
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rh-catalysed single-carbon insertion to 1,3-dienes†
Pau
Sarró‡
 ac,
Norman
Díaz‡
ac,
Josep
Esteve Guasch
ac,
Wei Jie
Teo
a and
Marcos G.
Suero
*ab
ac,
Norman
Díaz‡
ac,
Josep
Esteve Guasch
ac,
Wei Jie
Teo
a and
Marcos G.
Suero
*ab
aInstitute of Chemical Research of Catalonia (ICIQ-CERCA), The Barcelona Institute of Science and Technology, Av. Països Catalans 16, 43007 Tarragona, Spain. E-mail: mgsuero@iciq.es
bICREA, Pg. Lluis Companys 23, 08010 Barcelona, Spain
cDepartament de Química Analítica i Química Orgànica, Universitat Rovira i Virgili, Calle Marcel·lí Domingo, 1, Tarragona, 43007, Spain
First published on 19th June 2025
Abstract
Herein, we report the first catalytic single-carbon insertion to 1,3-dienes with Rh(II)-carbynoids. The skeletal editing process is based on the catalytic generation of a Rh-carbynoid that promotes the insertion of a cationic monovalent carbon unit (:+C–R) into the C(sp2)–C(sp2) bond of the 1,3-diene, leading to a pentadienyl cation. Regioselective attack on the latter species leads to the formation of multi-substituted 1,3-dienes or 1,4-dienes with a broad range of carbon and heteroatomic nucleophiles.
Since the discovery of the Diels–Alder reaction over a century ago,1 1,3-dienes have become one of the most important building blocks in the synthesis of complex natural products, drug molecules and polymers.2 Transition-metal and photoredox catalysis have played a central role in the discovery and development of both efficient stereoselective syntheses and chemical transformations of 1,3-dienes based on 1,2- and 1,4-difunctionalizations (Scheme 1A).3 Such processes rely on π-bond activations of the C(sp2)–C(sp2) double bonds that can occur with excellent diastereo-, regio- and enantiocontrol.4 However, catalytic processes that can functionalize 1,3-dienes through σ- and π-bond activations of the 1,3-diene C(sp2)–C(sp2) bonds are unexplored and remain limited to cross metathesis of 1,3-dienes and alkenes (Scheme 1A).5
 | ||
| Scheme 1 Peripheral and skeletal editing of 1,3-dienes. | ||
Over the recent years, the discovery and development of single-carbon insertion reactions in unsaturated6 and saturated7 systems have received enormous attention. These types of skeletal manipulations involving the insertion of a single-carbon atom are of high interest since they provide new retrosynthetic logic and disconnection approaches.8 Given the interest of our group in developing a general catalytic carbyne transfer platform, we recently questioned whether a novel catalytic cationic monovalent carbon insertion (:+C–R) of 1,3-dienes could be within reach. If successful, this previously unknown transformation would complement cross metathesis as a skeletal editing process for 1,3-dienes, while providing a novel single-carbon insertion logic.
Herein, we disclose the first single-carbon insertion of 1,3-dienes induced by a catalytically generated Rh-carbynoid (Scheme 1B). These species were responsible for the generation of a transient pentadienyl cation that underwent regioselective attack by a wide range of nucleophiles, leading to 1,3- or 1,4-dienes.
Our group is interested in developing a general catalytic carbyne transfer platform using the (photo)catalytic activation of a novel class of diazomethyl-substituted hypervalent iodine reagents.9 In 2019, we reported the first catalytic generation of Rh(II)-carbynoid species Rh![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–I(III)(E) [I(III) = I(III)(Ar)(X); E = ester]10 using dirhodium carboxylate complexes.11 We disclosed that Rh(II)-carbynoids promoted the skeletal manipulation of alkenes by inserting a cationic monovalent carbon unit (:+C–R) into the alkene C(sp2)–C(sp2) bond, resulting in the generation of allylic cations that were trapped by a broad range of nucleophiles in an inter- and intramolecular fashion. Moreover, we recently showed that such single-carbon insertion could occur with excellent regio- and enantiocontrol using chiral dirhodium catalysts.12 Experimental evidence supported the transient generation of a chiral intimate allyl cation–nucleophile pair.13
C–I(III)(E) [I(III) = I(III)(Ar)(X); E = ester]10 using dirhodium carboxylate complexes.11 We disclosed that Rh(II)-carbynoids promoted the skeletal manipulation of alkenes by inserting a cationic monovalent carbon unit (:+C–R) into the alkene C(sp2)–C(sp2) bond, resulting in the generation of allylic cations that were trapped by a broad range of nucleophiles in an inter- and intramolecular fashion. Moreover, we recently showed that such single-carbon insertion could occur with excellent regio- and enantiocontrol using chiral dirhodium catalysts.12 Experimental evidence supported the transient generation of a chiral intimate allyl cation–nucleophile pair.13
Recently, we hypothesized that single-carbon insertion into 1,3-dienes mediated by Rh-carbynoids could be of interest, considering that a pentadienyl cation may be generated. While these types of cations are known to undergo Nazarov 4π-conrotatory electrocyclizations that ultimately lead to the formation of 2-cyclopentenones,14 studies on their behavior towards nucleophilic attack remain largely unexplored.15 We envisioned that a cyclopropanation reaction between a 1,3-diene and Rh-carbynoid int-1 would generate a cyclopropyl–I(III)int-3, placing the vinyl group and hypervalent iodine moiety in a syn disposition.10 Analogous to previous results from our group, this diastereoselectivity may be explained based on int-2, where the non-reactive double bond prevents steric clashes with the ester group, as seen in int-2* (purple ball). Then, a disrotatory ring-opening would lead to a pentadienyl cation int-4 with three available electrophilic positions (α, γ, δ) (Scheme 2) that could lead to three different types of 1,3 and 1,4-dienes 3.
 | ||
| Scheme 2 Mechanistic proposal. | ||
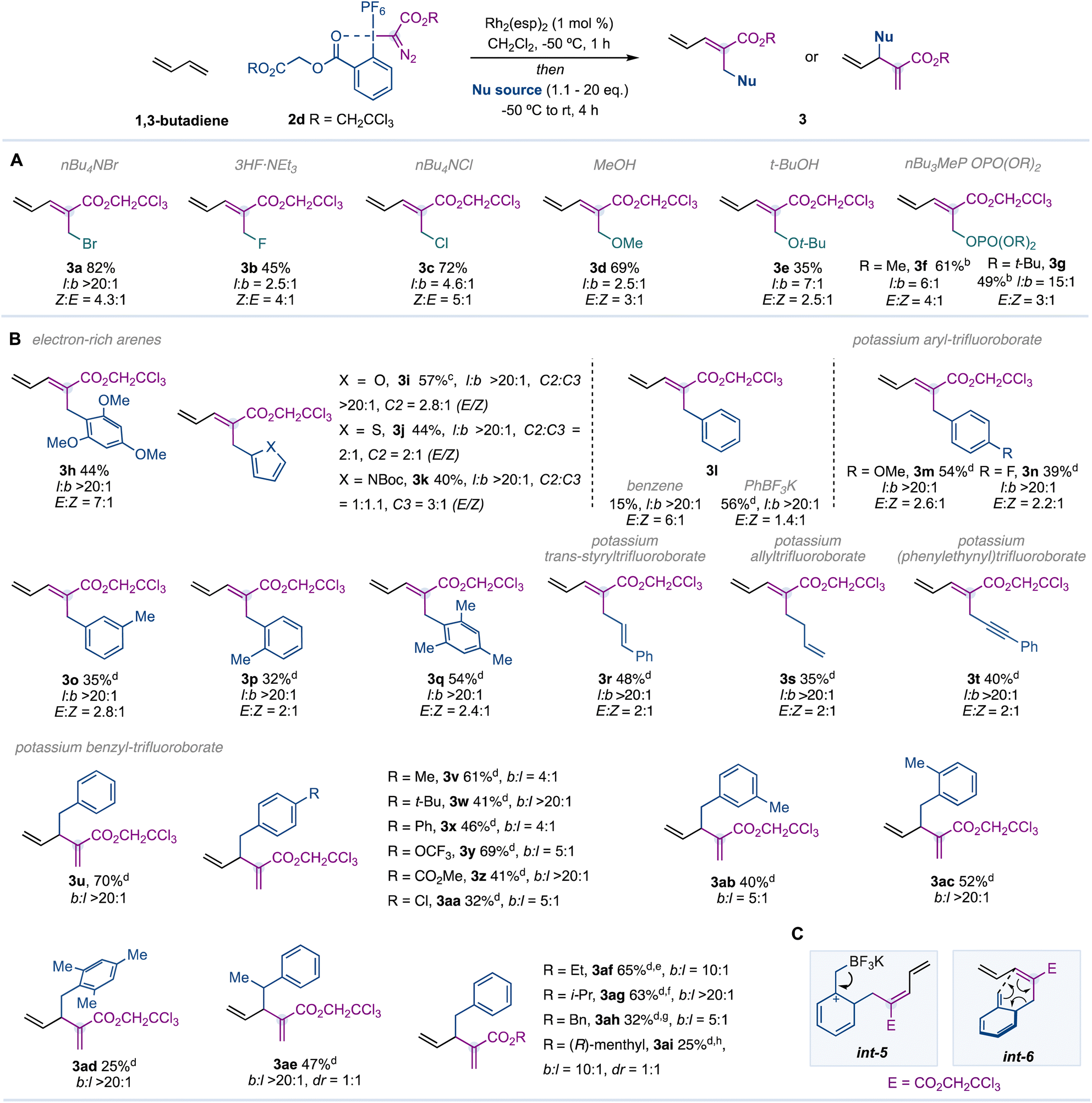
Initial experiments were carried out with 1,3-butadiene (1a, 5.0 eq.) – a feedstock chemical produced on a >10 million ton scale per year16 – hypervalent iodine reagent172a, Rh2(OAc)4 or Rh2(HFIB)4 (1.0 mol%) as catalysts, Bu4NBr (1.1 eq.) as the nucleophile and CH2Cl2 as the solvent (Table 1, entries 1,2). Unfortunately, we did not observe the formation of compound 3a. However, sterically demanding dirhodium catalysts such as Rh2(TPA)4, Rh2(Adc)4 or Rh2esp2 (Du Bois catalyst)18 provided 3a in good yields (entries 3–5, 73–80% yield) and with a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Z:E ratio. With the aim of improving the diastereoselectivity of the reaction, we explored a range of ester substituents on the hypervalent iodine reagent (2b–d; R = i-Pr, Bn, CH2CCl3) and found a superior Z:E ratio with a trichloroethyl substituent (entries 6–8). Finally, we observed that while an excess of 1,3-butadiene was necessary for the efficiency of the reaction (entry 9), a higher amount of Bu4NBr provides higher diastereoselectivity at the cost of yield (entry 10).
:1 Z:E ratio. With the aim of improving the diastereoselectivity of the reaction, we explored a range of ester substituents on the hypervalent iodine reagent (2b–d; R = i-Pr, Bn, CH2CCl3) and found a superior Z:E ratio with a trichloroethyl substituent (entries 6–8). Finally, we observed that while an excess of 1,3-butadiene was necessary for the efficiency of the reaction (entry 9), a higher amount of Bu4NBr provides higher diastereoselectivity at the cost of yield (entry 10).
|
|
||||
|---|---|---|---|---|
| Entry | 2 | Catalyst | Yield 3a–db [%] | Ratio Z:Ec |
| a Reactions were carried out with 1,3-butadiene (0.5 mmol), Rh catalyst (1.0 mol%) and reagent 2 (0.1 mmol) in CH2Cl2 (1.5 mL) at −50 °C for 60 min. Bu4NBr was added neat, and the tube was kept in the cooling bath and slowly warmed to rt over 4 h. b Yield reported on the basis of 1H-NMR analysis of the crude reaction mixture using CH2Br2 as the internal standard. c Ratio of diastereoisomers was determined using 1H-NMR analysis of the crude reaction mixture. d Isolated yield. e Using 0.1 mmol of 1,3-butadiene. f Using 0.2 mmol of Bu4NBr. esp = α, α, α′, α′-tetramethyl-1,3-benzenedipropanoate. HFIB = heptafluorobutyrate. TPA = triphenylacetate. Adc = 1-adamantylcarboxylate. | ||||
| 1 | 2a | Rh2(OAc)4 | 0 | — |
| 2 | 2a | Rh2(HFIB)4 | 0 | — |
| 3 | 2a | Rh2(TPA)4 | 73 | 3:1 |
| 4 | 2a | Rh2(Adc)4 | 74 | 3:1 |
| 5 | 2a | Rh2esp2 | 80 | 3:1 |
| 6 | 2b | Rh2esp2 | 75 | 3:1 |
| 7 | 2c | Rh2esp2 | 73 | 3:1 |
| 8 | 2d | Rh2esp2 | 76(82)d | 5:1 |
| 9 | 2d | Rh2esp2 | 48 | 5:1e |
| 10 | 2d | Rh2esp2 | 52 | 8:1f |

We next turned our attention to evaluate a range of heteroatomic nucleophiles under the optimized reaction conditions (Table 2A). We were delighted to observe that alternative halide sources (3b,c), alcohols (3d,e), or phosphates (3f,g) were well tolerated. We noticed that while diastereoselectivities were maintained (E:Z ratios), regioselectivities (linear:branched ratios) were superior for sterically demanding nucleophiles (see 3avs.3c, 3dvs.3e, 3fvs.3g). Unfortunately, amines such as morpholine, p-anisidine and dibenzylamine did not work under the optimised reaction conditions. Electron-rich (hetero)aromatic rings, such as 1,3,5-trimethoxybenzene (3h), furan (3i), thiophene (3j) and N-Boc-protected pyrrole (3k), led to the corresponding products in moderate yields with excellent regioselectivity. In contrast with such results, benzene provided 3l in poor yield. Then, we tested a range of phenyl derivatives and found that while organoboron compounds (PhBPin or PhB(OH)2), organosilicon (PhTMS) and organotin (PhSnBu3) provided poor efficiency (≤20% yield), the Molander potassium phenyltrifluoroborate19 provided 3l in 41% yield. The addition of tetrabutylammonium bisulfate (TBAHSO4) as a phase transfer agent to the reaction mixture increased the efficiency of the process (3l, 56%).20
| a Reactions were carried out with 1,3-butadiene (1.0 mmol), Rh2(esp)2 (1.0 mol%) and reagent 2d (0.2 mmol) in CH2Cl2 (3.0 mL) for 1 h at −50 °C. Nucleophile (1.1–20 mmol) was added neat, and the tube was kept in the cooling bath and slowly warmed to rt over 4 h. Yields are reported on the basis of the isolated pure product using flash column chromatography. b Nucleophile was added in CH2Cl2 (2.0 mL) dropwise over 10 min at −50 °C. c NaHCO3 (0.4 mmol) was added from the beginning. d nBu4NHSO4 (1.0 equiv.) was added together with the nucleophile at −50 °C. e Reagent 2a (0.2 mmol) was used. f Reagent 2b (0.2 mmol) was used. g Reagent 2c (0.2 mmol) was used. h Reagent 2e (0.2 mmol) was used. |
|---|
|
We then observed that alternative para-, meta- and ortho-substituted aryltrifluoroborates were well tolerated (3m–q). Vinylic, allylic and alkynyl Molander salts were also effective and provided the corresponding 1,3-dienes with excellent regioselectivity (3r–t).
In contrast with these observations, benzyl trifluoroborate salts provided 1,4-dienes from a presumable attack on the γ-position (3u–ai). It is interesting to see that substitutions on the phenyl ring (3v–ad), benzylic position (3ae) or the use of alternative reagents (3af–ai) did not prevent the attack on the γ-position (Table 2B). A reaction mechanism that could explain the preferred formation of the 1,4-diene may involve an ortho-selective electrophilic aromatic substitution of the Molander salt with the pentadienyl cation int-4 at the alpha position. Elimination of BF3 in int-5 would lead to int-6, which may undergo a 3,3-sigmatropic rearrangement, leading to the corresponding 1,4-diene (Table 2C).21 However, we cannot rule out the possibility of a direct attack of the benzyl nucleophile on the γ-position.
We next turned our attention to exploit our Rh-catalysed single carbon-insertion with substituted 1,3-dienes and tributylmethylphosphonium dimethylphosphate as the nucleophile (Scheme 3A). Under the optimized reaction conditions, isoprene led to a mixture of allylic phosphates 4a. We noticed that the major isomers found come from a preferred insertion into the more substituted double bond, consistent with prior observations reported for other metallocarbenes.22 In contrast, reactions carried out with 1,3-dienes substituted at C1 provided 1,3-dienes 4b,c with excellent levels of diastereo- and regioselectivity (l:b > 20:1; E:Z > 20:1) and cyclic dienes such as 1,3-cyclohexadiene provided 4d with good regioselectivity.
 | ||
| Scheme 3 1,3-Diene scope, NMR studies and enantioselective single-carbon insertion. a Reactions carried out with diene 1 (1.0 mmol), Rh2(esp)2 (1.0 mol%) and reagent 2d (0.2 mmol) in CH2Cl2 (3.0 mL) for 1 h at −50 °C. Then, nBu3MePOPO(OMe)2 (0.6 mmol) in CH2Cl2 was dropwise added over 10 min at −50 °C and the tube was kept in the cooling bath and slowly warmed to rt over 4 h. Yields are reported on the basis of the isolated pure product using flash column chromatography. b Reaction carried out with 1,3-diene 1c (0.2 mmol), Rh2(esp)2 (1.0 mol%) and reagent 2d (0.1 mmol) in CD2Cl2 (1.5 mL) for 1 h at −50 °C. Then, int-7 was observed by 1H NMR. After this, a solution of the nucleophile (0.3 mmol) in CD2Cl2 (1.0 mL) was added dropwise over 10 min at −50 °C. Then, int-8 and int-9 were observed by 1H NMR. c Reactions carried out with 1,3-dienes 1 (0.2 mmol), Rh2(S-NTTL)4(AcOEt)2 (5.0 mol%) and reagent 2c (0.24 mmol) in CH2Cl2:PhCl (3.0 mL, 1:1) for 1.5 hours at −60 °C. Then, nucleophile (0.6 mmol) was added in CH2Cl2 dropwise over 10 min at −60 °C and the tube was allowed to warm to rt over 1 h. Enantiomeric ratios (e.r.) were determined by supercritical fluid chromatography mass spectrometry (SFC-MS) analysis on a chiral stationary phase of the isolated pure product by using flash column chromatography. The absolute configuration of 1,3-diene products 5 was assigned by analogy to that confirmed for styrenes.12 | ||
A reaction carried out between 1-phenyl-1,3-butadiene and reagent 2d with Rh2(esp)2 at −50 °C in CD2Cl2 allowed us to detect and characterize the cyclopropyl–I(III)–PF6int-7 (Scheme 3B). The relative configuration assigned using NOESY experiments showed that the styryl and I(III) moieties were in a relative cis disposition. Addition of 3.0 equiv. of dimethylphosphate and benzyl Molander salt at −50 °C promoted the formation of cyclopropyl–I(III)–OPO(OMe)2int-8 and cyclopropyl–I(III)–F3BBn int-9, as observed by 1H NMR. As previously observed, a downfield chemical shift of the proton o-H to Ar–I(III) was observed. This was diagnostic to invoke the formation of int-8. With this information, we wondered whether analogue int-8 could evolve through an SN1-like SNi mechanism as previously observed for cyclopropyl–I(III)–OPO(OMe)2 intermediates derived from styrenes.12
Under the optimized reaction conditions previously developed using [Rh2(S-NTTL)4](AcOEt)2 (S-NTTL = N-naphthaloyl-(S)-tert-leucinate), benzylester reagent 2c, and CH2Cl2:PhCl (1:1) as the solvent, we found that substituted sorbic acid esters could provide the desired 1,3-dienes 5a–c with high asymmetric induction (Scheme 3C). It is worth highlighting that the single-carbon insertion occurred with excellent site-selectivity towards the remote double bond to the ester group. Analogous to alkenes, the excellent enantiocontrol could be explained based on the enantioselective formation of cyclopropyl–I(III)–PF6 that, upon anion exchange, evolves into cyclopropyl–I(III)–dialkylphosphate and subsequently to the final products 5a–c through an SN1-like SNi mechanism. Unfortunately, such excellent levels of enantiocontrol were not observed in other substrates (5d).
Conclusions
In conclusion, we have developed a catalytic single-carbon insertion to 1,3-dienes with Rh(II)-carbynoids. We have demonstrated that this process can transform simple 1,3-dienes into complex 1,3- and 1,4-substituted dienes via σ- and π-bond activation. The value of the constructive scission of this kind of C(sp2)–C(sp2) bonds is exemplified in its versatile nucleophilic substitution, as well as in the enantiomeric control achieved for some examples. This reaction adds to the new methodologies concerning skeletal editing processes that involve single-carbon insertion into C(sp2)–C(sp2) bonds.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
P. S., N. D., J. E. G., W. J. T. & M. G. S. planned the experiments. P. S., N. D., J. E. G. & W. J. T. performed the experiments. All authors contributed to the analysis and interpretation of the data. M. G. S. directed the project and wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
European Research Council (ERC-CoG 2019, 865554), the Agencia Estatal de Investigación (AEI, 10.13039/501100011033) of the Ministerio de Ciencia, Innovación y Universidades (PID2019-104101GB-I00, PID2022-140286NB-I00, Severo Ochoa Excellence Accreditation “CEX2024-001469-S), the ICIQ Foundation, the ICREA Foundation, and the CERCA Programme are gratefully acknowledged for financial support. The authors thank the European Union for Marie Skłodowska-Curie Individual Actions (101028657 to W. J. T.) and AEI for predoctoral FPI fellowships (BES-2017-080163 to P. S. and PREP2022-000211 to N. D).Notes and references
- (a) M. C. Kloetzel, Org. React., 1948, 4, 1–59 CAS; (b) K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS.
- (a) G. J. P. Perry, T. Jia and D. J. Procter, ACS Catal., 2020, 10, 1485–1499 CrossRef CAS; (b) R. G. Soengas and H. Rodríguez-Solla, Molecules, 2021, 26, DOI:10.3390/molecules26020249; (c) A. M. Canfield, D. Rodina and S. M. Paradine, Angew. Chem., Int. Ed., 2024, 63, e202401550 CrossRef CAS PubMed.
- (a) J. E. Baeckvall, Acc. Chem. Res., 1983, 16, 335–342 CrossRef CAS; (b) Y. Huang, M. Fañanás-Mastral, A. J. Minnaard and B. L. Feringa, Chem. Commun., 2013, 49, 3309–3311 RSC; (c) E. McNeill and T. Ritter, Acc. Chem. Res., 2015, 48, 2330–2343 CrossRef CAS PubMed; (d) M. Holmes, L. A. Schwartz and M. J. Krische, Chem. Rev., 2018, 118, 6026–6052 CrossRef CAS PubMed; (e) Z. Gao and S. P. Fletcher, Chem. Commun., 2018, 54, 3601–3604 RSC; (f) G. J. P. Perry, T. Jia and D. J. Procter, ACS Catal., 2020, 10, 1485–1499 CrossRef CAS; (g) P. Chen, L. Tian, X. Ji, G.-J. Deng and H. Huang, Org. Lett., 2025, 27, 2163–2169 CrossRef CAS PubMed.
- (a) X. Wu and L.-Z. Gong, Synthesis, 2019, 51, 122–134 CrossRef CAS; (b) M. M. Parsutkar and T. V. RajanBabu, J. Am. Chem. Soc., 2021, 143, 12825–12835 CrossRef CAS PubMed; (c) A. Flaget, C. Zhang and C. Mazet, ACS Catal., 2022, 12, 15638–15647 CrossRef CAS PubMed; (d) D. Singh and T. V. RajanBabu, Angew. Chem., Int. Ed., 2023, 62, e20221600 Search PubMed; (e) S. Chen, Y.-N. Wang, J. Xie, W. Li, M. Ye, X. Ma, K. Yang, S. Li, Y. Lan and Q. Song, Nat. Commun., 2024, 15, 5479 CrossRef CAS PubMed.
- (a) S. J. Connon and S. Blechert, Angew. Chem., Int. Ed., 2003, 42, 1900–1923 CrossRef CAS PubMed; (b) P. Liu, X. Xu, X. Dong, B. K. Keitz, M. B. Herbert, R. H. Grubbs and K. N. Houk, J. Am. Chem. Soc., 2012, 134, 1464–1467 CrossRef CAS PubMed; (c) S.-X. L. Luo, J. S. Cannon, B. L. H. Taylor, K. M. Engle, K. N. Houk and R. H. Grubbs, J. Am. Chem. Soc., 2016, 138, 14039–14046 CrossRef CAS PubMed.
- (a) M. Mortén, M. Hennum and T. Bonge-Hansen, Beilstein J. Org. Chem., 2015, 11, 1944–1949 CrossRef PubMed; (b) N. R. Candeias, R. Paterna and P. M. P. Gois, Chem. Rev., 2016, 116, 2937–2981 CrossRef CAS PubMed; (c) M. Ni, J. Zhang, X. Liang, Y. Jiang and T.-P. Loh, Chem. Commun., 2017, 53, 12286–12289 RSC; (d) G. S. Fleming and A. B. Beeler, Org. Lett., 2017, 19, 5268–5271 CrossRef CAS PubMed; (e) F. Huang, Z. Liu, Q. Wang, J. Lou and Z. Yu, Org. Lett., 2017, 19, 3660–3663 CrossRef CAS PubMed; (f) S. Peeters, L. N. Berntsen, P. Rongved and T. Bonge-Hansen, Beilstein J. Org. Chem., 2019, 15, 2156–2160 CrossRef CAS PubMed; (g) K. Luo, S. Mao, K. He, X. Yu, J. Pan, J. Lin, Z. Shao and Y. Jin, ACS Catal., 2020, 10, 3733–3740 CrossRef CAS; (h) R. Stenner, J. W. Steventon, A. Seddon and J. L. R. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1419–1428 CrossRef CAS PubMed; (i) B. D. Dherange, P. Q. Kelly, J. P. Liles, M. S. Sigman and M. D. Levin, J. Am. Chem. Soc., 2021, 143, 11337–11344 CrossRef CAS PubMed; (j) J. Kim and E. J. Yoo, Org. Lett., 2021, 23, 4256–4260 CrossRef CAS PubMed; (k) D. Ma, B. S. Martin, K. S. Gallagher, T. Saito and M. Dai, J. Am. Chem. Soc., 2021, 143, 16383–16387 CrossRef CAS PubMed; (l) L. Zhang, B. M. DeMuynck, A. N. Paneque, J. E. Rutherford and D. A. Nagib, Science, 2022, 377, 649–654 CrossRef CAS PubMed; (m) X.-L. Jiang, Q. Liu, K.-F. Wei, T.-T. Zhang, G. Ma, X.-H. Zhu, G.-X. Ru, L. Liu, L.-R. Hu and W.-B. Shen, Commun. Chem., 2023, 6, 35 CrossRef CAS PubMed; (n) B. W. Joynson, G. R. Cumming and L. T. Ball, Angew. Chem., Int. Ed., 2023, 62, e202305081 CrossRef PubMed; (o) C.-Y. Shi, G.-Y. Zhu, Y. Xu, M.-Y. Teng and L.-W. Ye, Chin. Chem. Lett., 2023, 34, 108441 CrossRef CAS; (p) S. Park, C.-E. Kim, J. Jeong, H. Ryu, C. Maeng, D. Kim, M.-H. Baik and P. H. Lee, Nat. Commun., 2023, 14, 7936 CrossRef CAS PubMed; (q) B. W. Joynson, G. R. Cumming and L. T. Ball, Angew. Chem., Int. Ed., 2023, 62, e202305081 CrossRef PubMed; (r) H. Guo, S. Qiu and P. Xu, Angew. Chem., Int. Ed., 2024, 63, e202317104 CrossRef CAS PubMed; (s) S. Liu, Y. Yang, Q. Song, Z. Liu, Y. Lu, Z. Wang, P. Sivaguru and X. Bi, Nat. Chem., 2024, 16, 988–997 CrossRef CAS PubMed; (t) D. Ly, J. Bacsa and H. M. L. Davies, ACS Catal., 2024, 14, 6423–6431 CrossRef CAS PubMed; (u) F.-P. Wu, C. C. Chintawar, R. Lalisse, P. Mukherjee, S. Dutta, J. Tyler, C. G. Daniliuc, O. Gutierrez and F. Glorius, Nat. Catal., 2024, 7, 242–251 CrossRef CAS PubMed; (v) S. Timmann, T.-H. Wu, C. Golz and M. Alcarazo, Chem. Sci., 2024, 15, 5938–5943 RSC; (w) P. Kafle, D. Herndon, I. Sharma, chemrXiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-6dncp.
- (a) T. Courant, M. Pasco and T. Lecourt, Org. Lett., 2018, 20, 2757–2761 CrossRef CAS PubMed; (b) S. Stockerl, T. Danelzik, D. G. Piekarski and O. García Mancheño, Org. Lett., 2019, 21, 4535–4539 CrossRef CAS PubMed; (c) S. F. Musolino, Z. Pei, L. Bi, G. A. DiLabio and J. E. Wulff, Chem. Sci., 2021, 12, 12138–12148 RSC; (d) J. Kim and E. J. Yoo, Org. Lett., 2021, 23, 4256–4260 CrossRef CAS PubMed; (e) F. Wang, Y. Nishimoto and M. Yasuda, J. Am. Chem. Soc., 2021, 143, 20616–20621 CrossRef CAS PubMed; (f) F. Wang, J. Yi, Y. Nishimoto and M. Yasuda, Synthesis, 2021, 53, 4004–4019 CrossRef CAS; (g) K. Saito, H. Aoyama, M. Sako, M. Arisawa and K. Murai, J. Org. Chem., 2022, 87, 16947–16951 CrossRef CAS PubMed; (h) F. Wang, Y. Nishimoto and M. Yasuda, Angew. Chem., Int. Ed., 2022, 61, e202204462 CrossRef CAS PubMed; (i) Y. Nishimoto and M. Yasuda, Synlett, 2024, 35, 367–378 CrossRef CAS; (j) H. L. Schmitt, D. Martymianov, O. Green, T. Delcaillau, Y. S. Park Kim and B. Morandi, J. Am. Chem. Soc., 2024, 146, 4301–4308 CrossRef CAS PubMed; (k) V. G. Alfonso, K. Vega-Hernández and M. G. Suero, J. Am. Chem. Soc., 2025, 147, 57–62 CrossRef PubMed.
- (a) J. Jurczyk, J. Woo, S. F. Kim, B. D. Dherange, R. Sarpong and M. D. Levin, Nat. Synth., 2022, 1, 352–364 CrossRef CAS PubMed; (b) B. W. Joynson and L. T. Ball, Helv. Chim. Acta, 2023, 106, e202200182 CrossRef CAS; (c) Z. Liu, P. Sivaguru, Y. Ning, Y. Wu and X. Bi, Chem.–Eur. J., 2023, 29, e202301227 CrossRef CAS PubMed; (d) H. Fujimoto and M. Tobisu, Chem.–Eur. J., 2024, 2, e202400005 CAS.
- Z. Wang, A. G. Herraiz, A. M. Del Hoyo and M. G. Suero, Nature, 2018, 554, 86–91 CrossRef CAS PubMed.
- (a) Z. Wang, L. Jiang, P. Sarró and M. G. Suero, J. Am. Chem. Soc., 2019, 141, 15509–15514 CrossRef CAS PubMed; (b) L. Jiang, P. Sarró, W. J. Teo, J. Llop and M. G. Suero, Chem. Sci., 2022, 13, 4327–4333 RSC.
- (a) M. P. Doyle, W. R. Winchester, J. A. A. Hoorn, V. Lynch, S. H. Simonsen and R. Ghosh, J. Am. Chem. Soc., 1993, 115, 9968–9978 CrossRef CAS; (b) H. M. L. Davies and K. Liao, Nat. Rev. Chem., 2019, 3, 347–360 CrossRef CAS PubMed; (c) R. Hrdina, Eur. J. Inorg. Chem., 2021, 2021, 501–528 CrossRef CAS.
- W. J. Teo, J. Esteve Guasch, L. Jiang, B. Li and M. G. Suero, J. Am. Chem. Soc., 2024, 146, 21837–21846 CrossRef PubMed.
- V. Lanke and I. Marek, Chem. Sci., 2020, 11, 9378–9385 RSC.
- (a) T. S. Sorensen, Can. J. Chem., 1964, 42, 2768–2780 CrossRef CAS; (b) P. H. Campbell, N. W. K. Chiu, K. Deugau, I. J. Miller and T. S. Sorensen, J. Am. Chem. Soc., 1969, 91, 6404–6410 CrossRef CAS; (c) T. N. Grant, C. J. Rieder and F. G. West, Chem. Commun., 2009, 5676–5688 RSC; (d) S. Chen, A. B. Keto, A. El-Hawli, L. R. Gahan, J. M. White, E. H. Krenske and B. L. Flynn, Chem.–Eur. J., 2024, 30, e202402779 CrossRef CAS PubMed.
- (a) M. Harmata, S. K. Ghosh, X. Hong, S. Wacharasindhu and P. Kirchhoefer, J. Am. Chem. Soc., 2003, 125, 2058–2059 CrossRef CAS PubMed; (b) H. Zhang, K. Hayashi, H. Tanimoto, T. Morimoto, Y. Nishiyama and K. Kakiuchi, Tetrahedron, 2014, 70, 8600–8605 CrossRef CAS; (c) J. R. Stepherson, C. E. Ayala, T. H. Tugwell, J. L. Henry, F. R. Fronczek and R. Kartika, Org. Lett., 2016, 18, 3002–3005 CrossRef CAS PubMed.
- (a) N. L. Morrow, Environ. Health Perspect., 1990, 86, 7–8 CrossRef CAS PubMed; (b) M. A. Tasdelen, Polym. Chem., 2011, 2, 2133–2145 RSC; (c) D. Cespi, F. Passarini, I. Vassura and F. Cavani, Green Chem., 2016, 18, 1625–1638 RSC.
- (a) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed; (b) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2024, 124, 11108–11186 CrossRef CAS PubMed.
- (a) C. G. Espino, K. W. Fiori, M. Kim and J. Du Bois, J. Am. Chem. Soc., 2004, 126, 15378–15379 CrossRef CAS PubMed; (b) D. N. Zalatan and J. Du Bois, J. Am. Chem. Soc., 2009, 131, 7558–7559 CrossRef CAS PubMed.
- S. Darses and J.-P. Genet, Chem. Rev., 2008, 108, 288–325 CrossRef CAS PubMed.
- J. Y. Hamilton, D. Sarlah and E. M. Carreira, J. Am. Chem. Soc., 2013, 135, 994–997 CrossRef CAS PubMed.
- D. W. Lee, R. K. Pandey, S. Lindeman and W. A. Donaldson, Org. Biomol. Chem., 2011, 9, 7742–7747 RSC.
- (a) A. J. Anciaux, A. J. Hubert, A. F. Noels, N. Petiniot and P. Teyssié, J. Org. Chem., 1980, 45, 695–702 CrossRef CAS; (b) M. P. Doyle, R. L. Dorow, W. E. Buhro, J. H. Griffin, W. H. Tamblyn and M. L. Trudell, Organometallics, 1984, 3, 44–52 CrossRef CAS; (c) A. Demonceau, E. A. Dias, C. A. Lemoine, A. W. Stumpf and A. F. Noels, Tetrahedron Lett., 1995, 36, 3519–3522 CrossRef CAS; (d) M. Garbo, C. Besnard, L. Guénée and C. Mazet, ACS Catal., 2020, 10, 9604–9611 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc03161c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |