
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isolation of a Lewis acid-base stabilized stannanone
Mike
Jörges
 ,
Daniel
Knyszek
,
Manoj
Kumar
,
Varre S. V. S. N.
Swamy
and
Viktoria H.
Gessner
*
,
Daniel
Knyszek
,
Manoj
Kumar
,
Varre S. V. S. N.
Swamy
and
Viktoria H.
Gessner
*
Faculty of Chemistry and Biochemistry, Ruhr-University Bochum, Universitätsstrasse 150, 44801 Bochum, Germany. E-mail: viktoria.gessner@rub.de
First published on 7th October 2025
Abstract
A diylide-substituted stannanone, stabilized by the Lewis acid SnCl2 (formally written as Y2Sn![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O → SnCl2) has been successfully isolated and characterized by spectroscopic, crystallographic and computational methods. The nature of the ylide substituent proved critical for its successful isolation. While oxidation of a stannylene with a cyano-substituted ylide yielded only a dimeric stannoxane, incorporation of a thiophosphinoyl moiety provided sufficient steric bulk and additional stabilization through PS coordination, enabling the isolation of the monomeric stannanone. Computational studies revealed a strongly polarized Sn–O bond with negligible π-contribution to the bonding interaction and high opposing charges, resulting in a short Sn–O linkage but a high reactivity toward bond cleavage. This study highlights the challenge associated with stabilizing formal multiple bonds with the heavier atoms and underscores the importance of substituent design in achieving such stabilization.
O → SnCl2) has been successfully isolated and characterized by spectroscopic, crystallographic and computational methods. The nature of the ylide substituent proved critical for its successful isolation. While oxidation of a stannylene with a cyano-substituted ylide yielded only a dimeric stannoxane, incorporation of a thiophosphinoyl moiety provided sufficient steric bulk and additional stabilization through PS coordination, enabling the isolation of the monomeric stannanone. Computational studies revealed a strongly polarized Sn–O bond with negligible π-contribution to the bonding interaction and high opposing charges, resulting in a short Sn–O linkage but a high reactivity toward bond cleavage. This study highlights the challenge associated with stabilizing formal multiple bonds with the heavier atoms and underscores the importance of substituent design in achieving such stabilization.
Introduction
The carbonyl moiety is one of the most ubiquitous functional groups in both nature and organic synthesis. Its highly stable yet polar CO linkage gives rise to a vast array of chemical reactivity and enables numerous transformations, including many large-scale processes. By contrast, much less is known about the corresponding compounds of the heavier group 14 elements owing to the pronounced lability of E = O bonds with E = Si–Pb.1–3 The weaker π overlap, combined with the greater electronegativity difference between oxygen and the tetrel atom imparts a pronounced ylidic character to the E+–O− linkage, leading to an enhanced reactivity and reduced stability relative to singly bonded analogues.4 Therefore, heavier carbonyls tend to undergo oligo- and polymerization reactions, as exemplified by the differences between carbon and silicon dioxide, or by the polymer chemistry of silicones.5 Consequently, heavier carbonyls have long been considered elusive species.6–9
The rich chemistry of the CO moiety in organic compounds has spurred intense research efforts in recent years to isolate heavier carbonyl analogues and to elucidate their structural and chemical properties. After the first report of a donor–acceptor stabilized silacarbonyl compound (A, Fig. 1) by Driess in 2007, many examples of isolable acid/base coordinate silanones have been reported.10–13 Using the same approach, also isolable germanones could be generated,14 for instance Nagendran's amino-troponiminate-supported Lewis acid-base stabilized germanones B or Driess's Lewis base stabilized system C.14,15 Donor/acceptor free, three-coordinate silanones and germanones long remained inaccessible but could be isolated through kinetic stabilization by introduction of highly sterically demanding substituents such as in Tamao's germanone D,16 or through electronic stabilization by employing strong donor substituents as employed in the first room temperature stable silanone E reported by Kato et. al.17
 | ||
| Fig. 1 (A) Examples of isolated acid/base and three-coordinate silanones and germanones (Dipp = 2,6-diisopropylphenyl, DMAP = 4-dimethylaminopyridine), (B) reported compounds with short SnO bonds and structures of reported stannanones. (C) Structures of the first stannanone and Lewis acid–base stabilized stannanone reported in this work. | ||
Although many additional examples of isolable silanones18–24 and germanones25,26 have been reported until today, the situation is much different for their heavier analogues. Owing to the increasing electronegativity difference and weaker π-overlap when descending the group, examples of isolated compounds with Sn–O or Pb–O multiple bonds are extremely rare. In 2008, the Hahn group reported the heavier carbon monoxide congeners F, featuring a formal E = O double bond with tin and lead.27 However, the reported SnO bond length of 2.079 (2) Å was only slightly shorter than the Sn–O single bonds in F, arguing for only a weak π-interaction. Until today, this compound remains the only isolated species with a partial SnO multiple bond character. In the preparation phase of this manuscript, Matsuo and coworkers reported the structural characterization of the three-coordinate stannanone I with a short SnO bond of 1.864 (6) Å but failed to isolate the compound due to its high reactivity.28 Further attempts to access stannanones led to structures with Sn–O single bonds or completely different reaction products through intramolecular rearrangement reactions.29 For example, Aldridge and Mo et al. independently reported the synthesis of oxylstannylenes G (ref. 30) and H,31 respectively, through oxidation of stannylenes with nitrous oxide followed by migration of one substituent. Notably, both compounds featured shorter Sn–O bond lengths (approx. 2.027 Å) compared to F.
In recent years, our group has showcased the potential of ylide groups in isolating reactive main group compounds32 and promoting small molecule activation.33,34 Their strong donor ability has proven highly effective in stabilizing low-valent and cationic species.35–38 Building on this insight, we hypothesized that ylide-functionalization could enable the stabilization of a stannanone. Here, we report on the isolation of a diylidylstannanone stabilized by a thiophosphinoyl-tethered ylide and SnCl2 as additional Lewis acid.
Results and discussion
We began our studies with the oxidation of the recently reported diylidylstannylenes (YPS)2Sn and (YTS)2Sn (Fig. 2).33,39 However, treatment of the compounds with nitrous oxide repeatedly resulted in the formation of complex product mixtures including considerable amounts of free ylide. Therefore, we turned our attention towards monoylidylstannylenes of the type YSnR with a variable R substituent. The variable R group appeared advantageous for tuning ligand properties to enable a sufficient stabilization of a stannanone. | ||
| Fig. 2 Diylidylstannanones tested in the synthesis of the corresponding stannanones (Pip = piperidyl). | ||
We initially started oxidation experiments using the ylidylstannylenes YCNSnR [RCl (1a), N(SiMe3)2 (1b)] containing a cyano-substituted ylide (Scheme 1).40 We quickly had to rule out the chlorostannylene due to its poor solubility, which hindered further reactions. In contrast, the amido system 1b readily reacted with N2O to form a new product as confirmed by 31P{1H} NMR spectroscopy, which we could identify as the cyclic stannoxane 2-O (Scheme 1). A similar reactivity was observed with sulfur,41 for which the corresponding heavier carbonyl analogues are usually more easily accessible.42–48 Crystallization and single-crystal X-ray diffraction (XRD) analysis of the products 2-E revealed dimeric structures in the solid-state (Fig. 3, SI) with Sn–O (average: 2.002 (5) Å) and Sn–S bond lengths (average: 2.423 (1) Å) in the range of typical single bonds.49 For both the oxygen and sulfur compound, the isomer syn-2-E with both ylide groups facing the same side of the central four-membered ring was observed.
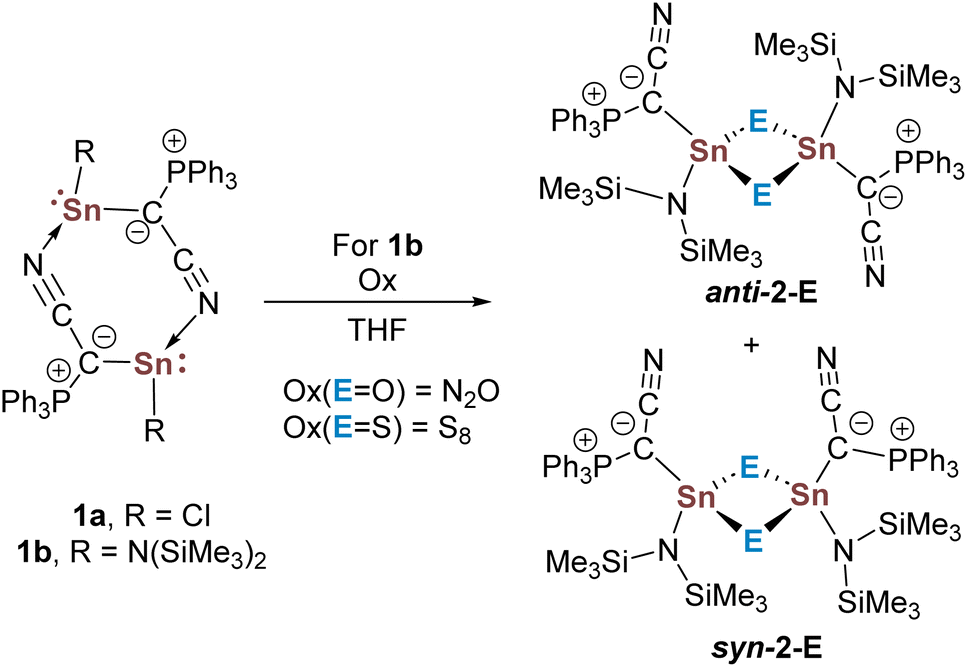 | ||
| Scheme 1 Formation of the cylic heavier stannoxane derivatives syn-2-E through oxidation of the dimeric stannylenes 1. | ||
 | ||
| Fig. 3 Molecular structure of syn-2-S, with thermal ellipsoids drawn at the 50% probability level. Hydrogen atoms omitted for clarity. Important bond lengths and angles as well as crystallographic details are given in the SI. | ||
Density functional theory (DFT) calculations on the B86/def2-TZVPP level of theory confirmed the preferred formation of the syn-isomers, which are energetically favored by 5.5 kcal mol−1 in the case of syn-2-O and by 6.2 kcal mol−1 in the case of syn-2-S. In addition, the dimers are clearly favored over the monomeric heavier ketones by more than 50 kcal mol−1 in each case (see SI for details).
To prevent the dimerization of the formed stannanone to the cyclic stannoxane, we turned our attention to an ylide system with increased steric bulk. The thiophosphinoyl substituted ligands 3a and 3d previously reported by our group appeared to be suitable candidates owing to their steric demand and ability to readily form mono- and diylide-substituted stannylenes.39,50,51 Recognizing that solubility could remain a challenge for the isolated stannylenes, we sought to develop similar ylide systems with enhanced solubility. Therefore, we also targeted the synthesis of cyclohexyl-substituted ylides 3b-c and 3e (Scheme 2). Although the synthesis of the fully cyclohexyl-substituted ylide 3e proceeded as planned, its reduced acidity prevented deprotonation to the metalated ylide, even when using strong metal bases such as benzyl potassium. In contrast, the unsymmetrical ylides 3b and 3c and their metalated congeners were readily accessible,52 and allowed the selective synthesis of stannylenes 4b and 4c. Attempts to oxidize these complexes with N2O to obtain the desired heavier acyl chlorides initially appeared successful. However, the obtained products rapidly decomposed via disproportionation, consistently yielding products containing tin(IV) chloride species (see below).
 | ||
| Scheme 2 Synthesis of chlorostannylenes 4 and oxidation with nitrous oxide to the Lewis acid base-stabilized stannanone 5 (Pip = piperidyl). | ||
Since isolating a stannanone using ylides 3b–3e proved unsuccessful, we shifted our focus to ylide 3a and its corresponding stannylene 4a. Initial experiments showed the successful synthesis of a new compound, whose selectivity, however, proved to be highly sensitive to the reaction conditions. To circumvent solubility issues with 4a and prevent decomposition, a precise control of reaction time and choice of solvent system was found to be decisive. The in situ formation of 4avia deprotonation of 3a with BnK in toluene, followed by reaction with 1 equiv. SnCl2, and subsequent addition of THF prior to exposure to N2O proved to be the optimal reaction conditions. In contrast, excess SnCl2 led to the formation of SnCl3 species. Strict anhydrous conditions were critical, as even trace amounts of water in N2O prevented any selective product formation. Under the optimal conditions the product crystallized directly from the reaction mixture and could be isolated as colorless solid in yields between 85 and 93%. XRD analysis revealed the product to be the diylide-substituted stannanone 5 stabilized by SnCl2 as Lewis acid (Fig. 3). The formation of 5 indicates that the oxidation of 4a proceeds through the stannylene-stabilized chlorostannanone intermediate 5-Int, which undergoes exchange of an ylide and chloride ligand to yield 5. Consequently, stannanone 5 cannot be accessed through the oxidation of (YPS)2Sn (Fig. 2) in the presence of SnCl2. All oxidation attempts starting from the diylidylstannylene have proven to be highly unselective.
The stannanone proved to be stable at room temperature under an inert atmosphere for extended periods (at least 6 months) and also showed extended stability in non-protic solvents. Even when a solution of 5 in CDCl2 was heated for three days at 70 °C, no decomposition was observed. Nonetheless, attempts to scale up the reaction from mg scale in a J-Young NMR tube to 20 mL Schlenk tubes remained unsuccessful.
5 is characterized by two doublets in the 31P{1H} NMR spectrum at 29.8 (Ph2PS) and 41.3 ppm (Ph2PPip) with a small coupling constant of 2JPP = 3.5 Hz and the expected signals for the phenyl and piperidyl protons in the 1H NMR spectrum. Further analysis in solution was unfortunately hampered by the extremely low solubility in common aprotic solvents. However, the purity of the crystalline compound was unambiguously confirmed by elemental analysis. In the solid-state structure (Fig. 4), the Sn1–O1 bond length of 1.954 (3) Å is slightly shorter than the Sn2–O1 bond (1.984 (4) Å), suggesting that 5 is more accurately described as a SnCl2-stabilized stannanone, rather than as a heavier analogue of phosgene (form 5c, Fig. 4e). Importantly, both bonds are clearly shorter than the Sn–O bonds in compounds F–H, but longer than in Matsuo's Lewis acid base-free stannanone I (1.89 Å). The Sn1–O1–Sn2 angle in compound 5 of 134.9 (2)° is in line with analogous compounds, such as the respective Ge–O–Sn angle of 136.10 (3)° in the SnCl2-stabilized germanone B. Interestingly, the Sn–C distance (2.126 (3) Å) in the stannanone is shortened compared to the chlorostanylene precursor 4a (2.202 (2) Å) as a consequence of the oxidation of the tin center and an increased shift of electron density from the ylide ligand to tin. As a result, the electrostatic interactions in the ylide backbone are weakened, as expressed in a slight elongation of the P–C bonds (e.g. P1–C1: from 1.683 (2) Å in 4a to 1.700 (3) Å in 5).
 | ||
| Fig. 4 Molecular structure of 5 with thermal ellipsoids drawn at 50% probability level. Hydrogen atoms are omitted for clarity. Important bond lengths [Å] and angles [°]: Sn1–O1 1.955 (4), O1–Sn2 1.984 (4), C1–Sn1 2.126 (3), C1–P1 1.700 (3), C1–P2 1.723 (3), Sn1–O1–Sn2 134.9 (2), P–C–P 128.5 (2). | ||
To gain insights into the bonding situation, DFT calculations on the BP86/def2-TZVPP//BP86/def2-svp level of theory were performed for 5 and the SnCl2-free stannanone 5′ (see the ESI for details). The optimized structure of 5 closely matches the experimental values obtained from single crystal XRD analysis (Fig. 4 and Table 1) and confirms the shorter Sn–O bond to the ylide-substituted tin centre. The Sn1–O bond only shortens slightly in the Lewis acid-free analogue 5′, a trend also observed for Matsuo's stannanone I. Analysis of the frontier molecular orbitals shows that the highest occupied molecular orbital (HOMO) of 5 is primarily localized on the SnCl2 moiety, corresponding to the lone pair at the tin centre and confirming its role as a Lewis acid (Fig. 5). HOMO-1 and HOMO-2 are localized on the ylidic carbon atoms. No π-symmetric molecular orbital indicative for a multiple bond between tin and oxygen was found.
| 5 | 5′ | I-SnCl2 | I | |
|---|---|---|---|---|
| d(Sn1–O) [Å] | 1.96 | 1.89 | 1.93 | 1.89 |
| d(Sn2–O) [Å] | 2.10 | — | 2.14 | — |
| Sn1–O–Sn2 [°] | 126.3 | — | 140.2 | — |
| ρ(BCP Sn1–O) | 0.12 | 0.14 | 0.13 | 0.14 |
| ρ(BCP Sn2–O) | 0.08 | — | 0.08 | — |
| ε(BCP Sn1–O) | 0.03 | 0.09 | 0.03 | 0.01 |
| ε(BCP Sn2–O) | 0.03 | — | 0.02 | — |
| MBI (Sn1–O) | 0.87 | 1.61 | 0.94 | 1.52 |
 | ||
| Fig. 5 Display of the Kohn–Sham orbitals in (a) 5 and in (b) 5′ as well as calculated NPA charges and Mayer bond indices in (c) 5 and (d) 5′. Possible Lewis structures of 5. (e) Canonical structures of 5. | ||
Natural bond orbital (NBO) analysis yielded a highly ionic picture of the bonding situation with predominantly electrostatic interactions within the Sn1–O–SnCl2 scaffold. Orbital interactions within this linkage are exclusively represented in the second order perturbation theory analysis, which yielded additional stabilizing O → Sn donor–acceptor interactions from the oxygen centre to both tin centres. In contrast, intrinsic bond orbital (IBO) analysis showed σ-bond orbitals for the Sn–O bonds, but no π-bonding orbitals. Accordingly, low Mayer bond indices (MBI) of 0.87 for the Sn1–O1 bond and 0.65 for Sn2–O1 bond are obtained. Additionally, the Sn–O–Sn linkage features strongly opposing charges, with a higher positive charge at Sn1 (q = 2.18) than at Sn2 centre (q = 1.16), supporting the oxidation of the Sn1 center and the interpretation of 5 as an SnCl2 stabilized stannanone, heavier ylide 5a or as zwitterionic stannoxane represented by the Lewis structure 5b (Fig. 5e). A high bond polarization is also found in the SnCl2-free stannanone 5′. Despite a significantly increased MBI for the SnO bond of 1.61, the IBO analysis of 5′ also shows no π-bonding orbital and both the HOMO and HOMO-1 are localized on the oxygen atom.
Next, we also examined the electronic structure by quantum theory of atoms in molecules (QTAIM) calculations,53 which revealed bond critical points (BCPs) for the Sn1–O linkage with low ellipticity values of ε = 0.03 for 5 and ε = 0.09 for 5′, indicative of single bonds (Fig. 6). Interestingly, analysis of the Laplacian profile along both Sn–O bond paths in 5 shows only one pronounced valence-shell charge concentration (VSCC) minimum close to the electronegative oxygen atom and a mere shoulder for the Sn VSCC. Both VSCCs reside in the atom basin of the oxygen atom, indicating a dative O → Sn interaction for both Sn–O bonds as suggested by the NBO analysis.54
 | ||
| Fig. 6 Topological analysis of 5. (a) Contour plot of the Laplacian of electron density in 5. BCPs of the Sn1–O–Sn2 linkage are shown as blue dots with the respective values for the charge density ρ and ellipticity ε of the electron density. (b) 1D Laplacian profiles of the Sn1–O1 bond-path. BCPs are shown as red dots. | ||
To examine whether this bonding situation is unique to our diylidylstannanone, we conducted comparative electronic structure analyses of Matsuo's stannanone I (including its Lewis acid adduct with SnCl2) and the Lewis acid stabilized germanone B reported by Nagendran et al. (see SI).15 For compound I, a higher MBI of 1.52 was calculated; however, this value reduces to 0.94 upon SnCl2 coordination, mirroring the trend observed for compound 5 and 5′. Topological analysis of I further reveals bond parameters comparable to those of 5′, albeit with an even lower ellipticity for the Sn1–O1 bond of ε = 0.01. Overall, the calculations demonstrate that the π-contribution to the bonding between the tin and the oxygen centre in the stannanones is negligible. The Sn–O bond is best described by an ylidic interaction with a polar single bond which shortens relative to standard Sn–O single bonds due to additional electrostatic interactions (structure 5a and 5b, Fig. 4).
In line with the highly polar Sn–O bond, stannanone 5 displays pronounced sensitivity toward trace amounts of water. Initial screening reactions towards small molecules, which at first appeared selective based on NMR analysis, were later attributed to unintended side reactions with residual water present in the starting materials. Careful drying of the reactants eliminated these side reactions and completely suppressed the reactivity. In the absence of water, 5 was found to be stable in the solid state for several months without any detectable decomposition.
In general, 5 was found to be highly reactive towards various substrates (e.g. CO, CO2, S8, CS2), but usually reacted in an uncontrollable fashion producing multiple products. Attempts to remove (addition of bases such as NH3, dmap) or exchange the SnCl2 Lewis acid (e.g. with GeCl2, ZnF2, B(C6F6)3) always resulted in decomposition. The only transformation, from which a clean product could be isolated, was achieved upon treatment with AgBF4 as halide abstraction reagent. This reaction led to the formation of the bisylide-substituted tin chloride cation 6, whose BF4 salt was generated with high selectivity but could not be isolated in pure form due to Sn–C bond cleavage occurring during workup (Scheme 3 and Fig. 7). Interestingly, 6 was also formed in many screening reactions, in which water was present, and could be crystallized as its SnCl62− salt. For example, the reaction proceeded highly selectively and rapidly with benzaldehyde in CDCl3. In addition to compound 6, ylide-substitute SnCl3 was also obtained from some of the reaction attempts (see the ESI), further underscoring the tendency of the stannanone to undergo Sn–O bond cleavage. In general, the high sensitivity toward trace amounts of water and the formation of the Sn–O bond cleavage products support the interpretation of the Sn–O bond as a highly polar ylidic bond.
 | ||
| Scheme 3 Reaction of stannanone 5 with AgBf4 and H2O (Pip = piperidyl). | ||
 | ||
| Fig. 7 Molecular structure of the BF4− salt of 6. Thermal ellipsoids drawn at 50% probability level. Hydrogen atoms omitted for clarity. Important bond lengths and angles as well as crystallographic details are given in the SI. | ||
Conclusions
To conclude, we have synthesized the first isolable monomeric Lewis acid base-stabilized stannanone formed through an oxidation-induced rearrangement in an ylide-substituted chlorostannylene. Although the stannanone exhibits a short Sn–O bond length of 1.95 Å, calculational studies revealed that there is negligible π-contribution to the bonding. Overall, the Sn–O bond was found to be highly polar, with a significant electrostatic component that accounts for its shortened bond length. As a results of this bonding situation, the stannanone is extremely sensitive to trace amounts of water and often reacts an uncontrollable fashion. Experimental observations indicate that the Sn–O bond undergoes facile cleavage, as evidenced by the formation of a tin cation. These findings highlight the challenges associated with stabilizing compounds of heavier elements featuring multiple bonds, especially when such bonds involve elements from different periods of the periodic table with markedly different properties.Author contributions
V. H. G. designed and oversaw the project. M. J. planned the study and carried out most of the synthetic work, analysed the spectroscopic data. D. K. performed all computational studies. M. K. synthesized compound 2-O and helped with crystal refinement of 2-S. V. S. V. S. N. S. synthesized compound 2-S. The manuscript was written by M.J. and D. K. and finalized by V. H. G.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2481150 (4a-SnCl3), 2481151 (4c+N2O), 2481152 (6-SnCl6), 2481153 (6-BF4), 2481154 (2-S) and 2481155 (5) contain the supplementary crystallographic data for this paper.55a–fThe data that support the findings of this study are available in the supporting information (SI). Supplementary information: this includes experimental procedures, NMR and IR spectra as well as crystallographic and computational details. See DOI: https://doi.org/10.1039/d5sc06549f.
Acknowledgements
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy—EXC-2033–390677874—RESOLV, and the European Union (ERC, CarbFunction, 101086951). Views and opinions expressed are however those of the authors only and do not necessarily reflect those of the European Union or the European Research Council. Neither the European Union nor the granting authority can be held responsible for them. The authors would like to thank Dr Kai-Stephan Feichtner for help regarding sc-XRD data refinement and analysis.Notes and references
- Y. Xiong, S. Yao and M. Driess, Angew. Chem., Int. Ed., 2013, 52, 4302–4311 CrossRef CAS.
- R. Okazaki and N. Tokitoh, Acc. Chem. Res., 2000, 33, 625–630 CrossRef CAS PubMed.
- Y. K. Loh and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 8626–8648 CrossRef CAS.
- R. Jaquet, W. Kutzelnigg and V. Staemmler, Theor. Chim. Acta, 1979, 54, 205–227 CrossRef.
- S. S. Nekrashevich and V. A. Gritsenko, Phys. Solid State, 2014, 56, 207–222 CrossRef CAS.
- N. Tokitoh, T. Matsumoto and R. Okazaki, Bull. Chem. Soc. Jpn., 1999, 72, 1665–1684 CrossRef CAS.
- R. J. Glinski, J. L. Cole and D. A. Dixon, J. Am. Chem. Soc., 1985, 107, 5891–5894 CAS.
- R. Withnall and L. Andrews, J. Am. Chem. Soc., 1985, 107, 2567–2568 CAS.
- C. A. Arrington, R. West and J. Michl, J. Am. Chem. Soc., 1983, 105, 6176–6177 CAS.
- Y. Xiong, S. Yao and M. Driess, Dalton Trans., 2010, 39, 9282–9287 CAS.
- S. Yao, M. Brym, C. van Wüllen and M. Driess, Angew. Chem., Int. Ed., 2007, 46, 4159–4162 CrossRef CAS PubMed.
- Y. Xiong, S. Yao, R. Müller, M. Kaupp and M. Driess, J. Am. Chem. Soc., 2010, 132, 6912–6913 CrossRef CAS.
- Y. Xiong, S. Yao, R. Müller, M. Kaupp and M. Driess, Nat. Chem., 2010, 2, 577–580 CrossRef CAS PubMed.
- S. Yao, Y. Xiong and M. Driess, Chem. Commun., 2009, 42, 6466–6468 RSC.
- S. Sinhababu, D. Yadav, S. Karwasara, M. K. Sharma, G. Mukherjee, G. Rajaraman and S. Nagendran, Angew. Chem., Int. Ed., 2016, 55, 7742–7746 CrossRef CAS.
- L. Li, T. Fukawa, T. Matsuo, D. Hashizume, H. Fueno, K. Tanaka and K. Tamao, Nat. Chem., 2012, 4, 361–365 CAS.
- I. Alvarado-Beltran, A. Rosas Sanchez, A. Baceiredo, N. Saffon-Merceron, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2017, 56, 10481–10485 CAS.
- D. Wendel, D. Reiter, A. Porzelt, P. J. Altmann, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 17193–17198 CAS.
- A. Rosas-Sanchez, I. Alvarado Beltran, A. Baceiredo, N. Saffon-Merceron, D. Hashizume, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2017, 56, 15916–15920 Search PubMed.
- S. U. Ahmad, T. Szilvasi, E. Irran and S. Inoue, J. Am. Chem. Soc., 2015, 137, 5828–5836 CAS.
- R. S. Ghadwal, R. Azhakar, H. W. Roesky, K. Pröpper, B. Dittrich, S. Klein and G. Frenking, J. Am. Chem. Soc., 2011, 133, 17552–17555 CAS.
- R. Rodriguez, T. Troadec, D. Gau, N. Saffon-Merceron, D. Hashizume, K. Miqueu, J.-M. Sotiropoulos, A. Baceiredo and T. Kato, Angew. Chem., Int. Ed., 2013, 52, 4426–4430 Search PubMed.
- Y. Wang, M. Chen, Y. Xie, P. Wei, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, Nat. Chem., 2015, 7, 509–513 CAS.
- D. C. H. Do, A. V. Protchenko, M. A. Fuentes, J. Hicks, E. L. Kolychev, P. Vasko and S. Aldridge, Angew. Chem., Int. Ed., 2018, 57, 13907–13911 CrossRef CAS.
- M. K. Sharma, S. Sinhababu, P. Mahawar, G. Mukherjee, B. Pandey, G. Rajaraman and S. Nagendran, Chem. Sci., 2019, 10, 4402–4411 RSC.
- X.-X. Zhao, T. Szilvási, F. Hanush and S. Inoue, Chem. Eur J., 2021, 27, 15914–15917 CrossRef CAS PubMed.
- A. V. Zabula, T. Pape, A. Hepp, F. M. Schappacher, U. C. Rodewald, R. Pöttgen and F. E. Hahn, J. Am. Chem. Soc., 2008, 130, 5648–5649 CrossRef CAS PubMed.
- T. Tanikawa, Y. Ueno, S. Konaka, Y. Numata, M. Imai, K. Ota, H. Fueno, K. Tanaka, D. Hashizume and T. Matsuo, Eur. J. Inorg. Chem., 2025, 20, e202401847 CAS.
- N. Parvin, N. Sen, P. V. Muhasina, S. Tothadi, P. Parameswaran and S. Khan, Chem. Commun., 2021, 57, 5008–5011 RSC.
- Y. Wang, A. E. Crumpton, M. A. Ellwanger, C. McManus and S. Aldridge, Angew. Chem., Int. Ed., 2024, 63, e202402795 CrossRef CAS.
- B. Lei, F. Cao, M. Cheng, X. Wang and Z. Mo, J. Am. Chem. Soc., 2024, 146, 17817–17826 Search PubMed.
- A. Sarbajna, V. S. V. S. N. Swamy and V. H. Gessner, Chem. Sci., 2021, 12, 2016–2024 RSC.
- C. Mohapatra, L. Scharf, T. Scherpf, B. Mallick, K.-S. Feichtner, C. Schwarz and V. H. Gessner, Angew. Chem., Int. Ed., 2019, 58, 7459–7463 CrossRef CAS.
- R. J. Ward, M. Jörges, H. Remm, E. Kiliani, F. Krischer, Q. Le Dé and V. H. Gessner, J. Am. Chem. Soc., 2024, 146, 24602–24608 CrossRef CAS.
- T. Scherpf, K.-S. Feichtner and V. H. Gessner, Angew. Chem., Int. Ed., 2017, 129, 3323–3327 CrossRef.
- C. Mohapatra, H. Darmandeh, H. Steinert, B. Mallick, K.-S. Feichtner and V. H. Gessner, Chem. Eur J., 2020, 26, 15145–15149 CrossRef CAS PubMed.
- T. Stalder, F. Krischer, H. Steinert, P. Neigenfind and V. H. Gessner, Chem. Eur J., 2022, 28, e202104074 CrossRef CAS PubMed.
- I. Alvarado-Beltran, A. Baceiredo, N. Saffon-Merceron, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2016, 55, 16141–16144 CAS.
- M. Jörges, A. Kroll, L. Kelling, R. Gauld, B. Mallick, S. M. Huber and V. H. Gessner, ChemistryOpen, 2021, 10, 1089–1094 Search PubMed.
- V. S. V. S. N. Swamy, M. Kumar, F. Krischer, K.-S. Feichtner, B. Mallick and V. H. Gessner, Z. Anorg. Allg. Chem., 2024, 650, e202400079 CAS.
- N. Tokitoh, M. Saito and R. Okazaki, J. Am. Chem. Soc., 1993, 115, 2065–2066 CAS.
- M. Saito, N. Tokitoh and R. Okazaki, Organometallics, 1996, 15, 4531–4536 CrossRef CAS.
- M. Saito, N. Tokitoh and R. Okazaki, J. Am. Chem. Soc., 2004, 126, 15572–15582 CrossRef CAS.
- Y. Zhou and D. S. Richeson, J. Am. Chem. Soc., 1996, 118, 10850–10852 CrossRef CAS.
- P. B. Hitchcock, J. Hu, M. F. Lappert and J. Severn, Dalton Trans., 2004, 24, 4193–4201 RSC.
- P. B. Hitchcock, E. Jang and M. F. Lappert, J. Chem. Soc., Dalton Trans., 1995, 19, 3179–3187 RSC.
- P. B. Hitchcock, M. F. Lappert, L. J.-M. Pierssens, A. V. Protchenko and P. G. H. Uiterweerd, Dalton Trans., 2009, 23, 4578–4585 RSC.
- S. M. Mansell, C. A. Russell and D. F. Wass, Dalton Trans., 2015, 21, 9756–9765 RSC.
- P. Pyykkö and M. Atsumi, Chem. Eur J., 2009, 15, 12770–12779 CrossRef.
- M. Jörges, R. M. Gauld, H. Steinert, L. Kelling, V. S. V. S. N. Swamy, A. Kroll, B. Mallick and V. H. Gessner, Chem. Eur J., 2023, 29, e202203863 CrossRef.
- M. Jörges, F. Krischer and V. H. Gessner, Science, 2022, 378, 1331–1336 CrossRef.
- F. Krischer, M. Jörges, T.-F. Leung, H. Darmandeh and V. H. Gessner, Angew. Chem., Int. Ed., 2023, 62, e202309629 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- A. E. Ferao, A. G. Alcaraz, S. Z. Noguera and R. Streubel, Inorg. Chem., 2020, 59, 12829–12841 CrossRef.
- (a) CCDC 2481150, Experimental Crystal Structure Determination 2025 DOI:10.5517/ccdc.csd.cc2p8v3y ; (b) CCDC 2481151, Experimental Crystal Structure Determination 2025 DOI:10.5517/ccdc.csd.cc2p8v4z ; (c) CCDC 2481152, Experimental Crystal Structure Determination 2025 DOI:10.5517/ccdc.csd.cc2p8v50 ; (d) CCDC 2481153, Experimental Crystal Structure Determination 2025 DOI:10.5517/ccdc.csd.cc2p8v61 ; (e) CCDC 2481154, Experimental Crystal Structure Determination 2025 DOI:10.5517/ccdc.csd.cc2p8v72 ; (f) CCDC 2481155, Experimental Crystal Structure Determination 2025 DOI:10.5517/ccdc.csd.cc2p8v83 .
| This journal is © The Royal Society of Chemistry 2025 |