
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and antimicrobial applications of α-peptoid polymers
Jiayang
Xie
 a,
Weilong
Hu
b,
Xi
Feng
b,
Zixin
Liu
b,
Min
Zhou
*bc and
Runhui
Liu
*abc
a,
Weilong
Hu
b,
Xi
Feng
b,
Zixin
Liu
b,
Min
Zhou
*bc and
Runhui
Liu
*abc
aSuzhou Institute of Biomedical Engineering and Technology, Chinese Academy of Sciences, Suzhou 215163, China
bState Key Laboratory of Bioreactor Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: rliu@ecust.edu.cn; minzhou@ecust.edu.cn
cShanghai Frontiers Science Center of Optogenetic Techniques for Cell Metabolism, Frontiers Science Center for Materiobiology and Dynamic Chemistry, Engineering Research Center for Biomedical Materials of Ministry of Education, Key Laboratory for Ultrafine Materials of Ministry of Education, Key Laboratory of Specially Functional Polymeric Materials and Related Technology (Ministry of Education), School of Materials Science and Engineering, East China University of Science and Technology, Shanghai 200237, China
First published on 4th July 2025
Abstract
The rapid evolution of drug-resistant pathogens and the lag in antibiotic development pose a severe threat to global public health. Host defense peptides (HDPs) have emerged as promising candidates due to their broad-spectrum antimicrobial activity and low resistance tendency. However, their practical application is hindered by poor proteolytic stability and high costs. Peptoids are ideal HDP mimics, as their characteristic side chain relocation from α-carbons to backbone nitrogen atoms confers superior proteolytic resistance. Nevertheless, their solid-phase synthesis remains inefficient and difficult to scale up. Recent advances in polymer chemistry enable the efficient synthesis of α-peptoid polymers, offering a promising platform for antimicrobial materials development. This perspective summarizes the progress in α-peptoid polymers research, focusing on monomer synthesis, polymerization reaction, and antimicrobial applications. We discuss their potential in the antimicrobial field and propose perspectives on current challenges and future directions, aiming to inspire further advances in the development of α-peptoid polymer-based antimicrobials with clinical application potential.
 Jiayang Xie | Jiayang Xie received his PhD degree from the East China University of Science and Technology (ECUST) in 2025. He is now a postdoctoral fellow in Suzhou Institute of Biomedical Engineering and Technology, Chinese Academy of Sciences. His current research interests focus on peptide-mimicking antimicrobial polymeric materials. |
 Min Zhou | Min Zhou received his PhD degree from the ECUST in 2021. He is now an associate professor at ECUST. His current research interests focus on antimicrobial polymer biomaterials. |
 Runhui Liu | Runhui Liu obtained his PhD degree in organic chemistry in 2009 from Purdue University. Afterward, he took postdoctoral training at the California Institute of Technology and the University of Wisconsin–Madison during 2010–2014. At the end of 2014, he took a professor position at the School of Materials Science and Engineering, ECUST. His current research focuses on peptide polymer-based biomaterials for antimicrobial and tissue engineering applications. |
1. Introduction
The rapid evolution and widespread dissemination of drug-resistant pathogens, coupled with the scarcity of novel antibiotics, have created therapeutic challenges for numerous common infectious diseases, posing a significant threat to global public health security.1,2 The World Health Organization predicted that without effective interventions, annual deaths from drug-resistant microbial infections could reach 10 million by 2050.3 Host defense peptides, which are important components of the innate immune system in organisms, have been widely studied as promising candidates in the post-antibiotic era due to their broad-spectrum antimicrobial activity and low propensity to induce microbial resistance.4–6 However, the inherent limitations of HDPs such as susceptibility to hydrolysis by proteases, poor in vivo stability, and high production costs have hindered their clinical application.5,7,8To overcome the inherent shortcomings of natural peptides, researchers have developed a series of HDP mimics.9–19 Among them, peptoids are considered ideal HDP mimics as they fundamentally eliminate the enzymatic cleavage site of the peptide bond by relocating the side-chain modification site from the α-carbon of the conventional peptide to the backbone amide nitrogen atom,20–22 thereby exhibiting remarkable proteolytic stability.23,24 This structural alteration also abolishes inter- and intramolecular chain hydrogen bonding, simplifying peptoid properties to primarily depend on side-chain composition and sequence arrangement, which significantly reduces engineering and design complexity.25–27 However, the preparation of peptoids typically requires stepwise solid-phase synthesis, which is cumbersome, time-consuming, costly, and difficult to scale up (limited to milligram-scale production) while also imposing constraints on repeat unit length (rarely exceeding 50 residues).27–30 Recent advances in polymer chemistry have opened new avenues for peptoid preparation, as polymerization strategies enable the production of higher yields and higher-molecular-weight polypeptoids.29,31 This facile synthetic approach establishes polypeptoids as a highly tunable platform for rapid polymer prototyping, demonstrating great potential for applications in antimicrobial materials, biomedicine, and other fields.32–34
In this perspective, we review recent advances in α-peptoid polymers research, with a focus on three core aspects, including monomer preparation, polymerization reaction, and antimicrobial design and applications (Fig. 1). Through in-depth discussion of these critical areas, we provide a further outlook on the current challenges and future research directions in the field. By reviewing the existing research results, this work aims to offer perspectives that may contribute to the development of novel antimicrobial materials with both efficient antimicrobial activity and clinical translation potential.
 | ||
| Fig. 1 This perspective summarizes the recent advances in α-peptoid polymers research, focusing on monomer synthesis, polymerization reaction, and antimicrobial application. | ||
2. Synthesis of monomers
The monomer type determines the polymerization conditions, fundamental structures, and functions of α-peptoid polymers. Currently commonly used monomers include N-substituted N-carboxyanhydride (NNCA), N-substituted N-thiocarboxyanhydride (NNTA), and N-phenoxycarbonyl N-substituted glycines (NNPC), each possessing unique chemical structural characteristics (Fig. 2).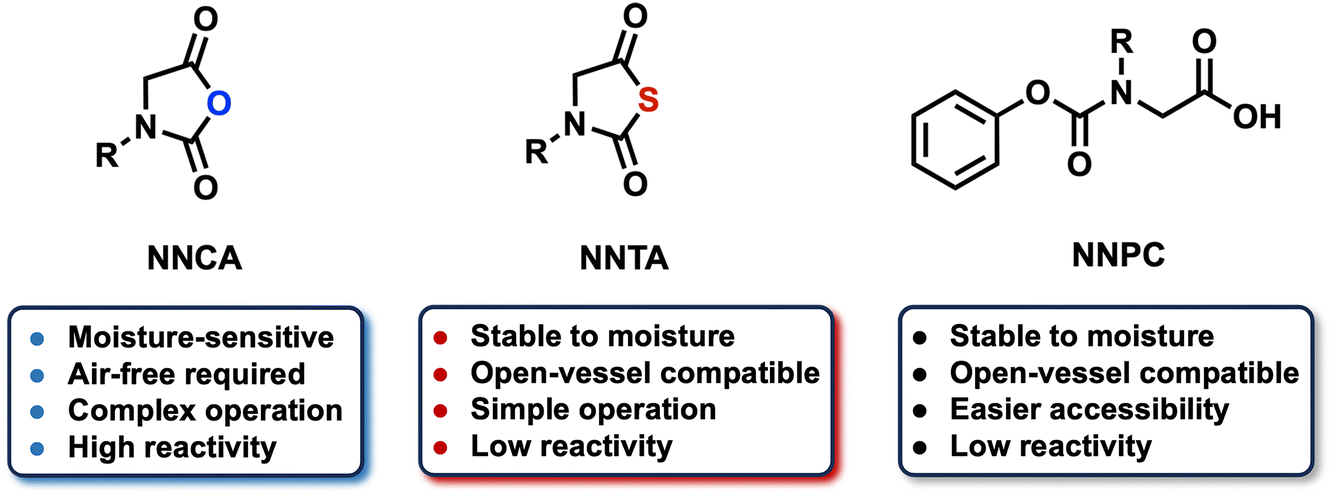 | ||
| Fig. 2 Types of monomers for α-peptoid polymers. | ||
2.1 Synthesis of NNCA
The preparation of NNCAs predominantly is based on the synthetic methods for N-carboxyanhydrides (NCAs). Hermann Leuchs prepared NCA monomers in 1906 through the reaction of N-alkoxycarbonyl amino acids with halogenating agents (e.g., PBr3, PCl3, PCl5, or SOCl2) under vacuum at 50–70 °C.35 Notably, this classic Leuchs method is also one of the most popular synthetic approaches for NNCA monomers (Fig. 3).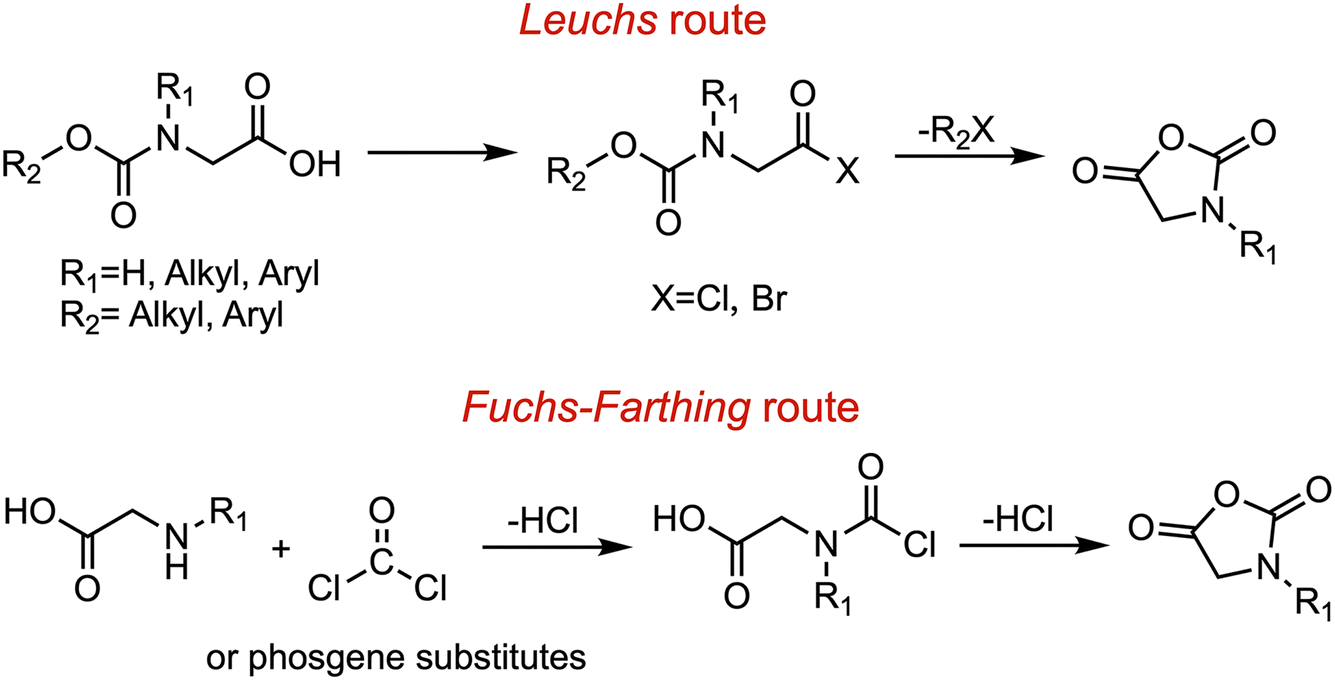 | ||
| Fig. 3 Synthesis of N-substituted N-carboxyanhydrides. | ||
The Fuchs–Farthing route represents an alternative method for the synthesis of NNCA monomers.36,37 This approach involves the reaction of N-substituted glycines with phosgene or non-gaseous phosgene substitutes, such as diphosgene or triphosgene, under heating conditions. Specifically, triphosgene first reacts with nucleophiles to generate phosgene in situ, which then reacts with the N-substituted glycine to form a highly reactive chloride intermediate. Subsequently, this intermediate undergoes an intramolecular cyclization to yield the NNCA monomer (Fig. 3). The phosgene generated during the reaction is rapidly consumed. However, this method is limited by the inevitable generation of hydrogen chloride (HCl) as a byproduct and incompatibility with acid-sensitive functional groups.
Acid scavengers such as triethylamine, (+)-limonene,38 and α-pinene39 can be employed to neutralize HCl generated during NNCA synthesis, thereby mitigating monomer decomposition and suppressing side reactions. However, these scavengers still exhibit limitations; for instance, the neutralization reactions of α-pinene are slow and incomplete.40 In recent years, advances have been made in HCl scavenging strategies. Barz and co-workers developed an efficient approach utilizing triethyloxonium tetrafluoroborate to covalently incorporate chloride into volatile byproducts, enabling effective chloride removal upon a single step.41 On the other hand, Lu et al. employed propylene oxide or epichlorohydrin as cost-effective HCl scavengers.40 This strategy not only prevents acid-catalyzed decomposition of NCAs and NNCAs under humid conditions but also facilitates ring closure by lowering the reaction energy barrier.
However, the use of highly toxic phosgene and its derivatives to synthesize NNCA has been mostly limited to laboratory-scale production. Additionally, NNCA monomers generally exhibit pronounced moisture sensitivity, necessitating rigorously anhydrous conditions throughout the synthesis process. Purification prior to use and storage is also essential, as impurities can significantly compromise the storage stability and polymerization reactivity of NNCA. Consequently, there is an urgent demand for more stable monomers and more facile synthetic approaches.
2.2 Synthesis of NNTA
NNTA is a thio analog of NNCA, where the oxygen in the five-membered ring is replaced by sulfur. The synthetic methodology developed by Kricheldorf et al. is the most commonly used approach in NNTA preparation, owing to its high yields and low requirement for toxic reagents such as the avoidance of phosgene derivatives.42 In this method, N-substituted glycines react with S-ethoxythiocarbonyl mercaptoacetic acid (XAA) in an alkaline aqueous solution to form the intermediate S-ethoxythiocarbonyl N-substituted glycines. Subsequently, in the presence of phosphine halides, the intermediates were further cyclized to yield the corresponding NNTA monomers (Fig. 4). | ||
| Fig. 4 Synthesis of N-substituted N-thiocarboxyanhydride according to the method developed by Kricheldorf. | ||
Compared with NNCA monomers, NNTA monomers are more stable, relatively insensitive to water, and easy to store. Furthermore, the synthesis and purification of NNTA monomers do not require strict anhydrous or oxygen-free operation, and can be performed in an open environment.43 The enhanced stability of NNTA arises from two factors: first, the sulfur atom in NNTA has lower electronegativity than the oxygen atom in NNCA, resulting in reduced nucleophilicity of the C5 carboxyl carbon; second, compared to CO2, carbonyl sulfide (COS) is more difficult to be released during the polymerization process.44
2.3 Synthesis of NNPC
NNPC are a class of protected amino acid derivatives that can be used in the synthesis of polypeptoids. Compared with NNCA and NNTA, NNPC offers the advantage of easier accessibility. This monomer can be prepared, purified, and stored long-term under ambient conditions while exhibiting good tolerance toward nucleophiles such as water and hydroxyl groups during polymerization.45Schlaad et al. extended the NPC synthesis approach to N-alkylglycines, synthesizing N-phenoxycarbonyl-sarcosine by a two-step reaction of sarcosine with tetrabutylammonium hydroxide and diphenyl carbonate (DPC) (Fig. 5A).46 Recently, Liu et al. reported a two-phase reaction system using methyl tert-butyl ether (MTBE) and water as solvents, which effectively avoids side reactions and enables the efficient synthesis of NPC and NNPC monomers.47 A variety of NNPC monomers have been prepared using this synthetic approach, including Sar-NPC, N-ethylglycine NPC (NEG-NPC), and N-butylglycine NPC (NBG-NPC) (Fig. 5B).45
 | ||
| Fig. 5 (A) The synthesis route of Sar-NPC used by Schlaad et al. (B) The synthesis route of Sar-NPC used by Liu et al. | ||
3. Polymerization of α-peptoid polymers
3.1 NNCA polymerization
NNCA ring-opening polymerization (ROP) is a facile and widely used method for synthesizing α-peptoid polymers, enabling the preparation of high-molecular-weight α-peptoid polymers with diverse structures and functionalities (Table 1). In this polymerization, primary amines are the most commonly employed nucleophilic initiators. Additionally, other initiators such as N-heterocyclic carbenes (NHCs), 1,8-diazabicycloundec-7-ene (DBU), and lithium hexamethyldisilazide (LiHMDS) can also be utilized for NNCA polymerization.| Monomer | Initiator | Catalyst | Topological structure | Characteristics |
|---|---|---|---|---|
| a In THF solvent, Li/Na/KHMDS can initiate the ROP of NNCAs to generate linear α-peptoid polymers. b In DMF solvent, LiHMDS can initiate the REP of NNCAs to generate cyclic α-peptoid polymers. | ||||
| NNCA | Primary amine | Linear | The polymerization proceeds via the normal amine mechanism (NAM) mechanism, exhibiting characteristics of living polymerization and enabling controlled synthesis of α-peptoid polymers. Polymerization requires purified monomers and anhydrous conditions | |
| Carboxylic acid | Carboxylic acid promotes proton transfer by acting as hydrogen bond donors and acceptors, thereby accelerating the polymerization rate and enabling the preparation of ultra-high molecular weight polysarcosine (chain lengths up to 8000) | |||
| 1,3-Bis[3,5-bis(trifluoromethyl)phenyl]urea (U–O) | U–O activates the carbonyl group on the NNCA ring through hydrogen-bonding interactions, thereby significantly accelerating the ROP rate of NNCAs. This catalytic strategy is particularly suitable for inactive NNCAs bearing bulky side chains | |||
| Alcohol | 1,1,3,3-Tetramethylguanidine (TMG) | TMG interacts with alcohols via hydrogen bonding, enhancing the nucleophilicity of the alcohol toward the ring-opening addition of NNCA monomers and thereby initiating chain growth | ||
| Li/Na/KHMDSa | Liner α-peptoid polymers were generated by ROP of NNCAs in THF. The rate of polymerization significantly increased compared to that initiated by primary amines, especially for NNCAs with bulky N-substitution group | |||
| NHC | Cyclic | In low dielectric constant solvents (such as THF and toluene), the quasi-living polymerization behavior is achieved by the ZROP mechanism. In polar solvents (such as DMF and DMSO), the polymerization is severely affected by side reactions | ||
| DBU | The polymerization proceeds via a ZROP mechanism to produce cyclic α-peptoid polymers. DBU has excellent robustness and availability | |||
| LiHMDSb | Cyclic α-peptoid polymers were generated by ring-expansion polymerization (REP) of NNCAs in DMF solvent | |||
| NNTA | Primary amine | Linear | By optimizing the polymerization conditions, controlled polymerization of NNTA can be achieved to obtain α-peptoid polymers | |
| Carboxylic acid | Controlled polymerization of NNTAs is achieved in polar amide solvents (DMAP, DMF and NMP) using acetic acid as catalyst | |||
| Rare earth borohydrides | Controlled ROP of NNTAs was achieved to prepare α-peptoid polymers | |||
| DBU | Cyclic | The polymerization proceeds via a ZROP mechanism with controlled molecular weight and narrow dispersion | ||
| NNPC | Primary/secondary amines | Tertiary amine base | Linear | Tertiary amine base accelerates the intramolecular condensation of NNPC and subsequent polymerization of NNCA |
 | ||
| Fig. 6 Mechanism of primary amine-initiated NNCA polymerization. | ||
Luxenhofer et al. demonstrated that the amine-initiated nucleophilic ROP of NNCAs follows pseudo-first-order kinetics, allowing precise control over the degree of polymerization by adjusting the monomer-to-initiator ratio while maintaining a narrow molar mass distribution.38 The remarkable robustness of NNCA living polymerization was confirmed by ≥10 repeated chain extensions without evidence of chain termination or chain transfer.49 Furthermore, well controlled block copolypeptoids such as poly(N-methylglycine)-b-poly(N-butylglycine) (PNMG-b-PNBG) can be prepared by sequential addition of monomers. This mechanism enables the synthesis of polymers with controlled molecular weights and predictable end groups.
The chemical structure of N-alkyl substituents in NNCA monomers directly affects their polymerization reactivity. To systematically investigate their relationship, Bonduelle et al. designed and synthesized 12 NNCA monomers with different N-alkyl groups, focusing on the steric and electronic effects of substituents on the ring-opening polymerization process.50 Their study revealed that bulky substituents slowed down ROP kinetics, whereas electron-donating groups accelerated the polymerization rate. These findings may provide new ideas for developing catalytic systems to further improve the preparation of peptoid polymers.
Conventional ring-opening polymerization of NNCA is typically constrained by monomer hydrolysis susceptibility. The presence of water or other trace impurities in monomers or solvents may lead to various side reactions, such as deactivation of the reactive end groups or the formation of cyclic structures, thereby significantly reducing reaction efficiency.51 Lu et al. developed an acetonitrile–water mixed solvent system that transforms water from a traditional limiting factor into a key promoter of polymerization and may provide a new strategy for overcoming the strict anhydrous requirements for NNCA polymerization (Fig. 7A).52 This system significantly enhances the efficiency and controllability of proline NCA (Pro-NCA) polymerization, enabling the synthesis of polyproline with a degree of polymerization up to 200 within merely 5 minutes. The resulting polymers possess well-defined end groups, predictable molecular weights, and narrow dispersity (Fig. 7B and C). Mechanistic studies revealed that water molecules serve as “proton carriers” that accelerate the proton shift in rate-determining step, substantially lowering the reaction energy barrier by 7.1 kcal mol−1 and thereby achieving exceptional polymerization efficiency (Fig. 7D).
 | ||
| Fig. 7 (A) ROP of Pro-NCA in mixed acetonitrile/H2O; (B) Mn and Đ of polyproline as a function of [Pro-NCA]0/[I]0 ratio; (C) conversion of Pro NCA ([Pro-NCA]0/[I]0 = 100/1); (D) plausible pathways for the ROP of Pro-NCA in dry acetonitrile (black) and mixed acetonitrile/H2O (red) revealed by density functional theory (DFT) calculations. Reproduced with permission from ref. 52. Copyright 2022, Oxford University Press. | ||
In addition to developing novel initiators, researchers are also dedicated to exploring catalytic polymerization strategies to achieve efficient and rapid polymerization of NNCAs. This approach can significantly enhance the preparation efficiency of α-peptoid polymers while utilizing existing initiators, even enabling the synthesis of ultra-high molecular weight polymers. Therefore, investigating catalytic NNCA polymerization is of significant importance for broadening its application scope.
Lu et al. proposed a polymerization strategy based on carboxylic acid catalysis, which successfully achieved the efficient synthesis of polysarcosine with ultra-high molecular weight and narrow distribution.53 Benzoic acid catalysis enabled full monomer conversion in 2.5 hours in a polymerization system with a monomer-to-initiator ratio of 2000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, with the apparent rate constant increasing by 15-fold compared to the catalyst-free group. Furthermore, employing pivalic acid as the catalyst facilitated rapid polymerization of N-nBu glycine NCA and enabled controlled synthesis of multiblock copolypeptoids. The authors proposed a possible catalytic mechanism wherein the carboxylic acid simultaneously interacts via hydrogen bonds with both the departing hydroxyl hydrogen and the leaving carboxyl oxygen in the rate-limiting transition state. This interaction avoids charge separation and high-tension conformations, thereby lowering the activation free energy and accelerating the polymerization process, ultimately achieving controlled synthesis of ultrahigh-molecular-weight polymers (Fig. 8).
:1, with the apparent rate constant increasing by 15-fold compared to the catalyst-free group. Furthermore, employing pivalic acid as the catalyst facilitated rapid polymerization of N-nBu glycine NCA and enabled controlled synthesis of multiblock copolypeptoids. The authors proposed a possible catalytic mechanism wherein the carboxylic acid simultaneously interacts via hydrogen bonds with both the departing hydroxyl hydrogen and the leaving carboxyl oxygen in the rate-limiting transition state. This interaction avoids charge separation and high-tension conformations, thereby lowering the activation free energy and accelerating the polymerization process, ultimately achieving controlled synthesis of ultrahigh-molecular-weight polymers (Fig. 8).
 | ||
| Fig. 8 Proposed mechanism for ROP of Sar-NCA catalyzed by carboxylic acids. Adapted with permission from ref. 53. Copyright 2024, American Chemical Society. | ||
NNCA monomers bearing bulky substituents typically exhibit slow reactivity, which severely limits the synthesis of α-peptoid polymers with diverse structures. Liu et al. found that 1,3-bis[3,5-bis(trifluoromethyl)phenyl]urea can activate the carbonyl group on the NNCA ring through hydrogen-bonding interactions, thereby significantly accelerating the ROP rate of NNCAs initiated by primary amines (Fig. 9).54 This urea-based catalytic system demonstrates broad compatibility with various NNCA monomers, enabling the efficient preparation of α-peptoid polymers with controlled molecular weights and narrow dispersity. Notably, it is particularly suitable for inactive NNCAs bearing bulky side chains, such as cyclohexyl-NNCA. This catalytic strategy provides a novel approach to expand the structural diversity of α-peptoid polymers and advance their functional exploration.
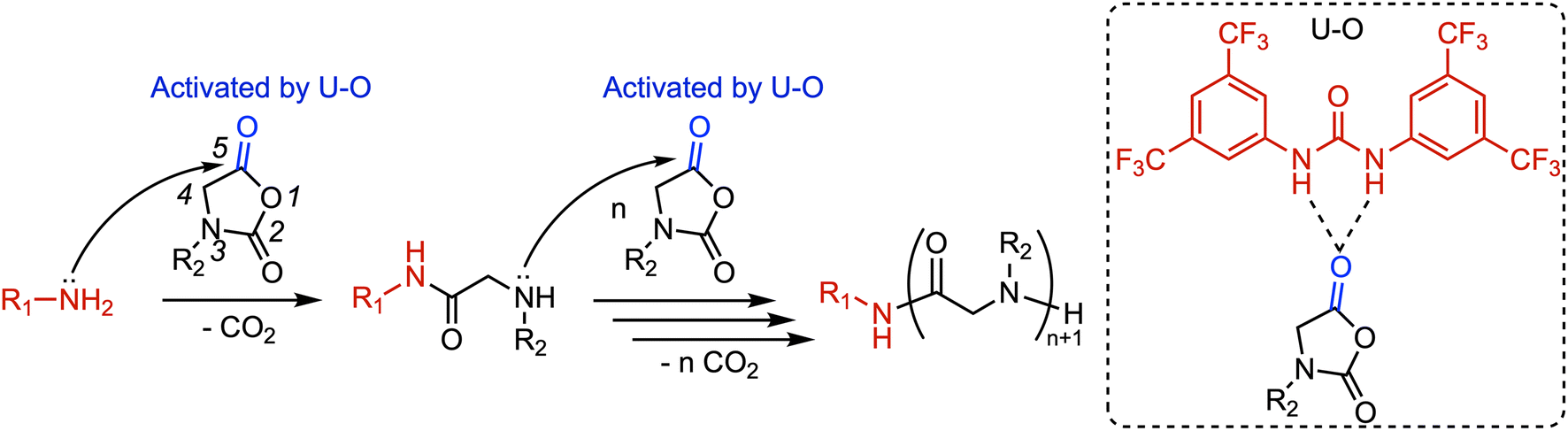 | ||
| Fig. 9 Mechanism of primary amine-initiated ROP of NNCA catalyzed by U–O. Reproduced from ref. 54 with permission from the Royal Society of Chemistry. | ||
 | ||
| Fig. 10 Mechanisms of alcohol-initiated ROP of NNCA catalyzed by TMG. | ||
 | ||
| Fig. 11 The Li/Na/KHMDS-initiated ring-opening polymerization of NNCA in THF. | ||
Kricheldorf et al. observed that base initiators including pyridine, tertiary amines, and imidazole, could initiate the polymerization of sarcosine NCA to yield cyclic PSRs.57,58 Zhang et al. selected NHCs as nucleophilic initiators to trigger the ROP of NNCAs, yielding cyclic α-peptoid polymers with controllable molecular weight and narrow distribution.59,60 Further studies revealed that the polymerization propagation rate strongly depended on the structure of the NHC and the choice of solvent.61 Kinetic studies have shown that in low dielectric constant solvents such as tetrahydrofuran (THF) and toluene, the propagating intermediate maintaining a cyclic structure with two chain ends in close contact due to coulombic attraction (Fig. 12). Moreover, the positively charged NHC chain end in the progating zwitterions could reduce the nucleophilicity and basicity of the anionic carbamate chain end through coulombic interactions, suppressing side reactions and thereby enabling quasi-living polymerization behavior. In contrast, in polar solvents such as N,N-dimethylformamide (DMF) and dimethyl sulfoxide (DMSO), the polymerization was significantly affected by extensive side reactions. In addition, the cyclic α-peptoid polymers could be converted into linear polymers through the addition of electrophiles.
 | ||
| Fig. 12 Mechanism of NHC-initiated NNCA polymerization. Adapted with permission from ref. 61. Copyright 2012, American Chemical Society. | ||
 | ||
| Fig. 13 Mechanism of DBU-initiated NNCA polymerization. Adapted with permission from ref. 64. Copyright 2016, American Chemical Society. | ||
 | ||
| Fig. 14 The LiHMDS-mediated REP of NNCA in DMF. | ||
3.2 NNTA polymerization
NNTA, the thio-analogue of NNCA, exhibits enhanced monomer stability but reduced polymerization reactivity compared to NNCA. Due to their low reactivity, NNTAs have traditionally been employed in the stepwise solid-phase synthesis of peptoids, with their polymerization studies very limited.42,66 | ||
| Fig. 15 Polymerization of NNTA initiated by primary amines. | ||
NNTA ring-opening polymerization initiated by primary amines is tolerant to small amounts of water and does not require strict anhydrous conditions. For this property, Ling et al. explored the relationship between water content and polymerization controllability by investigating the ROP of NNTA in systems with varying water content using benzylamine as the initiator.69,70 The study found that in the presence of small amounts of water (approximately 100–600 μg g−1), the polymerization exhibited excellent controllability. In contrast, in high-water-content systems (approximately 6000–14000 μg g−1), the degree of polymerization gradually deviated from the feed ratio. Mechanistic studies demonstrated that hydrogen sulfide (H2S), generated via hydrolysis of COS, was identified as the key factor suppressing both the yield and molar mass of the resulting polymers.70 Based on this finding, side reactions can be effectively prevented by simply purging the reaction solution with an inert gas such as argon to remove COS and H2S.
However, limited solvent compatibility is one of the major challenges in NNTA polymerization. Amide solvents such as N,N-dimethylacetamide (DMAc), DMF, and N-methylpyrrolidone (NMP) are commonly used solvents for biomacromolecules and poly(amino acid)s, but NNTA typically cannot achieve controlled polymerization in these solvents. Ling et al. addressed this issue by introducing stoichiometric acetic acid as a reaction promoter.71 In the presence of acetic acid, controlled polymerizations of NEG-NTA, Sar-NTA, and N-butyl glycine NTA (NBG-NTA) were successfully achieved in polar amide solvents (including DMAc, DMF, and NMP). Mechanism study revealed that acetic acid not only accelerates polymerization kinetics but also suppresses side reactions by decreasing the concentration of H2S dissolved in polar solvents. This work expands the solvent options for NNTA polymerization.
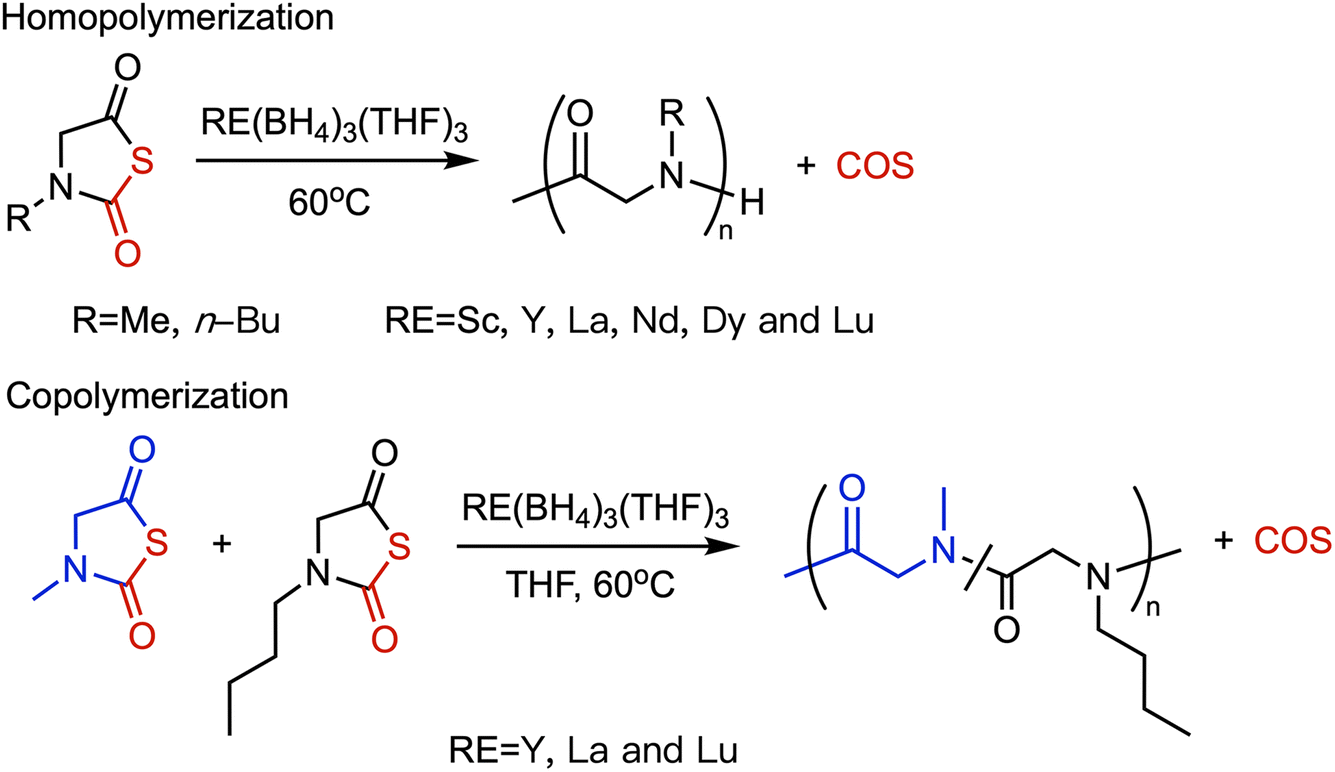 | ||
| Fig. 16 Polymerization of NNTA Initiated by RE(BH4)3(THF)3. Adapted with permission from ref. 72. Copyright 2014, American Chemical Society. | ||
3.3 NNPC polymerization
Schlaad et al. synthesized a series of polysarcosines with varying chain lengths using Sar-NPC as the precursor.46 The number-average molar mass of the obtained products closely matched the theoretical value with narrow molar mass distributions (Đ ≈ 1.1), indicating that the polymerization reaction proceeded in a controlled manner. The process specifically involved in situ conversion of Sar-NPC to Sar-NCA in DMSO solution at 60 °C, catalyzed by 2 vol% of tertiary amine base (DIPEA or TEA), followed by polymerization initiated by the primary amine. Mechanistic studies revealed that the tertiary amine base accelerates the intramolecular condensation of Sar-NPC and subsequent polymerization of Sar-NCA owing to the deprotonation of the Sar-NPC and the shift of the ammonium–amine equilibrium toward the amine (Fig. 17).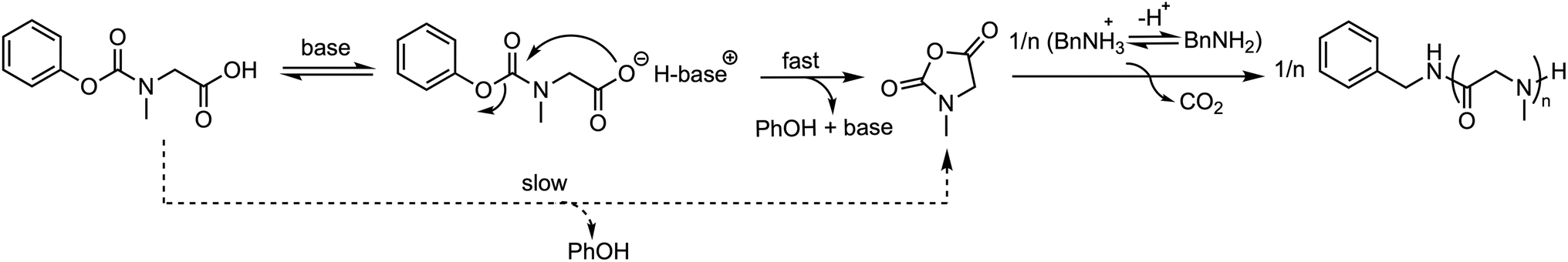 | ||
| Fig. 17 Polysarcosine were prepared by Sar-NPC polymerization in the presence of tertiary amine bases and primary amine initiators. | ||
Ling et al. successfully achieved well-controlled polymerization of NEG-NPC and NBG-NPC under acetic acid catalysis, yielding α-peptoid polymers with tunable molecular weights and low dispersity (Đ < 1.11) (Fig. 18A).45 In addition, kinetic studies and density DFT calculations confirmed that the side reaction during polymerization involves the backbiting reaction of oligomers to generate 1,4-dialkylpyrazine-2,5-diones (DAPs) (Fig. 18B).
 | ||
| Fig. 18 (A) NNPC polymerization in the presence of acetic acid. (B) Backbiting reaction of oligomer to form DAP. | ||
4. Antimicrobial applications of α-peptoid polymers
The antimicrobial activity of HDPs is closely related to their structural characteristics. Cationic groups or net positive charges serve as the critical element for exerting antimicrobial function, since the initial interaction between the HDPs and the negatively charged microbial cell membranes is primarily driven by electrostatic attraction.74,75 Meanwhile, the hydrophobic ratio plays a vital role in modulating both activity and selectivity:76,77 insufficient hydrophobicity weakens membrane interactions,78 whereas excessive hydrophobic content may induce mammalian cell cytotoxicity,79 or polymer aggregation, thereby reducing antibacterial efficacy.80 Additionally, the chain length also significantly affects their activity and cytotoxicity. Within a certain range, the antimicrobial efficacy increases with chain length, but excessively chain length may instead elevate hemolytic activity and cytotoxicity.81,82 In summary, the design of HDP mimics needs to synergistically modulate factors such as positive charge, hydrophobicity ratio, and chain length to achieve high antimicrobial efficacy while minimizing nonspecific damage to host cells.31,83,84Currently, by mimicking the critical amphiphilic structural features of HDPs, researchers have successfully designed and synthesized a variety of antimicrobial α-peptoid polymers. These polymers overcame the limitations of natural peptides, such as enzymatic degradation, poor stability, and high synthesis costs, while retaining potent antimicrobial activity (Table 2). Notably, some antimicrobial α-peptoid polymers have demonstrated excellent in vivo efficacy in animal infection models, including both localized and systemic infections, highlighting their great potential for treating drug-resistant microbial infections (Table 3).
| Polypeptoids structure | Antimicrobial activity | Biocompatibility | Ref. | ||
|---|---|---|---|---|---|
| Tested strains | MICa (μg mL−1) | Hemolysisa | Cytotoxicitya | ||
| a The MIC, hemolysis, and cytotoxicity values in this table are derived from literature, which are assay protocol dependent. The MIC is the minimum concentration of a compound to inhibit microbial growth. The HC50 is the concentration of a compound that causes 50% hemolysis. The IC50 is the concentration of a compound that causes 50% reduction in cell viability. N/A represents that no relevant data was reported in the literature. | |||||

|
S. aureus ATCC6538 | 3.9 | HC10 > 1000 μg mL−1 | N/A | 17 |
| E. coli ATCC25922 | 15.6 | ||||

|
S. aureus ATCC6538 | 2 | HC50 = 250 μg mL−1 | N/A | 85 |
| P. aeruginosa ATCC9027 | 7.8 | ||||
| E. coli ATCC25922 | 7.8 | ||||

|
S. aureus ATCC6538 | 7.8 | HC50 > 1000 μg mL−1 | N/A | 86 |
| P. aeruginosa ATCC9027 | 64 | ||||
| E. coli ATCC25922 | 256 | ||||
|
S. aureus strains | 6.25–12.5 | HC50 > 10000 μg mL−1 |
IC50 = 400 μg mL−1 | 18 |
| S. haemolyticus strains | 1.56–25 | ||||
| S. epidermidis strains | 3.13–6.25 | ||||
| E. faecium strains | 6.25–25 | ||||

|
S. epidermidis ATCC 49134 | 12.5 | HC50 > 200 μg mL−1 | IC50 > 200 μg mL−1 | 87 |
| S. aureus USA300 LAC | 12.5 | ||||
| E. coli ATCC25922 | 50 | ||||
|
P. aeruginosaO1 |
25 | ||||
| A. baumannii BAA747 | 100 | ||||

|
C. difficile | 62.5 | N/A | CC50 = 79 μg mL−1 | 88 |
| S. aureus | 31.25–62.5 | ||||
| L. monocytogenes | 15.6 | ||||
| B. fragilis | 7.8 | ||||
| E. faecalis | 31.25–62.5 | ||||
| E. coli | 62.5 | ||||
| P. aeruginosa | 31.25–62.5 | ||||
| A. baumannii | 31.25 | ||||
| H. pylori | 31.25 | ||||
| Polypeptoids | Infection model | Microbial species | Therapeutic doses | Therapeutic efficacy | Ref |
|---|---|---|---|---|---|
| PNAG25-ET-Cl | Rat full-thickness wound infection model | MRSA | 4 h after infection | Reduces bacterial load by ∼3 log, promotes wound healing, and reduces inflammatory infiltrate | 17 |
| 15 μL, 1 mg mL−1 | |||||
| PG42-BG10-(NH2)2 | Rat full-thickness wound infection model | MRSA | 6 h after infection | Reduces bacterial load by ∼2 log, promotes wound healing, and reduces inflammatory infiltrate | 85 |
| 15 μL, 2 mg mL−1 | |||||
| (PNAG60-g-NH2)-b-PNOG10 fiber-like micelles | Mice full-thickness wound infection model | S. aureus | 24 h after infection | Reduces bacterial load by ∼2.5 log, promotes wound healing, and reduces inflammatory infiltrate | 86 |
| 50 μL, 1 mg mL−1 | |||||
| HS(Naeg)20 | Mice full-thickness wound infection model | MRSA | 24 h after infection | Reduces bacterial load by ∼2 log | 18 |
| The treatments (15 μL, 1.56 mg mL−1) were applied every 4 h for a total of 3 times | |||||
| Mice full-thickness wound infection model | S. epidermidis | 24 h after infection | Reduces bacterial load by ∼2 log | ||
| The treatments (15 μL, 3.13 mg mL−1) were applied every 4 h for a total of 3 times | |||||
| Mice full-thickness wound infection model | S. haemolyticus | 24 h after infection | Reduces bacterial load by ∼2.4 log | ||
| The treatments (15 μL, 3.13 mg mL−1) were applied every 4 h for a total of 3 times | |||||
| Mice keratitis model | MRSA | 24 h after infection | Reduces bacterial load by ∼2.5 log | ||
| The treatments (10 μL, 1.56 mg mL−1) were applied every 5 min during the first hour and every 30 min for the next 7 h | |||||
| Mice peritonitis model | MRSA | 1 h after infection | Increases survival rate, reduces bacterial load for around 1.8 to 3.3 log CFU in all five organs, blood, and peritoneal fluid, and alleviates organ lesions | ||
| A single-dose (20 mg kg−1) was i.p. administered | |||||
| Mice peritonitis model | S. epidermidis | 1 h after infection | Increases survival rate, reduces bacterial load for around 2 to 3.7 log CFU in all five organs, blood, and peritoneal fluid | ||
| A single-dose (20 mg kg−1) was i.p. administered | |||||
| Mice peritonitis model | S. haemolyticus | 1 h after infection | Increases survival rate, reduces bacterial load for around 2.2 to 3.5 log CFU in all five organs, blood, and peritoneal fluid | ||
| A single-dose (20 mg kg−1) was i.p. administered |
4.1 Sulfonium-ion-bearing antibacterial α-peptoid polymers
Chen et al. employed a post-polymerization functionalization strategy to introduce cationic groups into α-peptoid polymers, thereby mimicking the key amphiphilic structure of HDPs with positive charge and hydrophobicity. Specifically, they synthesized α-peptoid polymers via ROP of N-allyl NCA using BnNH2 or methoxypolyethylene glycol amine as the initiators, followed by modification with ethyl thioalcohol and subsequent alkylation with functional epoxides to form antimicrobial polysulfonium derivatives (Fig. 19A).17 By optimizing the hydrophilic/hydrophobic balance, the chlorohydroxypropane-modified polysulfoniums demonstrated high antibacterial activity and selectivity against both E. coli and S. aureus (Fig. 19B). Furthermore, the poly(α-peptoid)s sulfoniums effectively inhibited biofilm formation, eradicated mature biofilms, and demonstrated excellent bactericidal efficacy and wound healing capacity in a MRSA mouse wound infection model (Fig. 19C and D). | ||
| Fig. 19 The antibacterial sulfonium-ion-bearing α-peptoid polymers. (A) Synthesis of the cationic poly(α-peptoid)s sulfoniums. (B) The MIC values against S. aureus and E. coli, as well as the HC10 values of poly(α-peptoid)s sulfoniums. (C) Schematic representation of the mouse skin MRSA infection model and in vivo antimicrobial activity. (D) Digital images and size changes of mouse skin wounds treated with saline, vancomycin and poly(α-peptoid)s sulfoniums at different time points. Adapted with permission from ref. 17. Copyright 2021, John Wiley and Sons. | ||
4.2 Copolypeptoids with antibiofilm activity
Sun et al. designed a novel cationic copolypeptoid antimicrobial agent by mimicking natural HDPs, synthesized via NNCA ring-opening polymerization followed by click chemistry modification (Fig. 20A).85 Specifically, the ROP of a variety of NNCA monomers was initiated by benzylamine, where the incorporation of allyl or propargyl groups allowed for the acquisition of positively charged structures through post-modification with the thiol-terminated amine, and other fragments provided hydrophobic groups. Through modulation of the hydrophilic/hydrophobic balance, the copolypeptoids exhibited broad-spectrum antimicrobial activity against both Gram-positive and Gram-negative bacteria (Fig. 20B), effectively inhibiting biofilm formation and eradicating mature biofilms while outperforming conventional antibiotics (Fig. 20C). Due to a membrane disruption mechanism similar to that of HDPs, resistance does not develop even with repeated use of the polymer (Fig. 20D). Moreover, the copolypeptoids exhibited excellent in vivo antibacterial efficacy, effectively eliminating bacteria and promoting wound healing. Notably, when coated onto magnetic nanospheres, the copolypeptoids demonstrated recyclable properties and enhanced antimicrobial activity as combined with near-infrared-induced photothermal therapy (Fig. 20E). | ||
| Fig. 20 Antimicrobial copolypeptoids with potent activity against drug-resistant bacteria and biofilms. (A) Synthesis of the cationic copolypeptoids. (B) The MIC/MBC values against S. aureus, P. aeruginosa, and E. coli, as well as the HC50 values of polypeptoids. (C) Antibiofilm activity of polypeptoids. (D) Potent activity against drug-resistant bacteria without visible resistance and cross-resistance after repeated usage. (E) Schematic representation of recycling antibacterial of polypeptoids coated FS nanospheres. Reproduced with permission from ref. 85. Copyright 2022, John Wiley and Sons. | ||
4.3 Self-assembled copolypeptoids for effective biofilm eradication
Sun et al. further developed a novel self-assembled cationic antimicrobial polypeptoid.86 Specifically, a cysteamine hydrochloride-modified diblock copolypeptoid, poly(N-allylglycine-g-NH2)-b-poly(N-octylglycine) (PNAG-g-NH2)-b-PNOG, was synthesized via ROP of NNCA and post-polymerization functionalization (Fig. 21A). This polymer could self-assemble into fiber-like and spherical micelles in aqueous solutions at different pH. The fiber-like micelles demonstrated high antimicrobial activity and selectivity, exhibiting superior efficacy to vancomycin in both inhibiting S. aureus biofilm formation and eradicating mature biofilms (Fig. 21B). Antimicrobial mechanism studies revealed that micelles exerted their bactericidal effects by disrupting bacterial cell membranes (Fig. 21C). Further in vivo experiments demonstrated that the polypeptoid micelles showed excellent antibacterial efficacy in a S. aureus infection mouse skin model (Fig. 21D). | ||
| Fig. 21 The antimicrobial copolypeptoid assemblies. (A) Self-assembly of the cationic micelles. (B) Antibiofilm activity of the fiber-like micelles. (C) Illustration of the proposed antibacterial mechanism. (D) The bacteria load in infected tissues after treatment with fiber-like micelles, spherical micelles and vancomycin. Reproduced with permission from ref. 86. Copyright 2022, John Wiley and Sons. | ||
4.4 HDP-mimicking α-peptoid polymers with potent in vivo efficacy
Liu et al. developed a novel strategy for the one-pot ring-opening polymerization of antibacterial α-peptoid polymers without requiring additional post-modification. They successfully synthesized an NNCA monomer with amino groups on its side chain (Nβ-Cbz-aminoethyl-NCA) for the first time and prepared a library of α-peptoid polymers with various C-terminal functional groups via one-pot polymerization (Fig. 22A).18 The optimal α-peptoid polymer exhibited potent activity against methicillin-resistant Staphylococcus aureus (MRSA) planktonic bacteria, persister cells, and biofilm. Moreover, S. aureus didn't acquire resistance upon polymer even after continuous treatment with polymer for 834 passages. The preferred molecule exhibited effective in vivo anti-infectious performance in the mouse wound model, the mouse keratitis model, and the mouse peritonitis model induced by MRSA (Fig. 22B). In addition, the α-peptoid polymers also displayed potent in vitro and in vivo antibacterial activity against various other drug-resistant Gram-positive bacteria.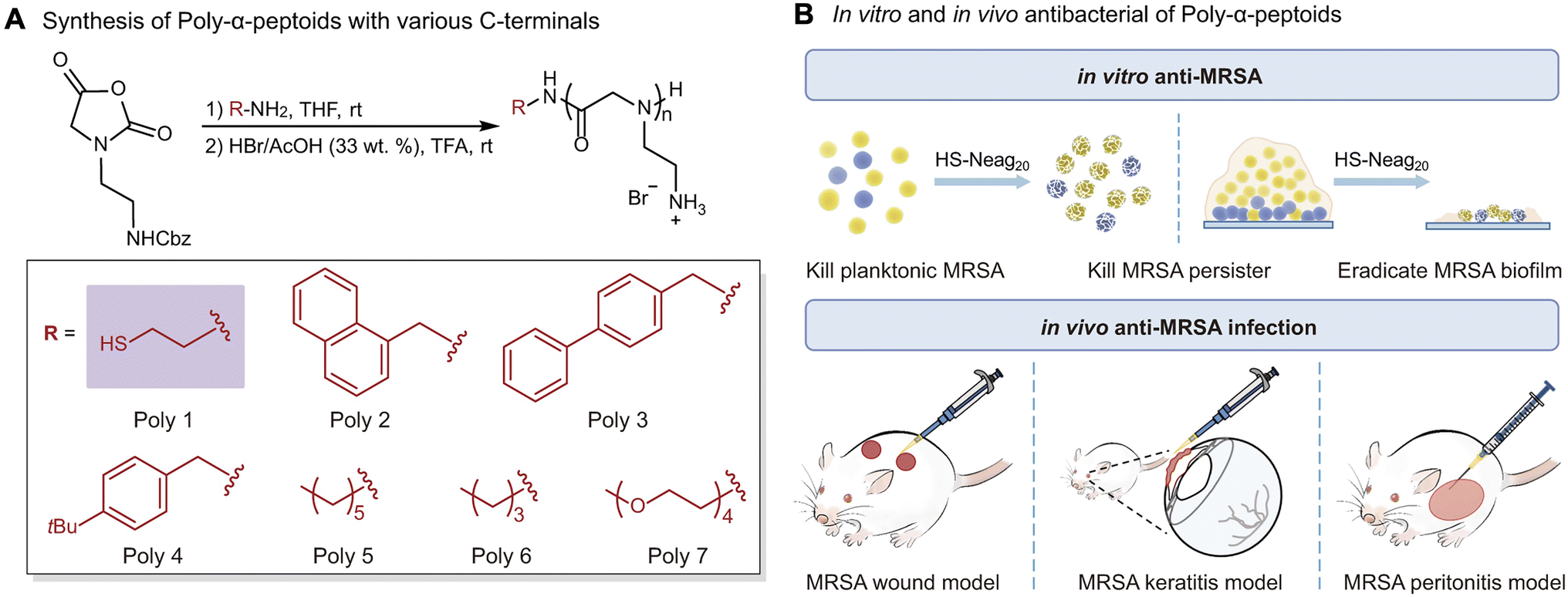 | ||
| Fig. 22 Design and synthesis of α-peptoid polymers with anti-MRSA activity. (A) Design and synthesis of antibacterial α-peptoid polymers with various C-terminal functional groups. (B) Schematic illustration on the antibacterial performance of α-peptoid polymers, in vitro and in vivo. Reproduced with permission from ref. 18. Copyright 2021, Springer Nature. | ||
This study designed a NNCA monomer bearing side-chain amino, overcoming the previous technical limitation in antimicrobial α-peptoid polymers synthesis that required post-polymerization functionalization to introduce cationic groups and providing a simplified route to obtain structurally controllable amphiphilic α-peptoid polymers. Furthermore, it also demonstrated the significant potential of HDP-mimicking α-peptoid polymers in the treatment of drug-resistant microbial infections through various animal models, especially systemic infection models.
4.5 HDP-mimicking cyclic α-peptoid polymers with broad-spectrum activity
Liu et al. further synthesized a series of amphiphilic cyclic α-peptoid polymers via copolymerization of cationic monomer Nβ-Cbz-aminoethyl-NCA and hydrophobic monomer N-pentafluorobenzyl-NCA, using DBU as the initiator.87 The optimal cyclic α-peptoid polymers, poly(Naeg0.7Npfbg0.3)20, exhibited broad-spectrum antimicrobial activity against drug-resistant bacteria, but low hemolysis and cytotoxicity (Fig. 23). The mode-of-action study revealed that its antibacterial activity was closely associated with bacterial membrane interaction. This study demonstrated the potential application of cyclic α-peptoids polymers in the antimicrobial field. | ||
| Fig. 23 Cyclic α-peptoid polymers displays potent activity against drug-resistant bacteria and low mammalian cell toxicity. Reproduced from ref. 87 with permission from the Royal Society of Chemistry. | ||
4.6 Star-like antibacterial α-peptoid polymers
Bonduelle et al. employed a dendritic macroinitiator (PAMAM) to synthesize star-like topology α-peptoid copolymers via ROP of N-protected-lysine-like NNCA and phenylalanine-like NNCA monomers.88 The star-like α-peptoid polymers featured cationic side chains and broad-spectrum antimicrobial activity against multiple bacterial strains, including Clostridium difficile, S. aureus, Listeria monocytogenes, and Enterococcus faecalis, with MIC values ranging from 7.8–62.5 μg mL−1 (Fig. 24). Furthermore, structure–activity relationship studies revealed that their antibacterial performance could be modulated by fine-tuning hydrophobic content and the number of arms. | ||
| Fig. 24 Design and synthesis of antibacterial star-like α-peptoid polymers. Reproduced from ref. 88 with permission from the Royal Society of Chemistry. | ||
5. Current challenges and future directions
HDP-mimicking peptoids represent a promising class of antimicrobial agents, offering a potential alternative to conventional antibiotics for treating drug-resistant microbial infections. However, peptoids are typically prepared by stepwise solid-phase synthesis, which is time-consuming, cumbersome, difficult to scale up, and expensive. The development of polymerization chemistry has opened up new avenues for the preparation of peptoid polymers, and a variety of HDP-mimicking polymers have been synthesized by solution polymerization. These antimicrobial α-peptoid polymers exhibit potent activity against bacteria, especially drug-resistant bacteria, and demonstrate excellent in vivo therapeutic efficacy in multiple animal infection models. Despite significant progress in the development of HDP-mimicking α-peptoid polymers, their clinical applications still face several key challenges, including:Currently, the diversity of monomers available for synthesizing HDP-mimicking α-peptoid polymers remains limited, with a particularly short of positively charged monomers. The existing cationic monomers are predominantly restricted to lysine-like structure,18,88 and this structural limit severely constrains the molecular diversity of antimicrobial α-peptoid polymers. To address this limitation, future research should pay attention the development of positively charged monomers, such as those introducing secondary amines, tertiary amines, or imidazole in their side chain, to diversify the structure of α-peptoid polymers and thus obtain antimicrobial agents with potent activity and high selectivity. Notably, while α-peptoid polymers have demonstrated considerable potential as antibacterial agents, their application in antifungal therapy remains largely unreported. The expansion of structural diversity will promote the development of antifungals and even broad-spectrum antimicrobials.
Although the polymerization strategy of α-peptoid polymers offers advantages over solid-phase synthesis in terms of yield and chain length, several aspects warrant further investigation, including the optimization of reaction condition and large-scale production processes. Future breakthroughs may arise from the development of novel controlled polymerization, such as exploring novel initiators and catalysts, to enable efficient and rapid synthesis of high-molecular-weight α-peptoid polymers under mild conditions.
In vivo studies on antimicrobial α-peptoid polymers are limited, with their biosafety, immunogenicity, pharmacokinetics, and antibacterial mechanisms in complex physiological environments still unclear, hindering clinical translation. The systematic investigation on the in vivo therapeutic potential of α-peptoid polymers need to be carried out in the future to promote their translation from laboratory research to clinical applications. Furthermore, innovative in vivo application strategies could be explored to enhance therapeutic efficacy against drug-resistant microbial infections, including investigating synergistic effects between α-peptoid polymers and existing antibiotics and optimizing targeted delivery systems to infection sites by utilizing nanoparticle-based drug delivery platforms or stimuli-responsive antimicrobial materials.89–91
Biodegradable antibacterial polymers represent promising materials for medical applications such as stents, implants, and wound dressings.92–95 Future research could focus on the development of degradation-tunable design strategies, such as constructing peptide/peptoid hybrid polymers with tunable degradation rates or incorporating chemically labile bonds into peptoid backbones that can be specifically cleaved by the infection microenvironment.96,97 By developing synthetic strategies and applications for biodegradable α-peptoid polymers, it is expected to maintain their antimicrobial activity while further improving other properties such as biocompatibility, thereby enhancing their potential for applications in medical devices and biomedicine.
It must be emphasized that the above challenges do not diminish the scientific value of antimicrobial α-peptoid polymers, but reveal the key scientific issues and technical bottlenecks that need to be address in this field (Fig. 25). α-Peptoid polymers, as representative natural peptide mimics, not only demonstrate unique advantages in terms of resistance to proteolysis, but also provide an expansive polymer prototype platform for developing novel antimicrobial agents with exceptional therapeutic potential. With continuous development of materials science, polymerization chemistry, and other disciplines, these biomimetic antimicrobial compounds are poised to achieve transformative breakthroughs across multiple fields, including novel antimicrobial materials and biomedical applications, thereby offering innovative solutions to address the global antibiotic resistance crisis.
 | ||
| Fig. 25 Outlook on future developments of antimicrobial α-peptoid polymers. | ||
Abbreviations
| HDP | Host defense peptide |
| NNCA | N-Substituted N-carboxyanhydride |
| NNTA | N-Substituted N-thiocarboxyanhydride |
| NNPC | N-Phenoxycarbonyl N-substituted glycine |
| NCA | N-Carboxyanhydride |
| HCl | Hydrogen chloride |
| XAA | S-Ethoxythiocarbonyl mercaptoacetic acid |
| COS | Carbonyl sulfide |
| DPC | Diphenyl carbonate |
| MTBE | Methyl tert-butyl ether |
| ROP | Ring-opening polymerization |
| NHC | N-Heterocyclic carbene |
| DBU | 1,8-Diazabicycloundec-7-ene |
| LiHMDS | Lithium hexamethyldisilazide |
| NAM | Normal amine mechanism |
| REP | Ring-expansion polymerization |
| DFT | Density functional theory |
| TMG | 1,1,3,3-Tetramethylguanidine |
| PEG | Poly(ethylene glycol) |
| ZROP | Zwitterionic ring-opening polymerization |
| THF | Tetrahydrofuran |
| DMF | N,N-Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| H2S | Hydrogen sulfide |
| DMAc | N,N-Dimethylacetamide |
| NMP | N-Methylpyrrolidone |
| DAP | 1,4-Dialkylpyrazine-2,5-diones |
| S. aureus | Staphylococcus aureus |
| E. coli | Escherichia coli |
| P. aeruginosa | Pseudomonas aeruginosa |
| S. haemolyticus | Staphylococcus haemolyticus |
| S. epidermidis | Staphylococcus epidermidis |
| E. faecium | Enterococcus faecium |
| A. baumannii | Acinetobacter baumannii |
| C. difficile | Clostridium difficile |
| L. monocytogenes | Listeria Monocytogenes |
| B. fragilis | Bacteroides fragilis |
| E. faecalis | Enterococcus faecalis |
| H. pylori | Helicobacter pylori |
| MRSA | Methicillin-resistant Staphylococcus aureus |
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
J. X.: visualization, investigation, writing – original draft, review & editing. W. H.: investigation. X. F.: investigation. Z. L.: investigation. M. Z.: investigation, writing – original draft, review & editing. R. L.: supervision, resources, funding acquisition, writing – review & editing, project administration.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. T2325010, 22305082, 52361165622, 22075078), Chinese Academy of Sciences Project for Young Scientists in Basic Research (Grant No. YSBR-111), the Science and Technology Innovation Action Plan Computational Biology Program (24JS2830400), the Fundamental Research Funds for the Central Universities (No. JKVD1251029, JKD01251701), Shanghai Frontiers Science Center of Optogenetic Techniques for Cell Metabolism (Shanghai Municipal Education Commission).References
- D. M. P. De Oliveira, B. M. Forde, T. J. Kidd, P. N. A. Harris, M. A. Schembri, S. A. Beatson, D. L. Paterson and M. J. Walker, Antimicrobial resistance in ESKAPE pathogens, Clin. Microbiol. Rev., 2020, 33, e00181–00119 CrossRef CAS.
- C. J. L. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. R. Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao, E. Wool, S. C. Johnson, A. J. Browne, M. G. Chipeta, F. Fell, S. Hackett, G. Haines-Woodhouse, B. H. K. Hamadani, E. A. P. Kumaran, B. McManigal, R. Agarwal, S. Akech, S. Albertson, J. Amuasi, J. Andrews, A. Aravkin, E. Ashley, F. Bailey, S. Baker, B. Basnyat, A. Bekker, R. Bender, A. Bethou, J. Bielicki, S. Boonkasidecha, J. Bukosia, C. Carvalheiro, C. Castaneda-Orjuela, V. Chansamouth, S. Chaurasia, S. Chiurchiu, F. Chowdhury, A. J. Cook, B. Cooper, T. R. Cressey, E. Criollo-Mora, M. Cunningham, S. Darboe, N. P. J. Day, M. De Luca, K. Dokova, A. Dramowski, S. J. Dunachie, T. Eckmanns, D. Eibach, A. Emami, N. Feasey, N. Fisher-Pearson, K. Forrest, D. Garrett, P. Gastmeier, A. Z. Giref, R. C. Greer, V. Gupta, S. Haller, A. Haselbeck, S. I. Hay, M. Holm, S. Hopkins, K. C. Iregbu, J. Jacobs, D. Jarovsky, F. Javanmardi, M. Khorana, N. Kissoon, E. Kobeissi, T. Kostyanev, F. Krapp, R. Krumkamp, A. Kumar, H. H. Kyu, C. Lim, D. Limmathurotsakul, M. J. Loftus, M. Lunn, J. Ma, N. Mturi, T. Munera-Huertas, P. Musicha, M. M. Mussi-Pinhata, T. Nakamura, R. Nanavati, S. Nangia, P. Newton, C. Ngoun, A. Novotney, D. Nwakanma, C. W. Obiero, A. Olivas-Martinez, P. Olliaro, E. Ooko, E. Ortiz-Brizuela, A. Y. Peleg, C. Perrone, N. Plakkal, A. Ponce-de-Leon, M. Raad, T. Ramdin, A. Riddell, T. Roberts, J. VictoriaRobotham, A. Roca, K. E. Rudd, N. Russell, J. Schnall, J. A. G. Scott, M. Shivamallappa, J. Sifuentes-Osornio, N. Steenkeste, A. J. Stewardson, T. Stoeva, N. Tasak, A. Thaiprakong, G. Thwaites, C. Turner, P. Turner, H. R. van Doorn, S. Velaphi, A. Vongpradith, V. Huong, T. Walsh, S. Waner, T. Wangrangsimakul, T. Wozniak, P. Zheng, B. Sartorius, A. D. Lopez, A. Stergachis, C. Moore, C. Dolecek, M. Naghavi and C. Antimicrobial Resistance, Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis, Lancet, 2022, 399, 629–655 CrossRef CAS.
- J. O'Neill, Tackling drug-resistant infections globally: final report and recommendations, 2016 Search PubMed.
- M. Zasloff, Antimicrobial peptides of multicellular organisms, Nature, 2002, 415, 389–395 CrossRef CAS.
- R. E. W. Hancock and H.-G. Sahl, Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies, Nat. Biotechnol., 2006, 24, 1551–1557 CrossRef CAS PubMed.
- N. Mookherjee, M. A. Anderson, H. P. Haagsman and D. J. Davidson, Antimicrobial host defence peptides: functions and clinical potential, Nat. Rev. Drug Discovery, 2020, 19, 311–332 CrossRef CAS PubMed.
- M. Sieprawska-Lupa, P. Mydel, K. Krawczyk, K. Wójcik, M. Puklo, B. Lupa, P. Suder, J. Silberring, J. Silberring, M. Reed, J. Pohl, W. Shafer, F. McAleese, T. Foster, J. Travis and J. Potempa, Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases, Antimicrob. Agents Chemother., 2004, 48, 4673–4679 CrossRef CAS.
- M. Mahlapuu, C. Bjorn and J. Ekblom, Antimicrobial peptides as therapeutic agents: opportunities and challenges, Crit. Rev. Biotechnol., 2020, 40, 978–992 CrossRef CAS PubMed.
- K. Kuroda and W. F. DeGrado, Amphiphilic polymethacrylate derivatives as antimicrobial agents, J. Am. Chem. Soc., 2005, 127, 4128–4129 CrossRef CAS.
- B. P. Mowery, S. E. Lee, D. A. Kissounko, R. F. Epand, R. M. Epand, B. Weisblum, S. S. Stahl and S. H. Gellman, Mimicry of antimicrobial host-defense peptides by random copolymers, J. Am. Chem. Soc., 2007, 129, 15474–15476 CrossRef CAS PubMed.
- N. P. Chongsiriwatana, J. A. Patch, A. M. Czyzewski, M. T. Dohm, A. Ivankin, D. Gidalevitz, R. N. Zuckermann and A. E. Barron, Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides, Proc. Natl. Acad. Sci. U.S.A., 2008, 105, 2794–2799 Search PubMed.
- P. Li, Y. F. Poon, W. Li, H.-Y. Zhu, S. H. Yeap, Y. Cao, X. Qi, C. Zhou, M. Lamrani, R. W. Beuerman, E.-T. Kang, Y. Mu, C. M. Li, M. W. Chang, S. S. J. Leong and M. B. Chan-Park, A polycationic antimicrobial and biocompatible hydrogel with microbe membrane suctioning ability, Nat. Mater., 2011, 10, 149–156 Search PubMed.
- K. Fukushima, S. Liu, H. Wu, A. C. Engler, D. J. Coady, H. Maune, J. Pitera, A. Nelson, N. Wiradharma, S. Venkataraman, Y. Huang, W. Fan, J. Y. Ying, Y. Y. Yang and J. L. Hedrick, Supramolecular high-aspect ratio assemblies with strong antifungal activity, Nat. Commun., 2013, 4, 2861 CrossRef.
- C. Ghosh, G. B. Manjunath, P. Akkapeddi, V. Yarlagadda, J. Hoque, D. S. S. M. Uppu, M. M. Konai and J. Haldar, Small molecular antibacterial peptoid mimics: The simpler the better, J. Med. Chem., 2014, 57, 1428–1436 CrossRef CAS.
- M. Xiong, M. W. Lee, R. A. Mansbach, Z. Song, Y. Bao, R. M. Peek Jr, C. Yao, L.-F. Chen, A. L. Ferguson, G. C. L. Wong and J. Cheng, Helical antimicrobial polypeptides with radial amphiphilicity, Proc. Natl. Acad. Sci. U.S.A., 2015, 112, 13155–13160 CrossRef CAS PubMed.
- Y. Shi, P. Teng, P. Sang, F. She, L. Wei and J. Cai, γ-AApeptides: Design, structure, and applications, Acc. Chem. Res., 2016, 49, 428–441 CrossRef CAS PubMed.
- J. Sun, M. Li, B. Zhang and X. Chen, High antibacterial activity and selectivity of the versatile polysulfoniums that combat drug resistance, Adv. Mater., 2021, 33, 2104402 CrossRef CAS PubMed.
- J. Xie, M. Zhou, Y. Qian, Z. Cong, S. Chen, W. Zhang, W. Jiang, C. Dai, N. Shao, Z. Ji, J. Zou, X. Xiao, L. Liu, M. Chen, J. Li and R. Liu, Addressing MRSA infection and antibacterial resistance with peptoid polymers, Nat. Commun., 2021, 12, 5898 CrossRef CAS PubMed.
- M. Zhou, L. Liu, Z. Cong, W. Jiang, X. Xiao, J. Xie, Z. Luo, S. Chen, Y. Wu, X. Xue, N. Shao and R. Liu, A dual-targeting antifungal is effective against multidrug-resistant human fungal pathogens, Nat. Microbiol., 2024, 9, 1325–1339 CrossRef CAS.
- R. J. Simon, R. S. Kania, R. N. Zuckermann, V. D. Huebner, D. A. Jewell, S. Banville, S. Ng, L. Wang, S. Rosenberg and C. K. Marlowe, Peptoids: a modular approach to drug discovery, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 9367–9371 CrossRef CAS.
- R. N. Zuckermann, Peptoid Origins, Biopolymers, 2011, 96, 545–555 CrossRef CAS PubMed.
- A. S. Knight, E. Y. Zhou, M. B. Francis and R. N. Zuckermann, Sequence programmable peptoid polymers for diverse materials applications, Adv. Mater., 2015, 27, 5665–5691 CrossRef CAS.
- S. M. Miller, R. J. Simon, S. Ng, R. N. Zuckermann, J. M. Kerr and W. H. Moos, Proteolytic studies of homologous peptide and N-substituted glycine peptoid oligomers, Bioorg. Med. Chem. Lett., 1994, 4, 2657–2662 CrossRef CAS.
- J.-K. Bang, Y. H. Nan, E. K. Lee and S. Y. Shin, A novel Trp-rich model antimicrobial peptoid with increased protease stability, Bull. Korean Chem. Soc., 2010, 31, 2509–2513 CrossRef CAS.
- J. Sun and R. N. Zuckermann, Peptoid polymers: a highly designable bioinspired material, ACS Nano, 2013, 7, 4715–4732 CrossRef CAS PubMed.
- B. Cai, Z. Li and C.-L. Chen, Programming amphiphilic peptoid oligomers for hierarchical assembly and inorganic crystallization, Acc. Chem. Res., 2021, 54, 81–91 CrossRef CAS.
- Z. Qiu, M. Zhang, D. Liu, X. Shen, W. Zhou, W. Liu, J. Lu and L. Guo, A review on the synthesis of polypeptoids, Catalysts, 2023, 13, 280 CrossRef CAS.
- A. S. Culf and R. J. Ouellette, Solid-phase synthesis of N-substituted glycine oligomers (α-peptoids) and derivatives, Molecules, 2010, 15, 5282–5335 CrossRef CAS PubMed.
- C. Secker, S. M. Brosnan, R. Luxenhofer and H. Schlaad, Poly(α-peptoid)s revisited: synthesis, properties, and use as biomaterial, Macromol. Biosci., 2015, 15, 881–891 CrossRef CAS PubMed.
- A. M. Clapperton, J. Babi and H. Tran, A field guide to optimizing peptoid synthesis, ACS Polym. Au, 2022, 2, 417–429 CrossRef CAS PubMed.
- Y. Wu, K. Chen, J. Wang, M. Chen, Y. Chen, Y. She, Z. Yan and R. Liu, Host defense peptide mimicking antimicrobial amino acid polymers and beyond: design, synthesis and biomedical applications, Prog. Polym. Sci., 2023, 141, 101679 CrossRef CAS.
- R. N. Zuckermann and T. Kodadek, Peptoids as potential therapeutics, Curr. Opin. Mol. Ther., 2009, 11, 299–307 CAS.
- A. M. Rosales, R. A. Segalman and R. N. Zuckermann, Polypeptoids: a model system to study the effect of monomer sequence on polymer properties and self-assembly, Soft Matter, 2013, 9, 8400–8414 RSC.
- M. Lin, D. Liu, Y. Gong, L. Shu, H. Wang, G. Zhang, J. Li, Z. Gao, J. Sun and X. Chen, Bioactive assembly cofactor-assisted ursolic acid helix for enhanced anticancer efficacy via in situ virus-like transition, J. Am. Chem. Soc., 2025, 147, 17010–17021 CrossRef CAS PubMed.
- H. Leuchs, Ueber die Glycin-carbonsäure, Ber. Dtsch. Chem. Ges., 1906, 39, 857–861 Search PubMed.
- F. Fuchs, Über N-Carbonsäure-anhydride, Ber. Dtsch. Chem. Ges., 1922, 55, 2943 Search PubMed.
- A. C. Farthing and R. J. W. Reynolds, Anhydro-N-carboxy-DL-β-phenylalanine, Nature, 1950, 165, 647 Search PubMed.
- C. Fetsch, A. Grossmann, L. Holz, J. F. Nawroth and R. Luxenhofer, Polypeptoids from N-substituted glycine N-carboxyanhydrides: Hydrophilic, hydrophobic, and amphiphilic polymers with poisson distribution, Macromolecules, 2011, 44, 6746–6758 CrossRef CAS.
- Q. Gao, H. Fu, H. Liu, P. Li, J. Shi, Y. Lin and Z. Song, Polymerization-induced self-assembly of N-substituted glycine N-carboxyanhydrides in selected solvents, Eur. Polym. J., 2025, 225, 113720 CrossRef CAS.
- Z.-Y. Tian, Z. Zhang, S. Wang and H. Lu, A moisture-tolerant route to unprotected α/β-amino acid N-carboxyanhydrides and facile synthesis of hyperbranched polypeptides, Nat. Commun., 2021, 12, 5810 Search PubMed.
- L. Capeloa, R. Miravet Marti, A. Duro-Castano, V. J. Nebot and M. Barz, Utility of triethyloxonium tetrafluoroborate for chloride removal during sarcosine N-carboxyanhydride synthesis: Improving NCA purity, Chem.–Eur. J., 2024, 30, e202304375 CrossRef CAS PubMed.
- H. R. Kricheldorf and K. Bösinger, Mechanismus der NCA-polymerisation, 3. Über die amin katalysierte polymerisation von sarkosin-NCA und -NTA, Die Makromolekulare Chem., 1976, 177, 1243–1258 CrossRef CAS.
- X. Tao, M. Li and J. Ling, α-Amino acid N-thiocarboxyanhydrides: A novel synthetic approach toward poly(α-amino acid)s, Eur. Polym. J., 2018, 109, 26–42 Search PubMed.
- A. Ma, X. Yu, M. Liao, W. Liu, S. Xuan and Z. Zhang, Research Progress in Polypeptoids Prepared by Controlled Ring-Opening Polymerizations, Macromol. Rapid Commun., 2023, 44, 2200301 CrossRef CAS.
- S. Xu, T. Bai and J. Ling, Understanding acid-promoted polymerization of N-phenoxycarbonyl N-substituted glycine: differences among methyl, ethyl, and butyl substituents, Macromolecules, 2025, 58, 4238–4249 Search PubMed.
- A. Doriti, S. M. Brosnan, S. M. Weidner and H. Schlaad, Synthesis of polysarcosine from air and moisture stable N-phenoxycarbonyl-N-methylglycine assisted by tertiary amine base, Polym. Chem., 2016, 7, 3067–3070 RSC.
- D. Zhang, S. Liu, Z. Cai, W. Luo, G. Liu and R. Liu, Facile synthesis of N-phenoxycarbonyl amino acids by a two-phase reaction for direct polymerization, Polym. Chem., 2024, 15, 97–105 RSC.
- T. Bai, B. Zheng and J. Ling, Density functional theory studies on the synthesis of poly(α-amino acid)s via the amine-mediated ring opening polymerizations of N-carboxyanhydrides and N-thiocarboxyanhydrides, Front. Chem., 2021, 9, 645949 CrossRef CAS.
- C. Fetsch and R. Luxenhofer, Highly defined multiblock copolypeptoids: Pushing the limits of living nucleophilic ring-opening polymerization, Macromol. Rapid Commun., 2012, 33, 1708–1713 CrossRef CAS.
- P. Salas-Ambrosio, A. Tronnet, M. Badreldin, S. Ji, S. Lecommandoux, S. Harrisson, P. Verhaeghe and C. Bonduelle, Effect of N-alkylation in N-carboxyanhydride (NCA) ring-opening polymerization kinetics, Polym. Chem., 2022, 13, 6149–6161 RSC.
- T. A. Bauer, L. Simić, J. F. R. Van Guyse, A. Duro-Castaño, V. J. Nebot and M. Barz, Polypept(o)ides – Origins, synthesis, applications and future directions, Prog. Polym. Sci., 2024, 158, 101889 Search PubMed.
- Y. Hu, Z. Tian, W. Xiong, D. Wang, R. Zhao, Y. Xie, Y. Song, J. Zhu and H. Lu, Water-assisted and protein-initiated fast and controlled ring-opening polymerization of proline N-carboxyanhydride, Natl. Sci. Rev., 2022, 9, nwac033 CrossRef CAS.
- S. Wang, M. Lu, S. Wan, C. Lyu, Z. Tian, K. Liu and H. Lu, Precision synthesis of polysarcosine via controlled ring-opening polymerization of N-carboxyanhydride: fast kinetics, ultrahigh molecular weight, and mechanistic insights, J. Am. Chem. Soc., 2024, 146, 5678–5692 CrossRef CAS PubMed.
- K. Chen, Y. Wu, X. Wu, M. Zhou, R. Zhou, J. Wang, X. Xiao, Y. Yuan and R. Liu, Facile synthesis of polypeptoids bearing bulky sidechains via urea accelerated ring-opening polymerization of α-amino acid N-substituted N-carboxyanhydrides, Polym. Chem., 2022, 13, 420–426 RSC.
- B. A. Chan, S. Xuan, M. Horton and D. Zhang, 1,1,3,3-Tetramethylguanidine-promoted ring-opening polymerization of N-butyl N-carboxyanhydride using alcohol initiators, Macromolecules, 2016, 49, 2002–2012 CrossRef CAS.
- Y. Wu, M. Zhou, K. Chen, S. Chen, X. Xiao, Z. Ji, J. Zou and R. Liu, Alkali-metal hexamethyldisilazide initiated polymerization on alpha-amino acid N-substituted N-carboxyanhydrides for facile polypeptoid synthesis, Chin. Chem. Lett., 2021, 32, 1675–1678 CrossRef CAS.
- H. R. Kricheldorf, C. Von Lossow and G. Schwarz, Tertiary amine catalyzed polymerizations of α-amino acid N-carboxyanhydrides: the role of cyclization, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4680–4695 CrossRef CAS.
- H. R. Kricheldorf, C. Von Lossow and G. Schwarz, Imidazole-initiated polymerizations of α-amino acid N-carboxyanhydrides – simultaneous chain-growth and step-growth polymerization, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5690–5698 CrossRef CAS.
- L. Guo and D. Zhang, Cyclic poly(α-peptoid)s and their block copolymers from N-heterocyclic carbene-mediated ring-opening polymerizations of N-substituted N-carboxylanhydrides, J. Am. Chem. Soc., 2009, 131, 18072–18074 CrossRef CAS PubMed.
- S. H. Lahasky, W. K. Serem, L. Guo, J. C. Garno and D. Zhang, Synthesis and characterization of cyclic brush-like polymers by N-heterocyclic carbene-mediated zwitterionic polymerization of N-propargyl N-carboxyanhydride and the grafting-to approach, Macromolecules, 2011, 44, 9063–9074 CrossRef CAS.
- L. Guo, S. H. Lahasky, K. Ghale and D. Zhang, N-heterocyclic carbene-mediated zwitterionic polymerization of N-substituted N-carboxyanhydrides toward poly(α-peptoid)s: Kinetic, mechanism, and architectural control, J. Am. Chem. Soc., 2012, 134, 9163–9171 CrossRef CAS.
- W. C. Shieh, S. Dell and O. Repic, Nucleophilic catalysis with 1,8-diazabicyclo 5.4.0 undec-7-ene (DBU) for the esterification of carboxylic acids with dimethyl carbonate, J. Org. Chem., 2002, 67, 2188–2191 Search PubMed.
- M. Carafa, E. Mesto and E. Quaranta, DBU-promoted nucleophilic activation of carbonic acid diesters, Eur. J. Org Chem., 2011, 2011, 2458–2465 Search PubMed.
- A. Li, L. Lu, X. Li, L. He, C. Do, J. C. Gamo and D. Zhang, Amidine-mediated zwitterionic ring-opening polymerization of N-alkyl N-carboxyanhydride: mechanism, kinetics, and architecture elucidation, Macromolecules, 2016, 49, 1163–1171 CrossRef CAS.
- P. Salas-Ambrosio, A. Tronnet, M. Since, S. Bourgeade-Delmas, J.-L. Stigliani, A. Vax, S. Lecommandoux, B. Dupuy, P. Verhaeghe and C. Bonduelle, Cyclic poly(α-peptoid)s by Lithium bis(trimethylsilyl)amide (LiHMDS)-mediated ring-expansion polymerization: simple access to bioactive backbones, J. Am. Chem. Soc., 2021, 143, 3697–3702 CrossRef CAS.
- H. R. Kricheldorf, ÜBer die polymerisation von α-aminosäure-N-carboxyanhydriden (1,3-Oxazolidin-2,5-dionen) und α-aminosäure-N-thiocarboxyanhydriden (1,3-Thiazolidin-2,5-dionen), Die Makromolekulare Chem., 1974, 175, 3325–3342 CrossRef CAS.
- H. R. Kricheldorf, M. Sell and G. Schwarz, Primary amine-initiated polymerizations of α-amino acid N-thiocarbonic acid anhydrosulfide, J. Macromol. Sci., Pure Appl. Chem., 2008, 45, 425–430 CrossRef CAS.
- X. Tao, B. Zheng, H. R. Kricheldorf and J. Ling, Are N-substituted glycine N-thiocarboxyanhydride monomers really hard to polymerize?, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 404–410 Search PubMed.
- B. Zheng, X. Tao and J. Ling, Water tolerated polymerization of N-substituted glycine N-thiocarboxyanhydride initiated by primary amines, Acta Polym. Sin., 2018, 72–79 Search PubMed.
- B. Zheng, T. Bai, X. Tao, H. Schlaad and J. Ling, Identifying the hydrolysis of carbonyl sulfide as a side reaction impeding the polymerization of N-substituted glycine N-thiocarboxyanhydride, Biomacromolecules, 2018, 19, 4263–4269 CrossRef CAS PubMed.
- B. Zheng, S. Xu, X. Ni and J. Ling, Understanding acid-promoted polymerization of the N-substituted glycine N-thiocarboxyanhydride in polar solvents, Biomacromolecules, 2021, 22, 1579–1589 Search PubMed.
- X. Tao, Y. Deng, Z. Shen and J. Ling, Controlled polymerization of N-substituted glycine N-thiocarboxyanhydrides initiated by rare earth borohydrides toward hydrophilic and hydrophobic polypeptoids, Macromolecules, 2014, 47, 6173–6180 CrossRef CAS.
- Z. Hu, S. Yan, J. Qi, X. Tao and S. Lin, Synthesis and characterization of cyclic polypeptoids based on controlled polymerizations of NNTA, Acta Polymerica Sinica, 2021, 52, 708–716 CAS.
- Y. Yang, Z. Cai, Z. Huang, X. Tang and X. Zhang, Antimicrobial cationic polymers: from structural design to functional control, Polym. J., 2018, 50, 33–44 CrossRef CAS.
- H. Etayash and R. E. W. Hancock, Host defense peptide-mimicking polymers and polymeric-brush-tethered host defense peptides: recent developments, limitations, and potential success, Pharmaceutics, 2021, 13, 1820 CrossRef CAS PubMed.
- K. Lienkamp, A. E. Madkour, A. Musante, C. F. Nelson, K. Nuesslein and G. N. Tew, Antimicrobial polymers prepared by ROMP with unprecedented selectivity: a molecular construction kit approach, J. Am. Chem. Soc., 2008, 130, 9836–9843 CrossRef CAS.
- E. F. Palermo, K. Lienkamp, E. R. Gillies and P. J. Ragogna, Antibacterial activity of polymers: discussions on the nature of amphiphilic balance, Angew. Chem., Int. Ed., 2019, 58, 3690–3693 CrossRef CAS PubMed.
- E. F. Palermo and K. Kuroda, Chemical structure of cationic groups in amphiphilic polymethacrylates modulates the antimicrobial and hemolytic activities, Biomacromolecules, 2009, 10, 1416–1428 Search PubMed.
- C. Ergene, K. Yasuhara and E. F. Palermo, Biomimetic antimicrobial polymers: recent advances in molecular design, Polym. Chem., 2018, 9, 2407–2427 RSC.
- Y. Chen, M. T. Guarnieri, A. I. Vasil, M. L. Vasil, C. T. Mant and R. S. Hodges, Role of peptide hydrophobicity in the mechanism of action of α-helical antimicrobial peptides, Antimicrob. Agents Chemother., 2007, 51, 1398–1406 CrossRef CAS PubMed.
- B. Deslouches, S. M. Phadke, V. Lazarevic, M. Cascio, K. Islam, R. C. Montelaro and T. A. Mietzner, De nova generation of cationic antimicrobial peptides: influence of length and tryptophan substitution on antimicrobial activity, Antimicrob. Agents Chemother., 2005, 49, 316–322 CrossRef CAS PubMed.
- Z. Liu, A. Brady, A. Young, B. Rasimick, K. Chen, C. Zhou and N. R. Kallenbach, Length effects in antimicrobial peptides of the (RW)n series, Antimicrob. Agents Chemother., 2007, 51, 597–603 CrossRef CAS PubMed.
- W. Shen, P. He, C. Xiao and X. Chen, From antimicrobial peptides to antimicrobial poly(α-amino acid)s, Adv. Healthcare Mater., 2018, 7, 1800354 Search PubMed.
- P. Salas-Ambrosio, A. Tronnet, P. Verhaeghe and C. Bonduelle, Synthetic polypeptide polymers as simplified analogues of antimicrobial peptides, Biomacromolecules, 2021, 22, 57–75 CrossRef CAS.
- B. Zhang, M. Zhang, M. Lin, X. Dong, X. Ma, Y. Xu and J. Sun, Antibacterial copolypeptoids with potent activity against drug resistant bacteria and biofilms, excellent stability, and recycling property, Small, 2022, 18, 2106936 CrossRef CAS.
- J. Sun, X. Ma, R. Li, M. Lin, L. Shu and X. Chen, Antimicrobial nanostructured assemblies with extremely low toxicity and potent activity to eradicate Staphylococcus aureus biofilms, Small, 2023, 19, 2204039 CrossRef CAS.
- W. Zhang, S. Deng, M. Zhou, J. Zou, J. Xie, X. Xiao, L. Yuan, Z. Ji, S. Chen, R. Cui, Z. Luo, G. Xia and R. Liu, Host defense peptide mimicking cyclic peptoid polymers exerting strong activity against drug-resistant bacteria, Biomater. Sci., 2022, 10, 4515–4524 Search PubMed.
- P. Salas-Ambrosio, A. Tronnet, M. Badreldin, L. Reyes, M. Since, S. Bourgeade-Delmas, B. Dupuy, P. Verhaeghe and C. Bonduelle, Star-like poly(peptoid)s with selective antibacterial activity, Polym. Chem., 2022, 13, 600–612 RSC.
- N. Jiang and D. Zhang, Solution self-assembly of coil-crystalline diblock copolypeptoids bearing alkyl side chains, Polymers, 2021, 13, 3131 Search PubMed.
- S. M. Coulter, S. Pentlavalli, Y. An, L. K. Vora, E. R. Cross, J. V. Moore, H. Sun, R. Schweins, H. O. McCarthy and G. Laverty, In Situ forming, enzyme-responsive peptoid-peptide hydrogels: an advanced long-acting injectable drug delivery system, J. Am. Chem. Soc., 2024, 146, 21401–21416 CrossRef PubMed.
- T. Jian, M. Wang, J. Hettige, Y. Li, L. Wang, R. Gao, W. Yang, R. Zheng, S. Zhong, M. D. Baer, A. Noy, J. J. De Yoreo, J. Cai and C.-L. Chen, Self-assembling and pore-forming peptoids as antimicrobial biomaterials, ACS Nano, 2024, 18, 23077–23089 CrossRef CAS PubMed.
- I. Bano, M. Arshad, T. Yasin, M. A. Ghauri and M. Younus, Chitosan: a potential biopolymer for wound management, Int. J. Biol. Macromol., 2017, 102, 380–383 CrossRef CAS PubMed.
- X. Ding, A. Wang, W. Tong and F. Xu, Biodegradable antibacterial polymeric nanosystems: a new hope to cope with multidrug-resistant bacteria, Small, 2019, 15, 1900999 CrossRef PubMed.
- K. Skrlova, K. Malachova, A. Munoz-Bonilla, D. Merinska, Z. Rybkova, M. Fernandez-Garcia and D. Placha, Biocompatible polymer materials with antimicrobial properties for preparation of stents, Nanomaterials, 2019, 9, 1548 Search PubMed.
- S. Deng, A. Chen, W. Chen, J. Lai, Y. Pei, J. Wen, C. Yang, J. Luo, J. Zhang, C. Lei, S. N. Varma and C. Liu, Fabrication of biodegradable and biocompatible functional polymers for anti-Infection and augmenting wound repair, Polymers, 2023, 15, 120 Search PubMed.
- P. Zhou, T. Shen, W. Chen, J. Sun and J. Ling, Biodegradable polysarcosine with inserted alanine residues: Synthesis and enzymolysis, Biomacromolecules, 2022, 23, 1757–1764 CrossRef CAS.
- J. Zou, M. Zhou, Z. Ji, X. Xiao, Y. Wu, R. Cui, S. Deng and R. Liu, Controlled copolymerization of α-NCAs and α-NNTAs for preparing peptide/peptoid hybrid polymers with adjustable proteolysis, Polym. Chem., 2022, 13, 388–394 RSC.
| This journal is © The Royal Society of Chemistry 2025 |