
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the emission properties of luminescent 1,2,3-diazaborinates†
Leonie
Wüst‡
ab,
Johannes
Chorbacher‡
ab,
Timo
Keim
ab,
Tim
Wellnitz
ab,
Julian
Spieß
ab,
Nele
Wieprecht
ab,
Maximilian
Michel
ab,
Holger
Helten
 *ab and
Holger
Braunschweig
*ab
*ab and
Holger
Braunschweig
*ab
aJulius-Maximilians-Universität Würzburg, Institute of Inorganic Chemistry, Am Hubland, 97074 Würzburg, Germany. E-mail: holger.helten@uni-wuerzburg.de; h.braunschweig@uni-wuerzburg.de
bInstitute for Sustainable Chemistry & Catalysis with Boron, Am Hubland, 97074 Würzburg, Germany
First published on 30th June 2025
Abstract
B-Aryl-based 1,2,3-diazaborinates (DABates), derived from neutral 1,2,3-diazaborines (DABs), are a promising class of unexplored fluorophores which show an intriguing high optical performance. This work systematically investigates previously unexplored factors that govern their photoluminescence. We strategically designed and synthesized a diverse library of DABate derivatives, modifying the exocyclic boron and Nα-substituents, the Nβ-lone pair, and the fused π system. All compounds were fully characterized by conventional NMR, HRMS and XRD techniques, as well as UV-vis and fluorescence spectroscopy. Our results allow the identification of key molecular attributes which enhance or quench the fluorescence. Complementary supporting time-dependent DFT calculations provide further insights into the excitation process. In addition, we also performed modifications to render the DABates air- and moisture-stable, expanding potential future applications of this novel class of potent fluorophores beyond inert conditions.
Introduction
Fluorescence-based techniques, such as fluorescence spectroscopy,1–3 cell labelling,4–6 dye-assisted lasers7 and luminescence chemosensing,8–11 are indispensable tools of modern technology and depend on effective fluorophores. Although novel fluorophores often originate from a serendipitous discovery, subsequent modification and optimization of optical performance predominantly rely on the rational manipulation of key molecular attributes that promote fluorescence: as the fluorescence process is normally preceded by an excitation within the lower energetical region of UV or visible light, this criterion is met by the absorption region of larger aromatic π systems or non-bonding molecular orbitals.12 In this context, n → π* transitions usually have a small transition probability due to insufficient n/π* orbital overlap and can additionally result in increased spin–orbit coupling, which promotes intersystem crossing and thus non-radiative decay.13–15 In contrast, π → π* transitions usually feature considerably higher molar absorptivity due to conjugative effects along the main molecular axis. This can be further enhanced by extending the π system or combining electron-donating (D) and -accepting (A) substituents, where the latter supports the redistribution of electrons during the excitation in form of an intramolecular charge transfer (CT).12Rigid molecular geometries are advantageous for this process and can be achieved by incorporating a chelated sp3-hybridized boron atom, which effectively locks the configuration of the organic framework.16–18 Prominent examples for boron-containing fluorophores that utilize this effect are the well-established boron dipyrromethene (BODIPY) derivatives, dating back to early works of Treibs and Kreuzer.19 More recently, boron difluoride hydrazones (BODIHY), first synthesized by the group of Aprahamian,20 and boron difluoride formazanate dyes (BF2Fz) have emerged as other classes of fluorescent boron compounds, both of which are currently mainly advanced by Gilroy and coworkers (Fig. 1).21–28 While common BODIPY dyes also feature a BF2-group (R = F, F-BODIPY), other boron substituents such as carbocycles (R = Ar, C-BODIPY) or oxygen (R = OR, O-BODIPY) and backbone modifications are also reported and result in different optical properties.29,30
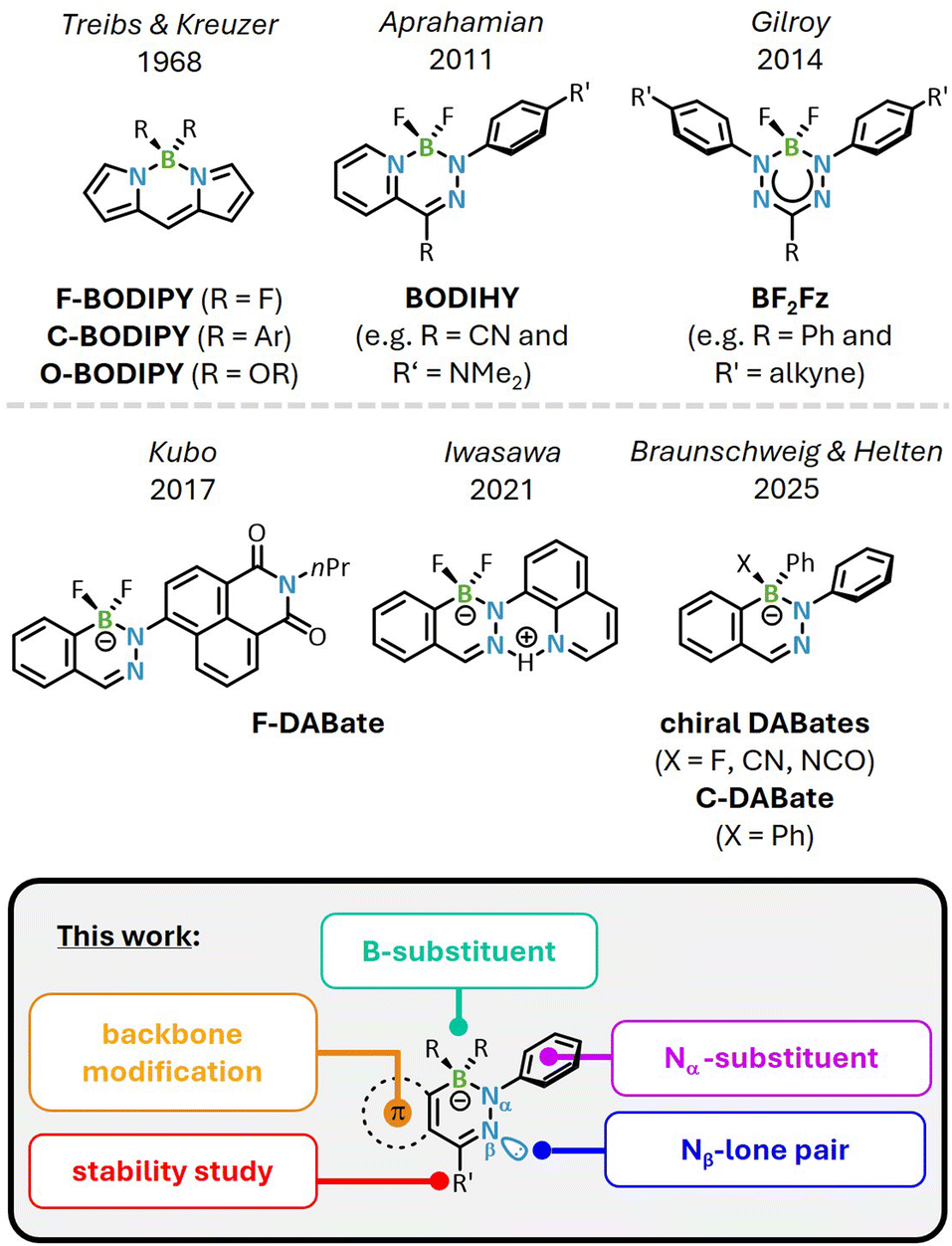 | ||
| Fig. 1 Basic scaffolds of established BODIPY, BODIHY and BF2Fz dyes (top), diazaborinates (DABates, middle) and investigated structure–properties relationships in this work (bottom). | ||
The incorporation of heavy atoms (S, Se, Te) in BODIPYs favors intersystem crossing (ISC) and a decreasing fluorescence quantum yield in accordance with heavy atom effects.31 Likewise, by structurally modifying the organic periphery of the BODIHY or BF2Fz dyes, their photoluminescence and redox properties can be tuned to enhance CT characteristics or show dual or aggregation-induced emission (AIE) behavior.21–24
Contrasting BODIHYs and BF2Fzs, the field of structurally related, luminescent 1,2,3-diazaborinates (DABates) is still in its infancy and limited to very few reported examples. While a comparatively larger number of O-DABates, mostly featuring B(OH)2-borates, is reported, they appear to be mostly non-emissive species.32–35 In 2017, Kubo and coworkers synthesized the naphthalimide-appended F-DABate with BF2-moiety as part of a fluoride (F−) anion sensing study, though the group only focused on changes in the absorption spectra upon anion addition as well as the luminescence of the neutral precursor (Fig. 1, middle).36 A few years later, the group of Iwasawa reported an F-DABate with a quinoline substituent, which was characterized as very weakly fluorescent in solution but showed an enhanced emission with AIE character in the crystalline solid state.37
Recently, our groups reported a synthetic access to B-aryl-substituted, neutral 1,2,3-diazaborines (DABs),38,39 which are isoelectronic BN-congeners of isoquinolines and can act as highly sensitive fluorescence “turn-on” chemosensors. The sensing process is based on the formation of chiral, emissive DABates upon nucleophilic attack of the respective anion (F−, CN−) at the boron center, with quantum yields reaching up to Φfl = 80% in a rigid environment. During our synthetic work on these neutral DABs, we also isolated an achiral, luminescent C-DABate, featuring a BPh2-unit.38 Intrigued by the optical performance of the borates formed, this work is aiming at systematically studying previously unexplored factors, which govern the photoluminescence of these DABates. For this purpose, we devised a strategic variety of system alterations on different positions of the DABate core, covering the exocyclic boron and Nα-substituents, the Nβ-lone pair as well as the fused π system (Fig. 1, bottom). In addition, we performed modifications to also render the DABates air- and moisture-stable to pave the way for potential applications beyond Schlenk conditions. Our photochemical and stability studies are rationalized and supported by XRD studies as well as TD-DFT calculations.
Results and discussion
Syntheses of DABate derivatives
In analogy to the previously reported preparation of the parent C-DABate 5Ph[Li] (parent compound in Scheme 1a, black),38 we developed a general synthetic procedure for achiral C-DABates. To install different exocyclic boron substituents, we reacted 1OTMS with a variety of organolithium reagents to obtain the C-DABates 5R[Li] (Scheme 1a, right-hand side, green). The lithium organyls were either used in solid, solvent-free form (R = Me, Thio) or generated in situ via prior transmetalation of a bromo precursor with n-butyllithium in a one-pot synthesis (R = Ph, Biph, Cbz, OMe, CF3). To prevent the formation of n-butylated side products (e.g.5nBu[Li]) with similar solubility as the products 5R[Li] due to a nucleophilic attack of excess transmetalation agent on 1OTMS, we used slightly substoichiometric amounts of n-BuLi in some cases. This results in lower yields of 5R[Li] but has proven to be the best method for synthesizing the C-DABates, since potential side products such as the monosubstituted, neutral DAB species 1R or remaining 1OTMS are usually easily removed by washing with n-pentane.38 We also found that the order of addition is crucial for the successful synthesis of 5R[Li]. The addition of 1OTMS to a solution of the respective lithium organyl leads to unselective reactions, presumably due to an additional attack of RLi on the electrophilic aldimine position of the DABate, as this order of addition generates an initial surplus of RLi during the addition of 1OTMS. Consequently, such side reactions can be prevented by slow addition of RLi to a solution of 1OTMS. While the reaction times and temperatures were varied slightly depending on the reactivity of the respective lithium organyl, the use of diethyl ether was essential in all cases. The diethyl ether-complexed DABates precipitate and can be isolated in high purity by filtration and washing. In contrast, the THF complexes of 5R[Li] show increased solubility even in non-polar solvents such as benzene, which prevents purification by washing. | ||
| Scheme 1 Rational synthetic manipulation of key attributes, based on the parent compounds 5Ph[Li] and 5Ph[TBA]. (a) Installing carbon-based, exocyclic boron substituents (R3) of varying electronic properties using different lithium organyls and a ketimine instead of an aldimine derivative for stability studies (Et2O, −78 °C → rt or rt, 16 h or 32 h); (b) installing fluoride-based, exocyclic boron substituents using [TBA]F (THF, 80 °C, 4 d); (c) salt metathesis to separate ion pairs (THF, sonification, rt, 1 h); (d) methylation of the Nβ-position with an excess of methyl triflate to a zwitterionic salt (MeCN, rt, 16 h); (e) synthesis of a derivative with a sterically demanding Nα-substituent (Et2O, rt, 16 h); (f) (CH2Cl2, rt, 16 h) and (g) (Et2O, rt, 16 h) synthesis of a derivative with a different backbone π system via its neutral phenyl-DAB (Ph = phenyl; o-Xyl = ortho-2,6-dimethylphenyl; Cbz = 9H-carbazole; OTf− = triflate; TBA = tetra-n-butylammonium). Respective changes on the parent compound 5Ph[Li] (black) are color coded. | ||
Taking these points into account, it was possible to prepare a wide range of different boron-substituted C-DAbates 5R[Li] in yields of 37–77%, with yield discrepancies mostly resulting from differences in solubility behavior, rather than unselective reactions. The substituents R3 include alkyl or aryl groups (R = Me, Ph), electron-donating or -accepting groups (R = p-C6H4-OMe, p-C6H4-CF3), the incorporation of a heavy atom (R = 2-thienyl) or additional fluorophores (R = 2,2′-biphenyl, p-C6H4-Cbz). Like the parent compound 5Ph[Li] and the anion sensing compounds from our previous work (cf.Fig. 1),38 all DABates show the expected high-field shift of the sp3-boron in the 11B NMR spectra compared to the precursor 1OTMS (δ(11B) = 27 ppm). In contrast to the sensing DABates, the individual diazaborinates 5R[Li] hardly differ in their 11B NMR resonances and are detected in the range from −3.1 to −7.1 ppm. For compounds 5Me[Li] and 5Thio[Li], with substituents that can be classified as comparatively electron-rich, a slightly more pronounced high-field shift is observed compared to the phenyl-substituted DABates. Similarly, the strongest high-field shift for the characteristic DABate aldimine proton (H-1) is also detected for 5Me[Li] (δ(H-1) = 6.92 ppm). Overall, however, no clear NMR spectroscopic trends can be determined for the different B-substitution patterns.
Inspired by the F-BODIPY family, as well as the F-DABates by Kubo and Iwasawa,36,37 we also synthesized the fluoro-substituted F-DABate 5F[TBA] from the reaction of 1OTMS with [TBA]F in good yields of 65% after reflux in THF for four days (Scheme 1b, upper right corner, green). Due to the strong electron-withdrawing effect of the fluoro substituents, compound 5F[TBA] exhibits a significantly low-field shifted 11B NMR resonance compared to the other DABates at +3.4 ppm.
As the lithium DABates feature a complexation of the Li+ cation by the Nβ-atom of the DABate (vide infra), we were also interested in an in-depth study of the influence of the electronic situation at Nβ on the photophysical properties. Our previous work demonstrated that the cation exchange TBA salt 5Ph[TBA] (Scheme 1c, upper right corner, blue) of the parent DABate 5Ph[Li] shows significantly different photophysical properties (vide infra).38 To further investigate the influence of the Nβ-lone-pair availability, we methylated the Nβ-position in a metathesis reaction of 5Ph[Li] with methyl triflate to the zwitterionic compound 5Ph[Me] (Scheme 1d, lower right corner, blue). 1H NMR spectroscopy reveals a strong electronic impact of the introduction of the positive charge on the BN2C3-ring, as the aldimine (H-1) resonance is experiencing a low field shift of ca. 1 ppm upon methylation (δ(H-1, 5Ph[Li]) = 7.11 ppm).
As the structural conformation of a fluorophores' adjacent aromatic group such as the DABates Nα-Ph substituent often strongly impacts the photoluminescence,40,41 we also synthesized compound 6Ph[Li] with an Nα-o-xylyl (2,6-dimethylphenyl) instead of the Nα-Ph substituent (Scheme 1e, upper left corner, purple). In 6Ph[Li], the free rotation of the Nα-aryl is effectively blocked by the ortho-methyl groups (cf. calculated rotational barrier of ca. 26 kcal mol−1 for Nα-xylyl vs. ca. 7 kcal mol−1 for Nα-phenyl, ESI Fig. S114 and S115†), leading to an almost perpendicular arrangement to the central DABate core (vide infra).
Lastly, direct alterations of a fluorophores' π system, e.g. by replacing a carboaromatic group with a thiophene-fused counterpart, often induces significant changes in the photophysical properties.42–45 While benzannulated DABs represent the currently predominant DAB derivatives, early reports by Gronowitz and Bugge inspired us to synthesize the thiophene-fused DABate 7Ph[Li] (Scheme 1g, lower left corner, yellow).46 Since direct borate formation from 3OTMS was not successful, we adapted our previously established procedure for the synthesis of neutral, aryl-substituted DABs to the thiophene-fused derivative. Thereby, we managed to isolate the intermediate product 3Ph and subsequently converted 3Ph to the corresponding DABate by treatment with PhLi, which allowed isolation of analytically pure 7Ph[Li] in good yield of 72%.
Solid-state structures
Single-crystal X-ray diffraction (XRD) experiments were performed on all DABates shown in Scheme 1. Most of the derivatives exhibit distinctive 2- or 3-fold whole-molecule disorders in the solid-state, which are likely a result of molecular horizontal mirror planes and limit a detailed bond parameter discussion to approximated values for the most relevant derivatives (Table 1). Visualized examples of these disorders can be found in the ESI appendix (Fig. S113†) to explain the restrictions on a detailed bond discussion. Fig. 2 depicts the solid-state molecular structures of 5Ph[TBA] (no Nβ → cation complexation), 5Ph[Li] (intact Nβ → Li+ coordination), 5Ph[Me] (methylated Nβ), and 6Ph[Li] (o-xylyl substituent at Nα).|
|
|||||
|---|---|---|---|---|---|
| No. | Modification | Nβ → Lia [Å] | B–Nαa [Å] | φ [°] | Nβ–Nα–C1–C2a [°] |
| a Due to the presence of major whole molecule disorders for most of the derivatives, only rounded values are discussed. For exact bond data on non-disordered derivatives see ESI. | |||||
 5Ph[Li]
5Ph[Li]
|
— | 2.02 | 1.58 | 33 | 36 |
 5Ph[TBA]
5Ph[TBA]
|
Cleaved Nβ-cation interaction | — | 1.59 | 10 | 16 |
 5Ph[Me]
5Ph[Me]
|
Nβ methylation | — | 1.60 | 40 | 38 |
 6Ph[Li]
6Ph[Li]
|
Nα xylyl substituent | 2.18 | 1.60 | 25 | 61 |
 7Ph[Li]
7Ph[Li]
|
Thiophene-fused backbone | 2.04 | 1.62 | 10 | 28 |
 8Ph[Li]
8Ph[Li]
|
Ketimine | 2.06 | 1.58 | 29 | 29 |
 8Ph[TBA]
8Ph[TBA]
|
Ketimine | — | 1.59 | 21 | 11 |
 5F[TBA]
5F[TBA]
|
Fluoro substituents at boron | — | 1.52 | 4 | 4 |

 | ||
| Fig. 2 Molecular structures of selected DABates (a) 5Ph[TBA], (b) 5Ph[Li], (c) 5Ph[Me], and (d) 6Ph[Li] in top view. Ellipsoids drawn at 50% probability (100 K). Complexing ether or tetrahydrofuran molecules and hydrogen atoms omitted for clarity. Phenyl substituents at the boron atoms rendered as wireframe. TBA counter ion omitted for 5Ph[TBA]. For selected bond parameters see Table 1. For the solid-state structures in side view of these compounds and the other DABates derivatives structures in side view of these compounds and the other DABates derivatives see the ESI.† | ||
In comparison to the other three derivatives, the TBA salt of 5Ph (Fig. 2a) with a free lone pair at the Nβ shows a significantly more planarized BN2C3 core as well as a nearly co-planarized Nα-phenyl substituent. In contrast, all DABates with a blocked lone pair, either by Li+ complexation or methylation, feature a strongly distorted BN2C3 half-chair core with the BR2 unit bending out of the remaining DABate plane. To quantify this distortion, we defined the angle φ (Table 1, inset) which reflects this twisted nature by high values of 25–44° for the parent compound 5Ph[Li], the Nβ-methylated 5Ph[Me], and Nα-o-xylyl derivative 6Ph[Li] (cf. TBA salt of the parent DAB with free Nβφ(5Ph[TBA]) = 10°). The Nβ–Nα–C1–C2 torsion angle serves as an additional indicator for the co-planarization of the respective Nα substituent. Comparison of this angle for all DABates gives the same trend as the bent angle φ: the derivatives 5Ph[Li] (parent DABate), 5Ph[Me] (Nβ methylated), and 6Ph[Li] (o-xylyl-Nβ) also exhibit the most twisted Nα substituents to accommodate the spatial requirements of the Li+ cation.
Here, the o-xylyl derivative 6Ph[Li] shows the highest angle of 61° and an elongated Nβ → Li bond due to the additional steric effects of the ortho-methyl groups. Interestingly, in direct comparison to the other Li+ coordinating DABates, the thiophene-fused compound 7Ph[Li] shows a relatively planarized geometry with bond parameters in a similar range as the TBA salt of the parent DAB 5Ph[TBA], despite the intact Nβ → Li coordination. F-DABate 5F[TBA] features the most planarized geometry of all DABates in this study, since the small F substituents in combination with the TBA counter cation minimize the steric constraints on the system. The B–Nα bond in 5F[TBA] of ca. 1.52 Å is also significantly contracted in comparison to the C-DABates, as the electron-withdrawing F-substituents retain a more Lewis-acidic boron center, which is compensated for by the non-bonding lone-pair of the adjacent Nα atom. In contrast, all C-DABates feature strikingly long B–Nα bonds of ca. 1.59–1.62 Å, despite their incorporation in a cyclic system (cf. 1.4210(19) Å for the neutral phenyl DAB 1Ph). While elongated B–Nα bonds in the B(sp3)-hybridized DABates compared to the neutral B(sp2)-DABs are generally to be expected due to the loss of π character, such drastic differences raise doubts as to whether the B–Nα bond can still be described as a single bond without dative participations. However, calculated NBO and Mulliken charges give unambiguous information that contradicts such a description (ESI, Table S4†). No additional information or trends can be extracted from the comparison of the other B-substituted derivatives 5R[Li]. All solid-state structures and bond parameters, if permitted by the absence of disorders, are provided in the ESI.†
Photophysical properties
We investigated the photophysical properties of all compounds by UV-vis absorption and fluorescence emission spectroscopy in solution and as PMMA films. For a more comprehensive comparison, we also include data for the parent compound 5Ph[Li] and its TBA congener 5Ph[TBA] in our discussion, which we have recently reported.38 The UV-vis spectra of the very low or non-emitting neutral DABs 1R–4R all show a maximum of the lowest-energy absorption band between 300–307 nm in THF, which demonstrates that the modification of the precursor has no major influence on the absorption properties (ESI, Table S3†). Compared to the neutral DABs, the DABates show a clear bathochromic shift of up to 111 nm. Depending on the respective variation of the parent structure, compounds 5R–8R (except from F-DABate 5F[TBA]) show a slight wavelength shift of the lowest-energy absorption bands from 390–415 nm (ESI, Table S4†). Particularly striking here is the hypsochromic shift of the F-DABate 5F[TBA], which shows a maximum at 355 nm, thus following the trend of the recently reported chiral DABate (Fig. 1, X = F) (369 nm) and 5Ph[TBA] (398 nm).38 Based on TD-DFT calculations of the vertical singlet excitations of the DABate compounds, we assign the lowest-energy absorptions to a π–π* process, which, except for 5Cbz with carbazole substituents, corresponds to a HOMO → LUMO excitation (vide infra).The unexpectedly pronounced fluorescence of the parent DABate 5Ph and the resulting sensor application of the precursor 1Ph has already been characterized in our recent work.38 This follow-up study aims to investigate the mechanism of the fluorescence and the influence of molecular modifications to 5Ph on the emission properties of this novel class of substances (vide supra). We began our investigations by varying the boron substituents. For this purpose, we attached substituents with electron-donating (5OMe[Li]) or -withdrawing (5CF3[Li]) properties, an extended π system (5Cbz[Li]), a rigid geometry (5Biph[Li]), a heteroaromatic ring (5Thio[Li]) or sterically varying (5F[TBA], 5Me[Li]) substituents to the boron atom. Except for 5F[TBA] and 5Me[Li], all differently substituted C-DABates show similar properties in solution and in the PMMA film (see ESI, Table S4 and Fig. S140†). They display a narrow emission curve in THF and the PMMA film, which is mirror-inverted to their lowest-energy absorption bands, with quantum yields of Φfl = 12–19% in THF and Φfl = 25–42% in the film (Fig. 3 and Table 2; for all data, see Table S4 in the ESI†).
 | ||
| Fig. 3 Normalized emission spectra of 5Ph[Li], 5CF3[Li], 6Ph[Li], 7Ph[Li], and 8Ph[Li] in THF. Inset: overview of structural factors influencing emission. | ||
| No. | λ abs,max [nm] | λ abs,max [nm] | λ abs,max [nm] | λ em,max [nm] | λ em,max [nm] | λ em,max [nm] | Φ fl , [%] | Φ fl , [%] | Φ fl , [%] | τ fl [ns] |
|---|---|---|---|---|---|---|---|---|---|---|
| a THF. b PMMA film. c Toluene. d Fluorescence quantum yields, determined using an integration sphere. | ||||||||||
 5Ph[Li]
5Ph[Li]
|
398 | 396 | 391 | 460 | 455 | 510 | 18 | 30 | — | 1.70; 2.57 |
 5Ph[TBA]
5Ph[TBA]
|
398 | 399 | 404 | 459 | 463 | 479 | 19 | 27 | 8 | 1.63; 2.57 |
 5Ph[Me]
5Ph[Me]
|
415 | — | 421 | — | — | — | — | — | — | — |
 5CF3[Li]
5CF3[Li]
|
395 | 391 | 388 | 453 | 445 | 529 | 19 | 42 | — | — |
 5Cbz[Li]
5Cbz[Li]
|
397 | 395 | 390 | 456 | 451 | 510 | 12 | 34 | — | 1.65; 2.34 |
 5F[TBA]
5F[TBA]
|
355 | 354 | 354 | 428 | 427 | — | — | 37 | — | — |
 6Ph[Li]
6Ph[Li]
|
390 | 387 | 393 | 501 | 483 | 504 | 7 | 3 | — | 2.16; 3.61 |
 7Ph[Li]
7Ph[Li]
|
410 | 410 | 411 | 465 | 477 | 490 | 63 | 28 | — | 5.63 |
 8Ph[Li]
8Ph[Li]
|
403 | 402 | 410 | 463 | 463 | 489 | 26 | 5 | — | 2.96 |
In toluene, however, only a quenched, bathochromically shifted and weak emission is present for all lithium DABates. As we have already shown, the counterion has a major impact on the properties in solution (see 5Ph[Li] and 5Ph[TBA]).38 We reasoned that the cleavage of the Nβ → Li coordination by formation of solvent-separated ion pairs [DABate][Li(thf)n] in THF is responsible for the emission in THF, whereas the Nβ → Li coordination should remain intact in toluene due to the absence of additional donor molecules. These observations suggest that the Nβ lone pair has an important influence on the emission behavior of the compounds. To verify this, we substituted the Nβ of 5Ph[Li] with a methyl group to form 5Ph[Me], which completely quenched the emission. This behavior of the DABates is therefore contrary to that of the related, parent CC-compound isoquinoline, which shows a clear increase in fluorescence when protonated at nitrogen.13
When comparing the compounds 5R (excluding the non-emissive compound 5Ph[Me]), it is striking that the DABates with small fluoro or methyl substituents at the boron 5F[TBA] and 5Me[Li] exhibit very low fluorescence in solution but with simultaneously intensified fluorescence compared to the other derivatives in this series in rigid PMMA film. This behavior indicates an aggregation-induced emission effect similar to the BODIHY or BF2Fz dyes.23–25 Thus, the lower steric demand of the boron substituents could enhance molecular motions, especially for the Ph group attached at the Nα position, which results in more efficient non-radiative relaxation processes. To prove this experimentally, we investigated the behavior of fluoro compound 5F[TBA] in DMSO, which led to an intensification of the emission and a decrease in intensity of the broad bathochromically shifted band of the dual emission (Fig. 4). However, since the compound is sensitive to hydrolysis, conventional AIE experiments, in which aggregation and thus hindered molecular motion through the hydrophobic character of the compound is forced by increasing the water content of a dissolved sample, cannot be performed. As 5F[TBA] does not dissolve in n-pentane, we tested samples with different THF/n-pentane ratios to investigate this effect. When n-pentane (up to 90%) was added to a THF solution of the F-DABatee, a clouding of the sample was observed and the fluorescence intensity increased ca. 94-fold relative to the pure THF solution (Fig. 4). Since molecular movements can also be restricted by cooling, which increases the viscosity, we monitored the emission intensity as a function of temperature in the range from 303 to 103 K in 2-MeTHF. By cooling to 103 K, an increase in intensity by a factor of 80 was observed (Fig. 4). Similar to the observations for the measurements in THF and DMSO, the intensity of the bathochromic band decreases with decreasing temperature until it is no longer visible beyond the glass transition temperature of 2-MeTHF (137 K) at 133 K (Fig. S129 in the ESI†). These observations suggest that a bulky residue on boron is favorable for efficient emission in solution but is not necessary in a rigid environment or in the solid state. To further explore the aggregation behavior of 5F[TBA], we prepared various PMMA films with different DABate contents (0.025–2.5 mg). With increasing contents of the PMMA matrices, a bathochromic shift of the bands from λem,max = 418 nm to 432 nm was observed (ESI, Fig. S132†). In solution, an increase in concentration led to quenching of the fluorescence. From these results it could be concluded that an aggregation effect such as π–π stacking may be present in the rigid films.
 | ||
| Fig. 4 Emission spectra of 5F[TBA] in different solvents (left), variable-temperature emission spectra of 5F[TBA] in 2-MeTHF from 303 to 103 K (middle), and emission spectra of 5F[TBA] in THF/n-pentane mixtures (conc. 5 × 10−5 M). Inset: structure of the compound and corresponding samples under UV light. | ||
The comparison of the XRD structures with the photophysical data suggests that the planarity of the backbone and the position of the freely rotatable Nα-Ph group plays a decisive role for the fluorescence behavior. For example, in the solid-state structure of the non-emissive, Nβ methylated DABate 5Ph[Me], a strong twisting of the backbone and the Nα-Ph group is observed. In contrast, the F-DABate 5F[TBA] is approximately planar in the solid state. This planar conformation is enforced by increasing the viscosity or in the rigid environment of the PMMA film, leading to a significant intensification of the emission (Fig. 4). To restrict rotation, we have installed the bulkier o-xylyl group at the Nα-position (6Ph[Li]). The result of this modification was a significantly lower emission intensity both in solution (Φfl = 7%) and in the PMMA film (Φfl = 3%) and a broadening and bathochromic shift of the fluorescence band (Fig. 3 and Table 2). This can be attributed to the almost perpendicular arrangement of the o-xylyl group to the DABate core and the associated low involvement in the emission process (vide infra).
In addition, we tested the modification of the π backbone by exchanging the benzannulated system with a fused thiophene system, resulting in 7Ph[Li] (Scheme 1). While similar fluorescence behavior of 7Ph[Li] is observed in the PMMA film in comparison to the benzannulated 5R[Li] compounds, a significantly increased quantum yield of up to 63% is found in solution (Table 2). This shows that the π backbone also has a significant influence on the emission behavior and thus provides a useful strategy for future, fluorescence enhancing modifications.
As a final modification, we prepared a DABate with a methyl group at the imine position (i.e.8Ph[Li] and 8Ph[TBA]) to achieve higher stability (vide infra). Here, both compounds show an increased quantum yield of Φfl = 26% in solution (compared to Φfl = 18% for the parent compound 5Ph[Li]), but a lower quantum yield in the PMMA film.
Computational studies
Density functional theory (DFT) calculations were performed on all DABates using the Gaussian 16 program (see ESI† for computational studies of all compounds).47 All calculations were carried out on the free DABates without inclusion of the corresponding countercations (ωB97X-D48/def2-SVP,49 see ESI†). In general, the calculated geometries exhibit less twisting and deplanarization compared to the solid-state structures (vide supra). This discrepancy is likely due to the absence of the Li+ coordination, which mimics the photoemissive situation upon THF solvation. Similar to the parent compound 5Ph−, the highest occupied molecular orbital (HOMO) of all DABate derivatives features a nodal plane between the Nα and the boron atom, with the orbital coefficient at the Nα resembling pz character (Fig. 5). Both the HOMO and the lowest unoccupied molecular orbital (LUMO) display π symmetry. The HOMO–LUMO energy gaps of all DABates (except 5Cbz−, vide infra) fall within the range of 7.2–7.5 eV. | ||
| Fig. 5 Exemplary frontier molecular orbitals (FMOs) of the parent compound 5Ph− (ωB97X-D/def2-SVP/isovalue 0.06 eÅ−3). | ||
The photoabsorption properties of all derivatives were also investigated using time-dependent (TD-DFT) methods (tHCTHhyb50/def2-TZVPP49,51) in a solvent model mimicking tetrahydrofuran solvation. All values are in good agreement with the experimental values (Table 3).
| No. | Transition | λ exp [nm] | λ calc [nm] | Main excitation |
|---|---|---|---|---|
 5Ph−
5Ph−
|
S0 → S1 | 398 | 401 | HOMO → LUMO |
 5F−
5F−
|
S0 → S1 | 355 | 364 | HOMO → LUMO |
 5Me−
5Me−
|
S0 → S1 | 404 | 415 | HOMO → LUMO |
 5Thio−
5Thio−
|
S0 → S1 | 388 | 392 | HOMO → LUMO |
 5CF3−
5CF3−
|
S0 → S1 | 395 | 404 | HOMO → LUMO |
 5OMe−
5OMe−
|
S0 → S1 | 398 | 401 | HOMO → LUMO |
 5Biph−
5Biph−
|
S0 → S2 | 404 | 409 | HOMO → LUMO |
 5Cbz−
5Cbz−
|
S0 → S3 | 397 | 402 | HOMO → LUMO+2 |
 6Ph−
6Ph−
|
S0 → S1 | 390 | 399 | HOMO → LUMO |
 7Ph−
7Ph−
|
S0 → S1 | 410 | 421 | HOMO → LUMO |
 8Ph−
8Ph−
|
S0 → S1 | 403 | 406 | HOMO → LUMO |
 5Ph[Me]
5Ph[Me]
|
S0 → S1 | 415 | 443 | HOMO → LUMO |
For all DABates, except for 5Cbz− with additional carbazole substituents, the primary electronic transitions correspond to HOMO(π) → LUMO(π*) excitations. In case of 5Cbz−, the presence of the incorporated carbazole units results in the LUMO and LUMO+1 being centered on the carbazole moiety. Consequently, the main electronic excitation is the HOMO(π) → LUMO+2(π*) transition, with the LUMO+2 resembling the LUMO situation of the other DABates (ESI, Fig. S124†). Except for 5Cbz− and 5Biph−, reminiscent of a borafluorene, all transitions correspond to S0 → S1 excitations. Geometry optimization of the first excited S1 state of the parent compound 5Ph− revealed a minor geometric change upon electronic excitation (Fig. 6), reminiscent of the phenomenon of planarity-induced CT.52 While the S0 ground state exhibits a bent DABate BN2C3 core and a consequently slightly out-of-plane twisted Nα–Ph substituent, the DABate in the optimized S1 geometry is fully planarized with a co-planar Nα–Ph substituent.
 | ||
| Fig. 6 Schematic depiction of the geometric changes during the excitation and relaxation of the model DABate 5Ph− (grey = planarized). | ||
To gain a deeper understanding of the photophysical differences observed for the key compounds 5Ph− (parent C-DABate), 6Ph− (Nα-o-xylyl), 7Ph− (thiophene-fused) and 5Ph[Me] (Nβ methylated), we analyzed the transition character of the first singlet excited state (S1), using the Multiwfn program (see ESI, section 4†).53,54 The visualized electron–hole distributions of 5Ph− and thiophene-fused 7Ph− revealed an overall dominant intramolecular CT character of 70–73%, with the Nα-Ph substituent acting as an electron donor for the DABate acceptor unit (Fig. 7). For this transition, the central BN2C3 ring appears to serve as a bridging connection with both electron and hole contributions that correspond to a local excitation (LE) of 39–43%. Interestingly, the boron atom is completely excluded from the transition in both cases, explaining the minor influence of the C-DABates exocyclic boron substituents on the photophysical properties. In 6Ph−, the Nα-xylyl substituent adopts a perpendicular orientation to the DABate, which effectively excludes the Nα substituent from the transition. This reduces the CT character to 63% with simultaneously high LE character of 44% for the transition from the BN2C3 unit to the benzannulated system and is experimentally reflected in the comparatively lower quantum yield of 6Ph− (vide supra). Similarly, in the Nβ-methylated 5Ph[Me], the Nα-substituent is also noticeably twisted out of the DABate plane. However, unlike 6Ph− with Nα-o-xylyl substituent, the hole remains located on the Nα-phenyl substituent, which likely contributes to the compounds non-emissive character. Unlike their distant CC-isoquinoline relatives,13 no relevant n → π* transitions involving the Nβ-lone pair are found for the DABates (vide supra), despite its drastic influence on the photoluminescence. This prompted us to further analyze the interfragment CT in search of an alternative explanation for the quenching effect of the Nβ-methylation or lithium coordination. Both modifications significantly lower the electron density in the central DABate unit, as evidenced by a substantially lower net amount of 0.18 e− transferred from the BN2C3 ring to the benzannulated system for the Nβ-methylated 5Ph[Me] (cf. 0.25 e− for parent compound 5Ph−, 0.27 e− for thiophene-fused 7Ph− and 0.30 e− for Nα-o-xylyl 6Ph−). Hence, the quenching is likely a result of electron deprivation in combination with a sterically hindered in-plane rotation of the Nα-Ph substituent in 5Ph[Me] and the DABate lithium complexes.
 | ||
| Fig. 7 Simultaneous isosurface of the electron (e−, green) and hole (h+, blue) distribution of 5Ph−, 6Ph−, 7Ph− and 5Ph[Me] for the S0 → S1 transition density analysis (isovalue 0.02 a.u.). | ||
Stability studies
Given that the C-DABates of this work represent a potent class of novel fluorophores, the question of their air- and moisture-stability seems consequential for practical use. We additionally investigated the photostability of selected compounds (5Ph[TBA], 5F[TBA], 7Ph[Li] and 8Ph[TBA]) by irradiation with an UV lamp at 254 and 365 nm. In THF solution, complete decomposition was observed after 90 min. In contrast, no noticeable change was detected when powders of the compounds were irradiated, rendering the DABates susceptible towards photolysis in solution but stable in the solid state. Following their synthesis under inert Ar atmosphere and Schlenk conditions, we noticed that solutions of all DABates their color from neon yellow to deep red within hours upon opening the reaction vessels to the atmosphere (Fig. 8a). This slow color change is accompanied by a rapid emission quenching as soon as the solutions are exposed to air. To understand the underlying degradation process, we separately reacted the isolated parent compound 5Ph[Li] with dry O2, as well as degassed H2O. While the addition of H2O resulted in a quenched emission, likely due to the formation of 5Ph[H] and LiOH in analogy to the non-emissive methylated compound 5Ph[Me] (vide supra), no color change to deep red was observed. In contrast, the reaction with dry O2 gas resulted in a deep red-violet reaction mixture after a few hours. While monitoring the O2 oxidation by 1H and 11B NMR spectroscopy revealed a highly unselective degradation reaction involving multiple species, one of which was confirmed as the neutral, monosubstituted DAB 1Ph, we managed to identify compound 9 as one of the other oxidation products via XRD analysis (Fig. 8b and ESI, Fig. S107†). | ||
| Fig. 8 (a) NMR samples of a DABate before (left, yellow solution) and after (middle and right, red solutions) atmosphere exposure. (b) Compounds 9 and 10 crystallized as part of the decomposition studies (for XRD structures see ESI, appendix Fig. S107 and S108†). | ||
The formation of 9 seems to be the result of an oxidative coupling of two DABates, which at some point of the reaction sequence rearomatized to the neutral DAB species by the loss of one of the exocyclic B–Ph substituents. A similar oxidative radical coupling is documented for structurally similar BODIHYs systems by Gilroy and coworkers.24 While 9 would be interesting for potential follow-up studies as an atropisomeric ligand comparable to other binaphthyls (e.g. (R/S)-BINAP55), we were not able to fully characterize or selectively synthesize 9 despite extensive efforts (ESI, Table S2†). However, we also obtained single crystals of compound 10 from a combined study with H2O and O2, which appears to be a final oxidation and hydrolysis product of this degradation pathway (Fig. 8b and ESI, Fig. S108†). The formation of 10 is reminiscent of the archetypal reaction of neutral DABs with reactive oxygen species (ROS) such as H2O2 and might indicate the formation of ROS during the decomposition process.56
Considering these results, we conceived stability enhancing modifications of our DABate systems. The formation of quenched 5Ph[H] as a result of hydrolysis seemed preventable by utilizing the TBA salts of our DABates. A hydrolysis study of 5Ph[TBA] under Ar atmosphere confirms this consideration, as no decomposition was observed via1H NMR spectroscopy even after 15 d in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 D2O/THF-d8 mixture (ESI, Fig. S109 and S110†). We then proceeded to address the problem of the observed oxidative coupling to species like 9 and 10 by synthesizing DABate 8Ph[TBA], featuring a ketimine (Nβ = C(CH3)) instead of an aldimine functionality (Nβ = C(H)) at the central BN2C3 ring (Scheme 1, upper right corner). A 1H NMR study of 8Ph[TBA] in a 1:1H2O/THF-d8 mixture left open to the atmosphere showed no significant decomposition over the course of 15 d (Fig. 9 and ESI, Fig. S111 and S112†). This confirms the air- and moisture-stability of 8Ph[TBA], which is additionally demonstrated by growing the single crystals of 8Ph[TBA] for XRD analysis from a THF/H2O mixture under aerobic conditions (ESI Fig. S106†).
:1 D2O/THF-d8 mixture (ESI, Fig. S109 and S110†). We then proceeded to address the problem of the observed oxidative coupling to species like 9 and 10 by synthesizing DABate 8Ph[TBA], featuring a ketimine (Nβ = C(CH3)) instead of an aldimine functionality (Nβ = C(H)) at the central BN2C3 ring (Scheme 1, upper right corner). A 1H NMR study of 8Ph[TBA] in a 1:1H2O/THF-d8 mixture left open to the atmosphere showed no significant decomposition over the course of 15 d (Fig. 9 and ESI, Fig. S111 and S112†). This confirms the air- and moisture-stability of 8Ph[TBA], which is additionally demonstrated by growing the single crystals of 8Ph[TBA] for XRD analysis from a THF/H2O mixture under aerobic conditions (ESI Fig. S106†).
 | ||
| Fig. 9 Stacked 1H NMR spectra of 8Ph[TBA] in a 1:1 D2O/THF-d8 mixture under air over a period of 16 d, showing no significant decomposition. | ||
Conclusion
In this work we have carried out a comprehensive study on the structural, photophysical and electronic behavior of a series of unprecedented diazaborinates (DABates). For this purpose, we have synthesized a diverse library of DABate derivatives with specific modifications, including the exocyclic boron and Nα-substituents, the Nβ-lone pair as well as the fused π system and the imine position. Our investigations showed that the lone pair at the Nβ position plays an important role for the planarity of the compounds in the solid state. While DABates with a free lone pair adopt a nearly planar arrangement and co-planarized Nα-Ph group, lithium coordination or methylation leads to twisting of the entire molecular framework. Our photophysical investigations revealed that the Nβ lone pair also has a major influence on the emission behavior. As shown by measurements in toluene, the fluorescence is quenched when the lithium is coordinated to the Nβ of the DABates. However, it reveals emission again when the counterion is exchanged for TBA, resulting in the formation of separated ion pairs. By selectively substituting this site with a methyl group, the emission is extinguished in all media, which confirmed our assumption. Varying the exocyclic boron substituents had no significant effect on the fluorescence when measured in the rigid PMMA film. Meanwhile, a strong decrease in emission intensity was observed in solution with smaller substituents (for Me and F). Viscosity- and temperature-dependent measurements of the F-DABate 5F[TBA] showed an increasing emission intensity due to a more rigid environment. This can be attributed to the restriction of molecular motions, which is more favorable for the small substituents, and thus points to an aggregation effect. By inhibiting the rotational freedom and preventing the possibility of coplanar adjustment of the Nα substituent via the incorporation of the o-xylyl substituent, the fluorescence was also reduced. Quantum chemical calculations confirmed the important influence of the co-planarization of the Nα-aryl substituent in the excited state. Further calculations revealed an overall dominant intramolecular CT character, with the planarized Nα-Ph substituent as an electron donor for the DABate acceptor unit. Modification of the π backbone with a thieno unit led to an increased quantum yield in solution, which provides a promising strategy for future tailoring the properties of DABate fluorophores. Finally, we investigated the stability of this novel class of compounds, which revealed that both the Nβ and aldimine positions play a significant role in determining overall stability. Guided by these principles, we were able to synthesize the first air- and moisture-stable C-DABate and are currently exploring the full potential of these and other further modified DABates.Data availability
The data supporting this work is available in the ESI.†Author contributions
L. W., J. C., H. H. and H. B. conceived the project. L. W. performed the synthesis and modification of all C-DABates except 7Ph[Li], with support of J. S. J. C. performed the synthesis of the F-DABate. T. K. performed the synthesis of the thiophene-fused compounds, including 7Ph[Li]. J. C. performed the photophysical experiments. L. W. performed the quantum chemical calculations. J. C. and L. W. performed the decomposition studies. T. W., L. W., N. W., and M. M. performed the XRD experiments. L. W., J. C., H. H., and H. B. discussed the results. L. W. and J. C. wrote the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conficts to declare.Acknowledgements
We are grateful to Dr Ivo Krummenacher for insightful discussions and feedback on the manuscript. We also thank Dr Krzysztof Radacki for help with X-ray diffraction experiments, Dr Krzysztof Radacki and Lukas Endres for help with quantum-chemical calculations and Lukas Swoboda for HRMS measurements.References
- R. Karoui and C. Blecker, Food Bioprocess Technol., 2011, 4, 364–386 CrossRef.
- A. Romani, C. Clementi, C. Miliani and G. Favaro, Acc. Chem. Res., 2010, 43, 837–846 CrossRef CAS PubMed.
- D.-E. Zacharioudaki, I. Fitilis and M. Kotti, Molecules, 2022, 27, 4801 CrossRef CAS PubMed.
- M. Streit, M. Budiarta, M. Jungblut and G. Beliu, Biophys. Rep., 2025, 5, 100200 CAS.
- N. Zhao, T. M. Williams, Z. Zhou, F. R. Fronczek, M. Sibrian-Vazquez, S. D. Jois and M. G. H. Vicente, Bioconjugate Chem., 2017, 28, 1566–1579 CrossRef CAS PubMed.
- T. Suzuki, T. Matsuzaki, H. Hagiwara, T. Aoki and K. Takata, Acta Histochem. Cytochem., 2007, 40, 131–137 CrossRef CAS PubMed.
- M. Shah, K. Thangaraj, M.-L. Soong, L. T. Wolford, J. H. Boyer, I. R. Politzer and T. G. Pavlopoulos, Heteroatom Chem., 1990, 1, 389–399 CrossRef CAS.
- E. Galbraith and T. D. James, Chem. Soc. Rev., 2010, 39, 3831–3842 RSC.
- W. Zhang, G. Li, L. Xu, Y. Zhuo, W. Wan, N. Yan and G. He, Chem. Sci., 2018, 9, 4444–4450 RSC.
- D. Wu, A. C. Sedgwick, T. Gunnlaugsson, E. U. Akkaya, J. Yoon and T. D. James, Chem. Soc. Rev., 2017, 46, 7105–7123 RSC.
- N. Kwon, Y. Hu and J. Yoon, ACS Omega, 2018, 3, 13731–13751 CrossRef CAS PubMed.
- D. M. Jameson, Introduction to fluorescence, CRC press, 2025 Search PubMed.
- E. Tervola, K.-N. Truong, J. S. Ward, A. Priimagi and K. Rissanen, RSC Adv., 2020, 10, 29385–29393 RSC.
- N. K. Joshi, H. C. Joshi, R. Gahlaut, N. Tewari, R. Rautela and S. Pant, J. Phys. Chem. A, 2012, 116, 7272–7278 CrossRef CAS PubMed.
- M. F. Anton and W. R. Moomaw, J. Chem. Phys., 1977, 66, 1808–1818 CrossRef CAS.
- D. Hu, R. Huang and Y. Fang, Precis. Chem., 2025, 3, 10–26 CrossRef CAS PubMed.
- D. Frath, J. Massue, G. Ulrich and R. Ziessel, Angew. Chem., Int. Ed., 2014, 53, 2290–2310 CrossRef CAS PubMed.
- J. Chorbacher, J. Klopf, A. Friedrich, M. Fest, J. S. Schneider, B. Engels and H. Helten, Angew. Chem., Int. Ed., 2025, 64, e202416088 CrossRef CAS PubMed.
- A. Treibs and F.-H. Kreuzer, Adv. Cycloaddit., 1968, 718, 208–223 CAS.
- Y. Yang, X. Su, C. N. Carroll and I. Aprahamian, Chem. Sci., 2012, 3, 610–613 RSC.
- F. L. Buguis, P. D. Boyle and J. B. Gilroy, Dyes Pigm., 2022, 198, 110002 CrossRef CAS.
- R. R. Maar and J. B. Gilroy, J. Mater. Chem. C, 2016, 4, 6478–6482 RSC.
- D. Cappello, F. L. Buguis, P. D. Boyle and J. B. Gilroy, ChemPhotoChem, 2022, 6, e202200131 CrossRef CAS.
- D. Cappello, D. A. B. Therien, V. N. Staroverov, F. Lagugné-Labarthet and J. B. Gilroy, Chem.–Eur. J., 2019, 25, 5994–6006 CrossRef CAS PubMed.
- D. Cappello, A. E. R. Watson and J. B. Gilroy, Macromol. Rapid Commun., 2021, 42, 2000553 CrossRef CAS PubMed.
- M. C. Chang and E. Otten, Chem. Commun., 2014, 50, 7431–7433 RSC.
- M.-C. Chang and E. Otten, Inorg. Chem., 2015, 54, 8656–8664 CrossRef CAS PubMed.
- M. C. Chang, A. Chantzis, D. Jacquemin and E. Otten, Dalton Trans., 2016, 45, 9477–9484 RSC.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed.
- O. M. Kovtun, Y. V. Zatsikha and Y. P. Kovtun, Chem. Heterocycl. Compd., 2023, 59, 357–367 CrossRef CAS.
- J. W. Campbell, M. T. Tung, K. N. Robertson, A. A. Beharry and A. Thompson, J. Org. Chem., 2023, 88, 10655–10661 CrossRef CAS PubMed.
- M. Z. H. Kazmi, J. P. G. Rygus, H. T. Ang, M. Paladino, M. A. Johnson, M. J. Ferguson and D. G. Hall, J. Am. Chem. Soc., 2021, 143, 10143–10156 CrossRef CAS PubMed.
- S. Shimo, K. Takahashi and N. Iwasawa, Chem.–Eur. J., 2019, 25, 3790–3794 CrossRef CAS PubMed.
- E. A. Sarina, M. M. Olmstead, D. Kanichar and M. P. Groziak, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 1085–1088 CrossRef CAS PubMed.
- M. P. Groziak, A. D. Ganguly and P. D. Robinson, J. Am. Chem. Soc., 1994, 116, 7597–7605 CrossRef CAS.
- Y. Satta, R. Nishiyabu, T. D. James and Y. Kubo, Tetrahedron, 2017, 73, 2053–2061 CrossRef CAS.
- S. Shimo, T. Nakamura, K. Takahashi, N. Toriumi and N. Iwasawa, ChemPhotoChem, 2022, 6, e202100195 CrossRef CAS.
- L. Wüst, J. Chorbacher, T. Wellnitz, S. Nees, H. Helten and H. Braunschweig, Chem. Sci., 2025, 16, 7284–7293 RSC.
- L. Wüst, L. Scheuring, T. Wellnitz, K. Radacki and H. Braunschweig, Chem. Sci., 2025, 16, 9934–9942 RSC.
- M. Kim, C. H. Ryu, D. K. You, J. H. Hong and K. M. Lee, ACS Omega, 2022, 7, 24027–24039 CrossRef CAS PubMed.
- U. P. Pandey and P. Thilagar, Adv. Opt. Mater., 2020, 8, 1902145 CrossRef CAS.
- J. Chorbacher, M. Maier, J. Klopf, M. Fest and H. Helten, Macromol. Rapid Commun., 2023, 44, 2300278 CrossRef CAS PubMed.
- S. Ji, J. Ge, D. Escudero, Z. Wang, J. Zhao and D. Jacquemin, J. Org. Chem., 2015, 80, 5958–5963 CrossRef CAS PubMed.
- K. Tanaka, H. Yamane, R. Yoshii and Y. Chujo, Bioorg. Med. Chem., 2013, 21, 2715–2719 CrossRef CAS PubMed.
- M. Maier, J. Chorbacher, A. Hellinger, J. Klopf, J. Günther and H. Helten, Chem.–Eur. J., 2023, 29, e202302767 CrossRef CAS PubMed.
- S. Gronowitz and A. Bugge, Acta Chem. Scand., 1965, 19, 1271–1285 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian Inc., Wallingford CT, 2019.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. D. Boese and N. C. Handy, J. Chem. Phys., 2002, 116, 9559–9569 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- G. Haberhauer, R. Gleiter and C. Burkhart, Chem.–Eur. J., 2016, 22, 971–978 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- Z. Liu, T. Lu and Q. Chen, Carbon, 2020, 165, 461–467 CrossRef CAS.
- R. Noyori and H. Takaya, Acc. Chem. Res., 1990, 23, 345–350 CrossRef CAS.
- J. P. M. António, J. I. Carvalho, A. S. André, J. N. R. Dias, S. I. Aguiar, H. Faustino, R. M. R. M. Lopes, L. F. Veiros, G. J. L. Bernardes, F. A. da Silva and P. M. P. Gois, Angew. Chem., Int. Ed., 2021, 60, 25914–25921 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General methods and materials, experimental procedures and characterization data, photophysical data, X-ray crystal structure determination and computational section. CCDC 2453528–2453546, 2453634 and 2453635. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc03814f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |