
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiselenide-enabled photocatalytic hydroazolation of gem-difluoroalkenes
Mohammed K.
Abd El-Gaber†
 abc,
Ryan M.
Herrick†
a,
Pranaya
Sudhakar
d,
Ashutosh
Rana
e,
Brent A.
Roach
e,
Jeffrey E.
Dick
ef and
Ryan A.
Altman
*ae
abc,
Ryan M.
Herrick†
a,
Pranaya
Sudhakar
d,
Ashutosh
Rana
e,
Brent A.
Roach
e,
Jeffrey E.
Dick
ef and
Ryan A.
Altman
*ae
aBorch Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University, West Lafayette, IN 47907, USA. E-mail: raaltman@purdue.edu
bMedicinal Chemistry Department, Faculty of Pharmacy, Assiut University, Assiut 71526, Egypt
cDepartment of Pharmaceutical Sciences, University of Tennessee Health Science Center, Memphis, TN 38163, USA
dDepartment of Biology, Purdue University, West Lafayette, IN 47907, USA
eJames Tarpo Jr and Margaret Tarpo Department of Chemistry, Purdue University, West Lafayette, IN 47907, USA
fElmore Family School of Electrical and Computer Engineering, Purdue University, West Lafayette, IN 47907, USA
First published on 21st October 2025
Abstract
Difluoromethylene-containing molecules and azoles, independently, have wide applications in materials science, pharmaceuticals, agrochemicals and as biological diagnostic probes. However, compounds bearing the N-α,α-difluoroalkyl azole [(azole)N–CF2R] motif remain scarce in academic and patent literature, presumably due to a lack of synthetic methods. Such compounds could be convergently accessed in a single step via the hydroazolation of gem-difluoroalkenes. However, most existing functionalization reactions of gem-difluoroalkenes proceed through a β-fluoride elimination pathway that generates monofluorinated derivatives. Herein, we report a photocatalytic hydroazolation of gem-difluoroalkenes to generate (azole)N–CF2R that employs an uncommon diselenide co-catalyst to avoid the defluorinative process, ultimately enabling facile access to underexplored medicinally and agriculturally-relevant chemical space.
Introduction
N-Heterocycles and fluorinated motifs are two frequently encountered substructures with applications in materials science, medicinal chemistry, agricultural chemistry, and as diagnostic probes.1–3 Strategies that enable access to novel and/or underrepresented combinations of these substructures have the potential to impact a variety of applied fields. One such combination, N-α,α-difluoroalkyl azoles [(azole)N–CF2R, 1, Scheme 1A], have displayed medicinal potential in neurodegenerative disease,4,5 oncology,6 and inflammation.7,8 Notwithstanding these examples, (azole)N–CF2R remain underutilized in medicinal and agricultural chemistry (<30 compounds with experimentally determined pharmacological activity for R = alkyl, SciFinder, Octcober 2025). This deficiency represents a missed opportunity to explore biologically relevant chemical space, as the perturbations of molecular physicochemical properties imparted by fluorine on (azole)N–CF2R containing compounds presumably mimic those observed for N-trifluoromethyl azoles [(azole)N–CF3], a more common N-fluoroalkyl azole substructure in medicinal chemistry.9–14 Specifically, (azole)N–CF3 possess decreased pKa, increased lipophilicity, and greater stability towards metabolic N-dealkylation relative to their non-fluorinated counterparts.11,15 Despite these attributes, the trifluoromethyl group precludes further growth of a ligand off the azole's nitrogen atom in (azole)N–CF3. In contrast, the (azole)N–CF2R substructure could benefit from fluorine-induced perturbations while also allowing for elaboration of the N-alkyl group. | ||
| Scheme 1 Accessing (azole)N–CF2R: azolation of gem-difluoroalkenes. | ||
The underutilization of the (azole)N–CF2R group can be partially attributed to the lack of viable synthetic methods to access this moiety. Existing methods for azole N-fluoroalkylation typically form N-fluoromethyl azoles16,17 or higher order N-perfluoroalkyl azoles (e.g., C2F4, C3F6)18,19 – few strategies exist for generating simple, hydrocarbon (azole)N–CF2R.20–22 To access this substructure, an attractive retrosynthetic disconnection across the N–CF2 bond might reveal gem-difluoroalkene (2)23–28 and azole (3) synthons, two readily accessible substrates (Scheme 1A). In the forward reaction, regioselective C–N bond formation would occur through attack of the azole at the electrophilic difluorinated carbon of 2.29,30 Indeed, azole nucleophiles do react with 3,3-difluoropropen-1-yl ammonium salt 4 to generate N-gem-difluoroallyl azoles (5) by an SN2′ process in the presence of stoichiometric NaH (Scheme 1B).31 However, the singular gem-difluoroalkene coupling partner prevents this reaction from serving as a convergent approach to generate a diverse array of (azole)N–CF2R.
In the absence of a quaternary ammonium leaving group, base-mediated nucleophilic addition of azoles to gem-difluoroalkenes proceeds through an unstable anionic intermediate 6 that readily loses a β-fluoride anion to form N-(α-fluorovinyl) azoles (7, Scheme 1B),29,32,33 not (azole)N–CF2R. Alternatively, single electron-mediated azolation of gem-difluoroalkenes represents a strategy that could circumvent the limitations of β-fluoride elimination and the necessity for specialized gem-difluoroalkene coupling partners (Scheme 1C). Specifically, the addition of azoles to diverse gem-difluoroalkenes under oxidative conditions generates a radical intermediate 8 that could be quenched by an appropriate radical source, thus avoiding anionic intermediate 6 and fluoride elimination.29 Such reactions of gem-difluoroalkenes have been accomplished using photocatalysts and electrolytic cells as single-electron oxidants to promote difunctionalization reactions that add azoles with O2,34,35 fluoride,36,37 alcohols, and an additional azole molecule (Scheme 1C).38 However, a simple hydrofunctionalization of gem-difluoroalkenes with azoles to form (azole)N–CF2R remains elusive.
To address this synthetic deficiency, we herein disclose a diselenide-mediated photocatalytic hydroazolation of gem-difluoroalkenes with both monocyclic and benzannulated azoles that delivers previously unreported (azole)N–CF2R. In this reaction, the diselenide co-catalyst promotes the desired hydrofunctionalization process and avoids undesired reactivity with gem-difluoroalkenes, similarly to a hydroalkoxylation reaction of gem-difluoroalkenes previously reported by our group.44 However, amongst a series of tested dichalcogenide co-catalysts, (PhSe)2 uniquely reversed selectivity for defluorinative azolation. Additionally, experimental data supports a mechanism initiated by direct photocatalyst oxidation of gem-difluoroalkenes, which contrasts the previous report.44
By merging two commonly exploited substructures found in biologically active compounds (azoles and fluoroalkyl groups), this approach opens numerous possibilities for expanding the synthetically accessible chemical space that could impact the development of therapeutics, biological probes, agrochemical agents, and materials.
Results and discussion
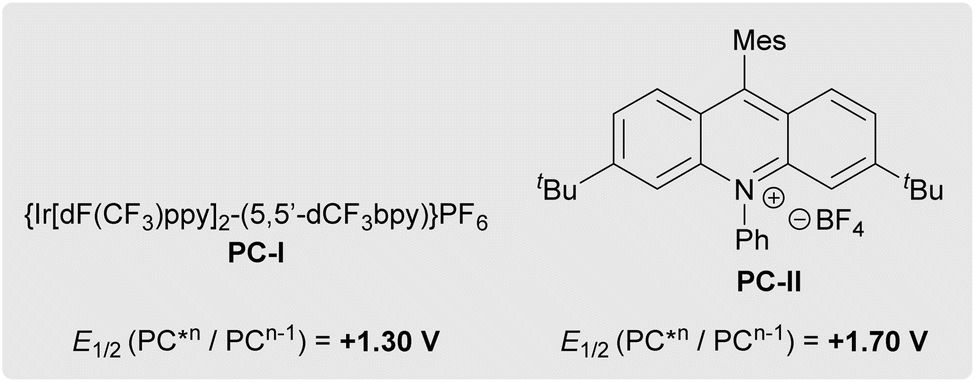
In the initial reaction design, we aimed to generate azole radicals that would react with gem-difluoroalkenes to afford carbon-centered radical intermediate 8, which, after quenching by a hydrogen atom transfer (HAT) step, would deliver the desired (azole)N–CF2R (11). This strategy was inspired by previous reports of gem-difluoroalkene hydrothiolation that are initiated by a photocatalyst-mediated oxidation/PCET of thiols that generates thiyl radicals.45–47 To test this hypothesis, photocatalysts spanning a range of excited state reduction potentials were reacted with benzimidazole and electron-rich gem-difluorostyrene 10a as model substrates on a 50 μmol scale (Table 1, entries 1–6). Conversion of the gem-difluorostyrene occurred only when photocatalysts with excited state reduction potentials greater than +0.45 V were employed; however, only trace product formed (entries 4–6). Instead, these reactions formed monofluorovinyl azole side-product 12, suggesting that the system lacked an adequate hydrogen atom source to quench presumed radical intermediate 8 (see Scheme S5 for a mechanistic proposal for the formation of side-products 12 and 13). Initial attempts to identify catalytic additives that could donate hydrogen atoms and generate desired product 11aa (e.g., arylamines, N-hydroxyphthalimide (NHP), silanes, and thiols) failed to increase selectivity for the desired (azole)N–CF2R 11aa (entries 7–10). However, dichalcogenide additives improved the reaction (entries 11–14), with the best yield and selectivity observed with (PhSe)2 (entry 14). Notably, (PhSe)2 was not consumed by a hydroselenolation side-reaction with gem-difluorostyrene 10a, which distinguishes the diselenide from a more common disulfide additive (entry 11).48,49 Reduction of (PhSe)2 loading from 20% to 5% further improved reaction performance (entry 15). Final optimization on a 0.50 mmol scale revealed optimal loadings of reagents [1.2 equiv. azole, 5% (PhSe)2, and 5% PC-I], solvent (DCE, Table S2), and concentration (0.25 M). Additionally, while the presence of H2O (up to 1 equiv.) had no deleterious effect on the reaction, O2 was detrimental. In parallel efforts, an alternate set of conditions derived from a previously reported gem-difluoroalkene hydroalkoxylation reaction44 were developed for the more strongly oxidizing PC-II [1.2 equiv. azole, 5% (PhSe)2, 3% PC-II, 0.25 M PhMe, Table S4] and would eventually be essential for certain substrates. Control experiments verified that photocatalyst, visible light, and (PhSe)2 were all necessary for successful hydroazolation of gem-difluorostyrenes (Table S1).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Photocatalyst | E 1/2 [PC*n/PCn−1] (V vs. Fc/Fc+)b | Additive | % Conv. 10a | % Yield 11aa | %Yield 12 | % Yield 13 |
| a Reaction conditions: gem-difluorostyrene 10a (50 μmol), benzimidazole (1.2 equiv.), additive, and photocatalyst (5 mol%) in DCE (0.25 mL) irradiated with a 40 W 427 nm LED under an atmosphere of N2. Conversion and yields were determined by 19F NMR using (trifluoromethyl)benzene as an internal standard. b Literature-reported reduction potentials were corrected to the Fc/Fc+ redox couple.43 c TRIP-SH = 2,4,6-triisopropylbenzenethiol, NHP = N-hydroxyphthalimide. d N.D. = not detected. | |||||||
| 1 | [Ir(dtbbpy)(ppy)2]PF6 | +0.28 (ref. 39) | None | <5 | 0 | 0 | N.D.d |
| 2 | Rose Bengal lactone | +0.28 (ref. 40) | None | <5 | 0 | 0 | N.D.d |
| 3 | Eosin Y (dibasic) | +0.45 (ref. 40) | None | <5 | 0 | 0 | N.D.d |
| 4 | 4CzIPN | +1.00 (ref. 40) | None | 67 | <1 | 66 | N.D.d |
| 5 | PC-I | +1.30 (ref. 41) | None | 69 | 5 | 64 | N.D.d |
| 6 | PC-II | +1.70 (ref. 42) | None | 66 | <1 | 64 | N.D.d |
| 7 | PC-I | +1.30 (ref. 41) | m-Anisidine (10%) | 61 | 3 | 56 | 0 |
| 8 | PC-I | +1.30 (ref. 41) | TRIP-SH (30%)c | 53 | 3 | 44 | 0 |
| 9 | PC-I | +1.30 (ref. 41) | NHP (50%)c | 72 | <1 | 66 | 0 |
| 10 | PC-I | +1.30 (ref. 41) | (TMS)3SiH (50%) | 70 | 8 | 56 | 0 |
| 11 | PC-I | +1.30 (ref. 41) | (PhS)2 (20%) | >99 | 26 | 42 | 17 (X = SPh) |
| 12 | PC-I | +1.30 (ref. 41) | (BnS)2 (20%) | 81 | 4 | 68 | 0 |
| 13 | PC-I | +1.30 (ref. 41) | (BnSe)2 (20%) | 82 | 21 | 44 | <1 (X = F) |
| 14 | PC-I | +1.30 (ref. 41) | (PhSe)2 (20%) | >99 | 81 | 4 | 2 (X = F) |
| 15 | PC-I | +1.30 (ref. 41) | (PhSe)2 (5%) | >99 | 93 | 0 | 0 |

A range of azoles successfully reacted with gem-difluorostyrene 10a using the optimized PC-I conditions (Table 2). Specifically, unsubstituted benzimidazole reacted to afford product 11aa in 87% yield. 5-Monosubstituted benzimidazoles bearing electron-withdrawing –Cl, –Br, –CO2Me, and –NO2 groups reacted with moderate to excellent yield (11ab–ae, 40–91%). A mixture of N-regioisomers was generated, but the 6-substituted products predominated, for which the major regioisomer was assigned by a combination of X-ray crystallography (see Data availability on p. 8) and 1H{19F} Nuclear Overhauser Effect (NOE) NMR. Other benzannulated azoles, such as indazoles (11af and 11ag, 92% and 61%) and benzotriazole (11ah, 96%) reacted with good yield and near exclusive regioselectivities. Notably, the high N2-regioselectivity for indazoles 11af and 11ag supports a process involving nucleophilic C–N bond formation from a neutral indazole molecule.50 Additionally, the FDA-approved drugs theophylline and triclabendazole were reacted with these conditions (11ai and 11aj, 43% and 86%). 7-Azaindoles also coupled successfully with slight alterations to the standard conditions, albeit in low yields and with long reaction times (11ak and 11al, 13% and 20%, 46 h and 44 h). Interestingly, these reactions exhibit exclusive N-regioselectivity for functionalization at the pyridyl nitrogen rather than the indole nitrogen, which is consistent with the observed inability of indoles to react in this system. However, 6-, 5-, and 4-azaindoles do not achieve net hydrofunctionalization and instead form monofluorovinyl azaindole products (ArCH = CFN(azaindole), 7).
| a Reaction conditions unless otherwise noted: gem-difluorostyrene 10a (0.50 mmol), azole (1.2 equiv.), 1,2-diphenyldiselane (5 mol%), and PC-I: {Ir[dF(CF3)ppy]2-(5,5′-dCF3bpy)}PF6 (5 mol%) in DCE (2.0 mL) irradiated with a 40 W 427 nm LED under an atmosphere of N2. Isolated yields represent an average of two independent reactions. Ratios in parentheses represent the major and minor N-regioisomeric ratio of the crude reaction mixture as determined by 19F NMR. b Structure of the major product was assigned by X-ray crystallography (see Data availability on p. 8). c Structure of the major product was assigned by 1H{19F} NOE. d Reaction contained N2-sparged H2O (0.50 equiv.). e With 7-azaindole (1.5 equiv.), 1,2-diphenyldiselane (15 mol%), and PC-I: {Ir[dF(CF3)ppy]2-(5,5′-dCF3bpy)}PF6 (1 mol%) for 46 h. f With 1,2-diphenyldiselane (25 mol%) and PC-II: 9-mesityl-3,6-di-tert-butyl-10-phenylacridinium tetrafluoroborate (3 mol%) in PhMe (2.0 mL) for 44 h. |
|---|
|
Reactions of monocyclic pyrazoles bearing electron-withdrawing groups (4-Br and 4-CO2Et, 11am and 11ao, 88% and 89% [H2O required for 11ao, see Notes section]), electron-donating groups (3,5-Me and 4-BPin, 11ap and 11ar, 91% and 88%), and sterically restrictive 3,5-disubstitution (11ap and 11aq, 91% and 87%) all furnished products in excellent yields. Importantly, the reaction's ability to tolerate bromide (11am, 88%), iodide (11an, 74% [H2O required, see Notes section]), and boronate ester (11ar, 88%) substituents enables further divergent functionalization by cross-coupling reactions. 4,5-Dichloro-1H-imidazole was successfully coupled (11as, 91%), and 1,2,3-1H-triazole reacted with excellent yield and exclusive N1–regioselectivity (11at, 90%). While the reaction demonstrated a broad scope of azole coupling partners, some poor-performing and unreactive substrates were identified (Table S5).
Using pyrazole as a model substrate, a wide range of gem-difluoroalkenes coupled in moderate to excellent yields under either PC-I or PC-II catalysis (11a–r, 24–88%, Table 3). To select the appropriate photocatalyst for the reaction of each gem-difluoroalkene, conditions derived for PC-I and PC-II were preemptively screened on a 50 μmol scale (Table S6) and successful reactions were repeated on a 0.50 mmol scale. The reaction tolerated a range of heterocyclic gem-difluorostyrenes (11b–f, 41–86%), including pyrazoles, benzothiophenes, benzofurans, indoles, and pyrroles. Additionally, though a rarely reported glucose-derived difluorinated enol substrate (10g) reacted sluggishly with pyrazole (5 days to achieve ∼50% yield on a 50 μmol scale), indazole successfully coupled with saccharide 10g to generate a single diastereomer at C2 (11g, 44%, determined by X-ray crystallography, see Data availability on p. 8). Surprisingly, the N1-substituted indazole regioisomer predominated in this reaction (13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 N1:N2, determined by 19F NMR and X-ray crystallography, see Data availability on p.8), which contrasts the coupling reactions of indazoles with gem-difluorostyrene 10a (11af and 11ag, Table 2) and gem-difluorostyrenes 10c, 10e, 10f, and 10j (Table S5) that display almost exclusive substitution at N2. Electronically neutral and rich gem-difluorostyrenes afforded moderate to high yields of N-α,α-difluoroalkyl pyrazoles (11a, 11j–p, 57–88%). However, gem-difluorostyrenes with electron-withdrawing substituents reacted in lower yields and with slower conversion (11q and 11r, 24% and 25%, 42 and 48 h). Notably, these substrates required the more strongly oxidizing acridinium-based photocatalyst (PC-II conditions) to generate detectable yields of the desired (azole)N–CF2R products, a correlation that was not apparent for electronically neutral and rich substrates. Interestingly, neither gem-difluorostyrenes bearing strongly electron-withdrawing substituents (e.g., 4-CN) nor aliphatic gem-difluoroalkenes reacted with azoles employing either PC-I or PC-II conditions (Table S6), which, supplemented by luminescence quenching studies and a comparison of substrate and photocatalyst redox potentials (see mechanistic discussion below), suggests that only gem-difluoroalkene substrates that can be oxidized by PC-I and PC-II will successfully react. Regardless, this limitation in substrate scope contrasts a previously reported hydroalkoxylation method that utilizes a similar co-catalytic system.44 Sterically hindered tetra-vinyl- and mono-ortho-substituted gem-difluorostyrene substrates reacted successfully (11h and 11o, 79 and 61%); however, a 2,6-dimethyl gem-difluorostyrene could not be coupled (Table S6). Notably, the reaction was successful on a gram scale, albeit with modification to the photochemical reactor (see SI, S30) and worsened conversion efficiency (11n, 70% on 4.6 mmol scale vs. 87% on 0.5 mmol scale).
:1 N1:N2, determined by 19F NMR and X-ray crystallography, see Data availability on p.8), which contrasts the coupling reactions of indazoles with gem-difluorostyrene 10a (11af and 11ag, Table 2) and gem-difluorostyrenes 10c, 10e, 10f, and 10j (Table S5) that display almost exclusive substitution at N2. Electronically neutral and rich gem-difluorostyrenes afforded moderate to high yields of N-α,α-difluoroalkyl pyrazoles (11a, 11j–p, 57–88%). However, gem-difluorostyrenes with electron-withdrawing substituents reacted in lower yields and with slower conversion (11q and 11r, 24% and 25%, 42 and 48 h). Notably, these substrates required the more strongly oxidizing acridinium-based photocatalyst (PC-II conditions) to generate detectable yields of the desired (azole)N–CF2R products, a correlation that was not apparent for electronically neutral and rich substrates. Interestingly, neither gem-difluorostyrenes bearing strongly electron-withdrawing substituents (e.g., 4-CN) nor aliphatic gem-difluoroalkenes reacted with azoles employing either PC-I or PC-II conditions (Table S6), which, supplemented by luminescence quenching studies and a comparison of substrate and photocatalyst redox potentials (see mechanistic discussion below), suggests that only gem-difluoroalkene substrates that can be oxidized by PC-I and PC-II will successfully react. Regardless, this limitation in substrate scope contrasts a previously reported hydroalkoxylation method that utilizes a similar co-catalytic system.44 Sterically hindered tetra-vinyl- and mono-ortho-substituted gem-difluorostyrene substrates reacted successfully (11h and 11o, 79 and 61%); however, a 2,6-dimethyl gem-difluorostyrene could not be coupled (Table S6). Notably, the reaction was successful on a gram scale, albeit with modification to the photochemical reactor (see SI, S30) and worsened conversion efficiency (11n, 70% on 4.6 mmol scale vs. 87% on 0.5 mmol scale).
| a Reaction conditions: gem-difluorostyrene 10a–r (0.50 mmol), azole (1.2 equiv.), 1,2-diphenyldiselane (5 mol%), and either PC-I: {Ir[dF(CF3)ppy]2-(5,5′-dCF3bpy)}PF6 (5 mol%) in DCE (2.0 mL) or PC-II: 9-mesityl-3,6-di-tert-butyl-10-phenylacridinium tetrafluoroborate (3 mol%) in PhMe (2.0 mL) irradiated with a 40 W 427 nm LED under an atmosphere of N2. Isolated yields represent an average of two independent reactions. b 15 mol% 1,2-diphenyldiselane was used. c Ratio represents the major and minor N-regioisomeric ratio of the crude reaction mixture as determined by 19F NMR. Structure of the major product was assigned by X-ray crystallography (see Data availability on p. 8). d Data in parentheses: gem-difluorostyrene 10n (4.6 mmol, 1.0 g), pyrazole (1.2 equiv.), 1,2-diphenyldiselane (5 mol%), and PC-II (3 mol%) in PhMe (18.5 mL) irradiated with a 40 W 427 nm LED under an atmosphere of argon. Isolated yield represents an average of two independent reactions. |
|---|

|
A combination of physicochemical data and literature precedent for photocatalyzed functionalization reactions of both nonfluorinated and gem-difluorinated alkenes supports a mechanism involving oxidation of gem-difluorostyrene 10 by excited state PC* to form radical cation 10˙+,35–37,51,52 nucleophilic attack by azole 14 to afford acidic radical cation 15, deprotonation to generate carbon-centered radical 16 and selenol 19,44,53,54 and subsequent hydrogen atom transfer to form (azole)N–CF2R 11 (Scheme 2A). Several routine experiments suggest a radical process. In support of the existence of radical 16, both PC-I and PC-II-catalyzed reactions of tetrasubstituted gem-difluorostyrene 10s produced cyclopropane ring opening product 11s (Scheme 2B). Notably, an azole-functionalized product bearing an intact cyclopropane ring was not observed. Additionally, light on/off experiments support a mechanism involving quenching of product-forming radical intermediates upon completion of a photocatalytic cycle, as reaction progression was not detected during dark periods (Scheme 2C).
 | ||
| Scheme 2 Mechanistic studies. aReaction conditions: gem-difluorostyrene 10s (0.50 mmol), pyrazole (1.2 equiv.), 1,2-diphenyldiselane (5 mol%), and either PC-I: {Ir[dF(CF3)ppy]2-(5,5′-dCF3bpy)}PF6 (5 mol%) in DCE (2.0 mL) or PC-II: 9-mesityl-3,6-di-tert-butyl-10-phenylacridinium tetrafluoroborate (3 mol%) in PhMe (2.0 mL) irradiated with a 40 W 427 nm LED under an atmosphere of N2 for 14 h. Isolated yield represents a single reaction. A single diastereomer was isolated, but the ratio in parentheses represents the crude reaction E/Z ratio as determined by 19F NMR. bReaction conditions: gem-difluorostyrene 10n (0.25 mmol), pyrazole (1.2 equiv.), 1,2-diphenyldiselane (5 mol%), (trifluoromethyl)benzene (9.6 mol%), and either PC-I: {Ir[dF(CF3)ppy]2-(5,5′-dCF3bpy)}PF6 (5 mol%) in DCE (1.0 mL) or PC-II: 9-Mesityl-3,6-di-tert-butyl-10-phenylacridinium tetrafluoroborate (3 mol%) in PhMe (1.0 mL) irradiated with a 40 W 427 nm LED under an atmosphere of N2. Conversion and yield were determined by 19F NMR using (trifluoromethyl)benzene as an internal standard. | ||
Alternatively, both the formation of intermediate 16 and a lack of reaction progression in the dark are also consistent with a mechanism involving attack of an azole-based radical into the neutral gem-difluoroalkene (Scheme S2). This alternative hypothesis is supported by (a) photophysical data that suggests oxidation of azoles is possible in this system: pyrazole, indazole, and benzimidazole quench the fluorescence of PC-II (but not PC-I) with comparable rate constants to gem-difluorostyrenes (see SI S33–42), and (b) measured oxidation potentials of these azoles (pyrazole: Ep = +1.6 V; indazole: Ep = +1.2 V; benzimidazole: Ep = +1.1 V) that are similar to the excited state reduction potentials of PC-I [E1/2(PC-I*III/PC-III) = +1.30 V]41 and PC-II [E1/2(PC-II*+/PC-II˙) = +1.70 V].42 However, though several radical traps did capture azoles using PC-II conditions, radical traps did not capture azole radicals using PC-I conditions, and for both catalyst systems, radical traps failed to inhibit the photocatalytic reactions (Table S7). All combined, this data suggests that azole radicals might form under the conditions, but that the reactions do not proceed by addition of azole radicals to gem-difluoroalkenes.
Notably, in this process, PhSe-containing intermediates facilitate a polar/radical crossover event by acting as (1) an oxidant to turn over ground-state photocatalyst (17 → 18), (2) a Brønsted base (18 → 19), and (3) a hydrogen atom source (19 → 17), respectively, as supported by a variety of physicochemical measurements. Specifically, blue light initiates the homolytic fragmentation of (PhSe)2 to form selenyl radical 17,55,56 which serves as an oxidant for reduced-state photocatalysts PC-I [E1/2(PC-IIII/PC-III) = −1.07 V]41 or PC-II [E1/2(PC-II+/PC-II˙) = −0.97 V]42 to regenerate ground-state photocatalysts PC-I/II and form selenolate 18. (PhSe)2 does not directly oxidize the reduced-state photocatalysts given its low measured reduction potential (Ep = −1.8 V). Subsequently, selenolate 18 (pKa = 4.60 in H2O)57 sequesters the proton from radical cation 15 to generate carbon-centered radical 16 and selenol 19. Finally, hydrogen atom abstraction from selenol 19 [BDE = 78 ± 4 kcal mol−1 (H–SePh)]58 by radical 16 [BDE = ∼85–96 kcal mol−1 (H–C)]59 affords (azole)N–CF2R product 11 and regenerates selenyl radical 17. Interestingly, multiple radical traps did not inhibit the reaction (Table S7), which could indicate that the sequence of 15 + 18 → 16 + 19 → 17 + 11 occurs rapidly within the solvent cage.
In contrast to a prior proposal suggesting that the diselenide mediates oxidation of the gem-difluorinated alkene (see further discussion in SI S49–51),44 experimental data supports an initiation step involving direct photocatalyst oxidation of gem-difluorostyrenes 10 (Table 4). More specifically, reactions proceed when the oxidation potential of a gem-difluorostyrene (Ep) is lower than the excited-state reduction potential of a photocatalyst (E1/2) and typically require the gem-difluorostyrene to quench the luminescence of the tested photocatalyst. For example, electron-rich gem-difluorostyrenes 10a (Ep = +1.0 V) and 10k (Ep = +1.0 V) quench the luminescence of both PC-I* [E1/2(PC-I*III/PC-III) = +1.30 V]41 and PC-II* [E1/2(PC-II*+/PC-II˙) = +1.70 V],42 and these reactions all generate products (entries 1–4). Furthermore, gem-difluorostyrene 10r (Ep = +1.5 V) only quenches the luminescence of PC-II* and thus only couples with pyrazole when reacted with PC-II (entries 7 and 8). Finally, gem-difluorostyrene 10t (Ep = +1.8 V) does not quench the luminescence of either PC-I* or PC-II* and does not react with any of the tested azoles (entries 9 and 10).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substratea | Photocatalyst | Luminescence quenchinga (M−1 s−1) | % Yieldb | % Conv. (time)b |
| a Experimental procedures and data for cyclic voltammetry and luminescence quenching studies can be found in the supporting information document (S31–44). Ep = anodic peak potential [vs. E1/2(Fc/Fc+)]; kq = bimolecular quenching rate constant. b Reaction conditions: gem-difluorostyrene (50 μmol), pyrazole (1.2 equiv.), 1,2-diphenyldiselane (5 mol%), and either PC-I: {Ir[dF(CF3)ppy]2-(5,5′-dCF3bpy)}PF6 (5 mol%) in DCE (200 μL) or PC-II: 9-mesityl-3,6-di-tert-butyl-10-phenylacridinium tetrafluoroborate (3 mol%) in PhMe (200 μL) irradiated with a 40 W 427 nm LED under an atmosphere of N2 at 30 °C. Conversion and yields were determined by 19F NMR using (trifluoromethyl)benzene as an internal standard. | |||||
| 1 |

|
PC-I | k q = 7.3 × 108 | 92 | >99 (14 h) |
| 2 | 10a (Ep = +1.0 V) | PC-II | k q = 8.3 × 109 | 31 | 35 (14 h) |
| 3 |

|
PC-I | k q = 1.2 × 109 | 70 | >99 (14.5 h) |
| 4 | 10k (Ep = +1.0 V) | PC-II | k q = 1.6 × 1010 | 94 | >99 (14.5 h) |
| 5 |
|
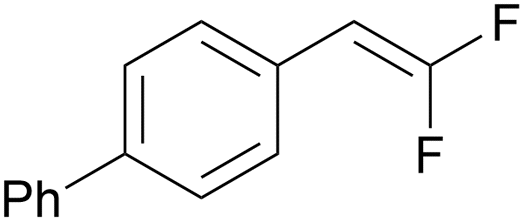
PC-I | no quenching observed | 93 | >99 (15.5 h) |
| 6 | 10n (Ep = +1.3 V) | PC-II | k q = 8.3 × 109 | 83 | >99 (15.5 h) |
| 7 |
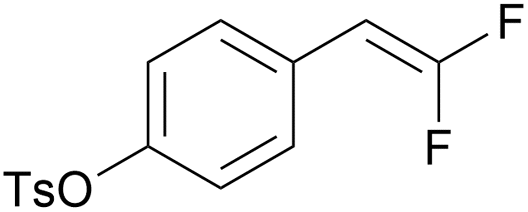
|
PC-I | No quenching observed | 0 | 0 (12.5 h) |
| 8 | 10r (Ep = +1.5 V) | PC-II | k q = 4.9 × 108 | 26 | 46 (12.5 h) |
| 9 |

|
PC-I | No quenching observed | 0 | 6 (14.5 h) |
| 10 | 10t (Ep = +1.8 V) | PC-II | No quenching observed | 0 | 18 (14.5 h) |
However, in contrast to this general correlation between luminescence of photocatalyst quenching and gem-difluorostyrene conversion, gem-difluorostyrenes 10n (Ep = +1.3 V) and 10o only quench the luminescence of PC-II* despite reacting successfully under both PC-I and PC-II catalysis (entries 5 and 6). In these reactions, only (PhSe)2 quenched PC-I* luminescence, though evidence for [(PhSe)2]•+–mediated gem-difluorostyrene oxidation was not found (see SI, S49–51). Therefore, for certain substrates, a plausible alternate mechanistic hypothesis for PC-I might involve an oxidative quenching cycle (Scheme S4) as opposed to a reductive quenching cycle (Scheme 2A). Specifically, PC-I* [E1/2(PC-I*III/PC-IIV) = −0.81 V]60 might first reduce selenyl radical 17, thus generating strong oxidant PC-IIV [Ep(PC-IIII/PC-IIV) = +1.56 V]60 which oxidizes gem-difluorostyrenes 10n and 10o.
Conclusion
In conclusion, the disclosed photocatalyst and diselenide co-catalyzed method couples azoles with gem-difluoroalkenes to deliver a range of N-α,α-difluorinated azoles in a single, convergent step. This strategy convergently generates the (azole)N–CF2R substructure and thus has the potential to increase the utilization of this underexplored fluorinated motif in medicinal and agricultural chemistry. Further, the optimization of two separate conditions for readily oxidizable azoles (PC-I conditions) or electron-deficient gem-difluoroalkenes (PC-II conditions) enables future practitioners to adapt the method to their substrate-dependent redox restrictions. Importantly, the (PhSe)2 co-catalyst reverses selectivity for defluorinative azolation and suppresses undesired side-reactions, perhaps by facilitating a radical-polar crossover event. Ongoing work aims to address limitations in scope.Notes
In the reactions generating examples 11an and 11ao, yields failed to exceed 10% after 24 h of irradiation in the absence of H2O, and the reactions predominantly formed monofluorvinyl azole[ArCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CFN(azole)] and vinyl gem-diazole [ArCHC{N(azole)}2]. The specific yield-enhancing role of H2O in these reactions is presently unclear and failed to extend to other poor-performing reactions (11ab, 11ag, and 11ai).
CFN(azole)] and vinyl gem-diazole [ArCHC{N(azole)}2]. The specific yield-enhancing role of H2O in these reactions is presently unclear and failed to extend to other poor-performing reactions (11ab, 11ag, and 11ai).
Author contributions
All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Data availability
CCDC 2414748 (11ab), 2414405 (11af), 2414406 (11ah), 2492669 (11al), and 2414370 (11g) contain the supplementary crystallographic data for this paper.61a–eThe data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental procedures and characterization data for the synthesized compounds as well as mechanistic experiments. See DOI: https://doi.org/10.1039/d5sc04074d.
Acknowledgements
Research reported in this publication was supported by the National Institutes of Health (R35 GM124661, R35 GM138133, CA023168), and the donors of the Steve and Lee Ann Taglienti Endowment and Ross-Lynn Research Scholar Fund. X-Ray crystallographic data was supported by the National Science Foundation through the Major Research Instrumentation Program (CHE 1625543). We thank Dr Huaping Mo for creating a protocol for 1H{19F} NOE experiments, Md. Arif Faisal and James Nguyen for assisting with electrochemical measurements, and Dr Andrew J. Intelli, Dr Jacob P. Sorrentino, Ryan T. Lee, and Dr Douglas L. Orsi for preparing several gem-difluoroalkene substrates. X-Ray crystallographic data was obtained by Dr Mattias Zeller and Denver Hopkins. This work was also supported in part by the Research instrumentation Center in the Department of Chemistry at Purdue University.References
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed.
- M. Shafiei, L. Peyton, M. Hashemzadeh and A. Foroumadi, Bioorg. Chem., 2020, 104, 104240 CrossRef CAS PubMed.
- N. Kitazawa, D. Shinmyo, K. Ito, N. Sato, D. Hasegawa, T. Uemura and T. Watanabe, WIPO, 2010098487A1, 2010 Search PubMed.
- C. Fischer, B. Munoz, S. Zultanski, J. Methot, H. Zhou and C. W. Brown, WIPO, 2008156580A1, 2008 Search PubMed.
- M. A. Estrada, J. Grina, P. Wehn and R. Xu, WIPO, 2020210139A1, 2020 Search PubMed.
- S. Patel, G. Hamilton, C. Stivala, H. Chen and G. Zhao, WIPO, 2017004500A1, 2017 Search PubMed.
- E. M. Bacon, G. Balan, C.-H. Chou, C. T. Clark, J. J. Cottell, M. Kim, T. A. Kirschberg, J. O. Link, G. Phillips, S. D. Schroeder, N. H. Squires, K. L. Stevens, J. G. Taylor, W. J. Watkins, N. E. Wright and S. M. Zipfel, WIPO, 2017007689A1, 2017 Search PubMed.
- A. Aliper, V. Aladinskiy and A. Zavoronkovs, US Pat., 20200270231A1, 2020 Search PubMed.
- B. Hakan, K. Henriksson, V. Hulikal and M. Lepisto, US Pat., 20090093485A1, 2009 Search PubMed.
- P. Samadder, T. Suchánková, O. Hylse, P. Khirsariya, F. Nikulenkov, S. Drápela, N. Straková, P. Vaňhara, K. Vašíčková, H. Kolářová, L. Binó, M. Bittová, P. Ovesná, P. Kollár, R. Fedr, M. Ešner, J. Jaroš, A. Hampl, L. Krejčí, K. Paruch and K. Souček, Mol. Cancer Ther., 2017, 16, 1831–1842 CrossRef CAS PubMed.
- M. K. Ameriks, G. Chen, C. Huang, B. N. Laforteza, S. Ravula, E. H. Southgate and W. Zhang, WIPO, 2019065613A1, 2019 Search PubMed.
- A. Bigot, J. H. Schaetzer, P. J. M. Jung, A. Stoller, J. D. H. Gagnepain, R. G. Hall, S. Rendine and N. Compagnone, WIPO, 2020030503A1, 2020 Search PubMed.
- J. A. Stafford, J. M. Veal, L. L. Trzoss, C. McBride, R. M. Pastor, S. T. Staben, C. Stivala and M. Volgraf, WIPO, 2020018970A1, 2020 Search PubMed.
- S. Schiesser, H. Chepliaka, J. Kollback, T. Quennesson, W. Czechtizky and R. J. Cox, J. Med. Chem., 2020, 63, 13076–13089 CrossRef CAS PubMed.
- K. Niedermann, N. Früh, R. Senn, B. Czarniecki, R. Verel and A. Togni, Angew. Chem., Int. Ed., 2012, 51, 6511–6515 CrossRef CAS PubMed.
- L. M. Yagupolskii, D. V Fedyuk, K. I. Petko, V. I. Troitskaya, V. I. Rudyk and V. V. Rudyuk, J. Fluorine Chem., 2000, 106, 181–187 CrossRef CAS.
- A. Budinská, J. Václavík, V. Matoušek and P. Beier, Org. Lett., 2016, 18, 5844–5847 CrossRef PubMed.
- L. Li, C. Ni, Q. Xie, M. Hu, F. Wang and J. Hu, Angew. Chem., Int. Ed., 2017, 56, 9971–9975 CrossRef CAS PubMed.
- F. X. Liu, W. Chen, L. Ma, K. Cheng, Z. Zhou and W. Yi, New J. Chem., 2023, 47, 12589–12594 RSC.
- M. Ziabko, B. Klepetářová and P. Beier, J. Org. Chem., 2023, 88, 6939–6946 CrossRef CAS PubMed.
- V. Motornov, A. Markos and P. Beier, Chem. Commun., 2018, 54, 3258–3261 RSC.
- D. J. Burton, Z. Y. Yang and W. Qiu, Chem. Rev., 1996, 96, 1641–1715 CrossRef CAS PubMed.
- M. J. Tozer and T. F. Herpin, Tetrahedron, 1996, 52, 8619–8683 CrossRef CAS.
- J. Ichikawa, J. Fluorine Chem., 2000, 105, 257–263 CrossRef CAS.
- G. Chelucci and D. Agraria, Chem. Rev., 2012, 112, 1344–1462 CrossRef CAS PubMed.
- M. Decostanzi, J. M. Campagne and E. Leclerc, Org. Biomol. Chem., 2015, 13, 7351–7380 RSC.
- X. J. Zhang, Y. M. Cheng, X. W. Zhao, Z. Y. Cao, X. Xiao and Y. Xu, Org. Chem. Front., 2021, 8, 2315–2327 RSC.
- J. P. Sorrentino and R. A. Altman, Synthesis, 2021, 53, 3935–3950 CrossRef CAS PubMed.
- X. Zhang and S. Cao, Tetrahedron Lett., 2017, 58, 375–392 CrossRef CAS.
- F. Ye, Y. Ge, A. Spannenberg, H. Neumann, L. W. Xu and M. Beller, Nat. Commun., 2021, 12, 1–9 CrossRef PubMed.
- Y. Xiong, X. Zhang, T. Huang and S. Cao, J. Org. Chem., 2014, 79, 6395–6402 CrossRef CAS PubMed.
- C. Liu, H. Zeng, C. Zhu and H. Jiang, Chem. Commun., 2020, 56, 10442–10452 RSC.
- X. Wu, G. Ma, X. Peng, Z. Ning, Z. Lin, X. Chen, Y. Tang and P. Feng, Org. Chem. Front., 2021, 8, 4871–4877 RSC.
- X. Han, X. Liu, C. Len, L. Liu, D. Wang, Y. Zhang, X. H. Duan and M. Hu, J. Org. Chem., 2023, 88, 12744–12754 CrossRef CAS PubMed.
- L. Wen, Z. Zou, N. Zhou, C. Sun, P. Xie and P. Feng, Org. Lett., 2024, 26, 241–246 CrossRef CAS PubMed.
- L. Wen, B. Li, Z. Zou, N. Zhou, C. Sun, P. Feng and H. Li, Org. Chem. Front., 2023, 11, 142–148 RSC.
- L. Wen, N. Zhou, Z. Zhang, C. Liu, S. Xu, P. Feng and H. Li, Org. Lett., 2023, 25, 3308–3313 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- T. Y. Shang, L. H. Lu, Z. Cao, Y. Liu, W. M. He and B. Yu, Chem. Commun., 2019, 55, 5408–5419 RSC.
- E. Tsui, A. J. Metrano, Y. Tsuchiya and R. R. Knowles, Angew. Chem., Int. Ed., 2020, 59, 11845–11849 CrossRef CAS PubMed.
- A. Joshi-Pangu, F. Lévesque, H. G. Roth, S. F. Oliver, L. C. Campeau, D. Nicewicz and D. A. DiRocco, J. Org. Chem., 2016, 81, 7244–7249 CrossRef CAS PubMed.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS.
- R. M. Herrick, M. K. Abd El-Gaber, G. Coy and R. A. Altman, Chem. Commun., 2023, 59, 5623–5626 RSC.
- V. V Levin and A. D. Dilman, J. Org. Chem., 2019, 84, 8337–8343 CrossRef PubMed.
- M. O. Zubkov, M. D. Kosobokov, V. V. Levin, V. A. Kokorekin, A. A. Korlyukov, J. Hu and A. D. Dilman, Chem. Sci., 2020, 11, 737–741 RSC.
- J. P. Sorrentino, R. M. Herrick, M. K. Abd El-Gaber, A. Z. Abdelazem, A. Kumar and R. A. Altman, J. Org. Chem., 2022, 87, 16676–16690 CrossRef CAS PubMed.
- A. Breder and C. Depken, Angew. Chem., Int. Ed., 2019, 58, 17130–17147 CrossRef CAS PubMed.
- T. M. Nguyen, N. Manohar and D. A. Nicewicz, Angew. Chem., Int. Ed., 2014, 53, 6198–6201 CrossRef CAS PubMed.
- G. Luo, L. Chen and G. Dubowchik, J. Org. Chem., 2006, 71, 5392–5395 CrossRef CAS PubMed.
- Y. Liang, N. Zhou, G. Ma, L. Wen, X. Wu and P. Feng, Mol. Catal., 2022, 528, 1–7 Search PubMed.
- R. Chen, D. Yin, L. Lu, X. T. Feng, Y. Dou, Y. Zhu and S. Fan, Org. Lett., 2023, 25, 7293–7297 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, J. Am. Chem. Soc., 2014, 136, 17024–17035 CrossRef CAS PubMed.
- X. Hu, G. Zhang, F. Bu and A. Lei, ACS Catal., 2017, 7, 1432–1437 CrossRef CAS.
- I. D. Lemir, W. D. Castro-Godoy, A. A. Heredia, L. C. Schmidt and J. E. Argüello, RSC Adv., 2019, 9, 22685–22694 RSC.
- A. Nomoto, Y. Higuchi, K. Yohsuke and A. Ogawa, Mini-Rev. Med. Chem., 2013, 13, 814–823 CrossRef CAS PubMed.
- B. Thapa and H. Bernhard Schlegel, J. Phys. Chem. A, 2016, 120, 8916–8922 CrossRef CAS PubMed.
- D. T. Leeck, R. Li, L. J. Chyall and H. I. Kenttämaa, J. Phys. Chem., 1996, 100, 6608–6611 CrossRef CAS.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boco Raton, 1st edn, 2007 Search PubMed.
- Q. Zhu, E. C. Gentry and R. R. Knowles, Angew. Chem., Int. Ed., 2016, 55, 9969–9973 CrossRef CAS PubMed.
- (a) CCDC 2414748: Experimental Crystal Structure, 2025, DOI:10.5517/ccdc.csd.cc2m1r3k.; (b) CCDC 2414405: Experimental Crystal Structure, 2025, DOI:10.5517/ccdc.csd.cc2m1d14; (c) CCDC 2414406: Experimental Crystal Structure, 2025, DOI:10.5517/ccdc.csd.cc2m1d25; (d) CCDC 2492669: Experimental Crystal Structure, 2025, DOI:10.5517/ccdc.csd.cc2pntpw; (e) CCDC 2414370: Experimental Crystal Structure, 2025, DOI:10.5517/ccdc.csd.cc2m1bxy.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |