
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular polymerization of permanently dipolar perylene diimide-based diazacoronenes†
Ani. N. Davis a,
Colette M. Sullivang,
Chengbin Fucd,
Rupam Roya,
A. M. Mahmudul Hasana,
Kaitlin Slickera,
Haoyuan Licdef,
Lea Nienhausghijk and
Austin M. Evans*ab
a,
Colette M. Sullivang,
Chengbin Fucd,
Rupam Roya,
A. M. Mahmudul Hasana,
Kaitlin Slickera,
Haoyuan Licdef,
Lea Nienhausghijk and
Austin M. Evans*ab
aGeorge and Josephine Butler Polymer Laboratory, Department of Chemistry, University of Florida, Gainesville, FL, USA. E-mail: AustinEvans@ufl.edu
bDepartment of Materials Science and Engineering, University of Florida, Gainesville, FL 32611, USA
cSchool of Microelectronics, Shanghai University, Shanghai 201800, China
dDepartment of Chemistry, College of Sciences, Shanghai University, Shanghai 200444, China
eKey Laboratory of Advanced Display and System Applications, Ministry of Education, Shanghai University, Shanghai 200072, China
fDepartment of Chemistry and Biochemistry, The University of Arizona, Tucson, AZ, USA
gDepartment of Chemistry, Rice University, Houston, TX, USA
hDepartment of Materials Science and NanoEngineering, Houston, TX 77005, USA
iDepartment of Physics and Astronomy, Rice University, Houston, TX 77005, USA
jRice Advanced Materials Institute, Rice University, Houston, TX 77005, USA
kDepartment of Chemical and Biomolecular Engineering, Rice University, Houston, TX 77005, USA
First published on 10th July 2025
Abstract
We demonstrate that ground-state dipoles guides the supramolecular assembly and resultant optoelectronic characteristics of perylene diimide-based diazacoronenes (PDACs). The synthetic difficulty of installing permanent ground-state dipoles on planar aromatic systems has largely constrained the exploration of dipole engineering in discotic molecules. Here, we synthesize a family of PDACs with ground-state dipoles between 1 and 6 Debye by installing functional groups on the diazacoronene. Systematically increasing the dipolar character of these PDACs led to red-shifted absorption (477 to 557 nm) and emission spectra (483 to 723 nm), which is consistent with their more negative electrochemical reduction potentials. Density functional theory revealed that sufficiently strong dipoles (PDAC-NMe2, 6.0 Debye) led to ground-state charge-transfer, which was confirmed by a combination of electrochemical and spectroscopic measurements. Molecular dynamics simulations predicted that PDACs with larger ground-state dipole moments have stronger intramolecular interactions and more well-defined assemblies. Variable-solvent, -concentration, and -temperature aggregation studies were consistent with this trend and, in all cases, revealed that supramolecular polymerization led to more extended electronic delocalization. Additionally, we observed that PDAC assemblies with larger ground state dipoles had enhanced emission lifetimes over their monomer counterparts (τ = 1.8 ns to 5.1 ns for PDAC-NMe2), whereas assemblies formed from molecules with smaller ground-state dipoles had virtually no change in their excited state lifetimes. Taken together, permanent ground-state dipoles are shown to be a powerful tool to control planar molecular assemblies and their optoelectronic characteristics.
Introduction
Supramolecular assemblies have distinct optoelectronic properties from their dissolved molecular counterparts due to electronic coupling between spatially co-located chromophores. Successfully engineering the spatial arrangement of electronically active molecules is a prerequisite for augmenting the properties of supramolecular assemblies or deploying them in various devices, including field-effect transistors,1–3 photodetectors,4–6 and light-emitting diodes.7–9 Conventionally, dipolar interactions such as H-bonding, dispersion forces, and certain π–π stacking interactions are engineered to tune these thermodynamically driven supramolecular assemblies.10Installing permanent ground state dipoles (µg > 0) is a complementary strategy to induce and control molecular assembly. Ground state dipole engineering is typically explored in systems with existing π–π stacking interactions, though the strategy would likely be advantageous when paired with other supramolecular interactions.11–13 Achieving ground-state dipolar character typically relies on ground-state charge-transfer or permanently zwitterionic systems. For example, it has been shown that installing asymmetric dipoles onto merocyanine dyes through different acceptor units alters the supramolecular structure of their solid-state assemblies.14–17 In these studies, the crystal packing of the different dipolar compounds varied from isolated dimers, to one-dimensional π stacks of antiparallel dimers, to extended staircase-like π–π stacking motifs. Out of the three different crystal packing systems, the staircase stacked materials were found to have the highest power conversion efficiency as solar cell active layers.14 Though ground-state dipolar character offers significant advantages in assembly engineering, it is difficult to install permanent dipoles on many supramolecular and optoelectronically active molecules such as discotic triphenylenes18–20 and coronenes21–23 or quadrupolar porphyrins24,25 and rylene (di)imides.26–35 The challenge with installing permanent dipoles on discotic molecules results from the halogenation or electrophilic aromatic substitution strategies used to functionalize them off of the aromatic core, which either generate electronically symmetric difunctionalized species or regiochemically impure mixtures that are challenging to separate.
The synthetic frustration with installing permanent dipoles in aromatic discotic molecules makes other synthetic strategies more widely explored for controlling discotic assembly. As one example, bay functionalizing perylene diimides (PDIs) influences their assembly and optoelectronic profiles.36 Lu and coworkers found that installing bromines at the bay position increased the π–π orbital overlap between H-aggregates, which led to improved photocatalytic performance.37 Side chain engineering is another well explored approach to guide assembly. For instance, the geometry of PDI assemblies can be tuned by modifying the N-imide solubilizing chain.38 Sánchez and coworkers demonstrated that altering the spacer length between assembly directing groups at the imide position alters the supramolecular assembly of PDI-based assemblies from chiral H-aggregates to J-type supramolecular polymers.39 Würthner and coworkers suppressed long-range assembly by installing asymmetric hydrogen-bonding interactions off of the imide position, which resulted in an anti-cooperative assembly mechanism that favored PDI dimer formation.26 Conversely, George and coworkers installed a rigid steroid side chain off of the imide position, creating a permanent dipole moment through PDI. This created a dipole-driven cooperative mechanism of assembly of the PDI molecules.29 These reports demonstrate that the nature of non-covalent interactions between discotic molecules dictates their assembly, influences their optoelectronic characteristics, and guides their application relevance.40 However, it is still difficult to leverage µg to control this assembly.
In this report, we demonstrate that permanently dipolar perylene diimide-based diazacoronenes (PDACs, Fig. 1) with higher µg have bathochromically-shifted absorption, emission, and reduced reduction potentials, as well as photoluminescence lifetimes that are enhanced by their assembly. This finding relies on a recent synthetic protocol developed by Goujon and coworkers that gives asymmetric rylenes from a 1,6-functionalized perylene tetraester without the need for chiral resolution or other separation chromatography.41 Using the 1,6-functionalized perylene tetraester as an entry point, we prepared bay-annulated permanently dipolar discotic PDACs with various electron-rich and electron-poor benzaldehyde precursors. By increasing the electron-donating character of the p-functionalized benzaldehydes, we systematically increase the magnitude of this permanent dipole (Fig. 1A). We studied the supramolecular polymerization of these PDACs through solvent-, concentration-, and temperature-dependent aggregation studies and characterized how assembly influenced their optoelectronic properties and excited-state lifetimes. We found that the molecules with larger µg had red-shifted absorption and emission wavelengths and increasingly negative reduction potentials. Additionally, we found that increased dipoles promoted supramolecular assembly and enhanced emission lifetimes in the assembled state, whereas smaller dipoles had little to no effect on the excited state lifetime in the assembled state. Collectively, these results establish permanent dipole installation as a mechanism to manipulate the optoelectronic behavior of discotic molecules and their assemblies.
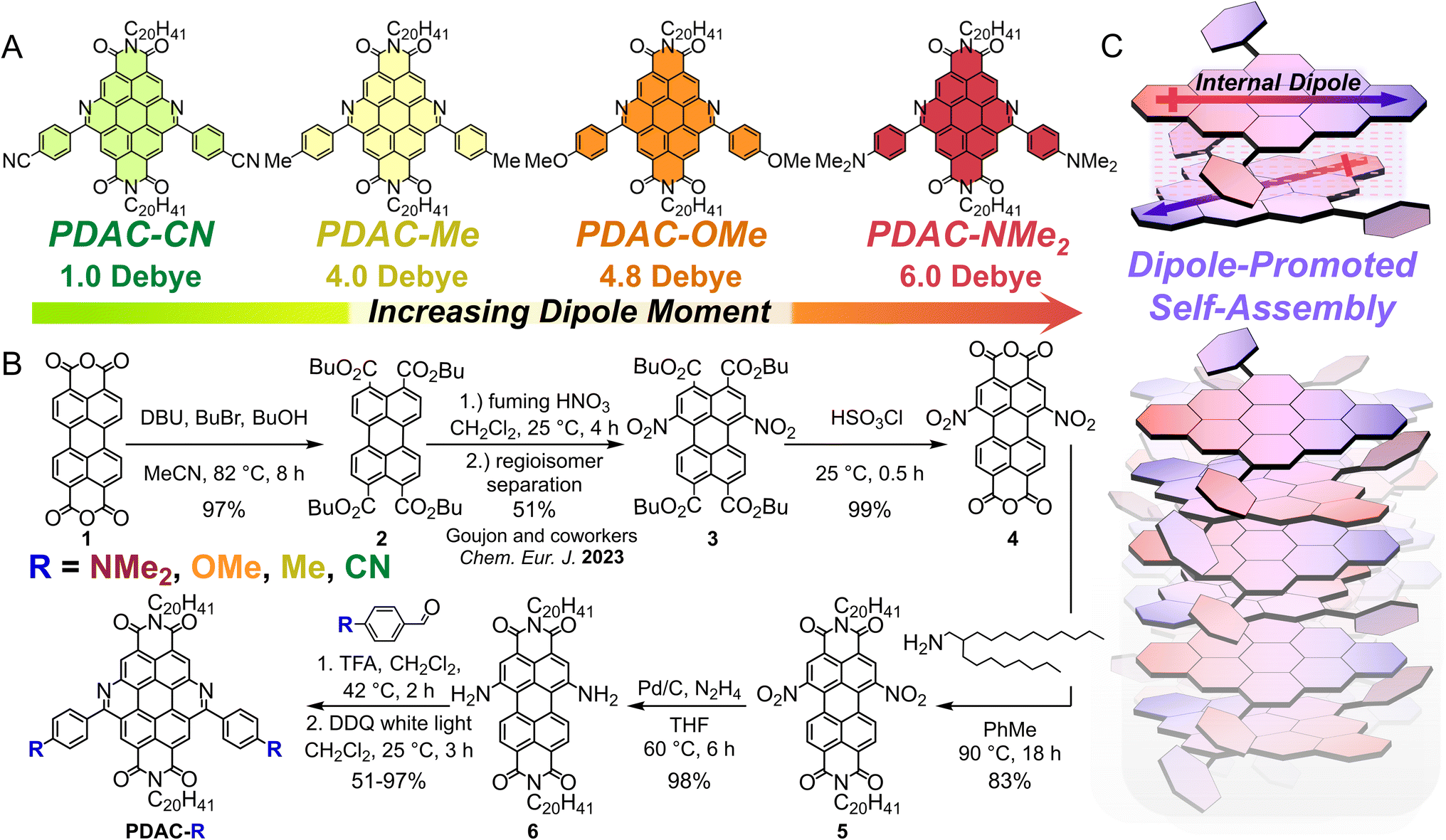 | ||
Fig. 1 Synthesis and assembly of PDACs with tunable µg. (A) Series of increasing internal dipole moments in PDAC molecules: PDAC-CN  , PDAC-Me , PDAC-Me  , PDAC-OMe , PDAC-OMe  , PDAC-NMe2 , PDAC-NMe2  . Dipoles were calculated using Multiwfn program (see Section E in ESI†). (B) Synthetic approach to prepare annullatively extended PDACs with various permanent dipoles. (C) Cartoon depiction of PDAC supramolecular assembly. . Dipoles were calculated using Multiwfn program (see Section E in ESI†). (B) Synthetic approach to prepare annullatively extended PDACs with various permanent dipoles. (C) Cartoon depiction of PDAC supramolecular assembly. | ||
Results and discussion
Functionalizing PDACs with increasingly electron-rich groups gives increasingly prominent ground-state dipoles. Density functional theory (DFT) calculations (see Section E in the ESI†) were first used to screen 5,13-p-functionalized diphenyl diazacoronene diimides (Fig. S26†) with –Me, –tBu, –CN, –CF3, –NO2, –OMe, –CN, and –NMe2 functional groups. The results revealed that electron-donating groups (e.g., –NMe2) resulted in large ground-state dipoles (6.0 Debye) and electron-withdrawing groups (e.g., –CF3) resulted in negligible ground-state dipoles (0.7 Debye) (Fig. S26†), consistent with the associated Hammett constant (σ) of each of these functional groups. The µg of these PDACs span the range available in highly polar bonds, such as S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in dimethylsulfoxide (µg = 3.9 Debye),42 to highly non-polar bonds, such as C–H in methane (µg = 0.3 Debye).43 From this selection of PDACs, we synthesized the –Me, –OMe, –CN, and –NMe2 PDAC derivatives (Fig. 1), which gave ground-state dipoles between 1 and 6 Debye.
O bond in dimethylsulfoxide (µg = 3.9 Debye),42 to highly non-polar bonds, such as C–H in methane (µg = 0.3 Debye).43 From this selection of PDACs, we synthesized the –Me, –OMe, –CN, and –NMe2 PDAC derivatives (Fig. 1), which gave ground-state dipoles between 1 and 6 Debye.
We synthesized the selected PDACs in an 8-step synthesis through a conserved asymmetric intermediate (see ESI† for more details, Fig. 1B).41 We began by esterifying the perylene dianhydride to the tetrabutylester through the addition of butanol and bromobutane in the presence of 1,8-diazabicycloundecene. Electrophilic aromatic substitution with fuming nitric acid installed nitro groups on the bay positions of the perylene tetrabutylester. The 1,6-nitro-functionalized perylene tetrabutyl ester was then regioselectively recrystallized from acetonitrile in large quantities (>4.5 g). The dianhydride was reformed from this tetrabutylester by dehydration with chlorosulfonic acid. A dipolar diimide was then prepared by reacting the 1,6-NO2 functionalized perylene dianhydride with 2-octyldodecylamine at 90 °C for 18 hours. Reduction of the nitro groups to amines was accomplished by hydrogenating with hydrazine over palladium on carbon. This yielded a 1,6-diamino perylene diimide (compound 6) that was subsequently annulated to produce permanently dipolar PDACs.
Performing a one-pot imine condensation followed by Mallory photocyclization with compound 6 and various p-functionalized benzaldehydes yielded a library of dipolar PDACs (Schemes S1–4†).44,45 We added trifluoroacetic acid (5 vol%) to compound 6 and p-functionalized benzaldehyde solution, which resulted in an immediate color change from blue to purple. This reaction was then subjected to white light irradiation (500 W) in a series of borosilicate NMR tubes, which was important to resolve challenges associated with optical penetration depth in these deeply colored solutions. After an hour of irradiation, 5.0 equivalents of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) was added, which led to an instantaneous color change that was dependent on the identity of the p-functionalized benzaldehyde. We found that more electron-donating substituents on the benzaldehyde led to higher Mallory photocyclization yields (e.g. 95% for PDAC-OMe), but electron-withdrawing substituents often suffered from lower yields (<10% for PDAC-CN), which were somewhat improved by increasing the reaction concentration by 10× (51% for PDAC-CN). This observation is expected based on the slow rate of Mallory photocyclization with electron-withdrawing substituents.46 Ultimately, this combined imine condensation and Mallory photocyclization produced four dipolar PDACs at sufficient scale for continued investigation.
Absorption and emission profiles reveal that installed permanent ground-state dipoles increase the HOMO energy of PDAC molecules. The optical absorption profiles of the monomeric PDACs were taken in chloroform at dilute concentrations to ensure that these molecules were unassembled (Fig. S12 and 13†). The monomeric absorption and emission spectra of PDAC-CN, PDAC-Me, and PDAC-OMe collected in dilute chloroform solutions are qualitatively similar to one another, with defined features, high-energy absorptions (∼350 nm) likely arising from the flanking phenyl units (Fig. 2A). However, as the µg strength is increased from 1 Debye (PDAC-CN) to 4.8 Debye (PDAC-OMe), the lowest energy absorption is red-shifted from a λmax, absorption of 477 nm to 498 nm (Fig. 2A and S1–3†). This red-shifted absorption is also accompanied by bathochromic photoluminescence shifts with increasing dipole strengths. Here, we observe the λmax, emission for PDAC-CN, PDAC-Me, and PDAC-OMe red-shifting to 2.57, 2.49, and 2.33 eV, respectively with corresponding quantum yields (ΦPL) of approximately 10 to 20% (Fig. 2B, S1–3 and Table S1†). DFT calculations (at the M06-2X/6-31G(d,p) level, see Section E in the ESI†) reveal that this shift arises from more electron-donating substituents lowering the excitation energy (S0 → S1) from 2.81 eV with PDAC-CN to 2.65 eV with PDAC-OMe (Fig. 2D and Table S4†). This experimental spectroscopic observation is consistent with the DFT-calculated HOMO–LUMO structures, which show that the HOMO and LUMO are colocalized on the diimide core (Fig. 2D and S25†). Within the optical absorption and emission profiles for PDAC-NMe2, we find two major spectral features: a high-energy λmax, absorption = 456 nm and λmax, emission = 505 nm pair similar to those observed in the other PDAC derivatives, and an additional broadened, low energy absorption/emission pair at λmax, absorption = 557 nm and λmax, emission = 723 nm (Fig. 2 and S4†). The origin of the broad, low-energy feature likely stems from an intramolecular charge-transfer state. In addition to this bathochromic shift and spectral broadening, we also observed a greatly reduced photoluminescence quantum yield (ΦPL = 1%, Fig. S6 and Table S1†). These spectroscopic observations are consistent with DFT calculations, which reveal that the HOMO and LUMO are on distinctive separate areas of the PDAC-NMe2 molecule and give the smallest HOMO–LUMO gap of the PDAC derivatives (4.04 eV, Table S4†). Together, these data lead us to conclude that PDAC-NMe2 has a significant ground-state donor–acceptor character, which is a manifestation of its substantial µg.
 | ||
| Fig. 2 Optoelectronic properties of dipolar PDACs. Monomeric PDACs were analyzed using (A) UV-Vis spectroscopy, (B) photoluminescence spectroscopy, (C) and cyclic voltammetry. UV-Vis and PL were measured at 10 µM in chloroform. PL experiments were performed with a 405 nm excitation. Cyclic voltammograms were measured at 0.5 mM in CH2Cl2 with 0.1 M nBu4NPF6 supporting electrolyte. (D) Computational characterization of the excitation energy between the ground state (S0) and excited singlet state (S1) of PDACs with the spatial electron density maps of model PDAC species that were extracted by density functional theory calculations at the M06-2X/6-31G(d,p) level (see ESI Section E† for more details). | ||
Our spectroscopic experiments were corroborated by electrochemical measurements in the monomer state that reveal PDACs functionalized with electron-donating units are less easily reduced than those functionalized with electron-withdrawing units (Fig. S7†). Conceptually, adding exogenous electron density to a less electron-rich substrate should be more thermodynamically favored.47 Cyclic voltammetry confirmed this expectation for the PDAC-CN, PDAC-Me, and PDAC-OMe molecules, which have reversible E1/2 = −0.96 V, −1.21 V, and −1.52 V vs. −0.27 V (Fc/Fc+), respectively (Fig. 2C and S9–11†). The least negative reduction potential for PDAC-CN is consistent with our understanding that adding electrons to an electron-deficient PDAC-CN should be facile. These trends in reduction potential are consistent with DFT calculations that demonstrate adding electron density to the PDAC diimide core lowers the LUMO energy (Fig. S25†). Similar to our optical spectroscopy experiments, PDAC-NMe2 is distinct from the other three PDAC derivatives. Reduction of this compound is electrochemically irreversible but chemically reversible, which we attribute to the reorganization induced by reduction in donor–acceptor complexes (Fig. 2C and S9†). These observations further substantiate that PDAC-NMe2 has a significant ground-state donor–acceptor character. In all PDAC systems, cyclic voltammetry shows that there are more than two electrochemically achievable reductions (Fig. 2C and S8–11†). This hints at the exciting possibility that assemblies composed of these materials might be desirable in electrochemical energy storage where material integrity and high gravimetric capacities are prized. This understanding motivated our exploration into µg promoted assembly with engineerable PDACs.
Supramolecular polymerization of each non-covalent PDAC aggregate was experimentally probed by variable-solvent, variable-concentration, and variable-temperature UV-Vis spectroscopy (Fig. 3A–D and S12–22†). Supramolecular assembly was driven by increased concentration, decreased temperature, and decreased solvent dipole, which collectively demonstrates that PDAC assemblies are thermodynamically favorable, entropically disfavored, and dipole promoted. In each case, differential absorption plots reveal that assembly suppresses monomeric absorption features (Fig. S1–4†) (e.g., PDAC-OMe, 333 K, λmax = 498 nm) and leads to the formation of red-shifted optical absorptions that we attribute to assembled aggregates (e.g., PDAC-OMe, 298 K, λmax = 523 nm) (Fig. S14–17†). Importantly, we observe well-defined isosbestic points in each variable-temperature assembly experiment, which indicates that the monomeric structures are forming defined aggregates as the temperature decreases. We attribute these red-shifted absorptions to more delocalized electronic environments associated with supramolecular polymerization. For all predicted dimers, we find an expected bathochromic shift for assembled PDAC spectra, and a decreased molar absorptivity compared to the simulated monomer spectra (Fig. S28–31†). These computational findings align well with our experimental spectroscopic experiments, illustrating that the PDACs co-facially assemble. We examined variable-solvent UV-Vis of a model compound (µg = 0) with identical branching side chains (Fig. S23†). We observed a hypsochromic shift with increasing non-polar conditions whereas the opposite trend was observed with the PDAC molecules. This can be attributed to a combination of two factors: increased solubility in hexanes and lack of steric bulk from flanking side groups. Importantly, it illustrates that the large alkyl chains are not the cause for the bathochromic shifts observed in PDAC aggregation. PDAC-CN and PDAC-Me both have complex assembly profiles with both hypsochromic and bathochromic shifts in assemblies (Fig. 3A, B, S16 and 17†). Plotting the temperature against the degree of aggregation for PDAC-CN reveals an isodesmic supramolecular assembly mechanism (Fig. S16 and Table S3, see Section D in the ESI†). All assemblies observed are not entirely consistent with either H- or J-aggregation, which we expect is likely due to the presence of phenyl flanking units that suppress perfectly aligned dipolar assemblies (Fig. S23†).48 We hypothesize that the limited µg of these PDAC-CN and PDAC-Me leads to unorganized aggregates rather than defined assemblies. To investigate the assembly of variably dipolar PDACs further, we performed molecular dynamics simulations to interrogate PDAC assembly. For PDAC-CN and PDAC-Me, we find that π–π stacked dimers are less likely to form than for PDAC-OMe or PDAC-NMe2. Specifically, we find that dimers with large π-surface separation (>7 Å) were found in PDACs with more limited dipoles (Fig. S42, 43 and Table S5†).
 | ||
Fig. 3 Variable-temperature PDAC assembly. Differential absorption assembly plots for (A) PDAC-CN (B) PDAC-Me (C) PDAC-OMe (D) PDAC-NMe2 PDACs were measured in conditions found to promote assembly (0.25 mM 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 vol hexanes:chloroform, 2 mM hexanes, 0.5 mM 95:5 vol hexanes:chloroform, 0.5 mM 92:8 vol hexanes:chloroform, respectively). The solutions were gradually heated from 298 K to dissipate the assemblies and return the PDACs to their monomeric states. In the plots, the monomer absorption spectrum (333 K) was subtracted from all of the other spectra to visualize the optical differences between the monomer (mon) and aggregated (agg) state through the course of the experiment (for more absorption spectra see ESI†). (E) Model of PDAC-OMe calculated head-to-tail dimer and (F) model of PDAC-NMe2 calculated rotational offset dimer found using molecular dynamic simulations in hexanes (see Section E in the ESI† for more details). :15 vol hexanes:chloroform, 2 mM hexanes, 0.5 mM 95:5 vol hexanes:chloroform, 0.5 mM 92:8 vol hexanes:chloroform, respectively). The solutions were gradually heated from 298 K to dissipate the assemblies and return the PDACs to their monomeric states. In the plots, the monomer absorption spectrum (333 K) was subtracted from all of the other spectra to visualize the optical differences between the monomer (mon) and aggregated (agg) state through the course of the experiment (for more absorption spectra see ESI†). (E) Model of PDAC-OMe calculated head-to-tail dimer and (F) model of PDAC-NMe2 calculated rotational offset dimer found using molecular dynamic simulations in hexanes (see Section E in the ESI† for more details). | ||
In contrast, PDAC-OMe and PDAC-NMe2 have more defined assemblies as a consequence of their larger µg. PDAC-OMe appears to undergo a two-stage assembly in both the variable-temperature and variable-concentration UV-Vis spectroscopy experiments (Fig. 3C and S20†). We hypothesize that this observation may be due to multiple types of stacking motifs emerging as they undergo assembly. Simulated molecular dynamics corroborate this observation as PDAC-OMe formed three different types of assemblies including head-to-head, head-to-tail, and rotationally offset dimers (Fig. 3E and S38–41†) with small (<3.6 Å) separations (Table S5†). Additionally, dipole vector calculations reveal that PDAC-OMe has an offset dipole, which could be contributing to the complex assembly observed (Fig. S26†). In contrast, the more dipolar PDAC-NMe2 forms defined rotationally offset assembly in molecular dynamics simulations (Fig. 3F, S36, 37 and Table S5†) with a pronounced, red-shifted absorption experimentally observed at 650–750 nm. The bathochromic shift of the charge-transfer feature implies that the charge-transfer state in the PDAC-NMe2 assemblies is distinct from the one that exists in the isolated PDAC-NMe2, which we attribute to electronic interactions between neighboring PDAC-NMe2 units (Fig. S11 and S13†). The strength of this interaction in this structure is evidenced by the observation that the highest temperature (40 °C) is needed to provoke disassembly of the PDAC-NMe2. When plotting the temperature-dependent UV-Vis spectra as degree of aggregation, the data fit the nucleation-elongation model associated with cooperative assembly (Fig. S14 and Table S2†). The change in supramolecular assembly from isodesmic for PDAC-CN (1.0 Debye) to cooperative for PDAC-NMe2 (6.0 Debye) follows the trend previously reported in investigations of PDI molecules with permanent ground state dipoles installed through side chains.29 Collectively, these observations indicate that the strong dipolar coupling in PDAC-NMe2 drives well-defined assembly into rotationally offset arrays of these planar molecules.
Larger µg enhance emission lifetimes in the assembled state while smaller dipoles have little to no effect on the excited state lifetime in the assembled state. To investigate the effect of permanent dipoles on PDACs, we measured photoluminescence emission decays of the monomeric PDACs and their assemblies (Fig. 4 and S24†). Briefly, by exciting the solutions with a 405 nm pulsed laser source at 1 MHz and utilizing a 425 nm long-pass filter, the isolated emission for the PDAC monomers and assemblies could be collected using a single photon avalanche diode. Consistent with the expectations of adding electron-rich units to aromatic chromophores, the monomeric lifetimes (τm) for PDAC-NMe2 (τm = 1.8 ns) and PDAC-OMe (τm = 3.6 ns) are significantly shorter than PDAC-Me (τm = 5.9 ns) and PDAC-CN (τm = 9.5 ns) (Fig. 4A and Table S1†). We also investigated the PDAC assembly lifetimes (τa) by placing the PDACs into solvent conditions in which they would partially assemble (50:50 vol hexanes:chloroform) and fully assemble (95:5 vol hexanes:chloroform) (Fig. 4B). We find that the assembly of more dipolar compounds leads to longer lifetimes with an increase from 3.6 ns to 5.0 ns for PDAC-OMe and an increase from 1.8 ns to 5.1 ns for PDAC-NMe2. In contrast, less polar PDAC-Me had a smaller relative change to their monomer-to-assembled excited state lifetimes of 5.9 ns to 6.6 ns while PDAC-CN had a shorter lifetime 9.5 ns to 7.8 ns (Table S1†). We suspect the defined dipolar assemblies of PDAC-OMe and PDAC-NMe2 result in the larger lifetimes in the assembled state. Fully describing how dipolar assembly influences excited-state lifetimes and how these impacts can be systematically engineered will require more intensive investigation. Collectively, these observations demonstrate that engineering permanent dipoles into planar chromophores offers a powerful strategy to tune ground- and excited-state optoelectronic character in molecular assemblies.
 | ||
| Fig. 4 Time-resolved photoluminescence spectroscopy of PDAC molecules and assemblies. (A) Fluorescence decays for monomeric PDACs in 0.01 mM solutions in chloroform. (B) Fluorescence decays for the PDAC compounds in decreasing solvent polarity (monomer: chloroform (pink); partially assembled: 50:50 vol hexanes:chloroform (yellow), and assembled: 95:5 vol hexanes:chloroform (blue)). All samples were recorded at 0.01 mM concentration with a 405 nm excitation pulse at a repetition rate of 1 MHz. | ||
Conclusion
We explored the impact of ground-state dipoles (µg) on the optoelectronic properties and supramolecular assemblies of permanently dipolar diazacoronenes. We found that increasing the permanent µg caused a significant bathochromic shift in absorbance and emissions, as well as a decrease in reduction potentials. This corresponded to the computationally determined decrease in HOMO–LUMO gap for increasingly dipolar PDACs. We found that PDACs with smaller µg had complex assembly profiles that arise from the interplay of non-covalent forces inherent to these mesogens. In contrast, stronger dipoles directed the formation of more defined dipolar assemblies. Additionally, we found that molecules with permanent dipoles had longer lifetimes in their assembled states, whereas molecules with smaller dipoles had minimal improvement between the monomer and assembled states. Overall, these findings show that permanent dipole moments can be used to guide supramolecular assembly. We expect that engineering the µg of discotic molecules will be a useful design consideration to achieve tailored molecular assemblies and their eventual deployment in devices.Data availability
The data supporting this article have been included as part of the ESI.† Primary data files are available on request.Author contributions
A. N. D. conceived the experiments, synthesized, and characterized monomer and assembly states of the PDAC molecules. C. M. S. characterized the fluorescence, quantum yields and lifetimes of the PDAC molecules. C. F. performed DFT calculations on PDACs and molecular dynamic simulations on PDAC assemblies. R. R. assisted with synthesis and optical characterization. A. M. M. H. assisted with electrochemical characterization. K. S. assisted with mass spectrometry of PDACs. A. N. D. and A. M. E. wrote the initial drafts of the manuscripts. H. L., L. N., and A. M. E. contributed to experimental design and supervised this work. All authors contributed to the writing of this manuscript.Conflicts of interest
The authors have no competing financial interests.Acknowledgements
We greatly thank the University of Florida for financially supporting this work. High-resolution mass spectrometry was performed at the Mass Spectrometry Research and Education Center at the Department of Chemistry at the University of Florida (NIH S10 OD021758-01A1 and NIH S10 OD030250-01A1). L. N. and C. M. S. acknowledge funding by the National Science Foundation under Grant No. DMR-2237977, the Camille and Henry Dreyfus Foundation (TC-23-050), the Alfred P. Sloan Foundation, and the Materials Characterization Laboratory (FSU075000MAC) at the FSU Department of Chemistry and Biochemistry. The computational work was supported by the Shanghai Technical Service Center of Science and Engineering Computing at Shanghai University. We also thank Prof. Antoine Goujon for guidance on the synthesis of the PDAC precursors.References
- M. Li, M. Rogatch, H. Chen, X. Guo and J. Tang, Supramolecular Design and Assembly Engineering toward High-Performance Organic Field-Effect Transistors, Acc. Mater. Res., 2024, 5, 505–517 CrossRef CAS.
- Y. Xiao, D. Zeng, L. M. Mazur, A. Castiglione, E. Lacaze, B. Heinrich, B. Donnio, D. Kreher, A.-J. Attias, J.-C. Ribierre and F. Mathevet, A new class of nanostructured supramolecular organic semiconductors based on intertwined multi-lamellar co-assemblies in π-conjugated liquid-crystalline side-chain polymers, Polym. J., 2017, 49, 31–39 CrossRef CAS.
- T. A. Barendt, M. L. Ball, Q. Xu, B. Zhang, B. Fowler, A. Schattman, V. C. Ritter, M. L. Steigerwald and C. Nuckolls, Supramolecular Assemblies for Electronic Materials, Chem.–Eur. J., 2020, 26, 3744–3748 CrossRef CAS PubMed.
- Y. Yao, Y. Chen, H. Wang and P. Samorì, Organic photodetectors based on supramolecular nanostructures, SmartMat, 2020, 1, e1009 CrossRef.
- S. B. Anantharaman, K. Strassel, M. Diethelm, A. Gubicza, E. Hack, R. Hany, F. A. Nüesch and J. Heier, Exploiting supramolecular assemblies for filterless ultra-narrowband organic photodetectors with inkjet fabrication capability, J. Mater. Chem. C, 2019, 7, 14639–14650 RSC.
- Y. Yao, Q. Ou, K. Wang, H. Peng, F. Fang, Y. Shi, Y. Wang, D. I. Asperilla, Z. Shuai and P. Samorì, Supramolecular engineering of charge transfer in wide bandgap organic semiconductors with enhanced visible-to-NIR photoresponse, Nat. Commun., 2021, 12, 3667 CrossRef CAS PubMed.
- C. Liu, Z. Cheng, X. Xu, N. Wang, X. Luo and X. Xin, Fluorescent Nanocomposite Prepared through Supramolecular Self-Assembly of a Tetraphenylethene Derivative and Polyoxometalate for Light-Emitting Diodes, ACS Appl. Nano Mater., 2024, 7, 11455–11464 CrossRef CAS.
- V. S. Sharma, A. S. Sharma, N. K. Agarwal, P. A. Shah and P. S. Shrivastav, Self-assembled blue-light emitting materials for their liquid crystalline and OLED applications: from a simple molecular design to supramolecular materials, Mol. Syst. Des. Eng., 2020, 5, 1691–1705 RSC.
- A. Liang, S. Dong, X. Zhu, F. Huang and Y. Cao, White light-emitting diodes based on an all-phosphorescent supramolecular polymer, Polym. Chem., 2015, 6, 6202–6207 RSC.
- C. Kulkarni, S. Balasubramanian and S. J. George, What Molecular Features Govern the Mechanism of Supramolecular Polymerization?, ChemPhysChem, 2013, 14, 661–673 CrossRef CAS PubMed.
- B. J. Eckstein, L. C. Brown, B. C. Noll, M. P. Moghadasnia, G. J. Balaich and C. M. McGuirk, A Porous Chalcogen-Bonded Organic Framework, J. Am. Chem. Soc., 2021, 143, 20207–20215 CrossRef CAS PubMed.
- B. J. Eckstein, H. R. Martin, M. P. Moghadasnia, A. Halder, M. J. Melville, T. N. Buzinski, G. J. Balaich and C. M. McGuirk, Influence of donor point modifications on the assembly of chalcogen-bonded organic frameworks, Chem. Commun., 2024, 60, 758–761 RSC.
- D. J. Pascoe, K. B. Ling and S. L. Cockroft, The Origin of Chalcogen-Bonding Interactions, J. Am. Chem. Soc., 2017, 139, 15160–15167 CrossRef CAS PubMed.
- A. Arjona-Esteban, J. Krumrain, A. Liess, M. Stolte, L. Huang, D. Schmidt, V. Stepanenko, M. Gsänger, D. Hertel, K. Meerholz and F. Würthner, Influence of Solid-State Packing of Dipolar Merocyanine Dyes on Transistor and Solar Cell Performances, J. Am. Chem. Soc., 2015, 137, 13524–13534 CrossRef CAS PubMed.
- F. Würthner, Dipole–Dipole Interaction Driven Self-Assembly of Merocyanine Dyes: From Dimers to Nanoscale Objects and Supramolecular Materials, Acc. Chem. Res., 2016, 49, 868–876 CrossRef PubMed.
- A. Liess, A. Lv, A. Arjona-Esteban, D. Bialas, A.-M. Krause, V. Stepanenko, M. Stolte and F. Würthner, Exciton Coupling of Merocyanine Dyes from H- to J-type in the Solid State by Crystal Engineering, Nano Lett., 2017, 17, 1719–1726 CrossRef CAS PubMed.
- A. Lv, M. Stolte and F. Würthner, Head-to-Tail Zig-Zag Packing of Dipolar Merocyanine Dyes Affords High-Performance Organic Thin-Film Transistors, Angew. Chem., Int. Ed., 2015, 54, 10512–10515 CrossRef CAS PubMed.
- N. Kim, J. S. Kang, T. Jun, J.-M. Suh, D.-H. Roh, W.-W. Park, O.-H. Kwon, T.-H. Kwon, M. H. Lim, D. Y. Ryu, M. Seo and B.-S. Kim, Tailoring Dynamic Chiral Supramolecular Assembly with Phototriggered Radical Anions of C3-Symmetric Triphenylene Triimides, Macromolecules, 2024, 57, 21–31 CrossRef.
- H. Zhang, J. Cheng, Q. Zhou, Q. Zhang and G. Zou, Impact of a chiral supramolecular nanostructure on the mechanical and electrical performances of triphenylene-based discotic physical gels, Soft Matter, 2020, 16, 5203–5209 RSC.
- I. Muñoz Resta, V. E. Manzano, F. Cecchi, C. C. Spagnuolo, F. D. Cukiernik and P. H. Di Chenna, Supramolecular Assembly of pH-Sensitive Triphenylene Derived π-Gelators and Their Application as Molecular Template for the Preparation of Silica Nanotubes, Gels, 2016, 2, 7 CrossRef PubMed.
- J. Yu, Y. Chen, Y.-H. Zhang, X. Xu and Y. Liu, Supramolecular Assembly of Coronene Derivatives for Drug Delivery, Org. Lett., 2016, 18, 4542–4545 CrossRef CAS PubMed.
- J. P. Hill, W. Jin, A. Kosaka, T. Fukushima, H. Ichihara, T. Shimomura, K. Ito, T. Hashizume, N. Ishii and T. Aida, Self-Assembled Hexa-peri-hexabenzocoronene Graphitic Nanotube, Science, 2004, 304, 1481–1483 CrossRef CAS PubMed.
- W. Jin, Y. Yamamoto, T. Fukushima, N. Ishii, J. Kim, K. Kato, M. Takata and T. Aida, Systematic Studies on Structural Parameters for Nanotubular Assembly of Hexa-peri-hexabenzocoronenes, J. Am. Chem. Soc., 2008, 130, 9434–9440 CrossRef CAS PubMed.
- E. Weyandt, L. Leanza, R. Capelli, G. M. Pavan, G. Vantomme and E. W. Meijer, Controlling the length of porphyrin supramolecular polymers via coupled equilibria and dilution-induced supramolecular polymerization, Nat. Commun., 2022, 13, 248 CrossRef CAS PubMed.
- H. Lee, H. Park, D. Y. Ryu and W.-D. Jang, Porphyrin-based supramolecular polymers, Chem. Soc. Rev., 2023, 52, 1947–1974 RSC.
- J. Gershberg, F. Fennel, T. H. Rehm, S. Lochbrunner and F. Würthner, Anti-cooperative supramolecular polymerization: a new K(2)-K model applied to the self-assembly of perylene bisimide dye proceeding via well-defined hydrogen-bonded dimers, Chem. Sci., 2016, 7, 1729–1737 RSC.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Perylene Bisimide Dye Assemblies as Archetype Functional Supramolecular Materials, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- A. S. Weingarten, A. J. Dannenhoffer, R. V. Kazantsev, H. Sai, D. Huang and S. I. Stupp, Chromophore Dipole Directs Morphology and Photocatalytic Hydrogen Generation, J. Am. Chem. Soc., 2018, 140, 4965–4968 CrossRef CAS PubMed.
- C. Kulkarni, K. K. Bejagam, S. P. Senanayak, K. S. Narayan, S. Balasubramanian and S. J. George, Dipole-Moment-Driven Cooperative Supramolecular Polymerization, J. Am. Chem. Soc., 2015, 137, 3924–3932 CrossRef CAS PubMed.
- A. M. M. Hasan, R. Roy, M. K. Shehab, A. N. Davis, K. Slicker, K. O. Kirlikovali, R. M. Pankow, O. K. Farha and A. M. Evans, Rapid Cathodic Coloration in Solution-Processable Electrochromic Polymers of Intrinsic Microporosity, J. Am. Chem. Soc., 2025, 147, 16331–16339 CrossRef CAS PubMed.
- A. N. Davis, K. Parui, A. M. M. Hasan, L. A. Pineda, J. D. Langhout, K. A. Treaster, M. M. Butala and A. M. Evans, Cross-linking organic cathodes enhances stability at the expense of ionic accessibility, J. Mater. Chem. A, 2024, 12, 28874–28881 RSC.
- A. K. Oanta, K. A. Collins, A. M. Evans, S. M. Pratik, L. A. Hall, M. J. Strauss, S. R. Marder, D. M. D'Alessandro, T. Rajh, D. E. Freedman, H. Li, J.-L. Brédas, L. Sun and W. R. Dichtel, Electronic Spin Qubit Candidates Arrayed within Layered Two-Dimensional Polymers, J. Am. Chem. Soc., 2023, 145, 689–696 CrossRef CAS PubMed.
- Z. Jin, Q. Cheng, S. T. Bao, R. Zhang, A. M. Evans, F. Ng, Y. Xu, M. L. Steigerwald, A. E. McDermott, Y. Yang and C. Nuckolls, Iterative Synthesis of Contorted Macromolecular Ladders for Fast-Charging and Long-Life Lithium Batteries, J. Am. Chem. Soc., 2022, 144, 13973–13980 CrossRef CAS PubMed.
- S. Jhulki, C. H. Feriante, R. Mysyk, A. M. Evans, A. Magasinski, A. S. Raman, K. Turcheniuk, S. Barlow, W. R. Dichtel, G. Yushin and S. R. Marder, A Naphthalene Diimide Covalent Organic Framework: Comparison of Cathode Performance in Lithium-Ion Batteries with Amorphous Cross-linked and Linear Analogues, and Its Use in Aqueous Lithium-Ion Batteries, ACS Appl. Energy Mater., 2021, 4, 350–356 CrossRef CAS.
- A. M. Evans, K. A. Collins, S. Xun, T. G. Allen, S. Jhulki, I. Castano, H. L. Smith, M. J. Strauss, A. K. Oanta, L. Liu, L. Sun, O. G. Reid, G. Sini, D. Puggioni, J. M. Rondinelli, T. Rajh, N. C. Gianneschi, A. Kahn, D. E. Freedman, H. Li, S. Barlow, G. Rumbles, J.-L. Brédas, S. R. Marder and W. R. Dichtel, Controlled n-Doping of Naphthalene-Diimide-Based 2D Polymers, Adv. Mater., 2022, 34, 2101932 CrossRef CAS PubMed.
- L. Rocard, A. Goujon and P. Hudhomme, Nitro-Perylenediimide: An Emerging Building Block for the Synthesis of Functional Organic Materials, Molecules, 2020, 25, 1402 CrossRef CAS PubMed.
- K. Zha, L. Li, J. Zhang, S. Tang, X. Li, J. Hai, D. Fan, M. Li, Y. Liu and Z. Lu, Investigation the influence of bay substitution with perylene diimide on the photocatalytic performance of perylene–diimide/TiO2 composites, J. Photochem. Photobiol., A, 2024, 451, 115517 CrossRef CAS.
- K. Balakrishnan, A. Datar, T. Naddo, J. Huang, R. Oitker, M. Yen, J. Zhao and L. Zang, Effect of Side-Chain Substituents on Self-Assembly of Perylene Diimide Molecules: Morphology Control, J. Am. Chem. Soc., 2006, 128, 7390–7398 CrossRef CAS PubMed.
- M. A. Martínez, A. Doncel-Giménez, J. Cerdá, J. Calbo, R. Rodríguez, J. Aragó, J. Crassous, E. Ortí and L. Sánchez, Distance Matters: Biasing Mechanism, Transfer of Asymmetry, and Stereomutation in N-Annulated Perylene Bisimide Supramolecular Polymers, J. Am. Chem. Soc., 2021, 143, 13281–13291 CrossRef PubMed.
- D. Sahoo, M. Peterca and V. Percec, Hierarchical Self-Organization and Disorganization of Helical Supramolecular Columns Mediated by H-Bonding and Shape Complementarity, J. Am. Chem. Soc., 2024, 146, 27299–27304 CrossRef CAS PubMed.
- A. Gapin, A. H. G. David, M. Allain, D. Masson, O. Alévêque, T. Ave, L. Le Bras, P. Hudhomme and A. Goujon, Synthesis of 1,6/7-(NO2)2-Perylenediimide and 1,6/7-(NH2)2-Perylenediimide as Regioisomerically Pure Materials, Chem.–Eur. J., 2023, 29, e202300652 CrossRef CAS PubMed.
- C. Di Mino, A. J. Clancy, A. Sella, C. A. Howard, T. F. Headen, A. G. Seel and N. T. Skipper, Weak Interactions in Dimethyl Sulfoxide (DMSO)–Tertiary Amide Solutions: The Versatility of DMSO as a Solvent, J. Phys. Chem. B, 2023, 127, 1357–1366 CrossRef CAS PubMed.
- P. Lazzeretti, R. Zanasi and W. T. Raynes, On the CH bond dipole moment in alkanes, J. Chem. Phys., 1987, 87, 1681–1684 CrossRef CAS.
- A. H. G. David, M. Roger, O. Alévêque, H. Melnychenko, L. Le Bras, M. Allain, A. Gapin, D. Canevet, O. Ségut, E. Levillain and A. Goujon, Intermolecular Anionic Mixed-Valence and π-Dimer Complexes of ortho-Pentannulated Bisazacoronene Diimide, Angew. Chem., Int. Ed., 2024, e202413616 Search PubMed.
- A. Gapin, E. Chatir, O. Alévêque, C. Pasgrimaud, A. H. G. David, A. De Maria, M. Legros, L. Le Bras, E. Levillain and A. Goujon, Synthesis of Electron-Deficient BisAzaCoroneneDiimide-Conjugated Polymers by Light-Locking Dynamic Covalent Bonds, J. Am. Chem. Soc., 2025, 147, 12218–12227 CrossRef CAS PubMed.
- A. Ghosh, D. Csókás, M. Budanović, R. D. Webster, I. Pápai and M. C. Stuparu, Synthesis of azahelicenes through Mallory reaction of imine precursors: corannulene substrates provide an exception to the rule in oxidative photocyclizations of diarylethenes, Chem. Sci., 2021, 12, 3977–3983 RSC.
- S. Jiao, X. Han, L.-L. Jiang, X. Du, Z. Huang, S. Li, W. Wang, M. Wang, Y. Liu and W.-L. Song, Functional Group-Driven Competing Mechanism in Electrochemical Reaction and Adsorption/Desorption Processes toward High-Capacity Aluminum-Porphyrin Batteries, Angew. Chem., Int. Ed., 2024, 63, e202410110 CrossRef CAS PubMed.
- N. J. Hestand and F. C. Spano, Expanded Theory of H- and J-Molecular Aggregates: The Effects of Vibronic Coupling and Intermolecular Charge Transfer, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc03568f |
| This journal is © The Royal Society of Chemistry 2025 |