
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterodimeric protein entangling motifs: systematic discovery, feature analysis, and topology engineering
Lianjie
Xu
a,
Xibao
Tian
a and
Wen-Bin
Zhang
 *ab
*ab
aBeijing National Laboratory for Molecular Sciences, Key Laboratory of Polymer Chemistry & Physics of Ministry of Education, Center for Soft Matter Science and Engineering, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, P. R. China. E-mail: wenbin@pku.edu.cn
bAI for Science (AI4S)-Preferred Program, Shenzhen Graduate School, Peking University, Shenzhen 518055, P. R. China
First published on 8th August 2025
Abstract
Synthesis of nontrivial protein topologies calls for genetically encoded protein entangling motifs, especially those of heterogeneous nature, to achieve structural complexity and functional relevance. Herein, we report the systematic discovery of heterodimeric entangling motifs using criteria like Gauss linking number, buried surface area and terminal distances. These motifs were analyzed to reveal their formation mechanisms (i.e., precursor cleavage, synergistic folding and segment piercing/wrapping) and biological significance (i.e., stability enhancement crucial for executing functions like regulation and catalysis). Six premium motifs were selected for experimental validation. Upon ring closure mediated by orthogonal split inteins, all six motifs led to protein hetero[2]catenanes with varying efficiency, providing versatile templates for making mechanically interlocked protein conjugates, such as Förster resonance energy transfer pairs and bispecific binders. The study not only helps untangle the influence of chain entanglements on protein properties but also provides a modular platform to enrich the toolbox of protein topology engineering.
Introduction
Proteins with nontrivial chemical topologies have drawn an ever-increasing interest, which is attributed not only to the aesthetic appeal of complex molecular topologies, but also to the advantageous topological effects in protein properties and functions.1–3 The formation of protein knots and links relies heavily on chain threading and entanglements, which are unfortunately kinetically unfavorable in protein folding and usually hard to design from scratch.4–6 In recent years, taking advantage of naturally intertwined symmetric protein motifs in combination with appropriate protein ligation tools, researchers have successfully prepared various protein topologies, including trefoil knot,7 Hopf links,8,9 pretzelanes,10 lassos11 and so on. While there are some reports on chain entanglements in homomeric protein assemblies,12–17 the investigation of intertwined heteromeric protein complexes is scarce, presumably due to the much smaller population of heteromeric proteins.18 For example, out of the about 900 entanglement-containing proteins that Cieplak and coworkers reported, only a few are heteromeric.19 The formation mechanism, structural abundance, and biological significance of these heteromeric entangling motifs remain a mystery. Discovering these heterogeneous entangling protein motifs is thus fundamental to understanding the relationship between primary sequences and structural entanglements involving multiple chains in distinct folds.18To date, rather limited heterodimeric intertwined motifs have been identified and used in protein topology engineering. They were either rationally engineered from known homodimeric precursors20 or developed by artificially splitting an entwined protein domain like lasso peptides,21 dihydrofolate reductase22 and green fluorescent protein.23 Notably, active templates were thus developed with the capacity of guiding chain entanglement upon reconstitution and catalyzing the covalent bond formation simultaneously, both of which are needed to achieve concatenation.24 The intrinsic asymmetry of these motifs can dramatically enhance the structural complexity of the resulting topological proteins, as evidenced by the successful synthesis of protein [n]catenanes (n = 3, 4, and 5) in radial configuration25 and both symmetric and asymmetric protein olympiadanes.26 It also provides a novel mechano-bioconjugation strategy for developing advanced protein therapeutics with multi-function integration possessing additional functional benefits such as aggregation resistance, prolonged circulation and enhanced antitumor efficacy.27 Nevertheless, both approaches are not particularly effective in developing new heterodimeric intertwined motifs, impeding the understanding of chain entanglements in biological systems.
Recent advancements in our research have led to a significant expansion of the symmetric entangling motif database through systematic screening and structural analysis of homomeric protein assemblies in the Protein Data Bank (PDB).16 Building on this progress, we further established deep learning frameworks capable of predicting entanglement features directly from amino acid sequences.28,29 Through this workflow, we successfully identified multiple novel entangling motifs of C2 or C3 symmetry within the vast genomic space.28,29 These achievements in searching and mining of homomeric entangling motifs provide a robust methodological foundation, which now motivates our exploration of heteromeric entangling motifs to unlock new opportunities for engineering topological complexity in protein architectures. It was found that AlphaFold sometimes mistakenly predict topological links in heterodimeric complexes, which suggests that the topological features associated with chain entanglements may not be well captured by current protein structure prediction methods.30 Thus, from a practical perspective, we want to systematically look into the entangling heterodimeric motifs in PDB to expand the design space of topological proteins. In this article, we report the systematic discovery as well as the feature analysis of intertwined heterodimeric motifs from PDB (Scheme 1). The formation mechanisms of chain entanglements in these motifs, as well as their biological implications, are discussed. We also demonstrate their utility in designing intriguing topological proteins by the synthesis of protein heterocatenanes based on the selected premium motifs.
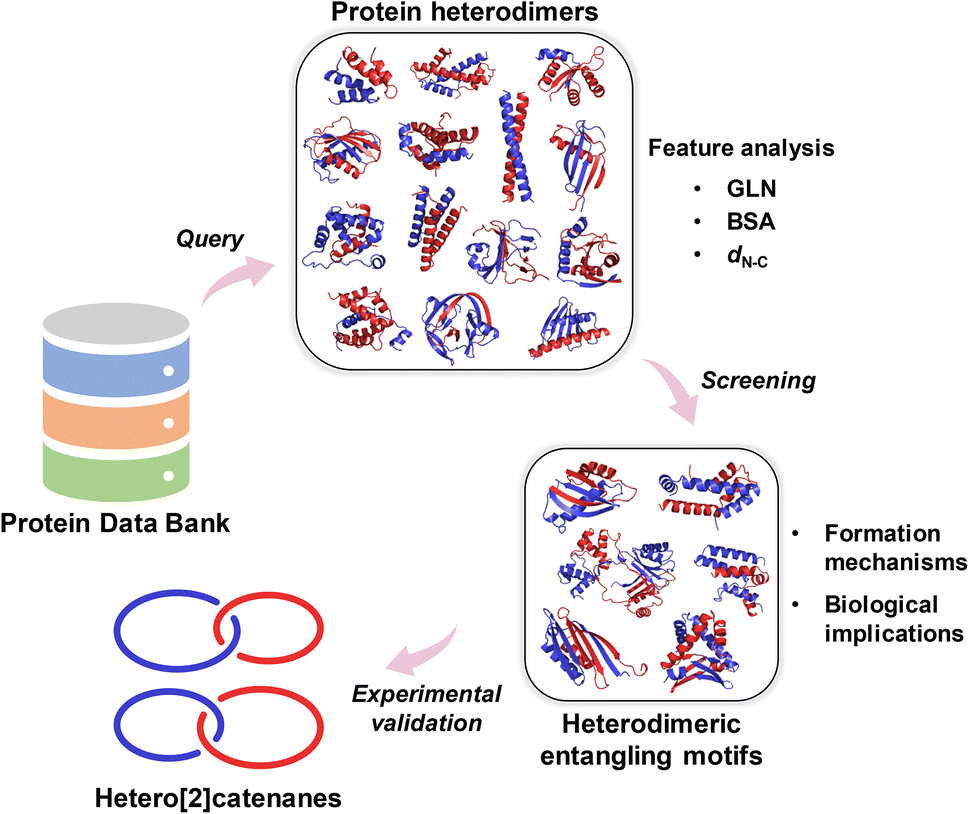 | ||
| Scheme 1 Workflow for the discovery of heterodimeric entangling motifs and their usage in protein topology engineering. The pipeline initiated with systematic searching of the Protein Data Bank to identify heterodimeric protein assemblies, followed by structure-based filtering and sequence clustering. Subsequent feature analysis of the non-redundant heterodimer database enabled screening of entangling protein motifs. Detailed investigation uncovered several formation mechanisms and biological implications of chain entanglements within protein heterodimers. A small set of promising heterodimeric entangling motifs were then engineered into protein heterocatenanes, highlighting their utility for constructing topological architectures. | ||
Results and discussion
Discovery and feature analysis of heterodimeric protein complexes
The workflow for the systematic discovery of intertwined heterodimeric protein motifs in PDB is as described previously for C2 assemblies (see Methods for more details).16 A total of 1709 protein heterodimers were collected after filtration and clustering (Table 1). Multiple parameters were calculated for each protein heterodimer, including the Gauss linking number (GLN) to measure the extent of chain intertwining, buried surface area (BSA) to estimate structural stability, and distance between N- and C-termini (dN–C) to evaluate the ease of chain cyclization. After automatic screening, manual curation was again conducted to select a group of heteromeric entangling motifs for protein topology engineering.| Entry | Operation | Number of entries |
|---|---|---|
| 1 | Downloaded | 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 741 741 |
| 2 | 30–400 a.a. | 7013 |
| 3 | Resolution ≤3.5 Å | 6932 |
| 4 | Clustering | 1709 |
| 5 | |GLN| ≥0.4 | 155 |
| 6 | BSA ≥600 Å2 | 143 |
| 7 | Manual curation | 20 |
The GLN calculation was adapted from the Gauss linking integral and has been applied to open curves including protein backbones, proving a convenient method to measure the extent of chain intertwining in a heterodimeric complex.15,31 A larger |GLN| value generally means a higher extent of chain intertwining. As shown in Fig. 1a, most of the heteromeric complexes have GLN around 0, and only 9.2% have |GLN| ≥0.4, which suggest that intertwined structures are rather rare in heteromeric complexes. Interestingly, highly intertwined motifs are significantly rarer in heterodimeric complexes as compared to homomeric assemblies, as represented by only 0.9% with a |GLN| ≥1 (Table S1). It implies that chain intertwining is more difficult to form in heterodimeric protein complexes than in homomeric assemblies. Unlike conventional synthetic polymers that tend to form entanglements as the chain gets longer, proteins mostly adopt well-defined folds and form entanglements through specific interactions that are chain-length-independent (Fig. 1b). Hence, special formation pathways may be required for two different protein chains to be significantly entangled. Typical examples of heterodimer proteins with varied GLN values are shown in Fig. 1c, providing an intuitive understanding of the correlation between the |GLN| value and extent of chain entanglement. The distribution of BSA of the heterodimeric proteins is shown in Fig. 1d. Notably, there is a moderate correlation between |GLN| values and BSA of heterodimeric complexes (Fig. 1e), suggesting that chain entanglements could also promote the stability of heteromeric complexes. We also calculated the terminal distance (dN–C) of each chain within the heteromeric complex (Fig. 1f). In general, motifs with smaller dN–C are easier to cyclize. However, this is not mandatory for topology synthesis. In many cases, chain cyclization can be achieved through orthogonal ligation tools such as SpyTag–SpyCatcher reactive pairs32 and split intein pairs.33 The use of flexible linkers of sufficient length and the reconstitution of the reactive partners can bring the termini closer with high specificity and coupling efficiency. Therefore, we do not impose a strict limit on dN–C and put more emphasis on the GLN and BSA cutoffs during the selection of promising motifs.
 | ||
| Fig. 1 Feature analysis of the collected 1709 heterodimeric protein complexes. (a) Histogram of GLN values of protein heteromeric complexes. (b) Distribution of |GLN| along with sequence lengths of the subunit pair of heteromeric complexes. (c) Typical examples of heterodimeric proteins with varied GLN values. (d) Histogram of BSA values of protein heteromeric complexes. (e) BSA of protein heteromeric complexes plotted as a function of |GLN|. The solid line is the linear least-squares regression fit. The Pearson correlation coefficient is 0.49. (f) Distribution of intra-subunit terminal distances of heterodimeric protein complexes. | ||
A quantitative criterion (i.e., |GLN| ≥0.4 and BSA ≥600 Å2) was applied to screen candidate intertwined motifs on the heterodimeric complexes. To facilitate their usage as entangling templates, we further selected a small set of premium motifs based on empirical criteria, such as the overall size, host strain, expression yield, and predicted topologies that can be formed upon cyclization. During manual curation, we also discarded the heterodimeric proteins that were particularly unqualified for protein topology engineering, e.g., loosely associated ones, heterodimeric coiled coils, reactive split inteins and so on (Fig. S1). The overview of screening results is summarized in Table 1, and the selected premium intertwined motifs are listed in Fig. S2. To evaluate the uniqueness of our collected motifs, we compared the 20 premium entangling heterodimers with the 1873 links in LinkProt and found no entries in common, which is probably due to the fact that protein links pose an additional requirement for covalent bond formation. We also compared our motifs with those discovered via the pulling-based method by Cieplak et al. and found only 5 motifs in common (i.e., 2BYK, 2ACM, 1B0N, 3A1G, and 4CZD).19 The small number of motifs in common is probably because the latter is mostly for homomeric complexes. We attribute this uniqueness to the different detection/screening method and the distinct research focus of our work. Therefore, we have convincingly developed a reliable platform to screen and discover those useful heteromeric entangling protein motifs.
Origins of chain entanglements in heterodimeric protein complexes
The scarcity of chain entanglements in protein complexes prompted us to interrogate the origins, or the formation mechanisms, of these highly intertwined heterodimers. Domain swapping is regarded as a major cause of chain entanglements in homomeric proteins but is hard to realize in heteromeric complexes, because it requires the exchange of identical domains from the subunits. Therefore, chain entanglements in heteromeric complexes must be formed via other routes. We conducted comprehensive investigation on the 155 protein heterodimers with genuine chain entanglements (entry 5 in Table 1) about their origin. Three formation mechanisms were proposed, including (1) cleavage of monomeric precursors, (2) mutual synergistic folding of multiple chains, and (3) direct interaction including segment piercing and wrapping (Fig. 2 and Table S2). | ||
| Fig. 2 Illustration of the mechanisms of chain entanglement formation within heterodimeric protein complexes. Three formation pathways were proposed, including the cleavage of monomeric precursors (a), mutual synergistic folding (b) and segment piercing or wrapping (c). | ||
The first mechanism, i.e., cleavage of monomeric precursors, is perhaps the most intriguing. Although there are numerous designed split proteins, most of them are not highly entangled.34 Herein, we identified several entangling heterodimers that come from the cleavage of monomeric precursors, accomplished through either auto-proteolysis or enzymatic cleavage. An example of the former is the SEA domain of mucin-1 (PDB ID: 2ACM), which undergoes spontaneous proteolysis attributed to folding-induced conformational stress at the Gly1097–Ser1098 loop (Fig. 2a).35 Similar autocatalytic cleavage was also observed in S-adenosylmethionine decarboxylases (PDB IDs: 1I7C, 1MHM, 3IWB and 5TVO),36 oxamate amidohydrolase HpxW (PDB ID: 5HFT),37 and ornithine acetyl transferase (PDB ID: 2YEP).38 There are also many examples of the latter. The Notch RR (PDB ID: 3I08), with the largest |GLN| among all the 155 entries, is formed via cleavage at the S1 site by furin-like protease during maturation (Fig. 2a).39 Human heparanase (PDB ID: 7RG8/5E8M) is activated as a heterodimer by stepwise proteolytic cleavage of a signal peptide and a linker segment from its precursor.40,41 Surprisingly, most of the cleavage sites in the above examples lie in the loops between adjacent β-strands, suggesting the importance of β-sheet structures in dictating chain entanglements. These naturally occurring intertwined split proteins would also shed light onto the topology engineering of transforming native single-domain linear proteins into their topological isoforms.22,23 It turned out that non-covalent entanglements are widespread among protein domains, which implies the vast design space of entangling motifs through chain rethreading.42,43
The second mechanism, i.e., mutual synergistic folding, involves the association and cooperative folding of two individual subunits that may otherwise be unstable or poorly folded in isolation. Therefore, preparing this class of heterodimers should usually resort to co-expression of two genes. One example is the heterodimeric complex of ACTR (activator for thyroid hormone and retinoid receptors) and CBP (cAMP responsive-binding protein) (PDB ID: 1KBH), which is archived in the Mutual Folding Induced by Binding (MFIB) database.44 Both ACTR and CBP are disordered on their own but tightly associate into a stable globular fold with substantial entanglement and high affinity (Kd = 34 nM) (Fig. 2b).45 Other examples include the BAG6 (Bcl-2-associated athanogene 6)–Ubl4a (ubiquitin-like protein 4a) complex (PDB ID: 4X86)46 and a sirohaem decarboxylase AhbA/AhbB (tend to precipitate when expressed individually, PDB ID: 4CZD).47 This class of heterodimeric entangling motifs are particularly suitable as templates for cellular synthesis of heterocatenanes since the undesired side products, i.e., cyclic monomers, are unstable and tend to form inclusion bodies facilitating the purification.48
The third mechanism is the direct contact-induced entanglement involving chain segment piercing through or wrapping around a folded domain. This pathway is straightforward but difficult to realize for two large folded domains. As aforementioned, the folded domain has to undergo significant conformational change and even unfolding in order to get deeply entangled with the other chain in direct contact. Nevertheless, it is easier to achieve via piercing or wrapping of much smaller segments. A representative case for segment piercing is the vasohibin (VASH) 2/vasohibin binding protein (SVBP) heterodimer (PDB ID: 6JZC), where SVBP, folded as a single α-helix, threads through the N-terminal loop region of VASH2 to form a three-helix bundle (Fig. 2c).49 Similar helix threading was also observed in the complex of C. elegans HMP-1/α-catenin and HMP-2/β-catenin (PDB ID: 5XA5), where the helical HMP-2 is inserted into the N-terminal four-helix bundle of HMP-1, and a protein rotaxane could be generated upon cyclization of HMP-1.50 On the other hand, segment wrapping-induced chain entanglement is usually realized by the winding of a small protein around the surface of a large domain. For example, in the AtaR–AtaT toxin–antitoxin complex (PDB ID: 6GTQ), the intrinsically disordered C-terminal region of AtaR wraps around the surface of AtaT to block all its functional hotspots for toxin neutralization (Fig. 2c).51 A similar association pattern is also adopted by some other antitoxin systems like RelE–RelB (PDB ID: 1WMI)52 and PaaA2–ParE2 (PDB ID: 5CZF)53 and the eukaryotic initiation factor 4E (eIF4E)–eIF4G complexes (PDB IDs: 1RF8 and 6FC0).54,55
Biological implications of chain entanglements in heterodimeric protein complexes
Although the formation mechanisms of protein chain entanglements can be deciphered by structure characterization, why such protein structures have evolved to embrace chain entanglements remain ambiguous. The collection of heterodimeric protein complexes (entry 4 in Table 1) offers a glimpse into the biological implications of chain entanglements within heterodimeric protein complexes. Enhancing stability is the most obvious and also widely acknowledged functional benefit of chain entanglements in protein structures, as has been extensively discussed in protein knots,56 complex lassos,57 as well as intertwined homomeric assemblies.14,58 This topological effect probably also applies to heterodimeric protein complexes. Significant chain entanglement could serve as an efficient approach to generating substantially extended binding interfaces, which is crucial in molecular functions like regulation, signaling and inhibition, especially for small-to-medium-sized proteins. For example, both BAG6 (81 a.a.) and Ubl4a (58 a.a.) are small intrinsically disordered proteins but are capable of synergetic folding and binding with a surprisingly high affinity (Kd = 2.2 ± 0.5 nM).46To obtain a more in-depth insight, we also analyzed the biological functions of the intertwined heterodimers in order to find out whether the entangled structures are enriched in certain functions. Out of the 1709 heterodimeric complexes, 1203 were successfully mapped to UniProtKB, matching 136 out of 198 annotation keywords of molecular function.59 The function annotations for the 100 entangling heterodimers (with |GLN| ≥0.4) are listed in Table S3. Surprisingly, similar to that observed previously in C2 assemblies, there is a strong preference for chain entanglements in DNA-binding proteins, with a total of 31 entries, accounting for 31% of all the annotated entangling heterodimers (Fig. 3a). The percentage of entangling heterodimers within each function type also exhibits such a preference. Among the top 10 molecular functions in terms of the percentage of intertwined heterodimers, 7 of them are associated with gene regulation, i.e., DNA-directed DNA polymerase, activator, DNA-binding, initiation factor, sigma factor, chromatin regulator, exonuclease and nuclease (Fig. 3b). The functional bias highlights the importance of chain entanglements within protein complexes in gene regulation and signaling. Interestingly, the preference of chain entanglements towards specific molecular functions was also observed for monomeric globular domains. It was revealed that proteins containing non-covalent lasso entanglements were enriched in lyase activity, transferase activity, catalytic activity on nucleic acid, hydrolase activity and so on, which agrees well with our observation in entangled heterodimers and may imply possible advantages of chain entanglements for enzymes.42 However, it was found that lasso entanglement-containing monomers are depleted in DNA-binding functions, while multimeric entangled protein complexes are somewhat enriched. This suggests that topological constraints may play distinct roles in different molecular functions.
 | ||
| Fig. 3 Biological implications of chain entanglements in heterodimeric complexes. (a) Top 10 function types in terms of the number of intertwined heterodimeric complexes; (b) top 10 function types in terms of the percentage of intertwined heterodimeric complexes with a minimum of total 10 entries; (c) structure of the CLOCK:BMAL1 transcriptional activator complex (PDB ID: 4F3L); (d) structure of sirohaem decarboxylase AhbA/B (PDB ID: 4CZD). | ||
The tight interacting pattern of entangled chains is likely to offer additional stabilizing effects crucially associated with the execution of various biological functions. A typical example is the mouse CLOCK:BMAL1 transcriptional activator complex (PDB ID: 4F3L), a tightly intertwined heterodimer (GLN = −0.885), that could bind E-box DNA with high affinity (Kd ∼ 10 nM) and play a crucial role in regulating the circadian clock (Fig. 3c).60 The interface-perturbing mutations revealed that the stability of the heterodimeric regulator is key to maintaining the circadian periodicity. We also noticed that 6 of the top 10 molecular functions in terms of the number of intertwined heterodimers are enzymes including transferase, hydrolase and so on, some of which are associated with nucleic acids. One class of enzymes with highly entangled structures, as aforementioned, are those generated from the cleavage of monomeric precursors. A notable case is the sirohaem decarboxylase AhbA/B from Desulfovibrio desulfuricans (PDB ID: 4CZD), where the two subunits possessing a near identical fold (with a sequence similarity of 39%) adopt a highly entangled association pattern (GLN = −0.797) much resembling that of a homodimer with C2 symmetry (Fig. 3d).47 Since both AhbA and AhbB are unstable when expressed individually, the domain swapping behavior may serve as a stabilizing strategy to form an active heterodimeric enzyme. It should be noted that the fundamental influence of chain entanglements on protein properties and functions, e.g., evolutionary advantages of entangled enzymes, remains an open question to be systematically investigated, especially with strictly controlled experimental systems. As topological differences are often mingled with constitutional differences, it is very difficult to reach a conclusion on the sole effects of entanglement or topology.
Selected premium intertwined heterodimeric motifs for heterocatenane synthesis
An important application of these intertwined heterodimeric motifs is to serve as templates to direct the synthesis of complex topological proteins. For a heterodimeric entangling motif, in the simplest case when the N- and C-termini of each subunit are covalently connected, a protein hetero[2]catenane could probably be formed. Based on our experience, we have selected a toolbox of premium intertwined heterodimeric motifs (entry 7, Table 1) with the calculated |GLN| typically around 1 for this purpose. Considering their small-to-medium size (subunits of 30–160 a.a.) and good expression yield by Escherichia coli, six motifs were further chosen for experimental validation. As shown in Fig. 4, these motifs feature not only a high extent of intertwining but also proximal termini (dN–C ≤ 30 Å) to facilitate ring closure. Following the assembly-reaction synergy, we inserted their gene sequences into the previously reported gene cassettes for protein hetero[2]catenane synthesis (Fig. 5a).61 Among all these designs, one ring (typically subunit A) is cyclized via Vidal intein and contains TEV site for subsequent proof of topology via controlled cleavage, while the other (typically subunit B) is cyclized via Npu DnaE intein and contains His-Tag for the ease of purification.62,63 For the heterodimer 2O97, it is known that HU predominantly forms the HUαβ heterodimer and HUαα dimer in E. coli64 and that the HUαβ heterodimer is much more stable than either the HUαα or Huββ homodimer.65 To prevent the contamination of HUαα homo[2]catenane, we intentionally placed the His-Tag at HUβ. The amino acid sequences of these designs are provided in SI sequence 1. | ||
| Fig. 4 Structures of the six selected premium intertwined heterodimeric motifs for experimental validation. | ||
 | ||
| Fig. 5 Experimental validation of selected heterodimeric intertwined motifs. (a) Illustration of the gene constructs and cellular synthesis of hetero[2]catenanes using heterodimeric intertwined motifs for entanglement formation and orthogonal split inteins for subunit cyclization; (b) SDS-PAGE analysis of TEVp-mediated cleavage of the purified major products to prove their catenane topology. | ||
The protein expression and purification followed previous protocols.61 The crude products after Ni-NTA affinity purification and the samples purified by SEC (Fig. S3) were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and liquid chromatography-mass spectrometry (LC-MS). Species with molecular weight close to the expected hetero[2]catenanes were observed in all constructs with varying amounts of single-ring side products. To prove the catenane topology, TEV protease (TEVp)-mediated cleavage experiments were conducted. Upon complete cleavage, the putative catenane bands in SDS-PAGE as well as the cyclic chain B bands in 4 of the 6 designs disappeared, leaving only cyclic chain A and linear chain B, as further confirmed by LC-MS (Fig. 5b and S4). The two exceptions are 2O97 and 3IWB. For the former, after complete cleavage, there remains a faint band with an apparent molecular weight close to the heterocatenane, which is confirmed by LC-MS to be the homo[2]catenane of HUβ. This is not surprising since HUβ is also capable of self-dimerization. For the latter, the situation is more complicated. The 3IWB heterocatenane is particularly resistant to TEVp cleavage even after long time incubation. We reasoned that it is probably caused by its tendency to oligomerize as shown in the SEC spectra, and the recognition site was hence too buried to be accessible by TEVp. We further quantified the heterocatenation efficiency (ε) by the molar percentage of heterocatenane in the purified mixture by densitometry analysis using ImageJ (Fig. S5). The results listed in Table 2 show that 2ACM and 4X86 have the highest selectivity and efficiency for heterocatenation. Therefore, all six motifs can be used to prepare hetero[2]catenanes with varying efficiency, and some of them exhibit comparable and even higher efficiency than the previously engineered heterodimeric motif derived from p53dim.20
| PDB ID | GLN | BSA (Å2) | d N1–C1 (Å) | d N2–C2 (Å) | Formation mechanism | Reported stability | Predicted −log(K) | Heterocatenation efficiency ε |
|---|---|---|---|---|---|---|---|---|
| 2ACM | −1.192 | 1997 | 28 | 26 | Cleavage of the monomeric precursor |
T
m ∼ 75 °C35 |
7.46 | 91% |
| 2O97 | 0.518 | 1807 | 19 | 18 | Mutual synergistic folding |
K
d ∼ 25 nM64 |
8.25 | 77% |
| 3A1G | 0.540 | 1604 | 24 | 30 | Mutual synergistic folding | Tight binding67 | 6.05 | 67% |
| 3IWB | −0.874 | 2239 | 20 | 20 | Cleavage of the monomeric precursor | — | 5.15 | 27% |
| 4CZD | −0.797 | 3341 | 20 | 9 | Mutual synergistic folding | Stable when coexpressed47 | 6.85 | 42% |
| 4X86 | 0.510 | 1267 | 30 | 22 | Mutual synergistic folding |
K
d ∼ 2.2 nM46 |
8.17 | 96% |
The distinct catenation efficiency further prompts us to interrogate the influence of various structural features on topology synthesis. The relevant parameters are also listed in Table 2. Among them, the terminal distances (dN1–C1 and dN2–C2) have little effect on ε since they all have dN1–C1 and dN2–C2 within 30 Å for convenient cyclization. While motifs with larger |GLN| may afford complex topologies like Solomon link, they do not necessarily bring about higher ε, presumably due to the higher kinetic barrier for their formation (Fig. S6a). As long as |GLN| goes above a certain critical value (e.g., >0.5), the extent of chain entanglement and ε are no longer strongly correlated, as reflected by 4X86 with the smallest |GLN|, yet the highest ε. Similarly, the BSA does not seem to be essential for efficient catenation (Fig. S6b).
Three factors, including the stability, chain organization, and tendency to self-association, seem to be more influential. First of all, to compare the relative stability of the six motifs, we used the AREA-AFFINITY web server to predict the binding affinity of these motifs, which are represented as −log(K) where K is the dissociation constant.66 The positive correlation between −log(K) and ε confirms the importance of higher stability in promoting nontrivial protein topologies (Fig. S6c), which is straightforward because the more stable the template is, the more likely the catenation occurs. Second, the structural features of chain organization, such as the distribution and orientation of N- and C-termini of both subunits and the simplicity of the fold, may play a significant role because it dictates the formation of an appropriate spatial relationship for ring closure. For example, the 4CZD motif contains two multi-domain subunits threaded together. The complex structure may hinder the correct folding and association of the two subunits fused with split inteins, leading to low ε. In contrary, both 2ACM and 4X86 have quite simple fold, which helps ensure robust and synergistic association of the two subunits and thus leads to high ε. Third, the tendency to self-association could lead to unintended oligomeric species as side products, as seen in the case of 3IWB. Therefore, a good entangling motif, as exemplified by 4X86, shall thus have stable, yet simple intertwined fold for robust assembly, possess favourable terminal orientation and distance for facile ring closure, and exhibit low tendency to self-associate for minimal side reactions. Some of these features are difficult to describe quantitatively and accurately at this stage, which necessitates manual curation and experimental validation to assess the usefulness of the heteromeric entangling motifs. Compared to homodimeric entangling motifs such as p53dim used in previous studies, these heterodimeric entangling motifs offer more design space of topological proteins. As shown above, heterocatenanes with varied structural features could be readily synthesized using heterodimeric entangling motifs. Although homodimeric motifs could be potentially engineered into heterodimeric version as shown in the case of X+/X−, the overall structural symmetry remains unchanged, which greatly limits the control over the geometric features of topological proteins.20 Moreover, more complex topological proteins such as higher-order hetero[n]catenanes, which are extremely difficult to design with homomeric entangling motifs, could in theory be realized using heterodimeric ones with mutual orthogonality. Another advantage of these heterodimeric entangling motifs is their robustness (as shown in Table 2), while p53dim has only moderate binding affinity (Kd ∼ 56 μM) and much lower thermal stability (Tm ∼ 37 °C).20
Making functional protein heterocatenanes via domain insertion
The high efficiency of both 2ACM and 4X86 motifs prompted us to explore their potential in making functional protein heterocatenanes via domain insertion. As a proof-of-concept, we fused a Förster resonance energy transfer (FRET) pair, cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP), onto the two subunits of 4X86, respectively, to generate a catenated CFP–YFP “fusion”. The CFP/YFP FRET pair was chosen due to its frequent usage and ease in the FRET assay. The CFP and YFP domains were inserted into the gene cassette to give the final construct VidC-CFP-4X86chA-VidN-L-NpuC-4X86chB-YFP-NpuN for cellular synthesis of the CFP/YFP catenane denoted as cat-CFP#YFP (Fig. 6a and SI sequence 2). The cat-CFP#YFP could be readily obtained with high ε (∼85%) via affinity chromatography and SEC separation (Fig. 6b). The catenane topology was proved by TEVp-mediated cleavage, as shown in SDS-PAGE and LC-MS results (Fig. 6c and S7a). The spatial proximity of CFP and YFP within the catenane scaffold was verified by the FRET signal (Fig. 6d). As the FRET signal is rather sensitive to the distance and orientation between CFP and YFP, we envisioned that topology transformation from heterocatenane to rotaxane via proteolytic digestion would release the conformational constraint on CFP, leading to context-dependent FRET signal modulation. Indeed, when the emission spectra were normalized at the CFP emission wavelength (475 nm), a ∼29% increase of YFP emission at 530 nm was observed upon TEVp-mediated cleavage (Fig. 6d and S7b). This is consistent with the relatively higher freedom of CFP in rotaxane form that allows it to diffuse and get closer to YFP. Hence, the heterocatenane form provides a unique platform to display various domains on the scaffold in minutely different poses. | ||
| Fig. 6 Design of a heterocatenane of CFP and YFP using the 4X86 motif. (a) Illustration of the construct and synthesis of cat-CFP#YFP; (b) SDS-PAGE analysis and SEC overlay of cat-CFP#YFP (E: elution from Ni-NTA resin; S: SEC-purified product); (c) TEVp-mediated cleavage of the purified product to prove its catenane topology; (d) normalized fluorescence emission spectra of cat-CFP#YFP before and after TEVp-mediated cleavage. | ||
To explore the generality of this approach, we further inserted various therapeutic protein domains into the heterocatenation scaffold. Specifically, mechanical conjugation of two antibody mimics via an intertwined motif leads to a unique bispecific antibody mimic. As an example, we fused the 4X86 subunits with two functional domains, one affibody that binds the human epidermal growth factor receptor (AffiEGFR)68 and the other affibody that binds human epidermal growth factor receptor 2 (AffiHER2),69 to give a bispecific affibody catenane (cat-bsAffi). To illustrate the topological effect, we also constructed a linear fusion of AffiEGFR–AffiHER2 (l-bsAffi) with a long, flexible linker in between two domains. To improve the catenation efficiency and facilitate purification of the target bispecific heterocatenane, we designed a coexpression system, where one gene in the pACYCDuet-1 vector encoding NpuC-AffiHER2-4X86chB-NpuN (with His-Tag) and another gene in the pET15b vector encoding VidC-AffiEGFR-4X86chA-VidN (with TEV site) were used to co-transform the BL21(DE3) competent cell (SI sequence 3). The higher copy number of pET15b over pACYCDuet-1 ensures higher expression level of VidC-AffiEGFR-4X86chA-VidN than NpuC-AffiHER2-4X86chB-NpuN, which could presumably promote the intertwined assembly of the two components to achieve high catenation efficiency.70
Both cat-bsAffi and l-bsAffi were well expressed and readily purified via affinity chromatography and SEC (Fig. 7a and S8a). Their structures were confirmed by LC-MS with expected molecular weights (Fig. S8b), and the catenane topology of cat-bsAffi was confirmed via proteolytic cleavage by TEVp (Fig. 7b). The bispecific activity of both cat-bsAffi and l-bsAffi was then assayed. It turned out that cat-bsAffi showed comparable activity as l-bsAffi, implying that the additional 4X86 domain and the catenane topology did not compromise the binding capability (with Kd values of 3.84 and 6.97 nM towards EGFR and HER2, respectively) of the two affibody domains (Fig. 7c). Therefore, the successful design and synthesis of an active bispecific affibody in catenane form provides a novel platform of bispecific antibody designs, proving the feasibility of harnessing heterodimeric entangling motifs to design protein heterocatenanes with multiple functions. While the intrinsic stability of affibodies may obscure observable stability improvements arising from catenation, the topological scaffold offers additional engineering flexibility for future applications requiring controlled domain orientation or protease-responsive activation, which may otherwise be difficult to achieve in linear counterparts. The availability of versatile heterodimeric entangling motifs in different geometries shall further allow the fine tuning of the binding capabilities towards synergistic enhancement or mutually exclusive activity, which are topics of further investigation.
 | ||
| Fig. 7 Design of a bispecific affibody catenane of AffiEGFR and AffiHER2 using the 4X86 motif. (a) SDS-PAGE analysis of l-bsAffi and cat-bsAffi; (b) TEVp-mediated cleavage of cat-bsAffi to prove its catenane topology; (c) binding characterization of cat-bsAffi and l-bsAffi towards EGFR and HER2, respectively. | ||
Conclusions
Creating topologically complex protein architectures is a fundamentally important topic far beyond the pursuit of structural aesthetics. Studying proteins with nontrivial topologies could not only help gain a deeper understanding of protein folding but also make full use of topology engineering for advanced biomaterials. On one hand, naturally occurring topological proteins are rare, and their functional benefits gained from unconventional topologies remain elusive due to the limited dataset and lack of strict topological control. On the other hand, the artificial design and synthesis of complex topological proteins, largely hindered by limited building blocks and reliable design methods, is still in its infancy. Therefore, there is an urgent need for researchers to enrich the toolkit for protein topology engineering and enhance the diversity of topological proteins, paving the way for practical applications.Here, we have systematically analyzed the chain entanglements within heterodimeric protein complexes and discovered a toolbox of heterodimeric entangling motifs that are suitable for protein topology engineering. The formation mechanisms and molecular functions of these entangling heterodimeric motifs were thoroughly analyzed, which revealed the biological implications of chain entanglements. These motifs, such as 2ACM and 4X86, are highly efficient in templating the cellular synthesis of protein heterocatenanes for function integration via mechanical interlocking. Notably, both 2ACM and 4X86 are human proteins and thus should have low immunogenicity, holding great promise in developing novel protein therapeutics. When used in combination, these motifs could further greatly expand the design space of topological proteins, leading to novel biomaterials with emergent properties such as switchable protein machines71,72 and advancing protein therapeutics.20,27,73,74 Although the study was conducted on natural heterodimeric proteins, the insights gained from this work should be broadly applicable to other topological proteins as well. For example, the entangled heterodimers generated from cleavage of monomeric precursors such as SEA domains are particularly instructive for creating topological isoforms of a single-domain protein via backbone rethreading. Therefore, our study not only establishes a platform for mining heteromeric entangling motifs for expanding the design space of protein topologies but also spurs the exploration of the evolutionary links underlying protein chain entanglements and functions. By going beyond the linear paradigm of protein backbones, the results are fundamental to understanding protein folding and offers topology as a powerful dimension in protein engineering to tailor protein, the workhorse of life, for practical and emergent biomedical applications.
It is crucial to acknowledge that our investigation remains constrained by the limited dataset of heterodimeric protein complexes currently available in PDB. This constraint becomes particularly evident when considering the vast space of potential interaction partners within the proteome that currently lack structural characterization, some of which may exhibit novel entanglement conformations. Although the preference of chain entanglement within certain protein families may imply its fundamental roles in regulating protein functions, the underlying relationships between chain entanglement and protein function remain to be illustrated on the basis of a larger dataset with significant entanglement, hopefully aided by the more powerful computational tools like deep learning.
To conclude, our work establishes a methodological paradigm that bridges bioinformatic analysis with experimental validation and protein engineering applications, offering a novel avenue for topological protein research. The entangling motifs discovered here proved useful in synthesizing protein hetero[2]catenanes and are currently inspiring the development of higher-order topological protein architectures with integrated functions.
Methods
Discovery and feature analysis of protein heterodimers
The analysis procedures and methods, including data query, calculation of parameters like GLN, BSA and dN–C, mapping of protein functions and selection of premium motifs, generally follow our previous work on homomeric protein assemblies with some minor adaptions as follows.16 (1) Instead of homomeric proteins, we queried heterodimeric protein complexes in PDB released on and before 2023/09/13 using Advanced Search Query Builder with the following criteria: entry polymer composition is “heteromeric protein”, number of protein instances (chains) per assembly equals 2 and oligomeric state is assigned as “hetero 2-mer”. (2) To remove redundancy, sequences of the two subunits in each heterodimer are concatenated for clustering using MMseqs2 with a sequence identity cutoff of 40%.75 (3) For terminal distance analysis, the dN–C,intra is conducted on both subunits within a heterodimeric complex, while the dN–C,inter is not calculated.DNA construction
All oligonucleotide primers were designed using DNAworks or IDT and then purchased from Azenta, Inc. Sequences of the primers are provided in the SI. Sequences encoding the Npu DnaE intein, Vidal intein, AffiHER2 and AffiEGFR were amplified by PCR from the corresponding plasmids previously constructed in our lab. Sequences encoding 2ACM, 2O97, 3A1G, 3IWB, 4CZD and 4X86 were obtained by assembly PCR. The complete genes of the constructs, including VidC-2ACMchA-VidN-L-NpuC-2ACMchB-NpuN, VidC-2O97chA-VidN-L-NpuC-2O97chB-NpuN, VidC-3A1GchA-VidN-L-NpuC-3A1GchB-NpuN, VidC-3IWBchA-VidN-L-NpuC-3IWBchB-NpuN, VidC-4CZDchA-VidN-L-NpuC-4CZDchB-NpuN and VidC-4X86chA-VidN-L-NpuC-4X86chB-NpuN (in pQE80L), VidC-AffiEGFR-4X86chA-VidN (in pET-15b) and NpuC-AffiHER2-4X86chB-NpuN (in pACYCDuet-1), were obtained via overlap PCR and then cloned into the bacterial pQE80L, pET-15b or co-expression pACYCDuet-1 vector using standard restriction enzyme digestion and T4 ligation protocols. The gene lin-AffiEGFR-FL-AffiHER2 in pET-15b was directly ordered from Azenta, Inc. All the DNA sequences were confirmed by sequencing.Protein expression and purification
A single colony was inoculated into 5 mL of LB medium with corresponding antibiotics and grown in a shaker (37 °C and 220 rpm). Overnight culture was inoculated in a ratio of 1:100 into 300 mL of 2xYT medium containing corresponding antibiotics. When OD600 reached 0.6 to 0.8, the culture was placed in a shaker with pre-set temperature. IPTG was then added at the concentration of 0.25 mM to induce target protein expression. The cultures were shaken for 12 hours at 16 °C before cells were harvested via centrifugation (4 °C, 5000g, and 15 min). Cell pellets were resuspended in buffer A (20 mM NaH2PO4, 500 mM NaCl, 20 mM imidazole, and 5% v/v glycerol, pH = 8.0) and lysed by ultrasonication. The supernatant was collected by centrifugation (4 °C, 12000g, and 30 min). The clear lysate was mixed with Ni-NTA resin (GE Healthcare, Inc.), equilibrated with buffer A and agitated on a rotator at 4 °C for 1 h. The sample was then loaded into an empty column and washed with buffer A for several column volumes. The target protein sample was then eluted with buffer B (20 mM NaH2PO4, 500 mM NaCl, 250 mM imidazole, and 5% v/v glycerol, pH = 8.0).
Protein characterization
SDS-PAGE was performed on 4–20% gels to analyze the composition of the crude expression products. Samples were mixed with 5× SDS-PAGE loading buffer (250 mM Tris–HCl, 50% glycerol, 10% SDS, 250 mM β-mercaptoethanol, and 0.05% bromophenol blue) and heated at 98 °C for 10 min. Size exclusion chromatography was performed on a Superdex 200 Increase 10/300 GL column in an ÄKTA FPLC system (GE Healthcare, Inc.) with Tris buffer (20 mM Tris and 150 mM NaCl, pH = 7.5) containing 1 mM DTT as the mobile phase (flow rate: 0.5 mL min−1). Protein concentrations were detected by UV absorption on NanoDrop microvolume spectrophotometers and fluorometer (IMPLEN P330). LC-MS with quadrupole rod SQ Detector 2 mass spectrometer (Water Corp.) was used to determine the molecular weights of all protein samples. Relative protein quantification from the SDS-PAGE results was conducted using ImageJ.Protease-mediated cleavage
The protein solution (10 μM) containing 1 mM DTT and 0.5 mM EDTA was mixed with TEV protease at a molar ratio of 20:1 at 30 °C until full digestion (checked by LC-MS). The completely digested products were collected and characterized by SDS-PAGE and LC-MS.
Förster resonance energy transfer analysis
Protein samples were prepared with 10 μM concentration in Tris buffer (20 mM Tris and 150 mM NaCl, pH = 7.5) and added to the black microplate. Fluorescence intensity was monitored at 475 and 530 nm using an EnSpire multimode plate reader (PerkinElmer Inc.) under 430 nm excitation. Fluorescence emission spectra of the samples were recorded from 450 nm to 600 nm under 430 nm excitation. The spectra were normalized at the intensity at 475 nm. All the measurements were conducted in triplicate.Biolayer interferometry analysis
The affinities of l-bsAffi and cat-bsAffi with human EGFR and human HER2 were determined using the biolayer interferometry assay on an Octet RED96 system (FortéBio). Human EGFR and HER2 proteins were purchased from ACROBiosystems as a white powder (100 μg) and dissolved to 8.5 μM in the running buffer (PBST buffer containing 0.1% BSA for titration) as a stock solution for testing. Both l-bsAffi and cat-bsAffi were diluted to various concentrations with the running buffer and loaded on a black polypropylene 96-well microplate. All experiments were conducted at 25 °C, and the running buffer was used as negative control. For the binding kinetics assay, the biotinylated receptor was first immobilized on the streptavidin probes and equilibrated with the running buffer until the signal was stable. Then, the flow of the probes was set as follows: (1) running buffer for 60 s, (2) receptor anchoring for 200 s, (3) running buffer for 60 s, (4) bsAffi sample binding for 200 s, (5) dissociation for 200 s, and (6) elution buffer (glycinate hydrochloric acid solution, pH 2.2) for 5 s and running buffer for 5 s 3 times, to detect the interaction between the bsAffi samples and the receptors. Finally, a 1:1 binding model was assumed for the binding kinetics analysis, and the BLI data were analyzed using Octet System Data Analysis software.
Author contributions
Conceptualization: L. X. and W.-B. Z. Methodology: L. X. Investigation: L. X. and X. T. Visualization: L. X. Supervision: W.-B. Z. Writing – original draft: L. X. Writing – review & editing: L. X. and W.-B. Z.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the SI.Protein sequences, supplemetary figures and tables have been included as part of the SI. See DOI: https://doi.org/10.1039/d5sc03953c.
Acknowledgements
We are grateful for the financial support from the National Key R&D Program of China (No. 2020YFA0908100), Shenzhen Medical Research Fund (No. B2302037), the National Natural Science Foundation of China (No. 22331003, 21925102, 22101010, 22201016, and 22201017), and Beijing National Laboratory for Molecular Sciences (BNLMS-CXXM-202006). The work was also supported by the High-performance Computing Platform of Peking University. L. X. acknowledges the Boya Postdoctoral Fellowship from Peking University.Notes and references
- L. Xu and W.-B. Zhang, Topology: a unique dimension in protein engineering, Sci. China: Chem., 2017, 61, 3–16 CrossRef.
- J. I. Sulkowska, On folding of entangled proteins: knots, lassos, links and θ-curves, Curr. Opin. Struct. Biol., 2020, 60, 131–141 CrossRef CAS PubMed.
- Z. Qu, S. Z. D. Cheng and W.-B. Zhang, Macromolecular topology engineering, Trends Chem., 2021, 3, 402–415 CrossRef CAS.
- P. Dabrowski-Tumanski and J. I. Sulkowska, Topological knots and links in proteins, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3415–3420 CrossRef CAS PubMed.
- L. A. Doyle, B. Takushi, R. D. Kibler, L. F. Milles, C. T. Orozco, J. D. Jones, S. E. Jackson, B. L. Stoddard and P. Bradley, De novo design of knotted tandem repeat proteins, Nat. Commun., 2023, 14, 6746 CrossRef CAS PubMed.
- M. Marenda, E. Orlandini and C. Micheletti, Discovering privileged topologies of molecular knots with self-assembling models, Nat. Commun., 2018, 9, 3051 CrossRef PubMed.
- N. P. King, A. W. Jacobitz, M. R. Sawaya, L. Goldschmidt and T. O. Yeates, Structure and folding of a designed knotted protein, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20732–20737 CrossRef CAS PubMed.
- L. Z. Yan and P. E. Dawson, Design and synthesis of a protein catenane, Angew. Chem., Int. Ed., 2001, 40, 3625–3627 CrossRef CAS PubMed.
- X.-W. Wang and W.-B. Zhang, Cellular synthesis of protein catenanes, Angew. Chem., Int. Ed., 2016, 55, 3442–3446 CrossRef CAS PubMed.
- X. Bai, Y. Liu, J. Lee, J. Fang, W.-H. Wu, J. Seo and W.-B. Zhang, Cellular synthesis of protein pretzelanes, Giant, 2022, 10, 100092 CrossRef CAS.
- Y. Liu, W.-H. Wu, S. Hong, J. Fang, F. Zhang, G. X. Liu, J. Seo and W. B. Zhang, Lasso proteins: modular design, cellular synthesis, and topological transformation, Angew. Chem., Int. Ed., 2020, 59, 19153–19161 CrossRef CAS PubMed.
- S. S. Mackinnon, A. Malevanets and S. J. Wodak, Intertwined associations in structures of homooligomeric proteins, Structure, 2013, 21, 638–649 CrossRef CAS PubMed.
- S. S. MacKinnon and S. J. Wodak, Landscape of intertwined associations in multi-domain homo-oligomeric proteins, J. Mol. Biol., 2015, 427, 350–370 CrossRef CAS PubMed.
- S. J. Wodak, A. Malevanets and S. S. MacKinnon, The landscape of intertwined associations in homooligomeric proteins, Biophys. J., 2015, 109, 1087–1100 CrossRef CAS PubMed.
- M. Baiesi, E. Orlandini, A. Trovato and F. Seno, Linking in domain-swapped protein dimers, Sci. Rep., 2016, 6, 33872 CrossRef CAS PubMed.
- L. Xu, P. Deng, H. Gao and W.-B. Zhang, Systematic discovery and feature analysis of intertwined symmetric protein motifs for topology engineering, Giant, 2024, 17, 100226 CrossRef CAS.
- P. Dabrowski-Tumanski, A. I. Jarmolinska, W. Niemyska, E. J. Rawdon, K. C. Millett and J. I. Sulkowska, LinkProt: a database collecting information about biological links, Nucleic Acids Res., 2017, 45, D243–D249 CrossRef CAS PubMed.
- J. A. Marsh and S. A. Teichmann, Structure, dynamics, assembly, and evolution of protein complexes, Annu. Rev. Biochem., 2015, 84, 551–575 CrossRef CAS PubMed.
- Y. Zhao, M. Chwastyk and M. Cieplak, Structural entanglements in protein complexes, J. Chem. Phys., 2017, 146, 225102 CrossRef PubMed.
- W.-H. Wu, X. Bai, Y. Shao, C. Yang, J. Wei, W. Wei and W.-B. Zhang, Higher order protein catenation leads to an artificial antibody with enhanced affinity and in vivo stability, J. Am. Chem. Soc., 2021, 143, 18029–18040 CrossRef CAS PubMed.
- C. D. Allen and A. J. Link, Self-assembly of catenanes from lasso peptides, J. Am. Chem. Soc., 2016, 138, 14214–14217 CrossRef CAS PubMed.
- J. Fang, T. Li, J. Lee, D. Im, L. Xu, Y. Liu, J. Seo and W.-B. Zhang, A single-domain protein catenane of dihydrofolate reductase, Natl. Sci. Rev., 2023, 10, nwad304 CrossRef CAS PubMed.
- Z. Qu, J. Fang, Y.-X. Wang, Y. Sun, Y. Liu, W.-H. Wu and W.-B. Zhang, A single-domain green fluorescent protein catenane, Nat. Commun., 2023, 14, 3480 CrossRef CAS PubMed.
- X. D. Da and W.-B. Zhang, Active template synthesis of protein heterocatenanes, Angew. Chem., Int. Ed., 2019, 58, 11097–11104 CrossRef CAS PubMed.
- F. Zhang, Y. Liu, Y. Shao and W.-B. Zhang, Active template synthesis of protein [n]catenanes using engineered peptide–peptide ligation tools, CCS Chem., 2024, 6, 377–389 CrossRef CAS.
- F. Zhang, Y. Liu, X.-D. Da and W.-B. Zhang, Toward selective synthesis of protein olympiadanes via orthogonal active templates in one step, CCS Chem., 2024, 6, 1047–1059 CrossRef CAS.
- Y. Liu, X. Bai, C. Lyu, J. Fang, F. Zhang, W.-H. Wu, W. Wei and W.-B. Zhang, Mechano-bioconjugation strategy empowering fusion protein therapeutics with aggregation resistance, prolonged circulation, and enhanced antitumor efficacy, J. Am. Chem. Soc., 2022, 144, 18387–18396 CrossRef CAS PubMed.
- P. Deng, L. Xu, Y. Wei, F. Sun, L. Li, W.-B. Zhang and H. Gao, Deep learning-assisted discovery of protein entangling motifs, Biomacromolecules, 2025, 26, 1520–1529 CrossRef CAS PubMed.
- P. Deng, Y. Zhang, L. Xu, J. Lyu, L. Li, F. Sun, W.-B. Zhang and H. Gao, Computational discovery and systematic analysis of protein entangling motifs in nature: from algorithm to database, Chem. Sci., 2025, 16, 8998–9009 RSC.
- Y. Hou, T. Xie, L. He, L. Tao and J. Huang, Topological links in predicted protein complex structures reveal limitations of AlphaFold, Commun. Biol., 2023, 6, 1098 CrossRef CAS PubMed.
- W. Niemyska, K. C. Millett and J. I. Sulkowska, GLN: a method to reveal unique properties of lasso type topology in proteins, Sci. Rep., 2020, 10, 15186 CrossRef CAS PubMed.
- C. Schoene, J. O. Fierer, S. P. Bennett and M. Howarth, SpyTag/SpyCatcher cyclization confers resilience to boiling on a mesophilic enzyme, Angew. Chem., Int. Ed., 2014, 53, 6101–6104 CrossRef CAS PubMed.
- A. Tavassoli and S. J. Benkovic, Split-intein mediated circular ligation used in the synthesis of cyclic peptide libraries in E. coli, Nat. Protoc., 2007, 2, 1126–1133 CrossRef CAS PubMed.
- S. S. Shekhawat and I. Ghosh, Split-protein systems: beyond binary protein–protein interactions, Curr. Opin. Chem. Biol., 2011, 15, 789–797 CrossRef CAS PubMed.
- B. Macao, D. G. A. Johansson, G. C. Hansson and T. Härd, Autoproteolysis coupled to protein folding in the SEA domain of the membrane-bound MUC1 mucin, Nat. Struct. Mol. Biol., 2005, 13, 71–76 CrossRef PubMed.
- S. Bale and S. E. Ealick, Structural biology of S-adenosylmethionine decarboxylase, Amino Acids, 2009, 38, 451–460 CrossRef PubMed.
- K. A. Hicks and S. E. Ealick, Biochemical and structural characterization of Klebsiella pneumoniae oxamate amidohydrolase in the uric acid degradation pathway, Acta Crystallogr., Sect. D: Struct. Biol., 2016, 72, 808–816 CrossRef CAS PubMed.
- A. Iqbal, I. J. Clifton, R. Chowdhury, D. Ivison, C. Domeneb and C. J. Schofield, Structural and biochemical analyses reveal how ornithine acetyl transferase binds acidic and basic amino acid substrates, Org. Biomol. Chem., 2011, 9, 6219 RSC.
- A. Bergmann, W. R. Gordon, D. Vardar-Ulu, S. L'Heureux, T. Ashworth, M. J. Malecki, C. Sanchez-Irizarry, D. G. McArthur, G. Histen, J. L. Mitchell, J. C. Aster and S. C. Blacklow, Effects of S1 cleavage on the structure, surface export, and signaling activity of human Notch1 and Notch2, PLoS One, 2009, 4, e6613 CrossRef PubMed.
- L. Wu, C. M. Viola, A. M. Brzozowski and G. J. Davies, Structural characterization of human heparanase reveals insights into substrate recognition, Nat. Struct. Mol. Biol., 2015, 22, 1016–1022 CrossRef CAS PubMed.
- C. Whitefield, N. Hong, J. A. Mitchell and C. J. Jackson, Computational design and experimental characterisation of a stable human heparanase variant, RSC Chem. Biol., 2022, 3, 341–349 RSC.
- V. Rana, I. Sitarik, J. Petucci, Y. Jiang, H. Song and E. P. O'Brien, Non-covalent lasso entanglements in folded proteins: prevalence, functional implications, and evolutionary significance, J. Mol. Biol., 2024, 436, 168459 CrossRef CAS PubMed.
- M. Baiesi, E. Orlandini, F. Seno and A. Trovato, Sequence and structural patterns detected in entangled proteins reveal the importance of co-translational folding, Sci. Rep., 2019, 9, 8426 CrossRef PubMed.
- E. Fichó, R. Pancsa, C. Magyar, Z. E. Kalman, É. Schád, B. Z. Németh, I. Simon, L. Dobson and G. E. Tusnády, MFIB 2.0: a major update of the database of protein complexes formed by mutual folding of the constituting protein chains, Nucleic Acids Res., 2025, 53, D487–D494 CrossRef PubMed.
- S. J. Demarest, M. Martinez-Yamout, J. Chung, H. Chen, W. Xu, H. J. Dyson, R. M. Evans and P. E. Wright, Mutual synergistic folding in recruitment of CBP/p300 by p160 nuclear receptor coactivators, Nature, 2002, 415, 549–553 CrossRef CAS PubMed.
- N. Kuwabara, R. Minami, N. Yokota, H. Matsumoto, T. Senda, H. Kawahara and R. Kato, Structure of a BAG6 (Bcl-2-associated athanogene 6)-Ubl4a (ubiquitin-like protein 4a) complex reveals a novel binding interface that functions in tail-anchored protein biogenesis, J. Biol. Chem., 2015, 290, 9387–9398 CrossRef CAS PubMed.
- D. J. Palmer, S. Schroeder, A. D. Lawrence, E. Deery, S. A. Lobo, L. M. Saraiva, K. J. McLean, A. W. Munro, S. J. Ferguson, R. W. Pickersgill, D. G. Brown and M. J. Warren, The structure, function and properties of sirohaem decarboxylase – an enzyme with structural homology to a transcription factor family that is part of the alternative haem biosynthesis pathway, Mol. Microbiol., 2014, 93, 247–261 CrossRef CAS PubMed.
- A. Mentes, C. Magyar, E. Ficho and I. Simon, Analysis of heterodimeric “mutual synergistic folding”-complexes, Int. J. Mol. Sci., 2019, 20, 5136 CrossRef CAS PubMed.
- C. Zhou, L. Yan, W.-h. Zhang and Z. Liu, Structural basis of tubulin detyrosination by VASH2/SVBP heterodimer, Nat. Commun., 2019, 10, 3212 CrossRef PubMed.
- X. Shao, H. Kang, T. Loveless, G. R. Lee, C. Seok, W. I. Weis, H.-J. Choi and J. Hardin, Cell–cell adhesion in metazoans relies on evolutionarily conserved features of the α-catenin·β-catenin–binding interface, J. Biol. Chem., 2017, 292, 16477–16490 CrossRef CAS PubMed.
- D. Jurėnas, L. V. Melderen and A. Garcia-Pino, Mechanism of regulation and neutralization of the AtaR–AtaT toxin–antitoxin system, Nat. Chem. Biol., 2019, 15, 285–294 CrossRef PubMed.
- H. Takagi, Y. Kakuta, T. Okada, M. Yao, I. Tanaka and M. Kimura, Crystal structure of archaeal toxin-antitoxin RelE–RelB complex with implications for toxin activity and antitoxin effects, Nat. Struct. Mol. Biol., 2005, 12, 327–331 CrossRef CAS PubMed.
- Y. G. J. Sterckx, T. Jové, A. V. Shkumatov, A. Garcia-Pino, L. Geerts, M. De Kerpel, J. Lah, H. De Greve, L. Van Melderen and R. Loris, A unique hetero-hexadecameric architecture displayed by the Escherichia coli O157 PaaA2–ParE2 antitoxin–toxin complex, J. Mol. Biol., 2016, 428, 1589–1603 CrossRef CAS PubMed.
- J. D. Gross, N. J. Moerke, T. v. d. Haar, A. B. Sachs, J. E. G. McCarthy and G. Wagner, Ribosome loading onto the mRNA cap Is driven by conformational coupling between eIF4G and eIF4E, Cell, 2003, 115, 739–750 CrossRef CAS PubMed.
- S. Grüner, R. Weber, D. Peter, M.-Y. Chung, C. Igreja, E. Valkov and E. Izaurralde, Structural motifs in eIF4G and 4E-BPs modulate their binding to eIF4E to regulate translation initiation in yeast, Nucleic Acids Res., 2018, 46, 6893–6908 CrossRef PubMed.
- S. E. Jackson, A. Suma and C. Micheletti, How to fold intricately: using theory and experiments to unravel the properties of knotted proteins, Curr. Opin. Struct. Biol., 2017, 42, 6–14 CrossRef CAS PubMed.
- W. Niemyska, P. Dabrowski-Tumanski, M. Kadlof, E. Haglund, P. Sulkowski and J. I. Sulkowska, Complex lasso: new entangled motifs in proteins, Sci. Rep., 2016, 6, 36895 CrossRef CAS PubMed.
- Y. Zhao and M. Cieplak, Stability of structurally entangled protein dimers, Proteins, 2018, 86, 945–955 CrossRef CAS PubMed.
- T. U. Consortium, UniProt: the universal protein knowledge base in 2021, Nucleic Acids Res., 2021, 49, D480–D489 CrossRef PubMed.
- N. Huang, Y. Chelliah, Y. Shan, C. A. Taylor, S.-H. Yoo, C. Partch, C. B. Green, H. Zhang and J. S. Takahashi, Crystal structure of the heterodimeric CLOCK:BMAL1 transcriptional activator complex, Science, 2012, 337, 189–193 CrossRef CAS PubMed.
- Y. Liu, Z. Duan, J. Fang, F. Zhang, J. Xiao and W.-B. Zhang, Cellular synthesis and X-ray crystal structure of a designed protein heterocatenane, Angew. Chem., Int. Ed., 2020, 59, 16122–16127 CrossRef CAS PubMed.
- P. Carvajal-Vallejos, R. Pallisse, H. D. Mootz and S. R. Schmidt, Unprecedented rates and efficiencies revealed for new natural split inteins from metagenomic sources, J. Biol. Chem., 2012, 287, 28686–28696 CrossRef CAS PubMed.
- A. J. Burton, M. Haugbro, E. Parisi and T. W. Muir, Live-cell protein engineering with an ultra-short split intein, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12041–12049 CrossRef CAS PubMed.
- J. Ramstein, N. Hervouet, F. Coste, C. Zelwer, J. Oberto and B. Castaing, Evidence of a thermal unfolding dimeric intermediate for the Escherichia coli histone-like HU proteins: thermodynamics and structure, J. Mol. Biol., 2003, 331, 101–121 CrossRef CAS PubMed.
- K. Arora, B. Thakur, A. Gupta and P. Guptasarma, HU-AB simulacrum: fusion of HU-B and HU-A into HU-B-A, a functional analog of the Escherichia coli HU-AB heterodimer, Biochem. Biophys. Res. Commun., 2021, 560, 27–31 CrossRef CAS PubMed.
- Y. X. Yang, J. Y. Huang, P. Wang and B. T. Zhu, AREA-AFFINITY: a web server for machine learning-based prediction of protein–protein and antibody–protein antigen binding affinities, J. Chem. Inf. Model., 2023, 63, 3230–3237 CrossRef CAS PubMed.
- K. Sugiyama, E. Obayashi, A. Kawaguchi, Y. Suzuki, J. R. H. Tame, K. Nagata and S.-Y. Park, Structural insight into the essential PB1–PB2 subunit contact of the influenza virus RNA polymerase, EMBO J., 2009, 28, 1803–1811 CrossRef CAS PubMed.
- M. Friedman, A. Orlova, E. Johansson, T. L. J. Eriksson, I. Höidén-Guthenberg, V. Tolmachev, F. Y. Nilsson and S. Ståhl, Directed evolution to low nanomolar affinity of a tumor-targeting epidermal growth factor receptor-binding affibody molecule, J. Mol. Biol., 2008, 376, 1388–1402 CrossRef CAS PubMed.
- M. Wikman, A. C. Steffen, E. Gunneriusson, V. Tolmachev, G. P. Adams, J. Carlsson and S. Stahl, Selection and characterization of HER2/neu-binding affibody ligands, Protein Eng., Des. Sel., 2004, 17, 455–462 CrossRef CAS PubMed.
- N. H. Tolia and L. Joshua-Tor, Strategies for protein coexpression in Escherichia coli, Nat. Methods, 2006, 3, 55–64 CrossRef CAS PubMed.
- C. Zong, M. J. Wu, J. Z. Qin and A. J. Link, Lasso peptide benenodin-1 is a thermally actuated [1]rotaxane switch, J. Am. Chem. Soc., 2017, 139, 10403–10409 CrossRef CAS PubMed.
- H. V. Schroder, Y. Zhang and A. J. Link, Dynamic covalent self-assembly of mechanically interlocked molecules solely made from peptides, Nat. Chem., 2021, 13, 850–857 CrossRef CAS PubMed.
- J. D. Hegemann, M. De Simone, M. Zimmermann, T. A. Knappe, X. Xie, F. S. Di Leva, L. Marinelli, E. Novellino, S. Zahler, H. Kessler and M. A. Marahiel, Rational improvement of the affinity and selectivity of integrin binding of grafted lasso peptides, J. Med. Chem., 2014, 57, 5829–5834 CrossRef CAS PubMed.
- Y. Liu, X. Tian, F. Zhang and W. B. Zhang, Probing the topological effects on stability enhancement and therapeutic performance of protein bioconjugates: tadpole, macrocycle versus figure-of-eight, Adv. Healthcare Mater., 2024, 13, 2400466 CrossRef CAS PubMed.
- M. Steinegger and J. Soding, MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets, Nat. Biotechnol., 2017, 35, 1026–1028 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |