
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structurally engineered CNT-confined MnxRu1−xO2 catalysts for efficient acidic oxygen evolution at low Ru loading
Xiaolin
Zheng†
a,
Xiaofei
Miao†
ab,
Zijie
Yang
a,
Zhaoyan
Luo
a,
Jun
Yu
a,
Huiqi
Li
*a and
Lei
Zhang
 *a
*a
aCollege of Chemistry and Environmental Engineering, Shenzhen University, Shenzhen, 518060, P.R. China. E-mail: huiqili@szu.edu.cn; lei.zhang@szu.edu.cn
bCollege of Biomedical Engineering, Shenzhen University, Shenzhen, 518060, P.R. China
First published on 18th September 2025
Abstract
Developing acidic oxygen evolution reaction (OER) catalysts with low noble metal loading and high activity remains a critical challenge for advancing proton exchange membrane water electrolyzers. Herein, we report structurally engineered MnxRu1−xO2 catalysts confined on carbon nanotubes (CNTs), enabling highly dispersed active sites and remarkable catalytic activity at low Ru content. The uniform nanoscale coating of MnxRu1−xO2 along CNT sidewalls promotes Mn–O–Ru interfacial bonding and establishes an electron-bridge for enhanced charge transfer. The optimized CNT-(Mn0.75Ru0.25)O2 catalyst delivers a low overpotential of 120 mV at 10 mA cm−2 and an exceptional mass activity of 5549 A gRu−1 at 270 mV—252 times that of commercial RuO2 (22 A gRu−1). Combined X-ray spectroscopy, in situ Raman spectroscopy, and differential electrochemical mass spectrometry reveal that the electron-rich Ru centers stabilized by Mn–O bridges accelerate charge transfer and suppress Ru dissolution during the OER. Moreover, the CNT substrate and Ru incorporation synergistically generate abundant oxygen vacancies, significantly enhancing the catalytic activity through an improved lattice oxygen-mediated mechanism. This work highlights the critical role of CNT confinement and interfacial electronic modulation in decoupling noble metal usage from performance, offering a versatile design strategy for next-generation acidic OER catalysts.
Introduction
Proton exchange membrane water electrolyzers (PEMWEs) offer a promising route for sustainable hydrogen production due to their high current densities, rapid system response, and superior energy efficiency.1–3 However, the harsh oxidative and acidic environment on the anode side necessitates the use of noble metal-based oxygen evolution reaction (OER) catalysts, primarily Ru and Ir, which significantly increases system cost and limits large-scale deployment.4–7Over the past decade, efforts to reduce noble metal usage in acidic OER catalysts have led to two competing trends: either maximizing intrinsic activity per noble metal atom (e.g., via single-atom or heteroatom doping strategies), or lowering overpotential at the cost of increased loading. Yet, these approaches often encounter an inherent trade-off between activity and noble metal content. Few systems can simultaneously deliver overpotentials below 150 mV at 10 mA cm−2 and mass activities above 3000 A gRu−1, as required for practical applications.8–13 In addition, many Ru-based materials undergo irreversible structural degradation due to the lattice oxygen-mediated mechanism (LOM), wherein lattice oxygen directly participates in O–O bond formation, accelerating metal dissolution and catalyst collapse.14–16
To overcome these limitations, a triple strategy is essential: (i) enhancing the atomic utilization of Ru via uniform dispersion, (ii) modulating the electronic structure to suppress the dissolution of active site Ru atoms, and (iii) improving catalyst activity through the introduction of abundant oxygen vacancies.17–20 Such an approach offers a promising pathway to break the traditional activity-cost-stability trade-off and enable the rational design of high performance and stable acidic OER catalysts.
Herein, we report structurally engineered MnxRu1−xO2 catalysts uniformly grown along carbon nanotube (CNT) sidewalls, forming a confined nanoscale interface that enables homogeneous Ru dispersion and C–Mn–O–Ru electronic coupling. Beyond physical confinement, CNTs play additional roles in modulating the catalyst structure and properties. During hydrothermal synthesis, CNTs react with KMnO4 to form chemically bonded MnO2 coatings, introducing oxygen vacancies and promoting uniform oxide dispersion. In addition, the CNTs enhance electrical conductivity, facilitate directional electron transfer from CNTs to Mn and then to Ru via Mn–O bridges, and mitigate structural degradation under acidic conditions.
These effects collectively contribute to the stabilization of Ru in a partially reduced state, suppressing over-oxidation and improving OER performance. As a result, the optimized CNT-confined (Mn0.75Ru0.25)O2 catalyst delivers a low overpotential of 120 mV at 10 mA cm−2 and a mass activity of 5549 A gRu−1 at 270 mV, outperforming commercial RuO2 and the majority of reported Ru-based acidic OER catalysts. Comprehensive spectroscopic and electrochemical analyses reveal that the synergy of CNT confinement and interfacial electronic modulation is key to achieving high activity, low Ru loading, and enhanced stability. This work provides a viable pathway toward the design of cost-effective, durable acidic OER catalysts for scalable hydrogen production.
Results and discussion
Materials synthesis and characterization
To illustrate the synthesis strategy and structural evolution of our catalyst, a schematic diagram is shown in Scheme 1. Pristine carbon nanotubes (CNTs) were oxidized using KMnO4 to form a MnO2 shell, followed by RuCl3 treatment to introduce Ru and generate the (MnxRu1−x)O2 [x = 0.7–0.9, based on the inductively coupled plasma (ICP) result of Ru to Mn] alloy on the CNT surface (see Methods). To validate the proposed catalyst design, the crystalline structure of the catalysts was first analyzed using X-ray diffraction (XRD, Fig. S1). All samples exhibit a characteristic peak near 12.5°, characteristic of the tetragonal α-MnO2 phase (PDF#44-0141), distinguishing it from other MnO2 phases.21 Meanwhile, there were no detectable peaks associated with RuO2 and metallic Ru, indicating successful incorporation of Ru into the MnO2 lattice. Transmission electron microscopy (TEM) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) were then employed to investigate the morphology and dispersion. As shown in Fig. 1a and S2, the oxide forms a continuous coating across the CNT surface, exhibiting nanoscale roughness and island-like surface features, with no apparent nanoparticle aggregation. This nanoscale confinement is critical for stabilizing the active phase and preventing particle growth, as observed in the previous studies.14,18,22 Elemental mapping (Fig. 1b) reveals a homogeneous distribution of Mn, Ru, and O across CNT surfaces, which further indicates the formation of an alloy rather than phase separation. High-resolution STEM images (Fig. 1c–e) exhibit distinct lattice fringes with spacings of 0.27 nm and 0.31 nm, corresponding to the (400) and (310) planes of α-MnO2, respectively. Moreover, intensity profiles (Fig. 1f–h and S3) show variations in atomic contrast consistent with Ru atoms substituting Mn within the oxide lattice. These structural features collectively indicate that Ru is incorporated into the MnO2 lattice, forming a homogeneous MnxRu1−xO2 solid solution. | ||
| Scheme 1 Schematic illustration of the synthesis of CNT-(Mn1−xRux)O2. KMnO4 oxidizes the CNT to form MnO2-coated CNT (CNT-MnO2), followed by RuCl3 treatment to generate Ru-substituted (Mn1−xRux)O2 domains confined on the CNT surface. The inset highlights the atomic-level incorporation of Ru into the MnO2 lattice, forming CNT-Mn–O–Ru bridges. | ||
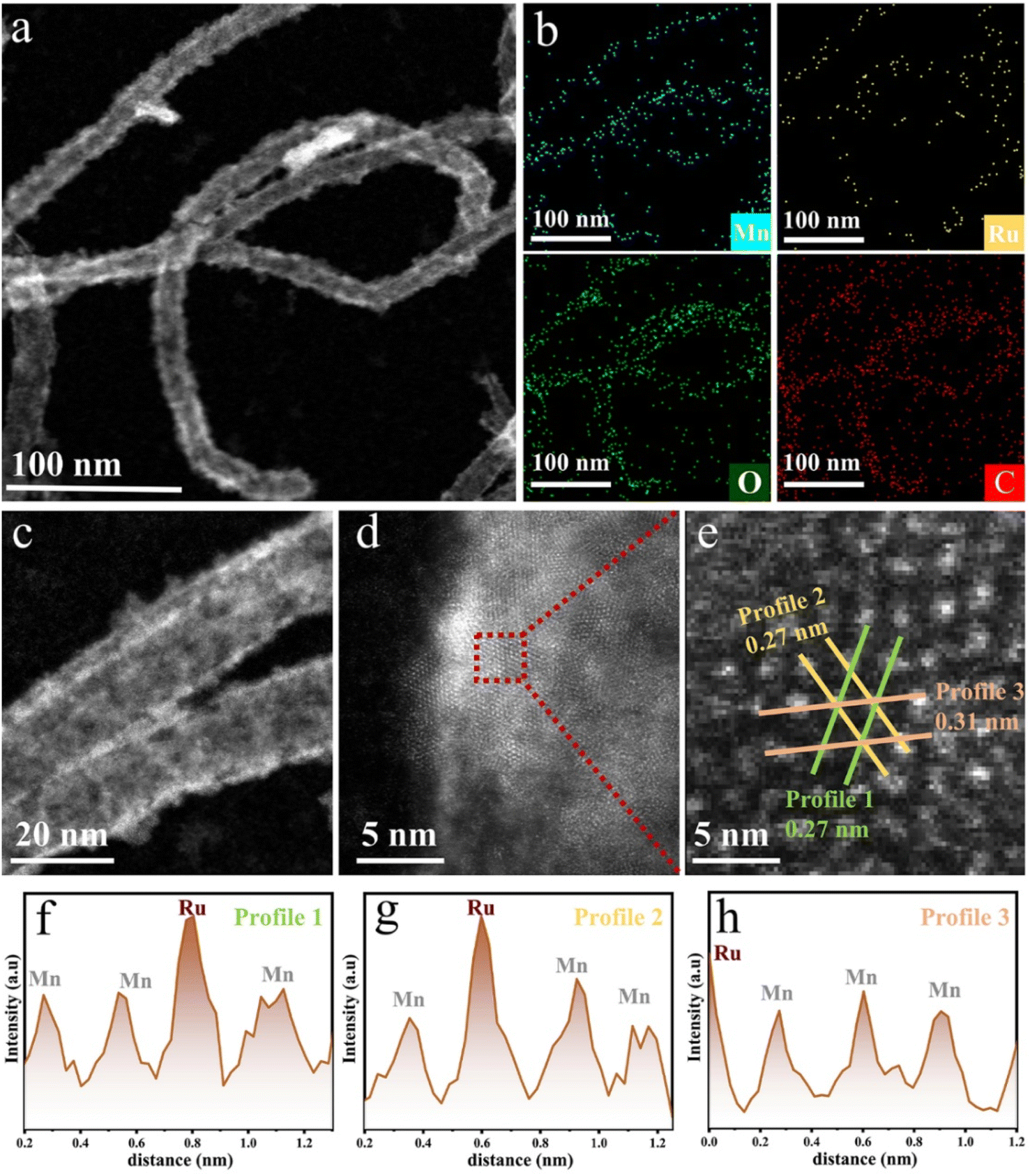 | ||
| Fig. 1 (a) HAADF-STEM image of CNT-(Mn0.75Ru0.25)O2. (b) EDS mapping of CNT-(Mn0.75Ru0.25)O2. (c) High-magnification STEM image showing the morphology from (a). (d) Atomic-resolution STEM image of a representative part selected from (c). (e) Enlarged view of the red-boxed region in (d). (f–h) The corresponding intensity profile of the line regions in (e). | ||
To further understand the electronic structure and coordination environment of CNT-(MnxRu1−x)O2 catalysts, X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS) analyses were conducted.23–25 The Mn 2p3/2 peak of CNT-MnO2 exhibits a 0.4 eV negative shift relative to that of MnO2, indicating that the introduction of CNTs promotes electron transfer from CNTs to Mn. For CNT-(Mn0.75Ru0.25)O2, the Mn 2p3/2 peak shows a 0.24 eV positive shift compared to CNT-MnO2, suggesting that Ru doping increases the oxidation state of Mn, leading to electron loss by Mn (Fig. 2a). When comparing (Mn0.75Ru0.25)O2 with commercial RuO2, the Ru 3p3/2 peak of (Mn0.75Ru0.25)O2 undergoes a 0.4 eV negative shift, indicating that Ru is in a partially reduced state. The opposite shifts between Mn and Ru confirm the existence of electron coupling through the oxygen bridge, with electrons transferring from Mn to Ru. Additionally, the Ru 3p3/2 peak of CNT-(Mn0.75Ru0.25)O2 shows a 0.2 eV negative shift compared to (Mn0.75Ru0.25)O2 (Fig. 2b). Taken together, these shifts confirm that in CNT-(Mn0.75Ru0.25)O2, electrons transfer from C to Mn and then to Ru through the Mn–O bridge. In addition, the progressive increase in work function across CNTs, MnO2, (MnxRu1−x)O2, and RuO2 enables spontaneous electron transfer along this sequence, which is consistent with the observed XPS peak shifts and supports the proposed C to Mn to Ru charge redistribution.26–28 This charge redistribution stabilizes Ru in a partially reduced state, which is expected to mitigate over-oxidation and dissolution under acidic OER conditions (Tables S1 and S2).29 This effect also can effectively optimize the electronic structure of active sites, thereby modulating both activity and stability.30–32
 | ||
| Fig. 2 (a) Mn 2p XPS spectra of CNT-(Mn0.75Ru0.25)O2, CNT-MnO2 and MnO2. (b) Ru 3p XPS spectra of CNT-(Mn0.75Ru0.25)O2, (Mn0.75Ru0.25)O2 and RuO2. (c) Ru K-edge XANES spectra of CNT-(Mn0.75Ru0.25)O2, RuO2 and Ru foil. (d) The Fourier-transformed Ru K-edge EXAFS spectra of CNT-(Mn0.75Ru0.25)O2, RuO2 and Ru foil. (e–g) The wavelet transforms of the Ru K-edge EXAFS signals of CNT-(Mn0.75Ru0.25)O2, RuO2 and Ru foil. | ||
The O K-edge soft X-ray absorption near-edge structure (XANES) spectrum of CNT-(Mn0.75Ru0.25)O2 (Fig. S4) exhibits a noticeable decrease in the intensity of the Ru–O hybridization peak and a negative energy shift relative to CNT-MnO2, indicating an increased electron density localized on oxygen atoms. Meanwhile, the incorporation of Ru into CNT-MnO2 increases the density of unoccupied states and lowers the t2g/eg ratio, which is known to enhance the adsorption of oxygenated intermediates. This electronic structure is consistent with the partial reduction of Ru species observed from XPS results.33,34 The Ru K-edge XANES spectra (Fig. 2c) further support this conclusion. A distinct negative shift in the absorption edge of CNT-(Mn0.75Ru0.25)O2 relative to commercial RuO2was observed, indicating a lower Ru oxidation state. Moreover, the corresponding white line intensity is also reduced, indicating a lower density of unoccupied Ru 4d states and weaker Ru–O hybridization. Fourier-transformed Ru K-edge EXAFS spectra (Fig. 3d, S5, S6 and Table S3) display coordination peaks at ∼1.44 Å (Ru–O), ∼2.42 Å (Ru–Ru/Mn), and ∼2.39 Å (Ru–Ru). As revealed by Ru K-edge EXAFS fitting results, the Ru–O bond length in CNT-(Mn0.75Ru0.25)O2 (1.95 Å) is slightly shorter than that in RuO2 (1.96 Å). This slight shortening can be rationalized by anisotropic Coulomb interactions induced by Mn incorporation into the Ru local environment, which has been reported as a structural signature of solid-solution formation in Ru–Mn oxides.53 Based on the comprehensive analysis above, the CNT-(MnxRu1−x)O2 catalyst is consistent with a solid solution structure. And the corresponding coordination number of Ru–O bonds in CNT-(Mn0.75Ru0.25)O2 (2.9) is lower than that in RuO2 (4.6) and Ru foil (4.6), indicating that the catalyst contains abundant oxygen vacancies. Wavelet transform (WT) analysis (Fig. 2e–g) further validates the coexistence of Ru–O, Ru–Mn, and Ru–Ru coordination environments.13 These XAS results and XPS analyses collectively confirm that the Ru sites in CNT-(Mn0.75Ru0.25)O2 are in an electron-rich state, which originates from CNT-to-Mn-to-Ru electron transfer and is stabilized by Mn–O bridges.
 | ||
| Fig. 3 (a) OER performance of the catalysts in 0.1 M HClO4 solution. (b) Mass activity at 1.5 V of the catalysts. (c) Comparison of OER overpotential and mass activity of previously reported electrocatalysts. (d) Tafel slope of the catalysts. (e) Durability test of CNT-(Mn0.75Ru0.25)O2 and RuO2. | ||
Electrocatalytic activity
The electrocatalytic performance of CNT-(MnxRu1−x)O2 catalysts was systematically evaluated for the acidic oxygen evolution reaction (OER) in the O2-saturated 0.1 M HClO4 electrolyte, using a standard three-electrode configuration. Linear sweep voltammetry (LSV) measurements were conducted with carbon paper (1 × 1 cm2) as the working electrode, a platinum wire as the counter electrode, and a reversible hydrogen electrode (RHE) as the reference electrode. All potentials were corrected for iR compensation to ensure accurate comparison. As shown in Fig. 3a and S7, CNT-(Mn0.75Ru0.25)O2 exhibits the lowest overpotential of 120 mV at 10 mA cm−2 and 270 mV at 400 mA cm−2, significantly outperforming both the CNT-free (Mn0.75Ru0.25)O2 (200 mV at 10 mA cm−2) and commercial RuO2 benchmark (230 mV at 10 mA cm−2). This enhancement demonstrates the critical role of the CNT support in dispersing the active sites and facilitating synergistic interactions between Mn and Ru. The inductively coupled plasma-optical emission spectrometry (ICP-OES) analysis confirms the low Ru content in the CNT-supported catalysts, with Ru loadings of 1.7 wt%, 3.2 wt%, 3.9 wt%, and 4.9 wt% for CNT-(Mn0.9Ru0.1)O2, CNT-(Mn0.8Ru0.2)O2, CNT-(Mn0.75Ru0.25)O2, and CNT-(Mn0.7Ru0.3)O2, respectively (Table S4). Notably, despite the low Ru content, CNT-(Mn0.75Ru0.25)O2 achieves a remarkable mass activity of 5549 A gRu−1 at 1.5 V vs. RHE, which is ∼252 times higher than that of commercial RuO2 (22 A gRu−1) and substantially higher than that of most reported Ru-based catalysts (Fig. 3b, c, S8 and Table S5). Although the Ru content in CNT-(Mn0.75Ru0.25)O2 is 3.9 wt%, the corresponding Ru loading on the carbon paper working electrode is only 0.077 mg cm−2. At this low loading, the CNT-(Mn0.75Ru0.25)O2 delivers an overpotential of 120 mV at 10 mA cm−2, along with outstanding mass activity (Table S6). This result highlights that the strategic incorporation of Ru into the MnO2 matrix, mediated by the CNT support, enables efficient atomic dispersion of Ru and optimizes electronic interactions, thereby achieving exceptional catalytic performance at minimal noble metal content.Kinetic analysis based on Tafel slopes further reveals that CNT-(Mn0.75Ru0.25)O2 exhibits the lowest slope of 95.9 mV dec−1, significantly lower than those of commercial RuO2 (123.1 mV dec−1) and (Mn0.75Ru0.25)O2 (125.0 mV dec−1, Fig. 3d and S9).35 This indicates that the CNT-Mn-Ru system effectively facilitates the reaction kinetics by modulating the electronic structure and lowering the energy barrier for the OER process. Electrochemical impedance spectroscopy (EIS) measurements (Fig. S10) reveal that CNT-(Mn0.75Ru0.25)O2 possesses the lowest charge transfer resistance (Rct) among all tested samples, further confirming the role of CNTs in providing a highly conductive network that accelerates electron transport.36–38 The electrochemical surface area (ECSA), determined from the dependence of double-layer capacitance (Cdl) on different scan rates (5 mV s−1 to 25 mV s−1), further supports the enhanced activity of CNT-(Mn0.75Ru0.25)O2. CNT-(Mn0.75Ru0.25)O2 exhibits an ECSA of 159.9 mF cm−2, which is 7.5 and 10.5 times higher than that of (Mn0.75Ru0.25)O2 (21.3 mF cm−2) and commercial RuO2 (15.2 mF cm−2), respectively (Fig. S11 and S12).34,39 This substantial increase reveals the dual role of CNTs: promoting high-density dispersion of active sites and maximizing their electrochemical accessibility. Long-term durability tests at a constant current density (10 mA cm−2, Fig. 3e) demonstrate that CNT-(Mn0.75Ru0.25)O2 retains stable operation for over 160 hours. To evaluate catalyst degradation, ICP-OES analysis of the post-electrolysis electrolyte revealed that only 4.9% of Ru was dissolved, substantially lower than the ∼19% reported for commercial RuO2 under similar acidic conditions, indicating enhanced resistance to Ru dissolution under acidic OER conditions.40 In contrast, commercial RuO2 suffers from rapid degradation, with significant activity loss observed within 3 hours. Meanwhile, CV tests at different scan rates were performed on the post-stability sample. The Cdl (Fig. S13) decreased by approximately 26% compared with the sample before the stability test. Combined with ICP and HAADF-STEM analysis, the decrease in double-layer capacitance supports the conclusion that catalysts agglomeration occurred during the stability test. To further understand the origin of performance decay after 160 h, TEM and HAADF-STEM images (Fig. S14) were recorded which showed that the homogeneous MnxRu1−xO2 solid solution structure is maintained. The initially uniform distribution became more aggregated on the CNT surface, indicating particle growth. XRD analysis (Fig. S15) reveals sharpened diffraction peaks, confirming increased crystallinity. This aggregation likely reduces the accessible active surface area, contributing to the observed decline in OER performance. Nonetheless, the well-dispersed (MnxRu1−x)O2 architecture before operation plays a key role in achieving high activity at low Ru content and future optimization will focus on mitigating particle growth to enhance durability.
Catalytic mechanism
To unravel the origin of the enhanced OER, we first examined the oxygen species in CNT-(Mn0.75Ru0.25)O2, (Mn0.75Ru0.25)O2, CNT-MnO2 and MnO2 catalysts by XPS. As shown in Fig. S16a, the O 1s spectra of all catalysts can be deconvoluted into three components corresponding to lattice oxygen (Olat), oxygen vacancies (Ov), and adsorbed oxygen species (Oads). Notably, the Ov percentage of CNT-(Mn0.75Ru0.25)O2 (60.4%) is significantly larger than that of other catalysts, indicating a higher concentration of oxygen vacancies in the CNT-Mn-Ru system. And the percentages of CNT-MnO2 (28.8%) and (Mn0.75Ru0.25)O2 (38.2%) are both larger than that of MnO2 (18.8%), indicating that CNTs and Ru can promote the generation of more oxygen vacancies (Fig. S16b). These results suggest that both the incorporation of Ru in MnO2 and the CNT support facilitates the generation of oxygen vacancies. The observed increase in Ov concentration can be rationalized by the following mechanism. Oxygen vacancies in metal oxides can be introduced through cation doping or anion substitution. Incorporation of cations with lower oxidation states or lower oxygen vacancy formation energies tends to promote the formation of oxygen vacancies via a charge compensation mechanism. In this work, the introduction of low-valent Ru3+ into the MnO2 lattice induces such a compensation process, thereby increasing the concentration of oxygen vacancies.41 This defect engineering not only enhances the intrinsic activity by providing additional active sites for O–O coupling, but also modulates the surface electronic structure to favor intermediate adsorption during the OER.42,43The dynamic structural evolution of the catalyst under OER conditions was further investigated by in situ Raman spectroscopy. Since the water band remains constant with increasing potential, all Raman spectra were normalized to the intensity of the water peak to minimize the effects of bubble interference and enable accurate comparison of intermediate-related features. As illustrated in Fig. 4a, an H-type flow cell was used to monitor the real-time structural evolution of catalysts under potential control. The vibrational spectra of RuO2 are observed in Fig. S17, showing characteristic Ru–O stretching modes.44 For CNT-(Mn0.75Ru0.25)O2, the Raman peaks at 510 cm−1 and 637 cm−1 are assigned to Mn–O/Ru–O stretching vibrations, while the peak at ∼580 cm−1 corresponds to the Mn3+–O band, consistent with previous reports.25,45–48 Additionally, carbon-related features are observed at ∼1350 cm−1 (D band), ∼1580 cm−1 (G band), and ∼2700 cm−1 (2 G band),49,50 corresponding to defect-induced vibrations, in-plane sp2 carbon stretching, and the second-order overtone, respectively. Notably, the intensities of Mn–O/Ru–O and Mn3+–O vibrational modes increase markedly from 0.8 V to 1.6 V vs. RHE, reflecting the potential-dependent accumulation of lattice oxygen intermediates (Fig. 4b). In addition, increasing peak intensity with the applied voltage suggests that the oxygen vacancies can participate in the progress of the reaction, whereas CNT-MnO2 shows negligible spectral variation over the same potential range (Fig. S18a). The corresponding contour plots (Fig. 4c and S18b) demonstrate reversible modulation of the characteristic peaks through CV cycling, indicating dynamic reconstruction of reversible oxygen vacancy changes of the CNT-(Mn0.75Ru0.25)O2 during the OER, which does not occur in CNT-MnO2. Notably, the CNT-(Mn0.75Ru0.25)O2 catalyst exhibits no significant red or blue shift of the Ru–O vibrational band at increasing potential, in contrast to the blue shift observed in CNT-MnO2. This spectral invariance suggests that the formation of higher-valence Ru species is effectively inhibited, likely due to directional electron transfer mediated by Mn–O bridges. These bridges facilitate electron transfer from CNTs to Ru centers, mitigating over-oxidation under acidic OER conditions and thereby enhancing catalyst durability.52 These observations reveal that the Mn–O–Ru interaction plays a key role in enabling dynamic surface restriction under OER conditions, which promotes the reversible binding of intermediates. Simultaneously, the CNT provides robust structural support, ensuring the stable dispersion of active sites throughout the reaction.51
 | ||
| Fig. 4 (a) Schematic of the in situ Raman spectroscopy setup. (b) In situ Raman spectra of CNT-(Mn0.75Ru0.25)O2 collected at increasing potentials from OCP to 1.6 V vs. RHE. (c) Corresponding Raman contour plots of CNT-(Mn0.75Ru0.25)O2 during CV cycling from 0.8–1.6 V vs. RHE at 1 mV s−1. (d) Schematic of the DEMS setup. (e) Mass spectrometry traces of 32O2, 34O2, and 36O2 signals during potential cycling of CNT-(Mn0.75Ru0.25)O2 in H216O–HClO4 electrolyte. (f) Comparison of the 34O2/32O2 peak area ratio of CNT-(Mn0.75Ru0.25)O2 and RuO2. | ||
To further probe the oxygen evolution pathway, we performed in situ differential electrochemical mass spectrometry (DEMS) measurements using 18O-labeled catalysts CNT-(Mn0.75Ru0.25)O2 and commercial RuO2 (Fig. 4d). Prior to DEMS analysis, the catalysts were pre-labelled by CV cycling in 0.1 M HClO4 prepared with H218O. After the labelling process, the evolved O2 during the OER was monitored in a 0.1 M HClO4 electrolyte prepared with H216O, allowing us to distinguish lattice oxygen (18O) and oxygen derived from water (16O). The evolution of 36O2 (18O18O), 34O2 (16O18O) and 32O2 (16O16O) was quantitatively tracked to probe the oxygen evolution mechanism of catalysts.52,53 As shown in Fig. 4e and S19, during the OER process of both catalysts, only 34O2 and 32O2 were detected, with no 36O2 observed, confirming that both catalysts follow a lattice oxygen-mediated mechanism (LOM). The ratio of 34O2 to 32O2 (Fig. 4f) for CNT-(Mn0.75Ru0.25)O2 was determined to be 2.5%, higher than that of commercial RuO2 (1.3%), indicating greater lattice oxygen participation in the CNT-(Mn0.75Ru0.25)O2. This phenomenon is attributed to the abundant oxygen vacancies on the CNT-(Mn0.75Ru0.25)O2, which can be labelled by 18O and then reversibly participate in the OER process. This trend aligns well with the higher oxygen vacancy concentration derived from XPS O 1s deconvolution and the potential-dependent reversibility observed in in situ Raman spectroscopy, suggesting that the CNT-confined Mn–O–Ru framework stabilizes Ru sites in a partially reduced state and inhibits over-oxidation.54,55
To further explore the reaction mechanism, density functional theory (DFT) calculations were performed (Fig. 5a, b and S20).58Fig. 5a presents the differential charge analysis results of CNT-(Mn0.75Ru0.25)O2 with Bader charge calculations showing that 0.62e− transfer from CNTs to (Mn0.75Ru0.25)O2. As shown in Fig. 5b, the rate-determining step (RDS) for both CNT-(Mn0.75Ru0.25)O2 and RuO2 involves the formation of the *OO intermediate. The energy barrier for the RDS of CNT-(Mn0.75Ru0.25)O2 is lower than that of RuO2 (110), indicating an improvement in the reaction kinetics. Combined with the experimental evidence from DFT calculations, it is shown that there exist more favorable oxygen evolution pathways between CNT-(Mn0.75Ru0.25)O2 and commercial RuO2. We believe that the classic LOM in RuO2 involves direct participation of lattice oxygen in the O–O formation process. However, the highly symmetric rutile structure of RuO2, while intrinsically active, renders it susceptible to excessive lattice oxygen activation under acidic OER conditions, promoting the formation of unstable RuO42− intermediates (Fig. S21).56,57 This over-oxidation pathway inevitably accelerates Ru dissolution and compromises catalyst stability. In contrast, as shown in Fig. 5c and d, the CNT-(MnxRu1−x)O2 adopts a structurally and electronically distinct configuration that enables a more favorable LOM process.58 The incorporation of Mn and the CNT support induces a highly dispersed architecture in which electron-rich Ru centers are stabilized by Mn–O bridges and abundant oxygen vacancies. These features synergistically enhance both activity and stability. On the one hand, the Mn–O bridges facilitate directional electron transfer from CNTs to Ru during the OER, serving as a protective pathway that mitigates over-oxidation of the Ru under acidic conditions.28 On the other hand, in situ Raman spectroscopy corroborated that CNT-(Mn0.75Ru0.25)O2 exhibits reversible, potential-dependent modulation of the Mn–O/Ru–O and Mn3+–O vibrational bands, while RuO2 shows limited dynamic flexibility.53 The enhanced and reversible Raman signals in CNT-(MnxRu1−x)O2 suggest that oxygen vacancies not only provide additional adsorption sites for intermediates but also enable structural adaptability during OER cycling, facilitating efficient formation and desorption of *O species.59–61
 | ||
| Fig. 5 (a) Charge distribution analysis of CNT-(Mn0.75Ru0.25)O2. Yellow region indicates charge accumulation, while the blue region indicates charge reduction. (b) The calculated reaction pathways of CNT-(Mn0.75Ru0.25)O2 and RuO2(110) for acidic OER at U = 1.23 V. Schematic illustration of the LOM pathway during acidic OER and (c and d) enhanced LOM pathway in CNT-(MnxRu1−x)O2 with improved oxygen vacancy dynamics and structural stability. | ||
Taken together, these results establish a coherent mechanistic framework: the synergistic combination of the CNT support, Mn incorporation, and Ru dispersion in CNT-(MnxRu1−x)O2 creates a unique Mn–O-bridge network that enables directional electron transfer, modulates surface electronic states, and tunes oxygen vacancy concentration. This CNT-confined Mn–O–Ru framework effectively improves the usage of Ru atoms, mitigates Ru over-oxidation and dissolution, and promotes a more stable and efficient OER process under acidic conditions. In contrast, RuO2 suffers from its symmetric crystal structure and strong lattice oxygen participation, which ultimately accelerates Ru leaching and deactivation.62,63
Conclusions
In summary, we report structurally engineered CNT-confined (MnxRu1−x)O2 catalysts that achieve outstanding acidic OER performance at low Ru loading. The uniform nanoscale coating of (MnxRu1−x)O2 along the CNT sidewalls not only promotes highly dispersed active sites and abundant oxygen vacancies, but also facilitates directional electron transfer through Mn–O bridges. This unique electronic architecture stabilizes Ru in a partially reduced state, effectively mitigating over-oxidation and dissolution pathways that typically plague Ru-based catalysts under acidic conditions. Combined spectroscopic and electrochemical analyses reveal that tuning the local electronic environment and oxygen vacancy concentration is essential for suppressing dissolution of Ru, thereby balancing catalytic activity and stability. This work highlights the importance of CNT confinement and Mn–O bridge-mediated electronic coupling in modulating Ru reactivity and durability. Overall, our findings establish a versatile design strategy for developing robust, high-performance acidic OER catalysts that decouple noble metal usage from activity, offering a promising pathway for advancing PEMWE technologies.Author contributions
L. Z., H. L. and X. Z. conceived the research and wrote the paper. X. Z. and X. M. performed catalysts synthesis and general characterization. Z. Y. performed DEMS, supervised by Z. L. J. Y. assisted X. Z. in the XAS measurements. L. Z. and H. L. supervised the research. All the authors contributed to the discussions and revisions of the manuscript.Conflicts of interest
The authors declare no competing interests.Data availability
The datasets are available from the corresponding author on reasonable request.Data for this article, including experimental precedure, supplementary tables and figures, characterization data of the products, etc., are available in the SI. See DOI: https://doi.org/10.1039/d5sc04431f.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 22472101), Guangdong Science and Technology Department Program (2025A1515010455, 2021QN02L252), Research Team Cultivation Program of Shenzhen University (023QNT007) and Scientific Foundation for Youth Scholars of Shenzhen University (868-000001033351).References
- H. Su, C. Yang, M. Liu, X. Zhang, W. Zhou, Y. Zhang, K. Zheng, S. Lian and Q. Liu, Nat. Commun., 2024, 15, 95 CrossRef PubMed.
- S. Kong, A. Li, J. Long, K. Adachi, D. Hashizume, Q. Jiang, K. Fushimi, H. Ooka, J. Xiao and R. Nakamura, Nat. Catal., 2024, 1–10 CAS.
- X. Ping, Y. Liu, L. Zheng, Y. Song, L. Guo, S. Chen and Z. Wei, Nat. Commun., 2024, 15, 2501 CrossRef CAS PubMed.
- A. Kumar, M. Gil-Sepulcre, J. Lee, V. Q. Bui, Y. Wang, O. Rüdiger, M. G. Kim, S. DeBeer and H. Tüysüz, Adv. Mater., 2024, 36, 2401648 CrossRef CAS PubMed.
- PGM prices and trading, https://matthey.com/products-and-markets/pgms-and-circularity/pgm-management, (accessed May 8, 2025).
- J. Zhao, Y. Guo, Z. Zhang, X. Zhang, Q. Ji, H. Zhang, Z. Song, D. Liu, J. Zeng, C. Chuang, E. Zhang, Y. Wang, G. Hu, M. A. Mushtaq, W. Raza, X. Cai and F. Ciucci, Nat. Nanotechnol., 2024, 57–66 CAS.
- L. Chong, G. Gao, J. Wen, H. Li, H. Xu, Z. Green, J. D. Sugar, A. J. Kropf, W. Xu, X.-M. Lin, H. Xu, L.-W. Wang and D.-J. Liu, Science, 2023, 380, 609–616 CrossRef CAS PubMed.
- L. Cao, Q. Luo, J. Chen, L. Wang, Y. Lin, H. Wang, X. Liu, X. Shen, W. Zhang, W. Liu, Z. Qi, Z. Jiang, J. Yang and T. Yao, Nat. Commun., 2019, 10, 4849 CrossRef PubMed.
- K. Wang, Y. Wang, B. Yang, Z. Li, X. Qin, Q. Zhang, L. Lei, M. Qiu, G. Wu and Y. Hou, Energy Environ. Sci., 2022, 15, 2356–2365 RSC.
- S. Zuo, Z.-P. Wu, D. Xu, R. Ahmad, L. Zheng, J. Zhang, L. Zhao, W. Huang, H. Al Qahtani, Y. Han, L. Cavallo and H. Zhang, Nat. Commun., 2024, 15, 9514 CrossRef CAS PubMed.
- Y. Xu, Z. Mao, J. Zhang, J. Ji, Y. Zou, M. Dong, B. Fu, M. Hu, K. Zhang, Z. Chen, S. Chen, H. Yin, P. Liu and H. Zhao, Angew. Chem., Int. Ed., 2024, 136, e202316029 CrossRef.
- W. Hu, B. Huang, M. Sun, J. Du, Y. Hai, W. Yin, X. Wang, W. Gao, C. Zhao, Y. Yue, Z. Li and C. Li, Adv. Mater., 2025, 37, 2411709 CrossRef CAS PubMed.
- C. Zhou, L. Li, Z. Dong, F. Lv, H. Guo, K. Wang, M. Li, Z. Qian, N. Ye, Z. Lin, M. Luo and S. Guo, Nat. Commun., 2024, 15, 9774 CrossRef CAS PubMed.
- H. Yan, Z. Jiang, B. Deng, Y. Wang and Z.-J. Jiang, Adv. Energy Mater., 2023, 13, 2300152 CrossRef CAS.
- J. Yang, K. An, Z. Yu, L. Qiao, Y. Cao, Y. Zhuang, C. Liu, L. Li, L. Peng and H. Pan, ACS Catal., 2024, 14, 17739–17747 CrossRef CAS.
- J. Chang, Y. Shi, H. Wu, J. Yu, W. Jing, S. Wang, G. I. N. Waterhouse, Z. Tang and S. Lu, J. Am. Chem. Soc., 2024, 146, 12958–12968 CrossRef CAS PubMed.
- Q. Ma and S. Mu, Interdiscip. Mater., 2023, 2, 53–90 Search PubMed.
- C. Li, D. Li, L. Li, H. Yang, Y. Zhang, J. Su, L. Wang and B. Liu, Adv. Mater., 2025, 2500416 CrossRef CAS PubMed.
- Y. N. Regmi, E. Tzanetopoulos, G. Zeng, X. Peng, D. I. Kushner, T. A. Kistler, L. A. King and N. Danilovic, ACS Catal., 2020, 10, 13125–13135 CrossRef CAS.
- J.-W. Zhao, K. Yue, H. Zhang, S.-Y. Wei, J. Zhu, D. Wang, J. Chen, V. Y. Fominski and G.-R. Li, Nat. Commun., 2024, 15, 2928 CrossRef CAS PubMed.
- C. Lin, J.-L. Li, X. Li, S. Yang, W. Luo, Y. Zhang, S.-H. Kim, D.-H. Kim, S. S. Shinde, Y.-F. Li, Z.-P. Liu, Z. Jiang and J.-H. Lee, Nat. Catal., 2021, 4, 1012–1023 CrossRef CAS.
- J. Kuang, B. Deng, Z. Jiang, Y. Wang and Z.-J. Jiang, Adv. Mater., 2024, 2306934 CrossRef CAS PubMed.
- Z. Tao, N. Lv, H. Zhao, X. Luo, Z. Li, J. Yu, L. Chen, X. Liu and S. Mu, Chem. Sci., 2024, 15, 16796–16803 RSC.
- J. Zhang, X. Cao, Y.-F. Jiang, S.-F. Hung, W. Liu, H. B. Yang, C.-Q. Xu, D.-S. Li, T. Zhang, Y. Li, J. Li and B. Liu, Chem. Sci., 2022, 13, 12114–12121 RSC.
- Y. Qin, Y. Liu, Y. Zhang, Y. Gu, Y. Lian, Y. Su, J. Hu, X. Zhao, Y. Peng, K. Feng, J. Zhong, M. H. Rummeli and Z. Deng, ACS Catal., 2023, 13, 256–266 CrossRef CAS.
- V. Barone, J. E. Peralta, J. Uddin and G. E. Scuseria, J. Chem. Phys., 2006, 124, 024709 CrossRef PubMed.
- H. Ago, T. Kugler, F. Cacialli, W. R. Salaneck, M. S. P. Shaffer, A. H. Windle and R. H. Friend, J. Phys. Chem. B, 1999, 103, 8116–8121 CrossRef CAS.
- X. Wu, C. Lin, W. Hu, C. Fu, L. Tan, H. Wang, F. Meharban, X. Pan, P. Fu, H.-D. Um, Q. Xiao, X. Li, M. Yamauchi and W. Luo, Small Struct., 2024, 5, 2300518 CrossRef CAS.
- E. Naranjo, B. Abd Alreda, M. Zaidi, K. M. Batoo, S. Hussain, A. A. Almulla, A. A. Al-Sattar Dawood, S. G. Al- Musawi, M. F. Ramadan, S. A. Hamood, S. H. Zain Al-Abdeen, R. S. Zabibah and A. Alsalamy, J. Phys. Chem. Solids, 2024, 187, 111856 CrossRef CAS.
- W. Wu, Y. Wang, S. Song, Z. Ge, C. Zhang, J. Huang, G. Xu, N. Wang, Y. Lu, Z. Deng, H. Duan, M. Liu and C. Tang, Angew. Chem., Int. Ed., 2025, 64, e202504972 CrossRef CAS PubMed.
- K. Wang, C. Ni, L. Jin, X. Qian, H. Xu, H. Chen and G. He, Chem. Sci., 2025, 16, 7467–7476 RSC.
- C. Zhu, Y. Wang, Z. Jiang, F. Xu, Q. Xian, C. Sun, Q. Tong, W. Zou, X. Duan and S. Wang, Appl. Catal., B, 2019, 259, 118072 CrossRef CAS.
- H. Wu, Z. Fu, J. Chang, Z. Hu, J. Li, S. Wang, J. Yu, X. Yong, G. I. N. Waterhouse, Z. Tang, J. Chang and S. Lu, Nat. Commun., 2025, 16, 4482 CrossRef CAS PubMed.
- G. Chen, R. Lu, C. Ma, X. Zhang, Z. Wang, Y. Xiong and Y. Han, Angew. Chem., Int. Ed., 2024, 63, e202411603 CrossRef CAS PubMed.
- S. Anantharaj, S. Noda, M. Driess and P. W. Menezes, ACS Energy Lett., 2021, 6, 1607–1611 CrossRef CAS.
- B. Zhang, J. Wang, G. Liu, C. M. Weiss, D. Liu, Y. Chen, L. Xia, P. Zhou, M. Gao, Y. Liu, J. Chen, Y. Yan, M. Shao, H. Pan and W. Sun, Nat. Catal., 2024, 7, 441–451 CrossRef CAS.
- A. C. Lazanas and M. I. Prodromidis, ACS Meas. Sci. Au, 2023, 3, 162–193 CrossRef CAS PubMed.
- J. Huang, Y. Gao, J. Luo, S. Wang, C. Li, S. Chen and J. Zhang, J. Electrochem. Soc., 2020, 167, 166503 CrossRef CAS.
- W. Zhao, F. Xu, L. Liu, M. Liu and B. Weng, Adv. Mater., 2023, 2308060 CrossRef CAS PubMed.
- Y. Qin, T. Yu, S. Deng, X.-Y. Zhou, D. Lin, Q. Zhang, Z. Jin, D. Zhang, Y.-B. He, H.-J. Qiu, L. He, F. Kang, K. Li and T.-Y. Zhang, Nat. Commun., 2022, 13, 3784 CrossRef CAS PubMed.
- G. Zhuang, Y. Chen, Z. Zhuang, Y. Yu and J. Yu, Sci. China Mater., 2020, 63, 2089–2118 CrossRef CAS.
- H. Li, X. Hu, L. Wang, L. Shi, T. T. Isimjan and X. Yang, Chem. Eng. J., 2024, 481, 148547 CrossRef CAS.
- X. Huang, C. Lee, Y. Li, J. Xu and D. Liu, ACS Appl. Nano Mater., 2024, 7, 9244–9251 CrossRef CAS.
- L. Wu, Q. Liang, J. Zhao, J. Zhu, H. Jia, W. Zhang, P. Cai and W. Luo, Chin. J. Catal., 2023, 55, 182–190 CrossRef CAS.
- D. Wang, F. Lin, H. Luo, J. Zhou, W. Zhang, L. Li, Y. Wei, Q. Zhang, L. Gu, Y. Wang, M. Luo, F. Lv and S. Guo, Nat. Commun., 2025, 16, 181 CrossRef CAS PubMed.
- Z. Lei, F. Shi and L. Lu, ACS Appl. Mater. Interfaces, 2012, 4, 1058–1064 CrossRef CAS PubMed.
- S. Pan, H. Li, D. Liu, R. Huang, X. Pan, D. Ren, J. Li, M. Shakouri, Q. Zhang, M. Wang, C. Wei, L. Mai, B. Zhang, Y. Zhao, Z. Wang, M. Graetzel and X. Zhang, Nat. Commun., 2022, 13, 2294 CrossRef CAS PubMed.
- J. Liu, T. Wang, M. Sun, M. Liao, S. Wang, S. Liu, H. Shi, Y. Liu, Y. Shen, R. Cao, Y. Huang, B. Huang and Q. Li, J. Am. Chem. Soc., 2024, 146, 33276–33287 CrossRef CAS PubMed.
- S. Kaushal, A. K. Sahu, M. Rani and S. R. Dhakate, Renewable Energy, 2019, 142, 604–611 CrossRef CAS.
- X. Feng, J. Liu, L. Chen, Y. Kong, Z. Zhang, Z. Zhang, D. Wang, W. Liu, S. Li, L. Tong and J. Zhang, J. Am. Chem. Soc., 2023, 145, 10259–10267 CrossRef CAS PubMed.
- M. Qi, X. Du, X. Shi, S. Wang, B. Lu, J. Chen, S. Mao, H. Zhang and Y. Wang, J. Am. Chem. Soc., 2025, 147(21), 18295–18306 CrossRef PubMed.
- X. Chen, X.-T. Wang, J.-B. Le, S.-M. Li, X. Wang, Y.-J. Zhang, P. Radjenovic, Y. Zhao, Y.-H. Wang, X.-M. Lin, J.-C. Dong and J.-F. Li, Nat. Commun., 2023, 14, 5289 CrossRef CAS PubMed.
- Z. Li, H. Sheng, Y. Lin, H. Hu, H. Sun, Y. Dong, X. Chen, L. Wei, Z. Tian, Q. Chen, J. Su and L. Chen, Adv. Funct. Mater., 2024, 2409714 CrossRef CAS.
- W. He, X. Tan, Y. Guo, Y. Xiao, H. Cui and C. Wang, Angew. Chem., Int. Ed., 2024, 63, e202405798 CrossRef CAS PubMed.
- L. Wu, W. Huang, D. Li, B. Zhao, H. Zhou and W. Luo, Angew. Chem., Int. Ed., 2025, e202420848 CAS.
- J. Fan, X. Zhang, M. Han, X. Xiang, C. Guo, Y. Lin, N. Shi, D. Xu, Y. Lai and J. Bao, Small, 2024, 20, 2303927 CrossRef CAS PubMed.
- Z.-X. Qian, G.-H. Liang, L.-F. Shen, G. Zhang, S. Zheng, J.-H. Tian, J.-F. Li and H. Zhang, J. Am. Chem. Soc., 2025, 147, 1334–1343 CrossRef PubMed.
- H. Liu, Q. Zhou, J. Yu, M. Nakabayashi, Y.-T. Lee, N. Shibata, Y. Li and J.-J. Delaunay, ACS Catal., 2025, 15, 8511–8521 CrossRef CAS.
- Y. Wang, X. Lei, B. Zhang, B. Bai, P. Das, T. Azam, J. Xiao and Z.-S. Wu, Angew. Chem., Int. Ed., 2024, 63, e202316903 CrossRef CAS PubMed.
- Y. Qin, X. Niu, R. Zhao, J. Sun, Z. Xu, Z. Guo, D. Liu, L. Guo, C. Liu, J. Zhang and Q. Wang, ACS Catal., 2024, 14, 12970–12981 CAS.
- T. Zhang, H.-F. Zhao, Z.-J. Chen, Q. Yang, N. Gao, L. Li, N. Luo, J. Zheng, S.-D. Bao, J. Peng, X. Peng, X.-W. Liu and H.-B. Yu, Nat. Commun., 2025, 16, 3327 CrossRef CAS PubMed.
- L. An, C. Wei, M. Lu, H. Liu, Y. Chen, G. G. Scherer, A. C. Fisher, P. Xi, Z. J. Xu and C.-H. Yan, Adv. Mater., 2021, 33, 2006328 CrossRef CAS PubMed.
- F.-Y. Chen, Z.-Y. Wu, Z. Adler and H. Wang, Joule, 2021, 5, 1704–1731 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |