
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrid dual-electrolyte electrochemical cells for glycerol oxidation upgradation
Genxiang
Wang
ab,
Junxiang
Chen
b,
Fen
Qiao
 a,
Junfeng
Wang
*a and
Zhenhai
Wen
*b
a,
Junfeng
Wang
*a and
Zhenhai
Wen
*b
aSchool of Energy and Power Engineering, Jiangsu University, Zhenjiang, 212013, China. E-mail: wangjunfeng@ujs.edu.cn
bCAS Key Laboratory of Design and Assembly of Functional Nanostructures, Fujian Provincial Key Laboratory of Materials and Techniques Toward Hydrogen Energy, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, China. E-mail: wen@fjirsm.ac.cn
First published on 19th June 2025
Abstract
Integrating the glycerol oxidation reaction (GOR) into aqueous electrochemical systems, such as fuel cells and electrolyzers, offers a promising strategy for utilizing the oxidized energy of glycerol to generate electricity and valuable chemicals. However, challenges remain in optimizing GOR selectivity, reducing electrolysis energy consumption, and enhancing fuel cell voltage and energy density. Recent advancements in GOR electrocatalysis have demonstrated its potential to efficiently convert glycerol into valuable products or electrical energy, even achieving dual functionality in some cases. Additionally, the integration of hybrid dual-electrolyte systems, where a pH gradient is established with a higher pH at the anode than at the cathode, has been shown to significantly improve the performance of GOR-based aqueous devices by harnessing electrochemical neutralization energy (ENE) and creating optimal reaction conditions for both the anode and cathode. In this perspective, we provide a comprehensive exploration of the electrochemical GOR and its integration into hybrid dual-electrolyte systems. Given the strong correlation between various factors and GOR performance in hybrid systems, we first provide a brief overview of GOR pathways, catalytic mechanisms, and key performance determinants (including potential, current density, electrolyte selection, and electrocatalyst design) to deepen the understanding of fundamental processes and guide catalyst design. We then highlight the integration of the GOR into aqueous advanced hybrid dual-electrolyte devices, emphasizing recent breakthroughs and issues warranting further research. Finally, we discuss the current challenges and future prospect concentrating on optimizing hybrid dual-electrolyte systems for large-scale application. This perspective aims to deepen the fundamental understanding of GOR application in hybrid dual-electrolyte systems, stimulate scientific curiosity, and guide future research in this emerging field.
 Genxiang Wang | Genxiang Wang received her PhD degree from University of Chinese Academy of Sciences in 2020. She is currently working as a lecturer at School of Energy and Power Engineering, Jiangsu University. Her current research interest mainly focuses on nano materials and catalysts for hydrogen production from water electrolysis coupled with electrooxidation of small molecules, and electrochemical reduction of CO2. |
 Junfeng Wang | Junfeng Wang received his PhD degree from Jiangsu University in 2002, where he has been serving as a faculty member since graduation. His research specializes in biomass energy conversion and green hydrogen production, dust pollution control, multiphase flow and heat transfer, and complex flow phenomena in energy and power engineering. His technical expertise includes particular expertise in electrostatic spraying, computational fluid dynamics (CFD), and advanced flow measurement technologies. |
 Zhenhai Wen | Zhenhai Wen received his PhD degree from the Chinese Academy of Sciences China in 2008. He joined the Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences in 2015. Research topics in his group mainly include the design and synthesis of functional nanostructures and exploration of their applications in electrochemical energy conversion and storage systems. |
1 Introduction
The unsustainable reliance on fossil-derived chemical production has intensified the search for alternative feedstocks and processes that simultaneously address energy storage and value-added synthesis.1–5 Among potential feedstocks, glycerol, as a biodiesel byproduct produced at 10 wt% yield, stands out due to unique structural features with triple hydroxyl functionality that enables diverse oxidation pathways with abundant type of oxidation products.6,7 Glycerol can be transformed into numerous value-added chemicals through diverse conversion pathways, such as reforming, hydrogenolysis, reduction and oxidation processes.8–11 Of particular significance is the electrochemical glycerol oxidation reaction (GOR), which has attracted substantial research interest for its dual capabilities in energy conversion and value-added production within aqueous electrochemical systems.7,12–14 As a green alternative to conventional thermochemical methods, the electrochemical GOR operates under mild conditions while offering precise reaction control. Its remarkably low thermodynamic potential (Eθ = 0.003 V), compared to the oxygen evolution reaction (OER, Eθ = 1.23 V), makes the GOR an excellent OER substitute in aqueous electrolysis devices, particularly when coupled with cathodic reactions like the hydrogen evolution reaction (HER), CO2 reduction reaction (CO2RR), and nitrogen/nitrate reduction reactions (N2RR/NO3−RR).6,15–19 This pairing not only significantly lowers the overall cell voltage but also generates valuable oxidation products at the anode. Moreover, glycerol's advantageous properties, including its non-toxic nature, high energy density (5.0 kW h kg−1) and renewable origin, position it as a promising candidate for dual-functional fuel cell application that combine power generation with chemical synthesis.20,21The conversion efficiency of the GOR, which fundamentally governs its practical application, is primarily dictated by the electronic and structural properties of electrocatalysts in this complex multi-electron transfer process.22 This process transforms hydroxyl into carboxyl groups and breaks C–C bonds, yielding a spectrum of C1 to C3 products with distinct economic values. Recent advances in catalyst engineering have led to remarkable improvements in GOR efficiency,22–26 particularly through the development of non-precious metal catalysts that have demonstrated exceptional activity (delivering 10 mA cm−2 at potentials less than the theoretical OER potential) and formate selectivity (>90%) over the past five years.27–31 Moreover, the application of advanced in situ characterization techniques and theoretical computation has significantly deepened our understanding of GOR reaction pathways and the effects of critical parameters such as pH, electrolyte type, and applied potential.25,32,33 These insights are now enabling the rational design of more efficient GOR systems.
Beyond addressing fundamental issues, integrating the GOR into electrochemical devices, especially electrolyzers, has markedly lowered energy consumption, increased reaction rates, and enabled dual-value production.6,34,35 This progress is evidenced by the evolution from traditional single-cell and H-cell designs to advanced flow reactors, where optimized electrolyte conditions and reactor designs have enabled remarkable performance improvements.36,37 For example, a GOR-assisted alkaline water splitting (HER‖GOR) flow cell configuration achieves 100 mA cm−2 at just 1.54 V, contrasting with the less than 50 mA cm−2 at 2.0 V in conventional H-type cells.36 To further reduce the input potential, our research group has proposed a hybrid alkali/acid-electrolyte system that leverages electrochemical neutralization energy (ENE) to further reduce operational potential.15,38,39 By pairing the GOR in an alkaline anolyte with the HER in an acidic catholyte, this configuration could drive 100 mA cm−2 at voltages below 1.0 V.15
The ENE arises from the concurrent electrochemical consumption of protons at the cathode and hydroxide ions at the anode, effectively harnessing acid–base neutralization energy (the electrochemical consumption 1 M OH− and 1 M H+ can provide an extra voltage of 0.828 V, i.e. 0.0591 × ΔpH, where ΔpH is the pH difference between the cathode and anode).40 This principle is not limited to the field of electrolysis, in direct glycerol fuel cell (DGFC), ENE enables a theoretical input voltage increase to 2.055 V (assuming glycerol fully oxidized to CO2 in an electrolyte with 1 M OH− and oxygen evolution reaction (ORR) in an electrolyte with 1 M H+).29,41,42 Critical to ENE utilization is maintaining pH-sensitive electrode reactions with the cathode consuming protons and the anode depleting hydroxide ions and proper electrolyte asymmetry with the anolyte being more alkaline than the catholyte. Also, as the H+ and OH− are consumed as the electrochemical reactions proceed, maintaining pH difference between two electrolytes by an external circulating supply is crucial for stable operation.40 Theoretically, the bigger the ΔpH, the more the harvested ENE for hybrid systems. However, the ENE collected by hybrid electrochemical systems fails to increase continuously with increasing ΔpH due to limitations such as ion transport efficiency and membrane tolerance.38,43 Therefore, making the quantitative effect of ΔpH manipulation on reaction kinetics is key to determining the optimal concentrations of dual electrolytes.
Besides, the hybrid dual-electrolyte configuration offers unique advantages beyond overall device performance by enabling both oxidation and reduction reactions to proceed in their respective optimal environments.32,40 For GOR-integrated systems, this means that the alkaline anolyte boosts glycerol oxidation and catalyst efficiency, particularly for non-precious metal catalysts, while the acidic catholyte is more conducive to supplying protons for electron–proton transfer in reduction reactions such as the ORR, HER, and CO2RR.44,45 This design principle has driven growing interest in GOR-based hybrid systems, with numerous studies confirming their superior energy efficiency and product value compared to conventional symmetric-electrolyte configurations.32,38 The ability to independently optimize reaction environments becomes increasingly valuable as more complex redox pairs are integrated. These advantages position hybrid dual-electrolyte systems as a key platform for developing next-generation electrochemical devices. Remarkably, while the ENE concept has been previously applied to other small molecule oxidation (e.g., urea,43 hydrazine,46 and glucose47) assisted water splitting systems, its integration with the GOR in hybrid dual-electrolyte systems represents a brand-new application expansion. Compared with the previous studies, the GOR-integrated hybrid dual-electrolyte technology represents a substantial advancement, combining both architectural and functional innovations apart from catalyst innovation. On one hand, the device architecture has evolved from simple H-type cells to optimized flow cells, enabling significantly enhanced reaction rates. On the other hand, this system combined with excellent catalysts uniquely achieves energy-efficient dual value-added production with near-unity conversion rates, marking a critical milestone toward practical implementation of dual-electrolyte devices. Therefore, the synergistic combination of the GOR with ENE utilization through dual-electrolyte design represents a promising direction for advancing both energy storage and chemical production technologies by employing abundant renewables and wastes (Fig. 1).
 | ||
| Fig. 1 Overview of GOR involved hybrid dual-electrolyte electrochemical cell application in energy conversion and storage technologies. | ||
Though there exist several reviews on the electrocatalytic GOR, which provides valuable insights into electrocatalyst design, reaction mechanism analysis and application, a timely and specialized perspective on GOR-integrated hybrid dual-electrolyte electrochemical devices remains essential to advance its practical application. In this perspective (Fig. 2), we review the latest advancements in GOR-involved hybrid dual-electrolyte electrochemical devices, which feature a pH gradient with a more alkaline anode than cathode, based primarily on recent discoveries from our research team. Given the close relationship between catalyst development, mechanistic studies, and the advancement of hybrid dual-electrolyte electrochemical devices, we first elucidate the fundamental principles of the electrocatalytic GOR by analyzing reaction pathways and mechanisms. We emphasize the critical role of in situ characterization techniques and theoretical calculations in driving progress in this field. Subsequently, we systematically examine the key factors governing GOR selectivity and activity, with particular focus on applied potential, current density, electrolyte pH, and electrocatalyst design. Subsequently, we discuss recent advances in GOR-integrated hybrid dual-electrolyte systems, emphasizing the critical role and effectiveness of hybrid dual-electrolyte configurations and the GOR. Finally, we outline the challenges and future prospect for GOR-based dual-electrolyte devices, including electrocatalyst development and device optimization, and industrial-scale implementation.
 | ||
| Fig. 2 Overview of this perspective. | ||
2 Pathway and mechanism investigation
This section begins by summarizing the proposed GOR pathway based on established research findings. We then present recent progress in theoretical calculations and in situ electrochemical techniques that have significantly advanced our understanding of GOR catalytic mechanisms.2.1 The possible GOR pathway
The glycerol electrooxidation follows complex pathways, yielding C3 (glyceraldehyde (GLAD), 1,3-dihydroxyacetone (DHA), glyceric acid (GLA), tartronic acid (TA), mesoxalic acid (MA), lactic acid (LA), hydroxypyruvic acid (HA), pyruvaldehyde, and 2-hydroxypropenal), C2 (glycolic acid (GA), oxalic acid (OA), and acetic acid (AA)), and C1 species (formate (FA) and CO2) products (Fig. 3).1,10,48 Initial two-electron (2e−) oxidation at a primary hydroxyl group (–OH) generates GLAD, which either oxidizes subsequent to GLA, TA, and MA, or undergoes C–C bond cleavage to form GA and formate.3,49 Subsequent oxidation of TA and MA yields C2 acids (GA and OA) and FA, ultimately decomposing to CO2, while secondary –OH oxidation produces DHA, which is converted to HA and MA before C–C cleavage. The aldehyde–ketone interconversion (GLAD ↔ DHA, 2-hydroxypropenal ↔ pyruvaldehyde) and dehydration reaction (forming 2-hydroxypropenal or pyruvaldehyde) followed by Cannizzaro rearrangement (yielding LA in alkaline media) further complicate product distribution.50,51 The above pathways are supported by in situ electrochemical characterization and product analysis, with pathway selectivity being critically dependent on both the specific atomic interactions between glycerol (O atoms of primary/secondary OH and primary/secondary C atoms) and the catalyst surface, as well as the reaction conditions (including electrolyte pH, potential, and current density) that govern the adsorption/desorption kinetics of reactants and intermediates.52–54 | ||
| Fig. 3 The possible pathway of the GOR.1,10,52 | ||
2.2 Theoretical calculation and advanced in situ characterization methods for GOR mechanism investigation
 | ||
| Fig. 4 (a) Optimized structures achieved by the end of AIMC, (b) catalyst-dependent distribution fractions of the atoms on the surface (c) and layer-dependent fraction of elements, (d) volcano plots (red line) of the GOR as a function of the C2H3O3 adsorption free energy. Reproduced from ref. 15, Copyright 2022, American Chemical Society. (e) Snapshots of GLA at different electrolysis times for the PE3 protocol (EH = 0.7 VRHE for 0.05 s, EL = 0.3 VRHE for 0.5). Data obtained through COMSOL multiple physical quantities. The catalyst of Pt@G is represented by seven round white dots. (f) Schematic of the pathways of the PE-based GEOR. Reproduced from ref. 55 with permission from Springer Nature, copyright 2024. | ||
The power of these combined methods is exemplified in studies of complex catalysts. For instance, the CoNiCuMnMo HEA investigation followed a cascading workflow that DFT initially identified electronic characteristics of individual sites, ML potentials then reconstructed multi-element interaction fields, and MC simulations ultimately mapped the 2 nm-scale spatial distribution of surface elements, pinpointing Mn, Mo, and Ni coordinated Mo sites as dominant active centers (Fig. 4b–d).15 In another research, FEM modeled elemental distribution within a high-entropy sulfide FeCoNiCrMnS2, guiding subsequent DFT, MC, and ML investigations into its electrocatalytic sites. These collaborative studies confirmed that GOR performance stemmed primarily from Ni and Co sites, with Cr and Mn optimizing their electronic structure.29 The above research methods provide valuable guidance for studying GOR mechanism over complex models. Besides, FEM via COMSOL Multiphysics elucidates the critical role of electric double layer (EDL) fields in governing GOR dynamics.55,59 By tracking local reactant/product concentration evolution on Pt catalyst surfaces, Zou et al. revealed that pulsed potential (PE) modes enhance GLAD selectivity by modulating EDL ion distribution (K+/OH−/H+/C2H5O2COO−).55 The simulations demonstrated that negative ion accumulation intensifies with rising potential, while potential reduction accelerates ion desorption and diffusion. Under constant-potential (CE) conditions, GLAD accumulated on the catalyst surface, blocking active sites and impeding further glycerol oxidation. Conversely, a pulsed potential mode alternating between high and low voltages reduces GLAD and GLA accumulation, with concentrations decreasing as low potential is applied for longer durations (Fig. 4e and f). This strategy balances reaction control, effectively mitigating catalyst deactivation.
The theoretical calculation methods mentioned provide useful guidance for how to reveal GOR catalytic mechanism over catalysts, especially these complex ones. The combination of DFT, ML, MC, and FEM not only validates the catalytic mechanism but also possibly generates transferable design principles for active catalysts. For example, the identified synergy between surface-segregated Mo and sublayer Mn in HEAs informs alloy composition optimization strategies, while pulsed potential parameters derived from FEM guide practical reactor operation protocols. By systematically connecting quantum interactions, nanoscale ordering, and macroscopic transport phenomena, the theoretical calculations provide a useful platform for understanding the GOR where dynamic interface evolution and multi-step pathways demand coordinated computational insights across spatial and temporal scales.
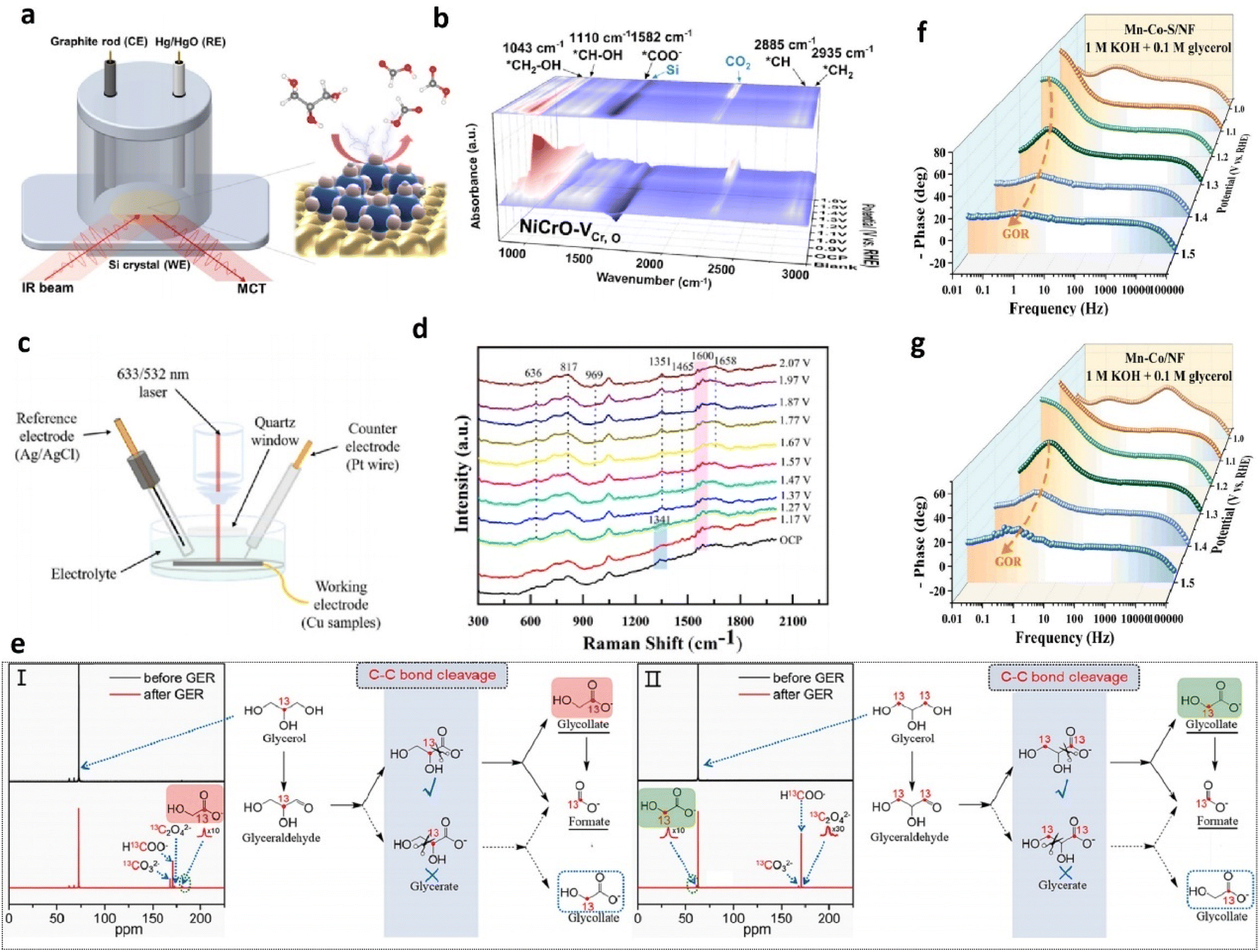 | ||
| Fig. 5 (a) Illustration of in situ ATR-SEIRAS. (b) In situ ATR-SEIRAS spectra of the GOR on NiCrO–VCr,O nanosheet electrodes at various potentials. Reproduced from ref. 63, copyright 2024, American Chemical Society. (c) Schematic diagram for in situ Raman measurements. (d) In situ electrochemical Raman spectra on Cu NPs in 0.1 M KOH with 0.1 M glycerol solution. Reproduced from ref. 70, copyright 2023, Elsevier. (e) 13C-NMR spectra for the electrooxidation of isotope-labelling (I) 2-13C glycerol and (II) 1, 3-13C glycerol. Reproduced from ref. 60, copyright 2023, Elsevier. Bode plots of Mn–Co–S/NF (f) and Mn–Co/NF (g) for the GOR. Reproduced with permission from ref. 66, copyright 2024, Elsevier. | ||
Aside from the techniques mentioned above, electrochemical impedance spectroscopy (EIS) conducted at various potentials examines interfacial electrochemical behavior of catalysts (which were termed as “in situ EIS” or “operando EIS” in some of reports), with Bode phase plots analyzing electrode/electrolyte interactions during the GOR process.28,32,66,68,72 The Bode phase plot tracks phase angle changes with frequency, where the phase angle peak at the high-frequency region indicates surface oxidation of catalysts, and the one at the low-frequency region is linked to the interfacial OER/GOR.66,73 The appearance of inflection points at the low-frequency region represents occurrence of the GOR or OER. The lower the potential for the first appearance of phase angle peak at the low-frequency region, the lower the onset potential for the GOR or OER. Moreover, a lower phase angle value indicates a faster interfacial charge transfer process and heightened reaction kinetics of the OER/GOR. For instance, Wang et al. investigated the interfacial behavior of their prepared sulfur doped manganese–cobalt hydroxide nanosheets on a nickel foam (Mn–Co–S/NF) electrode via EIS measurements.66 With the increase of the applied potential, Mn–Co–S/NF exhibits a much quicker decrease rate of the phase angle peak in the high frequency and lower potential for the first appearance of the phase angle peak in the low frequency than the controlled sample without introduced S (Mn–Co/NF) (Fig. 5f and g), confirming its faster oxidation rate of surface Co species and lower overpotential for the GOR.
The operando/in situ electrochemical techniques mentioned achieved multi-scale monitoring from molecular adsorption to interfacial kinetics, offering critical insights into GOR mechanisms. Nevertheless, limitations such as technique-specific detection biases, insufficient temporal resolution, and reliance on idealized conditions hinder holistic mechanistic understanding. To address these gaps, it is imperative to integrate tools like time-resolved in situ NMR (tracking liquid-phase product dynamics), online electrochemical mass spectrometry (EC-MS, capturing transient gas-phase intermediates), and multiscale computational models, with the aforementioned techniques to achieve full-spectrum reaction monitoring.
3 Key factors influencing the selectivity and activity of the GOR
The complexity of the GOR mechanism makes its selectivity and activity highly sensitive to operational parameters such as applied potential, current density, and electrolyte composition, as well as the choice of electrocatalyst. A deep understanding of these factors is essential for optimizing GOR performance across electrochemical systems. In this section, a summary of the influence of these factors based on the latest research is presented.3.1 Operating reaction conditions
Reaction conditions such as current density, applied potential, glycerol concentration, and electrolyte pH profoundly influence the GOR performance.33,54,55,74–77 Based on these previous studies, the impact rules on Pt-based electrocatalysts can be summarized as follows: the higher current densities and anodic potentials accelerated intermediate re-oxidation, favoring complete oxidation to CO2 over C3 products like GLYC or LAC. Slower anolyte flow rates extended intermediate residence times, enhancing C1 species formation, while lower glycerol concentrations and higher alkaline conditions favored deeper oxidation to C3 acids (e.g., GLA and TA) due to the significant reduced O–H bond activation energy.20,74 Overall, conditions including slower flow rates, reduced glycerol concentrations, elevated current densities, and higher anodic potentials collectively promote C–C bond cleavage while suppressing C3 product formation. This trend demonstrates broad applicability across both noble and non-noble metal catalyst systems.In addition, beyond its specific value, the potential mode also significantly influences GOR selectivity.76–78 Dynamic potential modulation represents a powerful strategy to enhance catalytic selectivity and activity.55,79,80 The programmed potential electrolysis employs an oxidation plateau with an extended plateau at one potential (tens of seconds) for primary reactions, then followed by a short pulse (about 0.5–1 s) to a more positive potential for oxidizing strongly adsorbed intermediates formed during the first potential stage, and an optional preconditioning phase with a short cathodic step at more negative potentials (less than 10 s) to facilitate organic adsorbate accumulation or adatom deposition for surface modification may also be included.80 Holade et al. pioneered pulsed glycerol electrolysis using Pd-based nano electrocatalysts, employing alternating potentials of 0.8 VRHE (20 s) for the GOR and 1.4 VRHE (1 s) for the regeneration of the electrode surface.79 Similarly, carbon-encapsulated Pt catalysts (C@Pt) under pulsed potentials (0.3 VRHE for 0.5 s and 0.7 VRHE for 0.05 s) achieve 81.8% GLA selectivity, a 2.2-fold improvement over constant potential operation at 0.7 VRHE (37.8%).55 This method mitigates over-oxidation of active sites under sustained anodic bias and accumulation of poisoning intermediates, thereby preserving active sites for sustained OH− adsorption and glycerol activation. Although demonstrated for noble metal catalysts, its applicability to non-noble metal catalysts remains to be established experimentally.
Additionally, electrolyte pH critically governs GOR performance as it influences catalyst species and modulates reaction activity and selectivity.78,81–86 Previous studies have shown that pH significantly affects the performance of Pt and Au electrodes, with both exhibiting higher activity in alkaline environments.83,86 Notably, while both Pt and Au show higher activity in alkaline media, Au displays distinct selectivity shifts, producing DHA, TA, and C1 products under alkaline conditions versus primarily TA, formate and CO2 in acidic environments. Pt electrodes, however, showed no pH dependence in selectivity.83 Then the pH dependence of oxidation activity for all potential GOR intermediates was examined over Pt catalysts to reveal its influence on GOR pathway.33 The study revealed that alkaline media enhanced glycerol oxidation and its adsorption and kinetics of oxidation products, leading to complete glycerol oxidation to CO2. However, further oxidation of certain intermediates like TA, formic acid, and OA was challenging, marking them as dead-end species in the alkaline GOR over Pt catalysts, which was thought to be a critical bottleneck restricting complete glycerol oxidation into CO2. Key intermediates from glycerol, such as GLAD and DHA, could be oxidized to CO2 with low overpotential, as can formate, irrespective of pH. Beyond the influence of noble metal-based catalysts, electrolyte pH critically modulates C–C bond scission in the non-noble metal-catalyzed GOR, as demonstrated by Mn-based electrocatalysts.49 MnOx (x ≈ 2) with adjacent Mn sites favored C3 product (GLAD and DHA) formation in neutral to mildly basic media (pH at round 7 to 10), while acidic or strongly alkaline environments promote geminal diol formation, accelerating C–C cleavage to formate as the primary product. The aforementioned studies offer insights into how pH adjustment can modulate selectivity in GOR processes. Notably, given the close interrelationship between pH and current density in reaction systems, solely correlating pH shifts with selectivity changes may be insufficient to fully capture the underlying mechanisms.
Beyond experimental studies, DFT calculations elucidate how pH governs GOR selectivity at the electronic level.32,86 Verma et al. employed DFT calculations to elucidate the pH-dependent catalytic behavior of Au in the GOR, systematically revealing why alkaline conditions significantly enhance both activity and selectivity.86 Their computational analysis demonstrated that the alkaline conditions facilitated generation of optimal surface hydroxyl coverage (Au(OH)ads), creating dual active sites where Au(OH)ads synergizes with metallic Au to stabilize key intermediates. This cooperative mechanism enables thermodynamically favored glyceric acid formation with low overpotential. Conversely, acidic media lacking OH− coverage force a kinetically constrained pathway dominated by dihydroxyacetone (DHA) production. Besides, the free energy changes associated with the C–C bond cleavage step against pH on Pt(111) surface was studied, specifically for CH2OHC*OC*Oads decomposing into CH2OHC*Oads and COads. It was found that free energy change values decreased with increasing pH, suggesting a higher barrier for C–C bond cleavage and higher selectivity to C3 product formation at lower pH levels. These computational insights confirm that pH universally steers reaction pathways by modulating intermediate adsorption strength and transition-state energetics, underscoring its critical role in determining GOR selectivity.
In summary, despite some catalyst-specific limitations in current studies, the operational principles for the GOR derived from the above research studies provide critical guidance for hybrid dual-electrolyte system optimization. The regulatory principles of current density, potential modulation, electrolyte flow rate, and glycerol concentration directly indicate operational parameter optimization. The influence mechanism of pH not only offers guidance for catalyst selection (alkaline conditions universally enhance catalytic activity for both noble and non-noble metals) but also directs target-product selectivity. Although alkaline electrolytes demonstrate clear advantages in hybrid systems, the dynamic equilibrium between their concentration and glycerol concentration requires further investigation to achieve synergistic optimization of selectivity and activity. Moving forward, it will be of great significance by synchronizing pulsed potential modes with pH adjustments in hybrid dual-electrolyte systems to decouple competing reactions, maximizing target-product selectivity.
3.2 Electrocatalysts
Electrocatalysts play a pivotal role in dictating the GOR performance in hybrid dual-electrolyte systems. Therefore, by employing various synthesis methods and state-of-the-art techniques, the development of efficient catalysts with desirable stability, activity and selectivity, while elucidating their catalytic mechanisms, has been at the forefront of research efforts (Fig. 6). GOR catalysts are broadly categorized into noble metal-based catalysts, such as Pt, Au, Pd, and non-noble metal-based materials like Ni, Co, Cu, Mn, etc. Both categories provide unique benefits in facilitating the GOR process, and have been widely used in various GOR-involved electrolysis systems. | ||
| Fig. 6 Overview of design strategies and involved-elements for GOR electrocatalysts. | ||
| Catalysts | Electrolyte | Durability test (current density/mA cm−2 or potential/VRHE) | Potential @ current density | FE (products) | References |
|---|---|---|---|---|---|
| SbOx–Pt/OMC | 0.5 M H2SO4 + 2 M glycerol | 120 h (0.92 VRHE) | 272.3 mA mgPt−1 @ ∼0.95 VRHE | 81.1%/(DHA) | 119 |
| Pt3Sn/hNCNC | 1 M KOH + 0.2 M glycerol | 2 h (0.75 VRHE) | 5.9 A gPt−1 @ ∼0.8 VRHE | (CO2) | 93 |
| PtZn-IMC@NC | 1 M KOH + 0.1 M glycerol | — | 1477 mA mgPt−1 @ ∼0.90 VRHE | (formate) | 42 |
| PdNi/C | 1 M KOH + 0.5 M glycerol | — | 211 mA gPd−1 @ 1.2 VRHE | 64% (GLA) | 143 |
| Pd-NC/NF | 1 M KOH + 0.1 M glycerol | 4 h (1.2 VRHE) | 250 mA cm−2 @ 1.22 VRHE | 42% (GLA) | 139 |
| Au/Ni(OH)2 | 3 M KOH + 0.3 M glycerol | 10 h (0.95 VRHE) | 317.7 mA cm−2 @ 0.95 VRHE | 77% (LA) | 125 |
| Au1Cu1(111) | 1 M KOH + 0.1 M glycerol | 24 h (1.23 VRHE) | 39 mA cm−2 @ 1.23 VRHE | 45% (GA) | 123 |
| RhCu-BMLs | 1 M KOH + 0.1 M glycerol | 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s (0.61 VRHE) 000 s (0.61 VRHE) |
579.3 A gRh−1 @ 0.61 VRHE | 78.7% (GLA) | 142 |
| Ru@MnO2 | 1 M KOH + 0.5 M glycerol | 24 h (10 mA cm−2) | 10 mA cm−2 @ 1.13 VRHE | 92% (formate) | 30 |
Pt-based materials are the most studied among noble metal catalysts for the GOR in DGFCs due to their high catalytic activity and low onset potential.20,41,89 Initial studies on single crystalline Pt catalysts revealed obvious crystalline facet-dependent reaction pathway,53,90 where Pt(111) facets produce multiple oxidation products (glyceraldehyde, glyceric acid, dihydroxyacetone) through facilitating dual Pt–C bonded enediol intermediates enabling diverse product formation, while Pt(100) facets exclusively generate glyceraldehyde via single Pt![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C interactions.53 The crystallographic effects, confirmed by combined experimental and computational analyses, originate from distinct coordination numbers of their Pt surface atoms. Besides, the GOR performance and reaction mechanisms of various polycrystalline Pt nanoparticles on conductive carbons (Pt/C) were carefully examined, which well revealed the reaction pathways and catalytic mechanism over Pt catalysts.83,91,92 Though Pt catalysts were confirmed to possess unique advantages of low overpotential and substantial C2 or C3 product yields at low overpotentials for the GOR, they faced issues with high costs, complex oxidation products, and CO poisoning, particularly in alkaline media, which led to subsequent modifications of Pt catalysts via strategies such as alloying,93,94 supporting,95,96 coating,97 modifying,97–99 forming intermetallics,42,100 and core–shell structures.101
C interactions.53 The crystallographic effects, confirmed by combined experimental and computational analyses, originate from distinct coordination numbers of their Pt surface atoms. Besides, the GOR performance and reaction mechanisms of various polycrystalline Pt nanoparticles on conductive carbons (Pt/C) were carefully examined, which well revealed the reaction pathways and catalytic mechanism over Pt catalysts.83,91,92 Though Pt catalysts were confirmed to possess unique advantages of low overpotential and substantial C2 or C3 product yields at low overpotentials for the GOR, they faced issues with high costs, complex oxidation products, and CO poisoning, particularly in alkaline media, which led to subsequent modifications of Pt catalysts via strategies such as alloying,93,94 supporting,95,96 coating,97 modifying,97–99 forming intermetallics,42,100 and core–shell structures.101
Among various modifications, bimetallic and multimetallic catalysts, such as those combining Pt with Au,102 Ru,91,103 Ag,104–106 Bi,107–110 Sn,111 Pb,112 In,112 Sb,113 Cu,114,115 Zn,42 Fe,116 and Co,117 have improved peak current density, selectivity, anti-poisoning capability, and reduced Pt dosage. Pt-based intermetallic catalysts have demonstrated improved GOR performance owing to the regulated electronic structure of Pt by introducing other metals.42,118 For instance, a N-doped carbon supported intermetallic PtZn catalyst (PtZn-IMC@NC) presented excellent activity for the GOR, delivering an ultrahigh oxidation peak current of 1477 mA mgPt−1 at 0.927 VRHE in alkaline medium with formate, DHA and GLAD as the main products, far higher than that of control samples (Fig. 7a–c).42 As DFT calculation indicated, the excellent activity of the prepared catalyst can be attributed to the highly ordered structure of the formed intermetallic PtZn that especially benefits Pt exposure. Also particularly noteworthy is the Bi-modification strategy, where Bi adatoms on Pt surfaces prevent formation of strongly adsorbed poison species and create geometrically blocked sites and electronic interfaces that fundamentally alter reaction pathways.98 In acidic media, Bi-modified Pt/C achieved near-perfect dihydroxyacetone (DHA) selectivity at reduced potentials through selective secondary alcohol oxidation,107 while under alkaline conditions, through selective activation of low-coordinated Pt atoms and stabilization of key reaction intermediates, optimal Bi decorated polycrystalline Pt favored glycerate production by suppressing C–C cleavage and CO formation.97,98,108 Subsequent single-crystal studies further unveiled fundamental facet effects that Bi decoration on Pt(111) enhanced both DHA selectivity and activity through enediol intermediate stabilization and CO poisoning resistance, while Bi decoration on Pt(100) maintained glyceraldehyde selectivity with compromised activity due to ineffective intermediate stabilization.109 Notably, the high selectivity of Bi modified Pt electrodes for DHA has only been shown under low current densities during catalytic mechanism research. As of now, there are no reports indicating that high-selectivity DHA production can be achieved at larger current densities (greater than 10 mA cm−2). Therefore, it is necessary to further explore whether Bi-modified Pt electrodes can maintain high DHA selectivity when operating at high current densities in acid electrolytes.
 | ||
| Fig. 7 (a) TEM image of PtZn-IMC@NC, (b) CVs of PtZn-IMC@NC and controlled samples in 1 M KOH containing 0.5 M glycerol solution, (c) the possible pathway of the GOR over PtZn-IMC@NC. Reproduced with permission from ref. 42. Copyright 2024, Wiley-VCH. (d) SEM image of the Au/Ni(OH)2 catalyst, (e) current density of Au/Ni(OH)2 and Au at 0.95 VRHE in the electrolytes with 0.3 M glycerol and different concentrations of KOH, (f) adsorption energies of glycerol in the form of alkoxide and the optimized geometries of (HOCH2)2CH2O−* for various catalysts. The color of each element is yellow for Au, blue for Ni, red for O, white for H, gray for C, and purple for K. (g) Schematic illustration of the adsorption configuration of glycerol alkoxide at the Au/Ni(OH)2 interface. Reproduced with permission from ref. 125. Copyright 2023, American Chemical Society. (h) TEM image of Bi1Pd NS, (i) potential-dependent FEs for different oxidation products on Bi1Pd NS. Reproduced with permission from ref. 138. Copyright 2024, Royal Society of Chemistry. (j) TEM image of hollow o-Pd2Sn NPs, (k) cyclic voltammetry curves of various catalysts in 1.0 M KOH solution containing 1.0 M glycerol (monoclinic Pd3Sn2 (m-Pd3Sn2)). Reproduced with permission from ref. 133. Copyright 2023, Wiley-VCH. | ||
Alternative modification approaches demonstrate comparable success. For example, amorphous antimony oxide on Pt nanoparticle surface (SnOx–Pt) supported by ordered mesoporous carbon (OMC) achieved a DHA selectivity of 81.1% in acid solution, maintaining 53.1% even after 120 h of constant electrolysis.119 As confirmed by the DFT calculation results, the SnOx improved the catalytic activity and stability of Pt by forming an ionic bond with O atoms of DHA, suppressing DHA interconversion reactions. Analysis of representative Pt-based catalyst systems above reveals that while significant advancements have been achieved in catalytic activity and stability, attaining high selectivity toward a single product remains challenging. The development of strategies that effectively balance selectivity with activity represents a critical requirement for functional device applications.
Aside from Pt-based materials, Au-based and Pd-based catalysts have been engineered into various structures to enhance GOR performance. Early studies on Au-based catalysts, primarily involving polycrystal Au or Au nanoparticles on carbon supports, showed high selectivity for C3 or C2 products.76,120 Subsequently, advanced nanostructuring of Au catalysts121,122 and the incorporation of secondary metals or metal compounds have created complex structures that significantly increase active surface area, adsorption of key intermediates, suppress C–C bond cleavage, and facilitate electron transfer.123–125 A self-supported nanostructured Au electrode achieved a GA selectivity of 41.2% in an alkaline electrolyte by adjusting Au(100) and (110) crystal facets, promoting OH adsorption for selective GA production.121 The selectivity to GA was further improved to 45% over a hollow spheroidal Au1Cu1 alloy catalyst, where Cu inclusion expanded the active surface area and enhanced the adsorption of glycerol and its intermediates, enhancing the capacity of Au to adsorb more –OH groups.123 Besides, a high selectivity of 77% to LA was also achieved at 0.95 VRHE with a current density of 326.2 mA cm−2 over a nickel hydroxide nanosheet-supported gold electrocatalyst (Au/Ni(OH)2) in alkaline media (Fig. 7d). The exceptional activity of Au/Ni(OH)2 was attributed to the interface enriching glycerol in the alkoxide form via a σ bond (between Au and glycerol alkoxide group) and hydrogen bond (between Ni(OH)2 and adjacent OH groups), subsequently oxidizing adsorbed alkoxides by OH* generated on the Au. The superior ability of Au/Ni(OH)2 to promote glycerol adsorption in its alkoxide was evidenced by a markedly higher current density at elevated KOH concentrations compared to Au alone and stronger adsorption energies (Eads) for glycerol alkoxide (Fig. 7e–g). While these Au-based catalysts have demonstrated favorable GOR selectivity and activity, further investigation into the relationship between catalyst structure and selectivity is needed to elucidate the origins of their activity and product specificity.
Similar to Pt and Au-based catalysts, previous studies confirmed that modifying Pd catalysts with other elements, tuning nano shapes, and alloying with other metals could enhance GOR performance by increasing Pd site exposure, improving poison resistance, and enhancing adsorption of OH species, whereas the structure–selectivity relationship remains insufficiently understood.79,126–132 Recent advances in nanosynthesis have expanded the diversity of Pd-based catalysts, such as single-atom modifications, intermetallic phases, and hollow architecture, enabling tailored activity and selectivity.133–140 For instance, a catalyst composed of Bi atoms dispersed on Pd nanosheets (Bi1Pd NS) has demonstrated over 90% selectivity for converting glycerol into C3 products with high partial current density and stability (Fig. 7h–i). The introduced Bi atoms modulated the electronic structure of Pd, lowering the adsorption energy of key C3 intermediates and alleviating product over-oxidation and catalyst poisoning.138 Another research study found that orthorhombic Pd2Sn intermetallic nanoparticles (o-Pd2Sn) with a hollow structure achieved exceptional mass activity of (12.9 A mgPd−1) and C–C cleavage capability (Fig. 7j and k),133 which is credited to the alloying effect of Pd and Sn, the ordered intermetallic phase, and the hollow structure that increase the reactive surface area and noble metal utilization rate. The above representative findings confirm advancement achieved in precise control over Pd-based catalyst structures to simultaneously address activity, selectivity, and stability challenges in the GOR, which give strategies for optimizing other noble metal catalysts.
Aside from these mainstream noble metal catalysts, materials based on Rh and Ru have also been tailored for the GOR through doping, complexing, or alloying with non-noble metals.17,30,72,141,142 For instance, RhCu bimetallenes (RhCu-BMLs) with optimal Rh/Cu molar ratios achieved a high mass activity of 579.3 A gRh−1 at 0.61 VRHE and selectivity to GLA with an FE of 78.7%, surpassing single-component Rh metallenes.142 The incorporated Cu atoms were found to weaken OH− adsorption, preventing the oxygen insertion reaction that resulted in TA production owing to the electronic effect.
In summary, the collective progress in noble-metal based catalysts for the GOR underscores the critical role of atomic-scale design strategies of electronic modulation, facet engineering, and interfacial synergy in balancing activity, selectivity, and stability. However, persistent gaps remain, including the scalability of high-selectivity for single C3 or C2 products to industrially relevant current densities and the need for universal descriptors linking catalyst structure to reaction pathways. Future efforts should prioritize operando mechanistic studies and scalable synthesis methods to bridge these gaps, advancing toward practical electrochemical glycerol valorization in various hybrid dual-electrolyte devices.
| Catalysts | Electrolyte | Durability test (current density/potential) | Potential (VRHE) @ current density (mA cm−2) | FE (products) | References |
|---|---|---|---|---|---|
| NiCrO–VCr,O | 1 M KOH + 0.1 M glycerol | 100 h (100 mA cm−2) | 1.37 V @ 10 | ∼95% (formate) | 63 |
| NiCo2O4 | 1 M KOH + 0.1 M glycerol | — | 1.219 @ 10 | 94% (formate) | 28 |
| NiCo2O4 | 1 M KOH + 0.1 M glycerol | 120 (1.42 VRHE) | 1.23 @ 10 | ∼70% (formate) | 57 |
| NiCo2O4/NF | 1 M NaOH + 0.1 M glycerol | 120 (1.4 VRHE) | 1.23 @ 10 | >97% (formate) | 60 |
| NiCo2O4/NF | 1 M KOH + 0.5 M glycerol | 12 h (10 mA cm−2) | 1.15 @ 10 | 91.2% (formate) | 163 |
| OX-Ni0.5Co0.5(OH)2 | 1 M KOH + 0.1 M glycerol | 20 h (10 mA cm−2) | 1.26 @ 10 | 91.% (formate) | 164 |
| NiCo2O4/NF | 1 M KOH + 0.1 M glycerol | 48 (1.4 V vs. RHE) | 1.42 @ 300 | 89.9% (formate), 7.62% (GLA) | 37 |
| CuCo2O4 | 0.1 M KOH + 0.1 M glycerol | 19 h (1.3 VRHE) | 1.38 @ 10 | 80.6% (formate) | 150 |
| NiOOH/Ni3S2/NF | 1 M KOH + 0.1 M glycerol | 24 h (10 mA cm−2) | 1.227 @ 10 | 97.7% (formate) | 69 |
| Ni0.33Co0.67(OH)2@HOS/NF | 1 M KOH + 0.1 M glycerol | 10 h (10 mA cm−2) | 1.30 @ 10 | 96% (formate) | 165 |
| Ni3N/Co3N-NWs | 1 M KOH + 0.1 M glycerol | 12 h (1.35 VRHE) | 1.26 @ 50 | 94.6% (formate) | 62 |
| Cu–NiCo/NF | 1 M KOH + 0.1 M glycerol | 40 h (1.45 VRHE) | 1.23 @ 10 | 93.8% (formate) | 144 |
| NiVRu/NF | 1 M KOH + 0.1 M glycerol | 50 h (50 mA cm−2) | 1.24 @ 10 | 97% (formate) | 72 |
| Rh0.3[Ni(OH)2]0.7/C | 1 M KOH + 0.5 M glycerol | 1 h (1.5 VRHE) | 1.29 @ 10 | 44.5% (TA) | 141 |
| A-NiFe-LDH | 1 M KOH + 0.1 M glycerol | — | 1.42 @ ∼200 | 99.03% (formate) | 151 |
| CoO/NF | 1 M KOH + 0.1 M glycerol | 108 h (∼200 mA cm−2) | 1.201 @ 10 | 91.3% (formate) | 166 |
| CoSe2/CC | 1 M KOH + 0.5 M glycerol | 90 h (1.48 VRHE) | 1.23 @ 10 | ∼88% (formate) | 147 |
| Mn–CoSe2/CFC | 1 M KOH + 0.1 M glycerol | 12 h (10 mA cm−2) | 1.27 @ 10 | >90% (formate) | 39 |
| Mn–CoN@C/NF | 1 M KOH + 0.3 M glycerol | 70 h (1.4 VRHE) | 1.19 @ 10 | 97.7% (formate) | 27 |
| CNs@CoP | 1 M KOH + 1 M glycerol | — | 1.6 @ 140 | >85% (formate) | 167 |
| Co–Mn–S/NF | 1 M KOH + 0.1 M glycerol | 60 h (10 mA cm−2) | 1.347 @ 100 | ∼90% (formate) | 66 |
| CoCuN0.6/CP | 1 M KOH + 0.1 M glycerol | 60 h (1.43 VRHE) | 1.07 @ 10 | 90% (formate) | 67 |
| CNs@CoP | 1 M KOH + 0.1 M glycerol | — | 1.6 @ 140 | 95.3% (formate) | 167 |
| Co(OH)2–SDS | 1 M KOH + 0.1 M glycerol | 20 h (1.42 VRHE) | 1.42 @ 27.73 | 83.3% (formate) | 148 |
| CoOxHy/CF | 0.4 M H3BO3 (80%) + 0.1 M Na2B4O7 (20%) + 0.05 M glycerol | 6 h (1.56 VRHE) | — | 43.2% (HPA), 12.8% (DHA) | 84 |
| MnO2–CuO/CF | 1 M KOH + 0.1 M glycerol | 30 h (1.3 VRHE) | 1.30 @ 10.7 | 60% (DHA) | 75 |
| MnO2 nanoneedles | 0.1 M Na2B4O7 + 0.1 M glycerol | 3 h (2.05 VRHE) | 2.05 @ ∼10 | 46% (DHA) | 78 |
| MnO2/CP | 0.005 M H2SO4 + 0.1 M glycerol | 865 h (10 mA cm−2) | 1.36 @ 10 | (Formate) | 156 |
| S–CuO/CF | 1 M KOH + 0.1 M glycerol | 10 h (1.35 VRHE) | 1.23 @ 100 | 95.7%/(formate) | 31 |
| CeVO4 | 1 M KOH + 0.2 M glycerol | — | 1.45 @ 10 | 91.5%/(formate) | 168 |
| HEA-CoNiCuMnMo/CC | 1 M KOH + 0.1 M glycerol | 12 h (10 mA cm−2) | 1.25 @ 100 | >90%/(formate) | 15 |
| FeCoNiCrMnS2/CC | 1 M KOH + 0.1 M glycerol | 12 h (100 mA cm−2) | 1.1 @ 10 | >90%/(formate) | 29 |
| (CoNiCuMnMo)Se/CF | 1 M KOH + 0.1 M glycerol | 25 h (10 mA cm−2) | 1.2 @ 10 | (Formate) | 161 |
Enhancements to Co-based catalysts for the GOR have been achieved by incorporating secondary metals (e.g., Ni, Cu, Mn, Fe),28,66,67 non-metal elements (e.g., S, B, Se),66,85,147 and organic compounds to form nano compounds or complex nanostructures.148,149 Notably, replacing Co with Ni in the Co3O4 spinel oxide to form NiCo2O4 has drawn considerable interest.28,37,60 A recent study detailed how NiCo2O4 nanosheets, with an optimal Ni:Co ratio of 2:1, managed competitive adsorption of OH* and glycerol, leading to high formate selectivity and low overpotentials (Fig. 8a–c).28 DFT calculations confirmed that the appropriate d-band center energy levels of NiCo2O4 for glycerol and C–O intermediate adsorption/desorption contributed to the good GOR performance. However, NiCo2O4 nanoneedle arrays exhibited different selectivity profiles, producing GA alongside formate (Fig. 8d and e).37 Moreover, the authors found that the GOR proceeded via direct oxidation at 1.00 VRHE, with active metal (oxy)hydroxides engaging in an indirect oxidation mechanism at elevated potentials (Fig. 8f). The improved performance was traced to the introduction of Ni into the Co3O4 structure, increasing oxygen vacancy concentration and promoting the rapid formation of active NiIII–OOH and CoIII–OOH sites for the GOR. Besides, other metals like Cu and Mn also promoted the formation of high-valance Co and Ni active sites and glycerol adsorption.27,144,150 For example, a Cu-doped NiCo alloy electrode confirmed surface NiIII–OOH and CoIII–OOH species as active sites, with Cu facilitating coupling of surface *O with reactive intermediates.144 A catalyst with carbon shell-encapsulated manganese-doped cobalt nitride nanoarrays on nickel foam (Mn–CoN@C/NF) exerted an industrial-level current density of 400 mA cm−2 at 1.37 VRHE for the GOR. The doped Mn species facilitated glycerol adsorption and increased Co3+ density in Mn–CoN via regulating the valence state of Co.27 Moreover, non-metal elements and organic molecules also enhanced the GOR performance.66,85,148 For instance, a sulfur-doped Mn–Co hydroxide nanosheet catalyst (Mn–Co–S) enhanced charge/mass transfer and increased high-valence Co active species by promoting surface reconstruction and forming a hierarchically porous structure with abundant oxygen vacancies, achieving 100 mA cm−2 at 1.347 VRHE with a FEformate exceeding 95%.66
 | ||
| Fig. 8 (a) The proposed synergistic mechanism of the GOR to formate catalyzed by NiCo2O4/NF with regulable Lewis and Brønsted acid sites, (b) SEM images of NiCo2O4 nanosheets on NF, (c) LSV curves for the GOR and OER. Reproduced with permission from ref. 28. Copyright 2023, Wiley-VCH. (d) SEM image of NiCo2O4 nanoneedles, (e) LSV curves of 1 M KOH with and without 0.1 M glycerol addition, (f) the scheme of the anode reactions at varied potentials. Reproduced with permission from ref. 37. Copyright 2024, Wiley-VCH. (g) Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of Ni3Sn. The arrangement of atoms is highlighted in the rectangular box, where the blue and orange spheres represent Ni and Sn atoms, respectively. (h) Adsorption configurations and adsorption energies of *OH at Ni(111), Ni3Sn(201) and Sn(200) surfaces. Adsorption configurations, adsorption energies, and bond lengths at Ni(111) and Ni3Sn(201), with a (i) mono-valent carbon-binding intermediate (CH2OH–CHOH–*CO) or (j) multi-valent carbon-binding intermediate (CH2OH–*CO–*CO). (k) Schematic illustrations of the GOR on different catalysts. Reproduced with permission from ref. 154. Copyright 2024, Wiley-VCH. (l) The TEM image of CoNiCuMnMo NPs, the LSV curves in 1 M KOH with and without 0.1 M glycerol addition (m), FEs for formate production at varied potentials (n) and the GOR stability test (o) of the CoNiCuMnMo-NPs/CC electrode. Reproduced with permission from ref. 15. Copyright 2022, American Chemical Society. | ||
Ni-based catalysts, like their Co counterparts, enhance GOR performance through the incorporation of various transition metals (such as Fe,151 Co,152 Cu,18,144 Cr,63 Mo,36,71 V/Ru,72 Pd153) in forms like oxides, hydroxides and alloys, which rapidly generated high-valance Ni sites and modulated metal coordination. For instance, introducing oxygen and cation vacancies into NiCrO nanosheets via chromium ion leaching significantly boosted co-adsorption of both OH− species and organic molecules, and intrinsic GOR activity.63 During the GOR process, the surface quickly reconstructed into NiOOH, achieving a FEformate of 96%. Besides, the selectivity of Ni-based catalysts can be adjusted towards C3 products by modifying the adsorption mode of key intermediates and controlling C–C bond cleavage propensity.154,155 Glycerol exhibits two adsorption modes on metal surfaces: mono-valent carbon–metal binding, which favors carbon chain preservation and produces GLA, and multi-valent carbon binding, which triggers C–C bond cleavage and forms C1/C2 products. In the context, Lyu et al. achieved a peak FEGLA of 62% ± 3% at 1.7 VRHE over an atomically ordered Ni3Sn intermetallic catalyst (Fig. 8g).154 Combined theoretical calculations and in situ characterization revealed that the introduced Sn, with its robust oxygen adsorption and weak carbon adsorption capability, effectively altered the adsorption of glycerol oxidation intermediates from multi-valent carbon binding to mono-valent carbon binding, and promoted *OH adsorption and subsequent nucleophilic attack, leading to a high selectivity to GLA production (Fig. 8h–k). This study offers crucial insights into screening secondary metals for regulating selectivity of non-noble metals, while whether the regulatory function of Sn or other elements with strong oxygen adsorption but weak carbon adsorption capabilities is applicable to metals like Co requires further validation. Not limited to the above incorporation of inorganic metals, introducing organic molecules also regulated the selectivity of Ni-based catalysts. Hybridizing phenanthroline with Ni(OH)2 can also modulate GOR selectivity by controlling electronic states to balance oxidative power and C–C bond cleavage activity, resulting in different oxidation products depending on the phenanthroline derivative used.155
Manganese oxides, with their diverse valence states and crystalline phases, are widely studied for the GOR, with their selectivity and activity influenced by phase structure, electrolytes, and potential.30,77,78,156 Under mild base or neutral media, MnO2 with various crystalline phase structures demonstrated high selectivity for C3 products, and increased potentials help preserve C–C bonds.77,78 For instance, α-MnO2, β-MnO2, and γ-MnO2 in 0.1 M Na2B4O7 with 0.1 M glycerol all presented comparable selectivity to C3 products (∼50% for DHA and ∼40% for GLDA), with γ-MnO2 exhibiting the best reaction rate.77 The preference for DHA over GLDA was ascribed to the surface's biased adsorption of secondary –OH group over primary ones. In situ Raman spectroscopy revealed that all three MnO2 catalysts underwent intricate surface reconstruction during the GOR process with δ-MnO2 and γ-MnOOH formed on the surface under high potentials, where δ-MnO2 hindered C–C bond breakage. Although high selectivity was achieved under mild base conditions, low reaction current density (<10 mA cm−2) at high potentials (>2 VRHE) limits large-scale applications. Studies that seek to enhance the reaction rate without compromising selectivity of these MnO2 catalysts would be highly valuable. In a separate study, γ-MnO2 nanosheets supported on carbon paper (γ-MnO2/CP) showcased excellent stability during the GOR in acid medium, with formate as the predominant product. This stability was credited to the reductive glycerol, which safeguarded the catalyst from deactivation by inhibiting the lattice oxygen mechanism that caused structural damage of the catalyst, and the oxidation of Mn–oxo (MnIVO) into MnO4−. However, it is yet to be determined if γ-MnO2/CP can maintain this ultralong stability in the commonly used 0.5 M H2SO4 electrolyte at industrial-scale current densities. Alongside single MnO2 catalysts, complexing manganese oxides with other active components has been shown to enhance GOR performance.30,75 For instance, Ru nanoparticles supported on defect-rich MnO2 (Ru@Mn2−x) catalysts prepared using a γ-ray reduction method, achieved excellent activity, delivering 10 mA cm−2 at just 1.13 VRHE with formate as the main product in alkaline solution. The outstanding performance of this catalyst was attributed to the synergistic effect of defect-rich MnO2 and Ru nanoparticles, which facilitated the glycerol adsorption and accelerated reaction kinetics via promoting electron transfer between the intermediate and the catalytic surface, thereby improving the bond-breaking process.30
Cu-based catalysts, including oxides, hydroxides, and related compounds have shown promise in the GOR, converting glycerol to C3 products like DHA in mild base media and to formate in strong alkaline environments.157–159 Cu-related catalysts exhibit selectivity and activity comparable to Ni and Co-based catalysts in alkaline electrolytes. Nonetheless, the notable degradation observed during prolonged stability assessments calls for further investigation to understand attenuation mechanism and improve durability.31,150,158,160 A case in point is the S-doped CuO nanorod array on copper foam (S–CuO/CF), which through rapid restructuring, optimized lattice oxygen deintercalation for efficient glycerol-to-formate conversion, achieving 100 mA cm−2 at 1.23 VRHE with a peak FEformate of 95%.31 Yet, the long-term stability of S–CuO/CF under high current densities is a concern due to lattice oxygen depletion, necessitating further research on the impact of the reduction of lattice oxygen on the durability of copper-based catalysts.
Beyond these catalysts, there is a rising trend in developing novel catalysts like high-entropy alloys and compounds, which also predominantly produce formate in the GOR.15,29,161,162 For instance, a self-supported electrode consisting of a CoNiCuMnMo high-entropy alloy (HEA) nanoparticles in situ grown on carbon cloth (CoNiCuMnMo-NPs/CC) for the GOR (Fig. 8l),15 attained a current density of 10 mA cm−2 at 1.25 VRHE with robust stability and boasted an FEformate over 90% within a potential window stretching from 1.27 to 1.47 VRHE (Fig. 8m–o). Combined with theoretical calculation and operando techniques, the synergistic effect among its constituents co-contributed to the exceptional performance.
In brief, non-precious Ni/Fe/Cu/Mn-based catalysts can achieve high activity and selectivity for formate production in alkaline media through electronic structure modulation via compositional and morphological design. Among these, Ni/Co-based catalysts demonstrate exceptional promise for scalable applications. By incorporating secondary components to promote in situ formation of high-valent MOOH species, these systems enable synergistic optimization of d-band center positioning, oxygen vacancy generation, and intermediate adsorption energetics, while maintaining cost-effectiveness and synthetic scalability. However, the MOOH-mediated process faces a critical challenge, namely, accelerated OER competes severely with the GOR at high current densities, drastically reducing conversion efficiency. Thus, suppressing OER competition under operational conditions emerges as the pivotal optimization target for practical implementation.
4 The GOR application in hybrid dual-electrolyte electrochemical devices
The investigation of reaction pathways, catalytic mechanisms, and influential factors in the GOR discussed earlier is all geared towards the effective utilization of the GOR in electrolytic devices. This section aims to summarize the research progress on hybrid dual-electrolyte GOR-integrated devices, including electrolysis systems and full cells. Subsequent analysis will focus on the reported hybrid dual-electrolyte electrolysis systems that incorporate the GOR with the HER (GOR‖HER) and the GOR with the CO2RR (GOR‖CO2RR), as well as GOR-involved fuel cells that encompass DGFC and zinc–air batteries. Emphasis will be placed on the pivotal role of the GOR, the catalysts involved, and the configuration of the dual-electrolyte's contribution to enhancing electrochemical performance.4.1 GOR involved hybrid dual-electrolyte electrolysis systems
| Anode: C3H8O3 + 11OH− → 3HCOO− + 8H2O + 8e− | (R1) |
| Cathode: 8H+ + 8e− → 4H2 | (R2) |
| Total reactions: C3H8O3 + 11OH− + 8H+ → 3HCOO− + 8H2O + 4H2 | (R3) |
A series of studies were conducted based on the alkali/acid-electrolyte HER‖GOR systems by developing various catalysts with main focus on the GOR side.15,39,151,158,161,163 The energy-saving dual production of the alkali/acid-electrolyte HER‖GOR electrolyzer was firstly achieved by assembling a commercial RuIr/Ti cathode in 0.5 M H2SO4 solution and a newly developed CoNiCuMnMo-NPs/CC anode in 1.0 M KOH containing 0.1 M glycerol solution, separated by a cation exchange membrane (CEM) (Fig. 9a and b).15 In this hybrid system, glycerol undergoes selective oxidation to formate at the alkaline anode with concomitant electron transfer through the external circuit, driving proton reduction to H2 at the acidic cathode. Charge balance is maintained via potassium ion (K+) migration through the CEM from the anolyte to catholyte compartment. As expected, the hybrid dual-electrolyte system leveraged both ENE and energy from glycerol oxidation to dramatically reduce the operational voltage. At 10 mA cm−2, the cell voltage dropped to 0.55 V, significantly lower than the traditional alkaline HER‖GOR (1.34 V) and HER‖OER (1.63 V) systems (Fig. 9c). This result suggested that the substitution of the OER with the GOR lowered the anodic potential by 290 mV and the ΔpH of the hybrid acid–alkaline electrolyte (ΔpH = 14) provided an additional 790 mV reduction, closing to theoretical 828 mV (0.0591 × ΔpH). Moreover, the electrolyzer also demonstrated excellent stability after periodic electrolyte replenishment to counteract consumption of proton/hydroxide and glycerol, sustaining constant electrolysis at 50 mA cm−2 for 300 h with near-unity FE for H2 production at the cathode and FEformate of around 92% at the anode (Fig. 9d). This work exemplifies the synergy between catalytic innovation and pH-gradient engineering, underscoring pH-asymmetric design as a viable pathway toward economically viable glycerol valorization. Building on this, various electrocatalysts like (CoNiCuMnMo)Se nanosheets, NiCo2O4 nanoneedles, Mn-doped CoSe2 nanonetworks, and Cu–Cu2O nanocluster compounds have been engineered for further developing the hybrid alkali/acid-electrolyte electrolysis systems.39,158,161,163 These studies effectively demonstrate the universality of hybrid alkali/acid-electrolyte HER‖GOR systems for various efficient catalysts to achieve energy-saving dual production. Nevertheless, there are still many aspects worthy of investigation like the stability of the hybrid electrolyzer at industrial-scale current densities (>200 mA cm−2), promoting GOR selectivity to other value-added products beyond formate, validating the feasibility for enhancing ENE by further elevation of ΔpH and direct use of crude glycerol as a reactant.
 | ||
| Fig. 9 Schematic illustration of the (a) asymmetric-electrolyte electrolyzer cell and (b) traditional alkaline electrolysis cell, (c) LSV curves of HER‖GOR and HER‖OER under different pH differences. (d) Long-term stability test of the assembled hybrid alkali/acid-electrolyte HER‖GOR at 50 mA cm−2. Reproduced with permission from ref. 15. Copyright 2022, American Chemical Society. (e) Graphical schematics illustrating AEM-based alkali–alkali and acid–alkali MEA devices, the acid–alkali MEA device possesses the ability to inhibit the crossover of product anions and the cleavage of C–C bonds (blue electrolyte: alkaline; red electrolyte: acidic), (f) LSV curves of the GOR in 1 M KOH with 1 M glycerol anolyte in an AEM-based MEA electrolyzer, total FEs of GOR liquid products in the anolyte and catholyte in the chronopotentiometry test at various current densities in AEM-based acid–alkali MEA (g) at 80 °C, comparison of (h) product crossover ratio and (i) total FE towards liquid production in different electrolyzers. Reproduced with permission from ref. 32. Copyright 2024, Royal Society of Chemistry. | ||
The above advancements in hybrid alkali/acid-electrolyte HER‖GOR electrolysis systems have seen the use of cation exchange membranes to separate acid catholytes and alkaline anolytes to harvest ENE, while a recent novel approach employed an anion exchange membrane (AEM) in a hybrid acid/alkali-electrolyte membrane electrode assembly (MEA) for HER‖GOR electrolysis, effectively harvesting ENE and preventing crossover of product anions and C–C bond cleavage during the GOR32 (Fig. 9e). The AEM-based acid–alkali MEA electrolyzer demonstrated superior performance with commercial Pt/C electrodes at 80 °C, exhibiting a low onset potential and achieving 200 mA cm−2 at 0.377 V. It maintained 55% FE for C3 products (GA and TA) at 1 A cm−2 with negligible crossover (Fig. 9f–h). Moreover, unlike the conventional AEM-based alkali–alkali MEA system which suffered severe product crossover (30% at 10 mA cm−2) and poor C3 and selectivity (7.1% at 0.45 A cm−2), this configuration achieved near-unity overall FE towards liquid products across current densities, effectively preventing C–C bond cleavage (Fig. 9g–i). Based on the comparison experiments, the enhanced performance stems from three synergistic factors: the ion regulation function of AEM efficiently minimizes anion product crossover by saturating ion-exchange capacity with OH− diffusion while electromigration directs anions toward the anode; concurrently, local pH control function of the proposed device, owing to the limited OH− provided by the cathode, leads to decreased local pH at the anode and thus higher selectivity to C3 products; and structural advantages provided by the acid–alkaline dual-electrolyte and MEA design effectively harvest ENE, enhance convective mass transfer, improve energy efficiency, and lower system resistance. This work opens new avenues for the application of hybrid acid/alkali-electrolyte systems in electrolysis. However, the broader application of the AEM-based acid–alkali MEA device, especially when equipped with non-noble metal catalysts or integrated into other coupled electrolysis or cell systems, requires further validation. Moreover, the stability of such a device under high current densities needs to be further confirmation, as stability is a crucial factor in assessing its potential for practical applications. Additionally, controlled experiments conducted on an AEM-based acid–alkali flow cell may be necessary to substantiate the proposed role of AEM within the MEA structure.
The aforementioned research efforts have enhanced the performance of hybrid alkali/acid-electrolyte HER‖GOR electrolysis systems through carefully designing catalysts and optimizing electrolyzer configurations, thereby lowering input voltage and increasing the selectivity of glycerol oxidation. Even so, these advancements represented only the beginning in achieving energy-saving dual-value-added production via hybrid dual-electrolyte HER‖GOR electrolysis systems. There are numerous scientific fundamental issues that warrant further exploration. Firstly, the majority of the aforementioned studies have primarily focused on developing GOR catalysts in alkaline medium with formate as the main product. Therefore, there is a demand for developing catalysts with high selectivity for other products and robust HER catalysts in acid medium. Secondly, it is crucial to conduct stability tests under industrial current densities to uncover the attenuation mechanisms of this type of electrolysis system. Thirdly, a comprehensive economical value comparison between the hybrid system and traditional system should be conducted to provide valuable insights for future research in this area.
| Anode: C3H8O3 + 11OH− → 3HCOO− + 8H2O + 8e− | (R4) |
| Cathode: 4CO2 + 4H2O + 8e− → 4CO + 8OH− | (R5) |
| Total reactions: 4CO2 + C3H8O3 + 3OH− → 4CO + 3HCOO− + 4H2O | (R6) |
 | ||
| Fig. 10 (a) Schematic illustration of electrolysis by coupling CO2 conversion with glycerol upgradation, (b) TEM image and the magnification of the TEM image (the inset) of NiSAs/FN-CNSs, (c) SEM image of CoSe2/CC, FECO and FEformate monitored in the cathode and anode at various cell potentials (d) and stability test at 2.2 V (e) over the NiSAs/FN-CNSs/CP‖CoSe2/CC with and without 2 M glycerol (NiSAs/FN-CNSs supported on carbon paper (NiSAs/FN-CNSs/CP, catholyte: 2 M KHCO3, anode: 2 M KOH + 2 M glycerol)). Reproduced with permission from ref. 147, copyright 2022, Elsevier. (f) Schematic of the flow cell setup used for co-electrolysis experiments with the eCO2R at the cathode and the GOR at the anode with BPM as the separator, (g) SEM image of Au-ND, (h) LSV curves for co-electrolysis using different anode electrodes with (0.5 M) and w/o glycerol in the anolyte, (i) stability test of co-electrolysis using the Au-ND/NiF anode catalyst at a constant current density of 175 mA cm−2. Reproduced with permission from ref. 171, copyright 2023, Cell Press. | ||
In the cathode, an atomically dispersed Ni–N single sites on F, N co-doped ultrathin nanoporous carbon nanosheet (NiSAs/FN-CNSs) catalyst (Fig. 10b) was developed for efficiently converting CO2 into CO with near-unity FECO and robust stability in a neutral electrolyte, while the anode featured a CoSe2 interconnected nanostructure on carbon cloth (CoSe2/CC) (Fig. 10c), which presents excellent GOR performance with peak FEformate of over 90% in alkaline medium. A flow electrolyzer pairing with the two catalysts facilitated electrolysis at merely 1.05 V thanks to the assistance of harvest ENE and GOR and achieved 10 mA cm−2 at only 1.24 V, 330 mV lower than the electrolyzer without addition of glycerol. It sustained high FEs of nearly 90% for both CO and formate across 1.2–2.5 V (Fig. 10d), and operated stably for over 400 h at 2.2 V with FECO and FEformate over 90% and a current density of ∼100 mA cm−2 (Fig. 10e). This work provides a strategy for practical CO2 electrolysis coupled with glycerol upgradation. Nevertheless, addressing carbonatization from CO2 and locally produced OH−, as well as accumulated alkali metal ions in the electrolysis system, is necessary for broader application and stability under high current densities, which requires further validation.
The dual-electrolyte structure can also be used to maintain a stable reaction environment without producing ENE by adjusting the membrane. Khan et al. proposed a bipolar membrane (BPM) involved co-electrolysis system to avoid carbonate formation and enhance CO2 utilization. The BPM not only prevented HCO3− and CO32− from reaching the anode but also supplied H+ to the cathode, reactivating carbonate and recycling CO2 on site for reduction (Fig. 10f).171 In their proposed BPM separated co-electrolysis electrolyzer, an Au nano-dendrite in situ grown on Ni foam (Au-ND/NF) electrode (Fig. 10g) was prepared as an efficient GOR catalyst with GA as the main product in 3 M KOH containing 0.5 M glycerol, and the cathodic CO2RR was catalyzed by a Cu sputtered on polytetrafluoroethylene (Cu/PTFE) gas diffusion layer (GDL) electrode in 1 M K2CO3. The assembled GOR-involved electrolysis system outperformed the OER-involved counterpart with an about 0.8 V voltage drop (Fig. 10h) and efficiently co-produced C2H4 and GA, maintaining stable electrolysis for 10 h at 175 mA cm−2 with no obvious degradation in voltage and FEC2H4 (Fig. 10i). A techno-economic analysis (TEA) indicated the potential for commercializing CO2 to C2H4 conversion with a competitive minimum selling price of ∼$1.1 per kgC2H4, providing a plausible strategy to upgrade glycerol and CO2 in CO2 electrolysis systems. Nevertheless, further efforts are needed to reduce the overall cell voltage, improve the product selectivity of both half-reactions, and enhance the stability of such electrolyzers. Besides, comparing the economic viability of systems with and without ENE is essential to guide research on optimizing stability, input voltage, CO2 utilization, and conversion efficiency.
Apart from joint production of CO or C2+ in the cathode and formate or other C2 and C3 products in the anode, CO2RR‖GOR hybrid electrolysis systems have also been demonstrated to co-produce formate at both the cathode and anode.159,165 For these systems, catalysts were developed to enable CO2-to-formate conversion in neutral media and glycerol-to-formate conversion in alkaline media. An established configuration achieved FEs approaching 90% for formate at both electrodes, confirming the viability of simultaneous formate production, a process with significant industrial promise enabled by effective electrocatalysts for formate synthesis on both sides.165 Additionally, it highlighted the advantage of simplified downstream purification, requiring isolation of only a single product. However, the harvested ENE has also not been discussed in these systems.
Despite progress in GOR-assisted CO2 electrolysis hybrid systems (Table 3), several fundamental scientific challenges must be addressed to achieve significant advancements for practical applications. Firstly, achieving high single-product selectivity in both the CO2RR and GOR under industrial-scale current densities remains a challenge. Precise tuning of selectivity and activity through catalyst design is a crucial research focus. Secondly, the hybrid system, which converts CO2 to CO or formate at the cathode and glycerol to formate at the anode, shows practical application potential in terms of conversion efficiency. However, issues such as carbonatation and accumulation of alkali metal salts in the cathode over long-term operation, and potential over-oxidation of formate at the anode, require further investigation. Thirdly, since the CO2RR in acidic media has demonstrated excellent selectivity for products like CO, formate, or C2+,172–174 exploring a hybrid system that combines the cathodic CO2RR in an acidic electrolyte with the anodic GOR in an alkaline environment is worthwhile. This approach can potentially avoid carbonation issues caused by the direct reaction between CO2 and locally produced OH−, and it also harvests elevated ENE, thereby reducing the required input electric energy.
| Electrodes (cathode‖anode) | Catholyte‖anolyte | Products (cathode(FE)‖anode(FE)) | Cell potential (V) | Current density (mA cm−2) | Cell type | Stability test | Ref. |
|---|---|---|---|---|---|---|---|
| HEA-CoNiCuMnMo‖RhIr/Ti | 0.5 M H2SO4‖1 M KOH + 0.5 M glycerol | H2‖formate (∼90%) | <1.0 V | 50 | Flow cell (CEM) | 300 h | 15 |
| Mn–CoSe2/CFC‖Mn–CoSe2/CFC | 0.5 M H2SO4‖1 M KOH + 0.5 M glycerol | H2‖formate (∼90%) | ∼1.2 | 100 | Flow cell (CEM) | 300 h | 39 |
| Pt/C‖Cu–Cu2O/CC | 0.5 M H2SO4‖1 M KOH + 0.5 M glycerol | H2‖formate (97%) | 0.59 | 10 | Flow cell (BPM) | 50 h | 158 |
| Pt/C‖NiCo2O4/NF | 0.5 M H2SO4‖1 M KOH + 0.5 M glycerol | H2‖formate (∼85%) | 0.36 | 10 | Flow cell (CEM) | 150 h | 60 |
| Pt/C‖NiFe-LDH/NF | 0.5 M H2SO4‖1 M KOH + 0.1 M glycerol | H2‖liquid products (∼100) | ∼0.9 | 100 | Flow cell | 550 h | 151 |
| NiSAs/FN-CNSs/CP | 2 M KHCO3‖2 M KOH + 2 M glycerol | CO (>90%)‖formate (∼90%) | 2.2 | ∼100 | Flow cell (CEM) | >400 h | 147 |
| Cu/PTFE‖Au-ND/NiF | 1 M K2CO3‖3 M KOH + 0.5 M glycerol | C2H4 (∼50%)‖glycolic acid (∼50%) | 3.95 | 175 | Flow cell (BPM) | 10 h | 171 |
| Bi/C-GDE‖Pt/C-PE | 0.5 M KCl + 0.45 M KHCO3‖1 M KOH + 1.0 M glycerol | Formate (85%)‖(formate (∼41%), DHA (4.1%)) carbonate (41.8%), MOX, GLAD | 4.51 | 98 | Micro flow cell (CEM) | — | 74 |
| Cu(OH)2/FC‖Cu(OH)2/FC | 0.1 M KHCO3‖1 M KCl + 0.6 M glycerol | Formate (89.2%)‖(formate (84.5%)) | 1.8 | 37.1 | H-type cell (CEM) | 72 h | 159 |
| BiOI/CP‖Ni0.33Co0.67(OH)@HOS/NF | 0.5 M KHCO3‖1 M KOH + 0.1 M glycerol | Formate (∼90%)‖(formate (∼90%)) | 1.8 | ∼10 | Flow cell (CEM) | 36 h | 165 |
| InBi NPs/GDE‖Ni2S3/NF | 0.5 M KHCO3‖1 M KOH + 0.05 M glycerol | Formate (89.2%)‖(formate (84.5%)) | 5–6.3 | 100 | Micro flow cell (BPM) | 24 h | 175 |
4.2 GOR involved fuel cells
Over the decades, research has predominantly concentrated on traditional DGFCs in alkaline medium, where factors such as operating conditions, noble metal catalyst species, and electrode structures have been systematically examined for their impact on cell performance.89,176 These studies provide precious insights for assembling DGFCs. Nevertheless, there remains significant room for improvement in cell performance, particularly in optimizing cell structure and enhancing the efficiency of the corresponding catalysts. In the subsequent section, a summary of the recently developed GOR-involved fuel cell systems that incorporate the hybrid alkali/acid-electrolyte configuration, including DGFCs and the anodic reaction in metal–air batteries during the recharging process, will be provided by analyzing the catalysts developed, their corresponding cell performances and some critical insights (Table 4).| Catalysts (cathode‖anode) | Electrolyte (catholyte‖anolyte) | Cathode reagent | Product (anode) | Peak power density (mW cm−2) | Open circuit voltage | Stability test | Cell type | Ref. |
|---|---|---|---|---|---|---|---|---|
| Pt/C‖Pt/C | 1 M H2SO4 + 1.0 M H2O2‖5 M NaOH + 1 M glycerol | H2O2 | — | 375/80 °C | 0.4 V | 0.5 h | MEA (CEM) | 41 |
| Pt/C‖Au–Ni/C | 1 M H2SO4 + 1.0 M H2O2‖5 M NaOH + 1 M glycerol | H2O2 | — | 142/80 °C | 1.49 V | 0.5 h | MEA (CEM) | 21 |
| Pt/C‖FeNiCoCrMnS2/CC | 2 M H2SO4‖4 M KOH + 0.1 M glycerol | Air | Formate | 50/80 °C | 1.29 V | 120 h | Flow cell (CEM) | 29 |
| PtZn-IMC@NC‖PtZn-IMC@NC | 2 M H2SO4‖6 M KOH + 0.1 M glycerol | Air | Formate, DHA, and GLAD | 257.3/80 °C | 1.99 V | 168 h | Flow cell (CEM) | 42 |
Reports indicate that hybrid alkali/acid-electrolyte direct liquid fuel cells, utilizing fuels like NaBH4, N2H4 and ethanol, exhibit higher output voltage and power density, leading to the proposal of similar hybrid alkali/acid-electrolyte DGFCs (AA-DGFCs).21,29,41,42 Two types of AA-DGFCs have been identified: one combines the acidic H2O2 reduction reaction with the alkaline GOR, yielding a theoretical open-circuit voltage (OCV) of 2.6 V (assuming full glycerol conversion to CO2), the corresponding reactions are as follows:
| Anode: C3H8O3 + 14OH− →14e− + 3CO2 + 11H2O | (R7) |
| Cathode: 7H2O2 + 14H+ + 14e− → 14H2O | (R8) |
| Fuel cell reaction: C3H8O3 + 14OH− + 7H2O2 + 14H+ → 3CO2 + 25H2O | (R9) |
The other pairs the acidic O2 reduction reaction with the alkaline GOR with a theoretical OCV of 1.97 V (assuming glycerol conversion to formate). The corresponding reactions are as follows:
| Anode: C3H8O3 + 11OH− → 3HCOO− + 8H2O + 8e− | (R10) |
| Cathode: 2O2 + 8H+ + 4e− → 4H2O | (R11) |
| Fuel cell reaction: C3H8O3 + 2O2 + 11OH− + 8H+ → 3HCOO− + 12H2O | (R12) |
Sangkheaw et al. initially explored the influence of operating conditions on the performance of AA-DGFCs.21,41 They analyzed factors such as electrolyte ratio, glycerol concentration, electrolyte flow rate, Nafion ionomer content in the microporous layer (MPL), anodic catalyst and temperature. Focusing on cathodic conditions in the AA-DGFC equipped with Pt/C as both the anode in 1.0 M glycerol containing 5 M NaOH alkaline electrolyte and cathode in H2SO4 with H2O2 catholyte, they found that the catholyte flow rate of 3 mL min−1, a 1:1 molar ratio of H2O2 to H2SO4 with a 0.1 M H2O2 concentration, and Pt/C catalysts supported on non-wetproof carbon cloth maximized the power density to 375 mW cm−2 and current density to 451 mA cm−2.41 Subsequent studies on anodic conditions revealed that the OCV was highly sensitive to these factors, the higher ratio of glycerol and NaOH, glycerol concentration, and temperatures leading to greater OCVs.21 The optimal conditions were determined as a 1:5 glycerol to NaOH ratio, 1.0 mL min−1 anolyte flow rate, 1 M glycerol concentration, operating temperature of 80 °C, and 20 wt% Nafion in the anode MPL, yielding a peak power density of 329 mW cm−2, double that of traditional alkaline systems, and an OCV of 1.67 V and an average current density of 501.7 mA cm−2 were obtained. Further screening of Au-based anodic catalysts (Au/C, Au–Ni/C and Au–Ag/C) identified Au–Ni/C as optimal, delivering 142 mW cm−2 under refined conditions. Notably, these studies did not examine the oxidized products and stability of the optimized AA-DGFC under operational conditions. Additionally, whether there remains potential to increase ENE in these systems requires further verification. Despite these limitations, the findings establish critical guidelines for optimizing AA-DGFC operations and advancing the technology.
Then our research group further verified and advanced the AA-DGFG by scrutinizing related catalysts and reaction impact factors. In one work, we designed a high-entropy sulfide electrode, i.e., FeCoNiCrMnS2 with a spherical structure with nodular rough surfaces in situ grown on carbon cloth (FeCoNiCrMnS2/CC) for the alkaline GOR (Fig. 11a),29 which required only 1.1 VRHE to achieve 10 mA cm−2 and obtained FEformate over 90% across 1.2–1.45 VRHE (Fig. 11b and c). The AA-DGFG assembled with this anode in 4 M KOH containing 0.1 M glycerol and commercial Pt/C for the cathodic ORR (oxygen is from air) in 2 M H2SO4 solution (Fig. 11d) attained a peak power density of 13.4 mW cm−2, 22.4, 33.6, and 50.1 mW cm−2 at room temperature, 40 °C, 60 °C and 80 °C, respectively (Fig. 11e). Critically, it demonstrated superior stability with continuous discharge at 10 mA cm−2 for 120 h, sustaining formate production at an average yield rate of 120 μmol h−1 cm−2 and FEformate over 80% (Fig. 11f). This work establishes non-noble metal catalysts for dual-purpose GOR systems enabling simultaneous chemical synthesis and energy generation.
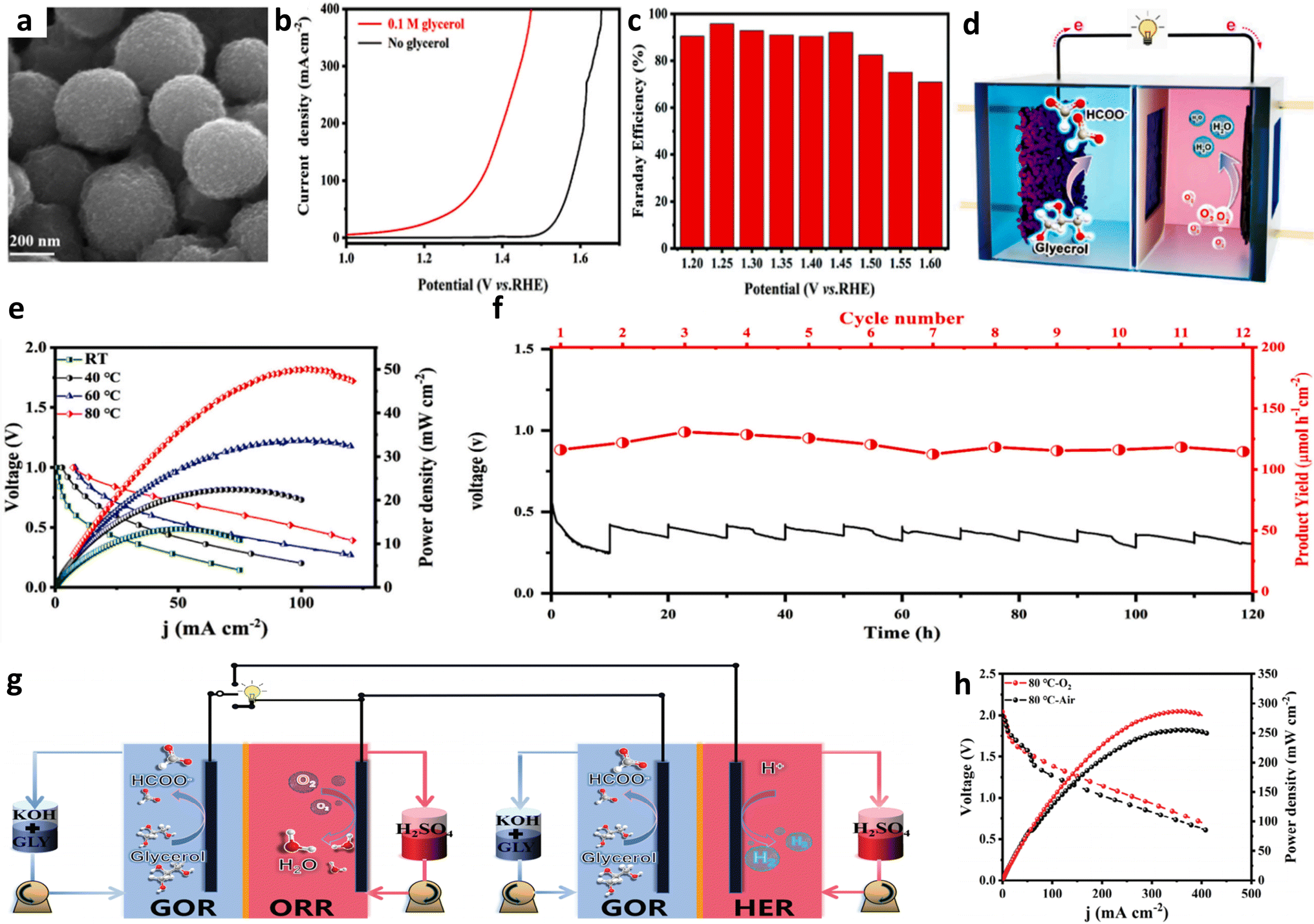 | ||
| Fig. 11 SEM image (a), LSV curves in 1 M KOH with and without 0.1 M glycerol addition (b), and FEformate production at varied potentials (c) of FeCoNiCrMnS2/CC, (d) schematic diagram of AA-DGFC. (e) Polarization curves (left y axis) and power density profiles at different temperatures, (f) long-term durability test at a current density of 10 mA cm−2. Reproduced with permission from ref. 29, copyright 2023, Elsevier. (g) Schematic diagram of AA-DGFC driving AA-GHEC, (h) polarization curves and power density profiles of PtZn-IMC@NC with air or O2 as the cathodic reagent. Reproduced with permission from ref. 42, copyright 2024, Wiley-VCH. | ||
To further enhance the AA-DGFG's energy output efficiency, a multi-functional PtZn-IMC@NC was prepared as a catalyst for the GOR, HER and ORR with excellent activity and stability.42 With PtZn-IMC@NC as both the cathode in 2 M H2SO4 and anode in 6 M KOH containing 1 M glycerol, we constructed the AA-DGFC and acid/alkali glycerol–hydrogen electrolytic cell (AA-GHEC), creating a self-powered integrated electrochemical device (Fig. 11g). The assembled AA-DGFC achieved a peak power density of 257.0 mW cm−2 at 80 °C with air as the reactant in the cathode (286.8 mW cm−2 with pure oxygen) (Fig. 11h), maintaining discharge for seven days at 10 mA cm−2 with discharge voltage at around 0.95 V and valuable products (DHA, GLAD and formate) produced in the anode. The assembled AA-GHEC also presented excellent performance, achieving 100 mA cm−2 at only 0.47 V with excellent stability and product (H2 and formate) selectivity, 610 mV less than that of water electrolysis. Thus, the output energy of AA-DGFC is sufficient to drive the operation of AA-GHEC, realizing self-sustained power generation and value-added product synthesis. This work provides an efficient alternative to the noble Pt/C catalyst and strategies for constructing efficient electrochemical energy storage and conversion systems.
Apart from application in AA-GHEC, the GOR can be used to reduce the charging voltage of the hybrid alkali/acid-electrolyte Zn–air battery. As there exist issues of poor reversibility between the OER and ORR, limited operating voltage, and carbonate salt formation in the traditional ZAB, we proposed a hybrid rechargeable Zn–air battery (h-RZAB) that couples the ORR in acid solution with the Zn oxidation reaction in alkaline solution during discharge and pairs Zn electroplating with the GOR in an alkaline electrolyte during charge (Fig. 12a). During the discharging process, the reactions are as follows:
| Anode: Zn + 4OH− → Zn(OH)42− + 2e− (Ea = −1.285 V) | (R13) |
| Cathode: O2 + 4H+ + 4e− → 2H2O (Ec = 1.265 V) | (R14) |
| Overall reaction: Zn + 4OH− + O2 + 4H+ → Zn(OH)42− + 2H2O (EDischarging = 2.55 V) | (R15) |
| Anode: Zn(OH)42− + 2e− → Zn2+ + 4OH− (Ea = −1.285 V) | (R16) |
| Cathode: C3H8O3 + 11OH− → 3HCOO− + 8H2O + 8e− (Ec = −0.671 V) | (R17) |
| Overall reaction: 4Zn(OH)42− + C3H8O3→ 4Zn + 8H2O + 3HCOO− + 5OH− (ECharging = 0.614 V) | (R18) |
 | ||
| Fig. 12 (a) Schematic diagram of the h-RZAB, TEM images of FeCo@PNC (b) and Ni(OH)2-HC (c), LSV cures of FeCo@PNC and Pt/C in 0.1 m HClO4 solution (d) and Ni(OH)2-HC in 1.0 m KOH solution with and without glycerol (e) and (f) voltage versus specific capacity curves, (g) discharging voltages at different current densities, (h) discharging polarization curves and corresponding power density curves, (i) charging and discharging voltages of h-RAZAB at different current densities. Reproduced with permission from ref. 177, copyright 2024, Wiley-VCH. | ||
The feasibility of the proposed battery was verified by two catalysts for the ORR and GOR. A N-doped porous carbon with a trace of Fe and Co decorating nanohybrid (FeCo@NPC) prepared for the ORR achieved a half-wave potential of 0.78 V (Fig. 12b and d), comparable to that of commercial Pt/C (0.82 V) in O2-saturated 0.1 M HClO4 solution. The Ni(OH)2 with a hollow octahedral cage structure (Ni(OH)2-HC) delivered 10 mA cm−2 at 1.35 VRHE in 1 M KOH solution with 0.1 M glycerol, 180 mV lower than that of for the OER (Fig. 12c and e). The h-RZAB assembled with these prepared catalysts, outperformed traditional ZABs, delivering an output voltage of 2.01 V at an operating current density of 10 mA cm−2 and energy density 1498 W h kgZn−1, compared to 1.31 V and 683 W h kgZn−1 for traditional ZABs (Fig. 12f). It also presented fast rate performance with a high operating voltage of 1.69 V at 100 mA cm−2 and a maximum power density of 562.7 mW cm−2, over twice that of the control ZAB (262.7 mW cm−2) (Fig. 12g–i). Moreover, thanks to incorporating the hybrid alkali/acid-electrolyte upon the discharging process and GOR upon the charging process, the h-RZAB delivered a high discharge voltage (∼2.0 V) above the charging voltage (∼1.5 V) until the current density exceeded 10 mA cm−2 (Fig. 12i), and the GOR product was formate with a FE of around 85%. Besides, the h-RZAB demonstrated long-term stability within 300 h and a round-trip energy efficiency over 100% as its charging voltage (1.89 V) was lower than its discharging voltage (1.96 V). This indicates that the decoupled h-RZAB markedly boosted electron utilization efficiency and upgraded chemicals. This research provides an ingenious approach to enhance the performance of rechargeable ZABs, opening up extensive opportunities for the development of other rechargeable metal–air batteries and the further optimization of catalyst performance.
The above proof-of-concept hybrid dual-electrolyte fuel cells fully underscore the beneficial impact of the GOR on enhancing cell performance, paving the way for future research. Key areas include developing highly selective GOR electrocatalysts to diversify oxidized products and boost the economic value of GOR-integrated fuel cells. Additionally, expanding the GOR's role in Zn–air batteries could lead to more efficient electrocatalysts and the development of other rechargeable metal–air batteries. Moreover, utilizing crude glycerol in these cells could eliminate costly purification steps, making it essential to assess their performance with crude glycerol for economic viability. In summary, there is substantial scope for research on GOR-involved fuel cells, with potential improvements in catalyst development, electrolyzer optimization, and economic benefits.
5 Conclusion and perspective
This perspective centers on GOR-involved hybrid dual-electrolyte electrochemical systems. It reviews advances in fundamental GOR science essential for system design (encompassing reaction pathway elucidation, advanced in situ/operando characterization techniques and theoretical methods for catalytic mechanism investigation, and performance-governing factors directly influencing GOR efficiency) and hybrid dual-electrolyte system development based on our group's research. Leveraging glycerol abundance, continuous ENE harvesting via facile external circulation, and optimized environments for value-added reactions, GOR-integrated hybrid systems demonstrate strong potential for large-scale implementation. However, while recent progress in GOR electrocatalyst design and mechanistic understanding has accelerated the preliminary development of such hybrid devices, significant challenges hinder their practical large-scale implementation. Further exploration is critically needed in electrocatalyst engineering, device integration/optimization (e.g., electrolyte, membrane, and device structure, etc.) and economic feasibility analysis for evaluating industrial-scale application. Based on the challenges encountered, future research could strategically focus on the following development pathways and optimization strategies for GOR-integrated hybrid systems.(1) For electrocatalyst engineering, although promising GOR catalysts have emerged, enhancing their applicability for hybrid systems remains crucial. Non-precious metal-based catalysts are particularly attractive for hybrid systems due to their cost efficiency, alkaline compatibility, and potential for generating valuable products with high selectivity. However, several challenges persist: (i) difficulty in maintaining high single-product selectivity (FE > 90%) at industrially relevant, ampere-level current densities (∼1 A cm−2), primarily due to the competing OER; (ii) development strategies often mimic the OER or other small molecule oxidations, lacking GOR-specific innovation; (iii) reliance on time-consuming trial-and-error processes. To address these limitations, future efforts should prioritize the development of catalysts specifically tailored for glycerol oxidation, rather than relying on those originally designed for the OER or other small-molecule oxidation reactions. The goal should be to achieve high glycerol conversion selectivity (faradaic efficiency > 90%) at industrially relevant current densities, while effectively suppressing the competing OER. To move beyond trial-and-error-based approaches, catalyst development must integrate in situ/operando characterization techniques, theoretical modeling (e.g., DFT), and artificial intelligence-assisted design. This combination will enable precise regulation of catalyst composition and nanostructure, mechanistic insight into reaction pathways, and efficient, targeted synthesis of high-performance materials.
(2) For the GOR-involved hybrid dual-electrolyte system development, though various hybrid electrolysis and fuel cell systems have been proposed, current devices have seen only conceptual validation. Beyond catalysts, key factors like electrolytes, membrane, reaction conditions and device structure require in-depth investigation.
(a) Given the intrinsic pH asymmetry in hybrid dual-electrolyte systems, in-depth investigation into charge carrier chemistry, including ion transport mechanisms, species crossover, and concentration gradients, is critical for improving reaction kinetics, operational stability, and ENE utilization. While conventional systems typically rely on standard acidic and alkaline electrolytes, research exploring the optimization of electrolyte composition and pH gradient to maximize ENE output remains scarce. Moreover, current devices often require frequent electrolyte replacement to sustain reaction rates and system stability, which increases operational complexity and cost. To address this, we recommend: (i) dynamic monitoring and selective replenishment of electrolytes to minimize waste and maintain performance; and (ii) investigating the reuse of industrial waste acids and bases as cost-effective electrolytes. This strategy offers a promising path toward converting industrial waste neutralization heat into useable electrical energy, thereby promoting sustainability in electrochemical system design.
(b) Membranes play a role as critical as electrodes in determining the long-term stability and overall efficiency of hybrid electrolyzers. However, their impact is often underestimated. Current commercial membranes, such as BPM, AEM, and CEM, suffer from challenges including product and ion crossover, salt crystallization, and increased internal resistance.32,38 These issues are frequently overlooked during device operation, leading to compromised performance and durability. Therefore, a systematic investigation into membrane behavior within hybrid systems is essential. Specifically, understanding their influence on system performance and their regulatory function in ion transport will be key to designing and developing next-generation high-performance functional membranes tailored for hybrid dual-electrolyte applications.
(c) Significant scope exists for refining reaction conditions and device structures to boost the performance. At present, most novel GOR-integrated hybrid electrolysis systems remain at the proof-of-concept stage, with limited optimization of key operational parameters such as temperature, electrolyte/reactant concentration, and electrolyte flow rate. This lack of optimization hinders the transition from lab-scale demonstrations to practical deployment. Furthermore, in situ studies of catalytic mechanisms are typically conducted under idealized conditions and often overlook the complexity of realistic two-electrode system environments. Developing model systems that enable real-time monitoring of catalytic behavior and reaction dynamics under practical operating conditions is crucial for optimizing performance and enhancing device viability.
Additionally, while ENE has been effectively harnessed in hydrogen production, its broader application in GOR-assisted processes, such as CO2 and nitrate reduction, remains underexplored. Leveraging ENE in these contexts could significantly improve energy efficiency and reduce system-wide consumption.
Additionally, GOR typically uses purified glycerol (∼$14,000 per ton, 99% purity) as the feedstock, significantly more expensive than crude glycerol (∼$200–250 per ton, 80% purity), leading to a major economic bottleneck.171,178,179 Opting for crude glycerol can markedly lower operational costs, necessitating catalysts resistant to its impurities for direct conversion.
Finally, research optimizing device structure is scarce. Apart from adopting MEA structures in more GOR-involved hybrid devices, developing multifunctional reactors incorporating optical or thermal field coupling holds promise for further improving system performance. Parallel efforts to integrate intelligent, online monitoring and feedback control systems will enable precise regulation of reaction conditions, facilitating the scalable and efficient implementation of GOR-based hybrid dual-electrolyte technologies.
(3) Conducting economic feasibility analysis for these GOR-integrated hybrid systems is crucial to evaluate their viability for industrial-scale application.180 However, despite the growing interest in value-added glycerol oxidation within electrolysis and fuel cell applications, current economic assessments remain scarce and are often overly simplistic, failing to capture the complexity of real-world deployment. Advancing the practical application of these systems requires a holistic focus on cost reduction and efficiency improvement. Future economic assessments must surpass simplistic evaluations by encompassing: (i) comprehensive costing of all device components (catalysts, reactants, electrolytes, membrane) and captured value (e.g., anode products and energy savings); (ii) specific attention to frequently overlooked catalyst production economics, requiring multidimensional evaluation (raw materials, preparation complexity, synthesis conditions, scalability) beyond mere performance or noble metal content; and (iii) system-level quantification linking technical performance to economic outcomes (e.g., impact of anode product value/separation costs and energy efficiency). The ultimate goal is an integrated, holistic assessment determining the net economic benefit and competitiveness of the entire GOR-integrated hybrid system.
In summary, GOR-involved hybrid electrochemical devices present a compelling nexus of challenges and opportunities. This perspective aims to deepen the mechanistic understanding of GOR and its system-level applications, identify critical bottlenecks, provide strategic guidance for future GOR-based hybrid dual-electrolyte systems, and ultimately enhance their role in improving electrochemical device performance.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
G. X. Wang, J. X. Chen, and F. Qiao searched for and collected the required publications for the perspective and co-wrote the manuscript. J. F. Wang contributed to revising and checking the manuscript, and provided constructive suggestions. Z. H. Wen proposed the concept, revised the manuscript, and provided constructive suggestions.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 22209183, 22225902, U22A20436, 52436005), the National Key Research & Development Program of China (2022YFE0115900, 2021YFA1501500), and Advanced Talents of Jiangsu University, China (Grant No. 23JDG027).References
- X. Hu, J. Lu, Y. Liu, L. Chen, X. Zhang and H. Wang, Environ. Chem. Lett., 2023, 21, 2825–2861 CrossRef CAS.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS.
- M. Simoes, S. Baranton and C. Coutanceau, Chemsuschem, 2012, 5, 2106–2124 CrossRef CAS.
- S. Shao, L. Ma, X. Li, H. Zhang and R. Xiao, Ind. Crops Prod., 2023, 192, 115912 CrossRef CAS.
- G. Hassan, M. A. Shabbir, F. Ahmad, I. Pasha, N. Aslam, T. Ahmad, A. Rehman, M. F. Manzoor, M. Inam-Ur-Raheem and R. M. Aadil, Food Chem., 2021, 363, 130352 CrossRef CAS PubMed.
- H.-H. Dong, Y.-X. Zhu, Y.-G. Li, J.-Y. Liang, Y. Tan, X.-Y. Zhang, H.-M. Jiang, L. Lin and Z.-M. Sun, Tungsten, 2024, 6, 696–710 CrossRef.
- L. Fan, B. Liu, X. Liu, N. Senthilkumar, G. Wang and Z. Wen, Energy Technol., 2020, 9, 2000804 CrossRef.
- M. H. Moklis, C. Shuo, S. Boonyubol and J. S. Cross, Chemsuschem, 2024, 17, e202300990 CrossRef CAS.
- L. Shen, X. Zhou, C. Zhang, H. Yin, A. Wang and C. Wang, J. Food Biochem., 2019, 43, e12931 Search PubMed.
- B. Katryniok, H. Kimura, E. Skrzyńska, J.-S. Girardon, P. Fongarland, M. Capron, R. Ducoulombier, N. Mimura, S. Paul and F. Dumeignil, Green Chem., 2011, 13, 1960–1979 RSC.
- Z. Zhao, J. Arentz, L. A. Pretzer, P. Limpornpipat, J. M. Clomburg, R. Gonzalez, N. M. Schweitzer, T. Wu, J. T. Miller and M. S. Wong, Chem. Sci., 2014, 5, 3715–3728 RSC.
- P. Prabhu, Y. Wan and J.-M. Lee, Matter, 2020, 3, 1162–1177 CrossRef.
- H. Kong, S. Gupta, A. F. Pérez-Torres, C. Höhn, P. Bogdanoff, M. T. Mayer, R. van de Krol, M. Favaro and F. F. Abdi, Chem. Sci., 2024, 15, 10425–10435 RSC.
- N. Tuleushova, Y. Holade, D. Cornu and S. Tingry, Electrochem. Sci. Adv., 2023, 3, e2100174 CrossRef CAS.
- L. Fan, Y. Ji, G. Wang, J. Chen, K. Chen, X. Liu and Z. Wen, J. Am. Chem. Soc., 2022, 144, 7224–7235 CrossRef CAS.
- S. Verma, S. Lu and P. J. A. Kenis, Nat. Energy, 2019, 4, 466–474 CrossRef CAS.
- J. Bai, H. Huang, F.-M. Li, Y. Zhao, P. Chen, P.-J. Jin, S.-N. Li, H.-C. Yao, J.-H. Zeng and Y. Chen, J. Mater. Chem. A, 2019, 7, 21149–21156 RSC.
- S. Li, P. Ma, C. Gao, L. Liu, X. Wang, M. Shakouri, R. Chernikov, K. Wang, D. Liu, R. Ma and J. Wang, Energy Environ. Sci., 2022, 15, 3004–3014 RSC.
- G. Wang, A. Chen, Y. Chen, F. Qiao, J. Wang, N. Yang, H. Zhang and Z. Wen, eScience, 2024, 100333 CrossRef.
- Z. Zhang, L. Xin and W. Li, Appl. Catal., B, 2012, 119–120, 40–48 CrossRef CAS.
- P. Sangkheaw, S. Therdthianwong, A. Therdthianwong, N. Wongyao and S. Yongprapat, Renewable Energy, 2020, 161, 395–407 CrossRef CAS.
- M. Kumar, B. Meena, A. Yu, C. Sun and S. Challapalli, Green Chem., 2023, 25, 8411–8443 RSC.
- J. Shi, J. Ma, E. Ma, J. Li, Y. Hu, L. Fan and W. Cai, Carbon Neutralization, 2024, 3, 285–312 CrossRef CAS.
- P. N. A. M. Othman, N. A. Karim and S. K. Kamarudin, Int. J. Energy Res., 2021, 45, 12693–12727 CrossRef CAS.
- L. S. Oh, M. Park, Y. S. Park, Y. Kim, W. Yoon, J. Hwang, E. Lim, J. H. Park, S. M. Choi, M. H. Seo, W. B. Kim and H. J. Kim, Adv. Mater., 2023, 35, e2203285 CrossRef.
- J. Yang, J. Yu, K. Zhang, F. Qiao and G. Yu, Adv. Mater. Technol., 2024, 9, 2301602 CrossRef CAS.
- J. Li, X. Meng, X. Song, J. Qi, F. Liu, X. Xiao, Y. Du, G. Xu, Z. Jiang, S. Ye, S. Huang and J. Qiu, Adv. Funct. Mater., 2024, 34, 2316718 CrossRef CAS.
- J. Ma, X. Wang, J. Song, Y. Tang, T. Sun, L. Liu, J. Wang, J. Wang and M. Yang, Angew. Chem., Int. Ed., 2024, 63, e202319153 CrossRef CAS.
- P. Wang, G. Wang, K. Chen, W. Pan, L. Yi, J. Wang, Q. Chen, J. Chen and Z. Wen, Nano Energy, 2023, 118, 108992 CrossRef CAS.
- H. Yu, M. Hu, C. Chen, C. Hu, Q. Li, F. Hu, S. Peng and J. Ma, Angew. Chem., Int. Ed., 2023, 62, e202314569 CrossRef CAS PubMed.
- R. Y. Fan, X. J. Zhai, W. Z. Qiao, Y. S. Zhang, N. Yu, N. Xu, Q. X. Lv, Y. M. Chai and B. Dong, Nano-Micro Lett., 2023, 15, 190 CrossRef CAS PubMed.
- C. Dai, Q. Wu, T. Wu, Y. Zhang, L. Sun, X. Wang, A. C. Fisher and Z. J. Xu, Energy Environ. Sci., 2024, 17, 6350–6359 RSC.
- T. Hamada, A. R. Circelli, H. Inoue, C. A. Randall and E. L. Clark, J. Phys. Chem. C, 2024, 128, 10790–10801 CrossRef CAS.
- N. Tuleushova, Y. Holade, D. Cornu and S. Tingry, Electrochem. Sci. Adv., 2022, 3, e2100174 CrossRef.
- M. Braun, C. S. Santana, A. C. Garcia and C. Andronescu, Curr. Opin. Green Sustainable Chem., 2023, 41, 100829 CrossRef CAS.
- S. Li, D. Liu, G. Wang, P. Ma, X. Wang, J. Wang and R. Ma, Nano-Micro Lett., 2023, 15, 189 CrossRef CAS.
- W. Luo, H. Tian, Q. Li, G. Meng, Z. Chang, C. Chen, R. Shen, X. Yu, L. Zhu, F. Kong, X. Cui and J. Shi, Adv. Funct. Mater., 2023, 34, 2306995 CrossRef.
- P. W. Cai, X. Hu, K. Chen, Z. W. Lu and Z. H. Wen, Sci. Bull., 2024, 69, 3571–3589 CrossRef CAS.
- L. Fan, Y. Ji, G. Wang, Z. Zhang, L. Yi, K. Chen, X. Liu and Z. Wen, J. Energy Chem., 2022, 72, 424–431 CrossRef CAS.
- Y. Ding, P. Cai and Z. Wen, Chem. Soc. Rev., 2021, 50, 1495–1511 RSC.
- J. Banjong, A. Therdthianwong, S. Therdthianwong, S. Yongprapat and N. Wongyao, Int. J. Hydrogen Energy, 2020, 45, 2244–2256 CrossRef CAS.
- P. Wang, K. Chen, J. Chen, G. Wang, W. Pan and Z. Wen, Adv. Funct. Mater., 2024, 34, 2408267 CrossRef CAS.
- G. Wang and Z. Wen, Nanoscale, 2018, 10, 21087–21095 RSC.
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC.
- T. Zhang, J. Zhou, T. Luo, J. Q. Lu, Z. Li, X. Weng and F. Yang, Chem.–Eur. J., 2023, 29, e202301455 CrossRef CAS.
- G. Wang, J. Chen, P. Cai, J. Jia and Z. Wen, J. Mater. Chem. A, 2018, 6, 17763–17770 RSC.
- D. Zheng, J. Li, S. Ci, P. Cai, Y. Ding, M. Zhang and Z. Wen, Appl. Catal., B, 2020, 277, 119178 CrossRef CAS.
- G. Dodekatos, S. Schünemann and H. Tüysüz, ACS Catal., 2018, 8, 6301–6333 CrossRef CAS.
- M. K. Goetz, E. Usman and K.-S. Choi, ACS Catal., 2023, 13, 15758–15769 CrossRef CAS.
- Z. Ke, N. Williams, X. Yan, S. Younan, D. He, X. Song, X. Pan, X. Xiao and J. Gu, J. Mater. Chem. A, 2021, 9, 19975–19983 RSC.
- L. Xu and S. Geng, Inorg. Chem., 2024, 64, 617–626 CrossRef PubMed.
- T. Li and D. A. Harrington, Chemsuschem, 2021, 14, 1472–1495 CrossRef CAS PubMed.
- A. C. Garcia, M. J. Kolb, C. van Nierop y Sanchez, J. Vos, Y. Y. Birdja, Y. Kwon, G. Tremiliosi-Filho and M. T. M. Koper, ACS Catal., 2016, 6, 4491–4500 CrossRef CAS.
- L. S. Ribeiro, E. G. Rodrigues, J. J. Delgado, X. Chen, M. F. R. Pereira and J. J. M. Órfão, Ind. Eng. Chem. Res., 2016, 55, 8548–8556 CrossRef CAS.
- W. Chen, L. Zhang, L. Xu, Y. He, H. Pang, S. Wang and Y. Zou, Nat. Commun., 2024, 15, 2420 CrossRef CAS.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- Y. Duan, M. Xue, B. Liu, M. Zhang, Y. Wang, B. Wang, R. Zhang and K. Yan, Chin. J. Catal., 2024, 57, 68–79 CrossRef CAS.
- X. Zi, Y. Zhou, L. Zhu, Q. Chen, Y. Tan, X. Wang, M. Sayed, E. Pensa, R. A. Geioushy, K. Liu, J. Fu, E. Cortés and M. Liu, Angew. Chem., Int. Ed., 2023, 62, e202309351 CrossRef CAS PubMed.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- G. Wu, X. Dong, J. Mao, G. Li, C. Zhu, S. Li, A. Chen, G. Feng, Y. Song, W. Chen and W. Wei, Chem. Eng. J., 2023, 468, 143640 CrossRef CAS.
- A. D. Handoko, F. Wei, Jenndy, B. S. Yeo and Z. W. Seh, Nat. Catal., 2018, 1, 922–934 CrossRef CAS.
- Y. Zhu, Q. Z. Qian, Y. X. Chen, X. Y. He, X. W. Shi, W. T. Wang, Z. Y. Li, Y. F. Feng, G. Q. Zhang and F. Y. Cheng, Adv. Funct. Mater., 2023, 33, e2300547 CrossRef.
- Z. Xia, C. Ma, Y. Fan, Y. Lu, Y.-C. Huang, Y. Pan, Y. Wu, Q. Luo, Y. He, C.-L. Dong, S. Wang and Y. Zou, ACS Catal., 2024, 14, 1930–1938 CrossRef CAS.
- R. N. S. Lima, J. Souza-Garcia and C. A. Angelucci, J. Phys. Chem. C, 2023, 127, 16374–16384 CrossRef CAS.
- Y. Zhang, S.-X. Guo, X. Zhang, A. M. Bond and J. Zhang, Nano Today, 2020, 31, 100835 CrossRef CAS.
- Y. Fang, C. Dai, X. Liu, Y. Wang, C. Ju, S. He, R. Shi, Y. Liu, J. Zhang, Y. Zhu and J. Wang, Nano Energy, 2024, 127, 109754 CrossRef CAS.
- K. Shi, D. Si, X. Teng, L. Chen and J. Shi, Chin. J. Catal., 2023, 53, 143–152 CrossRef CAS.
- N. Xi, Y. Zang, X. Sun, J. Yu, M. Johnsson, Y. Dai, Y. Sang, H. Liu and X. Yu, Adv. Energy Mater., 2023, 13, 2301572 CrossRef CAS.
- L. Xu, Y. Yang, C. Li, R. Ning, J. Ma, M. Yao, S. Geng and F. Liu, Chem. Eng. J., 2024, 481, 148304 CrossRef CAS.
- Y. Zhou, T. J. A. Slater, X. Luo and Y. Shen, Appl. Catal., B, 2023, 324, 122218 CrossRef CAS.
- Y. Li, X. Wei, L. Chen, J. Shi and M. He, Nat. Commun., 2019, 10, 5335 CrossRef PubMed.
- Q. Qian, X. He, Z. Li, Y. Chen, Y. Feng, M. Cheng, H. Zhang, W. Wang, C. Xiao, G. Zhang and Y. Xie, Adv. Mater., 2023, 35, 2300935 CrossRef CAS PubMed.
- W. Chen, C. Xie, Y. Wang, Y. Zou, C.-L. Dong, Y.-C. Huang, Z. Xiao, Z. Wei, S. Du, C. Chen, B. Zhou, J. Ma and S. Wang, Chem, 2020, 6, 2974–2993 CAS.
- K. Fernández-Caso, A. Peña-Rodríguez, J. Solla-Gullón, V. Montiel, G. Díaz-Sainz, M. Alvarez-Guerra and A. Irabien, J. CO2 Util., 2023, 70, 102431 CrossRef.
- Z. Huang, H. Ren, J. Guo, Y. Tang, D. Ye, J. Zhang and H. Zhao, Appl. Catal., B, 2024, 351, 123986 CrossRef CAS.
- Z. Zhang, L. Xin, J. Qi, D. J. Chadderdon, K. Sun, K. M. Warsko and W. Li, Appl. Catal., B, 2014, 147, 871–878 CrossRef CAS.
- G. S. Tran, T. G. Vo and C. Y. Chiang, ACS Appl. Mater. Interfaces, 2023, 15, 22662–22671 CrossRef CAS PubMed.
- G.-S. Tran, T.-G. Vo and C.-Y. Chiang, J. Catal., 2021, 404, 139–148 CrossRef CAS.
- Y. Holade, C. Morais, K. Servat, T. W. Napporn and K. B. Kokoh, ACS Catal., 2013, 3, 2403–2411 CrossRef CAS.
- Y. Holade, K. Servat, S. Tingry, T. W. Napporn, H. Remita, D. Cornu and K. B. Kokoh, Chemphyschem, 2017, 18, 2573–2605 CrossRef CAS PubMed.
- S. D. Mürtz, F. Musialek, N. Pfänder and R. Palkovits, ChemElectroChem, 2023, 10, e202201114 CrossRef.
- L. S. Oh, J. Han, E. Lim, W. B. Kim and H. J. Kim, Catalysts, 2023, 13, 892 CrossRef CAS.
- J. F. Gomes and G. Tremiliosi-Filho, Electrocatalysis, 2011, 2, 96–105 CrossRef CAS.
- X. Huang, Y. Guo, Y. Zou and J. Jiang, Appl. Catal., B, 2022, 309, 121247 CrossRef CAS.
- X. Huang, Y. Zou and J. Jiang, ACS Sustain. Chem. Eng., 2021, 9, 14470–14479 CrossRef CAS.
- A. M. Verma, L. Laverdure, M. M. Melander and K. Honkala, ACS Catal., 2021, 12, 662–675 CrossRef.
- C. Dai, L. Sun, H. Liao, B. Khezri, R. D. Webster, A. C. Fisher and Z. J. Xu, J. Catal., 2017, 356, 14–21 CrossRef CAS.
- P. Rodriguez, Y. Kwon and M. T. M. Koper, Nat. Chem., 2012, 4, 177–182 CrossRef CAS PubMed.
- Z. Zhang, L. Xin, J. Qi, D. J. Chadderdon and W. Li, Appl. Catal., B, 2013, 136–137, 29–39 CrossRef CAS.
- J. F. Gomes, F. B. C. de Paula, L. H. S. Gasparotto and G. Tremiliosi-Filho, Electrochim. Acta, 2012, 76, 88–93 CrossRef CAS.
- L. Huang, J.-Y. Sun, S.-H. Cao, M. Zhan, Z.-R. Ni, H.-J. Sun, Z. Chen, Z.-Y. Zhou, E. G. Sorte, Y. J. Tong and S.-G. Sun, ACS Catal., 2016, 6, 7686–7695 CrossRef CAS.
- Y. Liu, W. Yu, D. Raciti, D. H. Gracias and C. Wang, J. Phys. Chem. C, 2018, 123, 426–432 CrossRef.
- J. Jiang, L. Zhou, F. Xu, G. Chen, X. Liu, Z. Shen, L. Yang, Q. Wu, X. Wang and Z. Hu, Nano Res., 2023, 17, 4055–4061 CrossRef.
- Y. Li, J. Hong and Y. Shen, Int. J. Hydrogen Energy, 2024, 82, 448–455 CrossRef CAS.
- J. M. Sieben, A. A. Alvarez and M. D. Sanchez, ACS Appl. Energy Mater., 2024, 7, 6677–6686 CrossRef CAS.
- D. Lee, Y. Kim, Y. Kwon, J. Lee, T.-W. Kim, Y. Noh, W. B. Kim, M. H. Seo, K. Kim and H. J. Kim, Appl. Catal., B, 2019, 245, 555–568 CrossRef CAS.
- D. S. Ferreira, A. C. Gaiotti, H. R. Araujo, B. C. Batista, D. D. Reis, M. Janete Giz and G. A. Camara, J. Catal., 2023, 417, 445–452 CrossRef CAS.
- M. B. C. de Souza, V. Y. Yukuhiro, R. A. Vicente, C. T. G. Vilela Menegaz Teixeira Pires, J. L. Bott-Neto and P. S. Fernández, ACS Catal., 2020, 10, 2131–2137 CrossRef CAS.
- Y. Hai, Y. Chang, J. Jia, A. Xu and M. Jia, Int. J. Hydrogen Energy, 2023, 48, 14742–14748 CrossRef CAS.
- C. Y. Wang, Z. Y. Yu, G. Li, Q. T. Song, G. Li, C. X. Luo, S. H. Yin, B. A. Lu, C. Xiao, B. B. Xu, Z. Y. Zhou, N. Tian and S. G. Sun, ChemElectroChem, 2020, 7, 239–245 CrossRef CAS.
- Y. Zhou, Y. Shen and J. Xi, Appl. Catal., B, 2019, 245, 604–612 CrossRef CAS.
- X.-M. Yu, Y. Ke, X. Wang, H. Liu and H. Yuan, Chem. Mater., 2024, 36, 1737–1752 CrossRef CAS.
- B. Tam, M. Duca, A. Wang, M. Fan, S. Garbarino and D. Guay, ChemElectroChem, 2019, 6, 1784–1793 CrossRef CAS.
- T. Hamada, M. Chiku, E. Higuchi, C. A. Randall and H. Inoue, ACS Appl. Energy Mater., 2024, 7, 1970–1982 CrossRef CAS.
- C. C. Lima, M. V. F. Rodrigues, A. F. M. Neto, C. R. Zanata, C. T. G. V. M. T. Pires, L. S. Costa, J. Solla-Gullón and P. S. Fernández, Appl. Catal., B, 2020, 279, 119369 CrossRef CAS.
- Y. Zhou, Y. Shen, J. Xi and X. Luo, ACS Appl. Mater. Interfaces, 2019, 11, 28953–28959 CrossRef CAS.
- Y. Kwon, Y. Birdja, I. Spanos, P. Rodriguez and M. T. M. Koper, ACS Catal., 2012, 2, 759–764 CrossRef CAS.
- M. B. C. de Souza, R. A. Vicente, V. Y. Yukuhiro, C. T. G. V. M. T. Pires, W. Cheuquepán, J. L. Bott-Neto, J. Solla-Gullón and P. S. Fernández, ACS Catal., 2019, 9, 5104–5110 CrossRef CAS.
- A. C. Garcia, Y. Y. Birdja, G. Tremiliosi-Filho and M. T. M. Koper, J. Catal., 2017, 346, 117–124 CrossRef CAS.
- H. H. Lee, J. H. Choi, D. S. Kim, S. Jeon, E. A. Stach and H. K. Cho, EcoMat, 2024, 6, e12504 CrossRef CAS.
- H. R. Araujo, C. R. Zanata, E. Teixeira-Neto, R. B. de Lima, B. C. Batista, M. J. Giz and G. A. Camara, Electrochim. Acta, 2019, 297, 61–69 CrossRef CAS.
- Y. Kwon, T. J. P. Hersbach and M. T. M. Koper, Top. Catal., 2014, 57, 1272–1276 CrossRef CAS.
- S. Lee, H. J. Kim, E. J. Lim, Y. Kim, Y. Noh, G. W. Huber and W. B. Kim, Green Chem., 2016, 18, 2877–2887 RSC.
- F. Yang, J. Ye, Q. Yuan, X. Yang, Z. Xie, F. Zhao, Z. Zhou, L. Gu and X. Wang, Adv. Funct. Mater., 2020, 30, 1908235 CrossRef CAS.
- R. M. Castagna, J. M. Sieben, A. E. Alvarez and M. M. E. Duarte, Int. J. Hydrogen Energy, 2019, 44, 5970–5982 CrossRef CAS.
- J. Zhao, W. Jing, T. Tan, X. Liu, Y. Kang and W. Wang, New J. Chem., 2020, 44, 4604–4612 RSC.
- H. Du, K. Wang, P. Tsiakaras and P. K. Shen, Appl. Catal., B, 2019, 258, 117951 CrossRef CAS.
- J. Li, Y. Pan, H. Yan, X. Ding, Y. Liu, X. Feng and C. Yang, Ind. Eng. Chem. Res., 2025, 64, 5099–5113 CrossRef CAS.
- D. Kim, W.-G. Lim, Y. Kim, L. S. Oh, S. Kim, J. H. Park, C. Jo, H. J. Kim, J. Kang, S. Lee and E. Lim, Appl. Catal., B, 2023, 339, 123104 CrossRef CAS.
- Z. Zhang, L. Xin, J. Qi, Z. Wang and W. Li, Green Chem., 2012, 14, 2150 RSC.
- D. Kim, L. S. Oh, Y. C. Tan, H. Song, H. J. Kim and J. Oh, ACS Catal., 2021, 11, 14926–14931 CrossRef CAS.
- Y. Xie, L. Sun, X. Pan, Z. Zhou, Y. Zheng, X. Yang and G. Zhao, Carbon, 2023, 203, 88–96 CrossRef CAS.
- L. Shen, L. Sun, M. Douthwaite, O. Akdim, S. Taylor and G. J. Hutchings, ACS Catal., 2024, 14, 11343–11351 CrossRef CAS.
- L. Thia, M. Xie, Z. Liu, X. Ge, Y. Lu, W. E. Fong and X. Wang, ChemCatChem, 2016, 8, 3272–3278 CrossRef CAS.
- Y. Yan, H. Zhou, S.-M. Xu, J. Yang, P. Hao, X. Cai, Y. Ren, M. Xu, X. Kong, M. Shao, Z. Li and H. Duan, J. Am. Chem. Soc., 2023, 145, 6144–6155 CrossRef CAS PubMed.
- C. Coutanceau, A. Zalineeva, S. Baranton and M. Simoes, Int. J. Hydrogen Energy, 2014, 39, 15877–15886 CrossRef CAS.
- A. Zalineeva, S. Baranton and C. Coutanceau, Electrochim. Acta, 2015, 176, 705–717 CrossRef CAS.
- A. Serov, T. Asset, M. Padilla, I. Matanovic, U. Martinez, A. Roy, K. Artyushkova, M. Chatenet, F. Maillard, D. Bayer, C. Cremers and P. Atanassov, Appl. Catal., B, 2016, 191, 76–85 CrossRef CAS.
- N. Benipal, J. Qi, Q. Liu and W. Li, Appl. Catal., B, 2017, 210, 121–130 CrossRef CAS.
- M. S. Ahmad, S. Singh, C. K. Cheng, H. R. Ong, H. Abdullah, M. R. Khan and S. Wongsakulphasatch, Catal. Commun., 2020, 139, 105964 CrossRef CAS.
- Y. Holade, C. Morais, S. Arrii-Clacens, K. Servat, T. W. Napporn and K. B. Kokoh, Electrocatalysis, 2013, 4, 167–178 CrossRef CAS.
- A. C. Garcia, C. Morais, T. W. Napporn, K. B. Kokoh and G. Tremiliosi-Filho, ChemElectroChem, 2017, 4, 1314–1319 CrossRef CAS.
- B. Huang, Y. Ge, A. Zhang, S. Zhu, B. Chen, G. Li, Q. Yun, Z. Huang, Z. Shi, X. Zhou, L. Li, X. Wang, G. Wang, Z. Guan, L. Zhai, Q. Luo, Z. Li, S. Lu, Y. Chen, C. S. Lee, Y. Han, M. Shao and H. Zhang, Adv. Mater., 2023, 35, e2302233 CrossRef PubMed.
- L. Y. Jiang, F. M. Tian, X. Y. Chen, X. X. Ren, J. J. Feng, Y. Yao, L. Zhang and A. J. Wang, J. Colloid Interface Sci., 2023, 649, 118–124 CrossRef CAS PubMed.
- J. Li, L. Wu, J. Shu, H. Yang, Y. Cui, B. Hu and S. Li, Electrochim. Acta, 2023, 470, 143344 CrossRef CAS.
- M. Sheraz Ahmad, K. Hoong Ng, C.-L. Chen, F. Kabir, T. Witoon, T. Yeong Wu and C. Kui Cheng, Fuel, 2023, 333, 126471 CrossRef CAS.
- I. Terekhina, J. White, A. Cornell and M. Johnsson, ACS Appl. Nano Mater., 2023, 6, 11211–11220 CrossRef CAS.
- Z. H. Mao, L. Jia, X. N. Mao, X. Ding, B. B. Pan, T. R. Yan, J. Xu, L. Zhang, L. Wang, N. Han and Y. G. Li, J. Mater. Chem. A, 2024, 12, 24136–24143 RSC.
- I. Chauhan, H. Bajpai, B. Ray, S. K. Kolekar, S. Datar, K. K. Patra and C. S. Gopinath, ACS Appl. Mater. Interfaces, 2024, 16, 26130–26141 CrossRef CAS PubMed.
- M. Weber, P. Collot, H. El Gaddari, S. Tingry, M. Bechelany and Y. Holade, ChemElectroChem, 2018, 5, 743–747 CrossRef CAS.
- F. F. S. Xavier, A. C. Cunha, T. W. Napporn and P. Olivi, Int. J. Hydrogen Energy, 2023, 48, 31091–31100 CrossRef CAS.
- Q.-L. Hong, Y. Zhao, X. Ai, Y. Ding, F.-M. Li, P. Chen, P.-J. Jin and Y. Chen, Chem. Eng. J., 2024, 482, 148960 CrossRef CAS.
- J. White, I. Terekhina, E. Campos dos Santos, D. Martín-Yerga, L. G. M. Pettersson, M. Johnsson and A. Cornell, ACS Appl. Energy Mater., 2024, 7, 1802–1813 CrossRef CAS.
- C. Li, H. Li, B. Zhang, H. Li, Y. Wang, X. Wang, P. Das, Y. Li, X. Wu, Y. Li, Y. Cui, J. Xiao and Z. S. Wu, Angew. Chem., Int. Ed., 2024, 63, e202411542 CrossRef CAS PubMed.
- G. Zhao, G. Hai, P. Zhou, Z. Liu, Y. Zhang, B. Peng, W. Xia, X. Huang and G. Wang, Adv. Funct. Mater., 2023, 33, 2213170 CrossRef CAS.
- M. Fleischmann, K. Korinek and D. Pletcher, J. Electroanal. Chem. Interfacial Electrochem., 1971, 31, 39–49 CrossRef CAS.
- G. X. Wang, J. X. Chen, K. K. Li, J. H. Huang, Y. C. Huang, Y. J. Liu, X. Hu, B. S. Zhao, L. C. Yi, T. W. Jones and Z. H. Wen, Nano Energy, 2022, 92, 106751 CrossRef CAS.
- L. Li, Z. Zhang, H. Chen and F. Chen, Nano Res., 2022, 16, 4596–4602 CrossRef.
- S. Zhu, A. Xie, X. Tao, J. Zhang, B. Wei, Z. Liu, Y. Tao and S. Luo, J. Electroanal. Chem., 2020, 857, 113748 CrossRef CAS.
- X. T. Han, H. Y. Sheng, C. Yu, T. W. Walker, G. W. Huber, J. S. Qiu and S. Jin, ACS Catal., 2020, 10, 6741–6752 CrossRef CAS.
- Y. Song, X. Wan, Y. Miao, J. Li, Z. Ren, B. Jin, H. Zhou, Z. Li and M. Shao, Appl. Catal., B, 2023, 333, 122808 CrossRef CAS.
- R. Deng, Z. Wang, Y. Xie, X. Wu, K. Ma, B. Liu, X. Qin, H. Shu, F. Mo, Y. Li, H. Yang, W. Yu, Y.-w. Hu, J. Chen and W. Liu, Inorg. Chem. Commun., 2024, 167, 112687 CrossRef CAS.
- M. S. E. Houache, K. Hughes, A. Ahmed, R. Safari, H. Liu, G. A. Botton and E. A. Baranova, ACS Sustainable Chem. Eng., 2019, 7, 14425–14434 CrossRef CAS.
- N. Lyu, Y. Chen, A. Guan, R. Wei, C. Yang, Y. Huang, X. Lv, C. Hu, M. Kuang and G. Zheng, Small, 2024, 20, e2401872 CrossRef PubMed.
- J. Wu, X. Liu, Y. Hao, S. Wang, R. Wang, W. Du, S. Cha, X. Y. Ma, X. Yang and M. Gong, Angew. Chem., Int. Ed., 2023, 62, e202216083 CrossRef CAS PubMed.
- Y. Li, X. Wei, S. Han, L. Chen and J. Shi, Angew. Chem., Int. Ed., 2021, 60, 21464–21472 CrossRef CAS PubMed.
- C. Liu, M. Hirohara, T. Maekawa, R. Chang, T. Hayashi and C.-Y. Chiang, Appl. Catal., B, 2020, 265, 118543 CrossRef CAS.
- B. Liu, G. Wang, X. Feng, L. Dai, Z. Wen and S. Ci, Nanoscale, 2022, 14, 12841–12848 RSC.
- S. Zhou, Y. Dai, Q. Song, L. Lu and X. Yu, J. Electroanal. Chem., 2024, 957, 118133 CrossRef CAS.
- Z. Xi, H. Zhou, Y. Liu and C. Xu, Electrochim. Acta, 2023, 470, 143285 CrossRef CAS.
- H. Yao, Y. Wang, Y. Zheng, X. Yu, J. Ge, Y. Zhu and X. Guo, Nano Res., 2023, 16, 10832–10839 CrossRef CAS.
- G. Wang, J. Chen, Z. Wen and J. Li, Sci. China Mater., 2024, 67, 1791–1803 CrossRef CAS.
- X. Feng, K. Guo, C. Jia, B. Liu, S. Ci, J. Chen and Z. Wen, Acta Phys.-Chim. Sin., 2023, 2303050 Search PubMed.
- Y. Zou, W. D. Zhang, H. Xu, J. Yang, J. Liu, Z. G. Gu and X. Yan, J. Colloid Interface Sci., 2023, 650, 701–709 CrossRef CAS PubMed.
- Y. Pei, Z. Pi, H. Zhong, J. Cheng and F. Jin, J. Mater. Chem. A, 2022, 10, 1309–1319 RSC.
- H. Ren, Z. Huang, G. Cai, J. Guo, Y. Sun, W. Yao, D. Ye, H. Qian, J. Zhang and H. Zhao, J. Alloys Compd., 2024, 996, 174781 CrossRef CAS.
- J. Li, H. Li, K. Fan, J. Y. Lee, W. Xie and M. Shao, Chem Catal., 2023, 3, 100638 CrossRef CAS.
- K. Kumar, S. Jain, L. S. Panchakarla and S. S. Nagarkar, ACS Appl. Nano Mater., 2024, 7, 17578–17587 CrossRef CAS.
- J. Na, B. Seo, J. Kim, C. W. Lee, H. Lee, Y. J. Hwang, B. K. Min, D. K. Lee, H. S. Oh and U. Lee, Nat. Commun., 2019, 10, 5193 CrossRef PubMed.
- M. S. E. Houache, R. Safari, U. O. Nwabara, T. Rafaïdeen, G. A. Botton, P. J. A. Kenis, S. Baranton, C. Coutanceau and E. A. Baranova, ACS Appl. Energy Mater., 2020, 3, 8725–8738 CrossRef CAS.
- M. A. Khan, S. K. Nabil, T. Al-Attas, N. G. Yasri, S. Roy, M. M. Rahman, S. Larter, P. M. Ajayan, J. Hu and M. G. Kibria, Chem Catal., 2022, 2, 2077–2095 CrossRef CAS.
- J. E. Huang, F. Li, A. Ozden, A. Sedighian Rasouli, F. P. Garcia de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C. T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- H. Shen, H. Jin, H. Li, H. Wang, J. Duan, Y. Jiao and S. Z. Qiao, Nat. Commun., 2023, 14, 2843 CrossRef CAS PubMed.
- M. C. O. Monteiro, M. F. Philips, K. J. P. Schouten and M. T. M. Koper, Nat. Commun., 2021, 12, 4943 CrossRef CAS PubMed.
- B. van den Bosch, B. Rawls, M. B. Brands, C. Koopman, M. F. Phillips, M. C. Figueiredo and G. M. Gruter, Chempluschem, 2023, 88, e202300112 CrossRef CAS PubMed.
- A. Ilie, M. Simoes, S. Baranton, C. Coutanceau and S. Martemianov, J. Power Sources, 2011, 196, 4965–4971 CrossRef CAS.
- X. M. Yin, W. Sun, K. Chen, Z. W. Lu, J. X. Chen, P. W. Cai and Z. H. Wen, Adv. Sci., 2024, 11, e2402343 CrossRef PubMed.
- S. Rahim, C. S. Lee, F. Abnisa, M. K. Aroua, W. A. W. Daud, P. Cognet and Y. Peres, Sci. Total Environ., 2020, 705, 135137 CrossRef CAS PubMed.
- S. Angizi, E. Y. Kirici and D. Higgins, Trends Chem., 2024, 6, 14–21 CrossRef CAS.
- K. M. Ebeling, D. Bongartz, S. Mürtz, R. Palkovits and A. Mitsos, Ind. Eng. Chem. Res., 2024, 63, 8250–8260 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |