
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of symmetrical and unsymmetrical cyclic diborenes via NHC-directed C–H borylation†
Cornelius
Mihm
ab,
Sourav
Kar
 ab,
Andreas
Sachs
ab,
Dario
Duwe
ab,
Rian D.
Dewhurst
ab and
Holger
Braunschweig
*ab
ab,
Andreas
Sachs
ab,
Dario
Duwe
ab,
Rian D.
Dewhurst
ab and
Holger
Braunschweig
*ab
aInstitute for Inorganic Chemistry, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
bInstitute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 27th May 2025
Abstract
Cyclic diborenes are recognized for their diverse reactivity and properties arising partly due to their ring strain. However, progress in this field has been relatively slow due to the unavailability of suitable synthons for their synthesis. In this study, we describe the synthesis of both symmetrical and unsymmetrical cyclic diborenes using an N-heterocyclic carbene (NHC) as a chelating agent. The synthesis involves various haloboranes and diboranes that induce spontaneous and reductive C–H borylation of an aryl substituent of the NHC. Interestingly, the 1,1-(geminal) chelated isomer of the symmetrical diborane undergoes isomerization to form its 1,2-(vicinal) chelated isomer. Furthermore, the 1,1-(geminal) isomer exhibits a dihedral angle of 44°, making it the most twisted example of all established diborenes. Furthermore, the unsymmetrical cyclic diborene showed a polar B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) B double bond.
B double bond.
Introduction
Since the remarkable discovery of a neutral diborene by Robinson and co-workers in 2007,1 the chemistry of Lewis-base-stabilized diborenes has greatly expanded owing to their extensive reactivity and interesting properties.2–10 In this regard, the introduction of chelating ligands to diborenes has led to the isolation of various cyclic diborene species (I–VII; Fig. 1).7,11–15 These cyclic frameworks showed diverse reactivities owing to ring strain and/or their relatively sterically unencumbered environment.7,12–18 They can be classified into two groups, namely symmetrical (Fig. 1A) and unsymmetrical (Fig. 1B) cyclic diborene species. The latter offers polarized BB double bonds, which opens more reactivity opportunities. For example, unsymmetrical diborene V takes part in catenation and hydrophosphination,18,19VI acts as a highly efficient catalyst in hydroboration,7 and VII undergoes hydroboration, reaction with P4, and terminal alkyne activation.14 Interestingly, the unsymmetrical cyclic diborene VII was synthesized by reducing a cyclic diborane, which is a product of C–H borylation.14
 | ||
| Fig. 1 Examples of various symmetrical (A) and unsymmetrical (B) diborenes. Mes = mesityl, IiPr = 1,3-bis(diisopropyl)imidazol-2-ylidene; Dipp = 2,6-iPr2C6H3; IMeMe = [:C{N(CH3)C(CH3)}2]; CAAC = 1-(2,6-diisopropylphenyl)-3,3,5,5-tetramethylpyrrolidin-2-ylidene. | ||
The synthesis and chemistry of borylated organic species have been considerably advanced during the last few decades owing to the significance of the C–B bond in the functionalization of organic molecules.20–28 Although transition-metal-catalyzed borylation and diboration have emerged as promising synthetic routes, the environmental impact of the transition metals and the complexity associated with their removal have caused concern.21–28 Consequently, metal-free alternatives to catalytic borylation and diboration processes have been intensely investigated.29–41 Highly reactive boron reagents such as tetrachlorodiborane(4) and BH3 have been found to functionalize the double bond of alkenes in the absence of catalyst.42,43 Recent studies revealed that the introduction of Lewis bases such as amines, phosphines, or N-heterocyclic carbenes (NHC) activates boranes or diboranes for intra- or intermolecular borylation.29,31,32,35,41 Especially the intramolecular borylation of suitable Lewis base adducts of polyhalo-substituted boranes or diboranes may lead to the formation of chelated boranes or diboranes with labile halides, thus providing further reducing opportunities to form diborene species. For example, diborene VII was synthesized from a chelated diborane with labile bromides, which is the product of intramolecular C–H borylation of a CAAC-substituted tribromodiborane.14 In this work, we present the synthesis of cyclic dibromodiborane and diiodoborane species using an N-heterocyclic carbene (NHC) via spontaneous C–H borylation, which further underwent reduction to give symmetrical cyclic diborene. We also present the synthesis of an unsymmetrical cyclic diborene species utilizing a reductive C–H borylation process.
Results and discussion
In order to isolate diborene species utilizing the C–H borylation process, we have employed 1-(2,6-diisopropylphenyl)-3-(2-methylphenyl)-imidazolin-2-ylidene, [SIDippTol] (1) having both a carbene carbon center and an electrophilic sp2 C–H unit at the ortho position of the tolyl residue, in combination with various borane/diboranes. The room-temperature reaction of bis(dimethylsulfide)tetrabromodiborane, [B2Br4(SMe2)2],44 with four equivalents of 1 led to the formation of diborane 2 (Scheme 1). The single-crystal X-ray diffraction (SCXRD) analysis of 2 revealed the presence of two derivatized [SIDippTol] units symmetrically chelating the diboron unit and two terminal bromides in a staggered conformation (Fig. 2). The [SIDippTol] units were found to chelate the B2 unit in 1,2-(vicinal) fashion, forming six-membered rings on either side of the B2 unit. A single 11B NMR spectroscopic signal was noted for 2 at δ = −7.8 ppm and a highly symmetrical 1H NMR spectrum confirmed the symmetrical chelation of the derivatized [SIDippTol] units. The B–B bond distance of 2 (1.784(4) Å) is longer than that of diborane 1 (1.715(4) Å),44 but is comparable with that of the related chelated diborane [BH(hpp)]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 45 (1.772(3) Å) (hpp = 1,3,4,6,7,8-hexahydro-2H-pyrimidol[1,2-a]pyrimidinate) of Himmel and coworkers. Although the B–CTol bonds (B1–C2: 1.612(4) Å, B2–C4: 1.618(4) Å) are slightly longer than the B–CCarbene bonds (B1–C1: 1.579(4) Å, B2–C3: 1.584(4) Å), both are in the typical range of B–C single bonds. The formation of 2 is clearly the result of C–H borylation and presumably proceeds via the substitution of two SMe2 groups by two [SIDippTol] units. The observed product suggests that ortho-CH unit of the tolyl residue of the bis-[SIDippTol]-diborane adduct undergoes spontaneous C–H borylation with HBr elimination, promoted by the Brønsted basicity of the carbene SIDippTol. A similar reaction mechanism was observed during the synthesis of ItBu-chelated dibromoborane (ItBu = 1,3-di-tert-butylimidazol-2-ylidene).46
45 (1.772(3) Å) (hpp = 1,3,4,6,7,8-hexahydro-2H-pyrimidol[1,2-a]pyrimidinate) of Himmel and coworkers. Although the B–CTol bonds (B1–C2: 1.612(4) Å, B2–C4: 1.618(4) Å) are slightly longer than the B–CCarbene bonds (B1–C1: 1.579(4) Å, B2–C3: 1.584(4) Å), both are in the typical range of B–C single bonds. The formation of 2 is clearly the result of C–H borylation and presumably proceeds via the substitution of two SMe2 groups by two [SIDippTol] units. The observed product suggests that ortho-CH unit of the tolyl residue of the bis-[SIDippTol]-diborane adduct undergoes spontaneous C–H borylation with HBr elimination, promoted by the Brønsted basicity of the carbene SIDippTol. A similar reaction mechanism was observed during the synthesis of ItBu-chelated dibromoborane (ItBu = 1,3-di-tert-butylimidazol-2-ylidene).46
 | ||
| Scheme 1 Synthesis of symmetrical cyclic diborene species 3 and 5 involving NHC-directed spontaneous C–H borylation steps. | ||
 | ||
| Fig. 2 Crystallographically-determined molecular structures of 2, 4, 5 and 6 (ellipsoids shown at the 50% probability level). All hydrogen atoms and ellipsoids of ligand periphery are omitted for clarity. | ||
The presence of two terminal bromides and two neutral carbene donors in diborane 2 suggested that it could be reduced to a diborene under reducing conditions. Accordingly, room-temperature reaction of diborane 2 with 2.1 equivalents of KC8 in benzene and THF mixture afforded a red solid 3 in 65% yield (Scheme 1). The 1H and 13C NMR signal patterns of compound 3 are almost identical to those of diborane 2. However, the 11B NMR spectrum of 3 shows a broad chemical shift at δ = 31.7 ppm, significantly downfield of that of diborane 2. These NMR spectroscopic data suggested the elimination of two bromides and the formation of symmetrical diborene, while keeping the rest of the framework the same as diborane 2. Also, the 11B NMR spectroscopic signal of 3 is comparable with that of reported symmetrical cyclic diborenes II (δ11B = 29.3 ppm)11 and III (δ11B = 28.4 ppm),12 further supporting the identity of 3 as a diborene. After multiple attempts, we obtained a few crystals of 3 that upon SCXRD analysis confirmed the formation of symmetrical bicyclic diborene, where the [SIDippTol] groups chelate the B2 unit in 1,2-(vicinal) fashion on both sides of the BB bond (Fig. S27†). However, due to the poor data quality, the structural parameters of 3 cannot be discussed in detail.
In order to gain some insight into the structure and bonding of 3, we optimized its structure using the density functional theory (DFT) calculations at the ωB97X-D/Def2-SVP level of theory. The B–B bond in optimized 3 (1.611 Å) is slightly longer than those of reported symmetrical cyclic diborenes I (1.583(2) Å),11II (1.593(2) Å),11III (1.602(2) Å)12 and IV (1.585(3) Å).13 Further, the calculated Wiberg bond index (WBI) and Mayer bond order (MBO) for the B–B bond in 3, both 1.19, support the double bonding character. Also, the calculated 11B NMR chemical shift of 3 (δ = 29.3 ppm) is in line with that of the experimental one (δ = 31.7 ppm). Further, the highest occupied molecular orbital (HOMO) of 3 corresponds to the BB π-bonding orbital with some delocalization to the formally empty p orbital of the carbene carbon atoms (Fig. 3), which is also supported by NBO analysis (Fig. S33†). The LUMO mainly depicts the π-type orbitals between boron and carbene carbon atoms.
 | ||
| Fig. 3 Selected frontier molecular orbitals of diborenes 3, 5 and 7 and their HOMO–LUMO energy gaps. All hydrogen atoms and ellipsoids of ligand periphery are omitted for clarity. | ||
In order to determine which absorptions are responsible for the intense red color of diborene 3, we recorded its UV-vis absorption spectrum, revealing three discrete absorptions at λ = 516, 416, and 359 nm (Fig. S21†). Time-dependent (TD) DFT calculations (Table S1 and Fig. S39†) were then used to identify these bands. The absorption band at λ = 516 nm corresponds to the HOMO → LUMO transition, which is mainly the excitation of πB–B electrons to the πB–Carbene orbitals. The absorption bands at λ = 416 and 359 nm mainly correspond to the HOMO → LUMO+n (n = 1 and 3) and HOMO−1 → LUMO transitions, respectively (Fig. S32†).
In an endeavor to prepare the 1,1-(geminal) chelated isomer of diborene 3, we selected a route via the coupling of a probable 1,1-[SIDippTol]-chelated borylene, which may be generated from the corresponding 1,1-[SIDippTol]-chelated dihaloborane. Thereby, we treated boron triiodide with two equivalents of SIDippTol at room temperature in the absence of light (Scheme 1). Instantly, the reaction mixture showed a very high-field 11B NMR resonance at δ = −74.7 ppm, comparable with the chemical shifts of BI3 adducts of NMe3 (δ11B = −54.2 ppm)47 and PCy3 (δ11B = −74.1 ppm),48 which suggests the formation of an adduct between BI3 and SIDippTol (Scheme S1†). However, with time, this 11B chemical shift slowly disappeared, and simultaneously, the 11B signal at δ = −36.6 ppm became dominant. After 16 hours, a complete conversion to the latter 11B chemical shift was observed, which was isolated as a colorless solid 4 in 53% yield (Scheme 1). A SCXRD analysis of 4 revealed compound 4 as the 1,1-SIDippTol-chelated diiodoborane, where the boron atom is embedded in a five-membered ring spanned by the SIDippTol unit (Fig. 2). The B–CCarbene (1.615(6) Å) and B–CTol (1.594(5) Å) bond lengths are comparable with that of diborane 2 and fall in the range of single B–C bonds. The formation of geminally-chelated diiodoborane 4 is clearly the result of a spontaneous C–H borylation reaction, involving a BI3·SIDippTol adduct intermediate (Scheme S1†).
We then treated 1,1-SIDippTol-chelated diiodoborane 4 with two equivalents of KC8 and excess dimethyl sulfide in benzene. After 16 hours, the 11B NMR spectrum of the green reaction mixture revealed the formation of compound 5 with a broad resonance at δ = 20.3 ppm (Scheme 1), comparable to that of geminal-phosphine-chelated diborene IV (δ11B = 22.8 ppm).13 However, leaving 5 to sit in any common solvent leads to a color change from green to deep red, along with a new 11B NMR chemical shift at δ = 31.7 ppm (complete conversion needs 3 days at rt or 10 days at −30 °C), which corresponds to the 1,2-SIDippTol-chelated diborene 3. These observations suggest the identity of 5 as the 1,1-(geminal) isomer of diborene 3. As the conversion of 5 to 3 initiates within a short time, attempts to characterize the former by NMR spectroscopy were complicated by the presence of signals of the latter. However, a few single crystals of 5 were obtained by rapid crystallization in diethyl ether at −30 °C.
SCXRD analysis confirmed compound 5 as the 1,1-(geminal) isomer of diborene 3, in which each SIDippTol unit formed a five-membered ring through chelation with each boron center (Fig. 2). The B–B bond length of 1.640(3) Å in 5 falls in the longer range of reported BB double bonds (1.524(6)–1.679(9) Å).6,49,50 Further, the WBI and MBO of 1.29 and 1.25, respectively, for the B–B bond in 5 suggest its double bond character. Interestingly, the diborene unit in 5 is twisted (dihedral angle = 44°, Fig. 4), probably due to the steric overloading of the ligand periphery with both Dipp substituents. Notably, compound 5 is the most twisted diborene of all known diborenes by some margin. The only other example of a twisted diborene is VIII, having a dihedral angle of 18.1° (Fig. 4).49 Note that recently, we reported twisted congeners of diborene, in which the B–B π bond was completely disrupted and the species were biradicals (Fig. 4).51 The twisting around the diborene unit and the ring tension originating from the five-membered ring in geminal isomer 5 are the likely causes of its isomerization to vicinal isomer 3. The higher thermodynamic stability of the vicinal isomer 3 is also reflected by the fact that it is 8.74 kcal mol−1 more stable than the geminal isomer 5 (Gibbs free energies calculated at 298 K). This geminal-to-vicinal isomerization of diborene is the second example of its type, earlier observed only for phosphine-stabilized diborene IV.13 Despite the conspicuous twist in its geometry, the HOMO of geminal diborene 5 reflects π bonding between the two boron centers, with a partial delocalization across the spanned π-system like its vicinal isomer 3 (Fig. 3). However, the HOMO of geminal diborene 5 is less stable than that of its vicinal isomer 3 and the HOMO–LUMO gap of vicinal diborene 3 (6.09 eV) is higher than that of geminal diborene 5 (5.01 eV). This indicates a more reactive nature of the geminal isomer, which is in accord with the rapid isomerization to the vicinal isomer.
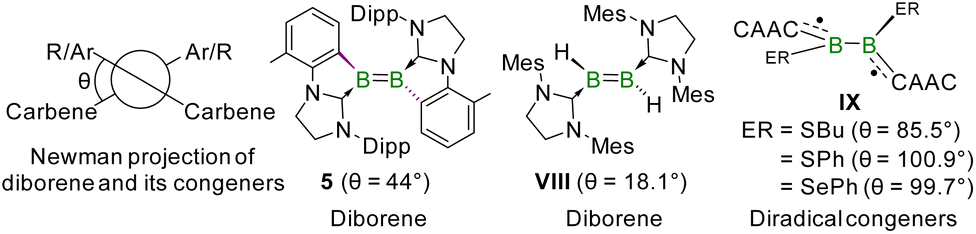 | ||
| Fig. 4 Dihedral angles (θ) between two Ar/R–B–carbene planes of selected diborenes and related diradical species. | ||
On the other hand, the presence of two iodides in cyclic diiodoborane 4 suggests that two-electron reduction of 4 in the presence of another appropriate Lewis base under suitable conditions may lead to the formation of another interesting class of compounds, i.e., a doubly base-stabilized borylene. Thus, we employed another Lewis base, namely IiPr (IiPr = 1,3-bis(diisopropyl)imidazol-2-ylidene). Indeed, the room-temperature reaction of diiodoborane 4 with 1.1 equivalents of IiPr, followed by the addition of two equivalents of KC8, led to the formation of doubly Lewis-base-stabilized borylene 6 in 71% yield (Scheme 1). The SCXRD analysis of 6 showed a doubly Lewis base-stabilized cyclic borylene, in which the SIDippTol unit chelates the boron center and the second base IiPr forms a dative bond to the boron atom (Fig. 2). Notably, neutral bis(NHC)-stabilized borylenes of this type remain quite rare in the literature.52–54 In 2020, we reported the first example of a doubly NHC-stabilized borylene, [(IiPr)2B{(C5H4)Mn(CO)3}], marking a significant advancement in the field.52 More recently, So and co-workers described a bis(IMeMe)-disilylamidoborylene capable of capturing and subsequently functionalizing CO2.53 Additionally, Driess and coworkers reported a bis(borylene)xanthene complex, in which the borylene center is stabilized by two IMeMe ligands.54 The Ccarbene–B bond distance in borylene 6 (C1–B1: 1.491(2) Å) falls within the range of B–C double bond distances, but is longer than that of a reported mixed CAAC/NHC-stabilized borylene (1.458(2) Å), suggesting lower π-acceptor ability of the SIDippTol ligand.55 Further, the WBI and MBO of the C(SIDippTol)–B bond (1.40 and 1.51, respectively) in borylene 6 indicate distinct multiple bonding. The NBO analysis also shows a π bond between the carbene carbon of SIDippTol and the boron atom (Fig. S36†). In contrast, the IiPr ligand acts more as a pure σ donor (C(IiPr)–B: 1.552(2) Å), which is also reflected in NBO analysis (Fig. S36†). Also, the WBI (1.06) and MBO (1.10) for the C(IiPr)–B support the σ bonding interaction. Furthermore, the NICS(1)zz and NICS(−1)zz values of −13.0 and −11.5, respectively, indicate the presence of π aromaticity in the five-membered BNC3 ring of 6. However, this π aromaticity is notably weaker than that of benzene, which exhibits significantly more negative NICS(1)zz and NICS(−1)zz values (−29.5 for both).
The 11B NMR spectrum of 6 showed a resonance at δ = −4.5 ppm, which is upfield of that of CAAC-stabilized borylene complexes bearing NHC (δ11B = 12.0 ppm)55 or pyridine (δ11B = 23.3 ppm)55 ligands, suggesting a higher electron density at the boron center of 6. This correlates well with the comparatively weaker π-acceptor strength of the SIDippTol ligand than CAAC, which is also reflected in the solid-state structure.
Further, the UV-vis absorption study of borylene 6 revealed two discrete absorptions at λ = 450 and 371 nm (Fig. S22†). TD-DFT calculations (Table S2 and Fig. S40†) revealed that the absorption band at λ = 450 nm corresponds to the HOMO → LUMO+2 transition, which is mainly the excitation of the π electrons associated with the C(NHC)–B–C(NHC) unit to the antibonding interaction in the N–C–N unit of the IiPr ligand. The other two absorption bands at λ = 371 nm can be assigned to the HOMO → LUMO+4 transition (Fig. S35†).
In an attempt to isolate a diborene in a similar manner to that of diborene 3, we explored the reaction of carbene 1 with another diborane tetrahalide, namely [B2Cl4(SMe2)2],44 however, no spontaneous C–H borylation was observed. The room-temperature reaction of [B2Cl4(SMe2)2] with 2.1 equivalents of SIDippTol in a benzene/THF mixture for 5 h, followed by a reaction with 4.2 equivalents of KC8 for 10 h led to the formation of the dark purple solid 7, which was identified as an unsymmetrical cyclic diborene in 49% yield (Scheme 2). Note that the same reaction with two equivalents of KC8 led to the formation of a dark green solution instantly, which showed a broad 11B NMR resonance at δ = 30.1 ppm, reminiscent of that of 1,2-dichlorodiborene [B2(IDipp)2Cl2] (δ11B = 26.8 ppm).56 Although this species could not be isolated due to its high solubility in all common solvents, the above 11B NMR study, as well as high-resolution HRMS measurement, suggest the formation of [B2Cl2(SIDippTol)2] (Scheme S2†). The further reduction of this green compound with two equivalents of KC8 led to the formation of diborene 7.
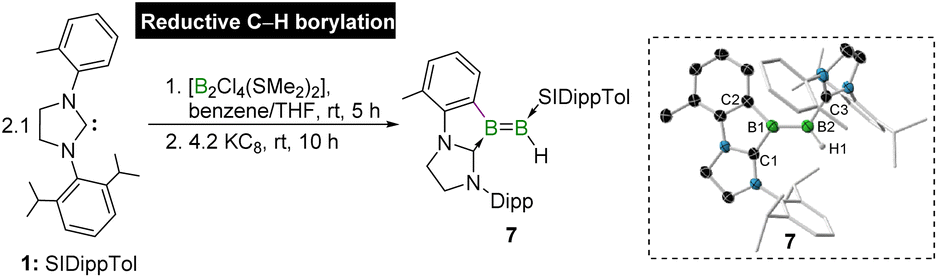 | ||
| Scheme 2 Synthesis of unsymmetrical cyclic diborene 7via NHC-directed reductive C–H borylation. Inset: crystallographically-determined molecular structure of 7 (ellipsoids shown at the 50% probability level). All hydrogen atoms and ellipsoids of ligand periphery are omitted for clarity. | ||
The 11B NMR signals of 7 were found at δ = 37.3 and 17.7 ppm, which fall in the range of 11B chemical shifts for diborene species.11,56,57 The difference of around 20 ppm in the values of 11B chemical shift typically suggests unsymmetrical substitutions at the boron atoms. Indeed, the SCXRD analysis of 7 showed a diborene species having substituents in an unsymmetrical pattern (Scheme 2, inset). One of the SIDippTol substituents chelates one of the boron atoms (B1) to form a five-membered ring, while the other SIDippTol unit is coordinated to the other (B2). The formation of diborene 7via two-electron reduction of the intermediate [B2Cl2(SIDippTol)2] can proceed through two possible pathways. One pathway involves reductive C–H borylation, where reducing conditions promote both the elimination of two chloride ligands from the diborene intermediate [B2Cl2(SIDippTol)2] and the intramolecular activation of an ortho C(sp2)–H bond of one of the SIDippTol ligands, leading to the formation of compound 7. Alternatively, diboryne formation could occur first, followed by spontaneous C–H borylation across the B![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) B triple bond. However, no evidence for the presence of a diboryne intermediate was found at any stage of the reaction. Thus, the formation of diborene 7 is most likely the result of a direct reductive C–H borylation process. The hydrogen atom abstracted by the C–H borylation is found in trans to the aryl substituent. The presence of an IR stretching band at 2432 cm−1 further supports the presence of the terminal B–H unit in diborene 7. The trans orientation is likely favoured due to the steric hindrance between the Dipp unit of the chelated SIDippTol ligand and the datively-bound SIDippTol ligand in its cis isomer. The B–B bond length of 1.588(4) Å in unsymmetrical diborene 7 is significantly shorter than that of symmetrical diborene 5, suggesting a stronger BB double bond in 7.
B triple bond. However, no evidence for the presence of a diboryne intermediate was found at any stage of the reaction. Thus, the formation of diborene 7 is most likely the result of a direct reductive C–H borylation process. The hydrogen atom abstracted by the C–H borylation is found in trans to the aryl substituent. The presence of an IR stretching band at 2432 cm−1 further supports the presence of the terminal B–H unit in diborene 7. The trans orientation is likely favoured due to the steric hindrance between the Dipp unit of the chelated SIDippTol ligand and the datively-bound SIDippTol ligand in its cis isomer. The B–B bond length of 1.588(4) Å in unsymmetrical diborene 7 is significantly shorter than that of symmetrical diborene 5, suggesting a stronger BB double bond in 7.
Further, the comparatively higher WBI (1.34) and MBO (1.23) of the B–B bond in the unsymmetrical diborene 7 suggest stronger multiple bonding than that of symmetrical diborenes 3 and 5. In addition, the HOMO of 7 shows significant π conjugation between the BB bond and the carbene carbon atom of the chelating SIDippTol ligand (Fig. 3), which is also reflected in NBO analysis (Fig. S38†). Whereas the other SIDippTol ligand functions mainly as a σ donor as shown by NBO analysis (Fig. S38†). Further, the natural population analysis of 7 showed a more negative natural charge (qB1 = −0.08) at the boron atom embedded in the five-membered ring than the other boron atom (qB2 = −0.05), suggesting the presence of comparatively higher electron density at B1. This polarization of the BB double bond of diborene 7 is also reflected in the experimental (δ = 37.3 and 17.7 ppm) and calculated (δ = 36.1 and 19.0 ppm) 11B NMR spectra.
In contrast to geminal diborene 5, the unsymmetrical diborene 7 does not undergo isomerization and remains thermally stable up to 80 °C. However, at elevated temperatures around 100 °C, slow decomposition of the compound is observed.
To determine the origins of the intense purple color of unsymmetrical diborene 7, we recorded its UV-vis absorption spectrum, which revealed two absorption bands at λ = 579 and 351 nm (Fig. S23†). The purple color of the compound likely arises from the absorption at 579 nm. TD-DFT calculations revealed that the absorption band at λ = 579 nm corresponds to the HOMO → LUMO transition, which mainly consists of the excitation of π(B–B) electrons to the π(B–CNHC) orbitals (Fig. 3).
Conclusions
In summary, we have successfully isolated and characterized symmetrical (3 and 5) and unsymmetrical (7) NHC-supported cyclic diborenes. Their syntheses involve spontaneous or reductive C–H borylation processes, leading to ring formation through chelation. The weaker π-acceptance and strong σ-donor ability of the SIDippTol ligand, along with the presence of C(sp2)–H bonds at the tolyl substituents, played a pivotal role in stabilizing these unique diborenes. The synthesis of the 1,1-(geminal)-chelated diborene demonstrates that the preparation of highly twisted diborenes is possible; however, it is highly reactive and prone to isomerization to its 1,2-(vicinal)-chelated isomer. In contrast, the presence of different substituents and the lack of symmetry introduce polarity in the BB double bond of 7. Computational studies provided valuable insights into the bonding and properties of these diborenes. Additionally, we were able to isolate a neutral bis(NHC)-stabilized borylene.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Supervision, funding acquisition, conceptualization: H. B.; conceptualization, investigation, writing – original draft: C. M.; formal analysis, visualization, writing – original draft: S. K.; investigation, formal analysis: D. D.; investigation: A. S.; writing – review & editing: R. D. D.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (grant numbers BR1149/22-2 and 466754611) is gratefully acknowledged. S. K. thanks the Alexander von Humboldt Foundation for a postdoctoral fellowship.Notes and references
- Y. Wang, B. Quillian, P. Wei, C. S. Wannere, Y. Xie, R. B. King, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, A Stable Neutral Diborene Containing a B=B Double Bond, J. Am. Chem. Soc., 2007, 129, 12412–12413 CrossRef CAS PubMed.
- H. Braunschweig and R. D. Dewhurst, Boron–Boron Multiple Bonding: From Charged to Neutral and Back Again, Organometallics, 2014, 33, 6271–6277 CrossRef CAS.
- P. Bissinger, A. Steffen, A. Vargas, R. D. Dewhurst, A. Damme and H. Braunschweig, Unexpected Luminescence Behavior of Coinage Metal π-Diborene Complexes, Angew. Chem., Int. Ed., 2015, 54, 4362–4366 CrossRef CAS PubMed.
- M. Arrowsmith, H. Braunschweig and T. E. Stennett, Formation and Reactivity of Electron-Precise B–B Single and Multiple Bonds, Angew. Chem., Int. Ed., 2017, 56, 96–115 CrossRef CAS PubMed.
- W. Lu, Y. Li, R. Ganguly and R. Kinjo, Alkene–Carbene Isomerization induced by Borane: Access to an Asymmetrical Diborene, J. Am. Chem. Soc., 2017, 139, 5047–5050 CrossRef CAS PubMed.
- R. Borthakur, K. Saha, S. Kar and S. Ghosh, Recent advances in transition metal diborane(6), diborane(4) and diborene(2) chemistry, Coord. Chem. Rev., 2019, 399, 213021 CrossRef CAS.
- J. Fan, J.-Q. Mah, M.-C. Yang, M.-D. Su and C.-W. So, A N-Phosphinoamidinato NHC-Diborene Catalyst for Hydroboration, J. Am. Chem. Soc., 2021, 143, 4993–5002 CrossRef PubMed.
- M. Härterich, B. Ritschel, M. Arrowsmith, J. Böhnke, I. Krummenacher, A. K. Phukan and H. Braunschweig, Hybrid Inorganic–Organic Cross-Metathesis between Diborenes and Acetylene, J. Am. Chem. Soc., 2021, 143, 18339–18345 CrossRef PubMed.
- A. Okorn, A. Jayaraman, L. Englert, M. Arrowsmith, T. Swoboda, J. Weigelt, C. Brunecker, M. Hess, A. Lamprecht, C. Lenczyk, M. Rang and H. Braunschweig, Synthesis and hydrogenation of polycyclic aromatic hydrocarbon-substituted diborenes via uncatalysed hydrogenative B–C bond cleavage, Chem. Sci., 2022, 13, 7566–7574 RSC.
- W. Lu and R. Kinjo, Complexation of asymmetric diborenes with magnesium bromide, Chem. Commun., 2018, 54, 8842–8844 RSC.
- P. Bissinger, H. Braunschweig, M. A. Celik, C. Claes, R. D. Dewhurst, S. Endres, H. Kelch, T. Kramer, I. Krummenacher and C. Schneider, Synthesis of cyclic diborenes with unprecedented cis-configuration, Chem. Commun., 2015, 51, 15917–15920 RSC.
- H. Braunschweig, I. Krummenacher, C. Lichtenberg, J. D. Mattock, M. Schäfer, U. Schmidt, C. Schneider, T. Steffenhagen, S. Ullrich and A. Vargas, Dibora[2]ferrocenophane: A Carbene-Stabilized Diborene in a Strained cis-Configuration, Angew. Chem., Int. Ed., 2017, 56, 889–892 CrossRef CAS PubMed.
- T. E. Stennett, J. D. Mattock, L. Pentecost, A. Vargas and H. Braunschweig, Chelated Diborenes and Their Inverse-Electron-Demand Diels-Alder Reactions with Dienes, Angew. Chem., Int. Ed., 2018, 57, 15276–15281 CrossRef CAS PubMed.
- T. E. Stennett, J. D. Mattock, I. Vollert, A. Vargas and H. Braunschweig, Unsymmetrical, Cyclic Diborenes and Thermal Rearrangement to a Borylborylene, Angew. Chem., Int. Ed., 2018, 57, 4098–4102 CrossRef CAS PubMed.
- W. Lu, A. Jayaraman, F. Fantuzzi, R. D. Dewhurst, M. Härterich, M. Dietz, S. Hagspiel, I. Krummenacher, K. Hammond, J. Cui and H. Braunschweig, An Unsymmetrical, Cyclic Diborene Based on a Chelating CAAC Ligand and its Small-Molecule Activation and Rearrangement Chemistry, Angew. Chem., Int. Ed., 2022, 61, e202113947 CrossRef CAS PubMed.
- M. Dömling, M. Arrowsmith, U. Schmidt, L. Werner, A. C. Castro, J. O. C. Jiménez-Halla, R. Bertermann, J. H. Müssig, D. Prieschl and H. Braunschweig, Spontaneous trans-Selective Transfer Hydrogenation of Apolar Boron-Boron Double Bonds, Angew. Chem., Int. Ed., 2019, 58, 9782–9786 CrossRef PubMed.
- U. Schmidt, L. Werner, M. Arrowsmith, A. Deißenberger, A. Hermann, A. Hofmann, S. Ullrich, J. D. Mattock, A. Vargas and H. Braunschweig, Trans-selective Insertional Dihydroboration of a cis-Diborene: Synthesis of Linear sp3–sp2–sp3-Triboranes and Subsequent Cationization, Angew. Chem., Int. Ed., 2020, 59, 325–329 CrossRef CAS PubMed.
- T. E. Stennett, A. Jayaraman, T. Brückner, L. Schneider and H. Braunschweig, Hydrophosphination of boron-boron multiple bonds, Chem. Sci., 2020, 11, 1335–1341 RSC.
- T. E. Stennett, R. Bertermann and H. Braunschweig, Construction of Linear and Branched Tetraboranes by 1,1- and 1,2-Diboration of Diborenes, Angew. Chem., Int. Ed., 2018, 57, 15896–15901 CrossRef CAS PubMed.
- H. C. Brown, Hydroboration—a powerful synthetic tool, Tetrahedron, 1961, 12, 117–138 CrossRef CAS.
- I. Beletskaya and A. Pelter, Hydroborations catalysed by transition metal complexes, Tetrahedron, 1997, 53, 4957–5026 CrossRef CAS.
- T. B. Marder and N. C. Norman, Transition metal catalysed diboration, Top. Catal., 1998, 5, 63–73 CrossRef CAS.
- H. Chen, S. Schlecht, T. Semple and J. F. Hartwig, Thermal, Catalytic, Regiospecific Functionalization of Alkanes, Science, 2000, 287, 1995–1997 CrossRef CAS PubMed.
- H. Braunschweig, R. D. Dewhurst and A. Schneider, Electron-Precise Coordination Modes of Boron-Centered Ligands, Chem. Rev., 2010, 110, 3924–3957 CrossRef CAS PubMed.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, C–H Activation for the Construction of C–B Bonds, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- J. F. Hartwig, Regioselectivity of the borylation of alkanes and arenes, Chem. Soc. Rev., 2011, 40, 1992–2002 RSC.
- E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Diboron(4) Compounds: From Structural Curiosity to Synthetic Workhorse, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS PubMed.
- S. K. Bose, L. Mao, L. Kuehn, U. Radius, J. Nekvinda, W. L. Santos, S. A. Westcott, P. G. Steel and T. B. Marder, First-Row d-Block Element-Catalyzed Carbon–Boron Bond Formation and Related Processes, Chem. Rev., 2021, 121, 13238–13341 CrossRef CAS PubMed.
- K.-S. Lee, A. R. Zhugralin and A. H. Hoveyda, Efficient C–B Bond Formation Promoted by N-Heterocyclic Carbenes: Synthesis of Tertiary and Quaternary B-Substituted Carbons through Metal-Free Catalytic Boron Conjugate Additions to Cyclic and Acyclic α,β-Unsaturated Carbonyls, J. Am. Chem. Soc., 2009, 131, 7253–7255 CrossRef CAS PubMed.
- F. Mo, Y. Jiang, D. Qiu, Y. Zhang and J. Yang, Direct Conversion of Arylamines to Pinacol Boronates: A Metal-Free Borylation Process, Angew. Chem., Int. Ed., 2010, 49, 1846–1849 CrossRef CAS PubMed.
- A. Bonet, H. Gulyás and E. Fernández, Metal-Free Catalytic Boration at the β-Position of α,β-Unsaturated Compounds: A Challenging Asymmetric Induction, Angew. Chem., Int. Ed., 2010, 49, 5130–5134 CrossRef CAS PubMed.
- A. Bonet, C. Pubill-Ulldemolins, C. Bo, H. Gulyás and E. Fernández, Transition-Metal-Free Diboration Reaction by Activation of Diboron Compounds with Simple Lewis Bases, Angew. Chem., Int. Ed., 2011, 50, 7158–7161 CrossRef CAS PubMed.
- M. Gao, S. B. Thorpe, C. Kleeberg, C. Slebodnick, T. B. Marder and W. L. Santos, Structure and Reactivity of a Preactivated sp2–sp3 Diboron Reagent: Catalytic Regioselective Boration of α,β-Unsaturated Conjugated Compounds, J. Org. Chem., 2011, 76, 3997–4007 CrossRef CAS PubMed.
- H. Braunschweig, A. Damme and T. Kupfer, Synthesis of a bicyclic diborane by selective boron carbon bond formation, Chem. Commun., 2013, 49, 2774–2776 RSC.
- J. M. Farrell and D. W. Stephan, Planar N-Heterocyclic Carbene Diarylborenium Ions: Synthesis by Cationic Borylation and Reactivity with Lewis Bases, Angew. Chem., Int. Ed., 2015, 54, 5214–5217 CrossRef CAS PubMed.
- M.-A. Légaré, M.-A. Courtemanche, É. Rochette and F.-G. Fontaine, Metal-free catalytic C–H bond activation and borylation of heteroarenes, Science, 2015, 349, 513–516 CrossRef PubMed.
- Q. Yin, H. F. T. Klare and M. Oestreich, Catalytic Friedel–Crafts C–H Borylation of Electron-Rich Arenes: Dramatic Rate Acceleration by Added Alkenes, Angew. Chem., Int. Ed., 2017, 56, 3712–3717 CrossRef CAS PubMed.
- J. Lv, X. Chen, X.-S. Xue, B. Zhao, Y. Liang, M. Wang, L. Jin, Y. Yuan, Y. Han, Y. Zhao, Y. Lu, J. Zhao, W.-Y. Sun, K. N. Houk and Z. Shi, Metal-free directed sp2-C–H borylation, Nature, 2019, 575, 336–340 CrossRef CAS PubMed.
- S. A. Iqbal, J. Pahl, K. Yuan and M. J. Ingleson, Intramolecular (directed) electrophilic C–H borylation, Chem. Soc. Rev., 2020, 49, 4564–4591 RSC.
- Y.-M. Tian, X.-N. Guo, H. Braunschweig, U. Radius and T. B. Marder, Photoinduced Borylation for the Synthesis of Organoboron Compounds, Chem. Rev., 2021, 121, 3561–3597 CrossRef CAS PubMed.
- O. Sadek, A. Le Gac, N. Hidalgo, S. Mallet-Ladeira, K. Miqueu, G. Bouhadir and D. Bourissou, Metal-Free Phosphorus-Directed Borylation of C(sp2)–H Bonds, Angew. Chem., Int. Ed., 2022, 61, e202110102 CrossRef CAS PubMed.
- G. Urry, J. Kerrigan, T. D. Parsons and H. I. Schlesinger, Diboron Tetrachloride, B2Cl4, as a Reagent for the Synthesis of Organo-boron Compounds. I. The Reaction of Diboron Tetrachloride with Ethylene, J. Am. Chem. Soc., 1954, 76, 5299–5301 CrossRef CAS.
- T. P. Fehlner, Reactions of Borane. II. 1 Absolute Rate of the Reaction of Borane with Ethylene in the Gas Phase, J. Am. Chem. Soc., 1971, 93, 6366–6373 CrossRef CAS.
- M. Arrowsmith, J. Böhnke, H. Braunschweig, A. Deißenberger, R. D. Dewhurst, W. C. Ewing, C. Hörl, J. Mies and J. H. Muessig, Simple solution-phase syntheses of tetrahalodiboranes(4) and their labile dimethylsulfide adducts, Chem. Commun., 2017, 53, 8265–8267 RSC.
- O. Ciobanu, P. Roquette, S. Leingang, H. Wadepohl, J. Mautz and H.-J. Himmel, Synthesis and Characterization of a New Guanidine–Borane Complex and a Dinuclear Boron(II) Hydride with Bridging Guanidinate Ligands, Eur. J. Inorg. Chem., 2007, 2007, 4530–4534 CrossRef.
- P. Bissinger, H. Braunschweig, A. Damme, R. D. Dewhurst, T. Kupfer, K. Radacki and K. Wagner, Generation of a Carbene-Stabilized Bora-borylene and its Insertion into a C–H Bond, J. Am. Chem. Soc., 2011, 133, 19044–19047 CrossRef CAS PubMed.
- J. M. Miller, Nitrogen-15 and Boron-11 Nuclear Magnetic Resonance Studies of Trimethylamine Adducts of the Mixed Trihalides of Boron, Inorg. Chem., 1983, 22, 2384–2388 CrossRef CAS.
- H. Braunschweig, K. Radacki and K. Uttinger, Syntheses of Mono- and Dinuclear Diiodoboryl Complexes of Platinum, Inorg. Chem., 2007, 46, 8796–8800 CrossRef CAS PubMed.
- Y. Wang, B. Quillian, P. Wie, Y. Xie, C. S. Wannere, R. B. King, H. F. Schaefer, P. v. R. Schleyer and G. H. Robinson, Planar, Twisted, and Trans-Bent: Conformational Flexibility of Neutral Diborenes, J. Am. Chem. Soc., 2008, 130, 3298–3299 CrossRef CAS PubMed.
- S. R. Wang, M. Arrowsmith, J. Böhnke, H. Braunschweig, T. Dellermann, R. D. Dewhurst, H. Kelch, I. Krummenacher, J. D. Mattock, J. H. Müssig, T. Thiess, A. Vargas and J. Zhang, Engineering a Small HOMO-LUMO Gap and Intramolecular B—B Hydroarylation by Diborene/Anthracene Orbital Intercalation, Angew. Chem., Int. Ed., 2017, 56, 8009–8013 CrossRef CAS PubMed.
- J. Böhnke, T. Dellermann, M. A. Celik, I. Krummenacher, R. D. Dewhurst, S. Demeshko, W. C. Ewing, K. Hammond, M. Heß, E. Bill, E. Welz, M. Röhr, R. Mitrić, B. Engels, F. Meyer and H. Braunschweig, Isolation of diborenes and their 90°-twisted diradical congeners, Nat. Commun., 2018, 9, 1197 CrossRef PubMed.
- U. Schmidt, F. Fantuzzi, M. Arrowmsith, A. Hermann, D. Prieschl, A. Rempel, B. Engels and H. Braunschweig, Tunable reduction of cymantrenylboranes to diborenes or borylene-derived boratafulvenes, Chem. Commun., 2020, 56, 14809–14812 RSC.
- J. Fan, A.-P. Koh, C.-S. Wu, M.-D. Su and C.-W. So, Carbon dioxide capture and functionalization by bis(N-heterocyclic carbene)-borylene complexes, Nat. Commun., 2024, 15, 3052 CrossRef CAS PubMed.
- J. Fan, S. Pan, S. Yao, C. Ding, G. Frenking and M. Driess, From Bis(borylene)-Substituted Xanthenes as Reactive Intermediates to Diboraoxirane Complexes, J. Am. Chem. Soc., 2025, 147, 6925–6933 CrossRef CAS PubMed.
- H. Braunschweig, I. Krummenacher, M.-A. Légaré, A. Matler, K. Radacki and Q. Ye, Main-Group Metallomimetics: Transition Metal-like Photolytic CO Substitution at Boron, J. Am. Chem. Soc., 2017, 139, 1802–1805 CrossRef CAS PubMed.
- A. Stoy, M. Arrowsmith, M. Eyßelein, T. Dellermann, J. Mies, K. Radacki, T. Kupfer and H. Braunschweig, NHC-Stabilized 1,2-Dihalodiborenes: Synthesis, Characterization and Reactivity Toward Elemental Chalcogens, Inorg. Chem., 2021, 60, 12625–12633 CrossRef CAS PubMed.
- M. Arrowsmith, J. Böhnke, H. Braunschweig, M. A. Celik, T. Dellermann and K. Hammond, Uncatalyzed Hydrogenation of First-Row Main Group Multiple Bonds, Chem.–Eur. J., 2016, 22, 17169–17172 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic, spectroscopic, X-ray crystallographic and computational details. CCDC 2447646–2447651. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc03150h |
| This journal is © The Royal Society of Chemistry 2025 |