
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the chemistry of higher acenes: from synthesis to applications
Hironobu
Hayashi
 *a and
Hiroko
Yamada
*b
*a and
Hiroko
Yamada
*b
aCenter for Basic Research on Materials, National Institute for Materials Science (NIMS), 1-2-1 Sengen, Tsukuba, Ibaraki 305-0047, Japan. E-mail: HAYASHI.Hironobu@nims.go.jp
bInstitute for Chemical Research, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan. E-mail: hyamada@scl.kyoto-u.ac.jp
First published on 4th June 2025
Abstract
This review explores the advancements in the chemistry of higher acenes and their derivatives, with a focus on their synthesis, characterization, and potential applications. Historically, higher acenes have presented challenges to study due to their inherent instability and reactivity under ambient conditions. However, innovative synthetic strategies, including on-surface synthesis and the precursor approach, have significantly contributed to the ability to synthesize higher acenes even at preparative scales while evaluating their magnetic and semiconducting properties. Furthermore, ethynylene-bridged acene oligomers and polymers, known for their extended π-conjugated systems, have shown promise not only as semiconducting materials but also as topological materials. As synthetic methods continue to evolve and characterization techniques become more sophisticated, higher acenes offer exciting opportunities for progress in the fields of organic chemistry and materials science, paving the way for advanced applications in organic electronics.
 Hironobu Hayashi | Hironobu Hayashi received his PhD degree in 2012 from Kyoto University, Japan. After working as a post-doctoral fellow at the University of Geneva, Switzerland (2012–2014), he was promoted to assistant professor at the Nara Institute of Science and Technology (NAIST) in 2014. He moved to the National Institute for Materials Science (NIMS) as a senior researcher in 2023, and was promoted to principal researcher in 2025. During 2021–2025, he was a researcher at PRESTO, JST. His current research interests include the synthesis of acene-based macrocycles and their application in organic devices. |
 Hiroko Yamada | Hiroko Yamada received her PhD degree in 1992 from Kyoto University. After post-doctoral fellowships at the Argonne National Laboratory, USA (1993), and Osaka University (1998–2003), she was promoted to associate professor at Ehime University (2003), and moved to the Nara Institute of Science and Technology (NAIST) in 2011. She was promoted to full professor at NAIST in 2012 and moved to the Institute for Chemical Research (ICR), Kyoto University in 2023. During 2006–2010, she was a researcher at PRESTO, JST. Her current research interests include the synthesis and morphology control of small molecular organic electronic materials for solution processing. |
1 Introduction
In recent years, the advancement of organic electronics has been truly remarkable. The unique characteristics of extremely lightweight and ultra-thin organic devices have paved the way for the creation of wearable technologies. In these devices, charge transport within organic semiconductors is facilitated through the overlapping of π-orbitals in conjugated molecules along the direction of carrier flow. Thus, organic semiconducting materials possessing a rigid and planar π-system hold great promise for providing optimal packing that enhances π-orbital overlap. Due to their extensive and robust π-systems, higher acenes and their derivatives emerge as promising candidates for efficient p-type semiconducting materials.1–5 Higher acenes, comprising multiple linearly fused benzene rings, represent a significant category of polycyclic aromatic hydrocarbons.6–8 Substantial research efforts have been directed towards their synthetic development, derivatization, and application as organic semiconductors. Organic field-effect transistors (OFETs) are preferred for rapid screening of charge carrier mobility in newly synthesized organic materials due to their straightforward fabrication and processing. A typical OFET device comprises an active layer (either an organic semiconductor film or a single crystal), three electrodes (gate, source, and drain), and a gate dielectric (insulator) (Fig. 1). The substrate and gate electrode are typically composed of highly doped silicon. Silicon oxide (SiO2) is used as the dielectric insulating layer, formed by the oxidation of the silicon surface. Organic semiconductors or single crystals are directly deposited onto the dielectric layer, while sometimes self-assembled monolayers are employed to modify the dielectric surface to improve its surface energy and roughness. In device operation for p-type molecules, as the gate voltage increases, holes accumulate at the interface between the organic semiconductor and the dielectric insulator, thus allowing the source–drain current to be modulated by the gate voltage. Charge carrier mobility is directly proportional to the channel current. | ||
| Fig. 1 (a) Bottom-gate, top-contact OFET device structure. (b) Image of an OFET using a single crystal. | ||
As described above, higher acenes possess extended π-conjugated systems with rigid and planar structures that facilitate enhanced charge delocalization. In device operation, π-orbital overlap in the active layer is crucial for achieving optimal performance. The rigid and planar backbone of higher acenes significantly contributes to efficient packing and π-orbital overlap, thereby enhancing device performance. This feature sets them apart from other organic materials by promoting superior charge transport properties, which are vital for organic electronics like OFETs and organic photovoltaics (OPVs). Additionally, the radical character of higher acenes, which varies depending on their length, opens up opportunities for their application in carbon-based spintronic devices. In terms of differentiation from other materials, higher acenes possess chemical reactivity that enables modifications to their backbone, significantly influencing the packing structure on the substrate and the processability of device fabrication. This adaptability is essential for achieving optimal device characteristics and tailor-made applications. Another advantage of higher acenes is their ability to achieve high purity materials, a feature that poses challenges for polymer-based organic semiconductors, which often suffer from impurities due to mixtures of varying polymer lengths. Pure materials lead to more uniform electronic properties and improved device reliability. Thus, the use of higher acenes in organic electronic devices offers numerous benefits, including enhanced device performance and the potential for solution-processed device fabrication. Their capacity for fine-tuning through molecular engineering enables the design of materials with specific electronic properties, further enhancing their versatility and appeal in advanced applications. For example, pentacene, which consists of five fused benzene rings, was found to function as an OFET when vacuum-deposited into thin films, exhibiting an exceptional hole mobility of 1.5 cm2 V−1 s−1 at the time,9 leading to explosive progress in research. Later, 6,13-ditriisopropylsilylethynylpentacene (TIPS-Pen),10 developed by Anthony and colleagues, emerged as a benchmark compound for organic semiconductor materials (Fig. 2). Unlike pentacene, which shows almost no solubility in any media, TIPS-Pen's high solubility—attributed to its TIPS groups—enables solution processing. The innovative fabrication of highly crystalline films has shown significantly enhanced hole mobility, reaching 11 cm2 V−1 s−1.11
 | ||
| Fig. 2 Pristine and functionalized pentacenes. | ||
Moreover, in recent years, higher acenes and their derivatives, including tetracene, pentacene, and hexacene, have been identified as efficient materials for exhibiting singlet fission phenomena.12–19 To thoroughly explore the intricacies of electron transfer dynamics, extensive research has been conducted on the synthesis of higher acene oligomers integrated with various substituents and linkers. Advancing these studies is anticipated to substantially boost power conversion efficiency of organic photovoltaic cells.
A defining characteristic of higher acenes is their remarkably narrow highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) gaps.2–4 As the acene chain length increases, the energy gap between the HOMO and LUMO diminishes significantly, leading to oxidative instability. This unique electronic feature is attributed to their zigzag-edged structure, in contrast to phenacenes—structural isomers of higher acenes—that exhibit stability against oxidation.20–26 This disparity stems from differences in electronic configurations between higher acenes and phenacenes, as illustrated in Fig. 3 and 4. According to Clar's aromatic sextet rule, structures that host the greatest number of aromatic sextets (resonating benzene rings) tend to be more stable. Phenacenes can have these aromatic sextets alternately without issue (Fig. 4). In contrast, attempts to arrange aromatic sextets in higher acenes result in unpaired electrons, rendering higher acenes unstable.
 | ||
| Fig. 3 Acenes and phenacenes. | ||
 | ||
| Fig. 4 Clar's aromatic sextet rule for nonacene and [9]phenacene. | ||
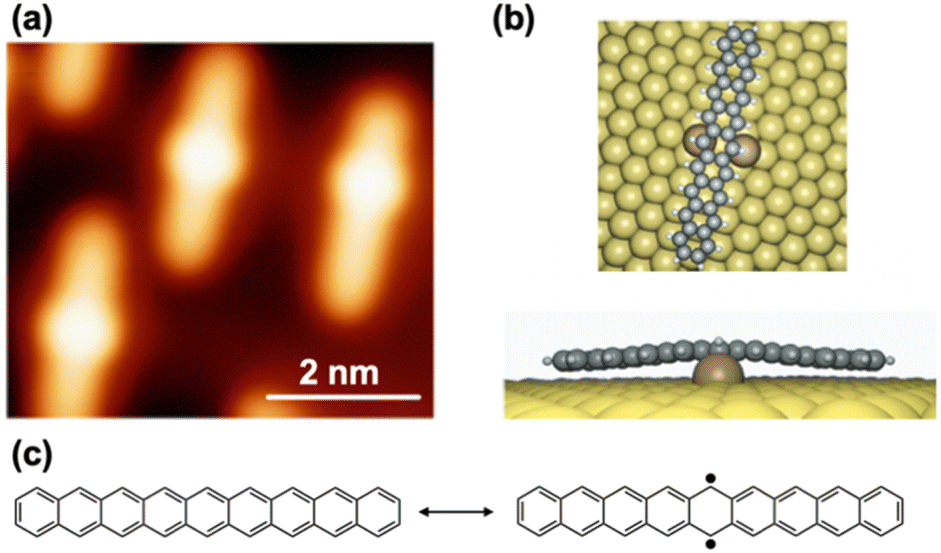
This zigzag-edged structure of higher acenes is strongly correlated with the magnetism observed in zig-zag type graphene nanoribbons (ZGNRs).27–33 Higher acenes are regarded as the narrowest ZGNRs, serving as excellent model systems for understanding the edge states, chemical reactivity, and electronic structures of ZGNRs (Fig. 5). Moreover, as demonstrated by the surface-assisted synthesis of armchair-type 7-GNRs (7-AGNRs) from brominated anthracene dimers,34 higher acenes have significant potential as key compounds in the bottom-up synthesis of AGNRs with narrow bandgaps.35–37 As such, higher acenes are deeply intertwined with the chemistry of GNRs and nanographenes. GNRs retain the electronic properties of graphene and are anticipated to possess superior charge transport properties compared to silicon semiconductors, positioning them as promising candidates for next generation semiconductors. Given these fascinating features of higher acenes, advancing their synthesis and exploring their applications in organic devices have become increasingly crucial.
 | ||
| Fig. 5 Higher acenes, which are deeply intertwined with GNR chemistry. | ||
Despite the intriguing electronic properties and structure of higher acenes, their stability decreases as the acene length increases. Furthermore, higher acenes exhibit poor solubility in common organic solvents owing to their uncomplicated structures. These challenges of poor solubility and instability associated with higher acenes have impeded the acquisition of experimental insights. Until recently, characterizing higher acenes, particularly verifying their formation, primarily relied on measuring absorption spectra under inert conditions at extremely low temperatures or exclusively using mass spectrometry.38–41 As a result, a definitive conclusion based on experimental evidence regarding the magnetism of higher acenes has remained elusive, leading to ongoing controversy for many years. Establishing the link between the length and magnetism of higher acenes is one of the most significant challenges in this research field, prompting numerous theoretical studies. In 2007, Hachmann et al. suggested that higher acenes exhibit a singlet polyradical nature in their ground state using an ab initio density matrix renormalization group algorithm.42 In 2016, Davidson and Yang applied the particle–particle random-phase approximation with the B3LYP functional, suggesting that the 1Ag ground states of acenes up to decacene are predominantly closed-shell, while undecacene and dodecacene display increasing polyradical character, trending towards open-shell states.43 In 2018, Malrieu et al. predicted spin-symmetry breaking in higher acenes beyond a certain length using density-functional theory (DFT), with the result dependent on the exchange–correlation potential.44 Heptacene (N = 7, where N represents the number of fused benzene rings) demonstrated the first symmetry breaking, with Clar sextets positioned on the outermost rings on its left and right sides, linked by tetra-methylene anthracene at the core (Fig. 6). Unpaired electrons, partially localized, extend across the central ring, with notable spin density observed up to N = 11. For 7 < N < 11, this configuration persists, with unpaired electrons distributed over extended tetra-methylene acenes. At N = 13, the acene can accommodate three Clar sextets, with one on each of the external rings and one on the central ring. In the case of N = 14, a naphthalene can be drawn in the central part. At N = 15, pentadecacene is expected to exhibit a tetraradical nature, with a Clar sextet occupying the central ring. Thus, various theoretical studies have been conducted concerning the magnetism of higher acenes, suggesting the need for further experimental investigations.
 | ||
| Fig. 6 Predicted spin density of higher acenes. Adapted with permission from ref. 44. Copyright 2018 American Chemical Society. | ||
To explore the intrinsic properties of higher acenes, their reactive nature must be managed. One promising approach involves functionalizing higher acenes with stabilizing and protective groups. As demonstrated by TIPS-Pen, introducing suitable substituents can effectively enhance the stability, solubility, and processability of higher acenes, making them more amenable to solution-based processing. Typically, alkylated silyl ethynyl groups or bulky substituents are introduced to the acene backbone. Specifically, in oligomer synthesis, a protection/deprotection strategy is commonly employed in combination with appropriate substituents. Another method involves the “precursor approach”, where entails synthesizing a higher acene precursor equipped with leaving groups that can be released in situ to generate higher acenes. Stable and soluble precursors can be transformed into the corresponding higher acenes under optimal conditions. Successful examples include thermally induced reactions, achieving quantitative conversion from precursors to higher acenes through simple thermal annealing, yielding only gaseous byproducts if the precursor purity is adequate (Fig. 7).45–51 Following the initial report of a thermal precursor for pentacene by Müllen et al.,50,51 several thermal precursors have been synthesized. The retro-Diels–Alder reactions of pentacene precursors occur quantitatively and can be executed in solution, powder, or film form. Remarkably, pentacene films after spin-coating and subsequent retro-Diels–Alder reactions through annealing showed OFET performance comparable to that of amorphous silicon. Chow et al. reported monoketone precursors that release a CO molecule via annealing and photoirradiation.45,47
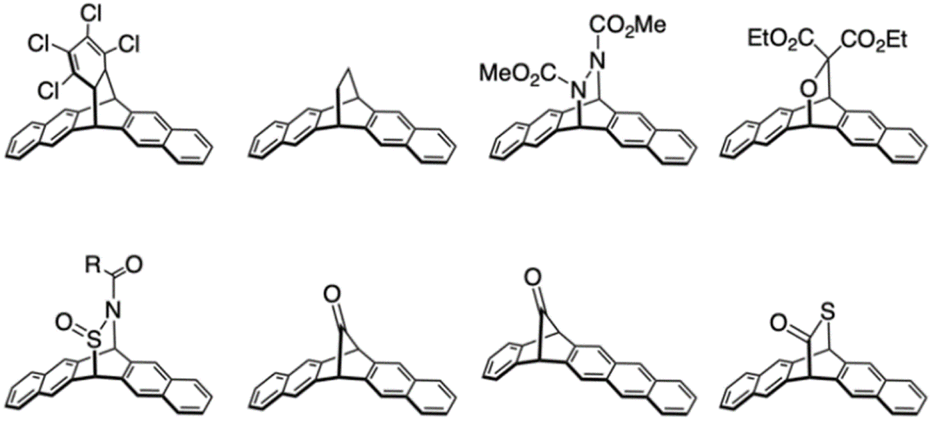 | ||
| Fig. 7 Representative examples of thermal precursors for pentacene. | ||
In addition, the Strating–Zwanenburg reaction,52 where α-diketone groups undergo visible-light-induced photodecarbonylation to produce higher acenes, exemplifies a precursor method. For the photochemical conversion of a pentacene precursor, Yamada et al. utilized the Strating–Zwanenburg reaction, converting a 6,13-α-diketone precursor of pentacene to pentacene upon exposure to visible light at approximately 460 nm, corresponding to the n–π* absorption of the diketone moiety (Fig. 8).53,54 This conversion is feasible solely through photoirradiation, with the precursor being thermally stable above 300 °C. Together with substitution of higher acene backbones, these precursor approaches propel contemporary higher acene chemistry forward.
 | ||
| Fig. 8 Visible-light-induced photodecarbonylation to generate pentacene. | ||
The challenges in synthesizing higher acenes, and evaluating their optical and physical properties can be effectively addressed using these precursors, which are optimized differently for bulk conditions versus “on-surface synthesis”. This review highlights recent advancements in the synthesis and exploration of the electronic structures of higher acenes under bulk conditions and within ultra-high vacuum (UHV) environments on metal surfaces. Given the compelling optical and electronic properties of higher acenes, it is essential not only to explore their synthesis and property evaluation but also to emphasize their material applications such as in OFETs. Indeed, higher acenes and their derivatives show promise in organic device applications. In this review, we present recent examples of their utilization in organic devices.
2 On-surface synthesis of higher acenes
In recent years, there has been increasing interest in studying higher acenes through on-surface synthesis. This method involves sublimating precursor molecules of higher acenes onto a metal surface and transforming them into higher acenes through external stimuli, such as annealing. Conducting these transformation reactions under UHV conditions effectively addresses the instability issues associated with higher acenes. Moreover, the structure, band gap, magnetism, and other properties of the resulting higher acenes can be evaluated at the single-molecule level through in situ observations with scanning tunneling microscopy (STM) and non-contact atomic force microscopy (nc-AFM).55,56 Thus, synthesizing suitable higher acene precursors, in conjunction with on-surface synthesis and probe techniques, is anticipated to provide definitive insights into the magnetism of higher acenes, which has predominantly been a subject of theoretical prediction until now.Heptacene and nonacene were synthesized from their respective photoconvertible precursors using on-surface synthesis. Two α-diketone moieties were introduced into the backbone of heptacene (7DK2) and nonacene (9DK2) (Fig. 9).57 The α-diketone precursors offer stability during sublimation and reactivity upon light exposure, which is crucial for on-surface synthesis since molecules must be evaporated onto a metal surface. Under UHV conditions, photoirradiation with visible light (470 nm) of 9DK2 on Au(111) resulted in a rod-like structure, indicating the formation of nonacene via photoinduced decarbonylation of 9DK2. Heptacene was synthesized using a similar approach. Detailed structural analysis was conducted using STM and nc-AFM with a CO-functionalized tip. In particular, the AFM images clearly revealed the presence of seven and nine benzene rings, along with the zig-zag edge carbon atoms with single hydrogen terminations, confirming the formation of pristine heptacene and nonacene.
 | ||
| Fig. 9 (a) Light-induced formation of heptacene and nonacene. STM and nc-AFM observations of (b) heptacene and (c) nonacene. Adapted with permission from ref. 57. Copyright 2019 Springer Nature. | ||
Notably, following photoconversion, lateral protrusions were observed near each nonacene, attributed to Au adatoms bound to the nonacene backbone (Fig. 10). This interaction between Au and nonacene, where Au atoms primarily occupy the center of the molecules, is consistent with the well-known increase in acene reactivity as their length grows, with the central rings being the most reactive. In fact, Au–acene interactions were also evident in heptacene, although less frequently (∼60% of the molecular species are bound to Au adatoms compared to ∼95% for nonacene). These Au–nonacene interactions suggest an enhanced open-shell character in nonacenes. Nonacene can be represented by either a closed-shell Kekulé structure or a non-Kekulé open-shell structure.58,59 In the non-Kekulé open-shell representation, an additional stabilizing aromatic π-sextet (totaling two π-sextets) is incorporated into the chemical structure, accompanied by unpaired electrons. This non-Kekulé open-shell structure aligns with the observed interactions between the central ring of nonacene and Au adatoms, indicating a shift from a purely closed-shell structure to an open-shell configuration. Theoretical calculations, along with experimental investigations of the HOMO–LUMO gap of nonacene via scanning tunnelling spectroscopy (STS), further support this open-shell character.
 | ||
| Fig. 10 (a) STM image of nonacene–Au interaction. (b) Side and top views of the corresponding DFT equilibrium geometry. (c) Closed-shell Kekulé (left) and one of the many possible non-Kekulé (right) Clar structures of nonacene. Adapted with permission from ref. 57. Copyright 2019 Springer Nature. | ||
The on-surface synthesis of nonacene, together with the experimental and theoretical evaluation of its open-shell character, has markedly advanced research on higher acenes. Further combined experimental and theoretical investigations are anticipated to reveal more details about the ground state nature of higher acenes. Therefore, developing precursors that produce even longer acenes is necessary. One distinct advantage of on-surface synthesis is leveraging the high catalytic activity of single-crystal metal surfaces.60 Substituents that are unreactive to external stimuli such as heat or light irradiation in bulk or solution states often undergo elimination reactions on the Au(111) surface. These substituents, which exhibit elimination reactions on the Au(111) surface, can serve as protective groups for unstable, higher acenes.
Echavarren et al. developed a valuable synthetic method for producing hydrogen-protected higher acenes.61–66 The synthetic process involves gold(I)-catalyzed cyclization of 1,7-enynes, obtained from a palladium-catalyzed Sonogashira cross-coupling between an aryl iodide and key precursors, which then form hydrogen-protected higher acenes through aromatization by eliminating a methanol molecule (Fig. 11). This strategy was applied to synthesize hydrogen-protected higher acenes from heptacene to undecacene (Fig. 12). The parent higher acenes could be generated either thermally, by annealing the sample at 520 K for 10 minutes, or using a microscope tip. Notably, the dehydrogenation process is highly efficient, achieving nearly a 100% conversion rate. The formation of the series of higher acenes and their definitive chemical structures were confirmed unambiguously by STM and nc-AFM with a CO-functionalized tip. Moreover, the series provided the opportunity to analyze the dependence of the STS-measured transport gap on the number of benzene rings (Fig. 12). In the range of 5–10 fused benzene units, both inverse proportionality and the exponential decay provided good fits. However, undecacene deviates towards larger gap values, indicating a saturation in further band gap reduction for longer higher acenes. This trend becomes even more evident when the gap is plotted as a function of inversed number of annulated rings. These findings could be rationalized by the expected increased contribution of the open-shell configuration to the overall electronic structure.43
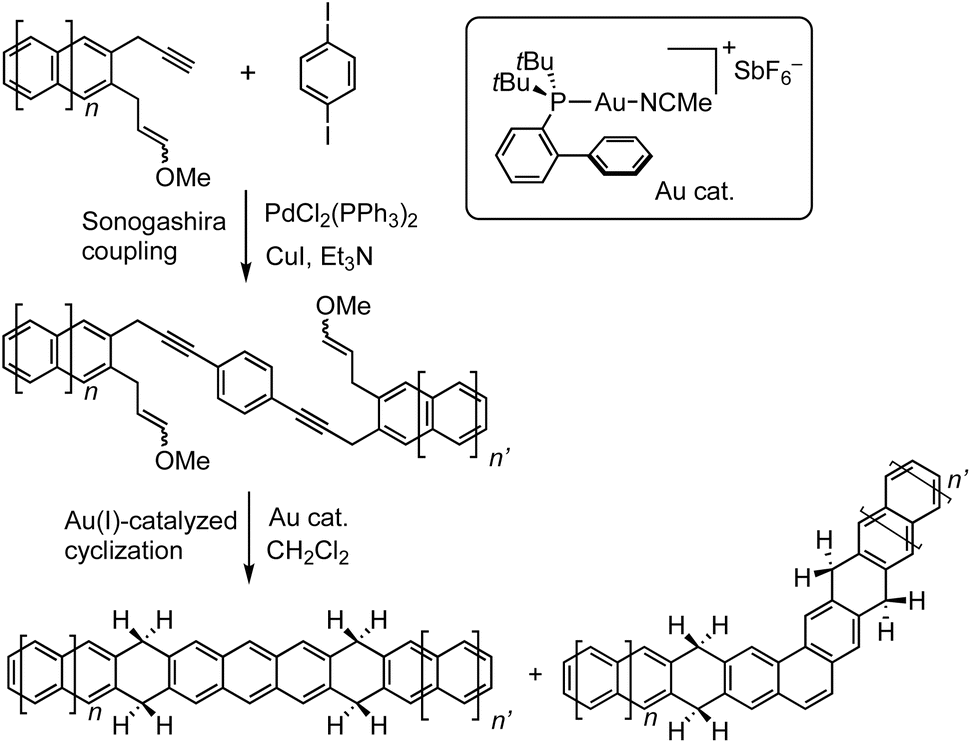 | ||
| Fig. 11 Synthesis of tetrahydroacenes. Adapted with permission from ref. 62. Copyright 2018 Wiley. | ||
 | ||
| Fig. 12 On-surface generation of undecacene. (a) High-resolution filled state STM image of undecacene. Laplace-filtered constant height, frequency shift nc-AFM images of (b) undecacene and (c) kinked acene. (d and e) STS measured transport gap for long acenes on Au(111). (d) Dependence of the gap on the number of fused benzene rings with the inverse proportionality fit (red curve) and exponential decay (blue curve). (e) Transport gap plotted as a function of the inverse number of rings with the linear fit. Adapted with permission from ref. 62. Copyright 2018 Wiley. | ||
Recently, Echavarren, Jelinek, and Godlewski et al. extended this synthetic strategy to octahydrotridecacene, synthesized through multiple Sonogashira cross-coupling reactions followed by a final four-fold gold(I)-catalyzed [4 + 2] cycloaddition (Fig. 13).63 The surface-assisted reaction of this precursor, achieved by annealing at 270 °C for 15 min yielded tridecacene. The formation of tridecacene was confirmed by STM observation. STS measurements and spatial orbital mapping, combined with theoretical modeling, indicated that tridecacene possesses an open-shell ground state. Importantly, an inelastic signal was detected, attributed to spin excitation from the singlet diradical ground state to the triplet excited state, with an estimated singlet–triplet gap of approximately 126 meV. This work provides experimental confirmation of tridecacene's magnetic character. Furthermore, the magnitude of the singlet–triplet band gap was found to be influenced by surface proximity and dynamic electron correlation.
 | ||
| Fig. 13 (a) Synthesis of the tridecacene precursor and (b) STM image of on-surface generated tridecacene. (c) Single point STS spectra acquired over tridecacene, exhibiting a symmetric step-like appearance (top) together with numerically differentiated d2I/dV2 spectra showing the presence of pronounced resonances centred at approximately ±126 meV. Adapted with permission from ref. 63. Copyright 2023 Wiley. | ||
Peña et al. introduced a different kind of precursor, specifically epoxy-protected higher acenes.67–70 Essentially, Diels–Alder reactions involving OTf and TMS-modified aromatic species and benzofuran create epoxy-protected higher acenes. This strategy was initially used to synthesize a tetracene precursor.67 Furthermore, repeated Diels–Alder reactions can extend the protected state of the fused benzene rings, producing decacene and dodecacene precursors (Fig. 14).69,70 The surface-assisted deoxygenation of pentaepoxy-protected precursors through annealing at 220 °C resulted in the formation of dodecacene. High-resolution STM images with a CO-functionalized tip revealed 12 lobes corresponding to the 12 benzene rings of dodecacene. STS experiments provided experimental insights into the electronic structure of dodecacene. Interestingly, while a progressive closing of the gap and its stabilization to about 1 eV was observed for decacene and undecacene, the energy gap of dodecacene unexpectedly increased again to 1.4 eV. This reopening of the gap was interpretated as a possible indication of an increased poly-radical (tetra-radical) character of dodecacene.44
 | ||
| Fig. 14 (a) Synthesis of dodecacene. (b) Development of the energy gap of acenes as a function of the number of benzene rings. Adapted with permission from ref. 70. Copyright 2023 American Chemical Society. | ||
Eimre, Yamada, Fasel, and Pignedoli et al. discovered that etheno-bridges can serve as protective groups for higher acenes.71 While bis-etheno-bridged undecacene precursors are thermally stable under bulk conditions, the surface-assisted reaction of these precursors on Au(111) effectively cleaves the etheno-bridges, yielding undecacene (Fig. 15). It is likely that acetylene molecules are removed as leaving groups via retro-Diels–Alder reactions. The on-surface reactions resulted not only in the formation of undecacene but also a variety of acene species, ranging from anthracene to decacene, including kinked acenes. The cleavage of different C(sp3)–C(sp2) bonds exhibited no clear selectivity, due to the similarity between the C(sp2) atoms in the etheno-bridge and those belonging to the acene backbone.
 | ||
| Fig. 15 (a) On-surface synthesis of undecacene from an etheno-bridged precursor. (b) Overview STM topography image after annealing the sample at 220 °C. (c) Table showing the statistics of the acene species found on the substrate after annealing at 220 °C. Adapted with permission from ref. 71. Copyright 2022 Springer Nature. | ||
Bettinger, Tonner-Zech, and Gottfried et al. employed etheno-bridged precursors to synthesize tridecacene and pentadecacene (Fig. 16).72,73 For tridecacene, STM tip manipulation initiated a three-dimensional conformational transformation of the precursor on Au(111), changing it from an edge-on structure to a stretched-out linear conformer. Subsequent repeated tip-assisted carbon–carbon bond dissociations removed the etheno-bridges, resulting in tridecacene. Notably, annealing at 490 K induced the edge-on to stretched-out transformation of the precursor without eliminating the etheno-bridges. STS measurements showed a HOMO–LUMO gap reduction to 1.09 eV, consistent with the gap reopening reported for dodecacene (Fig. 14).70 There was a discrepancy in the band gap between tridecacene derived from etheno-bridges versus epoxy-protected precursors. This difference was attributed to variations in the adsorption site of the molecule on Au(111),74 influenced by preparation steps like annealing and cleavage processes, along with the strong influence of the Au(111) surface on the transport gaps,75 altering the ground state electronic configuration.
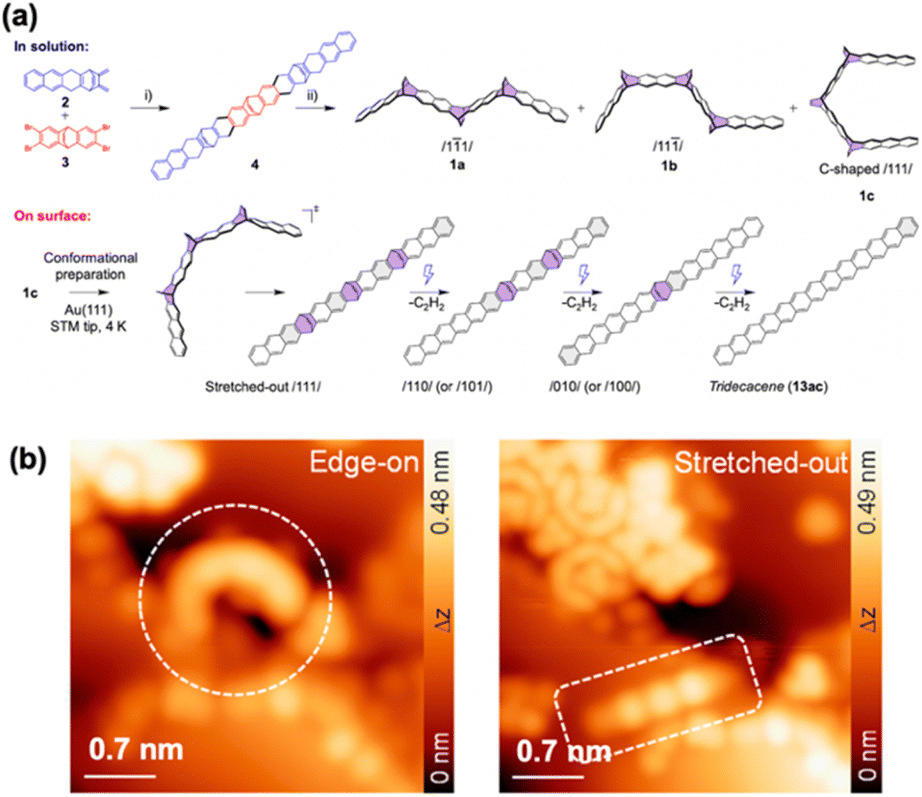 | ||
| Fig. 16 (a) Synthetic strategy for tridecacene. (b) Magnified constant-current STM image of edge-on and stretched-out conformations of a tridecacene precursor. Reproduced with permission from ref. 73. Copyright 2024 American Chemical Society. | ||
For pentadecacene, the trietheno-bridged pentadecacene precursor with a stretched-out conformation was converted to pentadecacene through repeating tip treatments applying a pulse voltage of 2.5–3.0 V (Fig. 17).72 The STS transport gap was approximately 1.12 eV, slightly larger than the 1.09 eV for undecacene and tridecacene. Surprisingly, the STS transport gap does not significantly change with length. Notably, a spin excitation feature near the Fermi energy, characteristic of a singlet open-shell electronic structure,76 was observed. Numerically obtained inelastic tunneling spectra (d2I/dV2) determined the singlet–triplet gap to be 124 meV, closely aligning with tridecacene's 126 meV.73
 | ||
| Fig. 17 (a) Synthetic strategy for pentadecacene. (b) dI/dV curves for pentadecacene; two resonance peaks are identified as the HOMO and LUMO. (c) Low-bias dI/dV curve (top) and its corresponding d2I/dV2 spectrum. Adapted with permission from ref. 72. Copyright 2025 American Chemical Society. | ||
Increasing the sublimation temperature to 750 K induced thermal cleavage of the etheno bridges during deposition. Importantly, directly generated pentadecacene reacted with Au atoms (Fig. 18). STM images showed a linear product with six noticeable edge indentations at the edges at low bias and pronounced protrusions at high bias, similar to adsorbed open-shell carbon nanostructures.57,77,78 The STM tip was found to eliminate the adatoms; interestingly, removing an Au adatom from one side left the opposite side unaffected, unlike typical acene metal complexes where adatoms are removed simultaneously.57 Removing a single Au adatom leaves an unpaired π-electron, evidenced by the Kondo effect.79,80 The observation of pentadecacene's spontaneous complexation with up to six Au atoms on the Au(111) surface reveals the high reactivity of the long acene, indicating substantial polyradical character contributing to the ground state of pentadecacene.42–44,81,82
 | ||
| Fig. 18 (a) (Left to right) STM, constant-current nc-AFM, and constant-height nc-AFM of the 6Au–pentadecacene complex. (b) Constant-height nc-AFM image with the chemical structure. (c) STM and constant-height nc-AFM image of the 5Au–pentadecacene complex, which was obtained by removing a single Au atom from the 6Au–pentadecacene complex. Reproduced with permission from ref. 72. Copyright 2025 American Chemical Society. | ||
As mentioned above, there have been numerous reports on the ground state and band gap of higher acenes. The use of various computational levels, methods, and basis functions has often led to different theoretical predictions for the electronic states of the same higher acenes. This discrepancy is especially pronounced for higher acenes that lie at the boundary between open-shell and closed-shell configurations, which may result in completely different interpretations. Therefore, it is necessary to reconcile the discrepancies between theoretical predictions and experimental measurements of the electronic states of higher acenes in order to provide clear guidance regarding the validity of computational methods. The synthesis of higher acenes through on-surface synthesis and the direct measurement of their electronic states and band gaps via STS, as mentioned above, have dramatically improved this situation. The synthesis and elucidation of the polyradical character of pentadecacene by Bettinger, Tonner-Zech, Gottfried and colleagues support previous predictions.44 However, it is also becoming apparent that the electronic states of higher acenes prepared by surface-assisted reactions need to consider the effects of the substrate, adsorption position, and even the synthesis method employed. Thus, the continued improvement of computational methods to accurately interpret experimental findings will become increasingly important.
Despite these successful on-surface syntheses of higher acenes and heteroatom-substituted ZGNRs,27 the investigation into higher heteroacenes has remained largely unexplored. Eimre, Yamada, Fasel, and Pignedoli et al. reported the on-surface synthesis of 6,12,19,25-tetraazaundecacene from a diethano-bridged precursor (Fig. 19).71 Interestingly, annealing the precursor on Au(111) did not yield 6,12,19,25-tetraazaundecacene directly; instead, it produced hydrogenated tetraazaundecacene and its analog with two edge-fused five-membered rings. The five-membered ring-fused tetraazaundecacene analog presumably forms through bond cleavage at the bridged head position of the bicyclo[2.2.2]octadieno unit, where the terminal part of the resulting fragment re-bonds to a neighboring nitrogen atom. These steps facilitate the removal of two protons from the ethano groups via dehydrogenative aromatization. To circumvent such side reactions (hydrogen passivation and/or five-membered ring formation), STM tip-induced deprotection with a voltage ramp (−1.0 to −3.0 V) yielded 6,12,19,25-tetraazaundecacene. STS measurements complemented by ab initio simulations revealed its considerable open-shell character on Au(111). Furthermore, the electronegative nitrogen atoms caused a noticeable shift in energy level alignment compared to the pristine undecacene.
 | ||
| Fig. 19 (a) On-surface synthesis of tetraazaundecacene and its analogues. (b) Successive STM topography images illustrating the cleavage process. (c) DFT-calculated equilibrium geometry of a cluster of tetraazaundecacene. (d) Experimental constant-height dI/dV maps and DFT-calculated molecular orbitals of tetraazaundecacene. Adapted with permission from ref. 71. Copyright 2022 Springer Nature. | ||
The success of on-surface synthesis and structure–property analysis using STM/nc-AFM for these higher acenes hints at the potential to synthesize cyclacene, a cyclic molecule that remains elusive (Fig. 20). Cyclacene is considered as the simplest zigzag-type carbon nanobelt, first proposed in 1954.83 Despite numerous synthesis attempts, success has been hindered by the strong ring strain, low stability, and high reactivity of cyclacene in solution. While Stoddart and co-workers managed to produce a precursor, they could not achieve cyclacene through reductive aromatization.84–86 In 1996, Vittal and co-workers made another attempt, and Wang and co-workers detected [8]cyclacene derivatives by mass spectrometry.87 The pursuit of cyclacene synthesis thus continues to captivate researchers globally.
 | ||
| Fig. 20 Attempts for cyclacene synthesis by (a) Sttodart and (b) Vittal. | ||
Recently, Peña, Gross, and colleagues explored cyclacene formation through on-surface synthesis.88 They synthesized epoxy-protected cyclacene precursors, akin to strategies used for higher acenes (Fig. 21). The precursors were deposited on a Cu(111) surface, and STM-based tip manipulation was used to remove oxygen atoms from the epoxy groups. Detailed characterization using AFM with CO-functionalized tips suggested that up to two oxygens per molecule could be removed, forming an oval-shaped diepoxy-[10]cyclacene. Although cyclacene was not fully synthesized, experimental results and DFT calculations predicted that further deoxygenation might be feasible for larger diepoxycyclacenes, as employing larger macrocycles could reduce the energy cost for the deoxygenation.
 | ||
| Fig. 21 Synthesis of a precursor of [10]cyclacene and its X-ray crystal structure. Zoomed-in AFM images of two molecules after tip manipulation. Adapted with permission from ref. 88. Copyright 2019 Wiley. | ||
Hayashi et al. extended the ethano-protection strategy to synthesize azacyclacenes.89 A dehydration condensation reaction between anthracene tethered with bis-diketone groups and benzene-1,2,4,5-tetraamine tetrahydrochloride yielded M[3 + 3] (Fig. 22). Although the isolated yield of M[3 + 3] was low (0.8%), the simplicity of the 1H NMR spectrum reflecting its highly symmetrical structure, along with HR-MALDI-TOF MS results, clearly indicated M[3 + 3] formation. Despite M[3 + 3] being unsuitable for sublimation on metal surfaces due to its large molecular weight, necessitating new deposition methods, this result offers a fresh perspective on azacyclacene synthesis.
 | ||
| Fig. 22 Structure of M[3 + 3] and its HR-MALDI-TOF MS spectrum. Adapted with permission from ref. 89. Copyright 2022 Wiley. | ||
These findings suggest that carefully designing precursors and employing appropriate methods are crucial for realizing pristine cyclacenes and azacyclacenes in the near future.
3 Organic synthesis of higher acenes
As noted earlier, on-surface synthesis is an excellent method for producing unstable higher acenes and analyzing their molecular structure and properties at the single-molecule level. However, large-scale synthesis, which is essential for practical applications, remains a significant challenging, although some GNRs, which were prepared through on-surface synthesis, were used for transistors.90–93 Thus, it is crucial to leverage insights gained from on-surface synthesis to develop organic synthesis methods that support large-scale production.Bettinger et al. have made substantial advancements in synthesizing higher acenes using α-diketone precursors.38–41 Converting these precursors to higher acenes in either an Ar matrix or poly(methyl methacrylate) (PMMA) has been proven effective. Acenes ranging from hexacene to nonacene had been synthesized, and their optical properties had been explored alongside theoretical calculations. A notable feature of acenes is their distinctive optical spectrum; for acenes up to heptacene, these spectra are characterized by a single very strong transition in the UV region (1Bb, β band) and weaker transitions extending into the visible range (1La, p band).94 The p band arises from an excited state predominantly involving the HOMO → LUMO transition, with its energy linked to the optical gap. Both characteristic bands shift bathochromically as the system extends. For octacene and nonacene, an additional strong band has been observed. DFT/MRCI computations suggest this is due to an excited state, labeled D2, with significant double excitation contributions.94 Theoretical analysis indicates that this state shifts to lower energies faster than the 1Bb and 1La states as the acene chain length increases.95
Further exploration of the optical properties of higher acenes led to the synthesis of undecacene.96 Here, the precursor was photo-converted to undecacene in a polystyrene matrix under cryogenic conditions (8 K) (Fig. 23). Photoirradiation resulted in a rapid decrease in n–π* transition intensity, alongside a strong signal at 354 nm and weak signals in the 600–800 nm range. These observations align with the β and p bands of heptacene, indicating stepwise photodecarbonylation.38 Although complete photoconversion took time, bands with maxima at 543 nm and 1007 nm increased, suggesting undecacene generation. Correlating with computed data, the strong band at 543 nm was attributed to the D2 state, while the weak band at 1007 nm was linked to the 1La state. Plotting the 1La state energies against 1/N (N = number of benzene rings) yielded a straight line, estimating an optical gap of 1.23 eV at infinite chain length in the acene series.
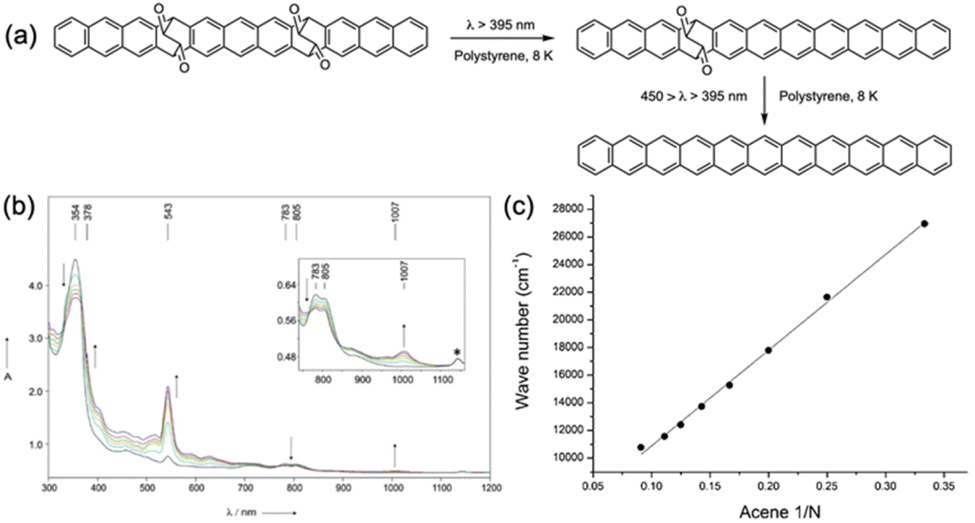 | ||
| Fig. 23 (a) Stepwise photochemical synthesis of undecacene. (b) Absorption spectra obtained after irradiation (450 nm > λ > 350 nm) of the undecacene precursor in polystyrene at 8 K. (c) Plot of the energy maximum of the p band (HOMO → LUMO) transition energy in the acene series. Adapted with permission from ref. 96. Copyright 2018 Wiley. | ||
In the early days of higher acene chemistry, these compounds were considered inherently unstable, posing significant challenges for isolating or bulk synthesizing higher acenes under ambient conditions. Higher acenes were noted for their tendency to undergo oxidation in solution and their proclivity toward dimerization (Fig. 24). However, in 2017, it was discovered that heptacene could remain in a solid state for extended periods, only gradually undergoing dimerization or oligomerization at room temperature.97 When diheptacene was heated in a 1-methylnaphthalene solution, heptacene was formed. Weak signals at 753, 682, and 623 nm, along with a shoulder at 792 nm on the long-wavelength side of the 753 nm signal, persisted at 230 °C for at least 40 minutes, indicating heptacene's unexpected stability.
 | ||
| Fig. 24 (a) Heptacene and its dimerization reaction. (b) Solid state 13C CP-MAS NMR spectrum. The sample was stored at room temperature in an ambient atmosphere for 1 month, and then heated at 300 °C for 12 min. (c) Absorption spectra of heptacene obtained by heating a solution of diheptacene in 1-methylnaphthalene. Adapted with permission from ref. 97. Copyright 2017 American Chemical Society. | ||
Jancarik et al. introduced a versatile strategy for synthesizing a wide variety of higher acenes, using (1S,4S)-7,7-dimethoxy-2,3,5,6-tetramethylenebicyclo[2.2.1]heptane as a pivotal compound (Fig. 25).98 This key compound serves as a precursor for protected higher acenes via Diels–Alder reactions, and the subsequent cleavage of the ketals leads to CO-bridged intermediates. The target acenes can be quantitatively produced by heating in the solid state at approximately 200 °C. This method enabled the synthesis of heptacene, dibenzopentacene isomers, and benzohexacene as notable examples.
 | ||
| Fig. 25 Synthetic strategy for higher acenes and higher acene derivatives employed in this study. Adapted with permission from ref. 98. Copyright 2019 Wiley. | ||
This strategy was further advanced to successfully synthesize nonacene in bulk.99 By performing repeated Diels–Alder reactions followed by oxidation with DDQ, isomers of ketal-protected nonacenes were obtained (Fig. 26). Cleaving the ketals produced bis-monoketone protected nonacenes. Thermogravimetric analysis (TGA) showed that complete conversion occurs below 190 °C in a single step. Remarkably, nonacene exhibited thermal stability up to nearly 500 °C. Additionally, storing nonacene at room temperature within a glovebox for two months did not lead to degradation or dimerization. These findings align with previous observations that both bulk heptacene and even thin-film heptacene remain stable for months at room temperature in a nitrogen-filled environment. This stability potentially paves the way for applications in OFETs and molecular spintronics.
 | ||
| Fig. 26 (a) Synthetic route for nonacene precursors. (b) Thermal decarbonylation of precursors at 350 °C for 20 min and 13C CP-MAS NMP spectra. Adapted with permission from ref. 99. Copyright 2022 Springer Nature. | ||
Research findings in bulk synthesis and on-surface synthesis of higher acenes have demonstrated that these compounds, previously considered unstable, can exhibit relatively long-term stability in oxygen-free or low-oxygen environments. Hayashi et al. discovered that the interior of a single crystal can serve as an isolated space, protecting higher acenes from external influences.100 By irradiating the interior of a single crystal of the photo-convertible heptacene precursor with a CW laser (470 nm), they observed an increase over time in the intensity of the absorption spectrum at 623, 682, 753, and 792 (shoulder) nm, indicating heptacene formation via decarbonylation of the precursor (Fig. 27). This result supports the notion that the interior of the single crystal functions as an isolated space. The generated heptacenes were shielded by precursor molecules or oxidized heptacenes located in the outer regions of the crystal, thereby enabling access to otherwise inaccessible compounds without requiring deoxygenated conditions.
 | ||
| Fig. 27 (a) Photoconversion reaction. (b) X-ray single crystal structure of the precursor and its optical image. (c) Transmission image of the single crystals of the precursor before (left), during (middle), and after (right) photoirradiation, and UV/Vis-NIR absorption spectra recorded during photoirradiation. Adapted with permission from ref. 100. Copyright 2020 Wiley. | ||
In higher acene research, the main focus has traditionally been on the synthesis and property evaluation of acenes with fixed lengths. Conversely, research on polyacenes, which consist of numerous fused benzene rings, has experienced limited progress. In 2023, Kitao and Uemura et al. introduced a strategy for synthesizing exceptionally long polyacenes using metal–organic frameworks (MOFs).101 MOFs are notable for their structural diversity, which allows precise control over pore size and shape at the molecular level, providing an ideal environment for encapsulating various guest species and controlling their assembly structures.102,103
In this study, monomers for polyacenes were introduced into the pores of the MOF via sublimation, forming host–monomer composites (Fig. 28).104 Polymerization was achieved by heating the composites at 250 °C for 24 hours in a sealed glass tube, resulting in the formation of polyacene precursor polymers. The spatial constraints of the MOF enabled highly regulated polycoupling reactions within one-dimensional nanochannels, leading to linearly extended polymeric precursors. The subsequent dehydro–aromatization reaction, performed by heating at 300 °C in an air atmosphere, produced bulk polyacenes. FTIR spectroscopy not only provided evidence for the presence of polyacenes but also offered quantitative structural information. The out-of-plane aromatic C–H vibration modes are classified as SOLO, DUO, TRIO, and QUATRO, based on the number of adjacent C–H groups.105 Only the SOLO and QUATRO modes were detected at 900 and 736 cm−1, respectively. Polyacene length was analyzed using IR spectroscopy, correlating the peaks of out-of-plane sp2 C–H vibration modes (SOLO and QUATRO) with the number of benzene rings. Combined with simulated IR analysis, a linear correlation between the relative peak area of the SOLO to QUATRO modes and the number of benzene rings estimated the mean number (±S.D.) of benzene rings in polyacene from two precursor polymers to be 17.8 ± 3.3 and 18.6 ± 3.5, respectively. The biradical character of polyacenes was evaluated using electron spin resonance (ESR) and a superconducting quantum interference device (SQUID). The ESR spectrum of polyacene revealed a signal with a g value of 2.003, attributable to a carbon-centered radical.106 Additionally, SQUID data showed a component with a steep decrease in magnetic susceptibility upon cooling from 70 to 20 K, consistent with the Bleaney–Bowers equation.107 This magnetic behaviour, characteristic of open-shell singlet biradical molecules, indicates the biradical nature of polyacene.108
 | ||
| Fig. 28 (a) Schematic of polyacene synthesis using an MOF. (b) FTIR spectra of tetracene, pentacene, hexacene, heptacene and polyacenes. (c) The relative peak area of SOLO to QUATRO vibration modes (ASOLO:AQUATRO) plotted against the benzene ring number for the acene series. (d) ESR spectrum of polyacene. No hyperfine splitting was observed, suggesting the delocalization of spin density along the polymer chains. (e) Temperature dependence of the magnetic susceptibility χ of polyacene. Reproduced with permission from ref. 101. Copyright 2023 Springer Nature. | ||
The bulk synthesis of higher acene opens the door for their application in devices such as OFETs and single crystal (SC) FETs. Here, the intrinsic properties of higher acenes, including their π-conjugation length and electronic properties, determine how effectively they can facilitate charge delocalization and transport. Additionally, the instability and insolubility of higher acenes must be overcome to enable their application in devices. The stacking mode and morphology of higher acenes is another vital factor. These factors greatly influence OFET characteristics. The crystalline order and grain size of the semiconductor layers can affect the degree of charge carrier mobility. Here, SCFETs can unveil the intrinsic charge transport properties of organic semiconducting materials due to ordered arrangement of molecules, absence of grain boundaries and minimal defects. The hole mobility obtained from SC FETs reflects the potential of the synthesized molecules.
SCs of hexacene were obtained by sublimating the monoketone precursor, with in situ formation during sublimation made possible through thermally triggered decarbonylation (Fig. 29).109 Hexacene SC FETs demonstrated a hole mobility of 4.28 cm2 V−1 s−1 and maintained functionality for more than 19 days. However, the mobility decreased by 32% even in a nitrogen atmosphere, suggesting potential surface oxidation of the single crystal. These results experimentally unveil the high charge transport potential of hexacene attributed to effective π-overlap, while also indicating the need to enhance stability for practical applications.
 | ||
| Fig. 29 X-ray crystallographic analysis of hexacene. (a) ORTEP drawing depicting two adjacent hexacene molecules. (b) Layered arrangement of hexacene molecules on the a–b plane. (c) Arrays of hexacene along the a-axis. SCFETs based on hexacene. (d) Output characteristics. Inset: a crystal across the electrodes (scale bar, 50 μm). (e) Transfer characteristics recorded at VDS = −80 V. (f) Time-dependent decay of performance under ambient conditions (red squares) and in a nitrogen atmosphere (blue diamonds). Reproduced with permission from ref. 109. Copyright 2012 Springer Nature. | ||
Hole mobility of the thin film of hexacene prepared by vacuum deposition was evaluated.110 Halide-substituted acenes, such as pentacene, have shown significantly superior hole mobility (>5 cm2 V−1 s−1) compared to the parent acene (1.4 cm2 V−1 s−1),111,112 as the bromine substituent improves crystal packing. The OFET properties of brominated hexacene were compared to those of parent hexacene (Fig. 30). The monoketone-type precursor of hexacene was synthesized, and its complete thermal conversion at 230 °C was confirmed by TGA, IR, and solid-state 13C NMR. OFET devices were fabricated by vacuum sublimation of hexacene/brominated hexacene to create thin films on an 1,1,1,3,3,3-Hexamethyldisilazane(HMDS)/SiO2/Si substrate, followed by the deposition of gold electrodes on top of the films. The best hole mobility achieved for brominated hexacene was 0.83 cm2 V−1 s−1, with an on/off ratio of 5.0 × 104 and a threshold of −52 V. In contrast, the best mobility for parent hexacene was 0.076 cm2 V−1 s−1, with an on/off ratio of 2.4 × 102 and a threshold of −21 V. Notably, the best hole mobility for spin-coated hexacene, prepared from a different precursor, was 0.035 cm2 V−1 s−1.113 Thus, brominated hexacene exhibited higher hole mobility, and better crystalline films of hexacene were obtained by thermal deposition. AFM confirmed that the smoother surface of brominated hexacene contributed to reduced energy loss during hole transport between the source and drain electrodes.
 | ||
| Fig. 30 (a) Synthetic route for brominated hexacene and pristine hexacene. (b) Vacuum-deposited thin-film OFETs based on brominated hexacene. Output (left) and transfer characteristics (right, VDS = −100 V). (c) Vacuum-deposited thin-film OFETs based on hexacene. Output (left) and transfer characteristics (right, VDS = −100 V). AFM images of thin films of (d) brominated hexacene and (e) pristine hexacene. Adapted with permission from ref. 110. Copyright 2018 Royal Chemical Society. | ||
Heptacene was expected to exhibit comparable or even better hole mobility than hexacene, due to its potentially smaller reorganization energy. Heptacene was synthesized from the corresponding monoketone precursor (Fig. 31).114 TGA indicated a thermal conversion temperature of 220 °C. Top-contact OFET devices were fabricated by thermal evaporation of heptacene on a HMDS/SiO2/Si substrate. Notably, the highest hole mobility was 2.2 cm2 V−1 s−1, with an on/off ratio of 5 × 103 and a threshold voltage of −56 V. Thus, heptacene thin films exhibited higher hole mobility than hexacene. However, high contact resistance was evident in the output characteristics of the OFET devices. Considering the HOMO level of heptacene (−5.03 eV) and the Au electrode (−4.76 eV),115 along with XRD analysis, the presence of a hole-injection barrier and the formation of small crystals in the heptacene thin film contributed to the high contact resistance. This is because holes injected from the top-contact Au electrodes find it more challenging to reach the channel region near the SiO2 gate insulator surface due to the presence of smaller crystals. These thin films certainly contain grain boundaries. Thus, using a single crystal of heptacene for OFET fabrication could lead to higher mobility, as observed with hexacene.109
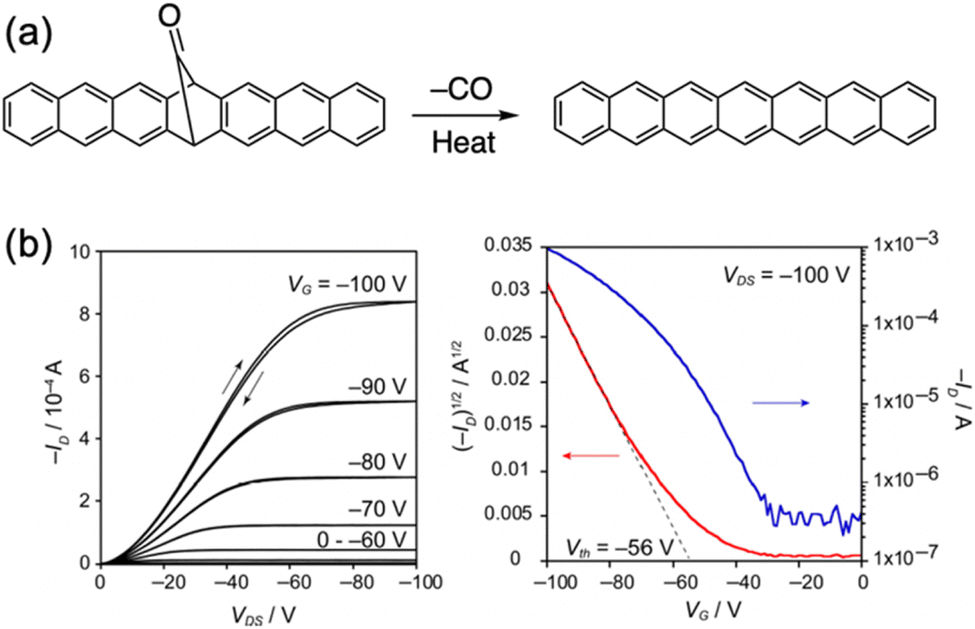 | ||
| Fig. 31 (a) Heptacene generation from its monoketone precursor. (b) Output (left) and transfer characteristics (right, VDS = −100 V) of heptacene thin films. Adapted with permission from ref. 114. Copyright 2021 Wiley. | ||
These results highlight the significant potential of higher acenes as organic semicondctors. Notably, the observed hole mobility of heptacene thin films, exceeding 2 cm2 V−1 s−1, is a particularly promising value. Here, analyzing the packing structure in the fabricated devices is crucial, as vacuum-deposited films inherently contain grain boundaries, which differ fundamentally from SC FETs. Pristine higher acenes have an inherent tendency to aggregate, which can complicate the preparation of films with fewer domains through vacuum deposition. Maximizing the potential of higher acenes necessitates providing feedback for molecular design to optimize the packing structure and morphology on the substrate, thereby facilitating further refinements.
4 Organic synthesis of oligoacene derivatives
For the application of acene-based compounds in organic devices, the overlap of π-electrons between molecules in the crystal packing is a crucial factor. The crystal packing geometry and orientation of acenes are particularly favorable for use in OFETs. Consequently, controlling factors such as the packing structure, orientation, and morphology on the substrate, through the introduction of substituents to the molecule is essential. Importantly, substitutions on higher acenes have the potential to create promising packing structures, due to the addition of other intermolecular interactions, that cannot be achieved with pristine higher acenes. Furthermore, soluble substituents impart solution-processability to higher acenes, which is advantageous for practical applications.Anthracene derivatives, known for their rigid and planar structure coupled with high air stability, serve as efficient p-type semiconducting materials.116 Pristine anthracene single crystals exhibit hole mobilities of 0.02 cm2 V−1 s−1.117 Additionally, extending the π-system at the 2,6-positions of anthracene through oligomerization or functionalization has yielded promising candidates for p-type FETs, with mobility reaching up to 34 cm2 V−1 s−1 (Fig. 32).118,119
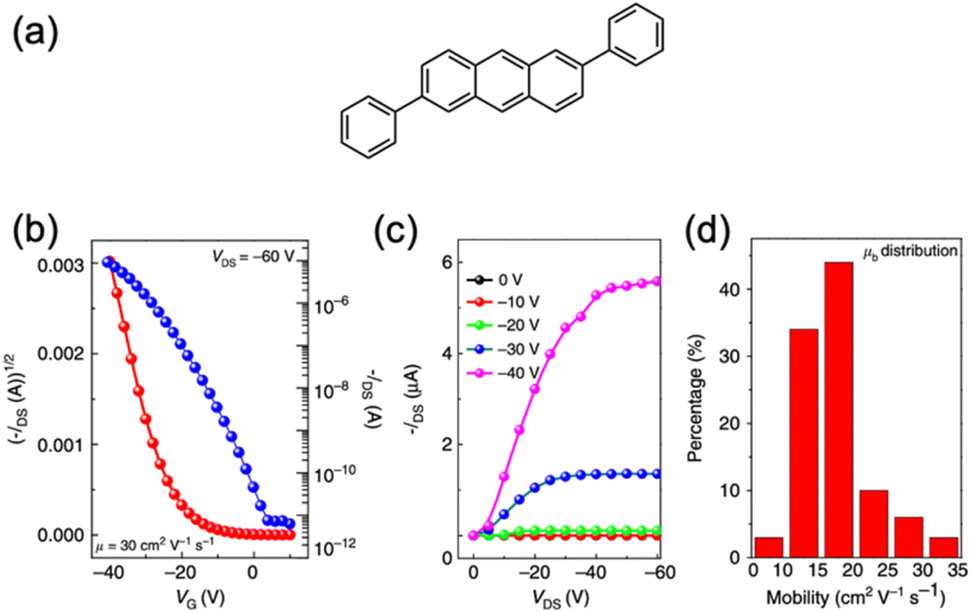 | ||
| Fig. 32 (a) Chemical structure of 2,6-diphenylanthracene. Typical (b) transfer and (c) out-put characteristics of the SCFET device based on 2,6-diphenylanthracene. (d) Mobility distribution. Reproduced with permission from ref. 119. Copyright 2015 Spring Nature. | ||
As described earlier, acenes longer than anthracene, possessing greater conjugation lengths and thus stronger intermolecular overlaps, also demonstrate efficient charge–transport properties. Charge carrier mobilities for tetracene-based OFETs range between 0.4 and 2.4 cm2 V−1 s−1.120,121 Pentacene, a benchmark compound among organic semiconductors, exhibits hole mobilities ranging from 5–40 cm2 V−1 s−1.11,122,123 Hexacene single crystals showed hole mobilities of up to 4.28 cm2 V−1 s−1.109 To achieve efficient charge transport, π-system elongation through oligomerization is a promising strategy. Among various approaches for π-extension, the incorporation of triple-bond systems remains a subject of intense research interest in the development of organic electronic devices.124–126 The introduction of TIPSethynyl groups to the pentacene backbone enhances not only stability but also solubility, thereby enabling solution processability and high hole mobility.11 Furthermore, ethynylene-bridged systems are widely employed in organic electronic materials. The availability of efficient synthetic protocols for ethynylene bridging facilitates the easy modification of effective π-conjugation lengths by controlling the number of acene–ethynylene repeating units, yielding shape-persistent, rod-like structures.
In the case of anthracene, its better stability and relatively easier chemical modification compared to higher acenes have led to numerous studies.116 It is noteworthy that oligomerization at the 9,10-positions results in large dihedral angles between adjacent anthracenes due to significant steric hindrance, which disrupts π-conjugation within the molecule. Nonetheless, effective conjugation extension at the 9,10-positions of an anthracene core can be achieved using suitable conjugated spaces such as ethynylene groups. Hu and co-workers synthesized di-anthrylene-ethynylene, where the ethynylene bridge between the anthracene units prevents repulsion between hydrogen atoms in adjacent anthracene rings, resulting in a rigid, planar “H”-type molecular structure (Fig. 33).127 SC FETs were fabricated using an organic mask ribbon technique.128–131 The shorter intermolecular distance (b = 6.04 Å) of this dimer along the b axis, compared to that of anthracene, coupled with its rigid “H”-type conjugation, effectively resulted in the best hole mobility of 0.82 cm2 V−1 s−1 with an on/off ratio of 5 × 105.
 | ||
| Fig. 33 (a) Chemical structure of di-anthrylene-ethynylene. (b) Single crystal X-ray structure. (c) Schematic of device fabrication by using the “organic ribbon mask” technique, and SEM images of devices with different channel lengths by using the technique. Adapted with permission from ref. 127. Copyright 2008 Wiley. | ||
As part of oligomerization efforts, a series of 9,10-ter-anthrylene-ethynylenes were synthesized and characterized.132–134 Specifically, the introduction of ethynylene-bridges along with solubilizing groups, such as alkyl and alkoxy chains, enabled their evaluation as organic semiconductors in solution processed OFET devices (Fig. 34). For examples, the impact of different alkyl chains (n-butyl, n-octyl, 2-ethyl-hexyl, and n-decyl) on OFET performance was studied.133 It was discovered that 9,10-ter-anthrylene-ethynylene with 2-ethyl-hexyl groups exhibited the best hole mobility (average hole mobility: 1.1 ± 0.1 × 10−2 cm2 V−1 s−1 with an on/off ratio of 105 and a threshold voltage ranged between −3 V and −10 V), while the variant with n-decyl groups demonstrated similar performance (average hole mobility: 1.2 ± 0.1 × 10−2 cm2 V−1 s−1 with an on/off ratio of 104). It is known that the initial monolayers of an organic film significantly influence conduction in OFET devices.135 Therefore, the two-dimensional organization of these molecules on the film substrate was investigated. The molecules were spontaneously physiosorbed on both highly ordered pyrolytic graphite (HOPG) and reconstructed Au(111) surfaces. STM images of the self-assembled structures at the liquid–solid interface revealed that molecules with n-butyl alkyl chains self-assembled into a quasi hexagonal packing on HOPG, whereas those with longer alkyl chains formed lamellar rows. This behavior is attributed to the insufficient van der Waals interactions between the shorter n-butyl chains to enable efficient interlocking. In contrast, due to stronger interactions with the molecular cores, the molecules arranged more closely to form lamellar rows on Au(111), irrespective of alkyl chain length; these layers are assumed to be tilted relative to the substrate.
 | ||
| Fig. 34 (a) Chemical structure of ethynylene-bridged anthracene trimers with solubilizing groups. (b) Output (left) and transfer (right) characteristics of the trimer with n-decyl groups. (c) STM image of the trimer with n-butyl groups at the n-tetradecane/HOPG interface. (d) Proposed packing model of the trimer with n-butyl groups. (e) STM image of the trimer with n-decyl groups. (f) Proposed model packing of the trimer with n-decyl groups. Adapted with permission from ref. 133. Copyright 2010 Royal Chemical Society. | ||
Hayashi et al. examined the impact of further oligomerization on OFET performance.136 Ethynylene-bridged anthracene oligomers ranging from monomers to tetramers were synthesized from anthraquinone. It was observed that the 3mer formed both block-shaped and needle-like crystals. In both crystal types, the anthracene backbones are densely packed through π–π interactions and CH–π interactions (Fig. 35). In the block crystal, the three anthracene planes are nearly coplanar along the linear molecular axis, forming a face-to-face π-stack structure with slip-stacked packing. Conversely, the needle-like crystals display anthracene units arranged in a twisted formation with a torsion angle of around 36°, also adopting a face-to-face packing structure. The corresponding transfer integral is calculated to be 18.8 meV, with intercolumn values between anthracene units being 5.2 meV and 7.8 meV. The 4mer consistently formed needle-like crystals; although a twisted structure was observed, the crystal quality was insufficient to obtain detailed information.
 | ||
| Fig. 35 (a) Ethynylene-bridged anthracene trimers and tetramers employed in this study. (b) Optical microscope image of needle crystals of the 3mer. (c) Top and side views of the crystal structure of the 3mer in needle crystals. (d) Packing structure and transfer integrals. Reproduced with permission from ref. 136. Copyright 2021 Wiley. | ||
The charge transport properties were evaluated using SC FETs. Crystals of the 3mer and 4mer were grown by drop-casting. The XRD patterns of the 3mer and 4mer crystals on the substrate suggested that the molecular orientations were favorable for SC FETs, with charge transport occurring through the π–π stacking of anthracene units (Fig. 36). Gold source and drain electrodes were deposited on a needle-type crystal by using the “gold layer glue technique”.137–143 The 3mer showed a hole mobility of up to 0.14 cm2 V−1 s−1 with an on/off ratio of 5.6 × 105 and a threshold voltage of −0.34 V, surpassing than that of the 4mer (3.3 × 10−2 cm2 V−1 s−1 with an on/off ratio of 4.4 × 105 and a threshold voltage of −6.3 V) (Fig. 35). Thus, the 3mer crystals exhibited slightly higher hole mobility than the 4mer, despite similarities in packing structure. This superior performance is likely related to the reduced number of defects in 3mer crystals, as 3mer achieves a favorable balance between effective π-conjugation and defect-less crystal formation, compared to the freer rotation of ethynylene units in the 4mer.
 | ||
| Fig. 36 (a and b) Experimental out-of-plane XRD and simulated powder XRD patterns of (a) 3mer and (b) 4mer single crystals on a Si/SiO2/OTS substrate. (c and e) Transfer characteristics of the device for (c) 3mer and (e) 4mer at a fixed source/drain voltage, VSD = −40 V. (d and f) Output characteristics of the device for (d) 3mer and (f) 4mer. Reproduced with permission from ref. 136. Copyright 2021 Wiley. | ||
Considering π-conjugation and effective molecular packing, π-extension—specifically using tetracene oligomers instead of anthracene—appears to be well-suited for OFET applications. However, the instability of higher acenes often requires multi-step synthesis involving protection/deprotection processes. Barlier et al. discovered a one-pot synthesis of ethynylene-bridged tetracene dimers from 5,12-naphthacenequinone (Fig. 37).144 The yields of monomers and dimers were highly dependent on the pH during reductive aromatization, as deprotection of the silyl group, competing with hydrolysis/reduction steps in a kinetically controlled process, promoted dimer formation.
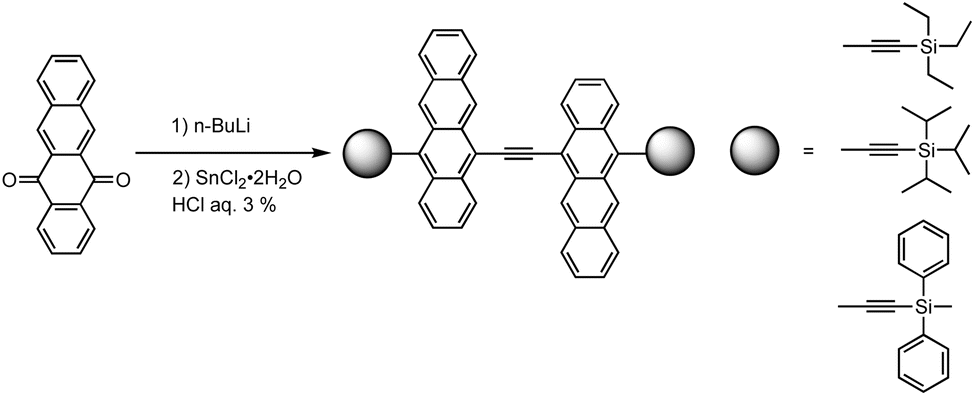 | ||
| Fig. 37 Ethynylene-bridged tetracene dimer formation. Adapted with permission from ref. 144. Copyright 2011 Royal Chemical Society. | ||
This dimerization reaction was modified to synthesize an ethynylene-bridged pentacene dimer.145 The key step involved successfully isolating the soluble and stable pentacene tetraol as an intermediate, providing additional insights into the dimerization mechanism (Fig. 38). Subsequent reductive aromatization of the tetraol with SnCl2 in 3 M HClaq. yielded an ethynylene-bridged pentacene dimer as deep-green crystals. X-ray single crystal analysis revealed that two pentacene moieties in the dimer exhibit a nearly coplanar arrangement. Each molecule effectively interacts with four different neighboring molecules, contributing to efficient charge transport through two-dimensional electronic interactions in the solid state. Transfer integrals between the HOMOs of stacked pentacene moieties were calculated to be 46.0 and 53.6 meV. To evaluate charge transport properties, crystalline films were obtained by a dip-coated method with a mixture of CS2/hexane (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) at a pull rate of 1.2 mm min−1 on Si/SiO2 substrates. Out-of-plane XRD analysis indicated edge-on packing of the dimer on the substrate. Hole mobility was measured in a bottom-gate, top-contact FET, exhibiting a value of up to 0.24 ± 0.05 cm2 V−1 s−1, comparable to the value obtained from single-crystal four probe measurements.
:1, v/v) at a pull rate of 1.2 mm min−1 on Si/SiO2 substrates. Out-of-plane XRD analysis indicated edge-on packing of the dimer on the substrate. Hole mobility was measured in a bottom-gate, top-contact FET, exhibiting a value of up to 0.24 ± 0.05 cm2 V−1 s−1, comparable to the value obtained from single-crystal four probe measurements.
 | ||
| Fig. 38 (a) Synthesis of ethynylene-bridged pentacene dimers. (b) Optical microscope image of dip-coated dimer films on a Si/SiO2 substrate. (c) Out-of-plane XRD patterns of dip-coated dimer films. (d) Representative transfer characteristics of the device at a fixed VSD = −60 V. (e) Representative output characteristics of the device. Reproduced with permission from ref. 145. Copyright 2018 Wiley. | ||
Further oligomerization of pentacene is expected to enhance electronic communication within the packing structure, improving charge transport properties. Although nucleophilic attacks on pentacene quinones followed by reduction is a straightforward approach for oligomerization, this method is challenging due to pentacene's reactive nature. Specifically, since TIPS-Pen easily decomposes after desilylation,146 protection/deprotection strategies have been employed to synthesize pentacene oligomers. Lehnherr et al. reported the synthesis of conjugated pentacene oligomers up to the tetramer using protected building blocks (Fig. 39).147 As the oligomer length increases, a corresponding red shift in the λmax is observed, indicating enhanced conjugation. Notably, trimers and tetramers exhibit remarkably low band gaps comparable to those of ethynylated heptacenes (ca. 1.3–1.4 eV).148–150 No emissions were observed for these oligomers. X-ray crystallographic analysis indicated that two pentacene moieties of the dimer adopt a pseudocoplanar arrangement featuring a two-dimensional slipped stacked configuration, forming a staircase arrangement along the crystallographic b axis. Cofacial π-stacking occurs within the staircase arrangement along the b axis and between adjacent staircases along the crystallographic a axis. This two-dimensional arrangement, coupled with the ability of the butadiynyl unit to facilitate electronic communication, offers potential for three-dimensional electronic interactions in the solid state.
 | ||
Fig. 39 (a) Pentacene oligomers employed in this study. Protected building blocks are also shown. (b) Absorption spectra of oligomers. R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si(nC6H13)3. Adapted with permission from ref. 147. Copyright 2012 Wiley. Si(nC6H13)3. Adapted with permission from ref. 147. Copyright 2012 Wiley. | ||
Thermal precursor approaches were explored to synthesize pentacene oligomers, circumventing the reactive nature of pentacene. It was discovered that oligomer precursors could be synthesized from BCOD-fused anthraquinone in a one-pot method, eliminating several steps, although the formation of the dimer was conceivable as seen in the previous report (Fig. 40).151 Notably, the achievable yield of the dimer via one-pot synthesis significantly reduces labour-intensive multi-step procedures required for dimer preparation. The obtained dimer serves as a crucial building block for further oligomerization, with bulky BCOD moieties enhancing solubility, which could be an important factor in this oligomerization. Despite being a common strategy for synthesizing ethynylene-bridged acene oligomers, this specific oligomerization beyond dimers from acenequinone had not been previously reported. This unexpected oligomerization, revealed through conventional reactions, provides valuable insights for the efficient synthesis of ethynylene-bridged compounds. Finally, the pentacene precursor oligomers were converted to the corresponding pentacene oligomers by heating (Fig. 41). The bulk powders of BCOD3mer and BCOD4mer were heated at 350 °C and 280 °C, respectively, for 2 hours under vacuum conditions. HR-MALDI-TOF-MS of the annealed solids showed parent ion peaks of Pen3mer and Pen4mer. Solutions of BCOD3mer or BCOD4mer in diphenyl ether were subjected to high temperatures via microwave irradiations under a N2 atmosphere. BCOD3mer was transformed into Pen3mer through retro-Diels–Alder reactions in diphenyl ether solution at 300 °C, and heating at 260–280 °C converted BCOD4mer to Pen4mer. The maximum absorption wavelengths of Pen3mer and Pen4mer were around 770–800 nm, with Pen4mer exhibiting a slight red-shift compared to Pen3mer, suggesting that the effective π-conjugation length is essentially achieved at the Pen4mer stage. Considering that crystalline dip-coated films of ethynylene-bridged pentacene dimers exhibited a hole mobility of 0.24 cm2 V−1 s−1,145 an improved charge transport performance is highly expected for Pen3mer.
 | ||
| Fig. 40 Unexpected oligomerization. Reaction yields depended on the lithiation time for TIPS ethynylene, and the yield corresponding to a 0.5-h-lithiation period is shown. | ||
 | ||
| Fig. 41 (a) Thermal conversion. (b) Representative normalized absorption spectra of BCDO3mer and BCOD4mer before and after heating in diphenyl ether. Heating conditions: 300 °C, 3 minutes for BCOD3mer, and 260 °C, 5 minutes and then 280 °C, 90 seconds for BCOD4mer under a N2 atmosphere. Adapted with permission from ref. 151. Copyright 2024 Wiley. | ||
Incorporating heteroatoms into higher acenes significantly enhances their stability and often modifies their optical and physical properties, as well as their molecular packing in the solid state.5,152–158 Among the doping options, nitrogen-doped higher acenes can act as p-type, n-type, or ambipolar organic semiconductors, depending on their structural configuration. Additionally, nitrogen-doped higher acenes were applied not only in OFETs but also in OPVs and organic light-emitting diodes (OLEDs).152–162 Here, again, silylethynylene groups often play a crucial role in creating soluble and stable organic semiconductors. Notably, silylethynylated nitrogen-doped pentacene (TIPS-TAP), where nitrogen atoms are incorporated into middle rings of the pentacene structure, has shown exceptional promise as a n-type organic semiconductor (Fig. 42).163–168 Initially, vacuum-deposited thin films of TIPS-TAP demonstrated n-type semiconductor behavior with field-effect mobilities ranging from 1.0 to 3.3 cm2 V−1 s−1.166,167 Then, solution-processed TIPS-TAP thin films achieved an electron mobility of 7.6 ± 1.6 cm2 V−1 s−1 with a maximum value of 11 cm2 V−1 s−1,164 whereas single crystal arrays displayed an electron mobility of 8.0 ± 2.2 cm2 V−1 s−1 with a maximum value of 13.3 cm2 V−1 s−1.163 The performance was significantly boosted by molecular engineering of TIPS-TAP through halogenation, optimizing the frontier molecular orbitals, molecular vibrations, and π–π stacking interactions. Solution-processed n-channel thin-film transistors using 4Cl-TAP, prepared via dip-coating, attained electron mobilities up to 27.8 cm2 V−1 s−1.165 The remarkably high electron mobility of 4Cl-TAP is attributed to the reduced reorganization energy and enhanced electron transfer integral, a result of modifying TIPS-TAP with four chlorine substituents.
 | ||
| Fig. 42 (a) Molecular structure of TIPS-TAP. (b) Transfer characteristics of the device for a thin-film transistor with TIPS-TAP at a fixed VSD = −60 V. (c) Molecular structure of 4Cl-TAP. (d) Transfer characteristics of the device for a thin-film transistor of 4Cl-TAP with the highest field effect mobility. Adapted with permission from ref. 166 for (b), and ref. 165 for (d). Copyrights 2011 Wiley for (b) and 2018 Wiley for (d). | ||
Similar to higher acenes composed solely of carbon and hydrogen, efforts have been made to synthesize longer acenes featuring extended π-conjugation in nitrogen-doped derivatives. Stability and solubility continue to be crucial concerns, although the introduction of nitrogen atoms enhances stability. Consequently, before synthesizing pristine nitrogen-doped higher acenes,89 research has prioritized those with silylethynyl groups (Fig. 43), which, as observed in carbon-based higher acenes, effectively enhance stability and solubility.169 While stabilizing higher acenes with a single pair of bulky silylethynyl substituents is feasible, this approach is less suitable for diazaacenes, wherein the pyrazine units occupy the central rings. Higher diazaacenes often undergo dimerization through [4 + 4] cycloaddition170 at the rings adjacent to the pyrazine units.171 Acknowledging the electronical stabilization provided by alkyne groups against cycloaddition type reactions, Bunz et al. developed a stabilization strategy for higher azaacenes by incorporating four silylethynyl substituents. While TIPS groups are not positioned particularly close to the acene core, they contribute significantly to packing and solubility.171,172 Intriguingly, the stability of azaheptacenes varies with the number of nitrogen atoms and TIPSethynyls' placement. UV-vis absorption spectra revealed specific p-band characteristics for DAH1, DAH2, TAH, and HAH with absorption onsets at 1074, 1064, 1152, and 1045 nm, respectively, demonstrating a red-shift relative to Hep173 (absorption onset: 950 nm). Further investigation using NMR and UV-vis spectroscopy assessed the impact of nitrogen atom loading on stability, with the following stability trend DAH1 > TAH > HAH ≈ DAH2 > Hep > DAH3. Remarkably, solutions of DAH1 remained unchanged after 7 days under ambient conditions, suggesting that their substitution patterns effectively prevent endoperoxide formation, dimerization, and Diels–Alder reactions. These findings indicate the importance of precise placement of silylethynyl substituents and the number of pyrazine units. Bottom-gate/top-contact OFETs based on DAH1 and TAH exhibited the best electron mobilities of 0.042 and 0.0031 cm2 V−1 s−1, respectively. DAH2 and Hep exhibited ambipolar transport characteristics, with DAH2 achieving an electron mobility of 0.005 cm2 V−1 s−1 and a hole mobility of 0.0017 cm2 V−1 s−1, while Hep showed an electron mobility of 0.023 cm2 V−1 s−1 and a hole mobility of 0.038 cm2 V−1 s−1. Taking advantage of their stability, further enhancements in thin film quality are likely to elevate charge transport properties.
 | ||
| Fig. 43 (a) Representative nitrogen-doped higher acenes with TIPSethynyl groups. The heptacene derivative (Hep) is also shown as a reference. (b) Time-dependent evolution of UV-vis absorption spectra of DAH1 (10−5 mol L−1 in dry CH2Cl2) under ambient light and atmospheric conditions. Adapted with permission from ref. 172. Copyrights 2024 American Chemical Society. | ||
From the perspective of enhancing stability, annulation proves to be an effective strategy. By incorporating multiple Clar-sextets through sandwiching azaacenes between aromatic units such as pyrene and coronene,174–178 the stability is improved owning to the cross-conjugated character of the annulated unit. In addition, benzannulation on both sides of higher azaacenes179 also contribute effectively to structural stabilization.
Mateo-Alonso and co-workers have demonstrated an elegant strategy for synthesizing molecular GNRs containing annulated azaacenes.180–186 These molecular GNRs are prepared through the formation of pyrazine rings via reaction between o-diamines and o-quinones, providing an efficient means of interconnecting building blocks (Fig. 44). With careful selection and placement of solubilizing groups that ensure both good solubility and stability, column chromatography can be used for purification. Remarkably, the longest molecular GNR (NR-147-Q) displayed an atomically precise core containing 920 sp2 atoms with a 35.8 nm long backbone (comprising 147 linearly fused rings).
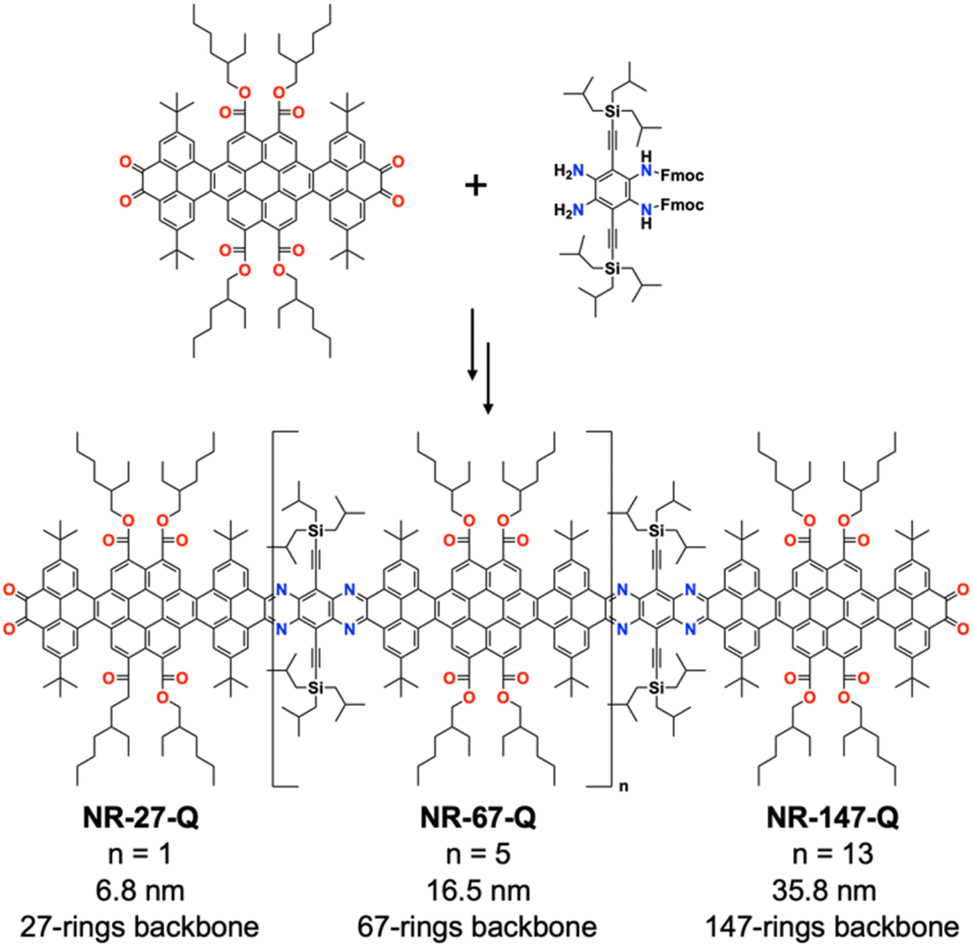 | ||
| Fig. 44 Synthesis of molecular GNRs. | ||
Thanks to their high solubility (NR-147-Q: ∼20 mg ml−1 in CHCl3) and stability, detailed structural characterization of the molecular GNRs was possible using NMR, along with optoelectronic and redox characterization (Fig. 45). NR-147-Q showed a molar absorption coefficient of 1845900 M−1 cm−1. Intriguingly, it also displayed fluorescence despite its large dimensions, and demonstrated a record fluorescence brightness value of 250500 M−1 cm−1, approximately 1 order of magnitude higher than that of carbon quantum dots and comparable to that of state-of-the-art inorganic quantum dots.187 The redox properties of NR-147-Q were determined by cyclic voltammetry, estimating the electrochemical LUMO level at −3.84 eV, with no oxidation observed in the solvent-supported electrolyte window. For assessing charge carrier mobility, optical pump-THz probe spectroscopy was employed. The maximum value of time-resolved THz photoconductivity, normalized to the absorbed photon density (Δσ/Nabs), in the molecular GNR thin films increased with the length of the GNRs, indicating enhanced charge charrier mobility in longer molecular GNRs. The intrinsic charge carrier mobility value of NR-147-Q was estimated to be ∼149 ± 32 cm2 V−1 s−1, consistent with those observed in other type of GNRs.188–192
 | ||
| Fig. 45 (a) UV-vis absorption and (c) fluorescence spectra of molecular GNRs in CHCl3. (b) 5 mM solutions of GNRs in CHCl3 under natural light and (d) UV light. (e) Cyclic voltammograms of the GNRs in n-Bu4NPF6/CH2Cl2. (f) Time-resolved THz photoconductivity (Δσ) of different GNRs normalized to absorbed photon density following resonant excitation. Adapted with permission from ref. 184. Copyrights 2023 Cell Press. | ||
As discussed, higher acenes have shown remarkable potential in various applications. Here, we present a table summarizing the transistor characteristics of higher acenes, azaacene, and other major organic molecules (Table 1). While mobility varies depending on the fabrication method, it is evident that some higher acenes exhibit not only high hole mobility but also exceptionally high electron mobility. By incorporating suitable substituents and optimizing transistor fabrication processes, as well as improving thin film and single crystal preparation, even higher mobility is expected in the future.
| Molecular structure | Maximum mobility (cm2 V−1 s−1) | Active layer | Ref. | Molecular structure | Maximum mobility (cm2 V−1 s−1) | Active layer | Ref. |
|---|---|---|---|---|---|---|---|
| a Single crystal. | |||||||
|
2.75 (p-type) | Thin film | 193 |

|
14.7 (p-type) | Thin film | 194 |

|
16 (p-type) | Thin film | 195 |

|
21 (p-type) | Thin film | 196 |
|
18 (p-type) | SCa | 197 |
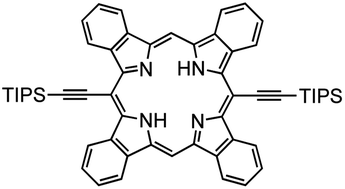
|
2.2 (p-type) | SCa | 137 |

|
4.1 (p-type) | Thin film | 198 |

|
0.02 (p-type) | SCa | 117 |

|
34 (p-type) | SCa | 118 and 119 |

|
0.82 (p-type) | SCa | 121 |
|
1.1–1.2 × 10−2 (average) (p-channel) | Thin film | 133 |

|
0.14 (p-type) | Thin film | 136 |
|
0.24 (p-type) | Thin film | 145 |

|
1.5 (p-type) | Thin film | 9 |
|
11 (p-type) | Thin film | 11 |
|
4.28 (p-type) | SCa | 109 |
|
0.83 (p-type) | Thin film | 110 |

|
2.2 (p-type) | Thin film | 114 |

|
0.038 (p-type) 0.023 (n-type) | Thin film | 172 |

|
3.5 (n-type) | Thin film | 198 |
|
12.6 (n-type) | SCa | 199 |

|
8.6 (n-type) | SCa | 200 |

|
3.0 (typical value) (n-type) | Thin film | 201 |

|
13.3 (n-type) | Thin film | 163 |

|
27.8 (n-type) | Thin film | 165 |

|
0.042 (n-type) | Thin film | 172 |

|
149 ± 32 (estimated by optical pump-THz probe spectroscopy) | Thin film | 184 | ||||
5 Conclusions
The synthesis and evaluation of electronic properties of higher acenes have been compelling research targets because of their extended π-systems and radical characteristics. Over the past two decades, the combination of on-surface synthesis and advanced observation techniques such as STM and nc-AFM has enabled the synthesis of higher acenes with various lengths, providing insights into their electronic properties. The precursor method for preparing higher acenes has been instrumental in advancing synthesis, while the development of pristine, functionalized, and heteroatom-doped higher acenes has expanded their applications in organic devices, including OFETs, OPVs, and OLEDs. Some higher acene derivatives have demonstrated exceptional charge transport properties, functioning as both p-type and n-type organic semiconductors. Effective stabilization strategies, combined with solubilizing groups as discussed, significantly contribute to the fabrication of solution-processed device. The next challenge involves developing strategies for producing large-scale crystalline films with minimal domains and grain boundaries by solution-processing, a crucial requirement for high-end practical applications.Looking ahead, higher acenes are prepared to increasingly interact with nanocarbon chemistry. As demonstrated in this review, higher acenes, with their rigid structures and extended π-conjugation, serve as ideal building blocks for nanocarbon materials such as GNRs, carbon nanotubes, and giant ladder-type polymers. Exploring the properties of higher acenes based on their length will deepen our understanding of the edge states in ZGNRs and could lead to the discovery of interesting nanocarbon materials suitable for applications in semiconducting and/or spintronics devices. For example, ethynylene-bridged higher acene oligomers and polymers are broadening their application scope. Recent studies highlight the significance of ethynylene-bridged acene systems not only as semiconducting materials but also as topological materials, due to their ability to transition between two resonant structures by tuning π-conjugation.202–204 Ecija et al. developed a strategy to synthesize polymers with engineered topologies by adjusting their π-conjugation (Fig. 46).202 The polymer features two resonant configurations, where altering the conjugation of the central acene rings from a quinoid to an aromatic structure simultaneously transforms the cumulene into an ethynylene bridge. These conjugation forms correlate with different topological classes, with transitions depending on the size of the repeating unit. It is fascinating to note how Peierls distortions and Clar's sextet formalism work synergistically, leading to an increase in Clar's sextets in the cumulenic form as the bond localization decreases. In pentacene polymers, each pentacene can host a single aromatic ring in a Kekulé representation. Alternatively, a second Clar sextet can form, leaving two electrons unpaired, thereby imparting the molecule with partial open-shell character. The unpaired π-electrons from quinoid rings are likely to be shared along the polymeric chain, favoring the cumulene over the ethynylene bridge—a competition making the topological inversion point. Analogous to polyacetylene, a π-bond is observed at the polymer's end in the quinoid–cumulene structure, leaving an unpaired electron, unlike the aromatic-ethynylene structure, which highlights their topologically non-trivial and trivial characters, respectively. These results indicate that higher acenes are poised to play a significant role in the field of quantum materials.
 | ||
| Fig. 46 (a) Schematic evolution of the bandgap of acene monomers and polymers with increasing size of acene monomers. The latter case shows a phase transition between trivial and non-trivial topological classes accompanied by transformation of the π-conjugation. (b) Band diagrams for ethynylene-linked acene polymers obtained from tight-binding calculations. (c) Chemical sketch of the resulting pentacene-based polymer. (d) Experimental determination of Egap with STS. (e) nc-AFM image of the pentacene-based polymer. (f) Constant-height nc-AFM (top) and STM (bottom) images of a H-terminated pentacene polymer. (g) STS spectra along the termination at the positions depicted by purple dots in (f). Adapted with permission from ref. 202. Copyright 2020 Spring Nature. | ||
Thus, while higher acenes possess a simple structure, they exhibit extraordinary potential. The history of higher acene research is long-standing, but recent advancements combining state-of-the-art synthetic205 and observational techniques, theoretical calculations, and device fabrication are pushing the field forward. The molecular structures of higher acenes obtained to date, as well as the resulting crystal and packing structures, are extensive. Therefore, the integration of machine learning is expected to accelerate the search for more optimal molecular structures in the future.206,207 In addition, the ongoing interaction with nanocarbon chemistry and device optimization heralds a new era of research on higher acenes, with much more to discover and develop.
Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.Author contributions
HH and HY contributed to this article through conceptualization and writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by JST PRESTO grant no. JPMJPR21AC (HH) and JSPS KAKENHI grant no. JP24K01576 (HH), JP20H05833 (HY), and JP25K01751 (HY).Notes and references
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- Z. Sun, Q. Ye, C. Chi and J. Wu, Chem. Soc. Rev., 2012, 41, 7857–7889 RSC.
- Q. Ye and C. Chi, Chem. Mater., 2014, 26, 4046–4056 CrossRef CAS.
- J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452–483 CrossRef CAS PubMed.
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed.
- L. Lerena, R. Zuzak, S. Godlewski and A. M. Echavarren, Chem. - Eur. J., 2024, 30, e202402122 CrossRef CAS PubMed.
- C. Tönshoff and H. F. Bettinger, Chem. - Eur. J., 2021, 27, 3193–3212 CrossRef PubMed.
- H. Yamada and H. Hayashi, Photochem. Photobiol. Sci., 2022, 21, 1511–1532 CrossRef CAS PubMed.
- Y.-Yi Lin, D. I. Gundlach, S. F. Nelson and T. N. Jackson, IEEE Trans. Electron Devices, 1997, 44, 1325–1331 CrossRef CAS.
- J. E. Anthony, J. S. Brooks, D. L. Eaton and S. R. Parkin, J. Am. Chem. Soc., 2001, 123, 9482–9483 CrossRef CAS PubMed.
- Y. Diao, B. C. K. Tee, G. Giri, J. Xu, D. H. Kim, H. A. Becerril, R. M. Stoltenberg, T. H. Lee, G. Xue, S. C. B. Mannsfeld and Z. Bao, Nat. Mater., 2013, 12, 665–671 CrossRef CAS PubMed.
- Z. Birech, M. Schwoerer, T. Schmeiler, J. Pflaum and H. Schwoerer, J. Chem. Phys., 2013, 12, 665–671 Search PubMed.
- M. Dvořák, S. K. K. Prasad, C. B. Dover, C. R. Forest, A. Kaleem, R. W. Macqueen, A. J. Petty, R. Forecast, J. E. Beves, J. E. Anthony, M. J. Y. Tayebjee, A. Widmer-Cooper, P. Thordarson and T. W. Schmidt, J. Am. Chem. Soc., 2021, 143, 13749–13758 CrossRef PubMed.
- L. Catti, H. Narita, Y. Tanaka, H. Sakai, T. Hasobe, N. V. Tkachenko and M. Yoshizawa, J. Am. Chem. Soc., 2021, 143, 9361–9367 CrossRef CAS PubMed.
- T. Kinoshita, S. Nakamura, M. Harada, T. Hasobe and G. Fukuhara, Chem. Sci., 2023, 14, 3293–3301 RSC.
- J. Zirzlmeier, D. Lehnherr, P. B. Coto, E. T. Chernick, R. Casillas, B. S. Basel, M. Thoss, R. R. Tykwinski and D. M. Guldi, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5325–5330 CrossRef CAS PubMed.
- M. W. B. Wilson, A. Rao, K. Johnson, S. Gélinas, R. Di Pietro, J. Clark and R. H. Friend, J. Am. Chem. Soc., 2013, 135, 16680–16688 CrossRef CAS PubMed.
- J. Lee, M. J. Bruzek, N. J. Thompson, M. Y. Sfeir, J. E. Anthony and M. A. Baldo, Adv. Mater., 2013, 25, 1445–1448 CrossRef CAS PubMed.
- S. N. Sanders, E. Kumarasamy, K. J. Fallon, M. Y. Sfeir and L. M. Campos, Chem. Sci., 2020, 11, 1079–1084 RSC.
- T. S. Moreira, M. Ferreira, A. Dall’armellina, R. Cristiano, H. Gallardo, E. A. Hillard, H. Bock and F. Durola, Eur. J. Org. Chem., 2017, 2017, 4548–4551 CrossRef CAS.
- F. B. Mallory, K. E. Butler, A. Bérubé, E. D. Luzik, C. W. Mallory, E. J. Brondyke, R. Hiremath, P. Ngo and P. J. Carroll, Tetrahedron, 2001, 57, 3715–3724 CrossRef CAS.
- F. B. Mallory, K. E. Butler, A. C. Evans, E. J. Brondyke, C. W. Mallory, C. Yang and A. Ellenstein, J. Am. Chem. Soc., 1997, 119, 2119–2124 CrossRef CAS.
- H. Okamoto, S. Hamao, R. Eguchi, H. Goto, Y. Takabayashi, P. Y.-H. Yen, L. U. Liang, C.-W. Chou, G. Hoffmann, S. Gohda, H. Sugino, Y.-F. Liao, H. Ishii and Y. Kubozono, Sci. Rep., 2019, 9, 4009 CrossRef PubMed.
- Y. Shimo, T. Mikami, H. T. Murakami, S. Hamao, H. Goto, H. Okamoto, S. Gohda, K. Sato, A. Cassinese, Y. Hayashi and Y. Kubozono, J. Mater. Chem. C, 2015, 3, 7370–7378 RSC.
- H. Okamoto, N. Kawasaki, Y. Kaji, Y. Kubozono, A. Fujiwara and M. Yamaji, J. Am. Chem. Soc., 2008, 130, 10470–10471 CrossRef CAS PubMed.
- H. Okamoto, N. Kawasaki, Y. Kaji, Y. Kubozono, A. Fujiwara and M. Yamaji, J. Am. Chem. Soc., 2008, 130, 10470–10471 CrossRef CAS PubMed.
- R. E. Blackwell, F. Zhao, E. Brooks, J. Zhu, I. Piskun, S. Wang, A. Delgado, Y.-L. Lee, S. G. Louie and F. R. Fischer, Nature, 2021, 600, 647–652 CrossRef CAS PubMed.
- P. P. Shinde, J. Liu, T. Dienel, O. Gröning, T. Dumslaff, M. Mühlinghaus, A. Narita, K. Müllen, C. A. Pignedoli, R. Fasel, P. Ruffieux and D. Passerone, Carbon, 2021, 175, 50–59 CrossRef CAS.
- Y. Fu, H. Yang, Y. Gao, L. Huang, R. Berger, J. Liu, H. Lu, Z. Cheng, S. Du, H. Gao and X. Feng, Angew. Chem., Int. Ed., 2020, 132, 8958–8964 CrossRef.
- J. Li, S. Sanz, N. Merino-Díez, M. Vilas-Varela, A. Garcia-Lekue, M. Corso, D. G. de Oteyza, T. Frederiksen, D. Peña and J. I. Pascual, Nat. Commun., 2021, 12, 5538 CrossRef CAS PubMed.
- P. Ruffieux, S. Wang, B. Yang, C. Sánchez-Sánchez, J. Liu, T. Dienel, L. Talirz, P. Shinde, C. A. Pignedoli, D. Passerone, T. Dumslaff, X. Feng, K. Müllen and R. Fasel, Nature, 2016, 531, 489–492 CrossRef CAS PubMed.
- P. Han, K. Akagi, F. Federici Canova, H. Mutoh, S. Shiraki, K. Iwaya, P. S. Weiss, N. Asao and T. Hitosugi, ACS Nano, 2014, 8, 9181–9187 CrossRef CAS PubMed.
- A. Kinikar, X. Xu, M. Di Giovannantonio, O. Gröning, K. Eimre, C. A. Pignedoli, K. Müllen, A. Narita, P. Ruffieux and R. Fasel, Adv. Mater., 2023, 35, 2306311 CrossRef CAS PubMed.
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470–473 CrossRef CAS PubMed.
- J. I. Urgel, M. Di Giovannantonio, G. Gandus, Q. Chen, X. Liu, H. Hayashi, P. Ruffieux, S. Decurtins, A. Narita, D. Passerone, H. Yamada, S. Liu, K. Müllen, C. A. Pignedoli and R. Fasel, ChemPhysChem, 2019, 20, 2360–2366 CrossRef CAS PubMed.
- J. I. Urgel, H. Hayashi, M. Di Giovannantonio, C. A. Pignedoli, S. Mishra, O. Deniz, M. Yamashita, T. Dienel, P. Ruffieux, H. Yamada and R. Fasel, J. Am. Chem. Soc., 2017, 139, 11658–11661 CrossRef CAS PubMed.
- J. Yamaguchi, H. Hayashi, H. Jippo, A. Shiotari, M. Ohtomo, M. Sakakura, N. Hieda, N. Aratani, M. Ohfuchi, Y. Sugimoto, H. Yamada and S. Sato, Commun. Mater., 2020, 1, 36 CrossRef.
- C. Tönshoff and H. F. Bettinger, Angew. Chem., Int. Ed., 2010, 49, 4125–4128 CrossRef PubMed.
- R. Mondal, C. Tönshoff, D. Khon, D. C. Neckers and H. F. Bettinger, J. Am. Chem. Soc., 2009, 131, 14281–14289 CrossRef CAS PubMed.
- R. Mondal, R. M. Adhikari, B. K. Shah and D. C. Neckers, Org. Lett., 2007, 9, 2505–2508 CrossRef CAS PubMed.
- R. Mondal, B. K. Shah and D. C. Neckers, J. Am. Chem. Soc., 2006, 128, 9612–9613 CrossRef CAS PubMed.
- J. Hachmann, J. J. Dorando, M. Avilés and G. K.-L. Chan, J. Chem. Phys., 2007, 127, 134309 CrossRef PubMed.
- Y. Yang, E. R. Davidson and W. Yang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E5098–E5107 CAS.
- G. Trinquier, G. David and J.-P. Malrieu, J. Phys. Chem. A, 2018, 122, 6926–6933 CrossRef CAS PubMed.
- T.-H. Chao, M.-J. Chang, M. Watanabe, M.-H. Luo, Y. J. Chang, T.-C. Fang, K.-Y. Chen and T. J. Chow, Chem. Commun., 2012, 48, 6148 RSC.
- H. Uoyama, H. Yamada, T. Okujima and H. Uno, Tetrahedron, 2010, 66, 6889–6894 CrossRef CAS.
- K.-Y. Chen, H.-H. Hsieh, C.-C. Wu, J.-J. Hwang and T. J. Chow, Chem. Commun., 2007, 1065–1067 RSC.
- A. Afzali, C. R. Kagan and G. P. Traub, Synth. Met., 2005, 155, 490–494 CrossRef CAS.
- A. Afzali, C. D. Dimitrakopoulos and T. L. Breen, J. Am. Chem. Soc., 2002, 124, 8812–8813 CrossRef CAS PubMed.
- A. R. Brown, A. Pomp, D. M. de Leeuw, D. B. M. Klaassen, E. E. Havinga, P. Herwig and K. Müllen, J. Appl. Phys., 1996, 79, 2136–2138 CrossRef CAS.
- P. T. Herwig and K. Müllen, Adv. Mater., 1999, 11, 480–483 CrossRef CAS.
- J. Strating, B. Zwanenburg, A. Wagenaar and A. C. Udding, Tetrahedron Lett., 1969, 10, 125–128 CrossRef.
- T. Aotake, S. Ikeda, D. Kuzuhara, S. Mori, T. Okujima, H. Uno and H. Yamada, Eur. J. Org. Chem., 2012, 2012, 1723–1729 CrossRef CAS.
- H. Yamada, Y. Yamashita, M. Kikuchi, H. Watanabe, T. Okujima, H. Uno, T. Ogawa, K. Ohara and N. Ono, Chem. - Eur. J., 2005, 11, 6212–6220 CrossRef CAS PubMed.
- F. J. Giessibl, Rev. Mod. Phys., 2003, 75, 949–983 CrossRef CAS.
- L. Gross, F. Mohn, N. Moll, P. Liljeroth and G. Meyer, Science, 2009, 325, 1110–1114 CrossRef CAS PubMed.
- J. I. Urgel, S. Mishra, H. Hayashi, J. Wilhelm, C. A. Pignedoli, M. Di Giovannantonio, R. Widmer, M. Yamashita, N. Hieda, P. Ruffieux, H. Yamada and R. Fasel, Nat. Commun., 2019, 10, 861 CrossRef PubMed.
- Physics and Chemistry of Graphene, ed. T. Enoki and T. Ando, Jenny Stanford Publishing, 2013 Search PubMed.
- S. Hauptmann, Zeitschrift für Chemie, 1973, 13, 200 CrossRef.
- L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, Nat. Nanotechnol., 2007, 2, 687–691 CrossRef CAS PubMed.
- L. Colazzo, M. S. G. Mohammed, R. Dorel, P. Nita, C. García Fernández, P. Abufager, N. Lorente, A. M. Echavarren and D. G. De Oteyza, Chem. Commun., 2018, 54, 10260–10263 RSC.
- R. Zuzak, R. Dorel, M. Kolmer, M. Szymonski, S. Godlewski and A. M. Echavarren, Angew. Chem., Int. Ed., 2018, 57, 10500–10505 CrossRef CAS PubMed.
- R. Zuzak, M. Kumar, O. Stoica, D. Soler-Polo, J. Brabec, K. Pernal, L. Veis, R. Blieck, A. M. Echavarren, P. Jelinek and S. Godlewski, Angew. Chem., Int. Ed., 2024, 63, e202317091 CrossRef CAS PubMed.
- R. Dorel, P. R. McGonigal and A. M. Echavarren, Angew. Chem., Int. Ed., 2016, 128, 11286–11289 CrossRef.
- R. Zuzak, R. Dorel, M. Krawiec, B. Such, M. Kolmer, M. Szymonski, A. M. Echavarren and S. Godlewski, ACS Nano, 2017, 11, 9321–9329 CrossRef CAS PubMed.
- L. Colazzo, M. S. G. Mohammed, R. Dorel, P. Nita, C. García Fernández, P. Abufager, N. Lorente, A. M. Echavarren and D. G. De Oteyza, Chem. Commun., 2018, 54, 10260–10263 RSC.
- J. Krüger, N. Pavliček, J. M. Alonso, D. Pérez, E. Guitián, T. Lehmann, G. Cuniberti, A. Gourdon, G. Meyer, L. Gross, F. Moresco and D. Peña, ACS Nano, 2016, 10, 4538–4542 CrossRef PubMed.
- J. Krüger, F. Eisenhut, J. M. Alonso, T. Lehmann, E. Guitián, D. Pérez, D. Skidin, F. Gamaleja, D. A. Ryndyk, C. Joachim, D. Peña, F. Moresco and G. Cuniberti, Chem. Commun., 2017, 53, 1583–1586 RSC.
- J. Krüger, F. García, F. Eisenhut, D. Skidin, J. M. Alonso, E. Guitián, D. Pérez, G. Cuniberti, F. Moresco and D. Peña, Angew. Chem., Int. Ed., 2017, 129, 12107–12110 CrossRef.
- F. Eisenhut, T. Kühne, F. García, S. Fernández, E. Guitián, D. Pérez, G. Trinquier, G. Cuniberti, C. Joachim, D. Peña and F. Moresco, ACS Nano, 2020, 14, 1011–1017 CrossRef CAS PubMed.
- K. Eimre, J. I. Urgel, H. Hayashi, M. Di Giovannantonio, P. Ruffieux, S. Sato, S. Otomo, Y. S. Chan, N. Aratani, D. Passerone, O. Gröning, H. Yamada, R. Fasel and C. A. Pignedoli, Nat. Commun., 2022, 13, 511 CrossRef CAS PubMed.
- Z. Ruan, J. Schramm, J. B. Bauer, T. Naumann, L. V. Müller, F. Sättele, H. F. Bettinger, R. Tonner-Zech and J. M. Gottfried, J. Am. Chem. Soc., 2025, 147, 4862–4870 CrossRef CAS PubMed.
- Z. Ruan, J. Schramm, J. B. Bauer, T. Naumann, H. F. Bettinger, R. Tonner-Zech and J. M. Gottfried, J. Am. Chem. Soc., 2024, 146, 3700–3709 CrossRef CAS PubMed.
- S. Mishra, M. Vilas-Varela, L. A. Lieske, R. Ortiz, S. Fatayer, I. Rončević, F. Albrecht, T. Frederiksen, D. Peña and L. Gross, Nat. Chem., 2024, 16, 755–761 CrossRef CAS PubMed.
- J. B. Neaton, M. S. Hybertsen and S. G. Louie, Phys. Rev. Lett., 2006, 97, 216405 CrossRef CAS PubMed.
- J. Li, S. Sanz, M. Corso, D. J. Choi, D. Peña, T. Frederiksen and J. I. Pascual, Nat. Commun., 2019, 10, 200 CrossRef PubMed.
- R. Zuzak, R. Dorel, M. Krawiec, B. Such, M. Kolmer, M. Szymonski, A. M. Echavarren and S. Godlewski, ACS Nano, 2017, 11, 9321–9329 CrossRef CAS PubMed.
- C. Berger, Z. Song, X. Li, X. Wu, N. Brown, C. Naud, D. Mayou, T. Li, J. Hass, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. De Heer, Science, 2006, 312, 1191–1196 CrossRef CAS PubMed.
- M. Gruber, A. Weismann and R. Berndt, J. Phys.:Condens. Matter, 2018, 30, 424001 CrossRef PubMed.
- H. O. Frota, Phys. Rev. B, 1992, 45, 1096–1099 CrossRef PubMed.
- C. U. Ibeji and D. Ghosh, Phys. Chem. Chem. Phys., 2015, 17, 9849–9856 RSC.
- D. E. Jiang and S. Dai, J. Phys. Chem. A, 2008, 112, 332–335 CrossRef CAS PubMed.
- E. Heilbronner, Helv. Chim. Acta, 1954, 37, 921–935 CrossRef CAS.
- P. R. Ashton, U. Girreser, D. Giuffrida, F. H. Kohnke, J. P. Mathias, F. M. Raymo, A. M. Z. Slawin, J. F. Stoddart and D. J. Williams, J. Am. Chem. Soc., 1993, 115, 5422–5429 CrossRef CAS.
- P. R. Ashton, G. R. Brown, N. S. Isaacs, D. Giuffrida, F. H. Kohnke, J. P. Mathias, A. M. Z. Slawin, D. R. Smith, J. F. Stoddart and D. J. Williams, J. Am. Chem. Soc., 1992, 114, 6330–6353 CrossRef CAS.
- F. H. Kohnke, A. M. Z. Slawin, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed., 1987, 26, 892–894 CrossRef.
- R. M. Cory, C. L. McPhail, A. J. Dikmans and J. J. Vittal, Tetrahedron Lett., 1996, 37, 1983–1986 CrossRef CAS.
- F. Schulz, F. García, K. Kaiser, D. Pérez, E. Guitián, L. Gross and D. Peña, Angew. Chem., Int. Ed., 2019, 131, 9136–9140 CrossRef.
- Y. S. Chan, H. Hayashi, S. Sato, S. Kasahara, K. Matsuo, N. Aratani and H. Yamada, Eur. J. Org. Chem., 2022, 2022, e202200621 CrossRef.
- J. P. Llinas, A. Fairbrother, G. Borin Barin, W. Shi, K. Lee, S. Wu, B. Yong Choi, R. Braganza, J. Lear, N. Kau, W. Choi, C. Chen, Z. Pedramrazi, T. Dumslaff, A. Narita, X. Feng, K. Müllen, F. Fischer, A. Zettl, P. Ruffieux, E. Yablonovitch, M. Crommie, R. Fasel and J. Bokor, Nat. Commun., 2017, 8, 633 CrossRef PubMed.
- G. Borin Barin, Q. Sun, M. Di Giovannantonio, C. Du, X. Wang, J. P. Llinas, Z. Mutlu, Y. Lin, J. Wilhelm, J. Overbeck, C. Daniels, M. Lamparski, H. Sahabudeen, M. L. Perrin, J. I. Urgel, S. Mishra, A. Kinikar, R. Widmer, S. Stolz, M. Bommert, C. Pignedoli, X. Feng, M. Calame, K. Müllen, A. Narita, V. Meunier, J. Bokor, R. Fasel and P. Ruffieux, Small, 2022, 18, 2202301 CrossRef CAS PubMed.
- P. B. Bennett, Z. Pedramrazi, A. Madani, Y.-C. Chen, D. G. de Oteyza, C. Chen, F. R. Fischer, M. F. Crommie and J. Bokor, Appl. Phys. Lett., 2013, 103, 253114 CrossRef.
- M. Ohtomo, H. Jippo, H. Hayashi, J. Yamaguchi, M. Ohfuchi, H. Yamada and S. Sato, ACS Appl. Mater. Interfaces, 2018, 10, 31623–31630 CrossRef CAS PubMed.
- N. Nijegorodov, V. Ramachandran and D. P. Winkoun, Spectrochim. Acta, Part A, 1997, 53, 1813–1824 CrossRef.
- H. F. Bettinger, C. Tönshoff, M. Doerr and E. Sanchez-Garcia, J. Chem. Theory Comput., 2016, 12, 305–312 CrossRef CAS PubMed.
- B. Shen, J. Tatchen, E. Sanchez-Garcia and H. F. Bettinger, Angew. Chem., Int. Ed., 2018, 57, 10506–10509 CrossRef CAS PubMed.
- R. Einholz, T. Fang, R. Berger, P. Grüninger, A. Früh, T. Chassé, R. F. Fink and H. F. Bettinger, J. Am. Chem. Soc., 2017, 139, 4435–4442 CrossRef CAS PubMed.
- A. Jancarik, G. Levet and A. Gourdon, Chem. - Eur. J., 2019, 25, 2366–2374 CrossRef CAS PubMed.
- A. Jančařík, J. Holec, Y. Nagata, M. Šámal and A. Gourdon, Nat. Commun., 2022, 13, 223 CrossRef PubMed.
- H. Hayashi, N. Hieda, M. Yamauchi, Y. S. Chan, N. Aratani, S. Masuo and H. Yamada, Chem. - Eur. J., 2020, 26, 15079–15083 CrossRef CAS PubMed.
- T. Kitao, T. Miura, R. Nakayama, Y. Tsutsui, Y. S. Chan, H. Hayashi, H. Yamada, S. Seki, T. Hitosugi and T. Uemura, Nat. Synth., 2023, 2, 848–854 CrossRef CAS.
- R. Kitaura, S. Kitagawa, Y. Kubota, T. C. Kobayashi, K. Kindo, Y. Mita, A. Matsuo, M. Kobayashi, H.-C. Chang, T. C. Ozawa, M. Suzuki, M. Sakata and M. Takata, Science, 2002, 298, 2358–2361 CrossRef CAS PubMed.
- A. A. Talin, A. Centrone, A. C. Ford, M. E. Foster, V. Stavila, P. Haney, R. A. Kinney, V. Szalai, F. El Gabaly, H. P. Yoon, F. Léonard and M. D. Allendorf, Science, 2014, 343, 66–69 CrossRef CAS PubMed.
- V. Guillerm, F. Ragon, M. Dan-Hardi, T. Devic, M. Vishnuvarthan, B. Campo, A. Vimont, G. Clet, Q. Yang, G. Maurin, G. Férey, A. Vittadini, S. Gross and C. Serre, Angew. Chem., Int. Ed., 2012, 51, 9267–9271 CrossRef CAS PubMed.
- A. Centrone, L. Brambilla, T. Renouard, L. Gherghel, C. Mathis, K. Müllen and G. Zerbi, Carbon, 2005, 43, 1593–1609 CrossRef CAS.
- R. Huang, H. Phan, T. S. Herng, P. Hu, W. Zeng, S. Q. Dong, S. Das, Y. Shen, J. Ding, D. Casanova and J. Wu, J. Am. Chem. Soc., 2016, 138, 10323–10330 CrossRef CAS PubMed.
- B. Bleaney and K. D. Bowers, Proc. R. Soc. A, 1952, 214, 451–465 CAS.
- J. J. Dressler, M. Teraoka, G. L. Espejo, R. Kishi, S. Takamuku, C. J. Gómez-García, L. N. Zakharov, M. Nakano, J. Casado and M. M. Haley, Nat. Chem., 2018, 10, 1134–1140 CrossRef CAS PubMed.
- M. Watanabe, Y. J. Chang, S. W. Liu, T. H. Chao, K. Goto, M. M. Islam, C. H. Yuan, Y. T. Tao, T. Shinmyozu and T. J. Chow, Nat. Chem., 2012, 4, 574–578 CrossRef CAS PubMed.
- M. Watanabe, T. Miyazaki, T. Matsushima, J. Matsuda, C. T. Chein, M. Shibahara, C. Adachi, S. S. Sun, T. J. Chow and T. Ishihara, RSC Adv., 2018, 8, 13259–13265 RSC.
- M. Watanabe, T.-H. Chao, C.-T. Chien, S.-W. Liu, Y. J. Chang, K.-Y. Chen and T. J. Chow, Tetrahedron Lett., 2012, 53, 2284–2287 CrossRef CAS.
- C.-T. Chien, M. Watanabe and T. J. Chow, Tetrahedron, 2015, 71, 1668–1673 CrossRef CAS.
- M. Watanabe, W.-T. Su, K.-Y. Chen, C.-T. Chien, T.-H. Chao, Y. J. Chang, S.-W. Liu and T. J. Chow, Chem. Commun., 2013, 49, 2240 RSC.
- T. Miyazaki, M. Watanabe, T. Matsushima, C. T. Chien, C. Adachi, S. S. Sun, H. Furuta and T. J. Chow, Chem. - Eur. J., 2021, 27, 10677–10684 CrossRef CAS PubMed.
- T. Matsushima, S. Hwang, A. S. D. Sandanayaka, C. Qin, S. Terakawa, T. Fujihara, M. Yahiro and C. Adachi, Adv. Mater., 2016, 28, 10275–10281 CrossRef CAS PubMed.
- M. Chen, L. Yan, Y. Zhao, I. Murtaza, H. Meng and W. Huang, J. Mater. Chem. C, 2018, 6, 7416–7444 RSC.
- A. N. Aleshin, J. Y. Lee, S. W. Chu, J. S. Kim and Y. W. Park, Appl. Phys. Lett., 2004, 84, 5383–5385 CrossRef CAS.
- J. Liu, H. Dong, Z. Wang, D. Ji, C. Cheng, H. Geng, H. Zhang, Y. Zhen, L. Jiang, H. Fu, Z. Bo, W. Chen, Z. Shuai and W. Hu, Chem. Commun., 2015, 51, 11777–11779 RSC.
- J. Liu, H. Zhang, H. Dong, L. Meng, L. Jiang, L. Jiang, Y. Wang, J. Yu, Y. Sun, W. Hu and A. J. Heeger, Nat. Commun., 2015, 6, 10032 CrossRef CAS PubMed.
- R. W. I. de Boer, T. M. Klapwijk and A. F. Morpurgo, Appl. Phys. Lett., 2003, 83, 4345–4347 CrossRef CAS.
- C. Reese, W.-J. Chung, M. Ling, M. Roberts and Z. Bao, Appl. Phys. Lett., 2006, 89, 202108 CrossRef.
- Y. Takeyama, S. Ono and Y. Matsumoto, Appl. Phys. Lett., 2012, 101, 083303 CrossRef.
- O. D. Jurchescu, M. Popinciuc, B. J. van Wees and T. T. M. Palstra, Adv. Mater., 2007, 19, 688–692 CrossRef CAS.
- K. Islam, H. Narjinari and A. Kumar, Asian J. Org. Chem., 2021, 10, 1544–1566 CrossRef CAS.
- M. Seri and A. Marrocchi, J. Mater. Chem. C, 2021, 9, 16164–16186 RSC.
- A. Broggi, I. Tomasi, L. Bianchi, A. Marrocchi and L. Vaccaro, ChemPlusChem, 2014, 79, 486–507 CrossRef CAS PubMed.
- L. Jiang, J. Gao, E. Wang, H. Li, Z. Wang, W. Hu and L. Jiang, Adv. Mater., 2008, 20, 2735–2740 CrossRef CAS PubMed.
- Y. Sun, L. Tan, S. Jiang, H. Qian, Z. Wang, D. Yan, C. Di, Y. Wang, W. Wu, G. Yu, S. Yan, C. Wang, W. Hu, Y. Liu and D. Zhu, J. Am. Chem. Soc., 2007, 129, 1882–1883 CrossRef CAS PubMed.
- Q. Tang, L. Li, Y. Song, Y. Liu, H. Li, W. Xu, Y. Liu, W. Hu and D. Zhu, Adv. Mater., 2007, 19, 2624–2628 CrossRef CAS.
- Q. Tang, H. Li, Y. Liu and W. Hu, J. Am. Chem. Soc., 2006, 128, 14634–14639 CrossRef CAS PubMed.
- Q. Tang, H. Li, M. He, W. Hu, C. Liu, K. Chen, C. Wang, Y. Liu and D. Zhu, Adv. Mater., 2006, 18, 65–68 CrossRef CAS.
- G. Romanazzi, A. Dell'Aquila, G. P. Suranna, F. Marinelli, S. Cotrone, D. Altamura, C. Giannini, L. Torsi and P. Mastrorilli, J. Mater. Chem., 2011, 21, 15186–15189 RSC.
- P. Keg, A. Dell'Aquila, F. Marinelli, O. L. Kapitanchuk, D. Fichou, P. Mastrorilli, G. Romanazzi, G. P. Suranna, L. Torsi, Y. M. Lam and S. G. Mhaisalkar, J. Mater. Chem., 2010, 20, 2448–2456 RSC.
- A. Dell'Aquila, F. Marinelli, J. Tey, P. Keg, Y. M. Lam, O. L. Kapitanchuk, P. Mastrorilli, C. F. Nobile, P. Cosma, A. Marchenko, D. Fichou, S. G. Mhaisalkar, G. P. Suranna and L. Torsi, J. Mater. Chem., 2008, 18, 786–791 RSC.
- A. Dodabalapur, L. Torsi and H. E. Katz, Science, 1995, 268, 270–271 CrossRef CAS PubMed.
- J. Zhu, H. Hayashi, M. Chen, C. Xiao, K. Matsuo, N. Aratani, L. Zhang and H. Yamada, Macromol. Chem. Phys., 2021, 222, 2100024 CrossRef CAS.
- J. Zhu, H. Hayashi, M. Chen, C. Xiao, K. Matsuo, N. Aratani, L. Zhang and H. Yamada, J. Mater. Chem. C, 2022, 10, 2527–2531 RSC.
- X. Wu, R. Jia, J. Pan, X. Zhang and J. Jie, Nanoscale Horiz., 2020, 5, 454–472 RSC.
- L. Jiang, H. Dong and W. Hu, J. Mater. Chem., 2010, 20, 4994 RSC.
- Y. Zhang, Q. Tang, H. Li and W. Hu, Appl. Phys. Lett., 2009, 94, 203304 CrossRef.
- Q. Tang, L. Jiang, Y. Tong, H. Li, Y. Liu, Z. Wang, W. Hu, Y. Liu and D. Zhu, Adv. Mater., 2008, 20, 2947–2951 CrossRef CAS.
- Q. Tang, Y. Tong, H. Li and W. Hu, Appl. Phys. Lett., 2008, 92, 083309 CrossRef.
- Q. Tang, Y. Tong, H. Li, Z. Ji, L. Li, W. Hu, Y. Liu and D. Zhu, Adv. Mater., 2008, 20, 1511–1515 CrossRef CAS.
- V. S. Barlier, C. W. Schlenker, S. W. Chin and M. E. Thompson, Chem. Commun., 2011, 47, 3754–3756 RSC.
- K. Kawano, H. Hayashi, S. Yoshimoto, N. Aratani, M. Suzuki, J. Yoshinobu and H. Yamada, Chem. - Eur. J., 2018, 24, 14916–14920 CrossRef CAS PubMed.
- D. Lehnherr, R. McDonald and R. R. Tykwinski, Org. Lett., 2008, 10, 4163–4166 CrossRef CAS PubMed.
- D. Lehnherr, A. H. Murray, R. McDonald and R. R. Tykwinski, Angew. Chem., Int. Ed., 2010, 49, 6190–6194 CrossRef CAS PubMed.
- M. M. Payne, S. R. Parkin and J. E. Anthony, J. Am. Chem. Soc., 2005, 127, 8028–8029 CrossRef CAS PubMed.
- D. Chun, Y. Cheng and F. Wudl, Angew. Chem., Int. Ed., 2008, 47, 8380–8385 CrossRef CAS PubMed.
- S. S. Zade and M. Bendikov, Angew. Chem., Int. Ed., 2010, 49, 4012–4015 CrossRef CAS PubMed.
- H. Hayashi, J. Zhu, N. Minamino, Y. Murakami, S. Kasahara, K. Matsuo, N. Aratani and H. Yamada, Eur. J. Org. Chem., 2024, 27, e202400770 CrossRef CAS.
- U. H. F. Bunz, Acc. Chem. Res., 2015, 48, 1676–1686 CrossRef CAS PubMed.
- W. Jiang, Y. Li and Z. Wang, Chem. Soc. Rev., 2013, 42, 6113 RSC.
- U. H. F. Bunz and J. Freudenberg, Acc. Chem. Res., 2019, 52, 1575–1587 CrossRef CAS PubMed.
- G. J. Richards, J. P. Hill, T. Mori and K. Ariga, Org. Biomol. Chem., 2011, 9, 5005 RSC.
- Q. Miao, Adv. Mater., 2014, 26, 5541–5549 CrossRef CAS PubMed.
- U. H. F. Bunz, J. U. Engelhart, B. D. Lindner and M. Schaffroth, Angew. Chem., Int. Ed., 2013, 52, 3810–3821 CrossRef CAS PubMed.
- U. H. F. Bunz, Chem. - Eur. J., 2009, 15, 6780–6789 CrossRef CAS PubMed.
- H. Hayashi, Y. Kato, A. Matsumoto, S. Shikita, N. Aizawa, M. Suzuki, N. Aratani, T. Yasuda and H. Yamada, Chem. - Eur. J., 2019, 25, 15565–15571 CrossRef CAS PubMed.
- M. Ganschow, S. Koser, S. Hahn, F. Rominger, J. Freudenberg and U. H. F. Bunz, Chem. - Eur. J., 2017, 23, 4415–4421 CrossRef CAS PubMed.
- P. Biegger, S. Stolz, S. N. Intorp, Y. Zhang, J. U. Engelhart, F. Rominger, K. I. Hardcastle, U. Lemmer, X. Qian, M. Hamburger and U. H. F. Bunz, J. Org. Chem., 2015, 80, 582–589 CrossRef CAS PubMed.
- V. Lami, D. Leibold, P. Fassl, Y. J. Hofstetter, D. Becker-Koch, P. Biegger, F. Paulus, P. E. Hopkinson, M. Adams, U. H. F. Bunz, S. Huettner, I. Howard, A. A. Bakulin and Y. Vaynzof, Sol. RRL, 2017, 1, 1700053 CrossRef.
- G. Xue, J. Wu, C. Fan, S. Liu, Z. Huang, Y. Liu, B. Shan, H. L. Xin, Q. Miao, H. Chen and H. Li, Mater. Horiz., 2016, 3, 119–123 RSC.
- X. Xu, Y. Yao, B. Shan, X. Gu, D. Liu, J. Liu, J. Xu, N. Zhao, W. Hu and Q. Miao, Adv. Mater., 2016, 28, 5276–5283 CrossRef CAS PubMed.
- M. Chu, J. X. Fan, S. Yang, D. Liu, C. F. Ng, H. Dong, A. M. Ren and Q. Miao, Adv. Mater., 2018, 30, 1803467 CrossRef PubMed.
- Z. Liang, Q. Tang, J. Xu and Q. Miao, Adv. Mater., 2011, 23, 1535–1539 CrossRef CAS PubMed.
- Z. Liang, Q. Tang, R. Mao, D. Liu, J. Xu and Q. Miao, Adv. Mater., 2011, 23, 5514–5518 CrossRef CAS PubMed.
- C. Wang, Z. Liang, Y. Liu, X. Wang, N. Zhao, Q. Miao, W. Hu and J. Xu, J. Mater. Chem., 2011, 21, 15201 RSC.
- B. D. Lindner, J. U. Engelhart, O. Tverskoy, A. L. Appleton, F. Rominger, A. Peters, H. Himmel and U. H. F. Bunz, Angew. Chem., Int. Ed., 2011, 50, 8588–8591 CrossRef CAS PubMed.
- B. Purushothaman, S. R. Parkin and J. E. Anthony, Org. Lett., 2010, 12, 2060–2063 CrossRef CAS PubMed.
- J. U. Engelhart, O. Tverskoy and U. H. F. Bunz, J. Am. Chem. Soc., 2014, 136, 15166–15169 CrossRef CAS PubMed.
- W. Zong, N. Hippchen, N. Zeitter, S. Maier, P. Ludwig, F. Rominger, J. Freudenberg and U. H. F. Bunz, J. Am. Chem. Soc., 2024, 146, 5793–5798 CrossRef CAS PubMed.
- N. Zeitter, N. Hippchen, P. Baur, T. V. Unterreiner, F. Rominger, J. Freudenberg and U. H. F. Bunz, Org. Mater., 2024, 6, 12–17 CrossRef CAS.
- A. H. Endres, M. Schaffroth, F. Paulus, H. Reiss, H. Wadepohl, F. Rominger, R. Krämer and U. H. F. Bunz, J. Am. Chem. Soc., 2016, 138, 1792–1795 CrossRef CAS PubMed.
- B. Gao, M. Wang, Y. Cheng, L. Wang, X. Jing and F. Wang, J. Am. Chem. Soc., 2008, 130, 8297–8306 CrossRef CAS PubMed.
- A. B. Marco, C. Gozalvez, M. Olano, X. Sun, A. Atxabal, M. Melle-Franco, L. E. Hueso and A. Mateo-Alonso, Phys. Chem. Chem. Phys., 2016, 18, 11616–11619 RSC.
- S. More, S. Choudhary, A. Higelin, I. Krossing, M. Melle-Franco and A. Mateo-Alonso, Chem. Commun., 2014, 50, 1976–1979 RSC.
- W. Chen, F. Yu, Q. Xu, G. Zhou and Q. Zhang, Adv. Sci., 2020, 7, 1903766 CrossRef CAS PubMed.
- M. Müller, H. Reiss, O. Tverskoy, F. Rominger, J. Freudenberg and U. H. F. Bunz, Chem. - Eur. J., 2018, 24, 12801–12805 CrossRef PubMed.
- B. Kohl, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2015, 54, 6051–6056 CrossRef CAS PubMed.
- M. Marongiu, T. Ha, S. Gil-Guerrero, K. Garg, M. Mandado, M. Melle-Franco, I. Diez-Perez and A. Mateo-Alonso, J. Am. Chem. Soc., 2024, 146, 3963–3973 CrossRef CAS PubMed.
- J. Mateos-Martín, K. Dhbaibi, M. Melle-Franco and A. Mateo-Alonso, Chem. - Eur. J., 2023, 29, e202302002 CrossRef PubMed.
- R. K. Dubey, M. Melle-Franco and A. Mateo-Alonso, J. Am. Chem. Soc., 2021, 143, 6593–6600 CrossRef CAS PubMed.
- R. K. Dubey, M. Marongiu, S. Fu, G. Wen, M. Bonn, H. I. Wang, M. Melle-Franco and A. Mateo-Alonso, Chem, 2023, 9, 2983–2996 CAS.
- W. Ran, A. Walz, K. Stoiber, P. Knecht, H. Xu, A. C. Papageorgiou, A. Huettig, D. Cortizo-Lacalle, J. P. Mora-Fuentes, A. Mateo-Alonso, H. Schlichting, J. Reichert and J. V. Barth, Angew. Chem., Int. Ed., 2022, 61, e202111816 CrossRef CAS PubMed.
- D. Cortizo-Lacalle, J. P. Mora-Fuentes, K. Strutyński, A. Saeki, M. Melle-Franco and A. Mateo-Alonso, Angew. Chem., Int. Ed., 2018, 57, 703–708 CrossRef CAS PubMed.
- P. Reineck, A. Francis, A. Orth, D. W. M. Lau, R. D. V. Nixon-Luke, I. Das Rastogi, W. A. W. Razali, N. M. Cordina, L. M. Parker, V. K. A. Sreenivasan, L. J. Brown and B. C. Gibson, Adv. Opt. Mater., 2016, 4, 1549–1557 CrossRef CAS.
- L. Yang, J. Ma, W. Zheng, S. Osella, J. Droste, H. Komber, K. Liu, S. Böckmann, D. Beljonne, M. R. Hansen, M. Bonn, H. I. Wang, J. Liu and X. Feng, Adv. Sci., 2022, 9, 2200708 CrossRef CAS PubMed.
- X. Wang, J. Ma, W. Zheng, S. Osella, N. Arisnabarreta, J. Droste, G. Serra, O. Ivasenko, A. Lucotti, D. Beljonne, M. Bonn, X. Liu, M. R. Hansen, M. Tommasini, S. De Feyter, J. Liu, H. I. Wang and X. Feng, J. Am. Chem. Soc., 2022, 144, 228–235 CrossRef CAS PubMed.
- X. Yao, W. Zheng, S. Osella, Z. Qiu, S. Fu, D. Schollmeyer, B. Müller, D. Beljonne, M. Bonn, H. I. Wang, K. Müllen and A. Narita, J. Am. Chem. Soc., 2021, 143, 5654–5658 CrossRef CAS PubMed.
- A. Narita, X. Feng, Y. Hernandez, S. A. Jensen, M. Bonn, H. Yang, I. A. Verzhbitskiy, C. Casiraghi, M. R. Hansen, A. H. R. Koch, G. Fytas, O. Ivasenko, B. Li, K. S. Mali, T. Balandina, S. Mahesh, S. De Feyter and K. Müllen, Nat. Chem., 2014, 6, 126–132 CrossRef CAS PubMed.
- W. Niu, J. Ma, P. Soltani, W. Zheng, F. Liu, A. A. Popov, J. J. Weigand, H. Komber, E. Poliani, C. Casiraghi, J. Droste, M. R. Hansen, S. Osella, D. Beljonne, M. Bonn, H. I. Wang, X. Feng, J. Liu and Y. Mai, J. Am. Chem. Soc., 2020, 142, 18293–18298 CrossRef CAS PubMed.
- H. Ebata, T. Izawa, E. Miyazaki, K. Takimiya, M. Ikeda, H. Kuwabara and T. Yui, J. Am. Chem. Soc., 2007, 129, 15732–15733 CrossRef CAS PubMed.
- H. Iino, T. Usui and J. I. Hanna, Nat. Commun., 2015, 6, 6828 CrossRef CAS PubMed.
- C. Mitsui, T. Okamoto, M. Yamagishi, J. Tsurumi, K. Yoshimoto, K. Nakahara, J. Soeda, Y. Hirose, H. Sato, A. Yamano, T. Uemura and J. Takeya, Adv. Mater., 2014, 26, 4546–4551 CrossRef CAS PubMed.
- H. Okamoto, S. Hamao, H. Goto, Y. Sakai, M. Izumi, S. Gohda, Y. Kubozono and R. Eguchi, Sci. Rep., 2014, 4, 5048 CrossRef CAS PubMed.
- Y. Shimo, T. Mikami, S. Hamao, H. Goto, H. Okamoto, R. Eguchi, S. Gohda, Y. Hayashi and Y. Kubozono, Sci. Rep., 2016, 6, 21008 CrossRef CAS PubMed.
- F. Zhang, Y. Hu, T. Schuettfort, C. Di, X. Gao, C. R. McNeill, L. Thomsen, S. C. B. Mannsfeld, W. Yuan, H. Sirringhaus and D. Zhu, J. Am. Chem. Soc., 2013, 135, 2338–2349 CrossRef CAS PubMed.
- J.-H. Dou, Y.-Q. Zheng, Z.-F. Yao, Z.-A. Yu, T. Lei, X. Shen, X.-Y. Luo, J. Sun, S.-D. Zhang, Y.-F. Ding, G. Han, Y. Yi, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2015, 137, 15947–15956 CrossRef CAS PubMed.
- T. He, M. Stolte and F. Würthner, Adv. Mater., 2013, 25, 6951–6955 CrossRef CAS PubMed.
- T. Okamoto, S. Kumagai, E. Fukuzaki, H. Ishii, G. Watanabe, N. Niitsu, T. Annaka, M. Yamagishi, Y. Tani, H. Sugiura, T. Watanabe, S. Watanabe and J. Takeya, Sci. Adv., 2020, 6, eaaz0632 CrossRef CAS PubMed.
- B. Cirera, A. Sánchez-Grande, B. de la Torre, J. Santos, S. Edalatmanesh, E. Rodríguez-Sánchez, K. Lauwaet, B. Mallada, R. Zbořil, R. Miranda, O. Gröning, P. Jelínek, N. Martín and D. Ecija, Nat. Nanotechnol., 2020, 15, 437–443 CrossRef CAS PubMed.
- S. Kawai, O. Krejčí, A. S. Foster, R. Pawlak, F. Xu, L. Peng, A. Orita and E. Meyer, ACS Nano, 2018, 12, 8791–8797 CrossRef CAS PubMed.
- A. Sánchez-Grande, B. de la Torre, J. Santos, B. Cirera, K. Lauwaet, T. Chutora, S. Edalatmanesh, P. Mutombo, J. Rosen, R. Zbořil, R. Miranda, J. Björk, P. Jelínek, N. Martín and D. Écija, Angew. Chem., Int. Ed., 2019, 58, 6559–6563 CrossRef PubMed.
- J. Freudenberg and U. H. F. Bunz, J. Am. Chem. Soc., 2024, 146, 16937–16949 CrossRef CAS PubMed.
- K. Kranthiraja and A. Saeki, Adv. Funct. Mater., 2021, 31, 2011168 CrossRef CAS.
- Y. Miyake, K. Kranthiraja, F. Ishiwari and A. Saeki, Chem. Mater., 2022, 34, 6912–6920 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |