
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntegrating a hydrogen-bonded complex as a secondary building unit to construct a multivariate framework for programmable drug delivery
Xujiao
Ma
,
Zhong
Zhang
,
Xianghui
Ruan
,
Jiarui
Cao
,
Ye
Yuan
 *,
Yajie
Yang
*,
Nan
Gao
* and
Guangshan
Zhu
*
*,
Yajie
Yang
*,
Nan
Gao
* and
Guangshan
Zhu
*
Key Laboratory of Polyoxometalate and Reticular Material Chemistry of Ministry of Education, Northeast Normal University, Changchun 130012, China. E-mail: Zhugs@nenu.edu.cn; gaon320@nenu.edu.cn; yangyajie@jlu.edu.cn; Yuany101@nenu.edu.cn
First published on 14th October 2025
Abstract
Porous organic frameworks (POFs) consisting of organic building blocks through covalent bonds are novel functional solids. However, the pure covalent bonding pattern leads to the inability to produce strong and selective interactions with organic molecules, restricting applications in biomedical, detection, and sensing fields. Herein, we report a hydrogen-bonded complex (HC) of 5-fluorouracil (5-FU) and p-aminobenzoic acid as a building block to construct a hybrid-bonded framework (F@POF) via a Schiff base reaction, integrating covalent and hydrogen-bonded motifs. The existence of multiple hydrogen bonds enabled 5-FU to exhibit 19-fold enhanced binding affinity versus physical encapsulation. Capitalizing on differential interaction modalities, cyclophosphamide (CTX) and methotrexate (MTX) were sequentially immobilized via van der Waals forces into the F@POF channels and dual hydrogen/π–π interactions on the pore surface, respectively, yielding a triple-drug co-doped multivariate porous framework, CMF@POF. The programmable release (5-FU > MTX > CTX) endowed CMF@POF with remarkable antitumor activity in in vivo mouse experiments, outperforming the classical CMF chemotherapy. This hydrogen-bond-guided engineering paradigm redefines multifunctional POFs for intelligent multidrug delivery, offering a transformative approach to optimize combination chemotherapy.
Introduction
Porous organic frameworks (POFs) are an emerging class of functional solids in recent decades, attracting extensive attention worldwide. Since the first report in the 1970s, POF materials have evolved into a large system including hyper-cross-linked polymers (HCPs),1,2 polymers of intrinsic microporosity (PIMs),3,4 covalent organic frameworks (COFs),5–7 covalent triazine frameworks (CTFs),8–10 conjugated microporous polymers (CMPs),11,12 porous aromatic frameworks (PAFs),13,14 and porous organic cages (CCs).15 This class of materials is generally composed of organic components through covalent bonds (C–C, B–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CN, etc.), similar to polymeric components and, has the full host–guest interactions of inorganic frameworks.16 On the basis of the low densities, high porosity, and tunable structure, POF materials have a wide range of applications in energy storage,17 catalysis,18,19 and molecular separation.20 However, the pure covalent bonding pattern of POFs reveals certain shortcomings in molecular recognition and binding affinity, limiting the applications in the biomedical, detection, and sensing fields.
C, CN, etc.), similar to polymeric components and, has the full host–guest interactions of inorganic frameworks.16 On the basis of the low densities, high porosity, and tunable structure, POF materials have a wide range of applications in energy storage,17 catalysis,18,19 and molecular separation.20 However, the pure covalent bonding pattern of POFs reveals certain shortcomings in molecular recognition and binding affinity, limiting the applications in the biomedical, detection, and sensing fields.
Recent research focuses on increasing the binding selectivity and ability of porous architectures to guest molecules by modifying functional groups, adjusting pore sizes, and altering structural compositions through in situ synthesis or post-modification. For example, researchers decorated carboxylate,21,22 alkoxy/thioether chain23,24 and ethylenediaminetetraacetic acid groups25 in the POF channels, which showed high selectivity and large capacity for mercury, cobalt, and the lanthanides. Nevertheless, these methods are generally applicable to the binding of metal ions through the coordination bonds of the organic ligands. For organic molecules, porous frameworks only produce weak interactions such as van der Waals forces, hydrogen bonds, or π–π interactions, and the bonding strength is generally weak. Although one can shrink the pore size to bring about multiple interactions with organic molecules, the pore channels are easily occupied and blocked, resulting in low adsorption capacity and slow diffusion kinetics of organic guests in porous frameworks.
Herein, a 5-FU-based hydrogen-bonded complex serving as the secondary building unit was impregnated into the POF network via a Schiff base reaction (Fig. 1). Since the lattice skeleton immobilizes the 5-FU molecule through multiple hydrogen bonds, its binding capacity for 5-FU is 19 times higher than that of conventional physical inclusion. According to the specific porous structure, different interactions including hydrogen bonds, π–π interactions, and van der Waals forces were incorporated into the porous organic framework, where the three drugs of CTX, MTX, and 5-FU were simultaneously loaded. The precise assembly enabled programmable drug delivery where CTX exhibited fast release (54 mg g−1 in <0.5 h), MTX reached an equilibrium concentration (8 mg g−1 in 1 h), and 5-FU molecules slowly diffused outwards reaching a high content (38 mg g−1 within 12 h). The order and dosage satisfy the requirements for the drug delivery process of CMF therapy (CTX, MTX, and 5-Fu combination drug therapy), which reveals a strong anticancer effect in breast cancer chemotherapy.
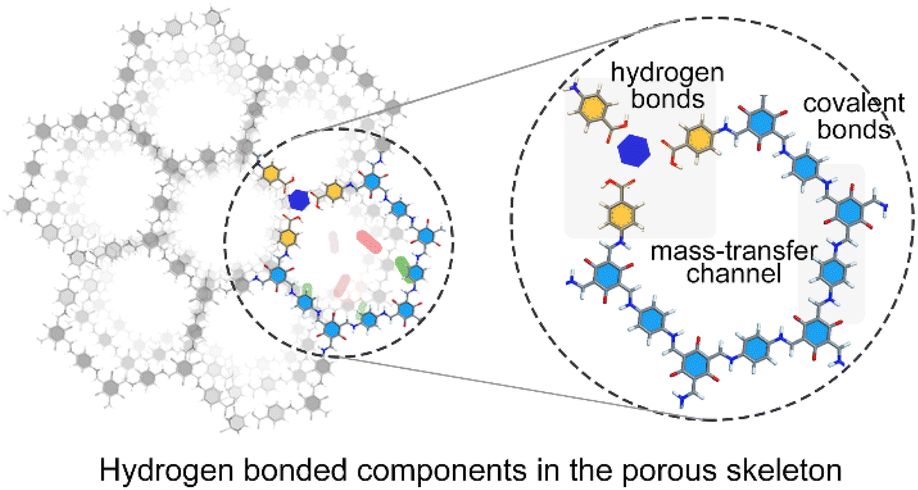 | ||
| Fig. 1 Schematic diagram illustrating the hydrogen-bonded organic molecules in the porous framework. | ||
Results and discussion
A hydrogen-bonded complex (HC) composed of 5-FU and p-aminobenzoic acid molecules in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio was synthesized as a secondary building unit (Fig. 2). As depicted in the 1H solution nuclear magnetic resonance (NMR) spectrum of HC, the peaks centered at 11.59 and 11.79 ppm are attributed to the active hydrogen of the N–H groups of 5-FU and the hydrogen atom of the –COOH groups of p-aminobenzoic acid, respectively. A significant shift from 11.43 to 11.59 ppm is observed, compared with 5-FU, indicating the formation of a hydrogen bond between the N–H groups and p-aminobenzoic acid (Fig. S1). Correspondingly, the hydrogen signal of the –COOH groups in p-aminobenzoic acid shifts to the low field (Fig. S1a). Those significant shifts suggests that there is a strong hydrogen bond interaction between 5-FU and p-aminobenzoic acid. 1H NMR titration experiments and Job's plot analysis (Fig. S1b and c) revealed that the addition of 1/3 equivalents of 5-FU to p-aminobenzoic acid led to the maximum shift of the –COOH's hydrogen (Hb), supporting a 3:1 binding stoichiometry for p-aminobenzoic acid and 5-FU in the HC. The association constants (1.1 × 104 M−1) for 5-FU's hydrogen (Ha) and –COOH's oxygen were determined by NMR titrations (Fig. S2). Moreover, the 19F NMR peaks of 5-fluorouracil (5-FU) exhibited significant shifts with increasing concentration of p-aminobenzoic acid, demonstrating the hydrogen-bonding interactions between the F atoms of 5-FU and the –COOH groups of p-aminobenzoic acid.26 Furthermore, the inflection point observed in the Job's plot analysis of the 19F NMR titration data further confirms a 3:1 binding stoichiometry (p-aminobenzoic acid:5-FU) (Fig. S3). Fourier transform infrared (FTIR) spectroscopic analysis of HC showed that the stretching vibration of –C–F changes from 1244 to 1249 cm−1 (Fig. S4), compared with 5-FU, which was attributed to the electron cloud on the F reducing the frequency of the tensile vibration through hydrogen interaction.27 The characteristic peaks of –NH2 stretching vibration showed no obvious changes, which is ascribed to the absence of –NH2 to participate in forming the hydrogen bonds. As confirmed by the FTIR and NMR spectra, the chemical structure of the HC unit is composed of one 5-FU molecule and three p-aminobenzoic acid molecules through multiple hydrogen bonds. DFT calculations further confirmed that 5-Fu and p-aminobenzoic acid could form a stable 1:3 HC. Significantly, utilizing the independent gradient model based on Hirshfeld partition (IGMH)28 analysis enables direct assessment of hydrogen bond strength within the HC, revealing the presence of multiple strong hydrogen bonds (X⋯H–O, where X = F, N, O)29 (Fig. S5).
:3 ratio was synthesized as a secondary building unit (Fig. 2). As depicted in the 1H solution nuclear magnetic resonance (NMR) spectrum of HC, the peaks centered at 11.59 and 11.79 ppm are attributed to the active hydrogen of the N–H groups of 5-FU and the hydrogen atom of the –COOH groups of p-aminobenzoic acid, respectively. A significant shift from 11.43 to 11.59 ppm is observed, compared with 5-FU, indicating the formation of a hydrogen bond between the N–H groups and p-aminobenzoic acid (Fig. S1). Correspondingly, the hydrogen signal of the –COOH groups in p-aminobenzoic acid shifts to the low field (Fig. S1a). Those significant shifts suggests that there is a strong hydrogen bond interaction between 5-FU and p-aminobenzoic acid. 1H NMR titration experiments and Job's plot analysis (Fig. S1b and c) revealed that the addition of 1/3 equivalents of 5-FU to p-aminobenzoic acid led to the maximum shift of the –COOH's hydrogen (Hb), supporting a 3:1 binding stoichiometry for p-aminobenzoic acid and 5-FU in the HC. The association constants (1.1 × 104 M−1) for 5-FU's hydrogen (Ha) and –COOH's oxygen were determined by NMR titrations (Fig. S2). Moreover, the 19F NMR peaks of 5-fluorouracil (5-FU) exhibited significant shifts with increasing concentration of p-aminobenzoic acid, demonstrating the hydrogen-bonding interactions between the F atoms of 5-FU and the –COOH groups of p-aminobenzoic acid.26 Furthermore, the inflection point observed in the Job's plot analysis of the 19F NMR titration data further confirms a 3:1 binding stoichiometry (p-aminobenzoic acid:5-FU) (Fig. S3). Fourier transform infrared (FTIR) spectroscopic analysis of HC showed that the stretching vibration of –C–F changes from 1244 to 1249 cm−1 (Fig. S4), compared with 5-FU, which was attributed to the electron cloud on the F reducing the frequency of the tensile vibration through hydrogen interaction.27 The characteristic peaks of –NH2 stretching vibration showed no obvious changes, which is ascribed to the absence of –NH2 to participate in forming the hydrogen bonds. As confirmed by the FTIR and NMR spectra, the chemical structure of the HC unit is composed of one 5-FU molecule and three p-aminobenzoic acid molecules through multiple hydrogen bonds. DFT calculations further confirmed that 5-Fu and p-aminobenzoic acid could form a stable 1:3 HC. Significantly, utilizing the independent gradient model based on Hirshfeld partition (IGMH)28 analysis enables direct assessment of hydrogen bond strength within the HC, revealing the presence of multiple strong hydrogen bonds (X⋯H–O, where X = F, N, O)29 (Fig. S5).
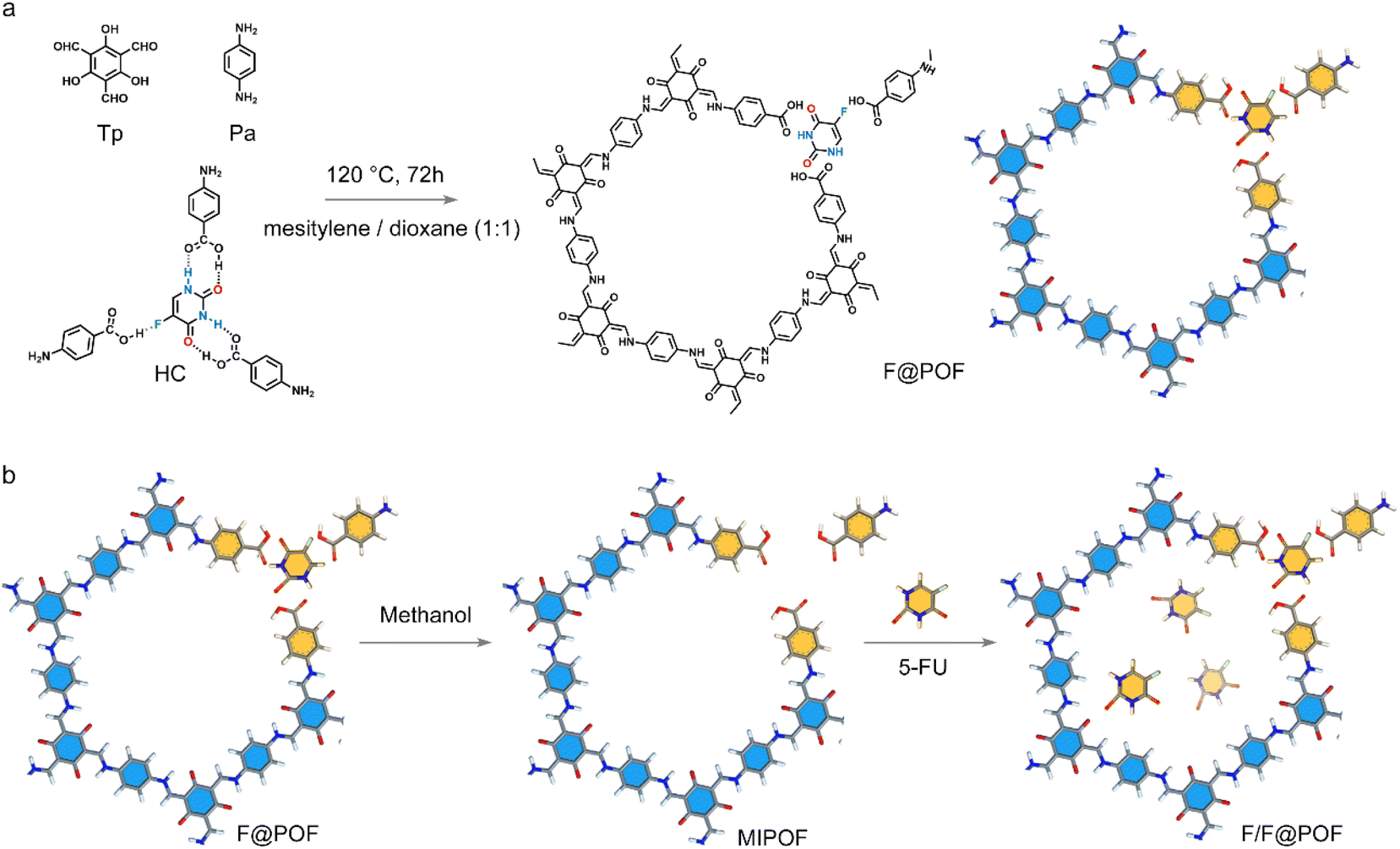 | ||
| Fig. 2 (a) HC unit integrated with the porous network to produce F@POF. (b) Synthesis schematic of F/F@POF by impregnation of F@POF into a 5-FU methanol solution. | ||
A classical POF material (TpPa-1) was selected as a scaffold, which was prepared using triformylphloroglucinol (TP) and p-phenylenediamine (PA) through a Schiff base crosslinking reaction. TpPa-1 possesses a well-defined hexagonal structure with a P6/m space group symmetry featuring a large specific surface area30 [∼764 m2 g−1 based on Brunauer–Emmett–Teller (BET) model] and uniform pore size distribution (1.50 nm) based on nonlocal density functional theory (Fig. S6 and S9). HC matches well with the size of TpPa-1 fragments, which facilitates the formation of extended TpPa-1-type networks (Fig. S7). Based on this, the 5-FU-based hydrogen-bonded complex coupled with TP and PA was cross-linked through a Schiff-base reaction to prepare the hydrogen-bonded POF material, F@POF (Fig. 2a), where the –NH2 molar ratios of HC and p-phenylenediamine were 25, 50 and 75%, denoted as F@POF-x (x = 25, 50, and 75) (Experimental Section for details, Table S1). In the FTIR spectra of F-POF-x, the –N–H stretching band of –NH2 in the PA and HC units disappeared at 3300–3500 cm−1, and a CC characteristic peak (1574 cm−1) appeared, confirming the completion of the Schiff base reaction (Fig. S8b). Additionally, the characteristic peaks at 1670 and 1170 cm−1 were shown in the FTIR spectrum of F@POF-x, assigned to the –CO and C–F of HC, demonstrating that the HC as building units were successfully combined into the POF skeleton. The powder X-ray diffraction (PXRD) patterns for F@POF-25 and F@POF-50 revealed the same peaks as the TpPa-1 (i.e., 2-theta = 4.7, 7.9, and 27.0°) which correspond to the crystal faces of the (100), (200), and (001) planes; and the diffraction peaks assigned to the HC disappeared from the F@POF pattern (Fig. 3a and S8). Therefore, F@POF inherits the parent structure of TpPa-1 with a hexagonal P6/m space group. However, as the molar ratio of HC increased to 75%, the periodic structure was destroyed, probably stemming from the flexibility of the hydrogen bond networks. The BET surface areas of F@POF-25 and F@POF-50 were 573 and 432 m2 g−1 as determined by the N2 adsorption experiment at 77 K (Fig. S9), lower than that of TpPa-1 (768 m2 g−1). Like TpPa-1, F@POF-x possesses a variety of pore sizes ranging from 1.5 to 7.5 nm (Fig. S9). The changes in the specific surface areas and pore volume distributions are ascribed to the electron-deficient –COOH group in the HC building block, reducing the activity of aniline for Schiff base polymerization.
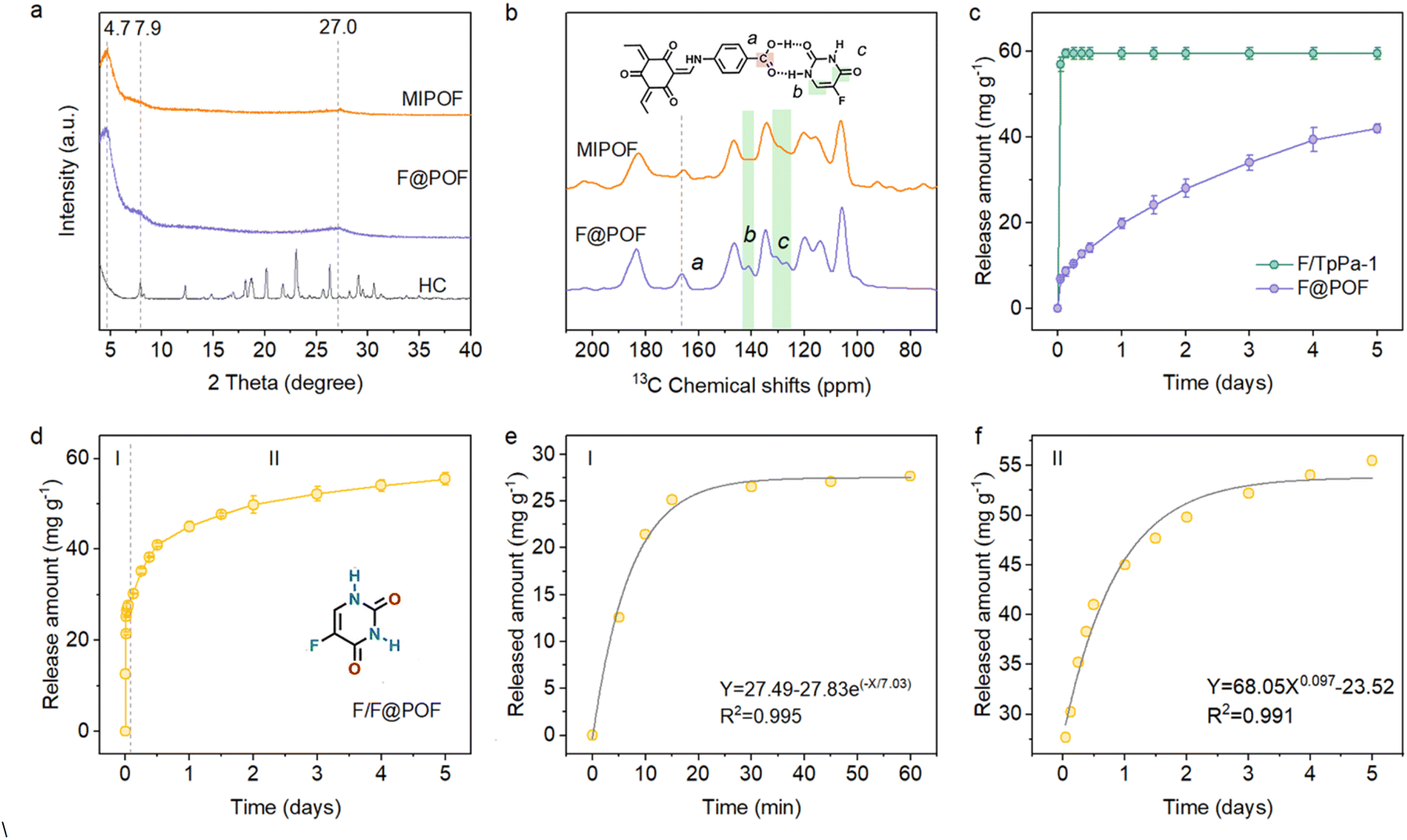 | ||
| Fig. 3 (a) PXRD patterns of the HC, F@POF, and MIPOF. (b) Solid-state 13C CP/MAS NMR spectra of MIPOF and F@POF. (c) Release kinetics of the 5-FU from F/TpPa-1 and F@POF in a PBS solution (pH = 7.4, 37 °C). (d) Release kinetics of 5-FU from F/F@POF in a PBS solution (pH = 7.4, 37 °C). (e) Stage I: first-order kinetic model fits the release of physically doped 5-FU from F/F@POF. (f) Stage II: Korsemeyor–Peppas kinetic model fits the release of hydrogen-bonded 5-FU from F/F@POF. | ||
To determine the incorporation of 5-FU in the F@POF skeleton, F@POF-50 is regarded as the main sample for structural characterization, namely F@POF. F@POF powder was stirred in methanol to release 5-FU, thereby obtaining a reference sample (Fig. 2b). Subsequent removal of 5-FU resulted in an increase in the pore volume from 0.609 to 0.683 cm3 g−1. Nonlocal density functional theory (NLDFT) pore size distribution analysis revealed the emergence of pores centered at ∼2.6 nm (Fig. S10), indicating the formation of complementary vacancies within the framework upon 5-FU extraction. Notably, structural integrity was maintained in the PXRD profiles (Fig. 3a), demonstrating that the framework skeleton remains stable after 5-FU removal. In selective adsorption experiments involving multiple pharmaceuticals (ibuprofen, floxuridine, tegafur), the material exhibited exceptional adsorption selectivity towards 5-FU (Fig. S11). We propose that 5-FU acts as a template molecule within the framework, generating size- and shape-matched binding cavities upon its removal. Consequently, we designate this sample as a molecularly imprinted POF (MIPOF). As depicted in the 13C solid-state NMR spectra, the reduced peak intensity of 5-FU in MIPOF was observed at 141.4 and 132.7 ppm, corresponding to the absence of carbon atoms in fluorouracil (Fig. 3b). In addition, there is an obvious shift (from 166.2 to 165.6 ppm) of the specific carbon signal assigned to the –COOH group in MIPOF after the release of 5-FU, confirming the hydrogen-bonding interactions between 5-FU and –COOH groups in the F@POF structure (Fig. 3b). In the FT-IR spectra, the CO stretching vibration band originating from p-aminobenzoic acid shifted from 1716 to 1706 cm−1 upon removal of 5-FU (Fig. S12), which indicates that the CO group acts as a hydrogen-bond acceptor in F@POF, forming interactions with 5-FU.
For comparison, a physically doped sample of F/TpPa-1 was prepared by impregnation of TpPa-1 in the 5-FU solution. The loading amount of 5-FU in the pore through van der Waals force was 60.1 mg g−1 as analysed by ultraviolet-visible (UV-vis) spectroscopy. The incorporation form of 5-FU was further determined by photoluminescence (PL) spectroscopy. In comparison to MIPOF and F/TpPa-1, the PL spectrum of F@POF exhibited a strong emission at 617 nm (Fig. S13). The enhanced fluorescence intensity was ascribed to the electron-donating property of 5-FU, sharing its electron cloud with the electron-accepting –COOH groups through the hydrogen bonds. A contrast sample with structural defect, denoted as D-POF, was prepared using triformylphloroglucinol (TP), p-phenylenediamine (PA) and p-aminobenzoic acid through a Schiff-base reaction (Fig. S14a). Compared with F@POF, the presence of a distinct additional peak at 3.95 ppm in both D-POF and F@D-POF (prepared by impregnating 5-FU into the D-POF) in the 1H NMR solid-state spectra, attributed to –NH2 (Fig. S14b), suggests that individual p-aminobenzoic acid molecules induce structural defects. These results validate that HC, as the secondary building unit, remains stable throughout the synthetic process, and structural defects similar to those in D-POF are absent in the skeletal structure of F@POF.
Moreover, thermogravimetric analysis reveals that F@POF exhibits an initial decomposition temperature (∼350 °C) comparable to that of TpPa-1, indicating that excellent thermal stability is maintained after the integration of HC into the framework (Fig. S15). The F element content in F@POF was determined based on energy dispersive X-ray spectrometry (EDX) and elemental analyses to be 0.47% (Fig. S16 and Table S2). Correspondingly, the actual capacity for fixed 5-FU was calculated to be about 41.0 mg g−1 in the F@POF powder. Based on the above characterization, it can be concluded that the hydrogen-bonded POF material was successfully prepared, and promises to realize the slow release of 5-FU under the restriction of hydrogen bonds.
Detailed studies on the effect of hydrogen bonds in F@POF on the release of drug molecules were carried out. The release of 5-FU from F@POF-25 and F@POF (F@POF-50) was evaluated in a phosphate-buffered saline (PBS) solution at physiological pH (7.4) and 25 °C by recording the absorbance via UV-vis spectrophotometry, and the corresponding concentration was determined based on the Beer–Lambert law (Fig. S17). F@POF (F@POF-50) exhibited the higher drug loading capacity (41.96 mg g−1), twice that of F@POF-25 (19.70 mg g−1) (Fig. S18), stemming from the more HC introduced in the skeleton. In the release profiles of 5-FU, fast release is observed in F/TpPa-1 as compared with F@POF, suggesting that the hydrogen-bonding interaction between the MIPOF and 5-FU suppresses the release of 5-FU (Fig. 3c). Therefore, F@POF (F@POF-50) was selected as the optimum material to utilize the releasing drug. Further, MIPOF (10 mg) is immersed in a 100 ppm 5-FU methanol solution (10 mL) for 30 min obtaining a sample F/F@POF, which concurrently possesses hydrogen-bonded 5-FU on the skeleton as well as adsorbed 5-FU in the pores, realizing controlled release of 5-FU with multivariate interactions (Fig. 2b). The 5-FU release process undergoes two stages (Fig. S19): (Stage I) in the first 1 h, ∼27 mg g−1 of 5-FU was rapidly released from the POF channels to the solution owing to the weak interaction between the physically doped 5-FU and the F@POF channels, and then (Stage II) ∼28 mg g−1 of 5-FU was slowly released for up to 5 days owing to the strong affinity (multiple hydrogen bonds) between the 5-FU and the F@POF skeleton (Fig. 3d).
Zero-order, first-order, and Korsemeyor–Peppas model fittings were adopted to investigate the bonding patterns (Fig. S20–S23). The initial release curve (Stage I) fits well with the first-order release model [R2 = 0.995]. The latter release curve (Stage II) matches the Korsemeyor–Peppas model (R2 = 0.991) (Fig. 3e and f). Based on the Arrhenius equation,31 the binding strength between the porous skeleton and the drug molecule is quantified by reaction rate (k) and affinity (Ea) between 5-FU and the porous skeleton, where the logarithm of the rate constant k has a negative correlation with Ea. The calculated k value of 0.14 min−1 in Stage I is higher than that for Stage II (0.0073 min−0.097). The affinity for Stage II (Ea2) is 19-fold higher than that for physical loading (Stage I; Ea1), which reduces the release kinetics of 5-FU from F@POF.32 The slow release is controlled by the multiple hydrogen bonds, which has not been encountered in previous carriers such as biocompatible polymers (PVP and polylactic acid), mesoporous silica, and metal–organic frameworks.33–35
The incidence of malignant tumor-triggered breast cancer has been increasing and it is a leading cause of death among women.36 The common treatment with a single drug possesses several limitations such as high metastasis, low response rate, and easy recurrence.37,38 Intravenous cyclophosphamide (CTX, 600 mg m−2), methotrexate (MTX, 40 mg m−2), and 5-fluorouracil (5-FU, 400 mg m−2), commonly named CMF chemotherapy, is used as a first-line therapy for advanced/metastatic breast cancer.39 The clinical practice is to first use the cell-cycle non-specific CTX, followed by MTX and 5-FU. Whether it is simultaneous intravenous injection of MTX and 5-FU, or the use of 5-FU followed by MTX, antagonistic drug reactions will occur, thereby affecting the treatment effect. Even if the injection procedure is strictly followed, the similar diffusion properties result in comparable release kinetics in the delivery process, leading to a complicated implementation process and severe dose-related side effects.40 Constructing an easy-to-use therapeutic system that can be easily and correctly administered in terms of sequence and dosage is a challenge in achieving programmable drug delivery for combination therapy.
Profiting from the porosity of F@POF as mentioned, CTX molecules with a three-dimensional size of 0.70 × 1.17 × 0.63 nm based on a CPK model were easily incorporated into the 1.5-nm porous channels through van der Waals forces, denoted as CF@POF (Fig. 4a and S24). With a large conjugated structure and several hydrogen-bonding components (–N–H and –COOH), methotrexate (MTX, CPK three-dimensional size of 0.82 × 2.35 × 0.78 nm)41 was immobilized on the inner surface of CF@POF through π–π/hydrogen-bonding interactions to afford CMF@POF (Fig. 4a and S24). By controlling the time and drug content of the impregnation progress, an ideal ratio of 5-FU, CTX, and MTX (400:600:40) in the POFs was realized. The loading content of CTX was 57.7 mg g−1, which was determined by the amount of the phosphorus (P) element of CMF@POF measured by inductively coupled plasma atomic emission spectrometry (ICP-AES) (Table S2). The amount of MTX loaded to CMF@POF was 8.6 mg g−1, as obtained from the absorbance analysed via ultraviolet-visible (UV-vis) spectroscopy.
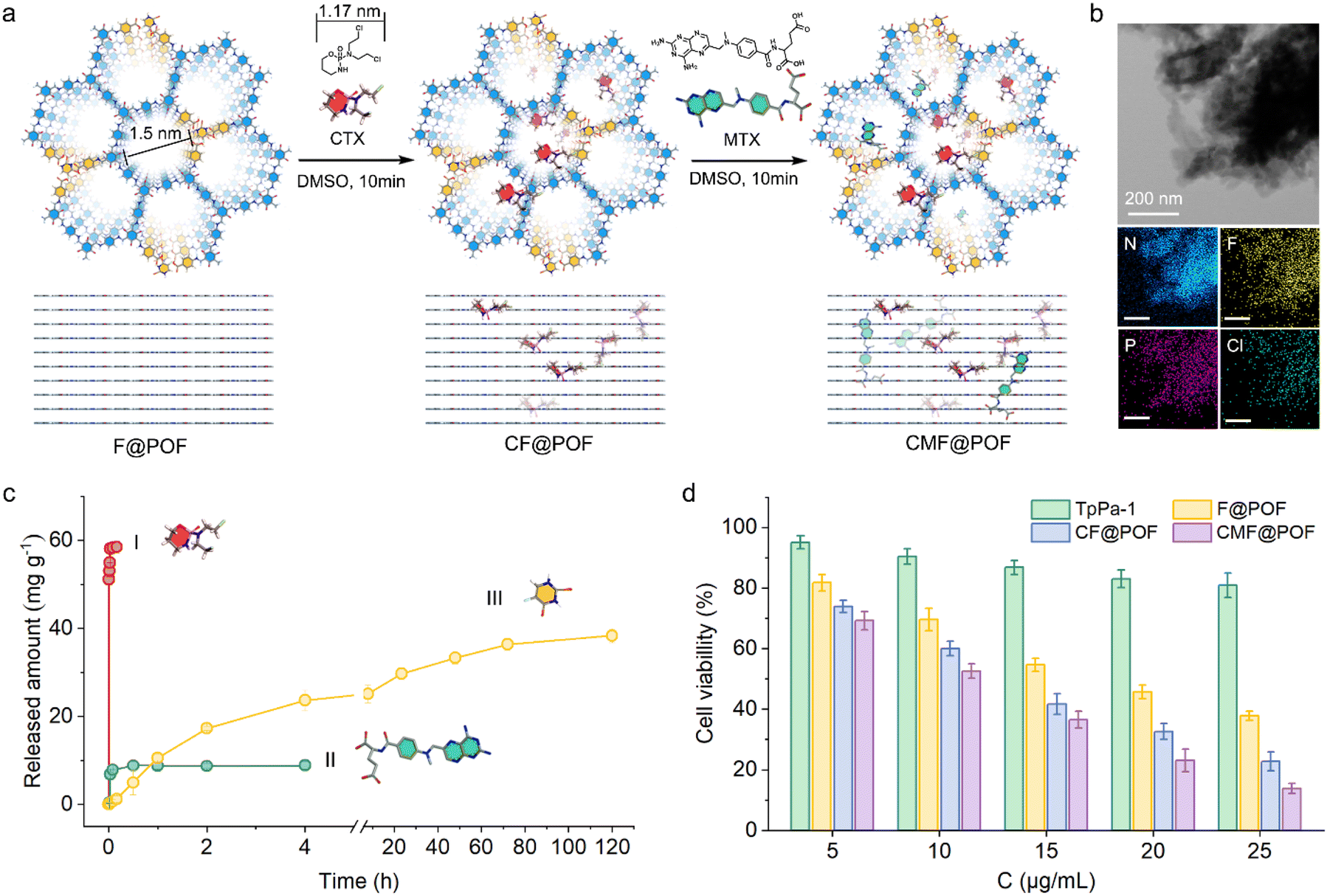 | ||
| Fig. 4 The programmed release of multiple drug molecules through multivariate interactions in porous networks. (a) Two drug molecules (CTX and MTX) are incorporated in the F@POF architecture. (b) Uniform distribution of F, P, and Cl in EDX spectra, indicating evenly doped 5-FU and CTX. (c) Release processes of three drug molecules in PBS solution (pH = 7.4, 37 °C). (d) MTT assay for the toxicity of all POFs to MCF-7 cell lines. | ||
The characteristic peaks of CF@POF and CMF@POF in the PXRD patterns at 2θ = 4.7°, 7.9°, and 27.0° indicated that the crystalline structure of the POF platform was maintained (Fig. S25). A new peak at 638 cm−1 appeared in the FT-IR spectra of CF@POF and CMF@POF, assigned to the C–Cl bonds of CTX (Fig. S26). The BET surface areas of drug-doped molecules decrease from 432 m2 g−1 (F@POF) to 101 (CF@POF) and 67 m2 g−1 (CMF@POF). As the POF cavities are occupied by guest molecules, the pore volume decreases from 0.609 (F@POF) to 0.0136 cm3 g−1 (CMF@POF) (Fig. S27 and S28). Scanning electron microscopy images showed that all POF samples comprise aggregated nanoparticles (Fig. S29). Each separate segment exhibited a sheet-like morphology with lengths in the range of 150–250 nm, contributing to drug delivery in vivo. Transmission electron microscopy and energy-dispersive X-ray (EDX) mapping demonstrated the even distribution of C, N, O, F, P, and Cl in the as-prepared CMF@POF, revealing the uniform distribution of CTX molecules in the F@POF structure (Fig. S30 and 4b), and confirming that 5-FU molecules are still stabilized on the porous skeleton after the encapsulation of CTX and MTX. CMF@POF feasibly simulated CMF treatment for programmable drug delivery of the three drugs owing to the advantage of the strong affinity of 5-FU in the F@POF platform (Fig. 4c). CTX preferentially desorbed from F@POF in simulated body fluids owing to weak van der Waals force and reaches 54 mg g−1 in <30 min. The CTX release curve was fitted to the first-order kinetic profile with an R2 value of 0.995 (Fig. S31–S33). MTX was released slowly owing to the combined effect of π–π and hydrogen bonding interactions and achieved an equilibrium concentration of 8 mg g−1 in 1 h. Its release isotherm satisfies the first-order kinetic curve (R2 = 0.844) (Fig. S34–S36). The release rates of CTX and MTX are 93.75% and 88.88%, respectively. 5-FU revealed delayed release initially, which reached a relatively high amount (38 mg g−1) in 5 days with a release rate of 92.6% (Fig. S37–S39). The trends and dosage for the three medicines are highly consistent with the release profiles of CMF therapeutics described above. Compared to most reported porous materials serving as combined drug delivery systems in recent years, which primarily rely on a single type of intermolecular interaction for drug loading/sustained release (Table S4), CMF@POF leverages multiple synergistic interactions to achieve programmed CMF therapy, demonstrating significant potential for practical anti-tumor treatment.
Due to the broad-spectrum anti-cancer nature of traditional CMF chemotherapy methods, we investigated the anti-cancer effects of CMF@POF against a range of cancer cells. Firstly, the toxicity of all POFs to the MCF-7 cell line (human breast cancer cells) was examined during the growth phase before the evaluation of anticancer efficacy to assess the superiority of multiple-drug release and cellular uptake.42 The cells were grown in Dulbecco's modified Eagle's medium in 96-well plates with a density of 105 cells per well. The samples (TpPa-1, F@POF, CF@POF, and CMF@POF) were added separately with original concentrations in the range of 10–50 μg mL−1. The medium was incubated for 24 h, and the culture solution was removed to eliminate the dead cells. MTT assay demonstrated the biocompatibility (cell viability of >80%) of the POF matrix against the cell lines (Fig. 4d). The synergistic anticancer effect of multiple drugs in the same load is better than that of a single drug. The cells were evaluated at a dose of 50 μg mL−1, and the cell viability was reduced to 37% after loading TpPa-1 with 5-FU. The anticancer activity of the three-drug system is higher with only 13% cellular survival (Fig. 4d). The MTT assay of the four POF samples was further validated via cellular migration studies performed on MCF-7 cells. As shown in Fig. S40, cellular migration gradually decreased with an increase in drug species and the controlled cells of CMF@POF exhibited distinct cellular migration after 24 h. Thus, CMF@POF is an effective drug delivery system that can inhibit cancer cell growth in combination with low-dose chemotherapy.
Then, 4T1 (mouse breast cancer cells), HeLa (human cervical cancer cells) and MG63 (human osteosarcoma cells) were also cultured to determine the broad-spectrum anti-cancer ability of CMF@POF. Through cell fluorescence imaging, it could be clearly seen that as the concentration of CMF@POF increased, the death trends of the three types of cancer cells were similar, especially when the concentration of CMF@POF reached 50 μg mL−1, all three cancer cell lines were almost completely killed (Fig. S41). The above results clearly demonstrate that CMF@POF has good anti-cancer ability. Finally, another cancer cell line, H22 (mouse hepatocarcinoma cell), was used to establish a xenograft tumor model using BALB/c mice to deeply explore the in vivo antitumor efficiency of CMF@POF. Fig. 5a shows that the tumor-bearing mice are randomly divided into four groups (n = 5 in each group). The mentioned drugs were intravenously injected into their tails: (i) saline (control group), (ii) TpPa-1, (iii) CMF@POF, and (iv) 5-FU + CTX + MTX on Day 0 and Day 6. Wild-type BALB/c mice were used to test the biocompatibility. No change in the body weight of the mice in the CMF@POF group indicated the excellent biocompatibility of CMF@POF. However, the mice in the 5-FU + CTX + MTX group lost weight, indicating that direct injection of anticancer drugs is biologically toxic, but CMF@POF can prevent this toxicity (Fig. 5b, S42 and S43). The tumor volume of the mice was enhanced in the control and TpPa-1 groups (Fig. 5c and d), while mice in the 5-FU + CTX + MTX group showed a certain degree of inhibition. Conspicuous tumor growth inhibition was observed in the CMF@POF group, which should be ascribed to the programmable drug release.
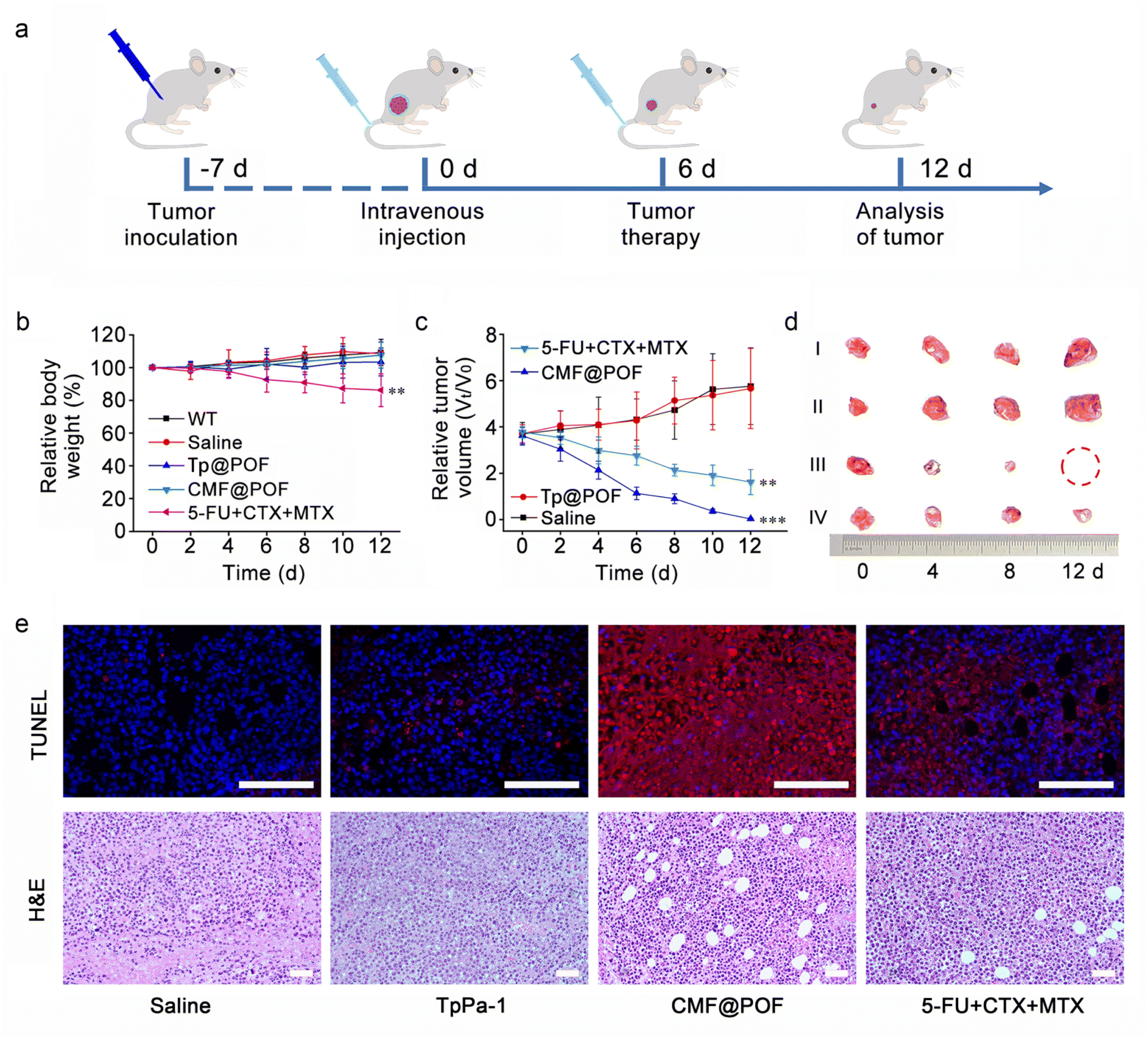 | ||
| Fig. 5 (a) Schedule of animal experiment procedures. (b) Relative body weight changes during various treatments using wild-type BALB/c mice. Statistical significance was assessed using ANOVA followed by a Bonferroni post-test (**p < 0.01). (c) Variation curves of relative tumor volume. Statistical significance was assessed using ANOVA followed by a Bonferroni post-test (**p < 0.01, ***p < 0.001). (d) Excised tumors after 12 days of different treatments. (i) Saline (control group), (ii) TpPa-1, (iii) CMF@POF, and (iv) 5-FU + CTX + MTX. (e) Histochemical analyses (TUNEL and H&E) of tumor tissues harvested from mice after different treatments on Day 8. The scale bar is 100 μm. | ||
Furthermore, terminal deoxynucleotidyl transferase-mediated nick end labeling (TUNEL) and hematoxylin and eosin (H&E) staining of tumor tissues were performed to verify the anticancer effect of CMF@POF. Fig. 5e shows that the strongest red fluorescence came from CMF@POF treated cells, demonstrating the most extensive cancer cell apoptosis among all groups. The pathological H&E staining indicated that tumor structures was severely damaged in the CMF@POF group after treatment, suggesting that CMF@POF can function as an effective antitumor agent through programmable drug release.
Conclusions
In conclusion, a hydrogen-bonded complex as a building unit was integrated into a porous skeleton to enrich the structural variety of POF structures. Accordingly, 5-FU, MTX, and CTX drugs were locked in the POF architecture through different binding patterns, which enabled a synergistic anticancer effect and exhibited outstanding antitumor activity. A clear correlation between the porous architecture and the guest molecules has been established, and a potential method has been provided for designing a carrier material for the programmable release of multiple molecules. This work opens up novel prospects for preparing other multiple-drug regimens to minimize the side effects and enhance anticancer efficacy.Ethical statement
All animal studies were conducted in accordance with the principles and procedures outlined in “Regulations for the Administration of Affairs Concerning Laboratory Animals”, approved by the National Council of China on October 31, 1988, and “The National Regulation of China for Care and Use of Laboratory Animals”, promulgated by the National Science and Technology Commission of China, on November 14, 1988. All animal studies were supervised by the Committee of Northeast Normal University Institutional Animal Care and Use. All operations related to mice strictly comply with the relevant requirements of the “Experimental Animal Environment and Facilities” (GB14925-2010) and the “Guidelines for Ethical Review of Experimental Animal Welfare” (GB/T 35892-2018).Author contributions
X. Ma and Y. Yuan conceived the project. X. Ma, Z. Zhang, X. Ruan and J. Cao performed the experimental studies. Y. Yuan, Y. Yang, N. Gao and G. Zhu supervised the research. X. Ma wrote the original draft of the manuscript, which was edited by all authors.Conflicts of interest
There are no conflicts to declare.Data availability
Extra data are available from the corresponding author upon request.The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: synthesis procedures, experiment details, and characterizations of materials, including SEM images, mapping images, PXRD patterns, TGA analysis, BET analysis, 1H NMR spectra, 19F NMR spectra, the cell fluorescence images and additional calculation results (PDF). See DOI: https://doi.org/10.1039/d5sc04021c.
Acknowledgements
This research was funded by the Science and Technology Research Project from the National Natural Science Foundation of China (22322501), the Education Department of Jilin Province (JJKH20250310KJ), the Science and Technology Development Plan Project of Jilin Province, China (20250101008JJ), the Fundamental Research Funds for the Central Universities (2412025QG002), the Natural Science Foundation of Jilin Provincial (3D5242487416), Liaoning Provincial Natural Science Foundation Joint Fund (Doctoral Research Start-up Project) (2023-BSBA-088 and 2023-BSBA-087), and China Postdoctoral Science Foundation (2024M750307).Notes and references
- L. Tan and B. Tan, Chem. Soc. Rev., 2017, 46, 3322–3356 RSC
.
- J. Huang and S. R. Turner, Polym. Rev., 2018, 58, 1–41 CrossRef
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC
- N. B. McKeown, Sci. China: Chem., 2017, 60, 1023–1032 CrossRef
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef PubMed
- T. Sun, Y. Liang and Y. Xu, Angew. Chem., Int. Ed., 2022, 61, e202113926 CrossRef PubMed
- R. Sun, X. Wang, X. Wang and B. Tan, Angew. Chem., Int. Ed., 2022, 61, e202117668 CrossRef PubMed
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC
- Y. Tian and G. Zhu, Chem. Rev., 2020, 120, 8934–8986 CrossRef CAS
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS PubMed
- X. Yang, Z. Ullah, J. F. Stoddart and C. T. Yavuz, Chem. Rev., 2023, 123, 4602–4634 CrossRef CAS
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS
- M. Yu, N. Chandrasekhar, R. K. M. Raghupathy, K. H. Ly, H. Zhang, E. Dmitrieva, C. Liang, X. Lu, T. D. Kühne, H. Mirhosseini, I. M. Weidinger and X. Feng, J. Am. Chem. Soc., 2020, 142, 19570–19578 CrossRef CAS
- Y. Yang, D. Deng, S. Zhang, Q. Meng, Z. Li, Z. Wang, H. Sha, R. Faller, Z. Bian, X. Zou, G. Zhu and Y. Yuan, Adv. Mater., 2020, 32, 1908243 CrossRef CAS
- Z.-J. Lin, J. Lü, L. Li, H.-F. Li and R. Cao, J. Catal., 2017, 355, 131–138 CrossRef CAS
- L. Liu, R. Yu, L. Yin, N. Zhang and G. Zhu, Chem. Sci., 2024, 15, 1924–1937 RSC
- Y. Yuan, Y. Yang, K. R. Meihaus, S. Zhang, X. Ge, W. Zhang, R. Faller, J. R. Long and G. Zhu, Nat. Chem., 2023, 15, 1599–1606 CrossRef CAS
- Y. Yang, M. Faheem, L. Wang, Q. Meng, H. Sha, N. Yang, Y. Yuan and G. Zhu, ACS Cent. Sci., 2018, 4, 748–754 CrossRef CAS PubMed
- L. Han, Y. Wu, K. Fang, S. Sweeney, U. K. Roesner, M. Parrish, K. Patel, T. Walter, J. Piermattei, A. Trimboli, J. Lefler, C. D. Timmers, X.-Z. Yu, V. X. Jin, M. T. Zimmermann, A. J. Mathison, R. Urrutia, M. C. Ostrowski and G. Leone, Nat. Commun., 2023, 14, 1 CrossRef CAS PubMed
- S.-Y. Ding, M. Dong, Y.-W. Wang, Y.-T. Chen, H.-Z. Wang, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2016, 138, 3031–3037 CrossRef CAS
- Y. Jiang, C. Liu and A. Huang, ACS Appl. Mater. Interfaces, 2019, 11, 32186–32191 CrossRef CAS
- T. Wang, Y. Zhang, B. Huang, B. Cai, R. R. Rao, L. Giordano, S.-G. Sun and Y. Shao-Horn, Nat. Catal., 2021, 4, 753–762 CrossRef CAS
- D. E. McCoy, T. Feo, T. A. Harvey and R. O. Prum, Nat. Commun., 2018, 9, 1 CrossRef CAS
- T. Lu and Q. Chen, J. Comput. Chem., 2022, 43, 539–555 CrossRef PubMed
- Y. He, Y. Zhao, X. Wang, Z. Liu, Y. Yu and L. Li, Angew. Chem., Int. Ed., 2023, 62, e202307160 CrossRef PubMed
- S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2012, 134, 19524–19527 CrossRef PubMed
- R.-A. Mitran, C. Matei and D. Berger, J. Phys. Chem. C, 2016, 120, 29202–29209 Search PubMed
- Z. Dong, Y. Sun, J. Chu, X. Zhang and H. Deng, J. Am. Chem. Soc., 2017, 139, 14209–14216 CrossRef PubMed
- S. Mitra, H. S. Sasmal, T. Kundu, S. Kandambeth, K. Illath, D. Díaz Díaz and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 4513–4520 CrossRef PubMed
- Y. Liu, X. Zhang, M. Zhou, X. Nan, X. Chen and X. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 43498–43507 CrossRef PubMed
- P. P. Bag, D. Wang, Z. Chen and R. Cao, Chem. Commun., 2016, 52, 3669–3672 RSC
- S. Loibl, P. Poortmans, M. Morrow, C. Denkert and G. Curigliano, Lancet, 2021, 397, 1750–1769 Search PubMed
- M. Heidari Khoee, S. Khoee and M. Lotfi, Anal. Biochem., 2019, 572, 16–24 CrossRef PubMed
- S. Lu, K. G. Neoh, C. Huang, Z. Shi and E.-T. Kang, J. Colloid Interface Sci., 2013, 412, 46–55 CrossRef PubMed
- F. Meric-Bernstam, J. Larkin, J. Tabernero and C. Bonini, Lancet, 2021, 397, 1010–1022 Search PubMed
- C.-F. Yen and H.-B. Lam, JAMA Dermatol., 2016, 152, 1281–1282 CrossRef
- Z. Zhang, Y.-W. Liu, H.-R. Tian, X.-H. Li, S.-M. Liu, Y. Lu, Z.-X. Sun, T. Liu and S.-X. Liu, Matter, 2020, 2, 250–260 CrossRef
- L. Bai, S. Z. F. Phua, W. Q. Lim, A. Jana, Z. Luo, H. P. Tham, L. Zhao, Q. Gao and Y. Zhao, Chem. Commun., 2016, 52, 4128–4131 CAS
| This journal is © The Royal Society of Chemistry 2025 |