
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrediction of enzyme function using an interpretable optimized ensemble learning framework
Saikat
Dhibar†
,
Sumon
Basak†‡
and
Biman
Jana
 *
*
School of Chemical Sciences, Indian Association for the Cultivation of Science, Jadavpur, Kolkata-700032, India. E-mail: pcbj@iacs.res.in
First published on 1st September 2025
Abstract
Accurate prediction of enzyme function, particularly for newly discovered uncharacterized sequences, is immensely important for modern biological research. Recently, machine learning (ML) based methods have shown promise. However, such tools often suffer from complexity in feature extraction, interpretability, and generalization ability. In this study, we construct a dataset for enzyme functions and present an interpretable ML method, SOLVE (Soft-Voting Optimized Learning for Versatile Enzymes), that addresses these issues by using only combinations of tokenized subsequences from the protein's primary sequence for classification. SOLVE utilizes an ensemble learning framework integrating random forest (RF), light gradient boosting machine (LightGBM) and decision tree (DT) models with an optimized weighted strategy, which enhances prediction accuracy, distinguishes enzymes from non-enzymes, and predicts enzyme commission (EC) numbers for mono- and multi-functional enzymes. The focal loss penalty in SOLVE effectively mitigates class imbalance, refining functional annotation accuracy. Additionally, SOLVE provides interpretability through Shapley analyses, identifying functional motifs at catalytic and allosteric sites of enzymes. By leveraging only primary sequence data, SOLVE streamlines high-throughput enzyme function prediction for functionally uncharacterized sequences and outperforms existing tools across all evaluation metrics on independent datasets. With its high prediction accuracy and ability to identify functional regions, SOLVE can become a promising tool in different fields of biology and therapeutic drug design.
Introduction
Enzymes, as biocatalysts, expedite biochemical reactions within cellular frameworks. Their functional categorization has extensive applications in biotechnology,1 healthcare,2 and metagenomics.3 In pharma companies, enzymes facilitate processes such as biosynthesis and polymer recycling.4 Many bacterial enzymes in the human gut microbiota require functional annotation, as alterations in bacterial colonies are associated with irritable bowel disease (IBD) and obesity.5 Functional annotations would assist medical science by underpinning species that produce the requisite enzymes, thereby aiding disease treatment. To accurately determine an enzyme's function using biochemical assays, wet labs require significant investments in costly reagents, extensive experimental time, and the expertise of skilled researchers.6 As of May 2024, UniProtKB/Swiss-Prot7 contains 283![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 902 manually annotated enzyme sequences, representing just 0.64% of the total 43.48 million enzyme sequences in the database. Thus, experimental methods become potentially unsustainable in the omics era, when large-scale genome projects continuously add new enzyme sequences to databases. Therefore, computational tools provide valuable guidance for experiments with models that are efficient, cost-effective, reproducible, and maintain high accuracy.
902 manually annotated enzyme sequences, representing just 0.64% of the total 43.48 million enzyme sequences in the database. Thus, experimental methods become potentially unsustainable in the omics era, when large-scale genome projects continuously add new enzyme sequences to databases. Therefore, computational tools provide valuable guidance for experiments with models that are efficient, cost-effective, reproducible, and maintain high accuracy.
Enzymes are classified using an ontological system known as the Enzyme Commission (EC) number, which organizes them based on the types of reactions they catalyze. This system is structured hierarchically into four levels, which we denote as L1, L2, L3, and L4. At the first level (L1), enzymes are divided into seven major classes: (i) oxidoreductases, (ii) transferases, (iii) hydrolases, (iv) lyases, (v) isomerases, (vi) ligases, and (vii) translocases. As the classification becomes more specific, the second level (L2) designates the subclass, the third level (L3) identifies the sub-subclass, and the fourth level (L4) specifies the substrate or substrate group upon which the enzyme acts. This tiered system ensures detailed and precise categorization of enzymes, from broad functional roles to specific substrate interactions. In silico methods can annotate a novel protein functionally with accurate EC number prediction at any level. Homology-based,8,9 physicochemical,10,11 structural,12,13 and sequence-derived14–17 properties have been explored in the last few decades—even specific methods have combined multiple properties for function predictions.14,18,19
Nonetheless, each of these methods exhibits distinct disadvantages alongside their advantages. For instance, BLAST identified adenylosuccinate lyase (involved in nucleotide biosynthesis), fumarase (involved in the citric acid cycle), and aspartate ammonia lyase (involved in amino acid metabolism) as homologs, albeit they perform dissimilar functions.20 As of March 2024, the Protein Data Bank (PDB) contains 103972 experimentally determined enzyme structures, representing only a tiny fraction of enzymes catalogued in UniProtKB. Although AlphaFold21 has enabled high-throughput structure prediction of proteins, classifying millions of them with structural information is still a computationally intense task. Sequence-based models primarily depend on manually selected intuitive descriptors, often sequence-length dependent. Several methods, such as pse-AAC, have been proposed to derive sequence-length-independent descriptors from sequence-length-dependent ones.11 Nevertheless, these methods usually necessitate manual intervention and may introduce errors through the standardization of dimensionality.
Computational approaches have demonstrated significant potential in elucidating both protein functions and their associated functional landscape.22–26 The first known use of machine learning (ML) for enzyme annotation dates back to 1997.27 Since then, numerous tools have been developed with improving accuracy.10,18,28–32 Several methods have been employed in enzyme function prediction models, including explainable artificial intelligence (XAI) tools such as k-Nearest Neighbor (kNN),31,33 Support Vector Machine (SVM),17,28,34 and more recently, neural networks such as N-to-1 neural networks,16 Artificial Neural Networks (ANNs),35 Convolutional Neural Networks (CNNs)36,37 and Recurrent Neural Networks (RNNs).19 These models were trained using six primary enzyme classes (L1), and while effective, many were developed prior to the inclusion of translocases as the 7th enzyme class in 2018,38 making them a bit outdated. Models developed after that, such as ECPred,39 ProteInfer,40 CLEAN,41 DeepEC,42 DeepECTransformer43 and ECPICK,36 were trained on seven primary classes. Despite the remarkable capabilities exhibited by these tools, the scope of improvement persists in feature extraction, model interpretability, and the adaptation of these methodologies to novel sequence datasets, particularly training ML models with minimal and low sequence similarity threshold datasets. Furthermore, a major limitation is their inability to reliably differentiate between enzyme and non-enzyme sequences, leading to the potential misassignment of an EC number to non-enzyme proteins when presented with novel sequences. A large-scale community-based Critical Assessment of protein Function Annotation (CAFA)44 revealed that nearly 40% of computational enzyme annotation is erroneous. Henceforth, the community requires novel tools capable of automating feature extraction, employing memory-efficient algorithms, and achieving highly accurate enzyme function predictions to accelerate biomedical research and drug development.
In this study, we have developed an XAI model, SOLVE (Soft-Voting Optimized Learning for Versatile Enzymes), designed to classify novel sequences as enzymes or non-enzymes and further determine whether they are mono- or multifunctional. The overview of the SOLVE method is demonstrated in Fig. 1. SOLVE identifies the L1 to L4 levels for both mono-functional and multi-functional enzymes. SOLVE operates on features extracted directly from the raw primary sequences of proteins. Unlike traditional approaches that depend on predefined biochemical features of protein sequences, our method captures the full spectrum of sequence variations, allowing the model to learn intricate patterns inherent in the protein sequences. Numerical tokenization enhances computational efficiency by reducing the dimensionality of the input space while preserving critical contextual sequence information. To our knowledge, no other contemporary study has successfully extended from enzyme–non-enzyme binary classification to L4 substrate binding multilabel multiclass prediction with such excellent to moderate accuracy. This method surpasses most existing algorithms in classification accuracy and offers a more interpretable model by directly linking specific subsequence patterns to enzyme activity, thereby providing novel insights into enzyme structure–function relationships.
 | ||
| Fig. 1 Overview of the model SOLVE. (A) Dataset curation process, (B) feature extraction process, (C) model training and prediction recording process and (D) Shapley analysis for feature interpretability. | ||
Results and discussion
Prediction performance in different levels of enzyme hierarchy: enzyme vs. non-enzyme
To optimize the performance of our model, we experimented with k-mer values ranging from 2 to 6 and evaluated the model's accuracy at each level of the hierarchy. Through systematic analysis, we found that 6-mers consistently yielded the best results across all levels. The box plot of accuracy scores presented in Fig. 2(A) shows that K-mer = 6 provides the best median accuracy scores consistently for enzyme versus non-enzyme prediction among all the other K-mer values. This result is consistent with some of the previous studies, where it is shown that a 6-mer provides the most optimal results in different bioinformatics classification problems.45 To further investigate why the 6-mer feature descriptors perform better compared with the 5-mer, we first extracted the feature vector embeddings from the 6-mer and 5-mer. Then, we reduced the feature vector dimension to 2-D using t-SNE, which is commonly used in different bioinformatics problems.41,46,47 Interestingly, we observed that when we projected the 6-mer feature vector in the t-SNE 2-D space, different enzyme functional classes were much more separated, and there was little overlap between them. However, using the 5-mer feature vector, different enzyme functional classes overlapped with each other. This essentially means the 5-mer feature descriptor can't differentiate different enzyme functional classes and therefore it fails to provide strong predictive performance for enzyme classification. However, 6-mer feature vectors capture some crucial functional patterns in enzyme sequences that enhance the predictive performance of ML models. The result is shown in Fig. S1. Furthermore, using 7-mers overloaded the memory, making it impossible to test beyond this value with our available resources. In our previous study, we tested beyond 6-mers and found that it provides worse performance on the independent dataset.48 These results indicate that local sequence patterns can be optimally captured using 6-mers, balancing computational efficiency and predictive performance. Throughout the manuscript, we have shown the performance of different ML models in all the enzyme hierarchy levels using these 6-mer feature descriptors. | ||
| Fig. 2 Performance of different ML models used in this study from stratified 5-fold cross-validation. Error bars indicate variance in performance across multiple runs. (A) The accuracy score of different K-mer feature descriptors is presented as a box plot, where the orange-colored horizontal line represents the median accuracy score. (B) Prediction performance of different ML models in predicting enzymes versus non-enzymes. (C) Performance of five ML models to predict mono and multi-functional enzymes. (D) Prediction performance of five ML models in predicting the main enzyme class. (E) Performance of different ML models in EC number level L2 prediction. (F) Performance of different ML models in EC number level L3 prediction. (G) Precision scores of five ML models in predicting enzyme substrate classes with 15 representative samples per class and in the unfiltered dataset, where even less than five samples for some classes are present. (H) Performance of five different ML models used in this study in multi-label multi-functional activity prediction. | ||
To check the performance of our method, we first took the EnzClass50 dataset of enzymes and non-enzymes, where each sample shares less than 50% sequence similarity among themselves. To check different model's performance in different enzyme hierarchy levels, we have used a stratified cross-validation method. This method provides a more reliable estimation of model's performance while reducing the risk of overfitting. Specifically, we have used 5-fold stratified cross-validation and presented the average performance metrics along with the variance across different test folds. Multiple ML models, as detailed in the Materials and methods section, were employed to determine the model that exhibits superior performance in distinguishing enzymes from non-enzymes. The performance of these models is presented in Fig. 2(B). The figure illustrates that the RF and LightGBM models demonstrated superior performance compared to the DT and KNN models used in this study. Notably, the prediction performance further improved when we employed SOLVE, which combines the predictions of both RF and LightGBM models. This ensemble classifier reached the highest performance in predicting enzymes, with precision, recall, and F1-scores of 0.97, 0.95, and 0.96, respectively. Additionally, the ROC curve is shown, which coherently demonstrates that SOLVE provides the highest accuracy compared to the other models. (see Fig. S2). It attained 0.98 accuracy in predicting enzymes and non-enzymes, surpassing its closest competitor, the RF model, which achieved 0.97 accuracy.
Prediction performance in multi-functional enzymes vs. mono-functional enzymes
Next, we advanced to the subsequent level of analysis to determine whether a particular enzyme sequence exhibits mono-functional or multi-functional enzyme activity. Protein sequences with a single EC number were classified as mono-functional enzymes, while those with multiple EC numbers were designated as promiscuous enzymes within the overall enzyme dataset. We evaluated the performance of various ML models on the test dataset using the same approach as previously described. The performance of these models in predicting mono-functional and multi-functional enzymes is presented in Fig. 2(C). As the figure indicates, SOLVE again outperformed the other ML models in predicting mono-functional and multi-functional enzymes. It reached precision, recall and F1-scores of 0.98, 0.98, and 0.99, respectively, notably superior to the other ML models used in this study. The confusion matrix that is shown reveals that the model correctly predicted 13958 mono-functional enzymes and 997 multi-functional enzymes (see Fig. S3(A)). The model misclassified only 1 sample as a multi-functional enzyme and four samples as mono-functional enzymes. This result further underscores the model's efficacy in predicting mono-functional and multi-functional enzymes.
Annotation of mono-functional enzymes
We then moved forward with predicting L1, the primary enzyme class, for mono-functional enzymes. To achieve this, we excluded all multi-functional proteins from the enzyme dataset and removed enzyme sequences with incomplete EC numbers at L4. This process resulted in 54232 samples belonging to one of the seven different enzyme classes. At this level, we found weights of 2:1.5:0.2 for the RF, LightGBM, and DT models in the ensemble learning framework, demonstrating the best performance. The performance of other weight ratios is systematically presented in Table S5. As shown in Fig. 2(D), SOLVE delivered the best performance in predicting the main enzyme class, achieving a precision, F1-score, and recall of 0.98, 0.97, and 0.97, respectively. The RF and LightGBM models also performed very well, although their performance was slightly inferior to SOLVE's. The confusion matrix for this hierarchy level illustrates that the true positive predictions for each enzyme class are significantly higher with SOLVE, further validating the superiority of this model for enzyme function prediction (Fig. S3(B)). We have also evaluated performance of different ML models in EC number level L2 and L3 predictions. The results shown in Fig. 2(E and F), demonstrate that SOLVE also provides highest performance in these levels compared with other ML models. In level 2 prediction, SOLVE delivers precision and F1-score of 0.91 and 0.90, respectively. It also attains precision and F1-score of 0.88 and 0.86, respectively, which is almost 3% better compared with the RF model.
The results of predicting enzyme substrate classes, i.e., the L4 level, are presented in Fig. 2(G). It is important to note that predicting enzyme function at L4 is particularly challenging due to the extreme sparsity of representative samples for each substrate class. This L4 level provides information about the particular substrate the enzyme acts or the precise chemical reaction it catalyzes. In some cases, only one or two samples are available for a given substrate class in the entire dataset. We first filtered the dataset to include only those substrate classes with at least 15 samples, removing other classes. We also evaluated our method's performance on the unfiltered dataset. As depicted in Fig. 2(G), even in the unfiltered dataset—where many substrate classes have very few samples—SOLVE achieves performance with a precision of 0.77. To address the issue of overfitting, we have presented the accuracy scores different folds from cross validation in Table S1, which depicts that model's performance is consistent across different folds. Additionally, we have conducted another analysis to observe how the performance at the L4 level increases and reaches a plateau when we take top K accuracy predictions other than a single prediction. The result shows that, given a considerable number of enzyme classes at L4, if one considers only the top 12 predictions, the accuracy goes above 90% (Fig. S4). This suggests that the prediction results presented in this study are not arbitrary, showing a strong correlation between the enzyme function and protein subsequence patterns. Experimentally, it is also helpful to identify the likely function of an uncharacterized enzyme within the top 10 or 20 predictions, helping to narrow down the vast range of possible enzyme functions. Hereby, we emphasize that there is room for significant improvement in substrate class prediction, and increasing the number of samples for rare substrate classes is crucial for training more effective ML models. Moreover, although we have constructed our dataset from the reviewed portion of the UniProt database, there can be some mis-annotations present in UniProt. To investigate how a certain portion of mis-annotation of enzyme EC numbers during training affects enzyme prediction performance, we have intentionally introduced errors into the dataset. Specifically, we have randomly selected between 10% to 50% of the training dataset and replaced their original EC numbers with arbitrary incorrect ones, and then we have checked the performance of SOLVE in different percentages of error. The results shown in Fig. S5 clearly depict that, even when the error in the training dataset is almost 30%, SOLVE provides an F1-score, which is just 8% lower compared to training with a clean dataset. We have also compared SOLVE's performance with different methods in a scenario where 20% errors in the training dataset are present. The results presented in Fig. S6 demonstrate that SOLVE outperforms all the other tools—CLEAN, DeepEC, and DeepECtransformer—in enzyme function prediction. SOLVE achieves 2% and 7% better F1-scores compared with CLEAN and DeepECtransformer, respectively.
Annotation of multi-label multi-functional enzymes
First, we excluded multi-functional enzyme sequences with incomplete EC numbers at the L4 level. It is important to note that an enzyme can exhibit promiscuous activity at any level of the enzyme hierarchy, from L1 to L4 of the EC number. Since L1 represents the main enzyme class, we initially predicted the multi-functional enzyme activity based on the primary enzyme function, and the results are presented in Table S4. The RF and LightGBM models demonstrated highly predictive metrics as tabulated, while the KNN and DT models performed poorly. Notably, when applying a soft voting ensemble with weights of 10:5:1 for RF, LightGBM, and DT, respectively, SOLVE outperformed others in predicting multi-functional enzymes at L1; other weight ratios with their performance are presented in Table S2. It attained a subset accuracy of 0.65, a micro F1-score of 0.97, and a micro precision of 0.97. Following the successful prediction of multi-functional enzymes at L1, we predicted multi-functional enzyme activity down to L4, corresponding to the substrate-binding class. We emphasize that predictions at this level are more challenging due to the dataset's many unique class combinations and the meager sample size for each unique substrate class. Consequently, few studies have attempted to predict multi-functional enzyme classes down to the last label.
Initially, we filtered our dataset to ensure at least five samples for each EC number present, a procedure commonly applied in other studies in this domain.49 This filtering step ensures that the ML models have some samples for each class combination during training. The results of multi-label, multi-functional enzyme prediction down to L4 are presented in Fig. 2(H). SOLVE reached a subset accuracy of 0.68, approximately 2% and 11% better than the RF and LGBM models, respectively. Additionally, SOLVE provided a micro F1-score of 0.89 and a micro precision of 0.90, outperforming the other models used in this study. Through rigorous testing with SOLVE, we found that a combination of RF, LGBM, and DT with an optimal ratio of 5:4:0.25 yields the best performance. The results for various other weighting ratios are provided in Table S3. We also tested the SOLVE performance under different data distribution scenarios to address the limitations of having only five representative samples per class. The subset accuracy drops from 0.68 to 0.62 when the minimum number of samples per class decreases from five to four (see Fig. S8). In the unfiltered dataset, many new class combinations that the model did not encounter during the training phase were present. Interestingly, even in this situation, SOLVE reached a subset accuracy of 0.51, a micro F1-score of 0.89, and a micro precision of 0.90. These results suggest that our model effectively recognizes the underlying patterns in enzyme sequences to predict specific catalytic activities, even with limited data.
Benchmarking SOLVE on an independent dataset to compare the performance among different methods
For the comparison of SOLVE with other existing tools, the final model was trained with all the manually reviewed UniProt sequences up to 2023 (UniEnz-ALL). To rigorously assess SOLVE's performance, we curated a dataset of enzyme and non-enzyme sequences from the reviewed portion of the UniProt database released after 2023, ensuring these sequences were not involved in SOLVE's development. The dataset comprises 505 enzyme and 694 non-enzyme sequences, against which we benchmarked SOLVE alongside state-of-the-art methods as mentioned in the Dataset preparation section. Fig. 3(A) clearly demonstrates the superiority of SOLVE in discriminating enzymes from the non-enzymatic ones. SOLVE achieved a precision of 0.84 and an F1-score of 0.82, suppressing the second-best model, ECPred, by 9% and 8%, respectively, in the independent dataset. Moreover, it is essential to note that CLEAN cannot discriminate enzymes from non-enzymes. Consequently, when presented with an unknown non-enzyme sequence, CLEAN indiscriminately assigns an EC number, resulting in wrong annotations and undermining the reliability of high-throughput enzyme function prediction. SOLVE also demonstrated notable improvements over DeepEC, delivering 17% and 18% greater precision and recall, respectively. We subsequently evaluated the predictive performance of SOLVE and other methods in different levels of enzyme EC number prediction using the Uniprot 2024 dataset (UniMono-2024). The results shown in Fig. 3(B) depict that SOLVE provided better performance compared to CLEAN in enzyme main class prediction, attaining an accuracy of 0.80 and an F1-score of 0.76, whereas CLEAN achieved 0.76 and 0.74, respectively. Additionally, DeepECtransformer and DeepEC lagged significantly, with accuracy scores of 0.67 and 0.58. SOLVE achieved improvements in F1-score of 4%, 12% and 20% over DeepECtransformer, Proteinfer and DeepEC, respectively. Furthermore, we examined the performance of SOLVE with contemporary methods at the L4 level. We have found that in this level of enzyme hierarchy, using combinations of 6-mer and 4-mer feature descriptors delivered better performance. So, at the L4 level, we have presented the performance of SOLVE with combinations of K-mer features along with other tools for enzyme EC number annotations in Fig. 3(C). The detailed ablation study of how different combinations of feature descriptors influence the prediction results is presented (Fig. S9). SOLVE consistently provided better performance than CLEAN and four other state-of-the-art methods, achieving an accuracy and F1-score of 0.48 and 0.36, representing 2% and 3% enhancement over CLEAN. The performance of SOLVE against CLEAN in L2 and L3 level prediction on this dataset is given in Table S6 and S7. Moreover, SOLVE delivered 11% and 14% better F1-score compared to recently developed tools DeepECtransformer43 and Proteinfer.40 For benchmarking SOLVE in multi-functional enzyme prediction, we have used thevUniMulti-2024 dataset. As demonstrated in Fig. 3(D), SOLVE demonstrated superiority in enzyme promiscuity predictions, providing a precision and F1-score of 0.89 and 0.73, respectively, which are almost 42% and 36% improvements over the second-best model CLEAN. It is important to note that we have trained mono-functional and multi-functional enzymes separately in our framework, whereas other methods used for benchmarking here trained these enzymes together. For this reason, we have also compared SOLVE's performance with the methods, which solely developed to predict promiscuous activity of enzymes such as mlHECNet and mlDEEPre.49,50 The results shown in Table S8 clearly depicts that SOLVE outperforms other multi-functional enzyme prediction tools also. We then evaluated the SOLVE's performance along with other tools in the dataset, where testing enzyme sequences share less than 40% sequence similarity with the training set ones. In such a low sequence similarity scenario SOLVE delivered a precision and F1-score of 0.8 and 0.78, respectively, as depicted in Fig. 3(E), whereas CLEAN achieved a precision and F1-score of 0.77 and 0.75 respectively. Next we evaluate SOLVE's performance on the New-392 dataset. Since SOLVE was trained using UniProt enzyme sequences up to 2023, we first removed all the 392 sequences from the training set before making predictions. DeepECtransformer,43 ProteInfer,40 DeepEC,42 and ECPred39 models are used for benchmarking. We have found that SOLVE outperformed all of these methods in performance indicators. SOLVE's precision and recall score is about 36% and 26% higher than ECPred and about 4% and 3% higher than DeepECtransformer (Fig. 3(F)). This demonstrates that our method, SOLVE, is more effective in predicting enzyme functions than these existing tools. However, comparative analysis showed that SOLVE underperforms slightly compared to CLEAN on New-392, with CLEAN exhibiting a marginal greater F1-score of 4%. In the end, we have tested SOLVE's generalization ability with other methods in five enzymes for which enzyme main class is known experimentally (NCBI-Bact dataset). SOLVE correctly characterized the enzyme class of 4 out of 5 sequences, whereas CLEAN correctly identified only one. The details of the predictions are given in Note S4 and Table S9, further underscoring SOLVE's superior efficacy in enzyme function annotation compared to CLEAN. Moreover, we have tested SOLVE's prediction performance in ‘UniEnz-2025’. Out of 10 enzymes, SOLVE accurately predicts the main enzyme class for 9 enzymes, as detailed in Table S10. | ||
| Fig. 3 Comparison of SOLVE with the state-of-the-art models for enzyme function prediction. (A) Prediction performance of SOLVE for predicting enzymes vs. non-enzymes in UniMono-2024. Three methods, CLEAN, DeepEC and ECPred, are taken for comparison, (B) prediction performance of SOLVE, CLEAN, DeepECtransformer, DeepEC in predicting the enzyme main class on the UniProt 2024 dataset, (C) prediction performance of SOLVE at the L4 level of the UniProt 2024 dataset compared with SOLVE, DeepECtransformer and DeepEC, (D) prediction performance of SOLVE in the Uniprot 2024 multi-functional dataset with three contemporary methods, (E) evaluation of SOLVE's performance on the 40% sequence similarity dataset and (F) prediction performance of different methods in the New-392 dataset for enzyme prediction at the L4 level. | ||
Feature importance analysis
Feature importance analyses are crucial as they provide insight into which features most influence model predictions, enabling better interpretability and trust in ML models. Many deep learning models lack interpretability, which in turn creates obstacles to understanding the actual biological phenomena that guide ML models' predictions, such as the CLEAN method. Understanding feature importance can significantly enhance model refinement and optimization by focusing on the most predictive variables, improving overall model accuracy and robustness. Over the last decade, many interpretable AI models have been developed to explain the ML model's outcomes.51,52 This study focused on interpreting our model by calculating the average feature importance with Shapley analyses, which provides critical insights about the underlying mechanism of the black-box ML model's decision process and is widely used in different domain problems.48,53–55 We selected two proteins from PDB (IDs: 5MO4 and 6OIM) and excluded these sequences from the training dataset. Our model accurately identified these enzymes' EC numbers.We computed the feature importance for each 6-mer amino acid subsequence and then highlighted the top 30 subsequence stretches that contributed most significantly to the model's predictions within the protein's tertiary structure (Fig. 4). Annotating these stretches on the protein structures revealed that our model effectively captures critical biological patterns within the protein's primary sequence. These top-performing features are located within these proteins' orthosteric binding sites and allosteric sites, as illustrated in Fig. 4. In recent years, numerous experimental and computational studies have focused on identifying allosteric sites and investigating the mechanism of signal transduction within proteins.56–60 ABL1 kinase is extremely important in signal transduction and is often targeted in cancer therapies, particularly for chronic myeloid leukemia (CML).61,62 For the ABL1 kinase, the asciminib drug binds to the myristoil pocket, which is also the allosteric site of this enzyme.63,64 In contrast, the nilotinib drug binds to the ATP-binding pocket of this enzyme. For the G12C mutant KRAS enzyme is an important target in cancer research.65 GDP binds to the orthosteric site of this KRAS enzyme, and a covalent inhibitor AMG 510 binds to the allosteric sites.66,67 The top 15 features are predominantly found within or adjacent to the allosteric site regions, while the subsequent top 15 features correspond to the orthosteric binding sites for both proteins.
 | ||
| Fig. 4 Important 6-mers extracted from feature importance analysis are mapped onto the 3D structure of two enzymes. (A) PDB ID: 4Q21 (KRAS protein) and (B) PDB ID: 5MO4 (ABL kinase protein). The top thirty important 6-mers calculated from SHAP analysis are shown in the left panel, and binding and allosteric sites are marked in red circles. The actual binding and allosteric sites of the two proteins are shown in the right panel. | ||
Conclusion
Prediction of enzyme function is a fundamental challenge in biology. Due to the time-consuming nature of experimental methods, new computational tools based on biochemical features, sequence similarity, and ML-based methods are necessary for high-throughput screening of enzyme functions. However, most of the previous enzyme annotation models needed improvements in feature extraction, interpretability, and their ability to adapt to unseen datasets. Most of them struggle to distinguish between enzyme and non-enzyme sequences, leading to misclassification and limited generalization.In this work, we developed SOLVE, a supervised ensemble learning framework augmented with a crucial focal loss penalty that iteratively learns the weights for hard-to-classify enzymes, boosting prediction accuracy for both mono- and multi-functional enzyme functions. This ensemble model leverages numerical tokenization of raw protein sequences, allowing it to capture key sequence patterns without relying on predefined biochemical features. This feature extraction strategy provides a route to predict enzyme functions based on their short subsequence pattern. SOLVE distinguishes enzymes from non-enzyme sequences, predicts mono- and multifunctional enzymes, and provides substrate-binding classifications down to the substrate-binding level with greater accuracy. SOLVE offers valuable insights into enzyme function and activity by pinpointing essential functional regions, such as those involved in oncogenic proteins such as KRAS. Our model's ability to directly link sequence patterns to enzymatic activity provides deeper insights into protein's functionality. These improvements may position SOLVE as a critical tool in accelerating drug discovery, genomics research, enzyme engineering and the functional annotation of novel enzymes. Moreover, this framework is not only limited to enzymatic activity prediction, but this method can be used to predict other functional proteins. User-friendly framework of SOLVE enables broader adaptation and streamlined enzyme EC number prediction by the research community. However, we acknowledge that there is room for improvement for enzyme function at the L4 level. Moreover, for feature extraction, incorporating protein structural features and information about the catalytic site of enzymes may significantly enhance the performance of this model. These points will be the main focus of our future work.
Materials and methods
Dataset preparation
Previous studies have compiled datasets for enzymes and non-enzymes, but the dataset preparation process has some drawbacks. These ambiguities include the following:(A) Prior studies have limited protein sequence lengths to between 50 and 1000 amino acids. While it is documented that feature extraction and training of ML models on protein sequences longer than 1000 amino acids are computationally intensive, excluding these sequences omits a significant portion of protein sequences present in the UniProt database.
(B) As noted earlier, datasets created before 2018 do not include translocase enzymes.38
This study aims to address these challenges in constructing enzyme and non-enzyme datasets. We utilized the publicly available UniProt database7 to assemble the datasets (Fig. 1(A)). Only reviewed sequences were included, as these are manually annotated and considered superior quality due to extensive curation, including experimental validation. To prepare the enzyme dataset, we first web-scraped all protein sequences containing an EC number from the UniProt database. For the non-enzyme dataset, we searched for protein sequences in UniProt that lack both EC numbers and catalytic activity. Then, we removed all protein sequences containing unnatural amino acids (e.g., X, J, B) from this filtered dataset to weed out unwanted noise and restricted sequence lengths to between 50 and 5000 amino acids. Our final dataset for enzymes and non-enzymes comprised 231907 enzyme sequences and 232474 non-enzyme sequences, and we refer to this dataset throughout the manuscript as UniEnz-ALL. At the L4 level of the enzyme dataset, there are 4835 different substrate classes. The number of samples per class varies widely, ranging from as few as 1 to over 2000 in some cases. It should be noted that we have collected all the sequences from UniProt up to the 2023 release, which is the most updated dataset for enzyme function prediction till now. Next, for multiple prediction tasks within the enzyme category, we have removed the sequences that have incomplete EC numbers and constructed subsequent datasets from this primary dataset. To assess the effectiveness of SOLVE, we subsequently deployed the CD-HIT68 program to ensure that the sequence similarity among the gathered sequences, both for enzymes and non-enzymes, remained at 50%. This implies that the training set and testing set share less than 50% sequence similarity, representing a challenge to any ML models. In this dataset, there are 74764 enzymes and 138256 non-enzyme sequences, which is referred to as EnzClass50. Throughout this manuscript, we have evaluated the performance of SOLVE in this EnzClass50 dataset at various enzyme hierarchy levels, and for benchmarking purposes, we have used the UniEnz-ALL dataset.
For comparison of our method with state-of-the-art methods, we have used several independent datasets. One is the New-392 dataset, which consists of 392 enzyme sequences with 177 different EC numbers created from Swiss-Prot released after 2022, and is used by other studies for comparison purposes. Another dataset is constructed by us from the UniProt 2024 release, which contains 504 mono-functional enzymes covering 205 different EC numbers. We refer to this dataset as UniMono-2024. For multi-functional enzyme prediction, we have curated another dataset from the UniProt 2024 release, which contains 39 samples, referred to as the UniMulti-2024 dataset. The dataset comprising enzyme sequences that share less than 40% sequence similarity with one another is described as EnzClass40. We have also extracted four bacterial enzymes from the NCBI database for which enzyme annotation at level 1 is known experimentally, and tested our model prediction ability on them. This dataset is called the NCBI-Bact dataset. We have further curated another dataset from UniProt containing experimentally verified enzyme sequences in 2025, which includes 8 enzyme sequences. We refer to this as the ‘UniEnz-2025’ dataset.
Feature extraction
In the literature, protein's raw sequences have been one-hot encoded40,42 to extract features and feed them into algorithms for classification. However, unlike protein structure prediction studies, the sliding window technique69 has received much less attention in enzyme classification. Recently, many language models have gained much attention for extracting meaningful representations from sequence information.70–73 Inspired by these methods, in this study, we have used a fine-tuned subsequence tokenization strategy to extract functional features from sequences, which was previously used in other bioinformatics problems;48,74,75 however, this strategy has not yet been used in enzyme prediction problems. Given a protein sequence, a window of a specified size (k) slides over the sequence, one position at a time, extracting k-length overlapping subsequences (k-mers). The feature extraction process is detailed in Fig. 1(B).Let S = s1s2s3…sn be a protein sequence of length n, where si represents the i-th amino acid in the sequence. We define k as the length of the k-mer subsequences. The sliding window technique involves extracting subsequences of length k from S, starting from each position i where 1 ≤ i ≤ n − k + 1. Mathematically, the i-th k-mer subsequence can be represented as:
| Sik= sisi+1si+2…si+k−1 | (1) |
For each starting position i, the subsequence Sik is generated by taking the amino acids from position i to i + k − 1, and the process continues until i = n − k + 1. The method can be represented mathematically as:
| {Sik}i=1n−k+1 = {sisi+1…si+k−1}i=1n−k+1 | (2) |
Once all k-mers have been generated, we proceed to tokenize each k-mer. Let  be the tokenizer function that maps each k-mer to a unique token:
be the tokenizer function that maps each k-mer to a unique token:
 | (3) |
 to encode all k-mer subsequences in the dataset. Considering E to be the encoding function that uses the tokenizer to encode each k-mer:
to encode all k-mer subsequences in the dataset. Considering E to be the encoding function that uses the tokenizer to encode each k-mer: | (4) |
Applying this encoding to all k-mers in the dataset:
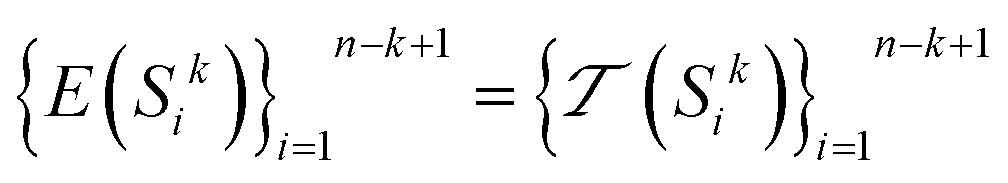 | (5) |
Once all possible k-mers have been assigned a numerical token, the features of every protein sequence are fed into the model along with their corresponding labels. This procedure not only automates feature extraction but also addresses the issue of dimensional non-uniformity and improves memory efficiency. By leveraging the sliding window technique for k-mer generation and subsequent tokenization, we aim to capture local sequence patterns crucial for enzyme classification, thereby improving the predictive performance of our model.
ML model selection and evaluation indices
We have used five different ML models in this study, namely, Random Forest (RF), Light Gradient Boosting Machine (LGBM), K-nearest neighbor (KNN), Decision Tree (DT), and SOLVE, to evaluate performance across different enzyme hierarchy levels. The brief descriptions of each ML model are given in Note S1. For all the predictions, we used evaluation indices—precision, recall, F1 score, sensitivity, and specificity—to evaluate the performance of the proposed method. For multi-functional enzyme prediction, we have used slightly different evaluation parameters: subset accuracy, micro F1, macro F1, micro precision, and macro precision.76 Micro-averaged evaluation parameters are calculated by aggregating all individual class predictions from the model, whereas macro-averaged evaluation parameters are calculated independently for each class and take the unweighted average. Subset accuracy is the most appropriate indicator to evaluate multi-label classification. All the mathematical expressions for each evaluation indicator and the tuned hyperparameters for each model are given in Note S2 and S3. As the enzyme function at the L4 level faces significant class imbalance and some classes are very rare, we implement a focal loss-inspired penalty into the SOLVE training framework, which adjusts the importance of classes during training to prioritize more on the difficult cases.77 Unlike traditional ML models with default hyperparameters, this approach updates the weight given to different classes dynamically throughout the training phase and boosts the prediction performance overall. The expression for the focal loss penalty function is given below. The details of how iterative training of SOLVE with focal loss penalty is conducted are given in Note S5. We analyzed prediction probabilities from SOLVE to understand their correlation with the prediction accuracy score, which provides researchers across disciplines access to the reliability of SOLVE's prediction (Fig. S7).| Lfocal = −α(1 − pt)γlogpt | (6) |
Author contributions
B. J. designed the research; S. D. and S. B. performed the research; S. D. and S. B. analyzed the data; S. D., S. B. and B. J. jointly wrote the paper.Conflicts of interest
The authors declare no competing interest.Data availability
All the data supporting the findings are included in the manuscript. Additionally, the datasets, all the codes and trained models can be accessed from Github via the following link: (https://github.com/saikat-ai/Enzyme_prediction).Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc04513d.
Acknowledgements
This research was supported by the Department of Science and Technology SERB grant CRG/2020/000756. All the authors acknowledge IACS for supercomputing facilities. S. D. acknowledges IACS for financial support.References
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed
.
- L. De Ferrari, S. Aitken, J. Van Hemert and I. Goryanin, Mach. Learn. Syst. Biol., 2010, 1–20 Search PubMed
- L. Liu, Y. Li, S. Li, N. Hu, Y. He, R. Pong, D. Lin, L. Lu and M. Law, J. Biomed. Biotechnol., 2012 DOI:10.1155/2012/251364
- J. L. Porter, R. A. Rusli and D. L. Ollis, ChemBioChem, 2016, 17, 197–203 CrossRef CAS PubMed
- S. Greenblum, P. J. Turnbaugh and E. Borenstein, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 594–599 CrossRef CAS PubMed
- J. L. Reymond, V. S. Fluxà and N. Maillard, Chem. Commun., 2009, 34–46 CAS
- UniProt Consortium, Nucleic Acids Res., 2021, 49, D480–D489 CrossRef PubMed
- S. F. Altschul, W. Gish, W. Miller, E. W. Myers and D. J. Lipman, J. Mol. Biol., 1990, 215, 403–410 CrossRef CAS PubMed
- S. F. Altschul, T. L. Madden, A. A. Schäffer, J. Zhang, Z. Zhang, W. Miller and D. J. Lipman, Nucleic Acids Res., 1997, 25, 3389–3402 CrossRef CAS PubMed
- C. Kumar, A. Choudhary and E. J. Bioinforma, Syst. Biol., 2012, 2012, 1–14 Search PubMed
- P. Chen, J. Li, L. Wong, H. Kuwahara, J. Z. Huang and X. Gao, Proteins:Struct., Funct., Bioinf., 2013, 81, 1351–1362 CrossRef CAS PubMed
- P. D. Dobson and A. J. Doig, J. Mol. Biol., 2005, 345, 187–199 CrossRef CAS PubMed
- R. Yang, C. Zhang, R. Gao and L. Zhang, Int. J. Mol. Sci., 2015, 16, 21191–21214 CrossRef CAS PubMed
- Y. D. Cai and K. C. Chou, J. Proteome Res., 2005, 4, 109–111 CrossRef CAS PubMed
- Y. H. Li, J. Y. Xu, L. Tao, X. F. Li, S. Li, X. Zeng, S. Y. Chen, P. Zhang, C. Qin, C. Zhang, Z. Chen, F. Zhu and Y. Z. Chen, PLoS One, 2016, 11, 1–14 Search PubMed
- V. Volpato, A. Adelfio and G. Pollastri, BMC Bioinf., 2013, 14, 1–7 CrossRef PubMed
- Y. C. Wang, Y. Wang, Z. X. Yang and N. Y. Deng, BMC Syst. Biol., 2011, 5, 1–11 CrossRef CAS PubMed
- C. Zhang, P. L. Freddolino and Y. Zhang, Nucleic Acids Res., 2017, 45, W291–W299 CrossRef CAS PubMed
- Y. Li, S. Wang, R. Umarov, B. Xie, M. Fan, L. Li and X. Gao, Bioinformatics, 2018, 34, 760–769 CrossRef CAS PubMed
- S. A. Benner, S. G. Chamberlin, D. A. Liberles, S. Govindarajan and L. Knecht, Res. Microbiol., 2000, 151, 97–106 Search PubMed
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed
- F. Morcos, B. Jana, T. Hwa and J. N. Onuchic, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20533–20538 CrossRef CAS PubMed
- X. Guan, Q. Y. Tang, W. Ren, M. Chen, W. Wang, P. G. Wolynes and W. Li, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, 1–11 Search PubMed
- S. Gelman, S. A. Fahlberg, P. Heinzelman, P. A. Romero and A. Gitter, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(48), e2104878118 CrossRef CAS PubMed
- S. Cocco, L. Posani and R. Monasson, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, 1–12 CrossRef PubMed
- A. J. M. Ribeiro, I. G. Riziotis, N. Borkakoti and J. M. Thornton, Biochem. J., 2023, 148, 1845–1863 CrossRef PubMed
-
M. desJardins, P. D. Karp, M. Krummenacker, T. J. Lee and C. A. Ouzounis, Proceedings, Fifth International Conference on Intelligent Systems for Molecular Biology ISMB, 1997, vol. 1997, pp. 92–99 Search PubMed
- C. Z. Cai, W. L. Wang, L. Z. Sun and Y. Z. Chen, Math. Biosci., 2003, 185, 111–122 Search PubMed
- C. Claudel-Renard, C. Chevalet, T. Faraut and D. Kahn, Nucleic Acids Res., 2003, 31, 6633–6639 CrossRef CAS PubMed
- W. Tian, A. K. Arakaki and J. Skolnick, Nucleic Acids Res., 2004, 32, 6226–6239 Search PubMed
- H. Bin Shen and K. C. Chou, Biochem. Biophys. Res. Commun., 2007, 364, 53–59 CrossRef PubMed
- A. Roy, J. Yang and Y. Zhang, Nucleic Acids Res., 2012, 40, 471–477 CrossRef PubMed
- E. Nasibov and C. Kandemir-Cavas, Comput. Biol. Chem., 2009, 33, 461–464 CrossRef CAS PubMed
- C. Chen, Y. X. Tian, X. Y. Zou, P. X. Cai and J. Y. Mo, J. Theor. Biol., 2006, 243, 444–448 CrossRef CAS PubMed
- K. Blekas, D. I. Fotiadis and A. Likas, J. Comput. Biol., 2005, 12, 64–82 CrossRef CAS PubMed
- S. R. Han, M. Park, S. Kosaraju, J. M. Lee, H. Lee, J. H. Lee, T. J. Oh and M. Kang, Briefings Bioinf., 2024, 25, 1–11 Search PubMed
- R. Semwal, I. Aier, P. Tyagi and P. K. Varadwaj, J. Biomol. Struct. Dyn., 2021, 39, 2733–2743 CrossRef CAS PubMed
- M. Ann Benore, Biochem. Mol. Biol. Educ., 2019, 47, 481–483 CrossRef CAS PubMed
- A. Dalkiran, A. S. Rifaioglu, M. J. Martin, R. Cetin-Atalay, V. Atalay and T. Doğan, BMC Bioinf., 2018, 19, 1–13 CrossRef PubMed
- T. Sanderson, M. L. Bileschi, D. Belanger and L. J. Colwell, eLife, 2023, 12, 1–21 CrossRef PubMed
- T. Yu, H. Cui, J. C. Li, Y. Luo, G. Jiang and H. Zhao, Science, 2023, 379, 1358–1363 CrossRef CAS PubMed
- J. Y. Ryu, H. U. Kim and S. Y. Lee, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 13996–14001 CrossRef CAS PubMed
- G. B. Kim, J. Y. Kim, J. A. Lee, C. J. Norsigian, B. O. Palsson and S. Y. Lee, Nat. Commun., 2023, 14, 1–11 CAS
- P. Radivojac, W. T. Clark, T. R. Oron, A. M. Schnoes, T. Wittkop, A. Sokolov, K. Graim, C. Funk, K. Verspoor, A. Ben-Hur, G. Pandey, J. M. Yunes, A. S. Talwalkar, S. Repo, M. L. Souza, D. Piovesan, R. Casadio, Z. Wang, J. Cheng, H. Fang, J. Gough, P. Koskinen, P. Törönen, J. Nokso-Koivisto, L. Holm, D. Cozzetto, D. W. A. Buchan, K. Bryson, D. T. Jones, B. Limaye, H. Inamdar, A. Datta, S. K. Manjari, R. Joshi, M. Chitale, D. Kihara, A. M. Lisewski, S. Erdin, E. Venner, O. Lichtarge, R. Rentzsch, H. Yang, A. E. Romero, P. Bhat, A. Paccanaro, T. Hamp, R. Kaßner, S. Seemayer, E. Vicedo, C. Schaefer, D. Achten, F. Auer, A. Boehm, T. Braun, M. Hecht, M. Heron, P. Hönigschmid, T. A. Hopf, S. Kaufmann, M. Kiening, D. Krompass, C. Landerer, Y. Mahlich, M. Roos, J. Björne, T. Salakoski, A. Wong, H. Shatkay, F. Gatzmann, I. Sommer, M. N. Wass, M. J. E. Sternberg, N. Škunca, F. Supek, M. Bošnjak, P. Panov, S. Džeroski, T. Šmuc, Y. A. I. Kourmpetis, A. D. J. Van Dijk, C. J. F. Ter Braak, Y. Zhou, Q. Gong, X. Dong, W. Tian, M. Falda, P. Fontana, E. Lavezzo, B. Di Camillo, S. Toppo, L. Lan, N. Djuric, Y. Guo, S. Vucetic, A. Bairoch, M. Linial, P. C. Babbitt, S. E. Brenner, C. Orengo, B. Rost, S. D. Mooney and I. Friedberg, Nat. Methods, 2013, 10, 221–227 CrossRef CAS PubMed
- Y. Ji, Z. Zhou, H. Liu and R. V. Davuluri, Bioinformatics, 2021, 37, 2112–2120 CrossRef CAS PubMed
- X. Ruan, S. Xia, S. Li, Z. Su and J. Yang, Sci. Rep., 2024, 14, 1–19 CrossRef PubMed
- A. Raza, J. Uddin, A. Almuhaimeed, S. Akbar, Q. Zou and A. Ahmad, J. Chem. Inf. Model., 2023, 63, 6537–6554 CrossRef CAS PubMed
- S. Dhibar and B. Jana, J. Phys. Chem. Lett., 2023, 14, 10727–10735 CrossRef CAS PubMed
- K. A. Khan, S. A. Memon and H. Naveed, Protein Sci., 2021, 30, 1935–1945 CrossRef CAS PubMed
- Z. Zou, S. Tian, X. Gao and Y. Li, Front. Genet., 2019, 10, 432910 Search PubMed
-
M. T. Ribeiro, S. Singh and C. Guestrin, NAACL-HLT 2016 – 2016 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies Proceedings of the Demonstration Session, 2016, pp. 97–101 Search PubMed
- S. M. Lundberg and S. I. Lee, Adv. Neural Inf. Process. Syst., 2017, 2017-Decem, 4766–4775 Search PubMed
- S. Dhibar and B. Jana, J. Chem. Theory Comput., 2024, 20, 7404–7415 CrossRef CAS PubMed
- A. Gupta, M. Kulkarni and A. Mukherjee, Patterns, 2021, 2, 100329 Search PubMed
- Q. Dickinson and J. G. Meyer, PLoS Comput. Biol., 2022, 18, 1–24 Search PubMed
- H. Tian, S. Xiao, X. Jiang and P. Tao, Nucleic Acids Res., 2023, 51, W427–W431 Search PubMed
- M. Leander, Y. Yuan, A. Meger, Q. Cui and S. Raman, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 25445–25454 Search PubMed
- S. Sarkar, S. Dhibar and B. Jana, Phys. Chem. Chem. Phys., 2024, 26(31), 21249–21259 RSC
- G. R. Bowman, E. R. Bolin, K. M. Hart, B. C. Maguire and S. Marqusee, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2734–2739 CrossRef CAS PubMed
- S. Sarkar, S. Dhibar and B. Jana, J. Phys. Chem. B, 2025, 129, 7745–7752 CrossRef CAS PubMed
- H. M. Kantarjian, F. Giles, A. Quintás-Cardama and J. Cortes, Clin. Cancer Res., 2007, 13, 1089–1097 CrossRef CAS PubMed
- J. Y. J. Wang, Mol. Cell. Biol., 2014, 34, 1188–1197 CrossRef PubMed
- J. Schoepfer, W. Jahnke, G. Berellini, S. Buonamici, S. Cotesta, S. W. Cowan-Jacob, S. Dodd, P. Drueckes, D. Fabbro, T. Gabriel, J. M. Groell, R. M. Grotzfeld, A. Q. Hassan, C. Henry, V. Iyer, D. Jones, F. Lombardo, A. Loo, P. W. Manley, X. Pellé, G. Rummel, B. Salem, M. Warmuth, A. A. Wylie, T. Zoller, A. L. Marzinzik and P. Furet, J. Med. Chem., 2018, 61, 8120–8135 CrossRef CAS PubMed
- A. A. Wylie, J. Schoepfer, W. Jahnke, S. W. Cowan-Jacob, A. Loo, P. Furet, A. L. Marzinzik, X. Pelle, J. Donovan, W. Zhu, S. Buonamici, A. Q. Hassan, F. Lombardo, V. Iyer, M. Palmer, G. Berellini, S. Dodd, S. Thohan, H. Bitter, S. Branford, D. M. Ross, T. P. Hughes, L. Petruzzelli, K. G. Vanasse, M. Warmuth, F. Hofmann, N. J. Keen and W. R. Sellers, Nature, 2017, 543, 733–737 Search PubMed
- L. Huang, Z. Guo, F. Wang and L. Fu, Signal Transduction Targeted Ther., 2021, 6, 1–20 CrossRef PubMed
- J. Canon, K. Rex, A. Y. Saiki, C. Mohr, K. Cooke, D. Bagal, K. Gaida, T. Holt, C. G. Knutson, N. Koppada, B. A. Lanman, J. Werner, A. S. Rapaport, T. San Miguel, R. Ortiz, T. Osgood, J. R. Sun, X. Zhu, J. D. McCarter, L. P. Volak, B. E. Houk, M. G. Fakih, B. H. O'Neil, T. J. Price, G. S. Falchook, J. Desai, J. Kuo, R. Govindan, D. S. Hong, W. Ouyang, H. Henary, T. Arvedson, V. J. Cee and J. R. Lipford, Nature, 2019, 575, 217–223 CrossRef CAS PubMed
- B. A. Lanman, J. R. Allen, J. G. Allen, A. K. Amegadzie, K. S. Ashton, S. K. Booker, J. J. Chen, N. Chen, M. J. Frohn, G. Goodman, D. J. Kopecky, L. Liu, P. Lopez, J. D. Low, V. Ma, A. E. Minatti, T. T. Nguyen, N. Nishimura, A. J. Pickrell, A. B. Reed, Y. Shin, A. C. Siegmund, N. A. Tamayo, C. M. Tegley, M. C. Walton, H. L. Wang, R. P. Wurz, M. Xue, K. C. Yang, P. Achanta, M. D. Bartberger, J. Canon, L. S. Hollis, J. D. McCarter, C. Mohr, K. Rex, A. Y. Saiki, T. San Miguel, L. P. Volak, K. H. Wang, D. A. Whittington, S. G. Zech, J. R. Lipford and V. J. Cee, J. Med. Chem., 2020, 63, 52–65 CrossRef CAS PubMed
- L. Fu, B. Niu, Z. Zhu, S. Wu and W. Li, Bioinformatics, 2012, 28, 3150–3152 CrossRef CAS PubMed
-
K. Chen, L. Kurgan and J. Ruan, Proceedings of the 2006 IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology : CIBCB '06, 2006, pp. 366–372 Search PubMed
- T. A. Adjuik and D. Ananey-Obiri, Int. J. Inf. Technol., 2022, 14, 3291–3299 Search PubMed
- D. Ofer, N. Brandes and M. Linial, Comput. Struct. Biotechnol. J., 2021, 19, 1750–1758 CrossRef CAS PubMed
- T. Bepler and B. Berger, Cell Syst., 2021, 12, 654–669.e3 CrossRef CAS PubMed
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss and V. Dubourg, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed
- D. Tobi and I. Bahar, BMC Bioinf., 2007, 8, 1–13 CrossRef PubMed
- K. L. Saar, A. S. Morgunov, R. Qi, W. E. Arter, G. Krainer, A. A. Lee and T. P. J. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(15), e2019053118 CrossRef CAS PubMed
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and É. Duchesnay, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed
- P. Yun, L. Tai, Y. Wang, C. Liu and M. Liu, IEEE Robot. Autom. Lett., 2019, 4, 1263–1270 Search PubMed
Footnotes |
| † Both authors contributed equally to this work. |
| ‡ Present institutional address: Department of Chemistry, Indian Institute of Technology Hyderabad, Sangareddy, Kandi, Telangana-502284, India. |
| This journal is © The Royal Society of Chemistry 2025 |