
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-dimensional analysis of single particles with sequential dual-nanopipette sensors†
Rui-Xue
Gao
a,
Si-Yu
Tian
a,
Ying
Chen
a,
Yu-Ting
Qi
a,
Limin
Xiang
ab,
Wei-Hua
Huang
 ac and
Xin-Wei
Zhang
*a
ac and
Xin-Wei
Zhang
*a
aCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, P. R. China. E-mail: xinweizhang@whu.edu.cn
bTaikang Center for Life and Medical Sciences, Wuhan University, Wuhan 430072, P. R. China
cDepartment of Hepatobiliary and Pancreatic Surgery, Zhongnan Hospital of Wuhan University, Wuhan 430071, P. R. China
First published on 30th June 2025
Abstract
Nanoparticles play crucial roles in a wide range of scientific and industrial fields. Analysing the physicochemical characteristics of nanoparticles is of paramount importance in identifying and classifying different particles. However, due to their small size and inherent heterogeneity, comprehensive analysis of the size and shape, especially the complex surface characteristics, of individual particles is still confronted with challenges. In this work, we report a facile strategy to construct a sequential dual-nanopipette sensor (SDNS) with two sensing interfaces for multi-parameter detection of nanoparticles at the single particle level. The collaboration of the two sensing interfaces enabled dual recognition of the same particle, which improved the detection accuracy effectively. With this platform, it was not only possible to perform concentration quantification and size characterization, but also to effectively classify the particles via dual determination of the surface charge or the biochemical components of the particles. As the sensing interfaces of SDNS can be varied to meet the detection requirements, this approach can be easily adjusted to analyse more kinds and dimensions of surface characteristics of single nanoparticles, which opens up new avenues for the exploration of biological or other kinds of nanoparticles as well as the deeper comprehension of particle functions and evolution.
Introduction
Nanoparticles are widely used in the fields of energy, biomedicine, and materials, and also occur naturally as organelles, vesicles, and lipoproteins in biological systems, playing important roles in pathophysiological processes.1–6 The functions of nanoparticles are closely related to their multiple physical and chemical properties, such as size, shape, surface charge, and chemical composition, which collectively govern their behaviour and efficacy.7,8 For instance, the size and surface charge of metal nanoparticles have an important effect on their catalytic activity and selectivity.9 In the extracellular environment, extracellular vesicles (e.g., exosomes and microvesicles) secreted by cells play crucial roles in intercellular communication.10 The complex surface protein composition of these vesicles can reflect their donor cell type, subcellular origin, and downstream function.11 Thus, the comprehensive and multi-dimensional analysis of nanoparticles' physicochemical properties can be used to shed light on their structure–function relationship, trace their sources, or study their evolution process. Conventional particle analysis relies on multiple instruments to measure different features of the particles separately, resulting in complex sample preparation and a lack of correlation between the information.12 And the measurement based on bulk solution also leads to neglecting of the population heterogeneity of particles.11,13,14 Therefore, it is highly desirable to perform multi-functional analysis of nanoparticles at the single particle level.Significant advancements in nanoparticle characterization techniques, like electron microscopy, nanoparticle tracking analysis, flow cytometry, surface-enhanced Raman scattering, etc., have enabled precise determination of size, composition, or surface properties at the single particle level.15–21 However, challenges persist in achieving reliable multi-dimensional analysis of individual particles, especially the simultaneous analysis of multiple surface features. Recently, the nanopore sensing technique, due to the advantages including being label-free and non-destructive, with high spatiotemporal resolution and high sensitivity, has evolved as a promising approach to investigate nanoparticles in aqueous solution or even cellular environments.22–30 During the nanopore measurements, a constant voltage is applied across the nanopore on a nanopipette or membrane to generate a steady ionic current. When the particle translocates through the nanopore, the ionic current will be partially blocked, resulting in a momentary drop, namely a resistive pulse signal (RPS).31–35 The signals can be used to analyse the concentration and size of these particles, as well as the surface biochemical features (charges, membrane proteins, etc.) through functionalizing the nanopore interface with sensing probes (e.g., charged polymer, antibody, or aptamer) to form a specific interaction between the particles and the nanopore wall.11,24,36–38 The interaction between the functionalized inner wall of the nanopore and the particle will modulate the translocation behaviour of the particles, which manifests as a significant prolongation of the duration of the pulse signals.39–43 But limited by the single interface of a regular nanopore, only one surface feature of a particle can be analysed at present. Combining multiple functionalized nanopores in series can provide extra sensing interfaces for different specific interactions, which is expected to be a potential approach to conducting multi-dimensional analysis of surface biochemical features at the single particle level. However, due to the narrow space and limited sensing area in nanopores, challenges still exist in the construction of nanopore sensors with multiple sensing interfaces.
Herein, we propose a simple and flexible strategy to construct a sequential dual-nanopipette sensor (SDNS) with double sensing interfaces for single nanoparticle multi-dimensional analysis. By modifying two nanopipettes with different sensing probes separately and assembling them in series, a sensor containing dual sensing interfaces was obtained. When particles sequentially passed through the two pores of the sensor, they interacted selectively with the two distinct sensing interfaces in turn, resulting in dual RPS providing more information than single RPS. The regular sizing and counting performance of SDNS were verified by using polystyrene nanoparticles (PS NPs) as a model of bio-nanoparticles. Furthermore, based on the discrepancy of the durations induced by the electrostatic or antigen–antibody interaction between particles and sensing interfaces, particles with different surface charges or biochemical characteristics could be distinguished with high accuracy. The established measurement strategy provided a simple but efficient tool for the multi-dimensional analysis of nanoparticles without labelling, which showed potential value for analysing practical samples such as exosomes and organelles.
Results and discussion
Fabrication and characterization of SDNS
To achieve multi-dimensional analysis for single nanoparticles, a sequential dual-nanopipette sensor (SDNS) containing double sensing interfaces was constructed by connecting two nanopipettes in series. The general fabrication process of SDNS is outlined in Fig. 1 and described in detail in the Experimental section. Two nanopipettes with distinct geometries—a longer capillary with a smaller diameter and another shorter capillary with a larger diameter—were fabricated via heat-pulling, as shown in Fig. S1.† Given their different capillary diameters, the smaller nanopipette can be inserted smoothly into the larger one with the aid of the microscope and micromanipulators. By further fixing the junction between them with epoxy resin, an SDNS was obtained (Fig. 2A-i) containing two pores (pore 1 on the inside as the first pore the particles will pass through, and pore 2 on the outside as the second).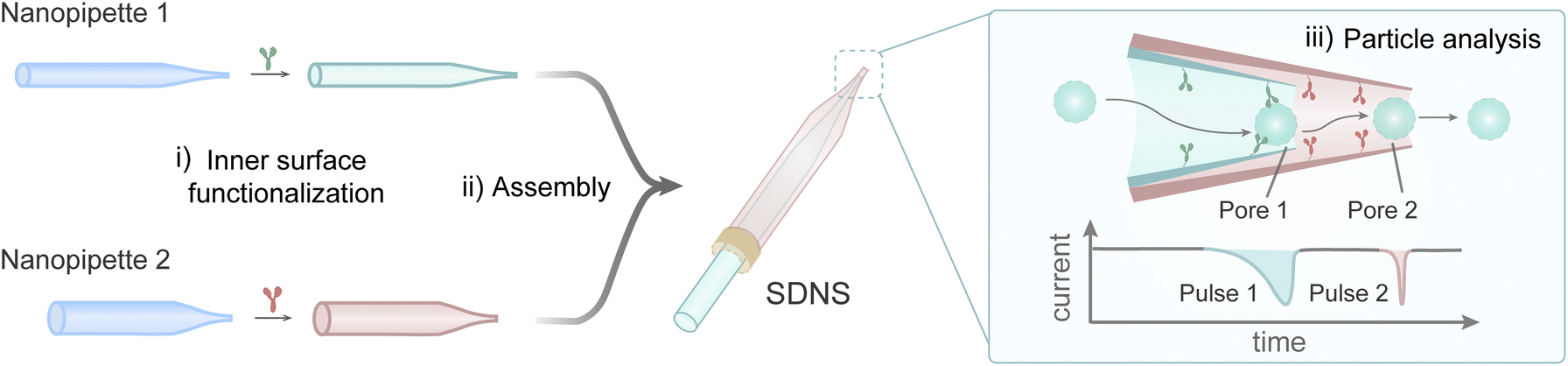 | ||
| Fig. 1 Schematic diagram of the main fabrication process of SDNS for single-particle analysis. | ||
 | ||
| Fig. 2 (A) (i) Photograph and (ii) SEM image of the SDNS. (B) Current–voltage (I–V) curves of pore 1, pore 2, and SDNS recorded in 0.1 M KCl. The data are represented as mean ± SD (n = 3). (C) Power spectral density for current traces of single pores and SDNS in 10 mM PBST. (D) Schematic illustration of the SDNS and the dual RPS. (E) Current trace of the 500 nm-radius PS NPs (dispersed in 10 mM PBST) recorded using the SDNS and (F) the enlarged view of dual RPS. Eapp = −100 mV, pressure = +10 hPa. | ||
The sidewall of SDNS was sculpted using a focused ion beam (FIB) along its axis to expose the profile inside (Fig. S2†). The SEM image of the sculpted SDNS indicated that the two nanopores were located on the same axis, and the pore-to-pore distance was about 50 μm (Fig. 2A-ii). The good coaxiality of the pores ensured that the nanoparticles could move smoothly along the axis and pass through pore 1 and pore 2 successively in sequence. The appropriate distance between them could prevent the interleaving of signals from different particles while ensuring the non-interference of pairwise pulse signals (Fig. S3†). By adjusting the apertures and cone angles of the nanopipettes, a series of sensors with appropriate apertures and pore-to-pore distances could be constructed for the analysis of particles with various sizes and concentrations (Fig. S4†).
Moreover, the results of current–voltage (I–V) measurements also further confirmed the formation of the dual-pore in-series structure in SDNS (Fig. 2B). From −1 V to +1 V, the current linearly increased with the voltage, and the reciprocal of the slope of the lines reflected the resistance of the dual pore system. In comparison to one single nanopipette, SDNS displayed a noticeably larger total resistance since the addition of a second pore to the electric circuit. Further calculation based on the I–V curves verified that the resistance of SDNS was about 15.3 MΩ, which was well matched with the total resistance of the two individual nanopipettes in series (RDual ≈ RInner + ROuter, RInner ≈ 8.9 MΩ, ROuter ≈ 6.5 MΩ). Meanwhile, the orifice radius of the glass nanopipettes could also be estimated (eqn (S1)†) as r1 ≈ 0.7 μm and r2 ≈ 1.0 μm by the resistances, which were consistent with the SEM results, providing us with a quick and non-destructive approach for determining apertures. And the power spectral density indicated that these sensors exhibited good noise performance (Fig. 2C). There was no increase in the noise of SDNS (Fig. 2C, grey curve) compared to single nanopipettes (Fig. 2C, blue curves), which ensured the feasibility of SDNSs to record small RPS.
The single-nanoparticle sensing ability of SDNS
We then initially verified the feasibility of SDNS to sense single particles and its capacity to carry out dual detection. Since PBST has good buffer capacity to maintain the pH stability of the solution and the Tween-20 in it can prevent the aggregation of particles, we used PBST as the electrolyte solution in RPS experiments. Polystyrene nanoparticles (PS NPs) with a 500 nm radius were used as a model and backfilled into the SDNS after dispersing in the PBST solution (Fig. S5†). Upon experimental optimization a continuous pressure of +10 hPa (Fig. S6†) was exerted in the sensor to drive PS NPs to pass through pore 1 and pore 2 in turn (Fig. 2D), which was confirmed by the fluorescence imaging of FITC-labelled PS NPs and the current fluctuation of this process at the same time (ESI movie 1†). The translocation results in a series of dual RPS on the current trace when a potential (Eapp = −100 mV) was applied across the sensor (Fig. 2E). The RPS appeared in pairs and consisted of a larger pulse (pulse 1) and a smaller one (pulse 2) with a short interval (∼0.12 s) (Fig. 2F). The distinct amplitudes of the pulses are due to the fact that two pores of different sizes (pore 1, r1 ≈ 0.7 μm; pore 2, r2 ≈ 1.0 μm) are blocked to different degrees by the same particle. To describe the dual RPS quantitatively, parameters like the duration of pulse 1 and pulse 2 (t1 and t2), blockage current (ΔI1 and ΔI2), and baseline current (I0) were all defined, see details in the ESI (Fig. S7†). In addition, when the pressure was removed, no signals were recorded no matter whether +100 mV or −100 mV was applied as the Eapp (Fig. S8†), which indicated that the pressure, rather than electroosmosis or electrophoresis, was the dominant driving force for the translocation of PS NPs. It should be noted that the appropriate pore aperture, pore-to-pore distance and particle concentration were all important factors in ensuring the single nanoparticle sensing ability of SDNS. The apertures, which matched the particle size, guaranteed the smooth particle translocation and excellent signal-to-noise ratio of the signal, while the appropriate pore-to-pore distance and particle concentration jointly ensured the independence and non-interference of the signals (Fig. S9†). Therefore, when sensing distinct particle samples, all of these aspects should be considered and adjusted properly.Nanoparticle concentration quantification and size characterization
The measurements of particle concentration and size are the basic functions of nanopore analysis. Herein, we also evaluated these two measurement performances of the SDNS and compared them with the classic single nanopore. For the evaluation of particle concentration measurement, a range of concentrations (from 2.28 × 106 to 114 × 106 particles per mL) of PS NP dispersions were tested using the SDNS. The results showed that, with the increase of particle concentrations, the frequency of particle translocation also gradually increased (Fig. 3A), which is consistent with previous reports.36,44 The corresponding linear curves are shown in Fig. 3B, and the linear relationship is expressed as the frequency of the pulses, f = 0.0057 + 0.0079 × Cparticle (R2 = 0.999), which indicated the good performance of SDNS in quantifying nanoparticle concentration. | ||
| Fig. 3 (A) Current traces for the presence of 500 nm-radius PS NPs with different concentrations in 10 mM PBST. (B) Event frequency (f) as a function of different concentrations of PS NPs. The data are represented as mean ± SD (n = 3). (C) Current trace for the presence of 400-, 500- and 600 nm-radius PS NPs in 10 mM PBST. (D) Enlarged views of three representative signals in the current trace (C) representing 400-, 500- or 600 nm-radius PS NPs translocated through the SDNS. (E) Histograms showing the distributions of the particle radius. (F) The relative standard deviation (RSD) of the radius distributions for mixed nanoparticles from single nanopore and SDNS. Eapp = −100 mV, pressure = +10 hPa. | ||
Moreover, the feasibility of SDNS to recognize the difference in particle size was also demonstrated by sensing the mixture of PS NPs with radii of 400 nm, 500 nm, and 600 nm. To ensure the smooth passage of the mixed particles and the high signal-to-noise ratio of the signals, the SDNS used here consisted of two nanopipettes with similar apertures around 0.9 μm. The current trace for the mixed particles in PBST is shown in Fig. 3C, in which three classes of pulse signals with different amplitudes (ΔI) existed (Fig. 3C and D). As a result, different current amplitudes indicated the presence of particles with different sizes. The relative current amplitude (ΔI/I0, which describes the degree of the nanoparticle blocking the nanopore) is proportional to the volume of the particle,45,46 and the radius of mixed nanoparticles could be obtained based on eqn (S2).† The radius distribution histogram validated the clear distinction between three kinds of particles (Fig. 3E), which well agreed with the results obtained by SEM (Fig. S10†). Compared to the single nanopore, the relative standard deviation (RSD) of the particle radius distribution acquired from SDNS was lower and much closer to the statistical results obtained from SEM (Fig. 3F and eqn (S3) and (S4)†). These results demonstrated that the random errors could be reduced effectually by averaging the signals from repeated measurements, so that the sizing results could be more accurate.
Classification of nanoparticles with different surface charges using polymer-functionalised SDNS
Surface charge is one of the most important surface properties of nanoparticles, influencing their further modification and application. Nanopores also enable surface charge analysis by exploiting the interaction between their inner wall and particles.39,42 As a charged particle travels through an oppositely charged nanopore, the electrostatic attraction between them will prolong the duration of the blocking signal, which can be used to determine the surface charge of the particle. Repeated detection of the same particle employing nanopores with opposite surface charge allows for a double test of the particle's surface charge, effectively improving the accuracy of the determination. Given that the dual-pore in-series structure of SDNS provides us with two pores that can easily be functionalized separately, dual recognition towards the same particle can be realized, which may make it suitable for identifying the surface properties of single particles.In order to obtain an SDNS consisting of contrary wall charges in two pores, we coated pore 1 with a positively charged polymer-polyethyleneimine (PEI), whereas pore 2 was bare and naturally negative-charged because the silanol groups on the glass surface are electronegative in neutral solution (Fig. 4A). SEM images and EDX mapping showed that a film rich in N element (the characteristic element of PEI) could be visibly seen on the orifice (Fig. S11†), indicating uniform coating of PEI on the surface of the pore. The charge of the pores could be further demonstrated by the I–V measurement (Fig. 4B). Because of the conical geometry and wall charge of nanopipettes, an asymmetrical ion transport phenomenon will occur inside the pore. This phenomenon can induce ionic current rectification (ICR) in the I–V curve, whose direction is strongly affected by the surface charge.47–49 As shown in Fig. 4B, the rectification factor (Rf = −lg(|I−1.0 V/I+1.0 V|)) of pore 2 and pore 1 were −0.14 and 0.32, indicating that they presented negative and positive charges, respectively. By assembling them together, a polymer-functionalized SDNS was successfully obtained.
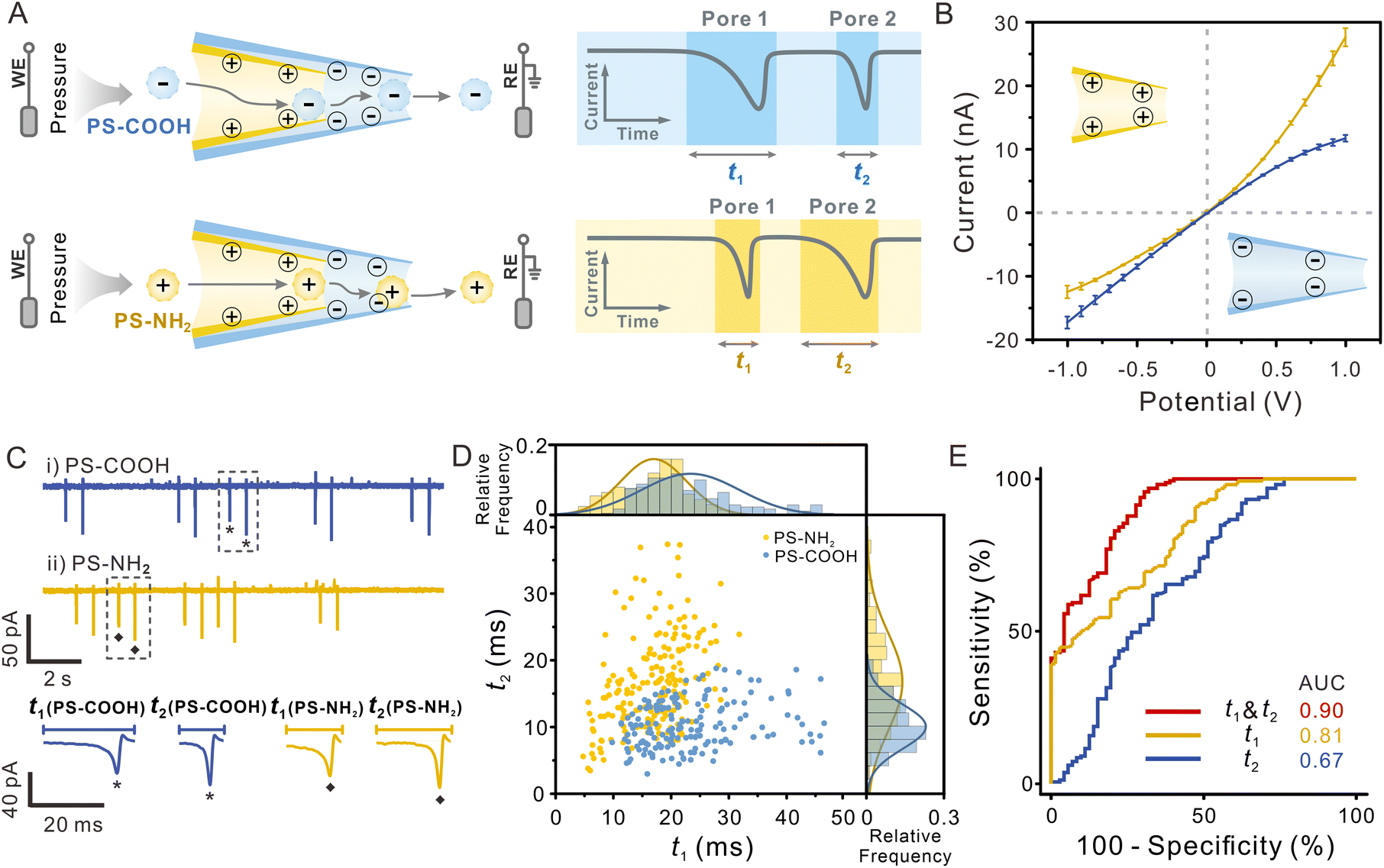 | ||
| Fig. 4 (A) Schematic illustration of the functionalized-SDNS distinguishing particles with different surface charges. (B) Current–voltage (I–V) curves of pore 1 (yellow) and pore 2 (blue) were recorded in 0.5 mM PBST (pH = 7.0). The data are represented as mean ± SD (n = 3). (C) Current traces for the presence of (i) PS-COOH and (ii) PS-NH2 in 0.5 mM PBST at −100 mV using the functionalized-SDNS and enlarged view of representative signals. Pressure = +5 hPa. (D) The duration distribution of two kinds of particles (PS-NH2, yellow; PS-COOH, blue) translocating through pore 1 and pore 2. (E) ROC curves showing the performance of the sensor for classifying PS-COOH and PS-NH2 (pore 1, yellow line; pore 2, blue line; dual-pore, red line). | ||
To investigate the capability of the polymer-functionalized SDNS to identify the surface charge of single particles, amino polystyrene particles (PS-NH2, zeta potential = 31.6 mV), and carboxy polystyrene particles (PS-COOH, zeta potential = −52.5 mV) were tested as the models (Fig. S12†). The electrolyte solution used here was 0.5 mM PBST, which had a lower ionic strength, to enhance the electrostatic interaction between the nanopores and particles. And a pressure (5 hPa, internal vs. external) was applied to drive particles passing through SDNS slowly without damaging the functionalized coating (Fig. S13†). Unlike the single-peak resistive pulses in Fig. 2 and 3, biphasic pulses were recorded on the current traces, with a normal current decrease followed by a current increase (Fig. 4C). According to previous studies, such signals were often observed when charged particles pass through charged nanopores under low electrolyte conditions.50–52 They were the consequence of the combined effect of an increased ion conductivity induced by the surface charge of the translocating particle and the current decrease due to the volume exclusion of electrolyte solution by the particle.
After comparison, we found that the pulses induced by PS-COOH and PS-NH2 exhibited relatively uniform pulse amplitudes (because of the similar particle sizes) but different durations. It showed that the duration of negatively charged PS-COOH (blue sphere) traveling through the positively charged pore 1 was much longer than that of positively charged PS-NH2 (yellow sphere) (t1(PS-COOH) > t1(PS-NH2)), while the case was opposite as they passed through the negatively charged pore 2, that is, t2(PS-COOH) < t2(PS-NH2). The charged inner wall of the pores exerted a pronounced impact on the translocation behaviour of charged particles, which could account for the results. As the PS-COOH entered pore 1, the particles experienced electrostatic attraction towards the positively charged PEI coating on the surface of the pore, resulting in a prolonged duration of pulse 1 and a shorter duration of pulse 2 because of the electrostatic repulsion between the particles and negatively charged pore 2. In contrast, for PS-NH2, the electrostatic attraction occurred when they approached pore 2, so only the duration of pulse 2 was prolonged. Considering that the charged pores produce more noticeable interaction with the opposite surface charged particles, distinguishing particles of unknown surface charge with SDNS containing dual recognition interfaces will achieve a better effect.
In the 2D scatter plot depicting the t1 and t2 of individual particles (Fig. 4D), two populations were observed, which indicated that PS-NH2 and PS-COOH could be discriminated from each other. While partial overlap between the two datasets still existed, this might arise from the low surface charge density of certain particles and their limited electrostatic interactions with the pore wall, it can likely be mitigated by further optimizing the surface charge density of the pore wall. Receiver operating characteristic (ROC) curves and area under the curve (AUC) were further used to evaluate the classification capacity (AUC = 0.90 for dual pores vs. 0.81 for only pore 1 vs. 0.67 for only pore 2), and it was evident that the performance of SDNS was superior to that of the single nanopore (Fig. 4E). The other two sets of polymer-functionalised SDNS also demonstrated similar discrimination performance in classifying PS-NH2 and PS-COOH, indicating the good reproducibility and reliability of this strategy (Fig. S14†).
Classification of nanoparticles with different surface proteins using antibody-modified-SDNS
The identification or classification of bioparticles is an important application of single-particle analysis. Nano-sized bioparticles often carry abundant surface proteins, lipids, glycans, etc. Obtaining the surface biochemical information of these particles will help us gain insight into their structure and function and fine-classify them more accurately. Through constructing the interaction between bioparticles and the inner wall of the nanopore, we can acquire the surface information of different nanoparticles by recording the duration. Considering the complexity of the bioparticle surface components, more dimensional measurements of surface molecules may effectively improve the reliability of identification and classification of particles, which can be realized by the dual nanopipettes of SDNS.Because the natural biovesicles are too complex and it is difficult to control the quantities of surface proteins, we modified PS NPs with two widely used tumour protein biomarkers (alpha-fetoprotein (AFP) and carcinoembryonic antigen (CEA)) as the simplified model of bioparticles (PS-AFP and PS-CEA, see the ESI and Fig. S15†). The PS-AFP and PS-CEA will then be tested by the functionalized SDNS with specific sensing probes on their inner walls. Transient immuno-binding will only occur between the sensing probes and the particles carrying specific biomarkers, leading to the difference in translocation behaviour of PS-AFP and PS-CEA (Fig. 5A). To achieve the effective identification of these two types of particles through dual biomarker recognition, we modified the surface of pore 1 and pore 2 in SDNS with the antibody of AFP or CEA (Fig. S16–S19,† see details in the Experimental section), respectively, and the uniform modification of antibodies on the respective pores could be observed more intuitively with fluorescence imaging (Fig. 5B).
 | ||
| Fig. 5 (A) Schematic illustration of the antibody-modified-SDNS distinguishing particles with different surface proteins. (B) Bright-field and fluorescence imaging of antibody-modified-SDNS. (C) Current traces of (i) PS-AFP and (ii) PS-CEA translocation in 10 mM PBST at −100 mV using the antibody-modified-SDNS and enlarged view of representative signals. Pressure = +10 hPa. (D) The duration distribution of two kinds of particles (PS-AFP, red; PS-CEA, green) translocating through pore 1 and pore 2. (E) ROC curves showing the performance of the sensor for classifying PS-AFP and PS-CEA (pore 1, green line; pore 2, pink line; dual-pore, red line). | ||
The antibody-modified SDNS was applied for the classification of PS-AFP and PS-CEA, and the typical current traces and representative signals are shown in Fig. 5C. Significant differences still existed between the durations of the two groups. The corresponding two-dimensional event scatter plots of duration 1 (t1) versus duration 2 (t2) shown in Fig. 5D visually displayed their distribution. As expected, the PS-AFP (pink sphere) translocated faster through pore 1 functionalized with anti-CEA than the PS-CEA (green sphere). When it passed through pore 2 modified with anti-AFP, its velocity was much slower than that of the PS-CEA. Distinct from the translocation dynamics of charged particles that were affected by the electrostatic interaction of the charged inner wall of the pores, the prolongation of the duration of PS-AFP/PS-CEA was mainly due to the specific interaction between the antigens on the particle surface and the corresponding antibodies on the pore wall. Instantaneous immunoreaction resulted in the particles being trapped transiently in the pore. Hence, the increase in duration only appeared when the antigen-modified particles passed through the pores functionalized with the specific antibodies. The combination of two antibody-modified pores in SDNS made it possible to identify dual biochemical characteristics on the surface of a single particle, which facilitated the precise discrimination of particles.
The ROC analysis confirmed the reliable differentiation of PS-AFP versus PS-CEA (AUC = 0.90) using t1 and t2 obtained by the antibody-modified SDNS (Fig. 5E). By contrast, when using the single pore individually, the AUC values were only 0.71 and 0.78, respectively. The above results demonstrated that SDNS exhibited a much lower misjudgement ratio. Consistent results were also carried out with different sets of antibody-modified SDNS (Fig. S20†), which further confirmed the reliability of the sensor. The dual-recognition strategy significantly improved the accuracy of classifying nanoparticles with different biochemical properties, indicating that the SDNS is a highly promising tool for classifying bioparticles efficaciously.
Conclusions
In summary, this work presented a facile and novel strategy for the orderly integration of dual sensing interfaces in a nanopore sensor to achieve multi-dimensional characterization of single particles. By assembling two nanopipettes in series, a sequential dual-nanopipette sensor (SDNS) containing two separate sensing interfaces could be easily constructed. Repetitive detection of the same particle using SDNS reduced the random error in particle sizing. More importantly, by performing dual recognition on single nanoparticles, the sensor exhibited higher accuracy in distinguishing particles with more physicochemical properties compared to single nanopores. By further increasing the number of functionalized nanopipettes, more abundant physical and biochemical features of single particles can be expected to be measured at the same time. The obtained multi-dimensional information for individual particles will facilitate the precise classification of complex particle samples, as well as the mechanism study of their structure and component heterogeneity, which may further advance the in-depth investigation of the function and evolution of nanoparticles in broad fields of medicine, biology, and environmental science.Data availability
All experimental and characterization data and detailed experimental procedures are available in the published article and the ESI.†Author contributions
W. H., X. Z. and R. G. designed the research. R. G. performed the research and wrote the manuscript. S. T. and R. G. analysed the data. Y. Q. and Y. C. provided technical support and suggestions. X. Z., W. H. and L. X. supervised the project and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the National Key Research and Development Program of China (Program No. 2023YFF0723100, 2022YFA1104800, 2022YFA1305400), National Natural Science Foundation of China (Grants 22090050, 22090051 and 22404126) and the China Postdoctoral Science Foundation (Grant BX20240268). We also thank the Core Facility of Wuhan University for SEM/EDS, FIB and DLS analysis.References
- K. Guo, L. Bao, Z. Yu and X. Lu, Chem. Soc. Rev., 2024, 53, 11100–11164 RSC.
- D. Wu, J. Zhou, M. N. Creyer, W. Yim, Z. Chen, P. B. Messersmith and J. V. Jokerst, Chem. Soc. Rev., 2021, 50, 4432–4483 RSC.
- P. J. Santos, P. A. Gabrys, L. Z. Zornberg, M. S. Lee and R. J. Macfarlane, Nature, 2021, 591, 586–591 CrossRef CAS PubMed.
- A. Saminathan, M. Zajac, P. Anees and Y. Krishnan, Nat. Rev. Mater., 2022, 7, 355–371 CrossRef.
- R. Kalluri and K. M. McAndrews, Cell, 2023, 186, 1610–1626 CrossRef CAS PubMed.
- Y. Han, E. J. Onufer, L. Huang, R. W. Sprung, W. S. Davidson, R. S. Czepielewski, M. Wohltmann, M. G. Sorci-Thomas, B. W. Warner and G. J. Randolph, Science, 2021, 373, eabe6729 CrossRef CAS PubMed.
- M. M. Modena, B. Rühle, T. P. Burg and S. Wuttke, Adv. Mater., 2019, 31, 1901556 CrossRef PubMed.
- J. Zhou, A. I. Chizhik, S. Chu and D. Jin, Nature, 2020, 579, 41–50 CrossRef CAS PubMed.
- T. W. van Deelen, C. Hernández Mejía and K. P. de Jong, Nat. Catal., 2019, 2, 955–970 CrossRef CAS.
- R. Kalluri and V. S. LeBleu, Science, 2020, 367, eaau6977 CrossRef CAS PubMed.
- R. P. Carney, R. R. Mizenko, B. T. Bozkurt, N. Lowe, T. Henson, A. Arizzi, A. Wang, C. Tan and S. C. George, Nat. Nanotechnol., 2025, 20, 14–25 CrossRef CAS PubMed.
- J. Penders, I. J. Pence, C. C. Horgan, M. S. Bergholt, C. S. Wood, A. Najer, U. Kauscher, A. Nagelkerke and M. M. Stevens, Nat. Commun., 2018, 9, 4256 CrossRef PubMed.
- D. G. Mullen and M. M. Banaszak Holl, Acc. Chem. Res., 2011, 44, 1135–1145 CrossRef CAS PubMed.
- J. Morla-Folch, A. Ranzenigo, Z. A. Fayad and A. J. P. Teunissen, Small, 2024, 20, 2307502 CrossRef CAS PubMed.
- L. Pascucci and G. Scattini, Biochim. Biophys. Acta, Gen. Subj., 2021, 1865, 129648 CrossRef CAS PubMed.
- V. Filipe, A. Hawe and W. Jiskoot, Pharm. Res., 2010, 27, 796–810 CrossRef CAS PubMed.
- R. A. Dragovic, C. Gardiner, A. S. Brooks, D. S. Tannetta, D. J. P. Ferguson, P. Hole, B. Carr, C. W. G. Redman, A. L. Harris, P. J. Dobson, P. Harrison and I. L. Sargent, Nanomedicine, 2011, 7, 780–788 CrossRef CAS PubMed.
- C. Gardiner, Y. J. Ferreira, R. A. Dragovic, C. W. G. Redman and I. L. Sargent, J. Extracell. Vesicles, 2013, 2, 19671 CrossRef PubMed.
- D. Choi, L. Montermini, H. Jeong, S. Sharma, B. Meehan and J. Rak, ACS Nano, 2019, 13, 10499–10511 CrossRef CAS PubMed.
- S. Stremersch, M. Marro, B.-E. Pinchasik, P. Baatsen, A. Hendrix, S. C. De Smedt, P. Loza-Alvarez, A. G. Skirtach, K. Raemdonck and K. Braeckmans, Small, 2016, 12, 3292–3301 CrossRef CAS PubMed.
- J. Wang, K. M. Koo, Y. Wang and M. Trau, Adv. Sci., 2019, 6, 1900730 CrossRef CAS PubMed.
- R. Yu, Y. Ying, R. Gao and Y. Long, Angew. Chem., Int. Ed., 2019, 58, 3706–3714 CrossRef CAS PubMed.
- J. Hu, N. Lin, L. Yuan, X. Lou and F. Xia, Acc. Chem. Res., 2025, 58, 834–846 CrossRef CAS PubMed.
- R. Maugi, P. Hauer, J. Bowen, E. Ashman, E. Hunsicker and M. Platt, Nanoscale, 2020, 12, 262–270 RSC.
- X. Zhang, D. Luo, Y.-W. Zheng, X. Li, J. Song, W. Zhao, H. Chen and J. Xu, ACS Nano, 2022, 16, 15108–15114 CrossRef CAS PubMed.
- L. Miao, B. Huang, H. Fang, J. Chai, Z. Liu and Y. Zhai, Adv. Mater., 2023, 2305159 CrossRef PubMed.
- S. Khatri, P. Pandey, G. Mejia, G. Ghimire, F. Leng and J. He, J. Am. Chem. Soc., 2023, 145, 28075–28084 CrossRef CAS PubMed.
- K. Chen, R. Yu, C. Zhong, Z. Wang, B. Xie, H. Ma, M. Ao, P. Zheng, A. G. Ewing and Y. Long, Angew. Chem., Int. Ed., 2024, 63, e202406677 CrossRef CAS PubMed.
- R. Pan, K. Hu, R. Jia, S. A. Rotenberg, D. Jiang and M. V. Mirkin, J. Am. Chem. Soc., 2020, 142, 5778–5784 CrossRef CAS PubMed.
- H. Ma, Y. Wang, Y. Li, B. Xie, Z. Hu, R. Yu, Y. Long and Y. Ying, J. Am. Chem. Soc., 2024, 146, 28014–28022 CAS.
- H. Bayley and C. R. Martin, Chem. Rev., 2000, 100, 2575–2594 CrossRef CAS PubMed.
- Y. Liu, C. Xu, P. Yu, X. Chen, J. Wang and L. Mao, Chemelectrochem, 2018, 5, 2954–2962 CrossRef CAS.
- R. Jia and M. V. Mirkin, Chem. Sci., 2020, 11, 9056–9066 RSC.
- L. Xue, H. Yamazaki, R. Ren, M. Wanunu, A. P. Ivanov and J. B. Edel, Nat. Rev. Mater., 2020, 5, 931–951 CrossRef CAS.
- W. Tang, J. P. Fried, R. D. Tilley and J. J. Gooding, Chem. Soc. Rev., 2022, 51, 5757–5776 RSC.
- G. S. Roberts, S. Yu, Q. Zeng, L. C. L. Chan, W. Anderson, A. H. Colby, M. W. Grinstaff, S. Reid and R. Vogel, Biosens. Bioelectron., 2012, 31, 17–25 CrossRef CAS PubMed.
- D. F. Cairns-Gibson and S. L. Cockroft, Chem. Sci., 2022, 13, 1869–1882 RSC.
- E. C. Yusko, B. R. Bruhn, O. M. Eggenberger, J. Houghtaling, R. C. Rollings, N. C. Walsh, S. Nandivada, M. Pindrus, A. R. Hall, D. Sept, J. Li, D. S. Kalonia and M. Mayer, Nat. Nanotechnol., 2017, 12, 360–367 CrossRef CAS PubMed.
- L. Chen, H. He and Y. Jin, Anal. Chem., 2015, 87, 522–529 CrossRef CAS PubMed.
- A. Arima, I. H. Harlisa, T. Yoshida, M. Tsutsui, M. Tanaka, K. Yokota, W. Tonomura, J. Yasuda, M. Taniguchi, T. Washio, M. Okochi and T. Kawai, J. Am. Chem. Soc., 2018, 140, 16834–16841 CrossRef CAS PubMed.
- Y. Zhou, D. Wang, C. Li, P. Hu and Y. Jin, Anal. Chem., 2019, 91, 7648–7653 CrossRef CAS PubMed.
- S. Pal, R. B, S. Jugade, A. Rao, A. Naik, B. Chakraborty and M. M. Varma, Sens. Actuators, B, 2020, 325, 128785 CrossRef CAS PubMed.
- Y. Horiguchi, N. Naono, O. Sakamoto, H. Takeuchi, S. Yamaoka and Y. Miyahara, ACS Appl. Mater. Interfaces, 2022, 14, 20168–20178 CrossRef CAS PubMed.
- Y. Liu, C. Xu, X. Chen, J. Wang, P. Yu and L. Mao, Electrochem. Commun., 2018, 89, 38–42 CrossRef CAS.
- W. Lan, D. A. Holden, B. Zhang and H. S. White, Anal. Chem., 2011, 83, 3840–3847 CrossRef CAS PubMed.
- S. R. German, T. S. Hurd, H. S. White and T. L. Mega, ACS Nano, 2015, 9, 7186–7194 CrossRef CAS PubMed.
- Z. Siwy, E. Heins, C. C. Harrell, P. Kohli and C. R. Martin, J. Am. Chem. Soc., 2004, 126, 10850–10851 CrossRef CAS PubMed.
- Z. S. Siwy, Adv. Funct. Mater., 2006, 16, 735–746 CrossRef CAS.
- Z. S. Siwy and S. Howorka, Chem. Soc. Rev., 2010, 39, 1115–1132 RSC.
- W. Lan, C. Kubeil, J. Xiong, A. Bund and H. S. White, J. Phys. Chem. C, 2014, 118, 2726–2734 CrossRef CAS.
- J. Menestrina, C. Yang, M. Schiel, I. Vlassiouk and Z. S. Siwy, J. Phys. Chem. C, 2014, 118, 2391–2398 CrossRef CAS.
- K. Chen, L. Shan, S. He, G. Hu, Y. Meng and Y. Tian, J. Phys. Chem. C, 2015, 119, 8329–8335 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02604k |
| This journal is © The Royal Society of Chemistry 2025 |