Adv. Funct. Mater., 2024, 34, 2401610 CrossRef CAS.
- W. Gou, S. Zhang, Y. Wang, X. Tan, L. Liao, Z. Qi, M. Xie, Y. Ma, Y. Su and Y. Qu, Energy Environ. Sci., 2024, 17, 6755–6765 RSC.
- L. Zeng, Z. Zhao, F. Lv, Z. Xia, S.-Y. Lu, J. Li, K. Sun, K. Wang, Y. Sun, Q. Huang, Y. Chen, Q. Zhang, L. Gu, G. Lu and S. Guo, Nat. Commun., 2022, 13, 3822 CrossRef CAS.
- M. Zhang, M. d. Respinis and H. Frei, Nat. Chem., 2014, 6, 362–367 CrossRef CAS PubMed.
- K. Fan, W. Xie, J. Li, Y. Sun, P. Xu, Y. Tang, Z. Li and M. Shao, Nat. Commun., 2022, 13, 7958 CrossRef CAS PubMed.
- N. Zhang and Y. Chai, Energy Environ. Sci., 2021, 14, 4647–4671 RSC.
- X. Luo, H. Zhao, X. Tan, S. Lin, K. Yu, X. Mu, Z. Tao, P. Ji and S. Mu, Nat. Commun., 2024, 15, 8293 CrossRef CAS.
- Z.-F. Huang, S. Xi, J. Song, S. Dou, X. Li, Y. Du, C. Diao, Z. J. Xu and X. Wang, Nat. Commun., 2021, 12, 3992 CrossRef CAS.
- J. Dai, Y. Tong, L. Zhao, Z. Hu, C.-T. Chen, C.-Y. Kuo, G. Zhan, J. Wang, X. Zou, Q. Zheng, W. Hou, R. Wang, K. Wang, R. Zhao, X.-K. Gu, Y. Yao and L. Zhang, Nat. Commun., 2024, 15, 88 CrossRef.
- S. Tang, M. Xie, S. Yu, X. Zhan, R. Wei, M. Wang, W. Guan, B. Zhang, Y. Wang, H. Zhou, G. Zheng, Y. Liu, J. H. Warner and G. Yu, Nat. Commun., 2024, 15, 6932 CrossRef CAS PubMed.
- Y. Liu, Z. Zhuang, Y. Liu, N. Liu, Y. Li, Y. Cheng, J. Yu, R. Yu, D. Wang and H. Li, Angew. Chem., Int. Ed., 2024, 63, e202411396 CAS.
- Y. Zhou, L. Zhang, M. Wang, Z. Zhu, N. Li, T. Qian, C. Yan and J. Lu, ACS Catal., 2024, 14, 7907–7916 CrossRef CAS.
- C. Guo, L. Shen, Y. Tang, M. Chen, X. Zhou, F. Ciucci and Z. Tang, Nano Res., 2025, 18, 94907623 CrossRef.
- X. Cheng, W. Shang, Y. Li, J. Hu, J. Guo, D. Cao, N. Zhang, S. Zhang, S. Song, T. Liu, W. Liu and Y. Shi, Nano Res., 2024, 17, 6826–6832 CrossRef CAS.
- Z. W. Chen, J. Li, P. Ou, J. E. Huang, Z. Wen, L. Chen, X. Yao, G. Cai, C. C. Yang, C. V. Singh and Q. Jiang, Nat. Commun., 2024, 15, 6932 CrossRef.
- L. Zhang, Y. Lei, Y. Yang, D. Wang, Y. Zhao, X. Xiang, H. Shang and B. Zhang, Adv. Sci., 2024, 2407475 CrossRef CAS.
- M. Wei, Y. Sun, J. Zhang, F. Ai, S. Xi and J. Wang, Energy Environ. Sci., 2023, 16, 4009–4019 RSC.
- J. Jiang, F. Sun, S. Zhou, W. Hu, H. Zhang, J. Dong, Z. Jiang, J. Zhao, J. Li, W. Yan and M. Wang, Nat. Commun., 2018, 9, 2885 CrossRef.
- J. Zhang, C. Chen, R. Zhang, X. Wang, Y. Wei, M. Sun, Z. Liu, R. Ge, M. Ma and J. Tian, J. Colloid Interface Sci., 2024, 658, 934–942 CrossRef CAS PubMed.
- Q.-L. Hong, B. Sun, X. Ai, X.-L. Tian, F.-M. Li and Y. Chen, Adv. Funct. Mater., 2024, 34, 2310730 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |