
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegulating the donor number of solvents for long-cycle anode-free lithium metal batteries†
Yining
Zhao‡
,
Shaopeng
Li‡
,
Lingtong
Zhu
,
Yahui
Li
,
Shu
Xu
,
Hui
Dou
 and
Xiaogang
Zhang
*
and
Xiaogang
Zhang
*
Jiangsu Key Laboratory of Materials and Technologies for Energy Storage, College of Material Science and Technology, Nanjing University of Aeronautics and Astronautics, Nanjing, 210016, P. R. China. E-mail: azhangxg@nuaa.edu.cn
First published on 8th July 2025
Abstract
Anode-free lithium-metal batteries (AFLMBs) are one of the up-and-coming high-energy-density battery systems because they eliminate the need for conventional graphite electrodes or excess lithium metal anodes (LMAs). However, AFLMBs have serious problems of low coulombic efficiencies (CEs) and poor cycle stability. Herein, this study proposes a local high-concentration electrolyte (LHCE) based on donor number (DN) modulation, which achieves stable dissolution of LiNO3 and an anion-enhanced solvation structure through the synergistic action of a high DN solvent (tetraglyme, G4) and an ultra-low DN diluent (1,1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoropropyl ether, TTE). The design addresses the core challenges of low CEs and poor cycling stability in AFLMBs due to the lack of a lithium source. Computational and experimental results show that regulating the DN value of the solvent significantly reduces the Li+ desolvation energy and forms an inorganic-rich solid electrolyte interphase (SEI), which guides the uniform deposition of Li. The electrolyte achieves a high CE (about 99.0%) and long cycle life (1400 h). Based on this, the AFLMB retains 86.3% capacity after 50 cycles, far exceeding the 25-cycle life of conventional carbonate electrolytes (CCEs).
1. Introduction
The growing demand for high-performance rechargeable batteries is driving the development of advanced battery technologies. In the pursuit of high energy density, the LMA is regarded as one of the ideal anode materials for next-generation lithium batteries owing to its ultra-high theoretical specific capacity (3860 mA h g−1) and extremely low electrochemical potential (−3.04 V vs. the standard hydrogen electrode).1–3 Nevertheless, lithium metal exhibits high reactivity and substantial volumetric fluctuations, which undoubtedly increases the cost of manufacturing, transportation and storage of lithium metal batteries.4 In addition, due to side reactions and unstable SEIs, the anode needs to provide excess lithium metal at the cost of compromising the energy density to maximize the CE and achieve a respectable cycle lifetime.5–8AFLMBs effectively address the shortcomings of traditional lithium metal batteries.9–12 AFLMBs are a special type of lithium metal battery in which the anode does not contain active material in the initial state.13 Due to the absence of surplus Li in the battery, the volume is reduced to a minimum and the energy density is increased to its maximum potential.14,15 However, the active Li lost during cycling cannot be replenished, thus leading to a rapid decline in battery capacity and a shortened cycle life.16,17 Currently, researchers are addressing this issue from several directions, such as collector modification,18–20 advanced electrolyte design,21–23 Li replenishment,24–26 and optimization of external conditions (pressure regulation, cut-off voltage and depth of discharge control).27,28
According to recent research, electrolyte optimisation to improve the cycle life of AFLMBs is a more practical and cost-effective strategy. Currently, in the solvation structure of CCEs, due to the strong ion–dipole interactions between Li+ and the solvent, EC dominates the inner layer of the solvation structure of Li+ and preferentially undergoes a reduction reaction at the anode, resulting in the formation of SEIs with organics as the main component. Organic-rich SEIs are plagued by poor conductivity and instability, leading to extremely low CE values and lithium dendrite growth.29,30 Many studies have shown that inorganic-rich SEIs have the advantages of high interfacial energy and strong mechanical properties, which are favorable for guiding the uniform deposition of lithium.31 Therefore, it is crucial to regulate the environment around Li+ to direct more anions into the solvation structure thereby generating inorganic-dominated SEIs. In this regard, tuning and balancing the DN values of lithium salts and solvents may be an effective way to regulate the solvation structure of Li+. DN values are used to describe the electron-donating capacity of a solvent or anion,32 which mainly affects the performance of the battery by influencing the solvation environment on the Li anode and the SEI components derived with it.
Herein, we elaborate a type of LHCE using lithium bis(fluorosulfonyl)imide (LiFSI) and lithium nitrate (LiNO3) as the lithium salts, where the DN values of NO3− and FSI− are 21.1 and 9.5 kcal mol−1, respectively. Meanwhile, G4 (DN = 16.6 kcal mol−1) is used as the main solvent, and TTE (DN = 1.9 kcal mol−1) as the diluent. By modulating the DN values of the solvents, not only the stable presence of a high concentration of LiNO3 in the electrolyte was achieved, but also the formation of an anion-enhanced solvation structure guided the formation of inorganic-rich SEIs, which helped to achieve uniform lithium deposition. Importantly, density functional theory (DFT) calculations show that adding diluents weakens the interaction between Li+ and solvent molecules, thereby accelerating the desolvation process of Li+. Although studies have been conducted to optimise electrolytes applied to conventional lithium-metal batteries using DN modulation, most of the strategies focus on single-salt systems or conventional solvents (e.g., DMC/DME) and are not designed for the unique requirements of AFLMBs. In this work, DN modulation was applied to AFLMBs through a combination of dual salts with high and low DN solvents to address the dual challenges of interfacial dynamics and SEI stability. This study highlights the importance of solvents with different electron-donating capabilities in stabilising the solvation structure through DN-directed solvation structure design, which prolongs the cycling life of AFLMBs.
2. Results and discussion
2.1. Rational design of the electrolyte based on the DN value
First, the choice of lithium salt is crucial for ensuring the stability of the anode. In this work, anion manipulation was accomplished by combining two of the most commonly used lithium salts: LiFSI and LiNO3. LiFSI is commonly utilized in the LHCE due to its high solubility and ionic conductivity.33 A previous study found that the addition of small amounts of LiNO3 to the 2 M LiFSI–DME electrolyte enhances the complete decomposition of FSI−.34 In addition, the reduction products of LiNO3 can reduce the nucleation overpotential and promote the uniform deposition of lithium metal.35–38 Compared with the conventional single-salt system, this stabilised solvation structure of double anions not only promotes the formation of a more stable SEI layer, but also significantly reduces the occurrence of side reactions. Nevertheless, the limited solubility and dissociation of LiNO3 in commonly used solvents have hindered its widespread usage as the primary salt.39–42In solvent screening, the DN value of the solvent is used as a key parameter for screening and optimising electrolytes. The DN value reflects the ability of the solvent molecules to provide electrons to Li+, which directly affects the solvation structure and SEI composition of Li+.43 Research indicates that solvents with lower DN values (such as DMC) have lower solubility for LiNO3, resulting in poor conductivity and suboptimal cycling performance. However, solvents with higher DN values (such as DME, THF and DEE), while effectively promoting the dissociation of LiFSI and LiNO3, exhibit overly strong solventisation capabilities. This results in the Li+ solvation shell primarily being composed of solvent molecules, effectively blocking direct interactions between anions (NO3− and FSI−) and Li+ as well as the LMA surface.44 Here, we chose G4 as the primary solvent. Compared to other ether solvents, G4 has a moderate DN (much larger than that of CCEs, but smaller than that of LiNO3) and abundant solvation sites (O atoms), resulting in high solubility (>4 M, 25 °C) for LiNO3.45 However, the ultra-low DN diluent TTE compresses the solvation structure and promotes the migration of anions (NO3− and FSI−) into the first solvated layer of Li+ through a weak electron donor effect (Fig. 1a and b). This synergistic design of high/low DN solvents achieves an anion-enhanced solvation structure in the electrolyte, resulting in a stable inorganic-rich SEI.
 | ||
| Fig. 1 (a) A LHCE with dual salts based on a DN modulation rule and the promoted stable SEI. (b) Schematic illustration of the strong solvation effect of G4 and the shielding effect of TTE. (c) Raman spectra. (d) 7Li NMR spectra. Radial distribution functions and corresponding coordination numbers of (e) 2.4F-1.6N and (g) 2.4F-1.6N–TTE electrolytes. (f) and (h) Corresponding MD-simulated snapshots of (e) and (g), respectively. | ||
First, Raman spectroscopy was performed on different electrolytes in order to investigate the coordination of NO3−, FSI− and G4 molecules with Li+. 1 M electrolytes of LiFSI and LiNO3 mixed in different molar ratios were prepared. As shown in Fig. S1,† “0.4F-0.6N” in the figure represents dissolving 0.4 M LiFSI and 0.6 M LiNO3 in G4 solvent. In pure G4 solvent, the signal with a Raman shift of 850 cm−1 is identified as the –CH2–O–CH3 stretching vibration of the free G4 molecule. The lithium salt dissolved in G4 solvent shows a new peak at 870 cm−1, which is attributed to the Li+-solvated G4. It's obvious that its signal weakens as the amount of LiNO3 increases, which suggests that the presence of NO3− in the solvation structure weakens the coordination of Li+-solvated G4. The effects of different double salt ratios on the lithium deposition morphology were systematically investigated by scanning electron microscopy (SEM). As shown in Fig. S2,† the deposition of Li is the most flat and dense in the range of 0.6F-0.4N. Meanwhile, the Li metal anode has the best cycling stability in the range of 0.6F-0.4N (Fig. S3†). Based on the above results, the optimum molar ratio of LiFSI and LiNO3 was determined to be 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4. It should be noted that the concentration involved in this work was limited to 4 M (2.4F-1.6N), and a further increase in concentration would lead to precipitation of lithium salts.
:4. It should be noted that the concentration involved in this work was limited to 4 M (2.4F-1.6N), and a further increase in concentration would lead to precipitation of lithium salts.
In order to form a LHCE, TTE was added as a diluent to 2.4F-1.6N. Fig. S4† shows the ionic conductivity of different electrolytes. For example, “1:2” indicates that the volume ratio of 2.4F-1.6N to TTE is 1:2. The data indicate that the conductivity exhibits a negative correlation with the concentration of lithium salt. Nevertheless, as the TTE increases, the conductivity also increases, indicating that the ionic conductivity of the electrolyte is significantly improved by the low viscosity of TTE. In order to further optimize the TTE addition, the effect of different ratios of TTE additions on the solvation structure was investigated by Raman spectroscopy. As shown in Fig. 1c, the peak of free G4 (850 cm−1) completely disappeared in 2.4F-1.6N because the G4 molecules are highly coordinated with Li+, forming contact ion pairs (CIPs) and aggregates (AGGs). With the addition of TTE, the peak of Li+-solvated G4 is red-shifted. This indicates that the shielding effect of TTE leads to the dissociation of G4 molecules from the solvation structure and an increase in the proportion of free G4 molecules. In addition, the characteristic peak of TTE at 660 cm−1 is not shifted in different solutions, suggesting that at the microscopic level of the solvation structure, TTE does not undergo direct coordination with Li+. Meanwhile, the slight blueshift of the peaks of NO3− and FSI− identifies more anion incorporation in the solvation structure of Li+. In terms of macroscopic environmental regulation, this can be reasonably understood as the strong electronegativity of the F atoms in the TTE molecule and Li+ ions may be attracted to each other through weak interactions, compressing the confined solvation structure and facilitating high anion aggregation. To further investigate the changes in the Li+-solvation structure, 7Li NMR spectroscopy was used to investigate the participation of anions in the solvation shell both before and after the introduction of TTE. As shown in Fig. 1d, the 7Li NMR spectra display a downfield shift when the salt concentration increases from 1 M (0.6F-0.4N) to 4 M (2.4F-1.6N), indicating a decrease in the electron cloud density around Li+ as the salt concentration increases. This is because some of the anions can be absorbed into the solvation structure as the free solvent decreases. Since NO3− has a weaker electron-donating ability than G4, it reduces the electron cloud density around Li+. In the 2.4F-1.6N–TTE electrolyte, the 7Li NMR signal experiences a downfield shift, suggesting that the interaction between Li+ and G4 is weakened, which is consistent with the findings from Raman spectroscopy. The above spectral analysis indicates that after the addition of TTE to the 2.4F-1.6N electrolyte, part of the force between Li+ and G4 in the solvation structure is shielded by TTE, and an anion-enhanced solvation structure is formed.
MD simulations further confirmed this conclusion. As shown in Fig. 1e–h, the radial distribution function (RDF, g(r), solid line) and coordination numbers (n(r), dashed line) of the electrolyte system before and after the addition of TTE were calculated. The RDFs of Li–O(NO3−), Li–O(FSI−), and Li–O(G4) in different electrolytes all exhibit strong peaks at 1.84, 1.94 and 1.86 Å, respectively, which indicates that NO3−, FSI−, and G4 tend to dominate the primary solvation sheath of Li+. The addition of TTE retained the CIPs and AGGs in the 2.4F-1.6N without changing the original solvation structure. In addition, the peaks in 2.4F-1.6N–TTE are higher than those in 2.4F-1.6N, suggesting that more anions are squeezed into the primary solvation shell due to the crowding effect of TTE. Notably, the 2.4F-1.6N–TTE does not show distinct Li–O(TTE) peaks. Also, the coordination number is zero from 0 to 3.5 Å, indicating that the interaction between TTE and Li+ is weak or negligible. An analysis was conducted on the distribution of the center of mass (CM) to get a precise understanding of the distribution between anions and solvents surrounding the core Li+. From the center of mass (CM) distribution (Fig. S5†), NO3− exhibits the strongest solvation ability and is positioned closest to Li+. Theoretical modeling findings provide a high level of concordance with the spectroscopic features, demonstrating that the addition of TTE does not change the original solvation structure.
2.2. Compatibility of electrolytes with the Li metal anode
The desolvation process of Li+ at the SEI/electrolyte interface is the rate-determining step in the lithium metal deposition process, which requires overcoming a large energy barrier. To examine the impact of various TTE additions on the interfacial dynamics at the anode, the electrochemical impedance spectroscopy (EIS) of Li‖Li symmetric batteries was performed at temperatures ranging from 298 K to 323 K. The temperature-dependent EIS (Fig. S6†) and fitting results (Fig. S7†) show that the desolvation energy of the 2.4F-1.6N–TTE (1:3) electrolyte is only 36.94 kJ mol−1, significantly lower than that of the 2.4F-1.6N electrolyte (65.85 kJ mol−1) (Fig. 2a). Meanwhile, the most probable solvation structures of the 2.4F-1.6N and 2.4F-1.6N–TTE electrolytes were extracted from MD simulations (Fig. S8†). These solvation structures were used to evaluate the Li+ desolvation energy, which is crucial for battery kinetics. As shown in Fig. 2b, DFT calculations indicate that the desolvation energy of Li+ is significantly reduced by the addition of TTE. Additionally, the high exchange current density (0.184 mA cm−2) extracted from the Tafel plots further demonstrated the ease of lithium plating/stripping in 2.4F-1.6N–TTE (1:3) (Fig. 2c). These results indicate that the Li+ desolvation energy was reduced by DN modulation, and the Li+ transfer kinetics at the anode interface was significantly improved. This advantage stems from the shielding of Li+–G4 interactions by TTE, which makes the anion-enhanced solvation structure easier to desolvate, which is particularly beneficial for AFLMBs with limited lithium sources. Ultimately, the volume ratio of 2.4F-1.6N to TTE is determined to be 1:3, which corresponds to an apparent concentration of 1.0 M, matching that of the lithium salt in the CCE (1.0 M LiPF6/EC-DMC (1:1 vol%)–30%FEC) used as control samples.
 | ||
| Fig. 2 (a) The activation energies for Li+ desolvation at the anode. (b) The desolvation energy of 2.4F-1.6N and 2.4F-1.6N–TTE electrolytes obtained from DFT calculations. (c) The exchange current density of different electrolytes. (d) The CE of Li metal plating and stripping was assessed using Li||Cu half cells at 1 mA cm−2 for 1 mA h cm−2. (e) Corresponding voltage-capacity profiles at the 200th and 400th cycles using different electrolytes. (f) The cycle life of electrolytes at different current densities and Li deposition capacities. (g) Cycling performance of Li||Li symmetric cells with different electrolytes at 1 mA cm−2 for 1 mA h cm−2. (h) Amplified voltage profiles of selected time in (g). | ||
Then, three different electrolytes, CCE, 2.4F-1.6N and 2.4F-1.6N–TTE, were used to assemble Li||Cu half-cells to investigate their electrochemical performance. As shown in Fig. 2d, the CCE displays obvious fluctuation after only 50 cycles. 2.4F-1.6N could be cycled stably for 300 cycles, but the average CE was only 97.6%. In contrast, 2.4F-1.6N–TTE offers a CE of ∼99.0% over 550 cycles with no noticeable fluctuation. The voltage–capacity curve (Fig. 2e) confirms that 2.4F-1.6N–TTE has good compatibility with the lithium anode for better reversibility. Simultaneously, the 2.4F-1.6N–TTE also showed durable cycling stability at various capacities and current densities (Fig. 2f and S9†). To further demonstrate its advantages, the optimized electrolyte (2.4F-1.6N–TTE) was compared with the LiFSI–LiNO3–DME–TTE electrolyte. Li||Cu half-cells employing the LiFSI–LiNO3–DME–TTE electrolyte exhibited a higher initial Li nucleation overpotential and a significantly shorter cycling lifespan (Fig. S10†). Li||Li symmetric cells were used to further test the stability of different electrolytes. As shown in Fig. 2g, the overpotential of the CCE gradually increased with cycling and caused a short circuit after 150 h. This occurs due to the inhomogeneous deposition of lithium metal, which generates numerous side reaction products from rapid interactions between the electrolyte and lithium dendrites, obstructing the interfacial Li+ diffusion and increasing voltage polarization. Compared with 2.4F-1.6N, the overpotential of the 2.4F-1.6N–TTE was significantly reduced and remained nearly constant for 1400 h. Even at 1000 h, the polarization voltage is only 60 mV (Fig. 2h), which is one-tenth of that of 2.4F-1.6N (620 mV). These results further confirm the superior performance of the 2.4F-1.6N–TTE electrolyte in reducing nucleation barriers, improving lithium deposition uniformity and enhancing cycling stability.
2.3. Interfacial properties of the lithium anode
In order to enhance the reversibility of LMBs and prevent the production of dead Li, the morphology of the Li deposition is essential. The morphology of 4 mA h cm−2 Li deposited in the CCE and 2.4F-1.6N–TTE was observed by SEM. As shown in Fig. 3a–c, selective deposition of Li was observed in the CCE, featuring a porous structure and a rough morphology with a thickness of ∼71.6 μm. Conversely, the lithium deposited in 2.4F-1.6N–TTE showed a flat and dense morphology with no dendrites or voids within the lithium metal. Its thickness was around ∼22.9 μm, almost matching the theoretical estimate of 19.4 μm (Fig. 3d–f). This dendrite-free lithium morphology reduced the contact area between the active lithium and the electrolyte, thereby mitigating side reactions and the generation of dead lithium on the surface of the lithium anode. In addition, to visualize the morphological evolution during lithium deposition/exfoliation, an in situ optical observation device was built (Fig. S11a†). In Li||Li symmetric cells with a current density of 5 mA cm−2 and a lithium deposition capacity of 5 mA h cm−2, the deposition morphology in the CCE is fluffy and porous. The inhomogeneous lithium deposition worsens and the porous lithium volume increases significantly with a longer deposition time (Fig. S11b†). In contrast, the nucleation of lithium deposited in 2.4F-1.6N–TTE was more uniform, forming a denser and smoother morphology (Fig. S11c†). Furthermore, when stripping Li at the same current density, dendrites remained on the surface of the Li anode in the CCE, leading to the formation of dead lithium. Conversely, Li deposits uniformly disappeared in 2.4F-1.6N–TTE. The morphological studies demonstrate that regulating the DN of solvents helps to construct an anion-derived SEI with a lower Li+ diffusion barrier, which effectively inhibits the growth of lithium dendrites. In order to comprehend the key factors influencing the distinct deposition behaviors of lithium, X-ray photoelectron spectroscopy (XPS) was used to explore the differences in the composition of the SEI in different electrolytes. In the Li1s and F1s spectra (Fig. S12† and 3g), the SEI layer produced by the CCE contains more LiF components compared to 2.4F-1.6N–TTE, which is due to the decomposition of FEC. Furthermore, many organic phases, such as carbonaceous compounds (ROCO2Li), are generated in the SEI, as a result of solvent decomposition. This SEI is bulky and does not work well to minimize side reactions between the electrolyte and lithium metal, leading to the rupture of SEI and growth of dendrites, thus offsetting the advantages of fluorinated compounds.46,47 Another notable difference in the SEI composition between the two electrolytes is observed in the N1s and S2p spectra. In the N1s spectrum (Fig. 3h), there are peaks attributed to Li3N and LiNxOy that are generated by the decomposition of LiNO3. In addition, in the S2p spectra (Fig. 3i), there are two peaks attributed to SO42−, and the presence of low oxidation state groups indicates that FSI− has been completely decomposed. In contrast, these components were not observed in the SEI of the CCE. This is mainly due to the DN modulation to form an anion-enhanced solvation structure, and the synergistic decomposition of the double salts further enriches the inorganic component of the SEI. | ||
| Fig. 3 Top views of Li deposited in (a) and (b) CCE and (d) and (e) 2.4F-1.6N–TTE electrolytes. The inset shows an optical image of Li deposited on copper foil. The cross-sectional SEM images on the deposited Li metal in electrolytes of (c) CCE and (f) 2.4F-1.6N–TTE. XPS spectra of (g) F1s, (h) N1s and (i) S2p on the Li-metal anode at 0.5 mA cm−2, 0.5 mA h cm−2 after 10 cycles. | ||
In the previous electrochemical tests, the 2.4F-1.6N–TTE showed superior cycling stability and higher CE, suggesting that in addition to F-containing substances (e.g., LiF), N-containing and S-containing SEI compositions should contribute to the construction of a stable SEI. Previous studies have shown that good SEI components, such as Li3N and LiNxOy, are favorable for increasing the conductivity of Li+ and can provide fast migration channels for Li+ thereby facilitating the diffusion of Li+ in the SEI.48,49 In addition, Li2SO4 can serve as a safeguard in the formation of a stable SEI and enhance the durability of the cycling process.50 In conclusion, the increase of the inorganic phase in the SEI can improve the mechanical strength, density, and flexibility of the SEI, which offers effective safeguarding for the lithium metal anode.51,52
In order to investigate the chemical composition and distribution of SEIs formed in different electrolytes, time-of-flight secondary ion mass spectrometry (TOF-SIMS) was performed on lithium foil after cycling. We observed stronger C− and F− signals and weaker O− signals in the SEI derived from the CCE compared to those from 2.4F-1.6N–TTE (Fig. S13†). Considering that the C− signal comes exclusively from solvent decomposition, this indicates that the solvent-dominated solvation structure formed in the CCE leads to severe solvent decomposition
TOF-SIMS three-dimensional mapping (Fig. 4a and b) revealed that F− is abundant in the SEIs formed in both electrolytes. However, along the thickness, the content of F− in the SEI from the CCE is significantly higher than that in 2.4F-1.6N–TTE, which is consistent with the XPS results. In addition, the distribution of C−, O−, and F− species detected on the surface of the SEI formed by the CCE was not uniform (Fig. S14a–c†). In the SEI formed in 2.4F-1.6N–TTE, however, the distribution of various elements was more uniform (Fig. S14d–f†). At the same time, trace amounts of Li3N− and S− species were also detected, which are characteristic ionic fragments of Li3N and LiFSI decomposition products, respectively. This suggests that the decomposition products of the double salts are jointly involved in the formation of the SEI.
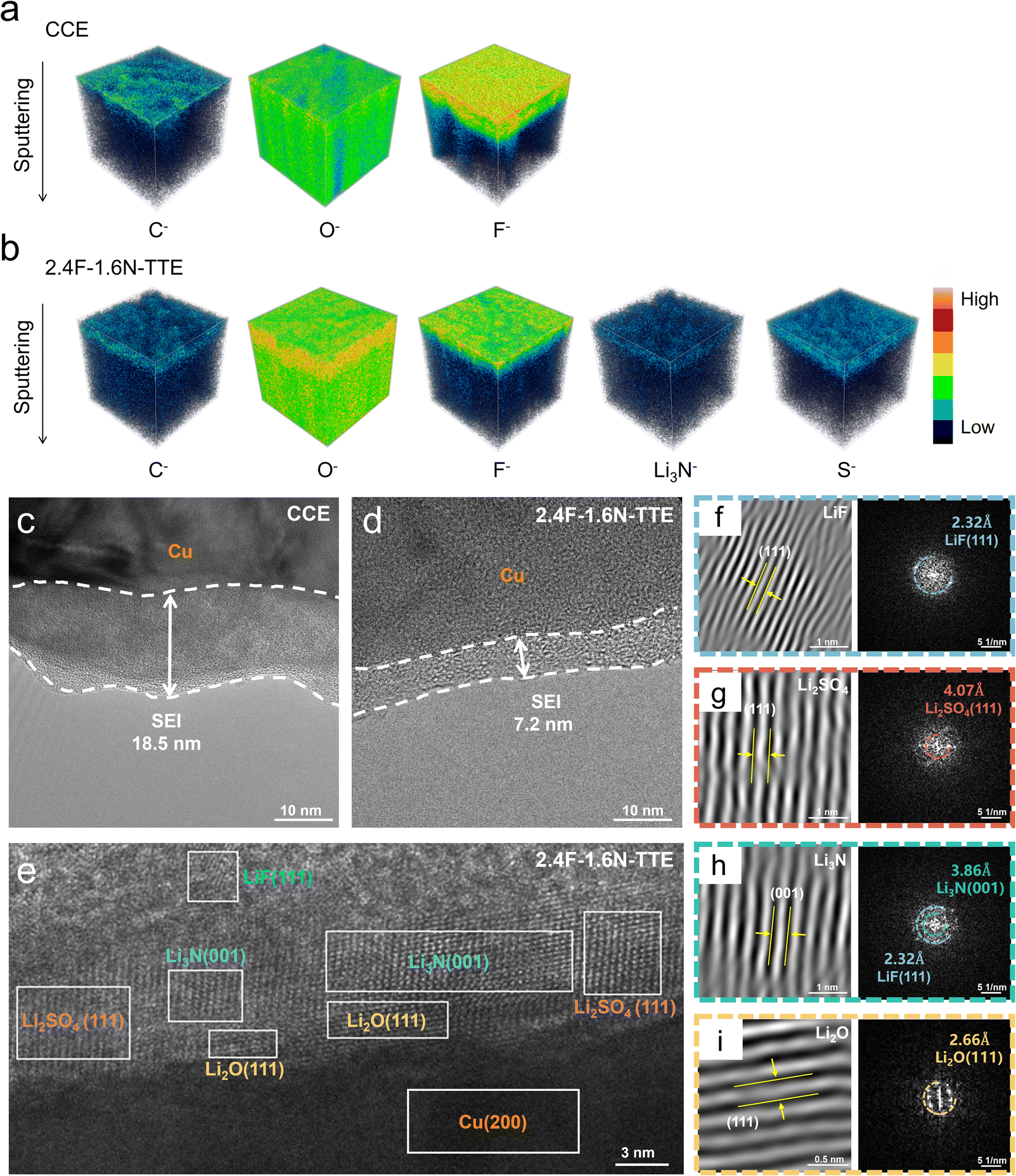 | ||
| Fig. 4 (a) TOF-SIMS three-dimensional depth-profiling images of the C−, O− and F− species in the SEI formed in the CCE. (b) TOF-SIMS three-dimensional depth-profiling images of the C−, O−, F−, Li3N− and S− species in the SEI formed in the 2.4F-1.6N–TTE. HRTEM characterization of the cycled Cu foil disassembled from the Li||Cu cells after 50 cycles at 0.5 mA cm−2 for 1 mA h cm−2 in different electrolytes: (c) CCE, (d) 2.4F-1.6N–TTE. (e) Magnified HRTEM image. The corresponding local fast Fourier transform images of (d): (f) LiF crystal, (g) Li2SO4 crystal, (h) Li3N crystal and (i) Li2O crystal. | ||
In order to investigate the refined structure of the SEI, the nanostructure of the SEI formed in different electrolytes was observed by using high-resolution transmission electron microscopy (HRTEM). Disassembling the Li||Cu half-cells after 50 cycles in different electrolytes, the formation of different thicknesses of the SEI on the Cu foil can be clearly observed. As shown in Fig. 4c, the SEI formed on the Cu foil cycled in the CCE had a thickness of approximately 18.5 nm and exhibited non-uniformity, which may impede interfacial transfer and lead to uneven Li deposition. In contrast, benefiting from the anion-enhanced solvation structure, the SEI formed in the 2.4F-1.6N–TTE electrolyte was thinner (7.2 nm) and more uniform (Fig. 4d). The high magnification images (Fig. 4e) reveal that the SEI layer induced by the 2.4F-1.6N–TTE electrolyte has a multilayer structure, consisting of an amorphous outer layer and a well-crystallized inner layer with a mixed layer including LiF, Li2O, Li2SO4 and Li3N. This particular SEI structure exhibits ultrafast Li+ conductivity, thus inhibiting the growth of Li dendrites. This was confirmed from the corresponding fast Fourier transform (FFT) and lattice fringe patterns. Based on the lattice distance, the darkest position was identified as the (2 0 0) planes of Cu with a spacing of 2.08 Å (Fig. S15†). The corresponding local FFT images displayed in Fig. 5f–i clearly identify the lattice spacings of crystalline components, in which the lattice spacings of LiF (1 1 1), Li2SO4 (1 1 1), Li3N (0 0 1) and Li2O (1 1 1) are 2.32, 4.07, 3.86 and 2.66 Å, respectively. The EDS mapping image (Fig. S16†) also shows a uniform distribution of F, S, N and O with the same profile.
 | ||
| Fig. 5 (a) Cycling performance of NCM622||Li cells using different electrolytes. Voltage profiles of NCM622||Li cells at selected cycles during cycling in (b) 2.4F-1.6N–TTE. (c) Histogram of transition metal dissolution detected on dismantled Li anodes after 100 cycles in Li||NCM622 cells with the investigated electrolytes. HRTEM characterization of NCM622 particles cycled in (d) 2.4F-1.6N–TTE and (g) CCE. SEM images of NCM622 particles cycled in (e) 2.4F-1.6N–TTE and (h) CCE. Schematics showing (f) the cell using 2.4F-1.6N–TTE and (i) the cell using CCE. (j) Cycling performance of NCM622||Cu cells using different electrolytes. (k) Voltage profiles of NCM622||Cu cells at selected cycles during cycling in different electrolytes. | ||
2.4. Performances of Li||NCM622 cells
Electrochemical tests and characterization were systematically performed to demonstrate the compatibility of the prepared LHCE with NCM622. The electrochemical performance of the Li||NCM622 cell was investigated with the NCM622 cathode (17.67 mg cm−2, 1C = 175 mA g−1) and Li metal anode (50 μm) in different electrolytes. As shown in Fig. 5a, the Li||NCM622 cell employing the 2.4F-1.6N–TTE electrolyte presents good cycling stability after 200 cycles at 0.5C. The initial discharge specific capacity was 161.46 mA h g−1, and the discharge-specific capacity after 200 cycles was 137.45 mA h g−1, with a capacity retention rate of 85.1% (Fig. 5b). In contrast, the capacity retention of the cell using the CCE drops rapidly to less than 60% after 140 cycles due to the buildup of organic byproducts from the decomposition of the electrolyte and the phase change of the cathode.In order to investigate the effect of the unique solvation structure in 2.4F-1.6N–TTE on the cathode–electrolyte interface (CEI), the NCM622||Li cell was disassembled after 100 cycles, and the composition of the CEI was characterized by XPS. As shown in Fig. S17,† for the NCM622 cathode cycled in the CCE, peaks attributed to LixFyPOz and LiF were observed in the F1s spectrum, which originated from the decomposition of LiPF6. The CEIs formed in 2.4F-1.6N–TTE are mainly composed of inorganic-rich phases such as LiF, Li3N, LiNO2, and Li2SO4, which are derived from the decomposition of the anions in the double salts. This inorganic-rich CEI contributes to fast diffusion kinetics. Meanwhile, S–Ox and N–Ox signals from the anion decomposition were observed in the CEI of 2.4F-1.6N–TTE in the O1s spectrum, indicating that S and N are bonded to O on the cathode surface for effective protection. In addition, the M–O signal can only be detected in the CCE, which is associated with a sustained corrosion. The dissolution–redeposition process of transition metal (TM) on the lithium anode was also analyzed by using EDS. As shown in Fig. 5c and S18,† there is a large attenuation of the TM elemental signal in the 2.4F-1.6N–TTE, suggesting that both LiFSI and LiNO3 significantly inhibit the corrosion of the cathode during electrochemical cycling. To understand the reasons why the prepared electrolyte inhibits TM dissolution and improves the cycling stability, the cycled NCM622 cathode was characterized by HRTEM. As can be seen in Fig. 5d, a CEI layer with a thickness of about 4.6 nm can be observed on the surface of the NCM622 cathode cycled in the 2.4F-1.6N–TTE electrolyte, which has an overall thinner and more uniform morphology. In contrast, the NCM622 cathode cycled in the CCE was covered by a CEI with a thickness of 10.3 nm (Fig. 5g). The CEI with good morphology generated in 2.4F-1.6N–TTE may be valuable for inhibiting side reactions between the cathode and the electrolyte. Furthermore, the surface and cross-sectional morphologies of the NCM622 cathode after long-term cycling in the CCE and 2.4F-1.6N–TTE were investigated. As shown in Fig. 5e and S19a–c,† the NCM622 electrode retained its structural integrity in the 2.4F-1.6N–TTE electrolyte, without experiencing serious cracking or pulverized disintegration. In sharp contrast, relatively large amounts of electrolyte decomposition byproducts were accumulated on the surface of the NCM622 cathode cycled in the CCE, which was associated with constant corrosive side reactions. Meanwhile, the cathode microstructure was degraded with fragmentation and intergranular cracks (indicated by arrows) (Fig. 5h and S19d–f†). In conclusion, an anion-enhanced solvation structure was successfully prepared by modulating the DN values of lithium salts and solvents, and the anion reduction generated an inorganic-rich interface. This not only effectively inhibits the growth of lithium dendrites, but also the thinner and denser CEI ensures good compatibility with the NCM622 cathode and inhibits microstructural degradation (Fig. 5f). In contrast, in the CCE, EC dominates the inner layers of the Li+ solvation structure because of the strong dipole–ion interaction between Li+ and the solvent. As a result, an organic-rich interface is formed, which cannot inhibit the growth of lithium dendrites. Meanwhile, as shown in Fig. 5i, an inhomogeneous CEI may lead to sustained electrochemical stress corrosion of the electrolyte and cathode materials, consistent with the degradation of electrochemical performance.53–55
In AFLMBs, the stability of the electrolyte directly determines the cycle life of the battery due to the absence of an active lithium source at the anode in the initial state. The anion-enhanced solvation structure formed by DN regulation significantly reduces the Li+ desolvation energy and reduces interfacial side reactions. This design is particularly important under lithium source-limited conditions: irreversible lithium loss in the AFLMBs can directly lead to a rapid decline in battery capacity compared to conventional Li metal batteries. As shown in Fig. 5j, the AFLMB using the 2.4F-1.6N–TTE electrolyte retains a specific discharge capacity of 130.8 mA h g−1 after 50 cycles, with a capacity retention rate of 86.3% (Fig. 5k). However, the AFLMB with the CCE can only retain its performance over 25 cycles due to uncontrolled side reactions that rapidly deplete the lithium or the electrolyte, leading to catastrophic capacity degradation. Furthermore, this study compares the optimized electrolyte (2.4F-1.6N–TTE) with both 2.4F-1.6N and LiFSI–LiNO3–DME–TTE electrolytes. AFLMBs using the latter two electrolytes exhibited rapid capacity decay during cycling (Fig. S20†). This difference is due to the formation of inorganic enriched SEIs under the DN regulation strategy, whose high mechanical strength and ionic conductivity effectively inhibit dendrite growth and dead lithium generation.
3. Conclusions
In summary, in this study, the donor number modulation strategy is applied to anode-free lithium metal batteries, and the synergistic design of solvents with high/low DN values is proposed to break through the solubility limitation of LiNO3 in conventional solvents. Comprehensive characterisation and theoretical tools, such as Raman, NMR, SEM, XPS, TOF-SIMS, in situ optical microscopy, HRTEM, MD and RDF, revealed that the anion-enhanced solvation structure is achieved by the combination of G4 and TTE, which generates high-quality SEI and CEI films and avoids the growth of anode lithium dendrites and the phase transition of anode materials. Consequently, the prepared 2.4F-1.6N–TTE electrolyte achieves a high CE value (∼99.0%) and ultra-long cycle life of the lithium anode. By comparing the electrochemical performance of AFLMBs based on this LHCE with that of a CCE, the critical role of the solvation structure and interfacial reaction mechanisms in the design and development of electrolytes is highlighted.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Yining Zhao: design and execution of the experiments and preparation of the article draft. Shaopeng Li: design of experiments, writing – reviewing and editing. Lingtong Zhu: validation, data curation, writing – reviewing and editing. Yahui Li: conceptualization, writing-reviewing and editing. Shu Xu: validation, data curation. Hui Dou: writing – reviewing and editing. Xiaogang Zhang: supervision, project administration, funding acquisition, writing – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2022YFE0109400), Leading Edge Technology of Jiangsu Province (BK20220009 and BK20232022), and Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). We acknowledge the facilities in the Center for Microscopy and Analysis at Nanjing University of Aeronautics and Astronautics. We acknowledge the technicians at Shenzhen HUASUAN Technology Co., Ltd for assistance with theoretical calculations.References
- S. Jiao, X. Ren, R. Cao, M. H. Engelhard, Y. Liu, D. Hu, D. Mei, J. Zheng, W. Zhao, Q. Li, N. Liu, B. D. Adams, C. Ma, J. Liu, J.-G. Zhang and W. Xu, Stable cycling of high-voltage lithium metal batteries in ether electrolytes, Nat. Energy, 2018, 3, 739–746 CrossRef CAS.
- K. Lee, S.-H. Kwon, J. Kim, E. Park, I. Kim, H. C. Ahn, A. Coskun and J. W. Choi, Fluorinated cyclic ether diluent for high-voltage lithium metal batteries, ACS Energy Lett., 2024, 9, 2201–2211 CrossRef CAS.
- W. Zhang, P. Sayavong, X. Xiao, S. T. Oyakhire, S. B. Shuchi, R. A. Vilá, D. T. Boyle, S. C. Kim, M. S. Kim, S. E. Holmes, Y. Ye, D. Li, S. F. Bent and Y. Cui, Recovery of isolated lithium through discharged state calendar ageing, Nature, 2024, 626, 306–312 CrossRef CAS PubMed.
- J. Zheng, M. S. Kim, Z. Tu, S. Choudhury, T. Tang and L. A. Archer, Regulating electrodeposition morphology of lithium: towards commercially relevant secondary Li metal batteries, Chem. Soc. Rev., 2020, 49, 2701–2750 RSC.
- F. Wu, J. Maier and Y. Yu, Guidelines and trends for next-generation rechargeable lithium and lithium-ion batteries, Chem. Soc. Rev., 2020, 49, 1569–1614 RSC.
- X. Zhang, Y. Yang and Z. Zhou, Towards practical lithium-metal anodes, Chem. Soc. Rev., 2020, 49, 3040–3071 RSC.
- J. Cao, Y. Shi, A. Gao, G. Du, M. Dilxat, Y. Zhang, M. Cai, G. Qian, X. Lu, F. Xie, Y. Sun and X. Lu, Hierarchical Li electrochemistry using alloy-type anode for high-energy-density Li metal batteries, Nat. Commun., 2024, 15, 1354 CrossRef CAS PubMed.
- M. Hu, Z. Tong, C. Cui, T. Zhai and H. Li, Facile, atom-economic, chemical thinning strategy for ultrathin lithium foils, Nano Lett., 2022, 22, 3047–3053 CrossRef CAS.
- Y. Tian, Y. An, C. Wei, H. Jiang, S. Xiong, J. Feng and Y. Qian, Recently advances and perspectives of anode-free rechargeable batteries, Nano Energy, 2020, 78, 105344 CrossRef CAS.
- W.-Z. Huang, C.-Z. Zhao, P. Wu, H. Yuan, W.-E. Feng, Z.-Y. Liu, Y. Lu, S. Sun, Z.-H. Fu, J.-K. Hu, S.-J. Yang, J.-Q. Huang and Q. Zhang, Anode-free solid-state lithium batteries: a review, Adv. Energy Mater., 2022, 12, 2201044 CrossRef CAS.
- W. Yao, P. Zou, M. Wang, H. Zhan, F. Kang and C. Yang, Design principle, optimization strategies, and future perspectives of anode-free configurations for high-energy rechargeable metal batteries, Electrochem. Energy Rev., 2021, 4, 601–631 CrossRef CAS.
- T. M. Hagos, H. K. Bezabh, C.-J. Huang, S.-K. Jiang, W.-N. Su and B. J. Hwang, A powerful protocol based on anode-free cells combined with various analytical techniques, Acc. Chem. Res., 2021, 54, 4474–4485 CrossRef CAS PubMed.
- Z. Tong, B. Bazri, S.-F. Hu and R.-S. Liu, Interfacial chemistry in anode-free batteries: challenges and strategies, J. Mater. Chem. A, 2021, 9, 7396–7406 RSC.
- P. Albertus, S. Babinec, S. Litzelman and A. Newman, Status and challenges in enabling the lithium metal electrode for high-energy and low-cost rechargeable batteries, Nat. Energy, 2018, 3, 16–21 CrossRef CAS.
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X. Q. Yang and J. G. Zhang, Pathways for practical high-energy long-cycling lithium metal batteries, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- D. Lin, Y. Liu and Y. Cui, Reviving the lithium metal anode for high-energy batteries, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- A. J. Louli, A. Eldesoky, R. Weber, M. Genovese, M. Coon, J. deGooyer, Z. Deng, R. T. White, J. Lee, T. Rodgers, R. Petibon, S. Hy, S. J. H. Cheng and J. R. Dahn, Diagnosing and correcting anode-free cell failure via electrolyte and morphological analysis, Nat. Energy, 2020, 5, 693–702 CrossRef CAS.
- S. Cho, D. Y. Kim, J.-I. Lee, J. Kang, H. Lee, G. Kim, D.-H. Seo and S. Park, Highly reversible lithium host materials for high-energy-density anode-free lithium metal batteries, Adv. Funct. Mater., 2022, 32, 2208629 CrossRef CAS.
- J. Wang, Y. Wang, X. Lu, J. Qian, C. Yang, I. Manke, H. Song, J. Liao, S. Wang and R. Chen, Ultra-sleek high entropy alloy tights: realizing superior cyclability for anode-free battery, Adv. Mater., 2024, 36, 2308257 CrossRef CAS.
- Y. Wang, Z. Qu, S. Geng, M. Liao, L. Ye, Z. Shadike, X. Zhao, S. Wang, Q. Xu, B. Yuan, X. Zhang, X. Gao, X. Jiang, H. Peng and H. Sun, Anode-free lithium metal batteries based on an ultrathin and respirable interphase layer, Angew. Chem., Int. Ed., 2023, 62, e202304978 CrossRef CAS PubMed.
- L. Su, H. Charalambous, Z. Cui and A. Manthiram, High-efficiency, anode-free lithium–metal batteries with a close-packed homogeneous lithium morphology, Energy Environ. Sci., 2022, 15, 843–854 RSC.
- M. Mao, X. Ji, Q. Wang, Z. Lin, M. Li, T. Liu, C. Wang, Y.-S. Hu, H. Li, X. Huang, L. Chen and L. Suo, Anion-enrichment interface enables high-voltage anode-free lithium metal batteries, Nat. Commun., 2023, 14, 1082 CrossRef CAS PubMed.
- R. Weber, M. Genovese, A. J. Louli, S. Hames, C. Martin, I. G. Hill and J. R. Dahn, Long cycle life and dendrite-free lithium morphology in anode-free lithium pouch cells enabled by a dual-salt liquid electrolyte, Nat. Energy, 2019, 4, 683–689 CrossRef CAS.
- Y. Qiao, H. Yang, Z. Chang, H. Deng, X. Li and H. Zhou, A high-energy-density and long-life initial-anode-free lithium battery enabled by a Li2O sacrificial agent, Nat. Energy, 2021, 6, 653–662 CrossRef CAS.
- X. Liu, Y. Tan, W. Wang, C. Li, Z. W. Seh, L. Wang and Y. Sun, Conformal prelithiation nanoshell on LiCoO2 enabling high-energy lithium-ion batteries, Nano Lett., 2020, 20, 4558–4565 CrossRef CAS PubMed.
- X. Su, C. Lin, X. Wang, V. A. Maroni, Y. Ren, C. S. Johnson and W. Lu, A new strategy to mitigate the initial capacity loss of lithium ion batteries, J. Power Sources, 2016, 324, 150–157 CrossRef CAS.
- C. Zhou, A. J. Samson, M. A. Garakani and V. Thangadurai, Communication—anode-free lithium metal batteries: a case study of compression effects on coin cell performance, J. Electrochem. Soc., 2021, 168, 060532 CrossRef CAS.
- A. J. Louli, M. Coon, M. Genovese, J. deGooyer, A. Eldesoky and J. R. Dahn, Optimizing cycling conditions for anode-free lithium metal cells, J. Electrochem. Soc., 2021, 168, 020515 CrossRef CAS.
- F. Ding, W. Xu, X. Chen, J. Zhang, M. H. Engelhard, Y. Zhang, B. R. Johnson, J. V. Crum, T. A. Blake, X. Liu and J.-G. Zhang, Effects of carbonate solvents and lithium salts on morphology and coulombic efficiency of lithium electrode, J. Electrochem. Soc., 2013, 160, A1894 CrossRef CAS.
- D. Aurbach, E. Zinigrad, Y. Cohen and H. Teller, A short review of failure mechanisms of lithium metal and lithiated graphite anodes in liquid electrolyte solutions, Solid State Ionics, 2002, 148, 405–416 CrossRef CAS.
- X. Lian, Z. Ju, L. Li, Y. Yi, J. Zhou, Z. Chen, Y. Zhao, Z. Tian, Y. Su, Z. Xue, X. Chen, Y. Ding, X. Tao and J. Sun, Dendrite-free and high-rate potassium metal batteries sustained by an inorganic-rich SEI, Adv. Mater., 2023, 36, 2306992 CrossRef PubMed.
- P. Zhou, Y. Xiang and K. Liu, Understanding and applying the donor number of electrolytes in lithium metal batteries, Energy Environ. Sci., 2024, 17, 8057–8077 RSC.
- Y. Zhao, Z. Hu, Z. Zhao, X. Chen, S. Zhang, J. Gao and J. Luo, Strong solvent and dual lithium salts enable fast-charging lithium-ion batteries operating from −78 to 60 °C, J. Am. Chem. Soc., 2023, 145, 22184–22193 CrossRef CAS PubMed.
- X.-Q. Zhang, X. Chen, L.-P. Hou, B.-Q. Li, X.-B. Cheng, J.-Q. Huang and Q. Zhang, Regulating anions in the solvation sheath of lithium ions for stable lithium metal batteries, ACS Energy Lett., 2019, 4, 411–416 CrossRef CAS.
- Z. Wen, W. Fang, F. Wang, H. Kang, S. Zhao, S. Guo and G. Chen, Dual-salt electrolyte additive enables high moisture tolerance and favorable electric double layer for lithium metal battery, Angew. Chem., Int. Ed., 2024, 63, e202314876 CrossRef CAS PubMed.
- J. Jiang, M. Li, X. Liu, J. Yi, Y. Jiang, C. Wu, H. Liu, B. Zhao, W. Li, X. Sun, J. Zhang and S. Dou, Multifunctional additives to realize dendrite-free lithium deposition in carbonate electrolytes toward low-temperature Li metal batteries, Adv. Energy Mater., 2024, 14, 2400365 CrossRef CAS.
- Q. Zhao, X. Liu, J. Zheng, Y. Deng, A. Warren, Q. Zhang and L. Archer, Designing electrolytes with polymer like glass-forming properties and fast ion transport at low temperatures, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 26053–26060 CrossRef CAS PubMed.
- D. Aurbach, E. Pollak, R. Elazari, G. Salitra, C. S. Kelley and J. Affinito, On the surface chemical aspects of very high energy density, rechargeable Li–sulfur batteries, J. Electrochem. Soc., 2009, 156, A694 CrossRef CAS.
- K. Wang, W. Ni, L. Wang, L. Gan, J. Zhao, Z. Wan, W. Jiang, W. Ahmad, M. Tian, M. Ling, J. Chen and C. Liang, Lithium nitrate regulated carbonate electrolytes for practical Li-metal batteries: mechanisms, principles and strategies, J. Energy Chem., 2023, 77, 581–600 CrossRef CAS.
- X. Li, R. Zhao, Y. Fu and A. Manthiram, Nitrate additives for lithium batteries: mechanisms, applications, and prospects, eScience, 2021, 1, 108–123 CrossRef.
- S. Zhang, G. Yang, Z. Liu, X. Li, X. Wang, R. Chen, F. Wu, Z. Wang and L. Chen, Competitive solvation enhanced stability of lithium metal anode in dual-salt electrolyte, Nano Lett., 2021, 21, 3310–3317 CrossRef CAS PubMed.
- S. H. Lee, J.-Y. Hwang, J. Ming, Z. Cao, H. A. Nguyen, H.-G. Jung, J. Kim and Y.-K. Sun, Toward the sustainable lithium metal batteries with a new electrolyte solvation chemistry, Adv. Energy Mater., 2020, 10, 2000567 CrossRef CAS.
- M. Baek, H. Shin, K. Char and J. W. Choi, New high donor electrolyte for lithium–sulfur batteries, Adv. Mater., 2020, 32, 2005022 CrossRef PubMed.
- P. Zhou, W. Hou, Y. Xia, Y. Ou, H.-Y. Zhou, W. Zhang, Y. Lu, X. Song, F. Liu, Q. Cao, H. Liu, S. Yan and K. Liu, Tuning and balancing the donor number of lithium salts and solvents for high-performance Li metal anode, ACS Nano, 2023, 17, 17169–17179 CrossRef CAS PubMed.
- Z. Wen, W. Fang, X. Wu, Z. Qin, H. Kang, L. Chen, N. Zhang, X. Liu and G. Chen, High-concentration additive and triiodide/iodide redox couple stabilize lithium metal anode and rejuvenate the inactive lithium in carbonate-based electrolyte, Adv. Funct. Mater., 2022, 32, 2204768 CrossRef CAS.
- J. Ko and Y. S. Yoon, Recent progress in LiF materials for safe lithium metal anode of rechargeable batteries: is LiF the key to commercializing Li metal batteries?, Ceram. Int., 2019, 45, 30–49 CrossRef CAS.
- M. Arakawa, S.-i. Tobishima, Y. Nemoto, M. Ichimura and J.-i. Yamaki, Lithium electrode cycleability and morphology dependence on current density, J. Power Sources, 1993, 43, 27–35 CrossRef CAS.
- N. A. Sahalie, A. A. Assegie, W.-N. Su, Z. T. Wondimkun, B. A. Jote, B. Thirumalraj, C.-J. Huang, Y.-W. Yang and B.-J. Hwang, Effect of bifunctional additive potassium nitrate on performance of anode free lithium metal battery in carbonate electrolyte, J. Power Sources, 2019, 437, 226912 CrossRef CAS.
- Q. Zhang, L. Xu, X. Yue, J. Liu, X. Wang, X. He, Z. Shi, S. Niu, W. Gao, C. Cheng and Z. Liang, Catalytic current collector design to accelerate LiNO3 decomposition for high-performing lithium metal batteries, Adv. Energy Mater., 2023, 13, 2302620 CrossRef CAS.
- B. Han, Z. Zhang, Y. Zou, K. Xu, G. Xu, H. Wang, H. Meng, Y. Deng, J. Li and M. Gu, Poor stability of Li2CO3 in the solid electrolyte interphase of a lithium-metal anode revealed by cryo-electron microscopy, Adv. Mater., 2021, 33, 2100404 CrossRef CAS PubMed.
- T. D. Pham, A. Bin Faheem, J. Kim, H. M. Oh and K.-K. Lee, Practical high-voltage lithium metal batteries enabled by tuning the solvation structure in weakly solvating electrolyte, Small, 2022, 18, 2107492 CrossRef CAS PubMed.
- C. Tian, K. Qin and L. Suo, Concentrated electrolytes for rechargeable lithium metal batteries, Mater. Futures, 2023, 2, 012101 CrossRef CAS.
- S. Lee, L. Su, A. Mesnier, Z. Cui and A. Manthiram, Cracking vs. surface reactivity in high-nickel cathodes for lithium-ion batteries, Joule, 2023, 7, 2430–2444 CrossRef CAS.
- Z. Wang, X. Che, D. Wang, Y. Wang, X. He, Y. Zhu and B. Zhang, Non-Fluorinated Ethers to Mitigate Electrode Surface Reactivity in High-Voltage NCM811-Li Batteries, Angew. Chem., 2024, 136, e202404109 CrossRef.
- Z. Wang, C. Chen, D. Wang, Y. Zhu and B. Zhang, Stabilizing interfaces in high-temperature NCM811-Li batteries via tuning terminal alkyl chains of ether solvents, Angew. Chem., 2023, 135, e202303950 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02399h |
| ‡ Y. Zhao and S. Li contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |