
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen-bond mediated electrocatalytic nitrate reduction to ammonia over metal–organic frameworks with industrial current density†
Xiao-Xue
Fu‡
ac,
Hui
Guo‡
ac,
Duan-Hui
Si‡
a,
Hong-Jing
Zhu‡
ac,
Yi-Ying
Lan
a,
Yuan-Biao
Huang
 abc and
Rong
Cao
*abc
abc and
Rong
Cao
*abc
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, 350002, China. E-mail: rcao@fjirsm.ac.cn
bFujian Science & Technology Innovation Laboratory for Optoelectronic Information of China, Fuzhou, 350108, China
cUniversity of Chinese Academy of Sciences, Beijing, 100049, China
First published on 24th June 2025
Abstract
Electrocatalytic reduction of the pollutant nitrate to ammonia (NO3RR) using clean energy is being considered as a viable alternative to the Haber–Bosch process for producing industrially valuable ammonia. However, the multi-electron–proton transfer process of the NO3RR to ammonia usually leads to poor selectivity and low current density, which still cannot meet the industrial requirements. Stabilizing the key intermediates during the reaction is particularly important for achieving high selectivity in the NO3RR towards the production of NH3. Herein, we develop a hydrogen bonding strategy to stabilize the key intermediates of the NO3RR, which involves the design and synthesis of trinuclear copper(I) cluster-based metal–organic frameworks (MOFs). The methyl groups in the copper-based MOFs (DiMe-Cu3-MOF) can regulate the electron density around the Cu3 site and stabilize the key intermediates, *NO2, through hydrogen bonding interaction with methyl groups. Thus, the DiMe-Cu3-MOF electrocatalyst delivers a high NH3 faradaic efficiency (95%) for the NO3RR with a high ammonia production of 401 μg cm−2 h−1, and the partial current density of ammonia reaches an industrial level value of −950.6 mA cm−2. Control experiments and theoretical studies demonstrated that the introduction of methyl groups into the DiMe-Cu3-MOF can facilitate atypical hydrogen bonding with the intermediates of the NO3RR and thus enhance the adsorption of intermediates and reduce the energy barrier of the conversion of NO3− to NH3. This work highlights the vital importance of adjusting the microenvironment through hydrogen bonding for enhancing the NO3RR performance.
Introduction
The removal of pollutants from aqueous solutions and the production of high-value compounds through green recycling are crucial for establishing a sustainable society with efficient resource recycling.1,2 Nitrates, originating from the long-term and continuous application of organic or synthetic fertilizers, pose a significant threat to groundwater and affect most aquifers in agricultural regions.3 Furthermore, excessive nitrate accumulation in the human body can be converted into carcinogenic N-nitroso substances, leading to serious diseases like methemoglobinemia.4,5 Physical methods such as electrodialysis, reverse osmosis, and membrane filtration techniques have been employed to separate nitrates from domestic sewage, inorganic fertilizers, nitrified soil organic matter and so on.6 Compared with physical approaches, the catalytic transformation of nitrate into useful chemicals such as ammonia not only promotes denitrification of wastewater and balances the disrupted nitrogen cycle, but also has the potential to become a promising pathway for renewable ammonia production.7,8 As is well known, the industrially important ammonia is conventionally produced using the Haber–Bosch process, which involves the mixing of hydrogen and nitrogen at high temperatures ranging from 400 to 650 °C and pressurizing them to 200 atm.9 In addition to the stringent conditions of high temperature and high pressure required to break the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond, the production and purification of H2 constitute the first step, which consumes a large amount of fossil fuels and results in energy waste and environmental pollution.10 Therefore, there is an urgent need for a clean, efficient, and sustainable ammonia production method to replace the energy-intensive and highly polluting Haber–Bosch process.11 An electrochemical reduction method using clean energy to convert toxic nitrates into ammonia offers a sustainable alternative to replace the Haber–Bosch process.12 The electrocatalytic process of nitrate can be performed under ambient conditions, owing to the comparatively low bond energy of N–O (204 kJ mol−1), thereby bypassing the energy-intensive N2 cleavage step (941 kJ mol−1) in N2 electroreduction.13 In this context, the electrochemical nitrate reduction reaction (NO3RR) represents a sustainable pathway to achieve carbon-free ammonia production, which can address water pollution issues and balance the nitrogen cycle.14–17
N bond, the production and purification of H2 constitute the first step, which consumes a large amount of fossil fuels and results in energy waste and environmental pollution.10 Therefore, there is an urgent need for a clean, efficient, and sustainable ammonia production method to replace the energy-intensive and highly polluting Haber–Bosch process.11 An electrochemical reduction method using clean energy to convert toxic nitrates into ammonia offers a sustainable alternative to replace the Haber–Bosch process.12 The electrocatalytic process of nitrate can be performed under ambient conditions, owing to the comparatively low bond energy of N–O (204 kJ mol−1), thereby bypassing the energy-intensive N2 cleavage step (941 kJ mol−1) in N2 electroreduction.13 In this context, the electrochemical nitrate reduction reaction (NO3RR) represents a sustainable pathway to achieve carbon-free ammonia production, which can address water pollution issues and balance the nitrogen cycle.14–17
The competing hydrogen evolution reaction (HER) usually occurs during the NO3RR in aqueous electrolytes.18 Moreover, despite the lower activation energy barrier of N–O, the complex reaction pathway of the NO3RR (NO3− + 9H+ + 8e− → NH3 + 3H2O) involves multiple electron and proton transfers and production of several intermediates, which frequently results in low activity and selectivity of the NO3RR, thereby posing a significant challenge for practical applications.19,20 Several electrocatalysts including metal-based electrocatalysts have been applied in the NO3RR; however, their activity and selectivity still need to be improved to meet the industrial requirements for ammonia production.21,22 Compared to other electrocatalysts, copper-based materials have been widely used in electrochemical reactions such as CO2 reduction and the NO3RR due to their suitable adsorption energy for intermediates.22–24 However, the various intermediates are usually detached from the copper active sites prior to completing the eight-electron transfer process required for ammonia generation.8,25 This is due to potential-dependent phase transitions, resulting in deactivation caused by changes in the oxidation state, morphology, crystal facets, and other factors. In addition, the process of reducing nitrate to ammonia involves eight electron transfer steps, and there may be multiple potential intermediates generated in the process, such as NO2−, NO, N2 and NH2OH.26,27 Therefore, stabilizing these intermediates in the reaction process to prevent them from dissociating from the active site will be beneficial for the formation of the final product.
As is well known, regulating the microenvironments and introducing suitable groups into electrode materials could reduce the energy barrier of the electrochemical reaction and thus enhance the faradaic efficiency (FE) and selectivity.28 Interestingly, the catalytic mechanism of one of the enzymes, copper nitrite reductase (CuNiR), involved in the biological reduction of NOx to ammonia, involves the binding of NO2− to the catalytic center T2Cu, replacing a soluble molecule.29 Simultaneously, hydrogen bonding forms between the residue coordinating with T2Cu and NO2−, leading to the formation of the subsequent intermediate in the reaction process.30,31 Inspired by this, introducing molecular interactions involving hydrogen bonding into the electrocatalytic NO3RR can effectively stabilize specific intermediates and direct the reaction along the expected pathway.32 Currently, there is limited research on the precise control of electrocatalytic processes through weak interactions, such as hydrogen bonding. Therefore, it is necessary to develop new catalysts to enhance the NO3RR.
Metal–organic frameworks (MOFs) have recently emerged as a promising class of crystalline porous polymers due to their large surface area, stability, and potential in various applications such as gas adsorption, heterogeneous catalysis, optoelectronic properties, and energy storage.33–36 The unique feature of flexible and designable structure endows MOFs be easily modified the microenvironment of the metal active sites through introducing functional groups. Thus, the hydrogen bonding interaction between the intermediates of the NO3RR process with the introduced functional groups could be formed. Consequently, the selectivity of the NO3RR towards the target products could be improved by reducing the reaction energy barrier. However, to the best of our knowledge, there is no report of using the hydrogen bonding strategy to enhance the NO3RR performance.
Based on this, three examples of copper(I) cyclic trinuclear unit (Cu-CTU)-based two-dimensional (2D) MOFs were designed and synthesized to investigate how the structure of the catalyst affects the microenvironment of the copper active center during the nitrate reduction process.37 As shown in Scheme 1, the DiMe-Cu3-MOF was synthesized by the reaction of the copper(I) cyclic trinuclear cluster Cu3(Me-PyCA)3 (Me-PyCA = 3,5-dimethyl-1H-pyrazole-4-carbaldehyde), 3,5-dimethyl-1H-pyrazol-4-amine (DiMe-PyA)35,38 and Cu2O. The copper center in DiMe-Cu3-MOF is surrounded by two methyl (–CH3) groups. Atypical hydrogen bonds C–H⋯Y (where Y is usually N and O) between the –CH3 groups and the nitrogen–oxygen intermediates adsorbed on the copper center during the NO3RR process may be formed. Thus, the intermediates could be stabilized and the energy barrier for ammonia generation would be lowered. For comparison, UniMe-Cu3-MOF was prepared by the reaction of the methyl-free copper(I) cyclic trinuclear cluster Cu3(PyCA)3,39 DiMe-PyA and Cu2O. The NonMe-Cu3-MOF containing no methyl group was also synthesized by imine condensation between Cu3(PyCA)3 and an amino copper(I) cyclic trinuclear complex, in situ generated from Cu2O and 1H-pyrazol-4-amine (PyA). The activity of DiMe-Cu3-MOF with atypical hydrogen bonds was higher than that of UniMe-Cu3-MOF and NonMe-Cu3-MOF, with the trend of DiMe-Cu3-MOF > UniMe-Cu3-MOF > NonMe-Cu3-MOF. It is worth noting that DiMe-Cu3-MOF exhibited a high faradaic efficiency (FENH3) of 95% and achieved a high NH3 partial current density (jNH3) of −950.6 mA cm−2.
 | ||
| Scheme 1 Schematic diagram of the syntheses of NonMe-Cu3-MOF, UniMe-Cu3-MOF, and DiMe-Cu3-MOF. | ||
Results and discussion
As shown in Scheme 1, DiMe-Cu3-MOF, in which all the Cu centers were surrounded by methyl groups, was successfully synthesized by reacting Cu3(Me-PyCA)3 clusters, Me-PyA, and Cu2O under solvothermal conditions. As a comparison, methyl-free NonMe-Cu3-MOF and UniMe-Cu3-MOF, in which only one half of the copper sites were near the methyl group, were also successfully synthesized using a similar method. The crystalline structure of the MOF was confirmed using Materials Studio software based on the powder X-ray diffraction (PXRD) patterns. Fig. 1c shows that DiMe-Cu3-MOF exhibited strong diffraction peaks at 4.7° and weaker reflections at approximately 8.1° and 26.1°, corresponding to the (100), (110), and (001) facets, respectively. Furthermore, the PXRD spectrum showed no presence of peaks corresponding to Cu2O, indicating the complete removal of Cu2O from the starting materials. DiMe-Cu3-MOF was crystallized with an AA-eclipsed stacking model in the P3 space group. The simulated PXRD pattern, with an unweighted-profile R factor (Rp) of 4.60% and a weighted-profile R factor (Rwp) of 6.13%, matched very well with the experimentally observed curve, indicating the effectiveness of the proposed model (Scheme 1). The Pawley refinement method was employed to conduct a full spectrum fitting of the experimental diffraction pattern, resulting in the determination of the unit cell parameters for DiMe-Cu3-MOF as follows: a = b = 22.0982 Å, c = 3.4883 Å, α = β = 90°, and γ = 120° (Scheme 1). These refinements indicate that DiMe-Cu3-MOF possesses a 1D hexagonal channel along the c-axis, with a theoretical pore size of approximately 1.91 nm and a π–π stacking distance of 3.49 Å between adjacent layers along the c-axis. The NonMe-Cu3-MOF and UniMe-Cu3-MOF exhibit similar PXRD patterns to DiMe-Cu3-MOF (Scheme 1 and Fig. 1a and b), suggesting that they are isomorphous materials.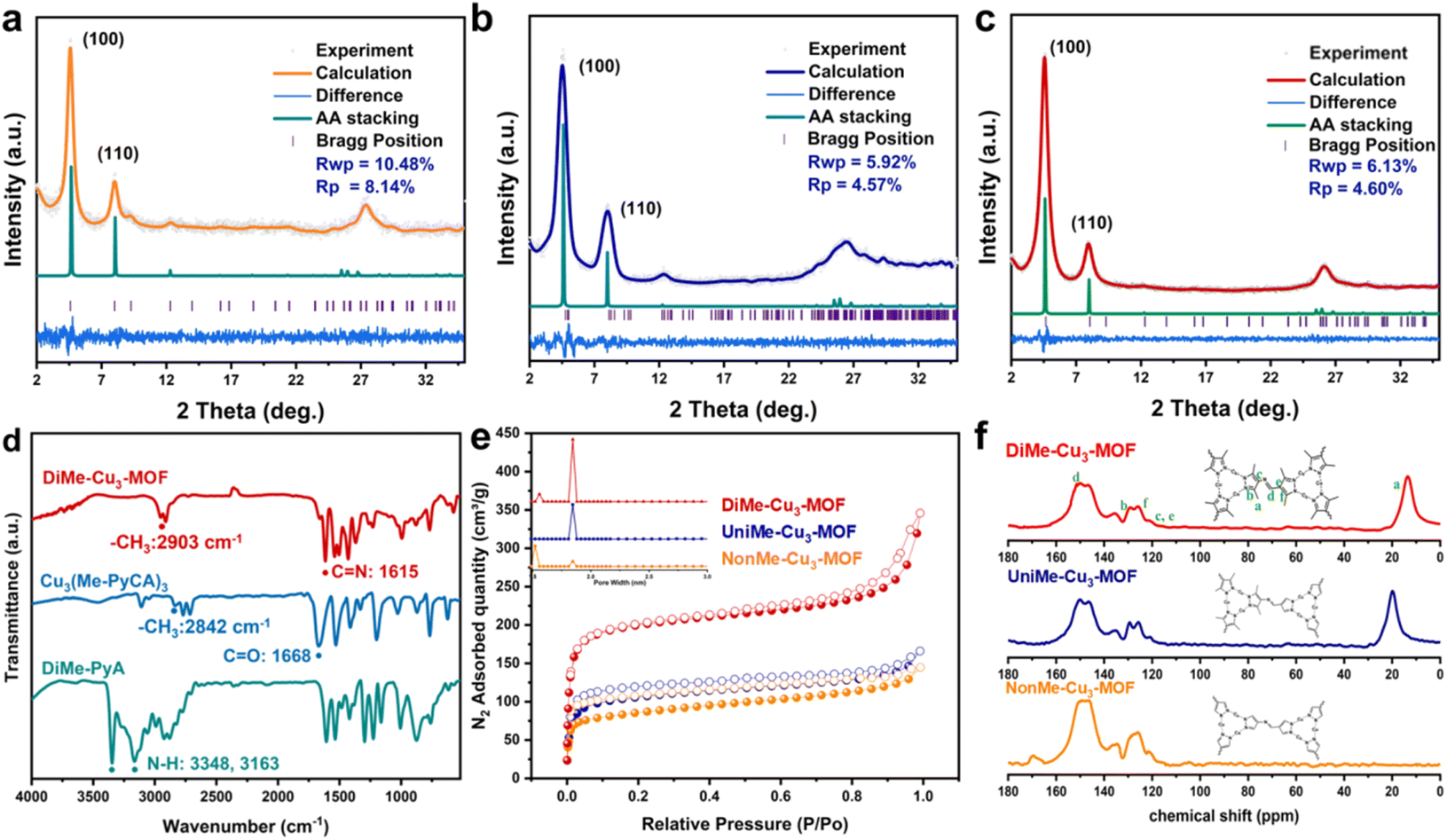 | ||
| Fig. 1 Experimental diffraction patterns, patterns simulated from Pawley refinements (black), and patterns simulated from the structural model (green) for (a) NonMe-Cu3-MOF, (b) UniMe-Cu3-MOF and (c) DiMe-Cu3-MOF. (d) The FT-IR spectra of DiMe-PyA, Cu3(Me-PyCA)3 and DiMe-Cu3-MOF. (e) N2 adsorption for NonMe-Cu3-MOF, UniMe-Cu3-MOF and DiMe-Cu3-MOF. (f) 13C CP-MAS NMR spectra of UniMe-Cu3-MOF, NonMe-Cu3-MOF and DiMe-Cu3-MOF. | ||
The successful synthesis of DiMe-Cu3-MOF, UniMe-Cu3-MOF, and NonMe-Cu3-MOF was further confirmed through Fourier transform infrared spectroscopy (FT-IR) and 13C solid-state cross-polarization magic angle-spinning nuclear magnetic resonance (CP-MAS NMR) spectroscopy. As shown in Fig. 1d and S1,† all three MOFs exhibited a newly formed C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N imine signal at approximately 1610 cm−1, while the –CHO peak (1668 cm−1) and –NH2 signals (3201 cm−1 and 3325 cm−1) in the reactant monomers disappeared.40 Furthermore, due to the increased conjugation (including planar conjugation and π–π stacking) of DiMe-Cu3-MOF, a strong inductive effect is generated on the –CH3 groups, leading to an increase in their bond strength. Consequently, the peak located at 2903 cm−1 for the –CH3 group in DiMe-Cu3-MOF shows a blue shift in comparison with the peak at 2842 cm−1 in Cu3(Me-PyCA)3. These results were supported by the 13C solid-state NMR spectrum (Fig. 1f), which showed a strong CN imine carbon resonance signal at ∼149 ppm and a prominent –CH3 signal at 19 ppm in DiMe-Cu3-MOF. Notably, the absence of methyl peaks was observed in NonMe-Cu3-MOF. Inductively coupled plasma (ICP) spectroscopy confirmed that DiMe-Cu3-MOF, UniMe-Cu3-MOF, and NonMe-Cu3-MOF had large Cu contents of 28.7 wt%, 32.7 wt%, and 41.1 wt%, respectively, indicating the integration of a large number of Cu catalytic sites into the MOF framework. N2 adsorption experiments were conducted to investigate the porosity of DiMe-Cu3-MOF, UniMe-Cu3-MOF, and NonMe-Cu3-MOF. The N2 isotherms of DiMe-Cu3-MOF at 77 K displayed type IV adsorption curves with hysteresis loops, indicating the presence of micropores. Further analysis of the pore size distribution revealed that DiMe-Cu3-MOF had micropores with an average diameter of approximately 1.85 nm (Fig. 1e and S2†), which closely corresponded to the theoretical value of 1.91 nm. The obtained DiMe-Cu3-MOF exhibited the highest Brunauer–Emmett–Teller (BET) surface area of 772 m2 g−1 among the three MOFs. The UniMe-Cu3-MOF had a BET surface area of 410 m2 g−1, sharing the same pore diameter of 1.85 nm, while the NonMe-Cu3-MOF still had a moderate BET surface area of 318 m2 g−1, along with a slightly smaller pore diameter of 1.83 nm. Such high porosity of the three materials is favorable for the accessibility and transformation of NO3−.
N imine signal at approximately 1610 cm−1, while the –CHO peak (1668 cm−1) and –NH2 signals (3201 cm−1 and 3325 cm−1) in the reactant monomers disappeared.40 Furthermore, due to the increased conjugation (including planar conjugation and π–π stacking) of DiMe-Cu3-MOF, a strong inductive effect is generated on the –CH3 groups, leading to an increase in their bond strength. Consequently, the peak located at 2903 cm−1 for the –CH3 group in DiMe-Cu3-MOF shows a blue shift in comparison with the peak at 2842 cm−1 in Cu3(Me-PyCA)3. These results were supported by the 13C solid-state NMR spectrum (Fig. 1f), which showed a strong CN imine carbon resonance signal at ∼149 ppm and a prominent –CH3 signal at 19 ppm in DiMe-Cu3-MOF. Notably, the absence of methyl peaks was observed in NonMe-Cu3-MOF. Inductively coupled plasma (ICP) spectroscopy confirmed that DiMe-Cu3-MOF, UniMe-Cu3-MOF, and NonMe-Cu3-MOF had large Cu contents of 28.7 wt%, 32.7 wt%, and 41.1 wt%, respectively, indicating the integration of a large number of Cu catalytic sites into the MOF framework. N2 adsorption experiments were conducted to investigate the porosity of DiMe-Cu3-MOF, UniMe-Cu3-MOF, and NonMe-Cu3-MOF. The N2 isotherms of DiMe-Cu3-MOF at 77 K displayed type IV adsorption curves with hysteresis loops, indicating the presence of micropores. Further analysis of the pore size distribution revealed that DiMe-Cu3-MOF had micropores with an average diameter of approximately 1.85 nm (Fig. 1e and S2†), which closely corresponded to the theoretical value of 1.91 nm. The obtained DiMe-Cu3-MOF exhibited the highest Brunauer–Emmett–Teller (BET) surface area of 772 m2 g−1 among the three MOFs. The UniMe-Cu3-MOF had a BET surface area of 410 m2 g−1, sharing the same pore diameter of 1.85 nm, while the NonMe-Cu3-MOF still had a moderate BET surface area of 318 m2 g−1, along with a slightly smaller pore diameter of 1.83 nm. Such high porosity of the three materials is favorable for the accessibility and transformation of NO3−.
The scanning electron microscopy (SEM) images showed that all three MOFs have a dendritic crystal morphology (Fig. S3†). High-resolution TEM (HR-TEM) images (Fig. 2a–c) show that the ordered micropore lattice spacing of NonMe-Cu3-MOF, UniMe-Cu3-MOF, and DiMe-Cu3-MOF is approximately 1.88 nm, 1.87 nm, and 1.85 nm, respectively, which is consistent with the pore size of the simulated structural model and N2 adsorption results (Fig. 1e). Furthermore, the absence of a Cu2O lattice within the three MOFs provides additional confirmation of the successful removal of Cu2O from the initial materials. Energy-dispersive X-ray spectroscopy (EDS) element mapping images show that Cu, C and N elements are uniformly distributed across the structures of NonMe-Cu3-MOF, UniMe-Cu3-MOF, and DiMe-Cu3-MOF (Fig. 2d–f).
 | ||
| Fig. 2 HR-TEM image and the lattice distance of (a) NonMe-Cu3-MOF, (b) UniMe-Cu3-MOF and (c) DiMe-Cu3-MOF. (d–f) HAADF-STEM image of (d) NonMe-Cu3-MOF, (e) UniMe-Cu3-MOF and (f) DiMe-Cu3-MOF and the corresponding C, N, and Cu EDS elemental mappings. | ||
X-ray absorption spectroscopy (XAS) was conducted to determine the oxidation state and coordination environment of copper species in the catalysts. In the Cu K-edge of X-ray absorption near edge structure (XANES) spectra for DiMe-Cu3-MOF, the absorption edge corresponding to the 1s → 4p transition is positioned between Cu foil and Cu2O, closer to Cu2O. At the same time, the absence of the 1s → 3d pre-edge due to the dipole-forbidden indicated the absence of Cu2+ in DiMe-Cu3-MOF (Fig. 3a). It suggests that the oxidation state of copper in the material is +1. Additionally, in the Cu K-edge extended X-ray absorption fine structure (EXAFS) of DiMe-Cu3-MOF (Fig. 3b), a clear signal corresponding to the Cu–N coordination path at 1.4 Å was observed, which differs from that of Cu2O. Furthermore, no signal corresponding to Cu–Cu at 2.23 Å was observed, indicating there is no Cu or CuOx in DiMe-Cu3-MOF. Through comprehensive resolution in both K and R space, the wavelet transform (WT) analysis of Cu K-edge EXAFS was performed to further explore the coordination structure of DiMe-Cu3-MOF. Fig. 3d illustrates the distinctive features of DiMe-Cu3-MOF compared to Cu2O, providing further evidence of the absence of Cu2O particles and a non-Cu–O coordination mode in DiMe-Cu3-MOF. The results indicated the dominance of Cu–N species and the absence of Cu and Cu2O particles in DiMe-Cu3-MOF. The EXAFS fitting spectrum (Fig. 3c and Table S1†) demonstrated a coordination number of 2.0 for the copper center in DiMe-Cu3-MOF, which is consistent with the simulated structure. In addition, the Cu K-edge XANES and EXAFS spectra of UniMe-Cu3-MOF and NonMe-Cu3-MOF are also characterized in Fig. S4 and S5 and Tables S2 and S3.† According to the analysis results based on the Cu K-edge XANES and EXAFS curves in Fig. 3a and b, the Cu species in UniMe-Cu3-MOF and NonMe-Cu3-MOF exhibited similar states and coordination environments to the Cu in DiMe-Cu3-MOF, which was coordinated by two N atoms (Fig. S4 and S5 and Tables S2 and S3†). High-resolution X-ray photoelectron spectroscopy (XPS) of Cu 2p reveals two peaks at 952.8 eV and 932.9 eV in DiMe-Cu3-MOF, corresponding to Cu 2p1/2 and Cu 2p3/2, respectively (Fig. 3e). Cu LMM Auger spectroscopy provided further elucidation of the valence state of the copper center, revealing a Cu+ peak with a kinetic energy of 914.5 eV in DiMe-Cu3-MOF (Fig. 3f). The XPS data for UniMe-Cu3-MOF and NonMe-Cu3-MOF showed similar results (Fig. S4 (e and f) and S5 (d and e)†) to those of DiMe-Cu3-MOF, confirming the previous conclusion from X-ray absorption near-edge structure (XANES) that copper in the three MOFs exhibits a Cu+ state.
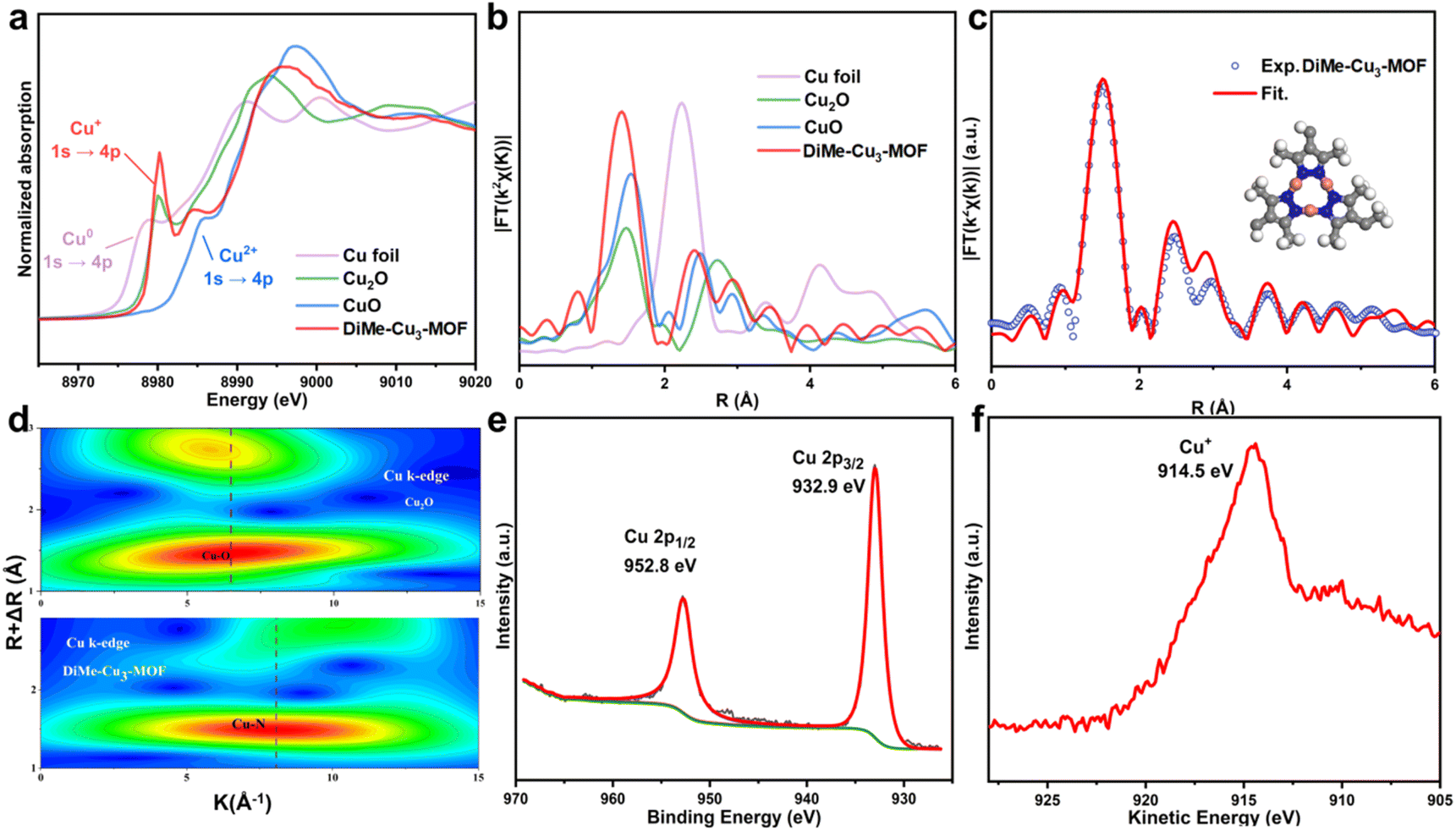 | ||
| Fig. 3 (a) Cu K-edge of XANES spectra of DiMe-Cu3-MOF, Cu foil, CuO and Cu2O. (b) Cu K-edge of EXAFS spectra of DiMe-Cu3-MOF, Cu foil, CuO and Cu2O. (c) The EXAFS fitting curves of DiMe-Cu3-MOF. (d) WT-EXAFS plots of DiMe-Cu3-MOF and Cu2O. (e) Cu 2p XPS spectra and (f) Cu LMM Auger spectra of DiMe-Cu3-MOF. | ||
In order to evaluate the influence of hydrogen bonding on the intermediates of the NO3RR, the activity of DiMe-Cu3-MOF, UniMe-Cu3-MOF, and NonMe-Cu3-MOF in the NO3RR was studied in a standard three-electrode H-cell. All the potentials mentioned in this work were referenced to Ag/AgCl. The ammonia generated after electrolysis was analyzed using ultraviolet-visible (UV-vis) spectrophotometry (Fig. S6†), and the accuracy of this method was verified by ion chromatography (IC) (Fig. S7†). There were significant differences in the electrochemical NO3RR between DiMe-Cu3-MOF and its corresponding control samples. As illustrated in Fig. 4a and S8,† the DiMe-Cu3-MOF, which features –CH3 groups in both Cu-CTUs, demonstrated a more positive onset potential, larger ammonia faradaic efficiencies (FENH3), and higher partial current densities when compared to the NonMe-Cu3-MOF (lacking –CH3 groups) and UniMe-Cu3-MOF (with only half of Cu-CTUs containing –CH3 groups). Especially, the current density for NonMe-Cu3-MOF was much lower than those of the other two catalysts, indicating its poor activity for the NO3RR. The ammonia yield and FENH3 of the NO3RR for the three catalysts are shown in Fig. 4b and c, respectively. DiMe-Cu3-MOF achieved an FENH3 of 95% at −1.55 V, with an ammonia yield of 401 μg h−1 cm−2 and a current density of −64 mA cm−2 (Fig. 4d). The ammonia yield of the NO3RR for the three materials increased in the order of NonMe-Cu3-MOF < UniMe-Cu3-MOF < DiMe-Cu3-MOF. The FENH3 of NonMe-Cu3-MOF was significantly lower than that of UniMe-Cu3-MOF and DiMe-Cu3-MOF within the potential range of −1.45 to −1.85 V. In contrast, the production of NO3− was determined by a typical colorimetric method to evaluate the corresponding electrocatalytic performance (Fig. S9†).41 The results show that NO2− content generated by NonMe-Cu3-MOF is higher than those of the other two catalysts, indicating that the NO2− intermediate escaped from the Cu active sites, which was not further transformed to NH3. It may be ascribed to the NO2− intermediate that cannot be stabilized by the Cu sites of NonMe-Cu3-MOF. This highlights the importance of introducing methyl groups into DiMe-Cu3-MOF to stabilize intermediates via hydrogen bonding. To further verify the industrial applicability of DiMe-Cu3-MOF, NO3RR tests were conducted under constant current electrolysis conditions. The results demonstrated that within the current density range of −400 to −1200 mA cm−2, DiMe-Cu3-MOF consistently maintains an FENH3 above 80%, with the partial current density for ammonia reaching −950.6 mA cm−2 (Fig. 4e). In addition, DiMe-Cu3-MOF has high stability, in which the current density remained consistent throughout a 12 hour testing period, as well as without any notable decline in Faraday efficiency (Fig. 4g). PXRD patterns, TEM images and Cu K-edge EXAFS and EXAFS showed no significant changes in DiMe-Cu3-MOF after 2 h of electrolysis compared to the corresponding freshly prepared material (Fig. S10–S12†).
 | ||
| Fig. 4 (a) LSV polarization curves of DiMe-Cu3-MOF, UniMe-Cu3-MOF, and NonMe-Cu3-MOF with and without 0.5 M K2SO4 and 50 mM KNO3 electrolytes, respectively. (b) Comparison of NH3 yields of DiMe-Cu3-MOF, UniMe-Cu3-MOF, and NonMe-Cu3-MOF. (c) The comparison of NH3 and NO3− faradaic efficiencies of DiMe-Cu3-MOF, UniMe-Cu3-MOF, and NonMe-Cu3-MOF. (d) The partial current densities of NH3 evolution for different catalysts with 50 mM KNO3. (e) NH3 and NO2− Faradaic efficiency and the partial current densities of NH3 of DiMe-Cu3-MOF under current-controlled conditions. (f) 1H NMR spectra of the products after 2 h electrolysis with the electrolytes containing 50 mM 14KNO3 or 15KNO3 as the feeding nitrate source using DiMe-Cu3-MOF. (g) Current densities of DiMe-Cu3-MOF during continuous electrolysis in 0.5 M K2SO4 and 50 mM KNO3 electrolytes at −1.45 V for 12 h (bottom panel) and the corresponding FENH3 (top panel). (h) Comparison of the NO3RR performance of the DiMe-Cu3-MOF with other catalysts reported in the literature. | ||
To confirm the source of the NH3 product for DiMe-Cu3-MOF, a series of control experiments and 15N isotope-labeled experiments were conducted.42,43 When nitrate was not added to the electrolyte or applied open circuit potential, no ammonia was produced (Fig. S13†). Additionally, by using 15NO3− as the nitrogen source, the origin of ammonia was traced to the nitrate in the electrolyte rather than environmental contamination. The typical double-peak 15NH3 signal effectively ruled out ammonia contamination and confirmed that ammonia was indeed produced through the electrocatalytic reduction of NO3− (Fig. 4f).
In order to determine whether the high activity of DiMe-Cu3-MOF originates from the effect of atypical hydrogen bonding in the NO3RR process, the activities are normalized by the number of redox-active copper sites, BET surface areas and the loading amount of catalysts on the electrode. As shown in Fig. S14 and S15 and Table S4,† DiMe-Cu3-MOF showed moderate intrinsic activity for the NO3RR towards NH3 from the ECSA-normalized I–V curves, indicating that the high performance of DiMe-Cu3-MOF is not from its high ECSA. Additionally, when the activity was normalized by the BET surface area, the DiMe-Cu3-MOF also showed moderate intrinsic activity for the NO3RR towards NH3 at moderate current density, which suggested that the high activity of DiMe-Cu3-MOF is not contributed by its high surface area (Fig. S16†). Meanwhile, different loading amounts on the electrode showed similar normalized current density and ammonia Faraday efficiency, indicating that the amount of the catalyst loading on the electrode had little effect on the catalytic activity (Fig. S17†). These results demonstrated that the high activity of DiMe-Cu3-MOF toward NH3 can be attributed to the effect of atypical hydrogen bonding. In order to elucidate the role of hydrogen bonds in the stabilization of intermediates in the NO3RR process, in situ Fourier transform infrared spectroscopy (in situ FTIR) measurements were carried out (Fig. S18†). As shown in Fig. 5a, b and S19,† the downward absorption band at 1380 cm−1 corresponds to the consumption peak of NO3−. With increasing potential, two distinct peaks corresponding to the vertical and bending adsorption of NO appear at ∼1730 cm−1 and 1649 cm−1, respectively, in DiMe-Cu3-MOF (Fig. 5a). As the applied potential increases, the intensity of these peaks decreases more noticeably. Additionally, as shown in Fig. 5b, with the increase of potential, the vertical and bending adsorption peaks of NO appear at ∼1778 cm−1 and 1643 cm−1 and their intensities increase significantly in NonMe-Cu3-MOF. However, after reaching the optimal point, the absorption peak decreases slightly. This indicates that NonMe-Cu3-MOF has a weaker ability to facilitate the conversion of NO2 into NO intermediates, which is different from DiMe-Cu3-MOF as shown in Fig. 5a, further illustrating the role of DiMe-Cu3-MOF in stabilizing NO2 intermediates. The trend of increasing absorption peaks with potential increase for NO2 (1550 cm−1) intermediates on NonMe-Cu3-MOF indicates the gradual accumulation of NO2 (Fig. 5b).44,45
 | ||
| Fig. 5 (a) Operando ATR-FTIR spectra collected at different applied voltages for DiMe-Cu3-MOF in 50 mM KNO3 and 0.5 M K2SO4 electrolytes and (b) Operando ATR-FTIR spectra collected at different applied voltages for NonMe-Cu3-MOF in 50 mM KNO3 and 0.5 M K2SO4 electrolytes. (c) Operando Raman spectra of DiMe-Cu3-MOF collected at different applied potentials in 50 mM KNO3 and 0.5 M K2SO4 electrolytes and (d) Operando Raman spectra of NonMe-Cu3-MOF collected at different applied potentials in 50 mM KNO3 and 0.5 M K2SO4 electrolytes. | ||
In situ Raman spectra were utilized further to investigate the catalyst surface during the reaction. The Raman peaks at 986 and ∼1046 cm−1 correspond to the characteristic vibration modes of SO24− and NO3− in the electrolyte, respectively. The DiMe-Cu3-MOF shows a Raman peak of the Cu–N bond at 454 cm−1 (Fig. 5c and S20†). The intensity of this peak initially increases and then decreases with the rising potential, suggesting an accumulation of the Cu–N bond at low potential, followed by the consumption of the intermediate of the NO3RR at high potential. The Raman peaks at 850, 1160, 1221 and 1534 cm−1 are related to the symmetric and antisymmetric stretching vibrations of chelated NO2−, *NO and *NHO intermediates, respectively.46,47 Therefore, the combination of catalytic performance, in situ FTIR and Raman spectroscopy results demonstrates that DiMe-Cu3-MOF exhibits stronger intermediate (*NO2) adsorption compared to other materials, leading to excellent NO3RR performance.
To study the effects of hydrogen bonding on the NO3RR and HER, DFT calculations were performed for the DiMe-Cu3-MOF and NonMe-Cu3-MOF. Two typical NO3RR pathways were considered: the NOH pathway (*NO3− → *NO2 → *NO2H → *NO → *NOH → *N → *NH → *NH2 → *NH3) and the NHO pathway (*NO3− → *NO2 → *NO2H → *NO → *NHO → *NH2O → *NH2OH → *NH2 → *NH3), respectively. According to in situ Raman results, the presence of the *NHO intermediate instead of the *NOH (1540 cm−1) intermediate was detected on both DiMe-Cu3-MOF and NonMe-Cu3-MOF, indicating that the nitrate was reduced to ammonia via the NHO pathway (Fig. 5c, d and S21†).48 As illustrated in Fig. 6a and c, the conversion from *NO to *NHO is identified as the rate-determining step (RDS). However, the difference in the Gibbs free energy (ΔG) for DiMe-Cu3-MOF (−0.14 eV) and NonMe-Cu3-MOF (−0.18 eV) for the conversion from *NO to *NHO is minimal. In the case where the difference in ΔG for the RDS is not significant, DiMe-Cu3-MOF and NonMe-Cu3-MOF exhibit pronounced performance distinctions, primarily attributed to the hydrogen bonding interaction between DiMe-Cu3-MOF and *NO2 during the NO3RR process. DFT calculations were employed to further elucidate how the hydrogen bonding interaction facilitates the performance of the NO3RR to ammonia. As shown in Fig. 6a, the ΔG of the conversion from the *NO2 intermediate to *NO2H over DiMe-Cu3-MOF (−0.98 eV) is lower than that of NonMe-Cu3-MOF (−0.66 eV, Fig. 6c), indicating thermodynamically favorable ammonia generation. This is primarily attributed to the hydrogen bonding interaction stabilizing the *NO2 intermediate in the reaction process of DiMe-Cu3-MOF, which has been experimentally confirmed by in situ FTIR spectroscopy. In addition, the free energy of the HER of DiMe-Cu3-MOF is higher than that of NonMe-Cu3-MOF, indicating that the introduction of the methyl group could impede the HER (Fig. 6d), which is consistent with the results of electrochemical experiments. Additionally, the noncovalent interaction (NCI) was further applied to illustrate the hydrogen bond.49 As shown in Fig. S22,† an obvious van der Waals interaction was observed between the methyl groups and intermediates, indicating that the existence of a hydrogen bond could stabilize the intermediates of the NO3RR. Based on the analysis of the NO3RR and HER, we propose that the formation of hydrogen bonding by adjusting the microenvironment of the active center can promote the NO3RR by stabilizing the *NO2 intermediate and reducing the energy barrier in the rate-limiting *NO2H generation step, thus promoting the NO3RR.
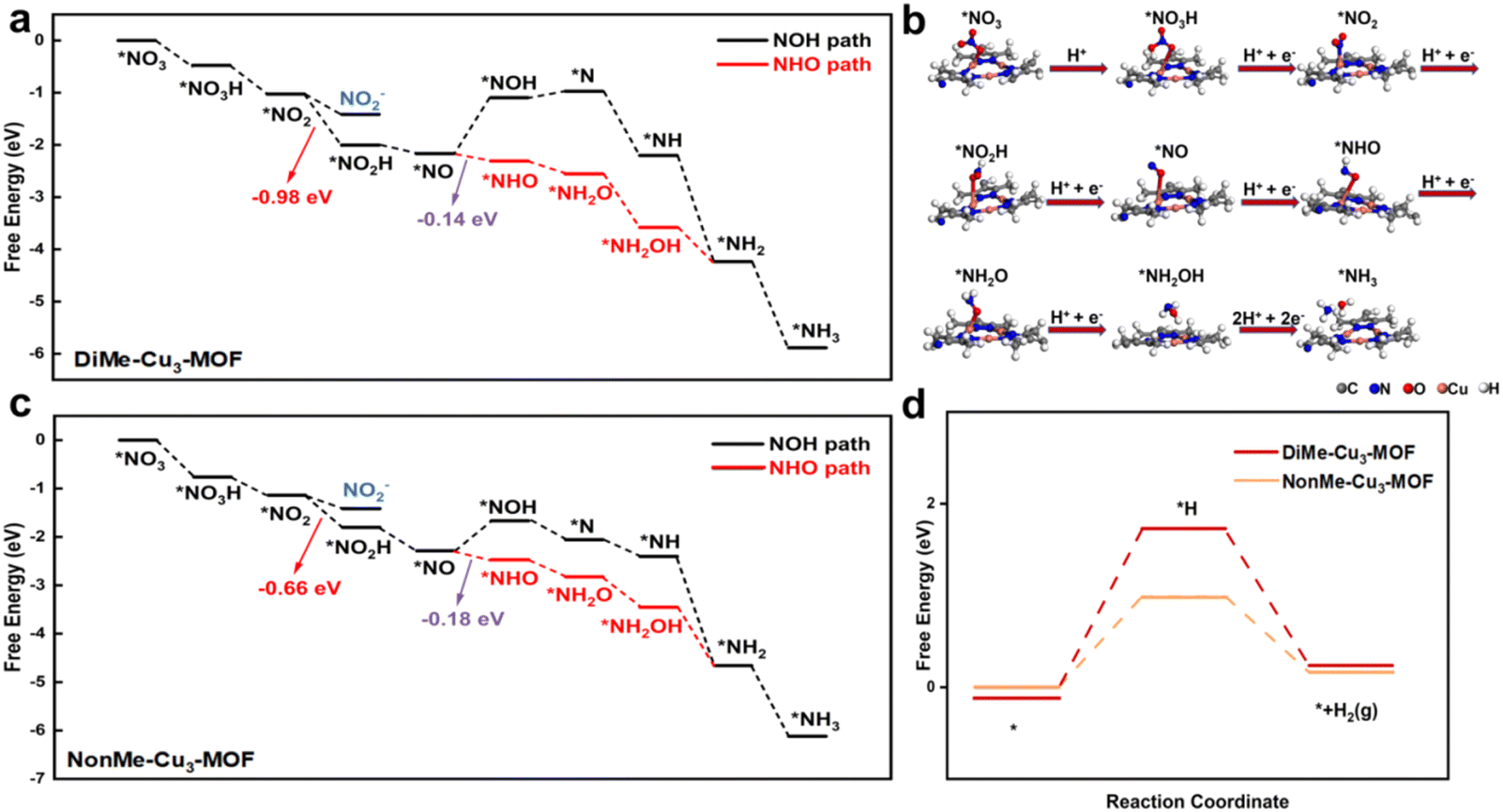 | ||
| Fig. 6 (a) Gibbs free energy diagram for the NO3RR on DiMe-Cu3-MOF. (b) The most probable pathway of the NO3RR on DiMe-Cu3-MOF (side view). (c) Gibbs free energy diagram for the NO3RR on NonMe-Cu3-MOF. (d) Gibbs free energy diagram for the HER on DiMe-Cu3-MOF and NonMe-Cu3-MOF. | ||
Conclusions
In conclusion, drawing inspiration from the natural nitrogenase, we have developed an effective strategy to promote the NO3RR on MOFs by introducing appropriate groups into copper-based electrocatalytic materials to stabilize the intermediate *NO2 through hydrogen bonding. By manipulating the microenvironment of Cu-CTU-based MOFs in the reduction process, we have constructed catalytic materials with trinuclear copper centers for nitrate. Therefore, the NO3RR activity of DiMe-Cu3-MOF, which can form hydrogen bonds with intermediates, is significantly higher than that of UniMe-Cu3-MOF, which can partially form hydrogen bonds, and NonMe-Cu3-MOF, which cannot form hydrogen bonds. The DiMe-Cu3-MOF electrocatalyst provides a high NH3 Faradaic efficiency of 95% at −1.55 V and reaches a current density of −950.6 mA cm−2. The NH3 production rate reaches a high activity of 401 μg cm−2 h−1 and maintains an FENH3 of about 80% at high current density (−1.2 A cm−2), surpassing all reported MOF/COF materials and most other copper-based catalysts. In situ infrared spectroscopy confirms the existence of hydrogen bonds between MOF materials and the key intermediate *NO2, and DFT calculations elucidate that the presence of hydrogen bonds reduces the energy barrier in the rate-limiting *NO2H generation step, thereby promoting NH3 production. This study provides new insights into the importance of stabilizing the key intermediate *NO2 and reducing the local binding energy of H2O to enhance the NO3RR.Data availability
https://app.globus.org/file-manager?origin_id=82f1b5c6-6e9b-11e5-ba47-22000b92c6ec%26origin_path=%2Ftmp%2FRaw+data%2F.Author contributions
Rong Cao and Yuan-Biao Huang proposed the research direction, were responsible for the infrastructure, guided the project, and revised the paper. Xiao-Xue Fu, Hui Guo and Hong-Jing Zhu realized the experiments, performed the characterization, and wrote the paper. Duan-Hui Si conducts the computational study. Yi-Ying Lan assisted with the experiments and characterization. All authors commented on the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge the financial support from the National Key Research and Development Program of China (2023YFA1507904 and 2021YFA1501500), NSFC (U22A20436, 22033008, 22220102005, and 22201286), and Fujian Science & Technology Innovation Laboratory for Optoelectronic Information of China (2021ZZ103). We thank the beamline BL14W1 station for XAFS measurements at the Shanghai Synchrotron Radiation Facility, China.References
- L. An, M. R. Narouz, P. T. Smith, P. De La Torre and C. J. Chang, Supramolecular enhancement of electrochemical nitrate reduction catalyzed by cobalt porphyrin organic cages for ammonia electrosynthesis in water, Angew. Chem., Int. Ed., 2023, 62, e202305719 CrossRef CAS PubMed.
- C. S. Gerke, Y. Xu, Y. Yang, G. D. Foley, B. Zhang, E. Shi, N. M. Bedford, F. Che and V. S. Thoi, Electrochemical C–N bond formation within boron imidazolate cages featuring single copper sites, J. Am. Chem. Soc., 2023, 145, 26144–26151 CrossRef CAS PubMed.
- H. Xu, J. Chen, Z. Zhang, C. T. Hung, J. Yang and W. Li, In situ confinement of ultrasmall metal nanoparticles in short mesochannels for durable electrocatalytic nitrate reduction with high efficiency and selectivity, Adv. Mater., 2023, 35, e2207522 CrossRef PubMed.
- Y. Wang, A. Xu, Z. Wang, L. Huang, J. Li, F. Li, J. Wicks, M. Luo, D. H. Nam, C. S. Tan, Y. Ding, J. Wu, Y. Lum, C. T. Dinh, D. Sinton, G. Zheng and E. H. Sargent, Enhanced nitrate-to-ammonia activity on copper-nickel alloys via tuning of intermediate adsorption, J. Am. Chem. Soc., 2020, 142, 5702–5708 CrossRef CAS PubMed.
- T. Mou, Y. Wang, P. Deak, H. Li, J. Long, X. Fu, B. Zhang, T. Frauenheim and J. Xiao, Predictive theoretical model for the selective electroreduction of nitrate to ammonia, J. Phys. Chem. Lett., 2022, 13, 9919–9927 CrossRef CAS PubMed.
- R. Jia, Y. Wang, C. Wang, Y. Ling, Y. Yu and B. Zhang, Boosting selective nitrate electroreduction to ammonium by constructing oxygen vacancies in TiO2, ACS Catal., 2020, 10, 3533–3540 CrossRef CAS.
- E. Pérez-Gallent, M. C. Figueiredo, I. Katsounaros and M. T. M. Koper, Electrocatalytic reduction of nitrate on copper single crystals in acidic and alkaline solutions, Electrochim. Acta, 2017, 227, 77–84 CrossRef.
- T. Ren, Y. Sheng, M. Wang, K. Ren, L. Wang and Y. Xu, Recent advances of Cu-based materials for electrochemical nitrate reduction to ammonia, Chin. J. Struct. Chem., 2022, 41, 2212089–2212106 CAS.
- D. Wang, N. He, L. Xiao, F. Dong, W. Chen, Y. Zhou, C. Chen and S. Wang, Coupling electrocatalytic nitric oxide oxidation over carbon cloth with hydrogen evolution reaction for nitrate synthesis, Angew. Chem., Int. Ed., 2021, 60, 24605–24611 CrossRef CAS PubMed.
- L. Liu, T. Xiao, H. Fu, Z. Chen, X. Qu and S. Zheng, Construction and identification of highly active single-atom Fe1-NC catalytic site for electrocatalytic nitrate reduction, Appl. Catal., B, 2023, 323, 122181 CrossRef CAS.
- J. Zhang, W. He, T. Quast, J. R. C. Junqueira, S. Saddeler, S. Schulz and W. Schuhmann, Single-entity electrochemistry unveils dynamic transformation during tandem catalysis of Cu2O and Co3O4 for converting NO3− to NH3, Angew. Chem., Int. Ed., 2023, 62, e202214830 CrossRef CAS PubMed.
- H. Zhang, C. Wang, H. Luo, J. Chen, M. Kuang and J. Yang, Iron nanoparticles protected by chainmail-structured graphene for durable electrocatalytic nitrate reduction to nitrogen, Angew. Chem., Int. Ed., 2023, 62, e202217071 CrossRef CAS PubMed.
- O. Q. Carvalho, R. Marks, H. K. K. Nguyen, M. E. Vitale-Sullivan, S. C. Martinez, L. Árnadóttir and K. A. Stoerzinger, Role of electronic structure on nitrate reduction to ammonium: a periodic journey, J. Am. Chem. Soc., 2022, 144, 14809–14818 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Combining theory and experiment in electrocatalysis: insights into materials design, Science, 2017, 355, eaad4998 CrossRef PubMed.
- Y. Feng, J.-T. Ren, M.-L. Sun and Z.-Y. Yuan, Valorization systems based on electrocatalytic nitrate/nitrite conversion for energy supply and valuable product synthesis, Chem. Sci., 2025, 16, 1528–1559 RSC.
- Y. Hu, J. Liu, W. Luo, J. Dong, C. Lee, N. Zhang, M. Chen, Y. Xu, D. Wu, M. Zhang, Q. Zhu, E. Hu, D. Geng, L. Zhong and Q. Yan, Alloying Pd with Ru enables electroreduction of nitrate to ammonia with ∼100% faradaic efficiency over a wide potential window, Chem. Sci., 2024, 15, 8204–8215 RSC.
- Y. Kim, J. Ko, M. Shim, J. Park, H.-H. Shin, Z. H. Kim, Y. Jung and H. R. Byon, Identifying the active sites and intermediates on copper surfaces for electrochemical nitrate reduction to ammonia, Chem. Sci., 2024, 15, 2578–2585 RSC.
- F. Sun, Y. Gao, M. Li, Y. Wen, Y. Fang, T. J. Meyer and B. Shan, Molecular self-assembly in conductive covalent networks for selective nitrate electroreduction to ammonia, J. Am. Chem. Soc., 2023, 145, 21491–21501 CrossRef CAS PubMed.
- L. Mi, Q. Huo, J. Cao, X. Chen, H. Yang, Q. Hu and C. He, Achieving synchronization of electrochemical production of ammonia from nitrate and ammonia capture by constructing a “two-in-one” flow cell electrolyzer, Adv. Energy Mater., 2022, 12, 2202247 CrossRef CAS.
- J. Cai, J. Huang, A. Cao, Y. Wei, H. Wang, X. Li, Z. Jiang, G. I. N. Waterhouse, S. Lu and S.-Q. Zang, Interfacial hydrogen bonding-involved electrocatalytic ammonia synthesis on OH-terminated MXene, Appl. Catal., B, 2023, 328, 122473 CrossRef CAS.
- S. Zhang, M. Han, T. Shi, H. Zhang, Y. Lin, X. Zheng, L. R. Zheng, H. Zhou, C. Chen, Y. Zhang, G. Wang, H. Yin and H. Zhao, Atomically dispersed bimetallic Fe–Co electrocatalysts for green production of ammonia, Nat. Sustain., 2022, 6, 169–179 CrossRef.
- R. Xu, D. H. Si, S. S. Zhao, Q. J. Wu, X. S. Wang, T. F. Liu, H. Zhao, R. Cao and Y. B. Huang, Tandem photocatalysis of CO2 to C2H4via a synergistic rhenium-(I) bipyridine/copper-porphyrinic triazine framework, J. Am. Chem. Soc., 2023, 145, 8261–8270 CrossRef CAS PubMed.
- C. Chen, X. Zhu, X. Wen, Y. Zhou, L. Zhou, H. Li, L. Tao, Q. Li, S. Du, T. Liu, D. Yan, C. Xie, Y. Zou, Y. Wang, R. Chen, J. Huo, Y. Li, J. Cheng, H. Su, X. Zhao, W. Cheng, Q. Liu, H. Lin, J. Luo, J. Chen, M. Dong, K. Cheng, C. Li and S. Wang, Coupling N2 and CO2 in H2O to synthesize urea under ambient conditions, Nat. Chem., 2020, 12, 717–724 CrossRef CAS PubMed.
- J.-M. Heng, H.-L. Zhu, Z.-H. Zhao, C. Yu, P.-Q. Liao and X.-M. Chen, Dicopper(I) sites confined in a single metal–organic layer boosting the electroreduction of CO2 to CH4 in a neutral electrolyte, J. Am. Chem. Soc., 2023, 145, 21672–21678 CrossRef CAS PubMed.
- X. Fu, J. Zhang and Y. Kang, Recent advances and challenges of electrochemical ammonia synthesis, Chem Catal., 2022, 2, 2590–2613 CrossRef CAS.
- H. Xu, Y. Ma, J. Chen, W. X. Zhang and J. Yang, Electrocatalytic reduction of nitrate – a step towards a sustainable nitrogen cycle, Chem. Soc. Rev., 2022, 51, 2710–2758 RSC.
- P. H. van Langevelde, I. Katsounaros and M. T. M. Koper, Electrocatalytic nitrate reduction for sustainable ammonia production, Joule, 2021, 5, 290–294 CrossRef.
- Q. J. Wu, D. H. Si, Q. Wu, Y. L. Dong, R. Cao and Y. B. Huang, Boosting electroreduction of CO2 over cationic covalent organic frameworks: hydrogen bonding effects of halogen ions, Angew. Chem., Int. Ed., 2023, 62, e202215687 CrossRef CAS PubMed.
- J. Y. Fang, Q. Z. Zheng, Y. Y. Lou, K. M. Zhao, S. N. Hu, G. Li, O. Akdim, X. Y. Huang and S. G. Sun, Ampere-level current density ammonia electrochemical synthesis using CuCo nanosheets simulating nitrite reductase bifunctional nature, Nat. Commun., 2022, 13, 7899 CrossRef CAS PubMed.
- M. M. M. Kuypers, H. K. Marchant and B. Kartal, The microbial nitrogen-cycling network, Nat. Rev. Microbiol., 2018, 16, 263–276 CrossRef CAS PubMed.
- M. Sundararajan, I. H. Hillier and N. A. Burton, Mechanism of nitrite reduction at T2Cu centers: electronic structure calculations of catalysis by copper nitrite reductase and by synthetic model compounds, J. Phys. Chem. B, 2007, 111, 5511–55177 CrossRef CAS PubMed.
- J. D. Yi, R. Xie, Z. L. Xie, G. L. Chai, T. F. Liu, R. P. Chen, Y. B. Huang and R. Cao, Highly selective CO2 electroreduction to CH4 by in situ generated Cu2O single-type sites on a conductive MOF: stabilizing key intermediates with hydrogen bonding, Angew. Chem., Int. Ed., 2020, 59, 23641–23648 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, The chemistry and applications of metal-organic frameworks, Science, 2013, 341, 1230444 CrossRef PubMed.
- W. X. Li, W. Fang, W. Chen, K. Dinh, H. Ren, L. Zhao, C. T. Liu and Q. Y. Yan, Bimetal-MOF nanosheets as efficient bifunctional electrocatalysts for oxygen evolution and nitrogen reduction reaction, J. Mater. Chem. A, 2020, 8, 3658–3666 RSC.
- J. Zhao, J. Luo, Z. Lin, X. Chen, G.-H. Ning, J. Liu and D. Li, Chiral copper(I)–organic frameworks for dye degradation and the enantioselective recognition of amino acids, Inorg. Chem. Front., 2022, 9, 4907–4912 RSC.
- Q.-J. Wu, D.-H. Si, Y.-L. Dong, Q. Chen, L.-L. Han, R. Cao and Y.-B. Huang, Steering CO2 electroreduction to hydrocarbons over 2D thiol-based conductive metal–organic framework, Sci. Bull., 2025, 70, 1107–1117 CrossRef CAS PubMed.
- Q. Chen, D. H. Si, Q. J. Wu, R. Cao and Y. B. Huang, Engineering copper-based covalent organic framework microenvironments to enable efficient CO2 electroreduction with tunable ethylene/methane switch, Adv. Funct. Mater., 2024, 34, 2315368 CrossRef CAS.
- R.-Q. Xia, J. Zheng, R.-J. Wei, J. He, D.-Q. Ye, M.-D. Li, G.-H. Ning and D. Li, Strong visible light-absorbing BODIPY-based Cu(I) cyclic trinuclear sensitizer for photocatalysis, Inorg. Chem. Front., 2022, 9, 2928–2937 RSC.
- R.-J. Wei, H.-G. Zhou, Z.-Y. Zhang, G.-H. Ning and D. Li, Copper(I)–organic frameworks for catalysis: networking metal clusters with dynamic covalent chemistry, CCS Chem., 2021, 3, 2045–2053 CrossRef CAS.
- R. J. Wei, P. Y. You, H. Duan, M. Xie, R. Q. Xia, X. Chen, X. Zhao, G. H. Ning, A. I. Cooper and D. Li, Ultrathin metal–organic framework nanosheets exhibiting exceptional catalytic activity, J. Am. Chem. Soc., 2022, 144, 17487–17495 CrossRef CAS PubMed.
- B. F. Rider and M. G. Mellon, Colorimetric determination of nitrites, Ind. Eng. Chem. Anal. Ed., 1946, 18, 96–99 CrossRef CAS.
- R. Y. Hodgetts, A. S. Kiryutin, P. Nichols, H.-L. Du, J. M. Bakker, D. R. Macfarlane and A. N. Simonov, Refining universal procedures for ammonium quantification via rapid 1H NMR analysis for dinitrogen reduction studies, ACS Energy Lett., 2020, 5, 736–741 CrossRef CAS.
- A. C. Nielander, J. M. McEnaney, J. A. Schwalbe, J. G. Baker, S. J. Blair, L. Wang, J. G. Pelton, S. Z. Andersen, K. Enemark-Rasmussen, V. Čolić, S. Yang, S. F. Bent, M. Cargnello, J. Kibsgaard, P. C. K. Vesborg, I. Chorkendorff and T. F. Jaramillo, A versatile method for ammonia detection in a range of relevant electrolytes via direct nuclear magnetic resonance techniques, ACS Catal., 2019, 9, 5797–5802 CrossRef CAS.
- L. Wu, J. Feng, L. Zhang, S. Jia, X. Song, Q. Zhu, X. Kang, X. Xing, X. Sun and B. Han, Boosting electrocatalytic nitrate-to-AMMONIA via tuning of N-intermediate adsorption on a Zn–Cu catalyst, Angew. Chem., Int. Ed., 2023, 62, e202307952 CrossRef CAS PubMed.
- K. Fan, W. Xie, J. Li, Y. Sun, P. Xu, Y. Tang, Z. Li and M. Shao, Active hydrogen boosts electrochemical nitrate reduction to ammonia, Nat. Commun., 2022, 13, 7958 CrossRef CAS PubMed.
- D. P. Butcher and A. A. Gewirth, Nitrate reduction pathways on Cu single crystal surfaces: effect of oxide and Cl−, Nano Energy, 2016, 29, 457–465 CrossRef CAS.
- S.-E. Bae, K. L. Stewart and A. A. Gewirth, Nitrate adsorption and reduction on Cu(100) in acidic solution, J. Am. Chem. Soc., 2007, 129, 10171–10180 CrossRef CAS PubMed.
- J. Yu, Y. Qin, X. Wang, H. Zheng, K. Gao, H. Yang, L. Xie, Q. Hu and C. He, Boosting electrochemical nitrate-ammonia conversion via organic ligands-tuned proton transfer, Nano Energy, 2022, 103, 107705 CrossRef CAS.
- Q. Yin, E. V. Alexandrov, D. H. Si, Q. Q. Huang, Z. B. Fang, Y. Zhang, A. A. Zhang, W. K. Qin, Y. L. Li, T. F. Liu and D. M. Proserpio, Metallization-prompted robust porphyrin-based hydrogen-bonded organic frameworks for photocatalytic CO2 reduction, Angew. Chem., Int. Ed., 2022, 61, e202115854 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02208h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |