
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface inclusion and dynamics of cucurbit[7]uril-based supramolecular complexes†
Carina
Santos Hurtado‡
a,
Guillaume
Bastien‡
a,
Doroteja
Lončarić
ab,
Martin
Dračínský
 a,
Ivana
Císařová
c,
Eric
Masson
*d and
Jiří
Kaleta
*a
a,
Ivana
Císařová
c,
Eric
Masson
*d and
Jiří
Kaleta
*a
aInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo nám. 2, 160 00 Prague 6, Czech Republic. E-mail: jiri.kaleta@uochb.cas.cz
bDepartment of Organic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 2030, 12840 Prague 2, Czech Republic
cDepartment of Inorganic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 2030, 128 40 Prague 2, Czech Republic
dDepartment of Chemistry and Biochemistry, Ohio University, Athens, Ohio 45701, USA
First published on 10th June 2025
Abstract
A supramolecular complex acting as a molecular rotor was assembled from cucurbit[7]uril and a rigid, rod-shaped molecular anchor. This rotor was then anchored to the facets of a hexagonal tris(o-phenylenedioxy)cyclotriphosphazene matrix, forming a regular 2D array. Semi-empirical calculations revealed very low rotational barriers for the macrocyclic units within this array, suggesting smooth rotational motion and promising potential for future applications in dynamic molecular systems.
Introduction
The development of two-dimensional (2D) stimuli-responsive systems, consisting of regular arrays of molecular motors, switches, and rotors, is a rapidly growing area of research.1–3 These 2D systems, which can reorient or isomerize as a response to external stimuli like light or electric fields, offer significant potential in molecular nanoelectronics,4–8 memory devices, optical filters,9 and heterogeneous catalysis.10 While synthesizing and characterizing functional molecules in solution – key building blocks for future smart materials – is a relatively well-established process, achieving precise control over the positioning of individual molecules within these arrays remains a major challenge.Three common approaches to developing such 2D materials include: (i) self-assembly, typically on metallic surfaces, either from solution or gas phase, (ii) the formation of Langmuir–Blodgett films at liquid–gas interfaces, and (iii) the intercalation of molecules into porous solid surfaces. Crucially, each of these methods requires carefully designed and fine-tuned stimuli-responsive molecules tailored for the specific approach.1
Herein, we are further exploring the third approach, which is based on the installation of molecules on the facets of the hexagonal form of a zeolite-like matrix called tris(o-phenylenedioxy)cyclotriphosphazene, often abbreviated as TPP (Fig. 1A–C).11–13 This porous material is interwoven with a network of parallel channels, approximately 5 Å in diameter, that connect two opposing crystal faces. These channels are spaced about 11 Å apart and are arranged in a trigonal pattern (Fig. 1C).14 We previously demonstrated that dipolar rotors,15 unidirectional light-driven molecular motors,16 and also azobenzene-based switches17 can form surface inclusions using sophisticated rod-like molecular anchors. Recently, we have shown that fullerene C60-based molecular rotors can also be installed on the facets of TPP nanocrystals using a molecular anchor.18
 | ||
| Fig. 1 Synthesis of molecular rod 1 and its reaction with CB[7] leading to supramolecular complex 1·CB[7]. The structure of the TPP-d12 molecule (A), the corresponding space-filling model (B) and a top view of a small fragment of hexagonal TPP (C). CB[7]: a schematic representation (D), top (E) and side (F) view on a space-filling model, and a chemical structure (G). Idealized graphical visualization of surface inclusion 1·CB[7]@TPP-d12: top (H) and side (I) view on a space-filling model. ORTEP representation of intermediates 6, 8, and 10 (thermal ellipsoids shown on 30% probability level). | ||
In this study, we demonstrate that 2D surface inclusions are not limited to traditional covalent molecules, but can also encompass supramolecular complexes, thus significantly expanding the available pool of guest molecules. To illustrate this, we designed and synthesized a model system featuring a rod-like molecule 1 with an adamantylated pyridinium at one terminus, which serves as a binding site for cucurbit[7]uril (CB[7], Fig. 1D–G). 1-(1-Adamantyl)pyridinium bromide was shown to have high affinity for CB[7], with a binding constant reaching 2.0 × 1012 M−1 in a 50 mM aqueous sodium acetate buffer.19 The other end contains a TPP binding site characterized by two aliphatic units, tert-butyl and bicyclo[1.1.1]pentyl (BCP), whose distinct 13C NMR signals allow the tracking of the degree of insertion into the matrix (Fig. 1). These two ends are separated by a bulky triptycene cage (∼9 Å diameter), acting as a stopper to prevent full insertion into the TPP matrix. When complexed with CB[7], the upper part of the system forms a molecular rotor with a diameter of ∼16 Å (ref. 20) enabling only the lower portion of the molecular rod 1·CB[7] to insert into the matrix (Fig. 1H and I). These regular arrays of molecular rotors could represent a significant advancement toward the development of 2D ferroelectric materials, which are appealing components of nanoelectronic devices.21–23
Results and discussion
The synthetic pathway towards rod 1 was designed to address its anticipated low solubility in common organic solvents. This approach involved constructing two reasonably soluble building blocks, 2 and 3,15 whose combination resulted in 1, which was easily separated from the crude reaction mixture by simple centrifugation (Fig. 1). The synthesis of building block 2 began with a Suzuki cross-coupling between boronic acid 4 and 1-bromo-4-iodobenzene (5), affording 6 in a 65% yield. A subsequent nucleophilic aromatic substitution with 1-chloro-2,4-dinitrobenzene (7) produced Zincke salt 8, which reacted with adamantan-1-amine (9) to give 10 in 88% yield. Finally, a Sonogashira cross-coupling between 10 and 11 (ref. 15) afforded building block 2 almost quantitatively.The structures of three intermediates, 6, 8, and also compound 10 containing the adamantylated pyridinium acting as a future CB[7] binding site, were independently confirmed by X-ray diffraction of the corresponding monocrystals (Fig. 1).
Supramolecular complex 1·CB[7] was quantitatively obtained in preparative quantities through a stoichiometric reaction between 1 and CB[7] in DMSO. Encapsulation was monitored using 1H NMR spectroscopy (Fig. 2). The attachment of CB[7] caused noticeable upfield shifts of both the hydrogen nuclei of the adamantyl unit (Ha–Hc) located inside the macrocycle's cavity and the hydrogens on the pyridine unit (Hd and He) situated close to the CB[7] carbonyl portal. In contrast, the chemical shift of the Hf hydrogens on the t-Bu group remained unchanged, supporting the proposed geometry of 1·CB[7], where CB[7] exclusively occupies the cationic binding site of the rod-shaped molecule. It is worth noting that 1·CB[7] was accompanied by a small but detectable amount of alkali or alkaline-earth metal-promoted trimers.24 Their presence does not interfere with subsequent mechanochemical synthesis of surface inclusions, as these species are stable only in non-aqueous environments. Detailed assignment of hydrogen and carbon atoms both in 1 and 1·CB[7] are shown in the ESI.†
 | ||
| Fig. 2 1H NMR spectra of CB[7] (A), 1 (B), 1 after addition of 0.6 equiv. (C) and 1.0 equiv. (D) of CB[7] recorded in DMSO-d6 at 25 °C. Green asterisks indicate alkali or alkaline-earth metal-promoted trimers. See Fig. 1 for atom labelling. | ||
The surface inclusion 1·CB[7]@TPP-d12 results from a mechanochemical reaction between solvent-free hexagonal form of TPP-d12 and 1·CB[7]. The mixture of the powdered materials was ball-milled using a planetary ball mill and subsequently annealed under argon atmosphere (see the ESI†). The 3 mol% guest loading that corresponds to statistical occupancy of 1/3 of portals to TPP channels,17 secures complete coverage of facets of TPP nanocrystals. The identity of 1·CB[7]@TPP-d12 was confirmed by solid-state 13C cross-polarized magic-angle spinning (ss 13C CP MAS) NMR spectroscopy. A comparison of the solution 13C NMR spectra of 1, CB[7], and the complex 1·CB[7] with their ss 13C CP MAS, provided insight into the intricate nature of the spectrum of the surface inclusion (Fig. 3).
 | ||
| Fig. 3 13C NMR spectra of 1 (A), CB[7] (C), and 1·CB[7] (E) recorded in DMSO-d6 solution at 25 °C. Solid-state 13C CP MAS NMR spectra of 1 (B), CB[7] (D), 1·CB[7] (F), and 1·CB[7]@TPP-d12 (H). Additionally, the solid-state 13C NMR of TPP-d12 (G) is shown. The carbon signals corresponding to 1 are highlighted in blue, those of CB[7] in red, and TPP-d12 in green. See Fig. 1 for carbon atoms labelling. | ||
While the 13C NMR spectrum of 1 in DMSO-d6 is complex (Fig. 3A), its corresponding ss CP MAS spectrum (Fig. 3B) shows five peaks in the aromatic region (∼150, 142, 133, 126, and 122 ppm), four in the triple-bond region (∼92, 88, 85, and 80 ppm), and six in the aliphatic region in (∼68 for adamantyl carbon Cd, ∼60 ppm for BCP methylenes Cg, ∼52 for triptycene bridgeheads Ce, ∼42 for adamantyl methylenes Cc, ∼36 for adamantyl methylenes Ca and tert-butyl quaternary carbon Ch, and ∼31 ppm for tert-butyl methyl groups Ci, adamantyl Cb and both BCP bridgeheads Cf). Subsequent addition of CB[7], whose 13C spectra both in solution (Fig. 3C) and ss CP MAS (Fig. 3D) consist of three peaks at ∼157 ppm (carbonyl group), ∼71 ppm (CH groups), and ∼53 ppm (CH2 groups), affords inclusion complex 1·CB[7]. In solution (Fig. 3E), the CB[7] carbon signals at the rim are doubled due to the pair of non-equivalent portals. The attachment of CB[7] also shields the adamantyl moiety, leading to the expected upfield shifts in its carbon signals. These trends are mostly preserved also in the ss CP MAS spectrum (Fig. 3F).
The solid-state 13C NMR spectrum of perdeuterated hexagonal TPP-d12 exhibits three resonances at ∼144, ∼124 and ∼110 ppm for the catechol units (Fig. 3G). The ss CP MAS spectrum shows no signals, as this material lacks hydrogen atoms.
The incorporation of 1·CB[7] into the TPP-d12 matrix, forming 1·CB[7]@TPP-d12, results in the shielding of carbon atoms associated with the TPP binding site (Fig. 1). Notably, the BCP and tert-butyl units provide the most significant indicators. The upfield shifts of their signals (the BCP methylene units Cg from 59.6 ppm in 1·CB[7] to 56.6 ppm upon surface inclusion, and methyl signals Ci shifting from 30.9 ppm to 27.0 ppm, Fig. 3H), indicate that the lower segment of the molecular rod is fully embedded within the matrix.13,15–18
Interestingly, the 13C resonances of the aromatic carbon atoms in 1·CB[7]@TPP-d12 are nearly invisible compared to those in neat 1·CB[7] (Fig. 3F and H). We speculate that this observation further supports the successful formation of the surface inclusion. These aromatic carbon atoms, which are primarily located at the TPP binding site of the molecular rod, are likely close to the neighboring CB[7] units in neat 1·CB[7], allowing efficient through-space polarization transfer during the CP MAS experiment. This mechanism is suppressed in 1·CB[7]@TPP-d12, as this region of the molecule is surrounded by the perdeuterated TPP-d12 matrix.
The successful formation of surface inclusion is also supported by a 31P CP MAS NMR experiment that clearly identifies the hexagonal form of TPP-d12.13,15–18 This polymorph would be otherwise readily converted to the monoclinic form if its empty channels were not stabilized by the guest molecules during the annealing process.25
The insertion of rod-shaped molecules into the TPP matrix is generally causing an expansion of the channels as they adapt to the inserted guest.17,18,26 This change is detectable by powder X-ray diffraction (PXRD), and analysis of 1·CB[7]@TPP-d12 (Fig. 4A) clearly shows noticeable increase of the in-plane spacing parameter a (11.62 ± 0.01 Å) and decrease of the layer spacing c (10.07 ± 0.01 Å) of the hexagonal TPP (compared to the guest-free material, where a = 11.454(5) Å and c = 10.160(5) Å).27 The fitting also produces estimates for the particle size in the powder (21 ± 2 Å) and strain in the material (3.3 ± 0.4%). Additionally, a Voigt parameter of 1 indicates that the Bragg peaks are Lorentzian in shape rather than Gaussian – a characteristic typically observed in surface inclusions.17,18
 | ||
| Fig. 4 1·CB[7]@TPP-d12: PXRD spectrum (A), and TEM visualization of nanocrystals (B). | ||
The morphology of 1·CB[7]@TPP-d12 was examined via transmission electron microscopy (TEM), revealing clusters of discoidal particles with an approximate diameter of 30–50 nm. These findings are consistent with previous observations,13,18 and align reasonably well with the values derived from PXRD fitting. Additionally, 1·CB[7]@TPP-d12 shows good thermal stability (melting of TPP at ∼251 °C and decomposition of 1 at ∼268 °C), that is comparable with structurally related systems15–17 as revealed by differential scanning calorimetry (DSC, see Fig. S7 in ESI† for details).
We then questioned whether the CB[7] units undergo rotation around the adamantyl headgroup. In DMSO-d6 solution, the equivalence of the equatorial Hx protons and the methylene Hy and Hz protons suggests fast, unrestricted rotation on the NMR timescale. However, the situation in the solid state remains uncertain, particularly given that the binding affinity of CB[7] towards the adamantyl unit is expected to increase significantly in the absence of solvent. Unfortunately, experimental probing of such dynamics in the solid state is highly challenging: techniques such as 2H NMR would require site-specific deuteration, and other established methods (e.g., dielectric spectroscopy, 1H T1 relaxation measurement, variable-frequency-MAS 13C NMR) either demand synthetically complex modifications or yield convoluted signals from multiple dynamic processes. Therefore, to gain insight into the rotational potential landscape of the system and evaluate its feasibility as a molecular rotor, we turned to computational modelling as a practical and informative alternative. Using the GFN2-xTB semi-empirical method,28–30 a binding affinity of 48 kcal mol−1 in the gas phase was determined, far greater than the affinity in aqueous solution of 1-(1-adamantyl)pyridinium (see above: 2.0 × 1012 M−1, i.e. 17 kcal mol−1). The presence of this very tight interaction then raises a question whether it allows or prevents the rotation of the macrocycle. As the resolution of the ss 13C CP MAS NMR spectra of 1·CB[7]@TPP-d12 is too low to test the respective equivalency of all CB[7] equatorial carbons CH, methylene carbons CH2 and carbonyl rim CO, we decided to assess CB[7] motion around the rod in silico using truncated rod 12.
Three motions of CB[7] using the GFN2-xTB semi-empirical method were scanned: rotation of both the adamantyl head group together with CB[7] around the rod (Fig. 5A), rotation of CB[7] around the adamantyl group and along its symmetry axis (Fig. 5B), and a rocking motion around the head (Fig. 5C).
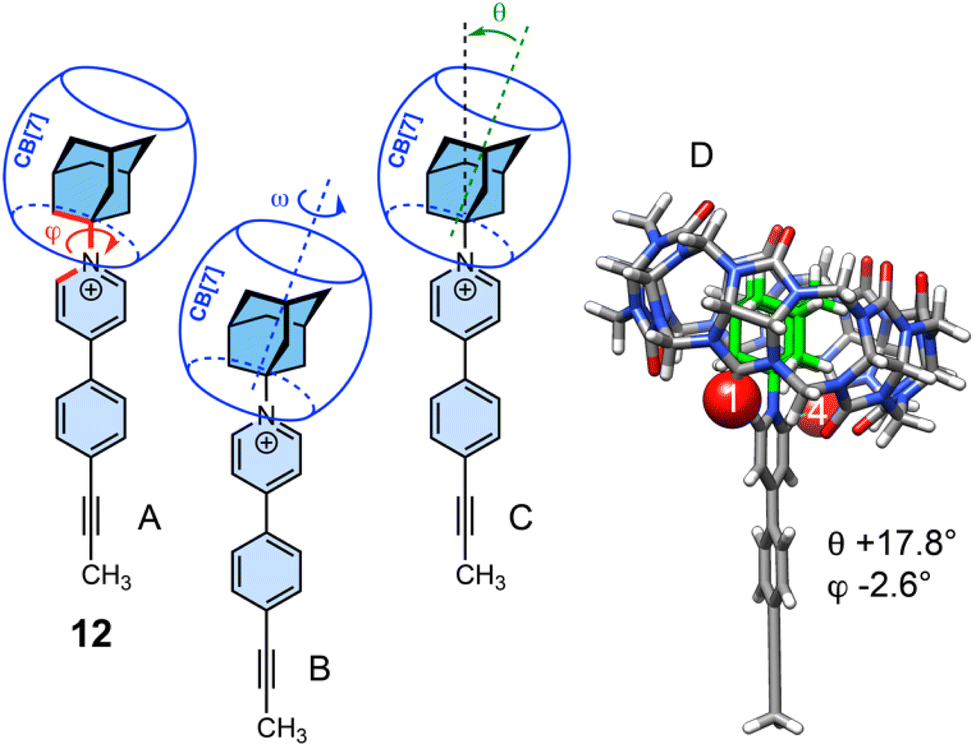 | ||
| Fig. 5 Three possible CB[7] motions in 1·CB[7]@TPP-d12: (A) rotation of the adamantyl head group around the rod (i.e. with varying C(2)(pyridinium)–N(pyridinium)–C(1)(adamantyl)–C(2)(adamantyl) dihedral angles φ; (B) rotation of CB[7] along its symmetry axis ω is a C(2)(adamantyl)–C(1)(adamantyl)–N(pyridinium)–C(carbonyl, CB[7]) dihedral angle); and (C) rocking motion of CB[7] around the adamantyl head group; the tilt angle θ is the angle between the N(pyridinium)–C(adamantyl) axis and a fitted plane through the 14 equatorial carbon atoms of CB[7]. (D) GFN2-xTB optimized structure of complex 12·CB[7]. Interactions between pyridinium hydrogen and the nearest oxygens of the CB[7] rim highlighted using a space-filling model. | ||
First, complex 12·CB[7] was optimized. It displays a near-zero dihedral angle φ (−2.6°) and a tilt angle θ of 17.8°. Scanning dihedral angle φ in the presence and absence of CB[7] returned contrasted energy profiles (Fig. 6). In the absence of CB[7], the periodicity is 6-fold, with energy minima obtained when φ and dihedral angle C(6)(pyridinium)–N(pyridinium)–C(1)(adamantyl)–C(2)(adamantyl) (180° + φ) are near 0°, 120° and 240°. Transition states are obtained when the pyridinium unit is almost perpendicular to one adamantyl C(1)–C(2) bond, i.e. at φ 27° (30° relative to the ground state) with a 60° periodicity; these are 1.45 kcal mol−1 less stable than the ground states. When encapsulated, as tilt angle θ is non-zero, the periodicity is reduced to 3-fold, and the energy profile features an intermediate at φ = 62° barely less stable than the ground state (ΔE = 0.10 kcal mol−1) and two transition states at φ = 27° and 87° (ΔE = 2.97 and 1.71 kcal mol−1, respectively). Relative energies are governed by the distance (i.e. the strength) of the CH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C interaction between the partially positive pyridinium hydrogens at positions 2 and 6, and the CB[7] rim oxygens. Distances of approximately 2.10 Å are measured in the ground state and in the intermediate, and up to 2.75 and 2.40 Å in the higher-energy and lower-energy transition states, respectively (Fig. 6). We note that the 7-fold symmetry of CB[7] is irrelevant when describing the rotational pathway, as the macrocycle is not frozen, and undergoes slight geometry adjustments during each step of the scan along dihedral angle φ. We also note that this mode of CB[7] motion, that is coupled to the rod motion, will operate with any rotation along the rod (along the C(pyridinium)–C(aryl) or C(aryl)–C
C interaction between the partially positive pyridinium hydrogens at positions 2 and 6, and the CB[7] rim oxygens. Distances of approximately 2.10 Å are measured in the ground state and in the intermediate, and up to 2.75 and 2.40 Å in the higher-energy and lower-energy transition states, respectively (Fig. 6). We note that the 7-fold symmetry of CB[7] is irrelevant when describing the rotational pathway, as the macrocycle is not frozen, and undergoes slight geometry adjustments during each step of the scan along dihedral angle φ. We also note that this mode of CB[7] motion, that is coupled to the rod motion, will operate with any rotation along the rod (along the C(pyridinium)–C(aryl) or C(aryl)–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C axis, for example).
C axis, for example).
 | ||
| Fig. 6 Relative stability of (A) rod 12, and (B) assembly 12·CB[7] along dihedral angle φ. Top views of assembly 12·CB[7], (C) at its most stable conformation, (D) at a transition state, (E) at an intermediate and (F) at a second transition state along the adamantyl rotation pathway. CB[7] carbonyl oxygens highlighted in red and numbered to highlight the pyridinium hydrogens–CB[7] CO distances along the rotation pathway. Relative energies in kcal mol−1. | ||
An energy profile for the rotation of CB[7] around the adamantyl group was determined, while the latter is allowed to wobble within its potential well (i.e. dihedral angle φ varies only slightly, and CB[7] motion is decoupled from the rod, see Fig. 7). Transition states are again detected once the pyridinium hydrogens at positions 2 and 6 find themselves between carbonyl groups of the CB[7] rim (see Fig. 7C). Due to the 7-fold symmetry of CB[7], the period of the energy profile is approximately 51° (360/7) and the activation barrier is again very low (1.82 kcal mol−1).
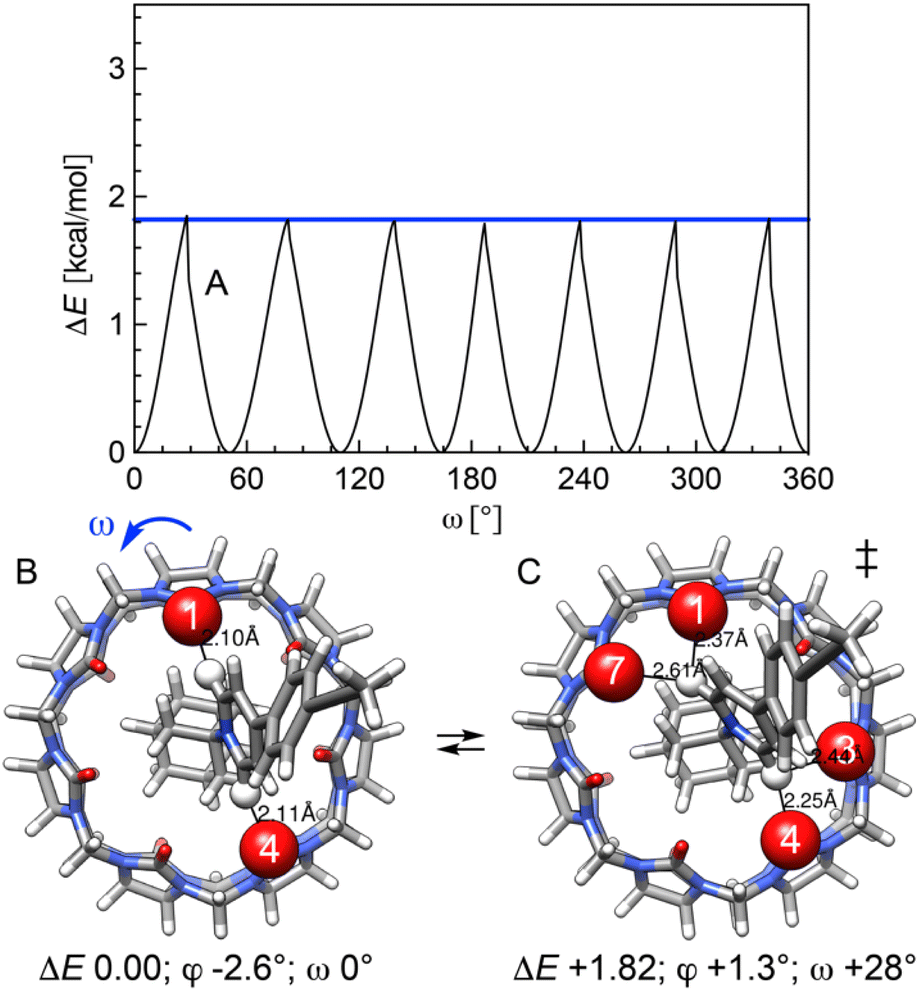 | ||
| Fig. 7 (A) Relative stability of assembly 12·CB[7] while rotating CB[7] along its symmetry axis. Top views of assembly 12·CB[7], at (B) its most stable conformation, and (C) a transition state along the CB[7] rotation pathway. See Fig. 5 for the description of the angles; relative energies in kcal mol−1. Interactions between pyridinium hydrogens and the nearest oxygens of the CB[7] rim highlighted using a space-filling model. | ||
Finally, the impact of the CB[7] tilt angle θ relative to the rod axis (Fig. 8A) was determined. Energies were scanned by varying the distance between the pyridinium C(4) atom and the nearest oxygen at the CB[7] rim. For each optimized assembly along the scan, the tilt angle was calculated using the guest N(pyridinium)–C(adamantyl) vector and a normal to the fitted plane across the 14 equatorial carbon atoms of CB[7] (see ESI section†). As the tilt angle narrows, hydrogen pyridiniums at positions 2 or 6 must relocate between two CB[7] rim carbonyls (Fig. 8), and the energy increases. To the contrary, when the tilt angle is forced to widen, the CH⋯OC distance is too short, and the interaction becomes repulsive (see Fig. 8A). The narrowest tilt angle that can be returned by freezing a single coordinate is 2.9°, with a 1.65 kcal mol−1 destabilization, i.e. other more favorable distortions take place instead of bringing CB[7] perfectly perpendicular to the rod axis.
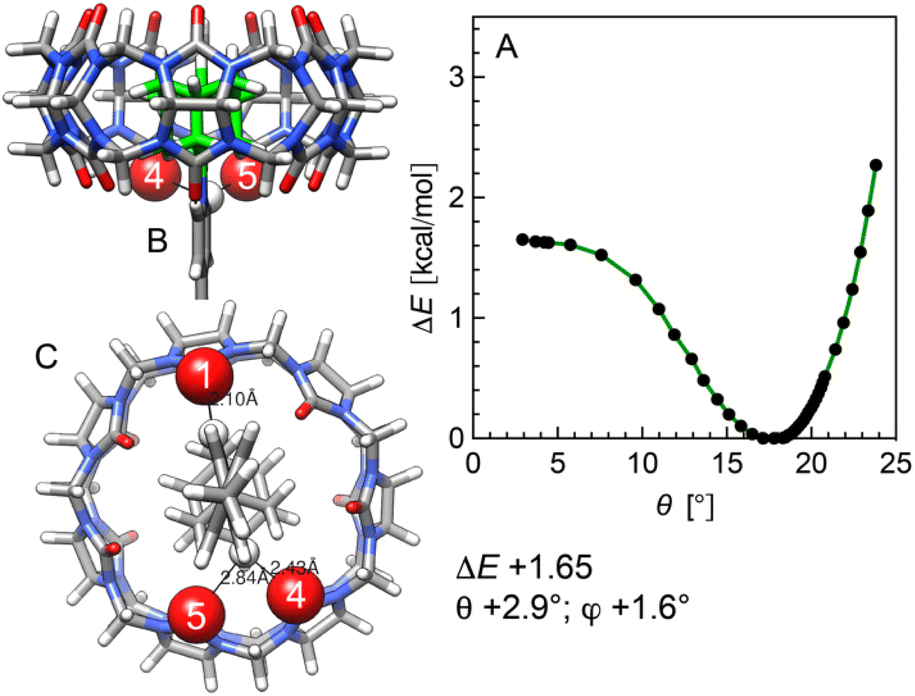 | ||
| Fig. 8 (A) Relative stability of assembly 12·CB[7] as a function of the CB[7] tilt angle θ relative to the rod axis. (B) Side and (C) top views of assembly 12·CB[7], with a tilt angles θ of 2.9°. See Fig. 5 for the description of the angles; relative energies in kcal mol−1. | ||
Conclusions
To conclude, we have demonstrated that even noncovalent supramolecular complexes can be arranged to 2D arrays on the surface of TPP nanocrystals using properly designed molecular anchors. The rod-shaped molecular anchor, featuring an adamantylated pyridinium-based headgroup, formed a host–guest complex with CB[7], while its lower section embedded into the porous matrix of the surface. The CB[7]-based unit functions as a molecular rotor. Despite CB[7] binding tightly to the head group in the solid state, it remains extremely mobile, and all motions described above can proceed concomitantly with very low energy barriers, approximately half of a C–C bond rotation in ethane.31,32Data availability
The data underlying this study are available in the published article and its ESI.†Author contributions
C. S. H., G. B., D. L., and M. D. carried out all experiments. I. C. determined all single-crystal X-ray diffraction structures. E. M. carried out all computational work, and wrote the corresponding sections. J. K. conceived the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences (RVO: 61388963), the Czech Science Foundation (grant number: 25-16074S), the Ministry of Education, Youth and Sports (grant number: LTAUSA19120), and the US National Science Foundation (grants CHE-2404391 and CHE-1905238). Calculations were performed at the Ohio Supercomputer Center (http://www.osc.edu/). We thank Prof. Zdeněk Sofer and Dr Iva Plutnarová for acquiring the powder X-ray patterns. We are also grateful to Prof. Charles T. Rogers and Dr Teddy Tortorici for interpreting the PXRD results and Dr Igor Rončević for performing preliminary calculations.References
- J. Kaleta, Molecular Switches and Motors in 2-D, in Molecular Photoswitches, ed. Z. L. Pianowski, Wiley-VCH GmbH, 2022 Search PubMed.
- R. Klajn, Pure Appl. Chem., 2010, 82, 2247–2279 Search PubMed.
- K.-Y. Chen, O. Ivashenko, G. T. Carroll, J. Robertus, J. C. M. Kistemaker, G. London, W. R. Browne, P. Rudolf and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 3219–3224 CrossRef CAS PubMed.
- D. Xiang, X. Wang, C. Jia, T. Lee and X. Guo, Chem. Rev., 2016, 116, 4318–4440 CrossRef CAS PubMed.
- H. Chen and J. F. Stoddart, Nat. Rev. Mater., 2021, 6, 804–828 CrossRef CAS.
- T. Li, V. K. Bandari and O. G. Schmidt, Adv. Mater., 2023, 35, 2209088 CrossRef CAS PubMed.
- P. T. Mathew and F. Fang, Engineering, 2018, 4, 760–771 CrossRef CAS.
- A. Köbke, F. Gutzeit, F. Röhricht, A. Schlimm, J. Grunwald, F. Tuczek, M. Studniarek, D. Longo, F. Choueikani, E. Otero, P. Ohresser, S. Rohlf, S. Johannsen, F. Diekmann, K. Rossnagel, A. Weismann, T. Jasper-Toennies, C. Näther, R. Herges, R. Berndt and M. Gruber, Nat. Nanotechnol., 2020, 15(1), 18–21 CrossRef PubMed.
- R. Feringa, H. S. Siebe, W. J. N. Klement, J. D. Steen and W. R. Browne, Mater. Adv., 2022, 3, 282–289 RSC.
- I. Vassalini and I. Alessandri, Catalysts, 2018, 8(12), 569 CrossRef.
- S. Bracco, A. Comotti, L. Ferreti and P. Sozzani, J. Am. Chem. Soc., 2011, 133, 8982–8994 CrossRef CAS PubMed.
- L. Kobr, K. Zhao, Y. Shen, A. Comotti, S. Bracco, R. K. Shoemaker, P. Sozzani, N. A. Clark, J. C. Price, C. T. Rogers and J. Michl, J. Am. Chem. Soc., 2012, 134, 10122–10131 CrossRef CAS PubMed.
- J. Kaleta, G. Bastien, J. Wen, M. Dračínský, E. Tortorici, I. Císařová, P. D. Beale, C. T. Rogers and J. Michl, J. Org. Chem., 2019, 84, 8449–8467 CrossRef CAS PubMed.
- P. Sozzani, S. Bracco, A. Comotti, L. Ferretti and R. Simonutti, Angew. Chem., Int. Ed., 2005, 44, 1816–1820 CrossRef CAS PubMed.
- J. Kaleta, P. I. Dron, K. Zhao, Y. Shen, I. Císařová, C. T. Rogers and J. Michl, J. Org. Chem., 2015, 80, 6173–6192 CrossRef CAS PubMed.
- J. Kaleta, J. Chen, G. Bastien, M. Dračínský, M. Mašát, C. T. Rogers, B. L. Feringa and J. Michl, J. Am. Chem. Soc., 2017, 139, 10486–10498 CrossRef CAS PubMed.
- C. Santos Hurtado, G. Bastien, M. Mašát, J. R. Štoček, M. Dračínský, I. Rončević, I. Císařová, C. T. Rogers and J. Kaleta, J. Am. Chem. Soc., 2020, 142, 9337–9351 CrossRef CAS PubMed.
- C. Santos Hurtado, G. Bastien, I. Rončević, M. Dračínský, E. Tortorici, C. Rogers, J. Michl and J. Kaleta, Chem. Commun., 2024, 60, 960–963 RSC.
- S. Liu, C. Ruspic, P. Mukhopadhyay, S. Chakrabarti, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2005, 127(45), 15959–15967 CrossRef CAS PubMed.
- K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44(2), 394–418 RSC.
- D. Thuau, M. Abbas, G. Wantz, L. Hirsch, I. Dufour and C. Ayela, Org. Electron., 2017, 40, 30–35 CrossRef CAS.
- A. Sultana, M. M. Alam, S. Fabiano, X. Crispin and D. Zhao, J. Mater. Chem. A, 2021, 9, 22418–22427 RSC.
- G. Zhao, H. He, S. Xinwei, X. Pinghua, Z. Ni, C. Junhao and D. Chungang, Adv. Electron. Mater., 2020, 6, 1900818 CrossRef.
- D. Lončarić, F. Movahedifar, J. R. Štoček, M. Dračínský, J. Cvačka, G. Shanshan, B. Bythell, I. Císařová, E. Masson and J. Kaleta, Chem. Sci., 2023, 14, 9258–9266 RSC.
- A. Comotti, R. Simonutti, S. Stramare and P. Sozzani, Nanotechnology, 1999, 10, 70–76 CrossRef CAS.
- K. Zhao, P. I. Dron, J. Kaleta, C. T. Rogers and J. Michl, Top. Curr. Chem., 2014, 354, 163–212 CrossRef CAS PubMed.
- A. Comotti, S. Bracco, L. Ferretti, M. Mauri, R. Simonutti and P. Sozzani, Chem. Commun., 2007, 350–352 RSC.
- S. Grimme, C. Bannwarth and P. Shushkov, J. Chem. Theory Comput., 2017, 13, 1989–2009 CrossRef CAS PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- S. Grimme, J. Chem. Theory Comput., 2019, 15, 2847–2862 CrossRef CAS PubMed.
- R. M. Pitzer, Acc. Chem. Res., 1983, 16, 207–210 CrossRef CAS.
- T. K. Brunck and F. Weinhold, J. Am. Chem. Soc., 1979, 101, 1700–1709 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures describing the synthesis of all compounds described; copies of 1H and 13C NMR spectra of all new derivatives; ORTEP views of a single molecule and packing in 3 crystal structures (6, 8, and 10); DSC data, and 31P solid-state NMR spectra of 1·CB[7]@TPP-d12. Optimized geometry of supramolecular complex 12·CB[7] single molecules shown in Fig. 5 (xyz). CCDC 2416417, 2416418 and 2416416. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc03152d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |