
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA weak acid-responsive porous polyelectrolyte membrane enables the high efficiency of a zinc anode interface†
Wenbin
Li
a,
Changhao
Wang
bc,
Wenxuan
Hu
a,
Yongkang
Wang
a,
Congcong
Li
a,
Xiao
Liu
a,
Linyan
Su
a,
Beibei
Yang
*a,
Yunsong
Li
*b,
Duan
Bin
 *a and
Hongbin
Lu
*a
*a and
Hongbin
Lu
*a
aCollege of Chemistry and Chemical Engineering, Nantong University, Nantong, 226019, China. E-mail: yangbeibei@ntu.edu.cn; luhb@ntu.edu.cn; dbin17@fudan.edu.cn
bZhejiang Laboratory, Hangzhou 311100, P. R. China. E-mail: ysli@zhejianglab.com
cSchool of Materials Science and Engineering, Northwestern Polytechnical University, Xi'an 710072, P. R. China
First published on 25th June 2025
Abstract
Polymer coatings are promising candidates as artificial interfacial materials due to their significant barrier properties and zincophilicity, whereas modified zinc anodes for high depth of discharge (DOD) remain a major challenge. Herein, we present a novel approach that combines electrostatic complexation with phase separation in a weak-acid solution, which induces the formation of acid-responsive porous polyelectrolyte membranes, leading to an extended lifespan of the zinc anode of up to 900 h at a high current density of 40 mA cm−2. Additionally, the Zn@PEI-PSSNa anode demonstrates excellent zinc utilization, operating for up to 200 h at an ultra-high DOD of 97.3%. The Zn@PEI-PSSNa‖Cu cell further exhibits long-term cycling stability with a super-high coulombic efficiency (CE) of 99.8% for nearly 4000 h. Experimental analysis and simulations indicate that the sulfonate and amino functional groups, along with the porous structure, could improve zincophilicity and facilitate the transport of Zn2+, thereby promoting uniform zinc deposition, accelerating the desolvation process and mitigating side reactions.
Introduction
With the rapid development of renewable energy such as solar energy and wind energy, the development of high-performance, safe and reliable energy storage devices is crucial to the stability and development of the modern energy system.1,2 Zinc-based aqueous batteries (ZABs) are very safe and environmentally friendly, and have excellent theoretical specific capacity (820 mAh g−1 or 5855 mAh cm−3) and low reoxidation potential3,4 (−0.76 V vs. standard hydrogen electrode), becoming a very promising energy storage system.5,6 However, major issues such as internal corrosion, hydrogen precipitation, and zinc dendrite formation frequently arise during the zinc plating/stripping process, significantly impeding the industrialization of ZABs.7To address the formidable challenges faced by ZABs, researchers have conducted extensive investigations in several directions, including the optimization of anode materials,8–10 the modification of electrolyte compositions,11,12 and the construction of artificial solid electrolyte interphases (SEI).13,14 Among these efforts, the development of artificial SEI has emerged as a focal point due to its precise regulation of zinc deposition behavior. At present, SEI construction techniques are mainly classified into three types including inorganic SEI,15 organic SEI16 and organic–inorganic composite SEI.17,18 Extensive studies have shown that these technologies effectively suppress dendrite growth, block side reactions and significantly improve battery performance.19 In particular, organic polymer-based artificial SEI have gained prominence due to their unique advantages, such as the designability of molecular chain segment functionalities and flexible network structures.20,21 Notably, in the design of segmental functional groups along the molecular chain, sulfonate (–SO3−) and amino (–NH2) groups have been demonstrated to exert positive effects on zinc anodes.22–24 Due to its strong hydrophilicity, the –SO3− group can effectively reduce the activity of water, thereby inhibiting the corrosion of the zinc anode and the side reaction of hydrogen evolution. Additionally, it enhances zinc ion transport efficiency due to its exceptional ionic conductivity.25 Conversely, the –NH2 group, with its reactive nature, forms a dense protective layer on the zinc anode surface, promoting uniform zinc ion deposition and effectively suppressing dendrite growth,26 thereby comprehensively enhancing the overall performance of the zinc anode. For instance, Xu et al. successfully achieved precise control over zinc ion deposition behaviour by constructing a SEI derived from amino sulfonate salts.27 This approach effectively suppressed undesirable side reactions and dendrite growth. Notwithstanding these advances, there are several significant issues that remain unresolved. Firstly, the compatibility between polymers and electrode interfaces is suboptimal, resulting in inadequate formation of tight and stable connections.28,29 This has a detrimental effect on both the efficiency of ion transport and the overall performance of the battery. Additionally, the structural characteristics of polymers have the potential to interfere with the normal insertion and extraction pathways of zinc ions, thereby affecting battery reaction kinetics and leading to reduced charge–discharge efficiency.30 Nevertheless, the development of a stable artificial polymer interface on zinc anodes remains a significant challenge, especially under high depth of discharge (DOD) and high plating current density conditions.
Building on the phenomenon that fluid protein particles form a complex, diffuse to create a protective film, and migrate towards the skin surface during skin formation, a homogeneous mixture of two polyelectrolytes based on polyethyleneimine (PEI) and sodium polystyrene sulfonate (PSSNa) was fabricated by dissolving them in water. On one hand, PEI is a cationic polymer with abundant amine groups, offering strong adhesion to various substrates and the ability to form compact and conformal layers.31 On the other hand, PSSNa is an anionic polyelectrolyte that can provide electrostatic interactions with PEI, and the presence of PSSNa endows the mixture solution with surface tension lower than that of water, which is the driving force for porous membranes.32 In the acidic environment, it was innovatively proposed to initiate the phase separation phenomenon by protonating the amine groups in the PEI-PSSNa solution from NH2 to –NH3+,33,34 which later combine with –SO3−, leading to the electrostatic complexation of PEI and PSSNa. Ultimately, a porous structure with a dynamic response mechanism was developed in ZABs. This structure promotes the uniform adsorption and deposition of zinc ions on the anode surface. By weakening the tip effect to inhibit dendrite growth, this structure can improve the cycling stability of the zinc anode. Meanwhile, the interconnected channel framework establishes directional transport pathways for zinc ions, enabling the battery to maintain excellent performance under high-current conditions. Furthermore, the porous structure effectively accommodates volumetric fluctuations of the zinc anode during electrochemical plating–stripping cycles, alleviating mechanical stress to preclude electrode disintegration and delamination. The stability of the symmetric cell is evident in its operation for over 900 hours at a current density of 40 mA cm−2 and an areal capacity of 2 mAh cm−2, accompanied by an ultra-high DOD of 97.3% at 6 mAh cm−2. In Zn‖Cu cell tests, it was found that the cell life was approximately 4000 hours, achieving a high CE of up to 99.83%.
Results and discussion
As demonstrated in Fig. 1a, upon the amalgamation of PEI and PSSNa in acidic aqueous media, the –NH2 of PEI underwent a transformation to a protonated –NH3+. This subsequently engaged in an electrostatic complexation phenomenon with the –SO3− present within PSSNa. The –NH2 was corroborated by the fact that the FT-IR characteristic peak corresponding to the protonated –NH3+ located at about 1600 cm−1, and the sulphonate group in PSSNa was observed at 1039 cm−1, as shown in Fig. S1†.32 The yield process of the porous membrane on the zinc surface was measured under different pH conditions, as displayed in Fig. 1b. Initially, the PEI-PSSNa solution was deposited onto the surface of the zinc foil. As shown in the Scanning Electron Microscope (SEM) image (Fig. 1d), when only using a PEI-PSSNa coating (deionized water), it showed a fragmented and non-uniform film-like structure and no distinctive features were observed on the zinc surface. Similarly, an alkaline KOH solution (pH ∼ 10) was applied to the zinc foil pre-coated with the PEI-PSSNa solution; it also presented a similar phenomenon with no distinctive features (Fig. 1c). Interestingly, the treatment of the PEI-PSSNa-coated zinc foil with 2 M ZnSO4 tended to form a uniformly porous surface structure (Fig. 1e), since PEI is a weak polyelectrolyte undergoing positive charge under weak acid conditions. To investigate the unique dynamic response mechanism of PEI-PSSNa coatings in an acidic environment, 2 M ZnSO4 electrolyte was gradually added dropwise to the PSSNa coatings during the process, which manifested the absence of a porous structure as displayed in Fig. S2.† It was concluded that the acidic medium is a necessary condition for forming an acid-responsive mechanism. As shown in Fig. S3,† the optical images obtained after inverting the PEI-PSSNa solution and the PSSNa solution clearly demonstrated the similarities and differences between the two sets of solutions. Evidently, while both solutions exhibited clarity and transparency, the PEI-PSSNa solution, in marked contrast to the PSSNa solution, not only displayed a faint yellowish tint but also demonstrated a significantly higher viscosity. In the design of the PEI-PSSNa coating, PEI is a weak polyelectrolyte bearing positive charge under acid conditions, and the PSSNa endows the mixture solution with surface tension lower than that of water as the driving force for surface spreading. As illustrated in Fig. 1f, when the PEI and PSSNa come into contact, amine protonation occurred and the pore structures of PEI-PSSNa were consistent with a membrane formed by weak-acid solution induced phase separation. The formation of these pores significantly increased the interfacial contact area between the electrode and the electrolyte, thereby enhancing the electrochemical reaction efficiency. Moreover, this porous structure effectively mitigated the current density on the electrode surface while simultaneously providing an efficient transport pathway for Zn2+ ions.35,36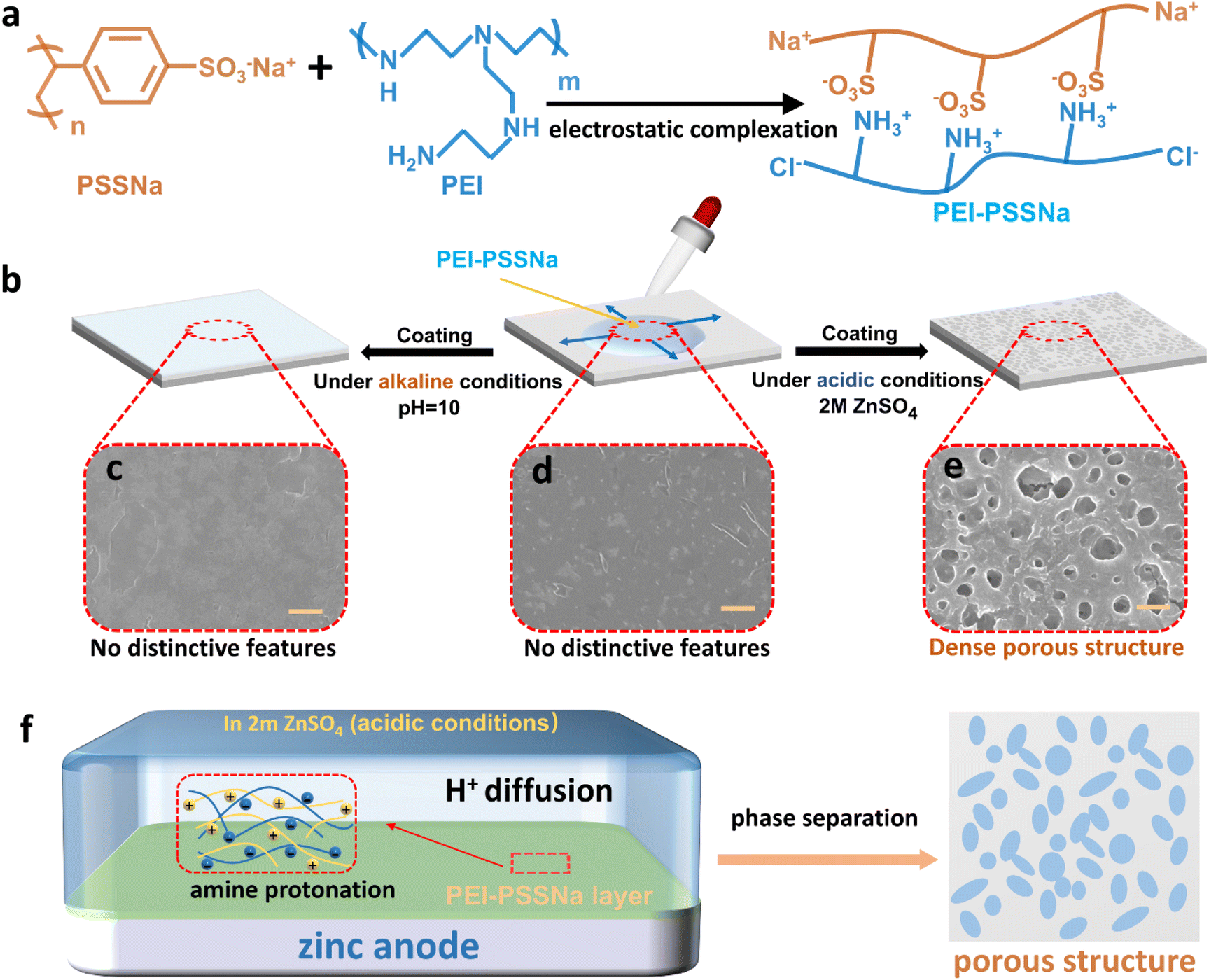 | ||
| Fig. 1 (a) Principle of PEI-PSSNa complexation by mixing PEI and PSSNa through electrostatic complexation. (b) Schematic representation of PEI-PSSNa coatings placed in a single thin film at different pH values. SEM images of PEI-PSSNa coatings under (c) alkaline, (d) neutral and (e) acidic conditions. (f) Formation mechanism of the PEI-PSSNa porous membrane under acidic conditions. | ||
To evaluate the impact of the PEI-PSSNa coating on the reversibility of Zn2+ plating/stripping, a series of galvanostatic charge–discharge cycling tests were conducted on symmetric cells. Notably in Fig. 2a, the Zn@PEI-PSSNa symmetric cell reached more than 900 h at a very high current density of 40 mA cm−2 and 2 mAh cm−2, which represented a significant performance improvement as listed in Table S1.† It was observed that while the Zn@PSSNa coating could effectively extend the life of the symmetric cell, the polarization increased significantly as the current rose. Owing to the larger ion size and lower potential of Na+ compared to Zn2+, the interface layer gradually evolves into an electrically responsive shield layer containing anion –SO3− and cation Na+, thereby suppressing the growth of zinc dendrites in the single PSSNa coating. It was also found that the symmetric cell assembled with bare Zn exhibited a short circuit at 130 h under operating conditions of 0.5 mA cm−2 and 0.5 mAh cm−2. In contrast, the PEI-PSSNa-modified symmetric cell achieved a prolonged cycle life exceeding 2500 h while maintaining exceptional cycling stability, as evidenced in Fig. 2b. As demonstrated in Fig. S4,† the bare Zn symmetric and the Zn@PSSNa symmetric cell exhibited a short circuit at the 85th and 340th cycles at a large current density of 5 mA cm−2 under a fixed capacity of 2.5 mAh cm−2. In contrast, the Zn@PEI-PSSNa symmetric cell demonstrated notable operational stability with a more durable cycle life up to 1200 h than its counterparts. As revealed in Fig. 2c, the multiplicative performance of the cell exhibited variation at distinct current levels when the real capacity was fixed at 2 mAh cm−2. In the multiplicity tests, the voltage polarization of the Zn@PEI-PSSNa symmetric cell was compared with that of the bare Zn. The results demonstrated that the polarization of Zn@PEI-PSSNa symmetric cells was significantly smaller than that of the bare Zn anode (Fig. S5†).
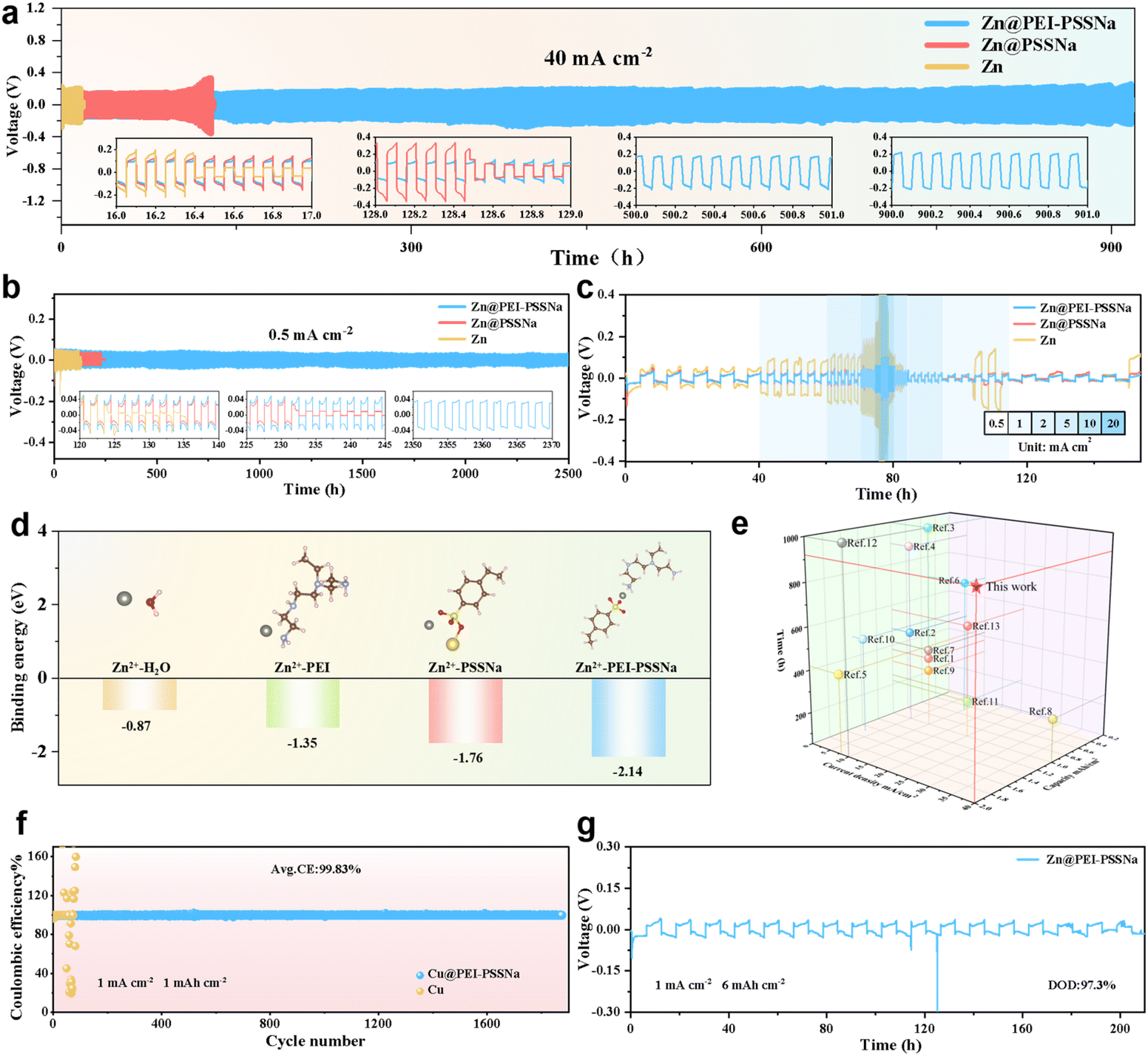 | ||
| Fig. 2 (a) Galvanostatic cycling performance at 40 mA cm−2 and 2 mAh cm−2. (b) Galvanostatic cycling performance at 0.5 mA cm−2 and 0.5 mAh cm−2. (c) Rate performance of Zn@PEI-PSSNa, Zn@PSSNa and Zn symmetrical cells. (d) Binding energy of samples. (e) Comparison of the cycle life and capacity of the zinc symmetric cell modified by the PEI-PSSNa coating under ultra-high current density with those of the zinc anodes optimized by previously reported strategies. (f) CE plots of Zn@PEI-PSSNa‖Cu and Zn‖Cu cells with an areal capacity of 1 mAh cm−2. (g) Test performance under a high DOD. | ||
Density functional theory (DFT) calculations revealed the Zn2+ binding energetics across interfaces (Fig. 2d). Due to the higher strong electronegativity of the SO3− group, the PSSNa (−1.76 eV) showed lower binding energy to Zn2+ as compared to PEI (−1.35 eV). Moreover, the PEI-PSSNa composite exhibited optimal Zn2+ affinity (−2.14 eV), attributed to synergistic interactions between protonated –NH3+ and –SO3− groups forming an ion-trapping network, surpassing individual polymers.37–39 Notably, Fig. 2e showed that the PEI-PSSNa-modified symmetric cell had remarkable competitiveness in cycle longevity and areal capacity under ultrahigh current density conditions. Remarkably, the Zn‖Cu cell assembled with Cu@PEI-PSSNa operated stably for nearly 2000 cycles (4000 h) at 1 mA cm−2 and 1 mAh cm−2, achieving an ultra-high CE of 99.83% (Fig. 2f). The corresponding voltage profiles of the selected Zn‖Cu cells, as depicted in Fig. S6,† revealed that during the initial cycling, the voltage hysteresis of the cells assembled with Cu@PEI-PSSNa is similar to that of the control samples. However, the control sample loses its operational capacity after 40 cycles, while the Cu@PEI-PSSNa‖Zn cell demonstrates significantly smaller voltage hysteresis and sustains long-term stability. Compared to cells with previously reported anode modification strategies (Fig. S7†), the PEI-PSSNa-modified cell exhibited substantial advantages in both CE and lifespan (Table S2†). This finding suggested that the presence of the coating led to a substantial enhancement in the reversibility of the electrode reaction.
To further explore the DOD of the Zn@PEI-PSSNa symmetric cell, constant-current charge/discharge tests were conducted on it under operating conditions of 6 mAh cm−2 (Fig. 2g). The optical images showed that the Zn@PEI-PSSNa electrode was almost completely consumed, but the bare Zn had a large amount of residue (Fig. S8†). Calculations indicated that the Zn@PEI-PSSNa symmetric cell was capable of operating for over 200 h at an ultra-high DOD of 97.3%, which was comparable to the previously reported strategies (Table S3 and Fig. S9†). The PEI-PSSNa-optimized zinc symmetric cells demonstrated superior long-term stability under ultrahigh depth-of-discharge conditions, attaining an exceptional DOD level. This improvement can be explained by the fact that the protonated –NH2 on the polymer molecular chain weakens the binding energy between Zn2+ and water molecules through coordination. This action accelerated the desolvation process and reduced the ionic mobility barriers, leading to more uniform and stable zinc deposition.40 To investigate the influence of sulfonate (–SO3−) groups from PSSNa and protonated amines (–NH3+) from PEI on the solvation environment of Zn2+, molecular dynamics (MD) simulations were conducted for the electrolyte/zinc anode interface with and without the PEI-PSSNa coating. As exhibited in Fig. S10,† the radial distribution function (RDF) results showed that the Zn–O(H2O) coordination peak remains sharply defined at 2.1 Å in the additive-containing system, indicating a well-structured hydration shell around Zn2+. Furthermore, the Zn2+ coordination number decreased from 5.75 to 5.11 upon the introduction of PEI-PSSNa, suggesting that –SO3− groups within the polymer can partially replace water molecules in the first solvation shell, thereby modulating the local solvation structure of Zn2+. It was also manifested that, the –SO3− of PSSNa also preferentially adsorbed Zn2+ and excluded competing ions, such as SO42−, via the electrostatic repulsion effect (Donnan effect), resulting in a uniform distribution of the electric field.41,42
SEM characterization revealed distinct electrode morphological evolution after 50 cycles (Fig. 3a and d). The PEI-PSSNa-modified electrode maintained a porous architecture enabling uniform Zn2+ deposition without dendrite formation, whereas bare Zn exhibited severe dendritic growth. After 30 days of immersion in a 2 M ZnSO4 electrolyte, the coated electrode maintained its structural integrity (Fig. 3b and e), in contrast to the bare Zn electrode, which exhibited accelerated surface degradation. In the case of prolonged immersion, scores of crystals adhered to the surfaces of the two electrodes, and the surface of the Zn@PEI-PSSNa electrodes was noticeably flatter, whereas the surface of the bare Zn electrodes had noticeably protruding crystals. In the Zn‖Cu system, the porous structure on the surface of the Cu@PEI-PSSNa electrode offered superior protection after 50 cycles (Fig. 3c), thereby preventing the occurrence of extensive dendrite growth, as was the case on the surface of bare copper (Fig. 3f). As shown in Fig. 3g, the electrodes of the symmetric cell after undergoing 50 cycles were analyzed using X-ray diffraction (XRD). On the Zn@PEI-PSSNa electrode, the characteristic peaks of Zn4SO4(OH)6·5H2O or other by-products were significantly weaker than those on the bare Zn electrode. In contrast, the PEI-PSSNa coating provided significant protection for the Zn electrode. As presented in Fig. 3h, it could be seen that there were only a few peaks on the Zn@PEI-PSSNa electrode, with a maximum value of merely 2.2 μm. However, the surface of the bare Zn electrode showed pronounced bulges and undulations (Fig. 3i), while the Zn@PEI-PSSNa electrode was substantially flatter. After undergoing a repeated zinc plating/striping process, the porous structure is still presented in the zinc surface, and the C, N, Na, O, S and Zn compositions were also indicated from the element analysis of the cycled Zn@PEI-PSSNa anode, indicating the good stability of the PEI-PSSNa coating (Fig. S11†). In addition, a comparison of the mechanical properties of bare Zn and PEI-PSSNa coated zinc anode is displayed in Fig.S12.† Under identical loading conditions, the indentation depth of PEI-PSSNa@Zn is significantly smaller than that of bare Zn, indicated that the PEI-PSSNa coating could withstand a stronger external force. As indicated by the recovery of deformation, the PEI-PSSNa@Zn displayed the best recovery with its deformation recovering closest to the pristine state, while the bare Zn showed almost no resilience. Based on the linear scanning voltammetry (LSV) results, the Zn@PEI-PSSNa electrode exhibited a higher hydrogen evolution overpotential (1.228 V) compared to the bare Zn electrode (1.154 V) at a current density of 50 mA cm−2, as shown in Fig. 3j. This result could be attributed to the fact that the robust coordination between the –SO3− and Zn2+ led to a partial substitution of the solvated shell layer of Zn2+.43,44 Consequently, this substitution brought about a reduction in the interfacial free water activity and simultaneously attenuated the hydrogen evolution process that was instigated by water decomposition. Zn@PEI-PSSNa exhibited an onset potential of 39 mV which was lower than that of the bare Zn (61 mV), indicating enhanced zinc nucleation with grain refinement under the lower overpotential conditions (Fig. 3k). In addition, the corrosion resistance of zinc metal was evaluated using Tafel plots as presented in Fig. 3l. The introduction of the PEI-PSSNa coating on the zinc anode surface resulted in a gradual decrease in corrosion current from 1.48 to 1.25 mA cm−2, indicating the enhanced corrosion resistance of the Zn@PEI-PSSNa electrode. Fig. 3m, n, and S13† showed the Nyquist plots corresponding to Zn@PEI-PSSNa, bare Zn, and Zn@PSSNa, respectively. Calculated in accordance with the Arrhenius equation, the corresponding activation energies (Ea) are presented in Fig. 3o and S14,† which clearly revealed that the activation energy of Zn@PEI-PSSNa (49.4 kJ mol−1) was considerably lower than those of Zn@PSSNa (56.7 kJ mol−1) and bare Zn (66.1 kJ mol−1). Noticeably, when compared to bare Zn, the Zn2+ transference number of the Zn@PEI-PSSNa electrode significantly increased from 0.39 to 0.53 (Fig. S15†), demonstrating that the PEI-PSSNa protective layer can promote the diffusion of Zn2+ to the anode surface. The reduction in activation energy and Zn2+ transference number indicated that the PEI-PSSNa porous polyelectrolyte membrane exerted an accelerating effect on the de-solvation and transport of Zn2+. To further investigate the nucleation behavior of bare Zn and Zn@PEI-PSSNa, chronoamperometry (CA) was conducted on the electrodes. The results showed that Zn@PEI-PSSNa exhibited a long-term 3D diffusion process and low current density, which corroborated that the PEI-PSSNa coating could promote the uniform deposition of ions (Fig. S16†).
 | ||
| Fig. 3 SEM images of (a) Zn@PEI-PSSNa and (d) bare Zn after 50 cycles in a symmetric cell at 1 mA cm−2 and 1 mAh cm−2. SEM images of (b) Zn@PEI-PSSNa and (e) bare Zn after 30 days of immersion in 2 M ZnSO4. Scanning electron microscopy images of (c) Zn@PEI-PSSNa and (f) bare Zn after 50 cycles in a Zn‖Cu cell at a current density of 1 mA cm−2 and 1 mAh cm−2. (g) Comparison of XRD images after 50 cycles at 1 mA cm−2 and 1 mAh cm−2. HRCM images of (h) Zn@PEI-PSSNa and (i) bare Zn after 50 cycles at 1 mA cm−2 and 1 mAh cm−2. (j) LSV testing of materials. (k) CV curve of the Zn‖Cu cell. (l) Tafel curves for samples. Nyquist plots of (m) Zn@PEI-PSSNa and (n) bare Zn at different temperatures. (o) Arrhenius curve corresponding to the sample. | ||
To gain insights into the deposition and stripping behaviors of Zn2+ on the electrode, we have conducted a comprehensive series of characterization experiments. The EDS mapping in Fig. 4a showed that all elements were evenly distributed in the Zn@PEI-PSSNa layer, and the cross-section of the PEI-PSSNa coating revealed a thickness of about 4.5 μm (Fig. 4b). As shown in Fig. 4c and d, the contact angles of the 2 M ZnSO4 electrolyte on Zn@PEI-PSSNa and bare Zn were 60.72° and 88.57°, respectively. This strongly suggested that the surface of Zn@PEI-PSSNa remarkably exhibited superior zincophilicity in virtue of enhanced Zn2+ affinity, thereby facilitating the uniform diffusion of Zn2+.45 Compared to bare Zn, the Zn@PEI-PSSNa interface presented a significantly lower interfacial resistance (Fig. 4e and S17†). Computational analyses in Fig. 4f also confirmed that PEI-PSSNa coatings had better ionic conductivity than PSSNa coatings. This combined improvement gave PEI-PSSNa-modified electrodes faster ion transport kinetics. To understand the coating composition, X-ray photoelectron spectroscopy (XPS) was used to analyze Zn@PEI-PSSNa electrodes after 50 cycles. High-resolution XPS spectra of the 50-cycle Zn@PEI-PSSNa electrode detected many C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, and CO bonds, revealing the internal structure of the PEI-PSSNa coating. In the N 1s orbitals, numerous Zn–N bonds were found to verify the good Zn2+ affinity (Fig. S18†). To track the changes of –SO3− groups and –NH3+ groups, we have measured the ex situ Raman spectra of the zinc plating/stripping process at different times, and the results are shown in Fig. S19.† As observed, the Raman signals at ∼638 cm−1 and 1605 cm−1 correspond to –SO3− and –NH3+ groups.46,47 The intensity of the two groups gradually increases during Zn dissolution on the Zn@PEI-PSSNa electrode, and then decreases with the Zn2+ deposition process. These results only indicate that the –NH3+ group may bind to the SO42− anions of electrolyte occurs the zinc plating/stripping process.
C, and CO bonds, revealing the internal structure of the PEI-PSSNa coating. In the N 1s orbitals, numerous Zn–N bonds were found to verify the good Zn2+ affinity (Fig. S18†). To track the changes of –SO3− groups and –NH3+ groups, we have measured the ex situ Raman spectra of the zinc plating/stripping process at different times, and the results are shown in Fig. S19.† As observed, the Raman signals at ∼638 cm−1 and 1605 cm−1 correspond to –SO3− and –NH3+ groups.46,47 The intensity of the two groups gradually increases during Zn dissolution on the Zn@PEI-PSSNa electrode, and then decreases with the Zn2+ deposition process. These results only indicate that the –NH3+ group may bind to the SO42− anions of electrolyte occurs the zinc plating/stripping process.
 | ||
| Fig. 4 (a) EDS mapping of coatings. (b) SEM image of the cross-section of the modified zinc anode. 2 M ZnSO4 electrolyte contact angle on (c) Zn@PEI-PSSNa and (d) bare Zn. (e) Nyquist plots of samples at room temperature. (f) Ionic conductivity of the sample. (g) In situ optical image of the sample at a current density of 10 mA cm−2. (h) Simulation of the Zn concentration field at different electrodes. (i) Simulation of the electric field on different electrodes. | ||
In situ optical microscopy observations under a current density of 10 mA cm−2 revealed stark contrasts in Zn deposition dynamics. Specifically, the bare Zn surface exhibited rampant growth of irregular zinc dendrites (Fig. 4g), whereas the PEI-PSSNa-modified electrode maintained a dendrite-free surface morphology, showing localized dendritic features only at unencapsulated cross-sectional regions. This suppression of dendrite proliferation originated from the protonation of –NH2 groups within the polyelectrolyte film under acidic conditions, generating a positively charged layer that synergized with the negatively charged PSSNa domains to establish an interfacial potential gradient. This gradient acted as a directional guide for Zn2+ ions, enforcing their uniform migration along the coating surface and effectively mitigating localized ion accumulation and subsequent dendritic growth.48–51 Furthermore, the electric and Zn2+ concentration field distributions at various electrodes were simulated using COMSOL Multiphysics. For bare Zn electrodes, the current density and Zn2+ concentration were highly localized at the surface tips, promoting the rapid formation and growth of Zn dendrites due to the tip effect (Fig. 4h and i). The PEI-PSSNa coating ensured the uniformity of the electric field strength around the zinc anode electrode, thereby facilitating a more uniform distribution of zinc ions within the electrolyte. Through software simulation, it was corroborated that a stable chemical structure, similar to the one presented in Fig. S20,† was formed on the electrode surface in the presence of the PSSNa coating and the PEI-PSSNa coating. As shown in Fig. S21,† the diffusion activation energy of Zn2+ on PEI-PSSNa was lower than that of PSSNa, demonstrating that the PEI-PSSNa coating could accelerate the Zn2+ interfacial dynamics.
To demonstrate the practicality of the PEI-PSSNa coating, Zn@PEI-PSSNa was assembled with the PTO cathode (Fig. S22†) to form a full battery, and a series of tests were conducted. As illustrated in Fig. 5a, the long cycle performance of the Zn@PEI-PSSNa‖PTO battery was compared with that of the bare Zn‖PTO battery at 5 A g−1. During the cycling process, the Zn@PEI-PSSNa‖PTO battery exhibited higher initial capacity and more stable CE. At the 600th cycle, the bare Zn‖PTO battery could not work normally due to the irreversible dendrite growth, while the Zn@PEI-PSSNa‖PTO still achieved a capacity retention of 71.2% up to 1300 cycles. It can be seen in Fig. 5b that the capacity retention of the Zn@PEI-PSSNa‖PTO battery was much higher than that of the bare Zn‖PTO battery when the current density was increased from 0.1 to 20 A g−1, which demonstrated the unique role of the PEI-PSSNa coating in improving the performance of the full battery. The CV curves showed that the Zn@PEI-PSSNa‖PTO battery possessed smaller overpotential gaps and higher peak current densities, which suggested that the Zn@PEI-PSSNa electrode possessed better redox kinetics (Fig. 5c).52,53Fig. 5d and e show the charging and discharging curves of the Zn@PEI-PSSNa‖PTO battery and bare Zn‖PTO battery at the specified number of cycles, respectively. In comparison, the Zn@PEI-PSSNa‖PTO battery shows better capacity retention and long-term cycling stability. Mechanistically, the negative –SO3− groups in PSSNa showed sufficiently high electronegativity to combine the Zn anode and PTO cathode, forming a “Zn–SO3−–PTO” sandwich architecture that lowered Zn2+ diffusion reaction energy, thereby accelerating Zn2+ diffusion kinetics. In addition, the amino protonated porous membrane based on –SO3− and –NH3+ groups played a vital role in inducing Zn2+ unform deposition and inhibiting dendrite growth and side reactions, thereby rationalizing the cycling performance leap. To further demonstrate the practicality of the PEI-PSSNa protective layer, a well-packaged pouch battery could sustain a stable open circuit voltage of 1.36 V and maintain a light bulb by assembling two batteries in series (Fig. 5f). The performance of the well-packaged pouch full AZBs composed of a limited zinc anode is also shown in Fig. 5g. When the N/P ratio was 3.6, the Zn/PTO battery with bare Zn showed stable capacity and cycling in the first 25 cycles at 0.5 A g−1, and then the capacity dropped sharply to almost zerocapacity at 60 cycles, probably due to the depletion of electrolyte. In sharp contrast, the Zn/PTO battery using PEI-PSSNa exhibited a maximum specific capacity of 251.2 mAh g−1 and retained 71.7% of the initial capacity after 150 cycles, revealing that the PEI-PSSNa protective layer can promote more stable zinc plating/stripping even with a limited zinc anode.
 | ||
| Fig. 5 (a) Comparative cycling performance of full batteries at a current density of 5 A g−1. (b) Multiplier performance test for full batteries. (c) CV curves for the full batteries at 1 mV s−1 sweep speed. Charge–discharge curves of the full batteries using (d) Zn@PEI-PSSNa and (e) bare Zn anode. (f) Test photo of the small pouch-cell and (g) cycling performance of the pouch cell at a high mass loading of the cathode material. | ||
Conclusions
In summary, we successfully demonstrated a novel technique for constructing a porous polyelectrolyte coating with a dynamically responsive mechanism on zinc anodes. In the electrostatic network of the PEI-PSSNa coating, the –SO3− groups aggressively penetrated the solvation shell of Zn2+, displacing reactive H2O molecules, while the –NH3+ moieties immobilized SO42− anions to accelerate the desolvation process. The modified cell not only could work stably for nearly 1000 h at 40 mA cm−2 and 2 mAh cm−2, but also operated for more than 200 h at an ultra-high DOD of 97.3%, which is far superior to most of the recently reported strategies for artificial interfacial layer construction. Furthermore, the Zn@PEI-PSSNa‖PTO battery demonstrated outstanding capacity retention and cycling stability at a current density of 5 A g−1. It was believed that this novel strategy could break through the limitations of the traditional polymers in zinc anode protection applications, and effectively broaden the scope of polyelectrolyte applications.Data availability
The data can be provided upon request. The data supporting this article have been included as part of the ESI.†Author contributions
Wenbin Li conducted the experiments and wrote the original draft; Changhao Wang and Yunsong Li performed the theoretical calculations; Wenxuan Hu, Yongkang Wang, Congcong Li, Xiao Liu, and Linyan Su carried out the formal analysis; Duan Bin and Beibei Yang were responsible for the conceptualization, as well as reviewing and editing the article, and securing the research funding; Hongbin Lu: assumed the supervisory work, and reviewed and edited the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the National Natural Science Foundation of China (22309091 and 22404085). This work was also funded by the Key Research Project of Zhejiang Lab (No. 3700-3AA240100).Notes and references
- K. Schulze, F. Kullmann, J. M. Weinand and D. Stolten, Joule, 2024, 8, 1936–1957 Search PubMed.
- X. Liu, W. Zhu, P. Deng and T. Li, ACS Nano, 2023, 17, 18657–18668 Search PubMed.
- X. Chen, W. Li, D. Reed, X. Li and X. Liu, Electrochem. Energy Rev., 2023, 6, 33 Search PubMed.
- J. Wei, P. Zhang, J. Sun, Y. Liu, F. Li, H. Xu, R. Ye, Z. Tie, L. Sun and Z. Jin, Chem. Soc. Rev., 2024, 53, 10335–10369 Search PubMed.
- W. Guo, T. Hua, C. Qiao, Y. Zou, Y. Wang and J. Sun, Energy Storage Mater., 2024, 66, 103244 Search PubMed.
- Z. Li, J. Tan, Y. Wang, C. Gao, Y. Wang, M. Ye and J. Shen, Energy Environ. Sci., 2023, 16, 2398–2431 Search PubMed.
- F. Liu, Y. Zhang, H. Liu, S. Zhang, J. Yang, Z. Li, Y. Huang and Y. Ren, ACS Nano, 2024, 18, 16063–16090 Search PubMed.
- Y. Zhang, X. Zheng, N. Wang, W.-H. Lai, Y. Liu, S.-L. Chou, H.-K. Liu, S.-X. Dou and Y.-X. Wang, Chem. Sci., 2022, 13, 14246–14263 Search PubMed.
- X. Cao, W. Xu, D. Zheng, F. Wang, Y. Wang, X. Shi and X. Lu, Angew. Chem., Int. Ed., 2024, 63, e202317302 Search PubMed.
- K. Qi, P. Liang, S. Wei, H. Ao, X. Ding, S. Chen, Z. Fan, C. Wang, L. Song, X. Wu, C. Wu and Y. Zhu, Energy Environ. Sci., 2024, 17, 2566–2575 Search PubMed.
- F. Chen, Y. Gao, Q. Hao, X. Chen, X. Sun and N. Li, ACS Nano, 2024, 18, 6413–6423 Search PubMed.
- M. Zhang, H. Wei, Y. Zhou, W. Wen, L. Zhang and X.-Y. Yu, Chem. Sci., 2024, 15, 18187–18195 Search PubMed.
- Z. Peng, H. Yan, Q. Zhang, S. Liu, S. C. Jun, S. Poznyak, N. Guo, Y. Li, H. Tian, L. Dai, L. Wang and Z. He, Nano Lett., 2024, 24, 9137–9146 Search PubMed.
- H. Yan, C. Han, S. Li, J. Liu, J. Ren, S. Yang and B. Li, Chem. Eng. J., 2022, 442, 136081 Search PubMed.
- L. Tao, X. Lu, K. Qu, Y. Zeng, M. B. Miller and J. Liu, Small, 2024, 20, 2311205 Search PubMed.
- M. Fayette, H. J. Chang, I. A. Rodrı́guez-Pérez, X. Li and D. Reed, ACS Appl. Mater. Interfaces, 2020, 12, 42763–42772 Search PubMed.
- J. Zhao, Y. Ying, G. Wang, K. Hu, Y. D. Yuan, H. Ye, Z. Liu, J. Y. Lee and D. Zhao, Energy Storage Mater., 2022, 48, 82–89 Search PubMed.
- G. Qu, H. Wei, S. Zhao, Y. Yang, X. Zhang, G. Chen, Z. Liu, H. Li and C. Han, Adv. Mater., 2024, 36, 2400370 Search PubMed.
- J. Yang, B. Yin, Y. Sun, H. Pan, W. Sun, B. Jia, S. Zhang and T. Ma, Nano-Micro Lett., 2022, 14, 42 Search PubMed.
- Z. Xu, M. Li, W. Sun, T. Tang, J. Lu and X. Wang, Adv. Mater., 2022, 34, 2200077 Search PubMed.
- X. Sun, X. Lv, M. Zhang, K. Shi, Z. Li, X. Pan, T. Lian, R. Chen, F. Wu and L. Li, ACS Nano, 2024, 18, 8452–8462 Search PubMed.
- K. Wang, Q. Li, G. Zhang, S. Li, T. Qiu, X.-X. Liu and X. Sun, Chem. Sci., 2024, 15, 230–237 Search PubMed.
- H. Ma, J. Yu, M. Chen, X. Han, J. Chen, B. Liu and S. Shi, Adv. Funct. Mater., 2023, 33, 2307384 Search PubMed.
- J. Zhang, Y. Wang, Z. Zhao, P. Li, G. Tang, W. Chen and Z. Peng, Adv. Energy Mater., 2024, 14, 2401560 Search PubMed.
- W. Xie, K. Zhu, W. Jiang, H. Yang, M. Ma, L. Zhao and W. Yang, ACS Nano, 2024, 18, 21184–21197 Search PubMed.
- C. Huang, J. Mao, S. Li, W. Zhang, X. Wang, Z. Shen, S. Zhang, J. Guo, Y. Xu, Y. Lu and J. Lu, Adv. Funct. Mater., 2024, 34, 2315855 Search PubMed.
- X. Xu, H. Su, J. Zhang, Y. Zhong, Y. Xu, Z. Qiu, H. B. Wu, X. Wang, C. Gu and J. Tu, ACS Energy Lett., 2022, 7, 4459–4468 Search PubMed.
- R. Qi, W. Tang, Y. Shi, K. Teng, Y. Deng, L. Zhang, J. Zhang and R. Liu, Adv. Funct. Mater., 2023, 33, 2306052 Search PubMed.
- B. Qiu, K. Liang, W. Huang, G. Zhang, C. He, P. Zhang and H. Mi, Adv. Energy Mater., 2023, 13, 2301193 Search PubMed.
- Y. Wang, H. Cui, R. Li, C. Yue, H. Pan, Z. Tang, X. Wang, Y. Lin, H. Li, C. Han, D. Nan, C. Zhi and H. Lv, Energy Storage Mater., 2024, 65, 103102 Search PubMed.
- J. Li, G. Van Ewijk, D. J. Van Dijken, J. Van Der Gucht and W. M. De Vos, ACS Appl. Mater. Interfaces, 2021, 13, 21844–21853 Search PubMed.
- S. Tang, J. Gong, Y. Shi, S. Wen and Q. Zhao, Nat. Commun., 2022, 13, 3227 Search PubMed.
- J. Schiebel, R. Gaspari, A. Sandner, K. Ngo, H. Gerber, A. Cavalli, A. Ostermann, A. Heine and G. Klebe, Angew. Chem., Int. Ed., 2017, 56, 4887–4890 Search PubMed.
- C. Zhao, J. Diao, Z. Liu, J. Hao, S. He, S. Li, X. Li, G. Li, Q. Fu, C. Jia and X. Guo, Nat. Commun., 2024, 15, 8835 Search PubMed.
- J. Liu, C. Ye, H. Wu, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2023, 145, 5384–5392 Search PubMed.
- S. Jiao, J. Fu, M. Wu, T. Hua and H. Hu, ACS Nano, 2022, 16, 1013–1024 Search PubMed.
- X. Li, W. Zhang, J. Yu, L. Wu, A. Zhang, J. Zhang, H. Wang, X. Liao and Y. Zhao, Adv. Funct. Mater., 2024, 34, 2316474 Search PubMed.
- X. Liang, R. Yang, Y. Zheng, F. Zhang, W. Zhang, C. Lee and Y. Tang, Adv. Funct. Mater., 2024, 2403048 Search PubMed.
- Y. Wang, T. Wang, Y. Mao, Z. Li, H. Yu, M. Su, K. Ye, D. Cao and K. Zhu, Adv. Energy Mater., 2024, 14, 2400353 Search PubMed.
- H. Du, X. Hu, H. Duan, L. Yu, F. Qu, Q. Huang, W. Zheng, H. Xie, J. Peng, R. Tuo, D. Yu, Y. Lin, W. Li, Y. Zheng, X. Fang, Y. Zou, H. Wang, M. Wang, P. S. Weiss, Y. Yang and C. Wang, ACS Cent. Sci., 2019, 5, 97–108 Search PubMed.
- W. Zhao, H. Gong, Y. Song, B. Li, N. Xu, X. Min, G. Liu, B. Zhu, L. Zhou, X. Zhang and J. Zhu, Adv. Funct. Mater., 2021, 31, 2100025 Search PubMed.
- Q. Dai, F. Xing, X. Liu, D. Shi, C. Deng, Z. Zhao and X. Li, Energy Environ. Sci., 2022, 15, 1594–1600 Search PubMed.
- D. Wang, D. Zhao, L. Chang, Y. Zhang, W. Wang, W. Zhang and Q. Zhu, Energy Storage Mater., 2025, 74, 103903 Search PubMed.
- H. Lin, L. Zeng, C. Lin, J. Wu, H. He, C. Huang, W. Lai, P. Xiong, F. Xiao, Q. Qian, Q. Chen and J. Lu, Energy Environ. Sci., 2025, 18, 1282–1293 Search PubMed.
- Y. Guo, C. Luo, M. Yang, H. Wang, W. Ma, K. Hu, L. Li, F. Wu and R. Chen, Angew. Chem., Int. Ed., 2024, 63, e202406597 Search PubMed.
- S. Jia, H. Pan, Q. Lin, X. Wang, C. Li, M. Wang and Y. Shi, Nanotechnology, 2020, 31, 365604 Search PubMed.
- X. Miao, Z. Wu, W. Hu, L. Guo and C. Nan, Adv. Energy Mater., 2025, 2500566 Search PubMed.
- C. Liao, R. Zou, J. Zhu, Z. Cui, M. Gao, L. Zhang, W. Wang and H. Chen, Small, 2024, 20, 2305085 Search PubMed.
- T. Wu, X. Zhang, Y. Wang, N. Zhang, H. Li, Y. Guan, D. Xiao, S. Liu and H. Yu, Adv. Funct. Mater., 2023, 33, 2210154 Search PubMed.
- K. Wang, T. Qiu, L. Lin, F. Liu, J. Zhu, X.-X. Liu and X. Sun, Chem. Sci., 2023, 14, 8076–8083 Search PubMed.
- Z. Ren, S. Gu, T. Li, L. Peng, C. Zou, F. Kang and W. Lv, J. Mater. Chem. A, 2024, 12, 32885–32894 Search PubMed.
- Z. Sang, J. Liu, X. Zhang, L. Yin, F. Hou and J. Liang, ACS Nano, 2023, 17, 3077–3087 Search PubMed.
- X. Shen, W. Chen, H. Wang, L. Zhang, B. Hao, C. Zhu, X. Yang, M. Sun, J. Zhou, X. Liu, C. Yan and T. Qian, Chem. Sci., 2024, 15, 10182–10192 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02326b |
| This journal is © The Royal Society of Chemistry 2025 |