
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling the role of cobalt in the product regulation for CO2 hydrogenation to light olefins over alumina-supported Co–Fe catalysts†
Zhihao
Liu‡
a,
Wenlong
Song‡
b,
Peipei
Zhang
c,
Jiaming
Liang
 *a,
Chengwei
Wang
a,
Chufeng
Liu
a,
Hanyao
Song
a,
Baojian
Chen
a,
Kangzhou
Wang
*d,
Guangbo
Liu
e,
Xiaoyu
Guo
a,
Yingluo
He
a,
Xinhua
Gao
b,
Jianli
Zhang
b,
Guohui
Yang
a and
Noritatsu
Tsubaki
*a
*a,
Chengwei
Wang
a,
Chufeng
Liu
a,
Hanyao
Song
a,
Baojian
Chen
a,
Kangzhou
Wang
*d,
Guangbo
Liu
e,
Xiaoyu
Guo
a,
Yingluo
He
a,
Xinhua
Gao
b,
Jianli
Zhang
b,
Guohui
Yang
a and
Noritatsu
Tsubaki
*a
aDepartment of Applied Chemistry, School of Engineering, University of Toyama, Gofuku 3190, Toyama 930-8555, Japan
bState Key Laboratory of High-efficiency Coal Utilization and Green Chemical Engineering, College of Chemistry and Chemical Engineering, Ningxia University, Yinchuan 750021, Ningxia, China
cCNOOC Institute of Chemical & Advanced Materials, Beijing 102209, China
dSchool of Materials and New Energy, Ningxia University, Yinchuan 750021, Ningxia, China
eKey Laboratory of Biofuels, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, China
First published on 10th July 2025
Abstract
CoFe-based catalysts for CO2 hydrogenation reactions have been widely studied, but the effects of cobalt on the product regulation remains largely over-looked. In the present study, we report a series of Na-decorated alumina-supported Co–Fe bimetallic catalysts with varying Co/Fe molar ratios for the direct conversion of CO2 to light olefins. In situ XRD and Mössbauer spectroscopy reveal that Co doping significantly promotes the reduction and carburization of Fe species, leading to the formation of active CoFe alloy carbides. Furthermore, the DFT results indicate that cobalt decreases the H2 adsorption energy, thereby regulating the surface C/H ratio and enhancing the tandem RWGS and Fischer–Tropsch reactions. These effects synergistically improve the formation and desorption of light olefins. As a result, the optimal Co1Fe2 catalyst (Co/Fe = 1/2) achieves a high STY of 315.1 g kgcat−1 h−1 for light olefins with a CO2 conversion of 51.9% at 320 °C. This study provides mechanistic insights into cobalt-assisted product selectivity control and offers a promising strategy for designing highly efficient CO2-to-light olefins catalytic systems.
Introduction
The excessive anthropogenic CO2 emissions caused by social development have resulted in significant environmental issues, including global warming and ocean acidification.1–3 In recent decades, the thermal catalytic conversion of CO2 with green H2 to produce high-value-added chemicals has attracted considerable interest.4 Among these, light olefins (including ethylene, propylene, and butene) are widely used in the production of plastics, fibers, and other chemical products. Traditionally, they are obtained by naphtha cracking. As the crisis of non-renewable energy sources has received increasing attention, the efficient catalytic CO2 hydrogenation to produce light olefins represents a potential strategy to mitigate the greenhouse effect and alleviate the market demand, given its status as an industrialized method today.Typically, the hydrogenation of CO2 to light olefins can be achieved through two distinct routes: a modified Fischer–Tropsch synthesis (CO2-FTS) route or a tandem methanol-mediated (MeOH) route.5,6 The FTS pathway can be divided into a two-stage cascade reaction, comprising a reverse water gas shift (RWGS) reaction and Fischer–Tropsch synthesis. Fe-based catalysts have been extensively investigated due to their cost-effectiveness and strong adaptability to reaction conditions.7–11 Nevertheless, the carbon efficiency remains low due to the restricted target selectivity of the FTS process and the constrained thermodynamic equilibrium CO2 conversion rate. To address this case, various strategies have been adopted to regulate the activity and product selectivity over Fe-based catalysts. The doping modification with alkaline promoters, such as Na and K, has been demonstrated to enhance the CO2 adsorption and facilitate the formation of active phases.12–14 Wang et al.12 reported that the presence of Na played an important role in regulating the electronic properties of iron carbide and adsorption behaviours of reactant molecules, resulting in a significant increase in total olefin selectivity from 27% to 76%. In addition, the incorporation of a second active metal (e.g., Co, Cu) was also a prevalent strategy to enhance the catalytic performance of Fe-based catalysts.15–17 Among them, cobalt is frequently employed to modify iron-based catalysts due to its high reducibility and the ability to dissociate CO and H2. However, the primary products of the individual Co-based catalysts in CO2 hydrogenation were mainly CH4 instead of C2+ hydrocarbons, due to the inability of cobalt in the RWGS reaction.18 As a result, the key to optimizing iron-based catalysts for efficient CO2 hydrogenation by cobalt doping lies in understanding how to regulate the structural evolution and surface chemistry properties of the iron species. Therefore, it is imperative to construct a cobalt–iron bimetallic catalyst that combines the high activity and reducibility of cobalt with the high olefin selectivity of iron.
Up to now, considerable efforts have been dedicated to developing efficient cobalt–iron bimetallic catalysts and exploring their synergistic effect for CO2 hydrogenation.10,19–24 By modifying the preparation methods, the active components and catalytic performance of catalysts with different composition and structure have been investigated. Zhang et al.19 reported a Na-modified CoFe alloy catalyst utilizing layered double hydroxide precursors that could directly convert CO2 into jet fuel comprising hydrocarbons. At 240 °C, the catalyst exhibited an unprecedented jet-fuel-range hydrocarbons selectivity of 63.5% with a CO2 conversion of 10.2%, and a combined selectivity of less than 22% for the unwanted CO and CH4. Liu et al.20 prepared a series of Na-promoted Co–Fe bimetallic catalysts with varying compositions or proximity and elucidated the structural evolution that occurred during the reduction and reaction. It was demonstrated that the incorporation of the Co element could enhance the reducibility of the catalyst and facilitate the formation of carbides, thereby exhibiting remarkable improvements for CO2 hydrogenation to olefins. Even though the doping of Co is believed to play a crucial role in determining the reduction and carburization of Fe-based catalysts and promoting cascade reactions between chain propagation, the current understanding is limited, and the contribution of Co in regulating the product distribution remains largely over-looked.
Herein, we prepared a series of Na-modified alumina-supported Co–Fe bimetallic catalysts with varying Co/Fe molar ratios and tested their catalytic performance for directly synthesis of light olefins from CO2 hydrogenation. The specific roles of Co doping on the structural evolution and surface chemical properties of iron-based catalysts were investigated by a variety of ex/in situ characterization techniques. It was observed that the reduction and carburization of Fe species was facilitated by the cooperation of Co. In addition, the augmented dissociation of H2 from Co doping helped to regulate the C/H ratio on the catalyst surface, accelerating transfer of reaction intermediates species and thus reinforcing the cascade reactions between RWGS and FTS. As a result, the optimal Co1Fe2 catalyst with the Co/Fe molar ratio of 1/2 exhibited more distinguished catalytic performance for CO2 hydrogenation to light olefins relative to other catalysts, and demonstrated no evident deactivation during the 100 h stability test. This work is expected to provide a valuable contribution for studying the specific promoting effects of cobalt on Co–Fe bimetallic catalysts.
Experimental
Catalyst preparation
Co(NO3)2·6H2O, Fe(NO3)3·9H2O, urea, and Na2CO3 were purchased from Fujifilm Chemical Reagent Co. Ltd. Al(NO3)3·9H2O was purchased from Sigma-Aldrich Chemical Reagent Co. Ltd. All reagents were of analytical grade and used as received without further purification.A series of precursor samples were synthesized by a facile hydrothermal method, as described in our previous work with minor modifications.18 Take the Co1Fe2 catalyst for example, 5 mmol of Co(NO3)2·6H2O, 10 mmol of Fe(NO3)3·9H2O, and 5 mmol of Al(NO3)3·9H2O were dissolved sequentially in 100 mL of deionized water with magnetic stirring. Subsequently, 0.1 mol of urea was added to aforementioned solution under constantly stirring and then the stirring was continued for 30 min. Subsequently, the suspension was transferred into a Teflon-lined stainless-steel autoclave and subjected to hydrothermal treatment at 120 °C for 10 h under autogenous pressure. The precipitate was filtered and washed three times with deionized water by centrifugation until the pH of the supernatant reached 7. The resulting sample was then dried at 80 °C overnight to obtain the precursor. The mixed metal oxides were obtained by calcining precursors in air at 400 °C for 4 h. Then, the Na-decorated catalyst was prepared by the incipient wetness impregnation method using Na2CO3. Specifically, a certain amount of Na2CO3 was dissolved in 0.3 mL of deionized water and slowly dropped onto 0.5 g of the calcined sample under ultrasonic conditions and continuously stirred with a glass rod. Once the surface was dry, the container was rinsed with 0.2 mL of water and the process was repeated, ensuring that all sodium ions were impregnated on the catalyst surface. This was followed by drying at 80 °C for 6 h and calcination at 550 °C for 2 h. The resulting product was denoted as Co1Fe2. The synthesis of Co0Fe, Co1Fe4, and Co2Fe1 catalysts was conducted using the same methodology as that employed for Co1Fe2, with the respective molar ratio of Co/Fe in the precursors. The amount of Al was kept constant, with the ratio of ([Co]2+ + [Fe]3+)/[Al]3+ set at 3.
The various Fe-based catalysts doped with typical metals (Cu, Mn, Zn, and Zr) were prepared using the same methodology as that employed for Co1Fe2, with the respective nitrates in the precursors. The obtained samples were denoted as Cu1Fe2, Mn1Fe2, Zn1Fe2, and Zr1Fe2, respectively.
Characterization
In order to ascertain the precise content of the Na promoter, the calcined samples were measured by inductively coupled plasma optical emission spectrometry (ICP-OES) analysis on a PerkinElmer 8300 instrument. The transmission electron microscope (TEM) and element mapping images were obtained using a FEI Tecnai F20 instrument operating at an accelerating voltage of 200 kV. The samples were dispersed in ethanol and dried on carbon-coated copper grids. The texture properties of the samples were determined by N2 physisorption measurements at −196 °C using a Micromeritics 3Flex instrument. The samples were first degassed at 250 °C for 2 h to purify. The pore structures and properties were determined using Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) method, respectively.The crystalline and phase of the samples were determined by X-ray diffraction (XRD) performed on a Rigaku D/MAX2200PC instrument with Cu Kα radiation (λ = 1.54 Å) at 40 kV and 40 mA. The intensity data were recorded over the 2θ range of 20–80° with a scanning step length of 5° min−1. The average particle sizes of the samples were calculated according to Scherrer equation. In situ XRD measurements were carried out using a Rigaku SmartLab instrument to investigate the phase evolution of catalysts during the reduction (atmospheric pressure and temperature increased from room temperature to 400 °C) and reaction (3 MPa and temperature increased from 50 °C to 320 °C) processes. Mo Kα radiation (λ = 0.71 Å) was employed due to its superior penetration through the stainless-steel walls of the in situ high-pressure reactor. This higher-energy X-ray source enabled reliable phase identification under operando conditions, particularly for catalysts undergoing reduction and reaction at elevated pressure and temperature. Specifically, 50 mg of the sample was placed in the chamber under reducing conditions (high-purity H2, 30 mL min−1) and gradually heated to 400 °C at a heating rate of 5 °C min−1 and maintained for 2 h. Subsequently, the reaction chamber was cooled to 50 °C naturally and the feed gas (CO2/H2/Ar = 27/68/5) was switched to 3 MPa. As the temperature increased to 320 °C, the XRD spectrum was recorded. The reaction intermediates and mechanism were identified through in situ diffuse reflectance infrared Fourier transform (DRIFT) measurement on a Bruker TENSOR II spectrometer. In detail, 50 mg of the 20–40 mesh sample was subjected to a 2 h pretreatment at 400 °C in a high-purity H2. Subsequently, the chamber was adjusted to the reaction temperature and pressure in an Ar atmosphere and the background spectrum was recorded. Subsequently, the feed gas (CO2/H2/Ar = 27/68/5) was introduced to 3 MPa, and the infrared signal of the catalyst surface was recorded at 10 min intervals.
H2 temperature-programmed reduction (H2-TPR) was conducted on an Autochem II 2920 instrument. In a typical process, 50 mg of the catalyst was loaded into a quartz tube and pretreated at 300 °C for 1 h in pure He flow. After cooling to 50 °C, a mixture of H2/He (1/9, v/v) was injected into the reactor with the temperature heated to 800 °C at a rate of 10 °C min−1. The hydrogen consumption signal was quantified using TCD. All temperature-programmed experiments were performed on the same instrument as H2-TPR. For CO2 temperature-programmed desorption (CO2-TPD) experiment, 50 mg of the sample was pretreated in H2 at 400 °C for 2 h and then cooled to 50 °C. Subsequently, CO2/He (1/9, v/v) was passed through for 1 h to saturate the sample with CO2. The sample was then flushed with He for 1 h. Finally, the catalyst was heated to 700 °C while recording the TCD signal. The CO-TPD and H2-TPD experiments were conducted using the same processing steps as CO2-TPD, with the exception of the introduction of alternative adsorption gases. For C3H6-TPD experiment, 50 mg of the spent catalyst was pretreated in He at 300 °C for 1 h and then cooled to 50 °C. Subsequently, pure C3H6 was passed through for 1 h to saturate the sample with C3H6. The sample was then flushed with He for 0.5 h. Finally, the catalyst was heated to 800 °C while recording the TCD signal. The 57Fe Mössbauer spectroscopy (MES) technique was employed to detect Fe species in spent catalysts. This was achieved by utilizing a MS500 spectrometer at room temperature, with 57Co(Rh) serving as the irradiation source.
Catalytic performance test
The catalytic performance of the catalysts was evaluated in a fixed-bed reactor with a 6 mm inner diameter. Firstly, 0.2 g pelletized precursor sample (20–40 mesh) mixed with 0.5 g quartz sand was packed into the reactor and in situ reduced at 400 °C for 8 h using a pure H2 flow (40 mL min−1, atmospheric pressure) prior to the reaction. After that, the reactor was cooled to reaction temperature and the reaction gas (CO2/H2/Ar = 27/68/5) was gradually introduced into the reactor until the pressure increased to the target condition. The reaction conditions were 320 °C, 3 MPa, 9000 mL gcat−1 h−1, and TOS = 8 h unless specified. During the reaction, the effluent gas was quantitatively analyzed utilizing two online gas chromatographs. One was equipped with a thermal conductivity detector (TCD) and an active charcoal column to analyze the inorganic components and CH4; the other was equipped with a flame ionization detector (FID) and a HP-PLOTQ column to detect light hydrocarbons. The heavy hydrocarbons were collected in a cold trap using 2 g of dodecane as a solvent and analyzed using an off-line gas chromatograph equipped with an FID and a DB-1 capillary column.The conversion of CO2, selectivity of CO and CiHx hydrocarbons (excluding CO), STY of C2–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) 4, and the probability of the chain growth value (α) were calculated as eqn (1)–(5).
4, and the probability of the chain growth value (α) were calculated as eqn (1)–(5).
 | (1) |
 | (2) |
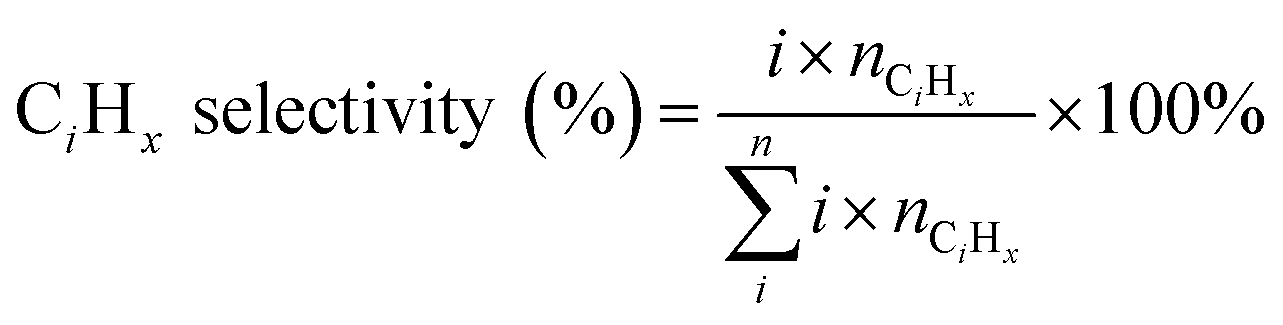 | (3) |
 | (4) |
 | (5) |
represent CO2 conversion and selectivity of CO and C2–C4, respectively; i and Wn represent the carbon number and weight fraction of a product containing n carbon atoms, respectively.
Results and discussion
Catalytic activity tests
Fig. 1a illustrated the XRD patterns of the prepared catalysts. After calcination, the diffraction pattern of the calcined Co0Fe catalyst indicated that α-Fe2O3 was supported on amorphous Al2O3 nanosheets.25 The addition of Co resulted in the appearance of diffraction peaks at 2θ = 30.3° and 43.4°, which could be attributed to the spinel CoxFe3−xO4 phase.26 Furthermore, the aforementioned diffraction peaks exhibited a shift towards higher 2θ with increasing Co addition (Fig. S1a†). This could be attributed to the isomorphous replacement of Fe3+ at the octahedral position in the Co–Fe bimetallic catalyst by Co3+, which had a smaller radius.20,26,27 This resulted in a contraction of the crystal structure and a shift of the diffraction peaks. Upon increasing the Co/Fe ratio to 1/2, the diffraction peaks belonging to Co3O4 emerged.28 In addition to its impact on the phase structure of the catalyst, the Co/Fe ratio also influenced the particle size. In accordance with the Scherrer equation, the particle size of the calcined catalyst exhibited a unidirectional decrease, from 22.1 nm for Co0Fe to 15.2 nm for Co2Fe1 (Table 1). In addition, the pore structure and performance of the catalysts were investigated using N2 physical adsorption measurements. As shown in Fig. 1b, all catalysts exhibited a type IV isotherm according to the IUPAC classification with a mesoporous structure. At medium relative pressure, the desorption branch rose faster than the adsorption branch due to N2 capillary condensation, resulting in a hysteresis loop. After the capillary condensation filled the mesopores, the catalyst continued to adsorb to form a multimolecular layer and the desorption isotherm continued to rise. Therefore, an H3 hysteresis loop was observed on the catalyst. The isotherm did not have an obvious saturated adsorption platform, indicating the irregular pore structure. Table 1 showed the specific surface area and pore structure parameters of different catalysts. The incorporation of cobalt increased the specific surface area and pore volume of the catalyst. Combined with the reduction in catalyst particle size after cobalt addition (Table 1), the proximity of cobalt to iron was more conducive to the distribution of iron species. | ||
| Fig. 1 (a) XRD patterns and (b) N2 adsorption–desorption isotherms of the prepared catalysts. | ||
| Catalysts | Crystal sizea (nm) | Surface area (m2 g−1) | Pore diameter (nm) | Pore volume (cm3 g−1) | Na contentb (wt%) |
|---|---|---|---|---|---|
| a Calculated from Fe2O3 (104) plane using the Scherrer equation. b Obtained from ICP-OES measurements. | |||||
| Co0Fe | 22.1 | 50.1 | 9.6 | 0.20 | 3.8 |
| Co1Fe4 | 17.3 | 108.1 | 10.3 | 0.41 | 3.7 |
| Co1Fe2 | 16.7 | 97.7 | 11.9 | 0.36 | 4.1 |
| Co2Fe1 | 15.2 | 93.2 | 14.3 | 0.46 | 4.1 |
The catalytic performance of a series of Fe-based catalysts doped with typical metals (Co, Cu, Mn, Zn, Zr, and Co) was investigated, and the results were showed in Fig. S2.† Among all tested catalysts, the Co-doping Co1Fe2 catalyst demonstrated the highest selectivity (44.3%) and STY (315.1 g kgcat−1 h−1) of light olefins, significantly outperforming the other catalysts. Moreover, it exhibited the lowest selectivity towards undesired by-products, with CO + CH4 accounting for only 21.4%, signifying a substantial advantage over Fe-based catalysts previously reported for CO2 hydrogenation to light olefins (Table S1†). This suggested a distinctive role for cobalt in facilitating superior product regulation, favoring the production of light olefins.
Fig. 2 and Table S2† illustrated the catalytic performance of the prepared catalysts with varying Co doping in the CO2 hydrogenation reactions. At 320 °C and 3 MPa, the Co-free catalyst (Co0Fe) showed a C5+ selectivity of up to 50.4% with a CO2 conversion of 36.8% (Fig. 2a). As the Co doping increased, the activity of the catalyst was enhanced, accompanied by a notable alteration in the product distribution towards light hydrocarbons. The CO2 conversion increased from 36.8% (for the Co0Fe catalyst) to 61.6% (for the Co2Fe1 catalyst), while the selectivity of C5+ in hydrocarbon products decreased from 50.4% to 25.2% with a marked increase in CH4 selectivity from 10.1% to 42.3%. Additionally, an increase in the Co/Fe molar ratio resulted in a notable volcano-shaped distribution in the space time yield (STY) of C2–C4, which initially rose from 163.0 g kgcat−1 h−1 for Co0Fe and reached its maximum of 315.1 g kgcat−1 h−1 for Co1Fe2, then declined precipitously to 36.1 g kgcat−1 h−1 for the Co2Fe1 catalyst.
 | ||
| Fig. 2 (a) The catalytic performance of the prepared catalysts. (b) The chain growth factors of the prepared catalysts. (c) The product distribution of the prepared catalysts (reaction conditions: 320 °C, 3 MPa, 9000 mL gcat−1 h−1, TOS = 8 h). | ||
The chain growth factors of catalysts with varying Co/Fe ratios revealed the alteration in product distribution after Co doping, as demonstrated in Fig. 2b. The Co0Fe catalyst, which lacked Co, exhibited the highest chain growth factor of 0.68. The chain growth factor demonstrated a decline with an increase in Co content, indicating that iron species was more advantageous than Co in promoting C–C coupling to C2+ hydrocarbons, as opposed to the production of CH4 in CO2 hydrogenation (Fig. 2c). Conversely, the Co1Fe2 catalyst exhibited the highest selectivity for C2–C4. This illustrated that the incorporation of cobalt into the Fe-based catalyst restrained the expansion of carbon chains, whereas the optimal amount of Co could effectively regulate the termination of carbon chains and enhance the selectivity of the target light olefins.29,30
The aforementioned results indicated that the Co1Fe2 catalyst demonstrated superior catalytic performance in comparison to other catalysts with varying Co/Fe ratios. As previously stated, the Na promoter modification has been demonstrated to enhance catalytic performance by modifying the electronic structure and adsorption properties of the catalyst surface.12,13 However, an excess of Na content may potentially result in the coverage of the catalyst surface. An investigation was conducted into the effect of Na content on the performance of the Co1Fe2 catalyst (Fig. S3†). The results indicated that the optimal Na content of 4% achieved the highest catalytic activity and STY of C2–C4. Moreover, the sensitivity of the Co1Fe2 catalyst to the reaction conditions, including temperature, pressure, and GHSV, was evaluated, and the results were summarized in Table S3.† As illustrated in Fig. 3a, when the temperature was varied from 280 °C to 320 °C, a notable enhancement in CO2 conversion was observed, rising from 31.6% to 51.9%. Concurrently, the light olefin selectivity exhibited a marked increase, rising from 35.7% to 44.3%. Jiang et al.31 reported that the addition of Co promoted the consumption of CO in the FT reaction after the RWGS, thereby enhancing the CO2 conversion of the Fe-based catalyst. It may be postulated that the rise in temperature served to further activate the synergistic effect between iron and cobalt, thereby promoting the efficient hydrogenation of CO2 to low-carbon olefins. Upon further elevation of the temperature to 380 °C, the performance of the catalyst did not exhibit a notable enhancement. Furthermore, an increase in pressure resulted in a slight increase in catalyst activity due to the molecule reduction reaction (Fig. 3b). An increase in space velocity resulted in a reduction of the effective residence time, and an increase in the number of reactant molecules entering the system. Concurrently, the adsorption sites and activation capacity of the catalyst surface were limited, resulting in a considerable portion of reactant molecules not participating in the reaction. Consequently, the CO2 conversion decreased with an increase in GHSV, while the distribution of catalyst products remained largely unchanged (Fig. 3c). Moreover, the STY of light olefins was sustained above 200 g kgcat−1 h−1 across almost all tested reaction conditions, indicating the stability of the catalysts and their adaptability to varying reaction conditions, which was conducive to large-scale industrial utilization. In addition, the catalytic activity and product distribution of the Co1Fe2 catalyst over a 100 h reaction period were illustrated in Fig. 3d. The catalytic performance of the Co1Fe2 catalyst remained largely stable after the initial few hours of the induction period. The CO2 conversion exhibited a slight decline, from 51.9% (8 h) to 48.2% (100 h), while the selectivity of light olefins in the hydrocarbons was consistently above 40% across the tested points.
 | ||
| Fig. 3 Effects of (a) reaction temperature, (b) pressure, and (c) GHSV on the catalytic performance of Co1Fe2. (d) The durability test of CO2 hydrogenation over the Co1Fe2 catalyst (reaction conditions: 320 °C, 3 MPa, 9000 mL gcat−1 h−1, TOS = 8 h unless specified). | ||
Structural evolution of the catalysts
The reduction behavior of the catalysts was investigated to monitor the dynamic structural evolution and to evaluate the contribution of Co throughout the reduction process. As illustrated in Fig. 4a, four reduction peaks were observed at 356, 458, 653 and 750 °C for Co0Fe catalysts. The former three peaks roughly corresponded to a stepwise reduction process from the initial α-Fe2O3 through Fe3O4, FeO, and then to metallic Fe, respectively, while the last one could be attributed to the reduction of the iron species that interacted strongly with the support.32 Following the introduction of Co, the position of the reduction peaks exhibited a notable shift towards lower temperatures. Moreover, the hydrogen consumption peak areas at elevated temperature were significantly reduced with increasing Co addition, indicating that the Co element may enhance the reducibility of the Co–Fe bimetallic catalysts. This could be assigned to the fact that Co was more readily reduced, thereby facilitating the dissociation of H2.33 | ||
| Fig. 4 (a) H2-TPR profiles of the prepared catalysts; (b) XRD patterns of the spent catalysts; in situ XRD patterns for the Co1Fe2 catalyst during the (c) reduction and (d) reaction process. “*” symbol represents the diffraction peaks of sample holder, which is unavoidable because of supporting the samples. | ||
This promotion effect was also reflected in the in situ XRD. As illustrated in Fig. S4a,† the Co0Fe catalysts did not exhibit notable alterations in the phase below 400 °C, remaining in the initial Fe2O3 (PDF # 24-0072) phase. After 0.5 h at 400 °C, the intensity of the diffraction peaks of Fe2O3 exhibited a gradual decline, accompanied by the gradual emergence of metal Fe (PDF # 01-1252). Following 2 h at 400 °C, the metal Fe had become the predominant phase, and the diffraction peaks belonging to iron oxides disappeared. In contrast, the diffraction peaks attributed to the CoFe alloy phase at 2θ = 15.1°, 16.2° and 24.1° appeared immediately at 400 °C in Co1Fe2 (Fig. 4c), which was earlier than that in the Co0Fe catalyst, indicating that the Co1Fe2 catalyst could be reduced to the final alloy phase at a lower temperature and in a shorter time than the Co0Fe catalyst. These results suggested that the incorporation of Co could facilitate the reduction of iron species and accelerate the structural transformations of iron species during the reduction process, which was in accordance with the findings of the H2-TPR experiments.
The structural evolution of the catalysts during the reaction process and the formation of active sites were characterized by in/ex situ XRD and STEM. Fig. 4c and S4a† illustrated the CoFe alloy and metallic Fe of the reduced Co1Fe2 and Co0Fe catalysts, respectively. Upon exposure to the reaction gas, the formation of carbides was observed at 320 °C in the Co1Fe2 catalyst (Fig. 4d), indicating the carburization of alloy species. As the reaction time increased, the diffraction peaks of the CoFe alloy phase diminished, while those of χ-(CoxFe1−x)5C2 (PDF # 20-0509) became more pronounced. Simultaneously, the characteristic peaks belonging to the FeCo alloy oxides reemerged, and the dual phases collaboratively catalyzed the conversion of CO2. In contrast, the formation of carbide peaks was not observed in Co0Fe until 1 h at 320 °C, indicating that the Co0Fe catalyst exhibited a lower carburization ability than the Co1Fe2 catalyst during the reaction (Fig. S4b†). It is widely acknowledged that metal carbides play a pivotal role in the synthesis of light olefins through CO2 hydrogenation.34–36 The presence of a greater number of metal carbides on Co1Fe2 with Co doping facilitated the formation of more active sites, thereby enhancing the overall activity and C2–C4 selectivity.
The XRD and TEM were employed to confirm the active phases and structures of the spent catalysts. As illustrated in Fig. 4b, the presence of Fe3O4 and Fe5C2 was observed in the spent Co0Fe catalyst, which were identified as the two crucial active sites for the catalytic CO2 hydrogenation to light olefins over iron-based catalysts.37 With the introduction of Co, the diffraction peaks of Fe5C2 shifted to a higher 2θ angle with an increase in the Co/Fe ratio (Fig. S1b†). This was consistent with the XRD patterns of the calcined catalysts. It was attributed to the doping of the Co with a small radius into the Fe5C2 lattice, forming the χ-(CoxFe1−x)5C2 structure, which narrowed the lattice.20,26,38 Additionally, the peaks of Fe3O4 were significantly weakened, accompanied by an increase in the peak belonging to iron carbide, which indicated that Co doping promoted the carburization of iron species. Furthermore, the microstructure of the spent catalysts was observed by HRTEM (Fig. 5). For the spent Co0Fe catalyst, Fe3O4 and Fe5C2 were distributed on the alumina matrix with fringe spacings of 0.250 and 0.205 nm (Fig. 5b), respectively, which matched well with the (311) and (510) lattice planes of Fe3O4 and Fe5C2, respectively. In the spent Co1Fe2 catalyst, the presence of alumina and metal carbide nanoparticles was evident. Concurrently, the d-spacing values of the lattice planes of the HRTEM images revealed the formation of the χ-(CoxFe1−x)5C2 phase with a lattice spacing of 0.202 nm (Fig. 5d). Furthermore, the images with EDS elemental mapping revealed the surface spatial distribution of Fe, Co, and Al elements (Fig. 5e). After the reaction, the distribution of Fe and Co elements was found to be uniform in the same position, confirming that Co penetrated into the Fe5C2 lattice to form Co–Fe alloy carbides. Concurrently, the crystallized carbides were observed to be surrounded by alumina, suggesting that a structure comprising Co–Fe carbides as the primary phase and alumina support was formed in the spent Co1Fe2 catalyst. Therefore, the addition of Co played a significant role in the structural evolution of iron species during the reduction and reaction of the catalyst. The strong ability of Co to decompose CO and H2 led to the promotion of the reduction and carburization of iron species, which in turn generated more active sites for catalytic CO2 hydrogenation reactions and promoted the production of light olefins.
 | ||
| Fig. 5 TEM and HRTEM images of the spent (a and b) Co0Fe and (c and d) Co1Fe2 catalysts; (e) elemental mapping of the spent Co1Fe2 catalyst. | ||
Structure–activity relationship
The traditional concept is that the conversion of CO2 to hydrocarbons requires two process pathways: the conversion of CO2 to CO is initiated by the RWGS reaction, followed by the hydrogenation of CO to hydrocarbons, which is a characteristic step of the Fischer–Tropsch process. This pathway requires Fe-based catalysts with bifunctional active sites, including iron oxide sites for RWGS and possibly iron carbide sites that promote C–C coupling to form hydrocarbons. In order to obtain accurate and detailed information about the composition of the iron-containing phase of the spent catalysts after 8 h of CO2 hydrogenation, 57Fe Mössbauer spectra were recorded (Fig. 6). For the spent Co0Fe catalyst, the raw spectra were deconvoluted into one doublet and five sextet subspectra. The sextets with hyperfine field (Hhf) of 187, 217, and 104 kOe were attributed to the typical iron carbide (A, B, and C) phases.39,40 The presence of sextets with hyperfine fields of 469 and 455 kOe, corresponding to Fe3O4 components (A and B), was also identified, along with double peaks IS = 0.24 mm s−1 and QS = 1.11 mm s−1, corresponding to Fe3+.41 This finding confirmed the in situ phase restructuring from metal to oxides and carbides as the actual active species during CO2 hydrogenation reactions. The detailed Mössbauer parameters exhibited in Table 2 demonstrated the presence of the primary iron phases in the spent Co0Fe1 and Co1Fe2. It was also noteworthy that the Hhf value of iron carbide changed following the addition of cobalt, particularly the Hhf value of iron carbide (III), which underwent a substantial reduction from 104 kOe (Co0Fe) to 93 kOe (Co1Fe2). This deviation indicated that the disorder in the local Fe–Co environment within the particles, proving the formation of mixed iron–cobalt carbides.20,26 | ||
| Fig. 6 57Fe Mössbauer spectra of the spent (a) Co0Fe and (b) Co1Fe2 catalysts. | ||
| Catalysts | Assignment | IS (mm s−1) | QS (mm s−1) | Hhf (kOe) | Area (%) | |
|---|---|---|---|---|---|---|
| Co0Fe | χ-Fe5C2 (A) | 0.04 | 0.03 | 187 | 11.5 | 36.0 |
| χ-Fe5C2 (B) | 0.12 | 0.04 | 217 | 11.5 | ||
| χ-Fe5C2 (C) | 0.29 | 0.05 | 104 | 13.0 | ||
| Fe3O4 (A) | 0.08 | 0.00 | 469 | 32.0 | 59.1 | |
| Fe3O4 (B) | 0.56 | 0.10 | 455 | 27.1 | ||
| Fe3+ | 0.21 | 1.00 | — | 4.8 | 4.8 | |
| Co1Fe2 | χ-(CoxFe1−x)5C2 (A) | 0.09 | 0.08 | 192 | 17.8 | 68.8 |
| χ-(CoxFe1−x)5C2 (B) | 0.12 | 0.07 | 218 | 23.6 | ||
| χ-(CoxFe1−x)5C2 (C) | 0.28 | 0.08 | 93 | 27.4 | ||
| Fe3O4 (A) | 0.03 | 0.15 | 462 | 10.4 | 16.9 | |
| Fe3O4 (B) | 0.69 | 0.04 | 455 | 6.5 | ||
| Fe3+ (spm) | 0.20 | 0.86 | — | 14.2 | 14.2 | |
Despite its high Fe3O4 content (Table 2), Co0Fe exhibited a low CO2 conversion of 36.8%. This suggested that only RWGS reaction could not limit the kinetics of CO2 hydrogenation reactions. For the second stage Fischer–Tropsch reaction, the CO-TPD results (Fig. S5†) demonstrated that the desorption peak shifted to lower temperatures following the addition of cobalt, indicating that the binding force between CO and the catalyst surface diminished. An increase in the desorption peak area was also observed, which was conducive to the rapid transfer and conversion of CO under reaction conditions. In addition, in order to determine the co-adsorption and reaction of CO and H2 on the catalyst surface, the adsorption energy of H2 on the carbide surface was calculated. As shown in Fig. S6 and S7,† the optimal configuration for the carbide surface to adsorb H2 after CO pre-adsorbed was determined. In this structure, the total adsorption energy of H2 on the (CoxFe1−x)5C2 surface was −0.02 eV (Fig. S8†), which was lower than that on the Fe5C2 surface of 0.17 eV (the positive adsorption energy could be attributed to the energy consumption required for surface reconstruction), suggesting that the surface H atoms were more likely to aggregate on the (CoxFe1−x)5C2 surface. This finding was consistent with the results of H2-TPD (Fig. 7a), which indicated a substantial enhancement in H2 adsorption following the incorporation of Co. This might be attributed to the pronounced H2 dissociation capacity exhibited by cobalt species. The lower C/H ratio on the surface of the Co1Fe2 catalyst was more conducive to the hydrogenation of CO. This process had been found to accelerate the transfer and conversion of reaction intermediates, thereby reinforcing cascade reactions between the RWGS reaction and FTS. Therefore, the Co1Fe2 catalyst exhibited higher activity for CO2 hydrogenation reactions. Furthermore, the H coverage on the catalyst surface might affect the formation of products. C3H6-TPD was performed using the spent Co0Fe and Co1Fe2 catalyst, and the results were shown in Fig. S9.† It was evident that the desorption peak position of C3H6 on the Co1Fe2 catalyst was at lower temperatures, and the peak area was larger. This indicated that light olefin products were more easily desorbed and generated on the Co1Fe2 catalyst without further secondary hydrogenation and coupling reactions. Furthermore, the H2/CO2 ratio of 2.5 in the feed gas was slightly lower than the commonly reported ratio of 3. This lower H2 partial pressure contributed to the suppression of the secondary hydrogenation of the primary olefin products, thereby enhancing the light olefin selectivity and improving the STY.
 | ||
| Fig. 7 (a) H2-TPD and (b) CO2-TPD profiles of the prepared catalysts. | ||
Reaction pathway
In situ DRIFTS experiments were conducted on Co0Fe and Co1Fe2 catalysts with the objective of elucidating the reaction intermediates and reaction mechanism (Fig. 8). Following the switching of the reaction gas at 320 °C for 10 min, signals attributable to carbonate (1440 cm−1) and bicarbonate (1349 and 1647 cm−1) species were observed on Co1Fe2 (Fig. 8b). This was attributed to the adsorption and interaction of CO2 and H2 on the catalyst surface. The bands at 1539 and 1774 cm−1 were attributed to the formate species, which were considered as the important intermediates for the formation of CO. In contrast, these intermediates were not detected until 30 min on Co0Fe (Fig. 8a). This finding was verified by the results of CO2-TPD (Fig. 7b). With the increase in cobalt doping, a shift in the CO2 desorption peaks towards higher temperatures was observed, accompanied by an enhancement in CO2 uptake. This indicated a heightened propensity for CO2 adsorption, consistent with the augmented initial reaction activity observed in the Co1Fe2 catalyst. Moreover, the signal associated with chemically adsorbed CO* on the Co1Fe2 catalyst became stronger and remained relatively stable (Fig. 8d), thereby confirming the hypothesis that cobalt doping increased the reaction activity and promoted the cascade reaction. The saturated C–H bond signal at 3014 cm−1 on the Co1Fe2 catalyst was discovered earlier and with higher intensity. This finding was consistent with the result that after adding cobalt, the selectivity of low-carbon alkanes increased significantly from 14.6% for Co0Fe to 24.6% for Co1Fe2. Furthermore, it had been demonstrated that the addition of cobalt served to regulate the production of low-carbon products. | ||
| Fig. 8 The in situ DRIFT spectra of the spent (a and c) Co0Fe and (b and d) Co1Fe2 catalysts. | ||
Conclusions
This work systematically studied the role of cobalt in the structural evolution and surface property regulation of Co–Fe bimetallic catalysts for CO2 hydrogenation to light olefins, and disclosed their structure–activity relationship. The results of in situ XRD demonstrated that the iron species in the Co–Fe catalyst was more readily reduced to alloys and further carburized into mixed metal carbides during the reduction and reaction process. The cascade reaction of RWGS and FTS was reinforced by the rapid adsorption and conversion of CO intermediates, thereby facilitating the conversion of CO2. Furthermore, the enhanced H coverage after Co doping helped regulate the C/H ratio on the catalyst surface, promoting the production and desorption of primary light olefins products. Consequently, the Co1Fe2 catalyst, with a Co/Fe molar ratio of 1/2, displayed remarkable performance in the CO2 hydrogenation to light olefins reaction, exhibiting a CO2 conversion of up to 51.9% at 320 °C and a STY of C2–4 of 315.1 g kgcat−1 h−1. This work provides a reference for understanding the effects of cobalt on product regulation in CO2 hydrogenation over Co–Fe catalysts.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Z. Liu and W. Song: writing–original draft, data curation, methodology. P. Zhang: supervision, validation. J. Liang: writing–review & editing, validation. C. Wang, C. Liu, H. Song, and B. Chen: supervision, validation. K. Wang: writing–review & editing, validation. G. Liu, X. Guo, Y. He, X. Gao, J. Zhang, and G. Yang: conceptualization, validation, supervision. N. Tsubaki: funding acquisition, writing–review & editing, validation, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Grant-in-Aid from Japan Society for the Promotion of Science (JSPS) (22H01864, 23H05404). Zhihao Liu acknowledges the financial support from China Scholarship Council (202308050122).Notes and references
- Y. Wang, J. Sun and N. Tsubaki, Acc. Chem. Res., 2023, 56, 2341–2353 CrossRef CAS PubMed.
- J. Rogelj, M. den Elzen, N. Höhne, T. Fransen, H. Fekete, H. Winkler, R. Schaeffer, F. Sha, K. Riahi and M. Meinshausen, Nature, 2016, 534, 631–639 CrossRef CAS.
- Y. Shi, W. Gao, G. Wang, J. Fan, C. Wang, F. Wang, Y. He, X. Guo, S. Yasuda, G. Yang and N. Tsubaki, Mater. Today Chem., 2023, 32, 101654 CrossRef CAS.
- Z. Liu, X. Gao, K. Wang, J. Liang, Y. Jiang, Q. Ma, T.-S. Zhao and J. Zhang, Chem. Eng. Sci., 2023, 274, 118692 CrossRef CAS.
- Y. Xu, P. Zhai, Y. Deng, J. Xie, X. Liu, S. Wang and D. Ma, Angew. Chem., Int. Ed., 2020, 59, 21736–21744 CrossRef CAS PubMed.
- S. Wang, L. Zhang, W. Zhang, P. Wang, Z. Qin, W. Yan, M. Dong, J. Li, J. Wang, L. He, U. Olsbye and W. Fan, Chem, 2020, 6, 3344–3363 CAS.
- C. Wang, Z. Jin, L. Guo, O. Yamamoto, C. Kaida, Y. He, Q. Ma, K. Wang and N. Tsubaki, Angew. Chem., Int. Ed., 2024, 136, e202408275 CrossRef.
- L. Wang, Y. Han, J. Wei, Q. Ge, S. Lu, Y. Mao and J. Sun, Appl. Catal., B, 2023, 328, 122506 CrossRef CAS.
- Z. Luo, F. Han, P. Zhang, Y. Zhao, S. Huang, Q. Guan and W. Li, Chem. Eng. J., 2024, 490, 151674 CrossRef CAS.
- J. Liang, J. Liu, L. Guo, W. Wang, C. Wang, W. Gao, X. Guo, Y. He, G. Yang, S. Yasuda, B. Liang and N. Tsubaki, Nat. Commun., 2024, 15, 512 CrossRef CAS.
- T. Liu, K. Wang, W. Zhang, W. Song, F. Bo, C. Li, Q. Ma, X. Gao, T.-S. Zhao and J. Zhang, J. Environ. Chem. Eng., 2024, 12, 113885 CrossRef.
- R. Wang, Y. Chen, X. Shang, B. Liang, X. Zhang, H. Zhuo, H. Duan, X. Li, X. Yang, X. Su, Y. Huang and T. Zhang, ACS Catal., 2024, 14, 11121–11130 CrossRef CAS.
- Y. Gu, W. Gao, W. Wang, Y. He, X. Guo, G. Yang, S. Yasuda, Z. Jin and N. Tsubaki, Mater. Today Chem., 2023, 33, 101707 CrossRef CAS.
- J. Zhu, M. Mu, Y. Liu, M. Zhang, G. Zhang, Z. Cheng, B. H. Yin, A. C. K. Yip, C. Song and X. Guo, Chem. Eng. Sci., 2023, 282, 119228 CrossRef CAS.
- B. O. Yusuf, I. Hussain, M. Umar, A. M. Alhassan, C. U. Aniz, K. R. Alhooshani, S. A. Ali, B. Ali and S. A. Ganiyu, J. Energy Inst., 2024, 117, 101849 CrossRef CAS.
- Y. Liu, Q. Cheng, S. Lyu, X. Li, S. Song, T. Ding, Y. Tian and X. Li, Chem. Eng. J., 2024, 494, 152936 CrossRef CAS.
- H. Yang, Y. Dang, X. Cui, X. Bu, J. Li, S. Li, Y. Sun and P. Gao, Appl. Catal., B, 2023, 321, 122050 CrossRef CAS.
- Z. Liu, X. Gao, B. Liu, W. Song, Q. Ma, T.-S. Zhao, X. Wang, J. W. Bae, X. Zhang and J. Zhang, Appl. Catal., B, 2022, 310, 121303 CrossRef CAS.
- L. Zhang, Y. Dang, X. Zhou, P. Gao, A. P. van Bavel, H. Wang, S. Li, L. Shi, Y. Yang, E. I. Vovk, Y. Gao and Y. Sun, Innovation, 2021, 2, 100170 CAS.
- N. Liu, J. Wei, J. Xu, Y. Yu, J. Yu, Y. Han, K. Wang, J. I. Orege, Q. Ge and J. Sun, Appl. Catal., B, 2023, 328, 122476 CrossRef CAS.
- L. Guo, X. Gao, W. Gao, H. Wu, X. Wang, S. Sun, Y. Wei, Y. Kugue, X. Guo, J. Sun and N. Tsubaki, Chem. Sci., 2023, 14, 171–178 RSC.
- F. Yuan, G. Zhang, M. Wang, J. Zhu, M. Zhang, F. Ding, Z. Cheng, C. Song and X. Guo, Ind. Eng. Chem. Res., 2023, 62, 8210–8221 CrossRef CAS.
- K. Y. Kim, H. Lee, W. Y. Noh, J. Shin, S. J. Han, S. K. Kim, K. An and J. S. Lee, ACS Catal., 2020, 10, 8660–8671 CrossRef CAS.
- A. S. Sandupatla, A. Banerjee and G. Deo, Appl. Surf. Sci., 2019, 485, 441–449 CrossRef CAS.
- G. Chen, R. Gao, Y. Zhao, Z. Li, G. I. N. Waterhouse, R. Shi, J. Zhao, M. Zhang, L. Shang, G. Sheng, X. Zhang, X. Wen, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2018, 30, 1704663 CrossRef.
- Q. Xu, X. Xu, G. Fan, L. Yang and F. Li, J. Catal., 2021, 400, 355–366 CrossRef CAS.
- W. Tu, C. Sun, Z. Zhang, W. Liu, H. S. Malhi, W. Ma, M. Zhu and Y.-F. Han, Appl. Catal., B, 2021, 298, 120567 CrossRef CAS.
- Z. Liu, X. Gao, K. Wang, T. Atchimarungsri, J. Tian, P. Reubroycharoen, Q. Ma, T.-S. Zhao, N. Tsubaki and J. Zhang, Fuel Process. Technol., 2023, 241, 107628 CrossRef CAS.
- R. Satthawong, N. Koizumi, C. Song and P. Prasassarakich, J. CO2 Util., 2013, 3, 102–106 CrossRef.
- M. K. Gnanamani, G. Jacobs, H. H. Hamdeh, W. D. Shafer, F. Liu, S. D. Hopps, G. A. Thomas and B. H. Davis, ACS Catal., 2016, 6, 913–927 CrossRef CAS.
- F. Jiang, B. Liu, S. Geng, Y. Xu and X. Liu, Catal. Sci. Technol., 2018, 8, 4097–4107 RSC.
- W. K. Jozwiak, E. Kaczmarek, T. P. Maniecki, W. Ignaczak and W. Maniukiewicz, Appl. Catal., A, 2007, 326, 17–27 CrossRef CAS.
- H.-Y. Lin and Y.-W. Chen, Mater. Chem. Phys., 2004, 85, 171–175 CrossRef CAS.
- M. Xu, X. Liu, G. Song, Y. Cai, B. Shi, Y. Liu, X. Ding, Z. Yang, P. Tian, C. Cao and J. Xu, J. Catal., 2022, 413, 331–341 CrossRef CAS.
- J. Zhu, P. Wang, X. Zhang, G. Zhang, R. Li, W. Li, T. P. Senftle, W. Liu, J. Wang, Y. Wang, A. Zhang, Q. Fu, C. Song and X. Guo, Sci. Adv., 2022, 8, eabm3629 CrossRef CAS PubMed.
- S. Najari, S. Saeidi, A. Sápi, Á. Szamosvölgyi, Á. Papp, A. Efremova, H. Bali and Z. Kónya, Chem. Eng. J., 2024, 485, 149787 CrossRef CAS.
- J. Wei, R. Yao, Q. Ge, D. Xu, C. Fang, J. Zhang, H. Xu and J. Sun, Appl. Catal., B, 2021, 283, 119648 CrossRef CAS.
- Z. Zeng, Z. Li, L. Kang, X. Han, Z. Qi, S. Guo, J. Wang, A. Rykov, J. Lv, Y. Wang and X. Ma, ACS Catal., 2022, 12, 6016–6028 CrossRef CAS.
- X. Li, K. Zhu, J. Pang, M. Tian, J. Liu, A. I. Rykov, M. Zheng, X. Wang, X. Zhu, Y. Huang, B. Liu, J. Wang, W. Yang and T. Zhang, Appl. Catal., B, 2018, 224, 518–532 CrossRef CAS.
- Y. Zeng, X. Li, J. Wang, M. T. Sougrati, Y. Huang, T. Zhang and B. Liu, Chem Catal., 2021, 1, 1215–1233 CAS.
- B. Liang, T. Sun, J. Ma, H. Duan, L. Li, X. Yang, Y. Zhang, X. Su, Y. Huang and T. Zhang, Catal. Sci. Technol., 2019, 9, 456–464 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc04407c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |